

Abstract
Background:
Anesthetic sevoflurane induces Tau phosphorylation and cognitive impairment in young mice. The underlying mechanism and the targeted interventions remain largely unexplored. We hypothesized that dexmedetomidine and clonidine attenuated sevoflurane-induced Tau phosphorylation and cognitive impairment by acting on alpha-2 adrenergic receptor.
Methods:
Six-day-old mice received anesthesia with 3% sevoflurane two hours daily on postnatal days 6, 9, and 12. Alpha-2 adrenergic receptor agonist dexmedetomidine and clonidine were used to treat the mice with and without the alpha-2 adrenergic receptor antagonist yohimbine. Mouse hippocampi were harvested and subjected to western blot analysis. The New Object Recognition Test and Morris Water Maze were used to measure cognitive function. We analyzed the primary outcomes by using two-way and one-way ANOVA, and Mann-Whitney U test to determine the effects of sevoflurane on the amounts of phosphorylated Tau, postsynaptic density-95 and cognitive function in young mice after the treatments with dexmedetomidine, clonidine and yohimbine.
Result:
Both dexmedetomidine and clonidine attenuated the sevoflurane-induced increase in phosphorylated Tau amount [94 ± 16.3% (dexmedetomidine plus sevoflurane) versus 240 ± 67.8% (vehicle plus sevoflurane), P < 0.001; 125 ± 13.5% (clonidine plus sevoflurane) versus 355 ± 57.6% (vehicle plus sevoflurane), P < 0.001; mean ± standard deviation], sevoflurane-induced reduction in postsynaptic density-95 [82 ± 6.6% (dexmedetomidine plus sevoflurane) versus 31 ± 12.4% (vehicle plus sevoflurane), P < 0.001; 95 ± 6.4% (clonidine plus sevoflurane) versus 62 ± 18.4% (vehicle plus sevoflurane), P < 0.001], and cognitive impairment in the young mice. Interestingly, yohimbine reversed the effects of dexmedetomidine and clonidine on attenuating the sevoflurane-induced changes in phosphorylated Tau, postsynaptic density-95 and cognitive function.
Conclusion:
Dexmedetomidine and clonidine could inhibit the sevoflurane-induced Tau phosphorylation and cognitive impairment via activation of alpha-2 adrenergic receptor. More studies are needed to confirm the results and to determine the clinical relevance of these findings.
Keywords: Anesthesia, dexmedetomidine, clonidine, Tau protein, synapse, cognition, PSD-95, alpha-2 adrenergic receptor
Introduction
In preclinical studies, anesthetics cause cell death, impair neurogenesis, and inhibit synapse formation in the brain tissues of young mice via impaired neurotrophin signaling, mitochondrial dysfunction, neuroinflammation, and interneuron phenotype changes. (reviewed in1) Tau, a microtubule-associated protein that is predominantly expressed inside neurons, is associated with microtubule assembly and function.2,3 Tau phosphorylation, aggregation, and spread4–7 underlie the neuropathogenesis of neurodegenerative diseases, including Alzheimer’s Disease. The inhalational anesthetic sevoflurane causes Tau phosphorylation in the hippocampus of five- to six-month-old mice8, while the intravenous anesthetics propofol9 and dexmedetomidine10 induce Tau phosphorylation in the hippocampus and cortex of adult mice and in SH-SY5Y cells. Anesthesia with 3% sevoflurane two hours daily for three days at postnatal days 6, 7, and 8 or postnatal days 6, 8 and 10 in mice induces Tau phosphorylation in the hippocampus and cognitive impairment in the young mice.11–13.
However, the underlying mechanisms through which sevoflurane induces Tau phosphorylation and cognitive impairment in young mice remain to be determined. Specifically, the involvement of alpha-2 adrenergic receptor in the sevoflurane-induced effects in young mice remains unknown. Moreover, whether anesthetic dexmedetomidine can mitigate the sevoflurane-induced Tau phosphorylation and cognitive impairment in young mice has not been determined.
Dexmedetomidine, a highly selective alpha-2 adrenergic receptor agonist, causes sedative effects by binding to alpha-2 adrenergic receptors in the locus coeruleus14 and is increasingly used in operating rooms and intensive care units.15 Dexmedetomidine decreases delirium in patients,16–19 although different reports also exist.20
Dexmedetomidine has been reported to induce Tau phosphorylation10,21 and apoptosis22 in rodents. However, dexmedetomidine could also attenuate neuroinflammation and cognitive impairment in adult mice induced by high molecular group box 1 protein (HMGB1) and surgery,23 mitigate autophagy in the hippocampus of offspring mice induced by sevoflurane in pregnant mice,24 and prevent propofol-induced apoptosis and cognitive impairment in neonatal rats25.
Therefore, the primary objective of the current study was to determine whether anesthetic dexmedetomidine can attenuate the sevoflurane-induced Tau phosphorylation and cognitive impairment in young mice. The another objective was to assess the role of alpha-2 adrenergic receptor in the sevoflurane-induced effects in young mice by employing the alpha-2 adrenergic receptor agonists dexmedetomidine14 and clonidine,26 and the alpha-2 adrenergic receptor antagonist yohimbine.27
We hypothesized that activation of the alpha-2 adrenergic receptor by dexmedetomidine and clonidine would attenuate sevoflurane-induced Tau phosphorylation and cognitive impairment while yohimbine would reverse the attenuation effects of dexmedetomidine and clonidine. These data would implicate that alpha-2 adrenergic receptors are one of the underlying mechanisms of anesthesia-induced Tau phosphorylation and demonstrate potential for targeted interventions for treating and preventing perioperative neurocognitive disorder in young patients.
Materials and Methods
Mice
All experimental procedures involving mice were approved by the Standing Committee on Animals at Massachusetts General Hospital, Boston, Massachusetts (Protocol number: 2006N000219) and conformed to National Institutes of Health (Bethesda, Maryland) guidelines. This manuscript was written according to applicable ARRIVE guidelines. Efforts were made to minimize the number of mice used in the studies. Adult mice (C57BL/6J) were purchased from Jackson Laboratory (Jackson Laboratory, Bar Harbor, ME). Mice were housed in the Massachusetts General Hospital animal facility. All mice were fed with standard rodent food and water and were housed (five mice per cage) in a controlled environment at 37 ° C with 12-hour light/dark cycles (lights on from 07:00 to19:00). The day of birth was designated as postnatal day zero (P0). The young mice were randomly allocated to different groups. Each group included a similar number of female and male mice. We randomly assigned mice to 10 groups: 1. control plus vehicle; 2. control plus dexmedetomidine; 3. control plus clonidine; 4. control plus yohimbine plus dexmedetomidine; 5. control plus yohimbine plus clonidine; 6. sevoflurane plus vehicle; 7. sevoflurane plus dexmedetomidine; 8. sevoflurane plus clonidine; 9. sevoflurane plus yohimbine plus dexmedetomidine; 10. sevoflurane plus yohimbine plus clonidine. Mice did not experience unexpected lethality and were euthanized according to institutional animal care and committee guidelines.
Anesthesia and treatment
Given the clinical observation that multiple exposures (e.g., three times) of anesthesia and surgery to children are associated with the increased risk of development of perioperative neurocognitive disorder, we treated the young mice with three times of sevoflurane to conceptually mimic the multiple exposures of anesthesia and surgery in children but also determine the role of anesthesia without surgery in the observed neurocognitive disorder in young mice as performed in our previous studies.11–13,28 Specifically, we anesthetized young mice with 3% sevoflurane plus 40% oxygen for 2 hours at postnatal days (P) 6, P9, and P12, which did not cause apparent changes in the values of pH, PO2 and PCO2 in the mice (Supplemental Table 1). Control mice received 40% oxygen at an identical flow rate in similar chambers and were separated from their mother as in our previous studies.11,28 We continuously monitored the concentrations of sevoflurane and oxygen using a gas analyzer (Dash 4000; GE Healthcare, Milwaukee, WI) during sevoflurane anesthesia. The anesthesia chamber temperature was monitored and controlled by a feedback-based system with a DC Temperature Control System (World precision instruments, Inc Sarasota, FL), which controls and automatically adjusts the temperature to keep rectal temperature at 37 °C (±0.5 °C) via a warming pad placed under the chamber. Dexmedetomidine (10 μg/kg, Product number: SML0956, Sigma, St. Louis, MO), clonidine (1 mg/kg, Product number: C7897, Sigma) or vehicle (saline) was injected intraperitoneally 30 minutes before sevoflurane anesthesia. Yohimbine (1 mg/kg, Product number: Y3125, Sigma) was injected intraperitoneally 10 minutes before the administration of dexmedetomidine or clonidine. We chose these doses of dexmedetomidine,29 clonidine30, and yohimbine29 based on reported effectiveness in activating or inhibiting the alpha-2 adrenergic receptor. Mice were decapitated for the harvest of the hippocampi immediately after the end of the control condition or sevoflurane anesthesia at postnatal day 12 for the measurement of Tau and phosphorylated Tau, and at postnatal day 37 for the measurement of PSD-95. We measured Tau phosphorylation at day 12 (end of the 3rd anesthesia) to determine the acute effects of sevoflurane on Tau phosphorylation as we did in the previous studies11. We measured the amount of postsynaptic density-95 (PSD-95) in the hippocampi of mice at P37 because sevoflurane induced cognitive impairment in the mice at P37. A separate cohort of mice were used for the New Object Recognition Test (NOR) and Morris Water Maze (MWM) test.
New Object Recognition Test (NOR)
The NOR was performed on postnatal day 29 and 30. In the first trial, a mouse was placed in a square arena (60 cm × 60 cm × 60 cm, with an even lighting of 20 lux) to habituate for 5 minutes. We used a black container to cover the mouse. Two identical objects (same shape and color) were placed in opposite corners of the upper half of the area and the mouse was then released from the container. We recorded mouse activity and the time spent interacting with the object for 10 minutes. We repeated the first trial 24 hours later, but one of the objects was replaced with a novel object (different shape and color). The interaction time with the novel object (novel time) and familial object (familiar time) was recorded separately. The discrimination index was defined as the ratio of novel time to the novel time plus familial time, which indicated new object recognition ability.
Morris Water Maze (MWM)
The MWM test was performed as described previously.11,28 In brief, postnatal day 31 mice were tested in the MWM in four trials per day for 7 days (postnatal day 31 to 37). Escape latency was recorded each day. We maintained mouse body temperature using a heating device as described in our previous studies.28 After every trial, each mouse was placed in a holding cage under a heat lamp for 5 minutes to dry the body before the mouse was returned to its home cage.
Brain tissue harvest, lysis, and protein quantification
Mice were decapitated on postnatal day 12 or day 37, and we harvested the hippocampi for western blot analysis. We homogenized harvested tissues on ice using an immunoprecipitation buffer (M-PER® Mammalian Protein Extraction Reagent, Cat# 78501, Thermo Scientific, Waltham, MA) plus protease inhibitor cocktail (Sigma; Cat# 11836170001). The lysates were collected and centrifuged at 4°C for 20 minutes at 12,000 rpm. The total amounts of protein were quantified by bicinchoninic acid protein assay kit (Pierce, Iselin, NJ).
Western blot analysis
Total Tau was detected by anti-Tau 46 antibody (Cat # T9450, 55 kDa, 1:2000, Sigma). AT8 antibody (Tau-PS202/PT205, Cat # MN1020, 55 kDa, 1:500, Thermo Scientific, Waltham, MA) was used to detect Tau phosphorylated at serine 202 and threonine 205 amino acid. PSD-95 antibody (95 kDa, Cat # GB11277, 1:1,000; Cell Signaling, Billerica, MA) was used to detect amount of PSD-95. Finally, anti-β-Actin antibody was used to detect nontargeted protein β-Actin (42 kDa, 1:5000, Sigma, St. Louis, MO) serving as a control for loading differences in total protein. Western blot quantification was performed using the standard methods as described by Xie et al31. Signal intensity was analyzed using the image analysis program Quantity One (Bio-Rad, Hercules, CA). We quantified western blots in two steps. First, we used β-Actin to normalize protein amounts (e.g., determining the ratio of Tau to β-Actin) and to decrease the influence of the differences in the protein amounts loaded. Second, we presented the changes in protein amount in the experimental group as a percentage of those in the control group; 100% of changes refers to the amounts of control condition for comparison to experimental conditions.
Statistical analyses
We present data obtained from the western blot, escape latency of MWW and NOR studies as mean ± standard deviation. Data from the platform crossing times of MWM are presented using median and interquartile range. Interaction between time and group factors was determined by a two-way ANOVA with repeated measurements (Greenhouse-Geisser correction) to analyze the difference in learning curves (based on escape latency) between mice in the control or vehicle group and mice in the sevoflurane anesthesia, dexmedetomidine, or clonidine with and without yohimbine group in the MWM experiments. A one-way ANOVA with Bonferroni’s multiple comparison test was used to determine the differences among groups in terms of total Tau, Tau-PS202/PT205 and PSD-95 amounts. Mann-Whitney U test was used to determine the difference between mice in the control or vehicle groups and mice in the sevoflurane anesthesia, dexmedetomidine, or clonidine with and without yohimbine treatment in terms of platform crossing numbers in MWM, and the ratio of novel time to the novel time plus familiar time. We performed these tests based on the distributional assumptions from our previous similar work. There were no missing data for the variables of the MWM (escape latency and platform crossing number) and NOR during the data analysis. P-values less than 0.05 were considered statistically significant and the significance testing was two-tailed. Statistical analysis was conducted using GraphPad Prism software (version 8.0; GraphPad Software, La Jolla, CA). There were 12 mice in each group for the behavioral studies and six mice in each group for the western blot study. The sample size was chosen empirically based on the previous studies.28
Results
Dexmedetomidine or clonidine mitigates sevoflurane-induced Tau phosphorylation and sevoflurane-induced reduction of PSD-95 in the hippocampi of young mice.
Treatment with dexmedetomidine alone did not apparently alter the amount of Tau-PS202/Tau-PT205 in the hippocampi of young mice; however, sevoflurane increased the amount of Tau-PS202/Tau-PT205 compared to control. Dexmedetomidine mitigated the sevoflurane-induced increases in Tau-PS202/Tau-PT205, as evidenced by less visibilities of bands representing the amounts of Tau-PS202/Tau-PT205 following sevoflurane plus dexmedetomidine treatment. There were no significant differences in β-Actin in the hippocampi of young mice among different conditions (Figure 1A). Quantification of the western blot corroborated these observations (Figure 1B). One-way ANOVA demonstrated that there was significant difference on the amounts of Tau-PS202/Tau-PT205 in the hippocampi of young mice (P < 0.001, Figure 1B), and dexmedetomidine mitigated the sevoflurane-induced increases in Tau-PS202/Tau-PT205: 109 ± 25.1% versus 266 ± 53.4%, P < 0.001 (Bonferroni’s multiple comparison test). Quantitative western blot analysis showed that there were no significant differences in Tau among the four conditions (P = 0.383, Figure 1C and 1D) and that dexmedetomidine mitigated the sevoflurane-induced increases in the ratio of Tau-PS202/Tau-PT205 to total Tau amounts in the hippocampi of young mice: 94 ± 16.3% versus 240 ± 67.8% (P < 0.001, Figure 1E).
Figure 1. Dexmedetomidine attenuates sevoflurane-induced Tau phosphorylation and sevoflurane-induced reduction in PSD-95 in the hippocampi of young mice.
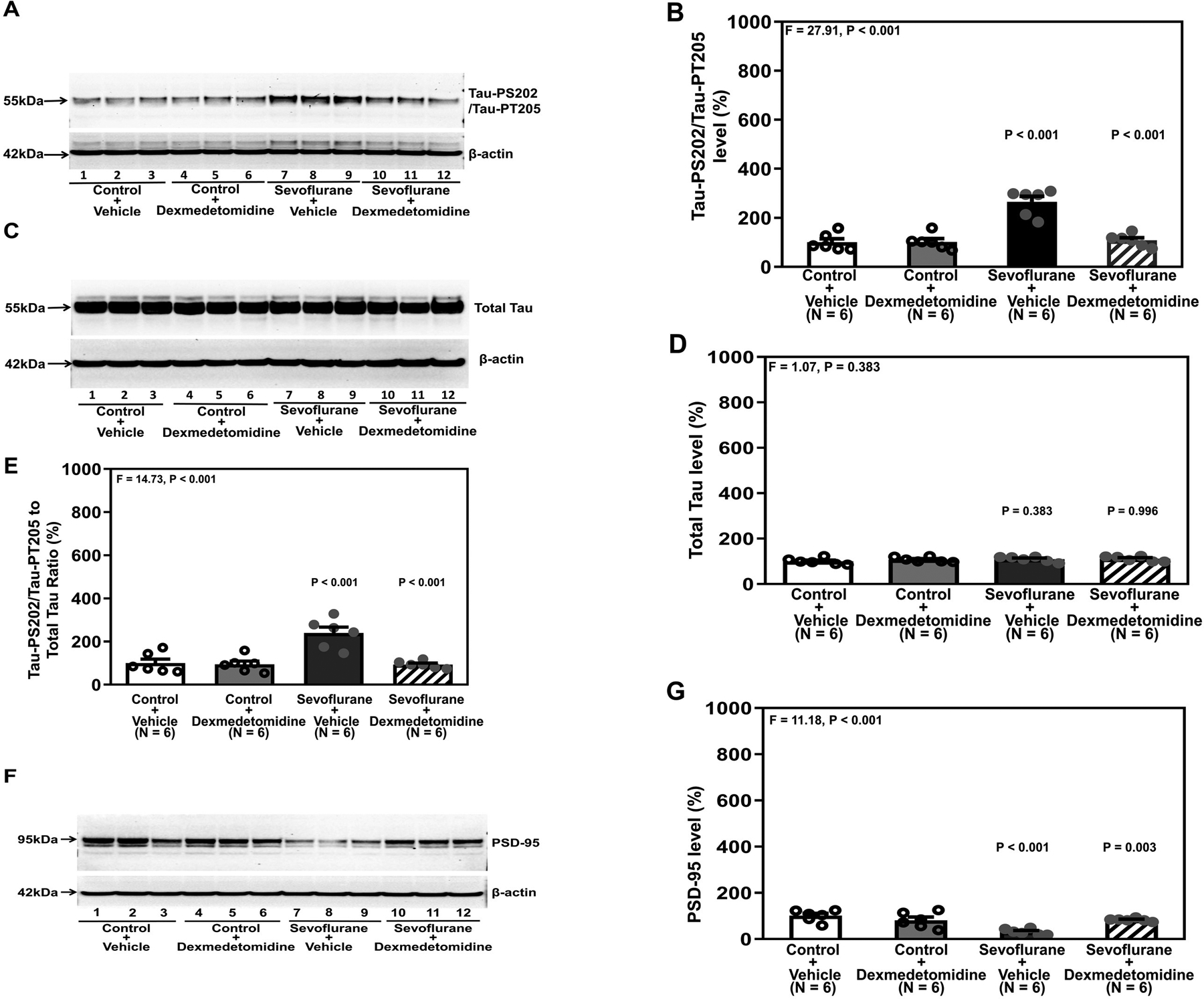
A. Tau-PS202/Tau-PT205 protein in the hippocampi of young mice after sevoflurane and dexmedetomidine treatment. B. Quantification of western blot in A. C. Total Tau protein after various treatments. D. Quantification of western blot in C. E. Quantification of the western blots in A and C presented as the ratio of Tau-PS202/Tau-PT205 to total Tau. F. PSD-95 in the hippocampi of young mice at P37 after dexmedetomidine and sevoflurane. G. Quantification of the western blot shown in F. There were no significant differences in β-Actin amounts among these treatments. P, postnatal; PSD, postsynaptic density; Tau-PS202, Tau phosphorylated at serine-202; Tau-PT205, Tau phosphorylated at threonine 205. A one-way ANOVA with Bonferroni’s multiple comparison test was used to analyze the data. N = 6 in each group.
Dexmedetomidine alone did not significantly alter PSD-95 amounts in the hippocampi of young mice. However, sevoflurane decreased PSD-95 amounts compared to control. Dexmedetomidine treatment mitigated the sevoflurane-induced decrease in PSD-95 (Figure 1F). Quantification of the western blot corroborated these observations that dexmedetomidine mitigated the sevoflurane-induced decrease in PSD-95 amount: 32 ± 12.4% versus 82 ± 6.6% (P = 0.003, Figure 1F).
To further determine the involvement of the alpha-2 adrenergic receptor in sevoflurane-induced Tau phosphorylation, we tested whether clonidine, another alpha-2 adrenergic receptor agonist, could mitigate sevoflurane-induced Tau phosphorylation and reduce PSD-95 in the hippocampi of young mice. Quantitative western blot showed that clonidine treatment attenuated the sevoflurane-induced increases in Tau-PS202/Tau-PT205: 304 ± 43.1% versus 107 ± 7.5% (P < 0.001, one-way ANOVA with Bonferroni’s multiple comparison test, Figure 2A and 2B). There were no significant differences in total Tau or β-Actin in the hippocampi of young mice among the different conditions (Figure 2C and 2D). Clonidine mitigated sevoflurane-induced increases in the ratio of Tau-PS202/Tau-PT205 to total Tau: 355 ± 57.6% versus 125 ± 13.5%, P < 0.001 (Figure 2E). Quantitative western blot (Figure 2F and 2G) also showed that sevoflurane anesthesia decreased PSD-95. Clonidine mitigated the sevoflurane-induced decreases in PSD-95: 95 ± 6.4% versus 62 ± 18.4%, P < 0.001 (Figure 2G).
Figure 2. Clonidine attenuates sevoflurane-induced Tau phosphorylation and sevoflurane-induced reduction in PSD-95 in the hippocampi of young mice.
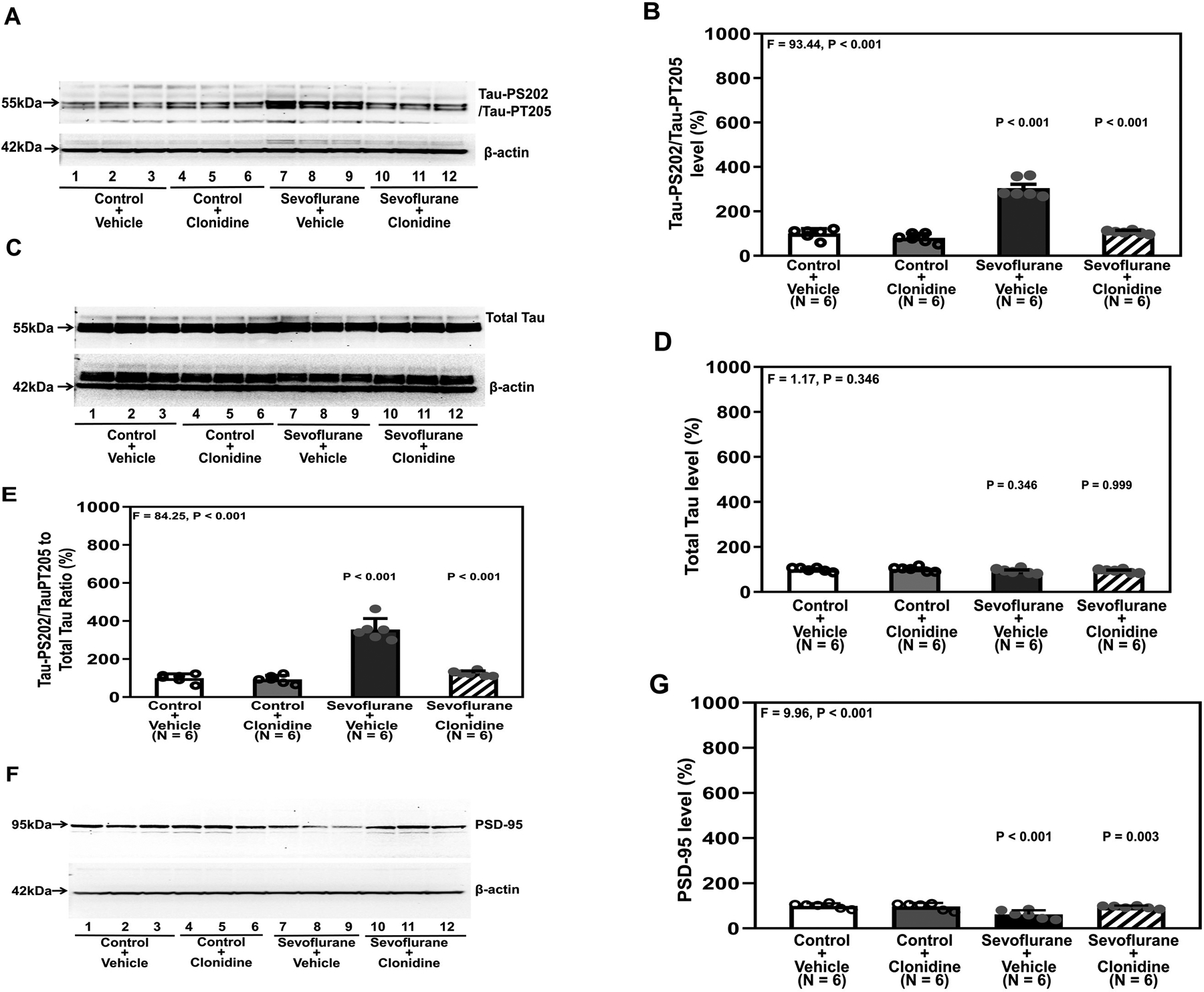
A. Tau-PS202/Tau-PT205 in the hippocampi of the young mice after clonidine and sevoflurane treatments. B. Quantification of the western blot in A. There were no significant changes of total Tau amounts in the different conditions (C and D). E. Quantification of the western blots in A and C presented as the ratio of Tau-PS202/Tau-PT205 to total Tau. F. PSD-95 in the hippocampi of young mice compared to control at P37 under sevoflurane and clonidine treatment. G. Quantification of the western blot in F. There were no significant differences in the β-Actin amounts among treatments. P, postnatal; PSD, postsynaptic density; Tau-PS202, phosphorylated at serine-202; Tau-PT205, Tau phosphorylated at threonine 205. A one-way ANOVA with Bonferroni’s multiple comparison test was used to analyze the data. N = 6 in each group.
Yohimbine inhibits the protective effects of dexmedetomidine or clonidine.
Sevoflurane plus vehicle or sevoflurane plus yohimbine plus dexmedetomidine induced Tau phosphorylation (Figure 3A and 3B). There were no significant differences in total Tau amounts in the hippocampi of the mice following these treatments (Figure 3C and 3D). Both sevoflurane plus vehicle and sevoflurane plus yohimbine plus dexmedetomidine increased the ratio of Tau-PS202/Tau-PT205 to total Tau in the hippocampi of young mice compared to control plus vehicle or control plus yohimbine plus dexmedetomidine (P < 0.001, one-way ANOVA, Figure 3E). Quantitative western blot showed that both sevoflurane plus vehicle and sevoflurane plus yohimbine plus dexmedetomidine decreased PSD-95 in the hippocampi of mice compared to control plus vehicle or control plus yohimbine plus dexmedetomidine (P < 0.001, one-way ANOVA, Figure 3F and 3G).
Figure 3. Yohimbine inhibits the protective effects of dexmedetomidine on attenuating sevoflurane-induced Tau phosphorylation and reduction in PSD-95 in the hippocampi of young mice.
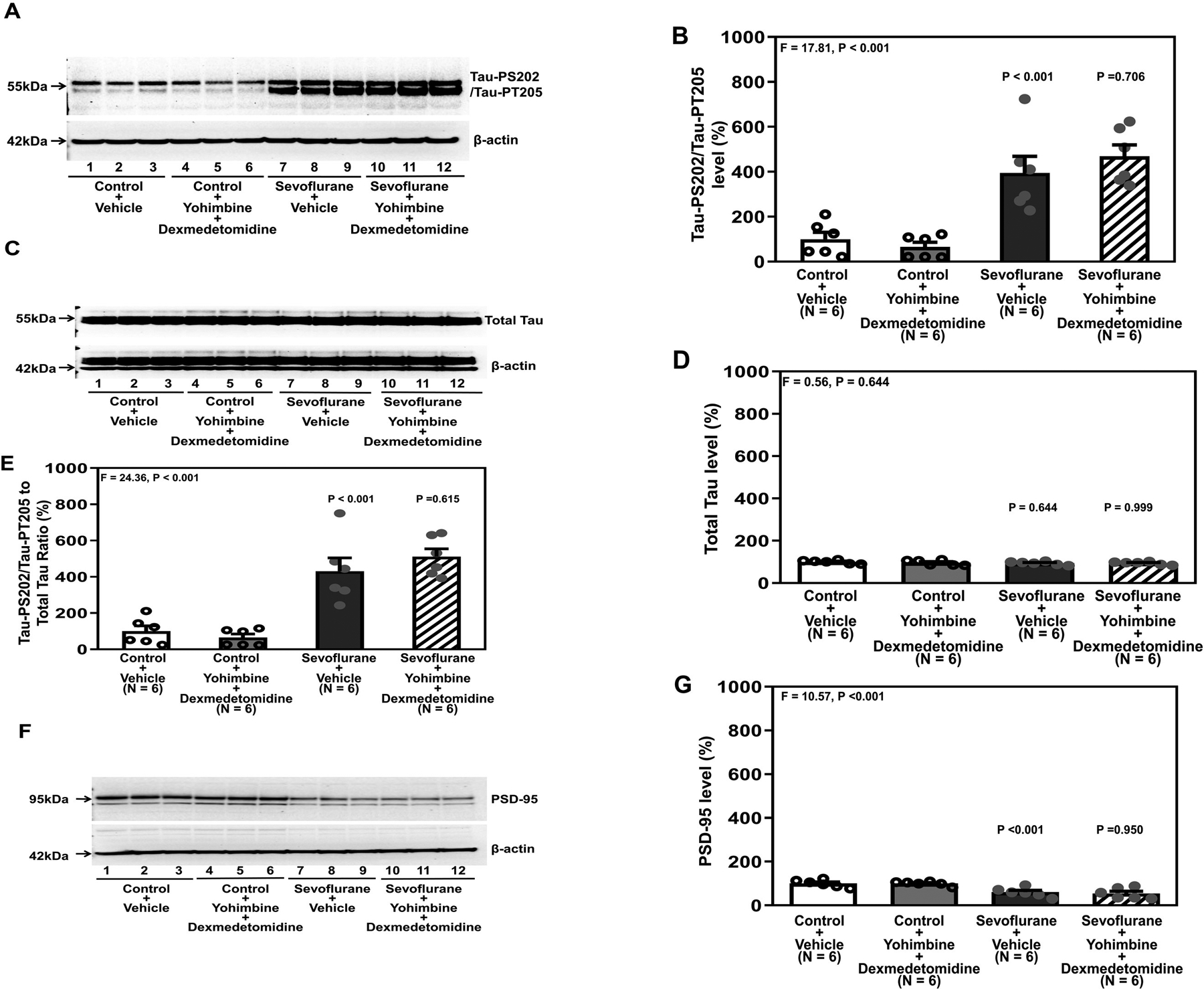
A. Tau-PS202/Tau-PT205 in the hippocampi of young mice under various treatment conditions. B. Quantification of the western blot in A. There were no significant changes in total Tau among the different conditions (C and D). E. Quantification of the western blots in A and C presented as the ratio of Tau-PS202/Tau-PT205 to total Tau in the hippocampi of young mice. F. PSD-95 in the hippocampi of young mice under various treatment conditions. G. Quantification of the western blot in F. There were no significant differences in the β-Actin amounts in the hippocampi of the young mice among the different conditions. P, postnatal; PSD, postsynaptic density, Tau-PS202, Tau phosphorylated at serine-202; Tau-PT205, Tau phosphorylated at threonine 205. A one-way ANOVA with Bonferroni’s multiple comparison test was used to analyze the data. N = 6 in each group.
Similarly, both sevoflurane plus vehicle and sevoflurane plus yohimbine plus clonidine increased Tau-PS202/Tau-PT205 amount (Figure 4A and 4B) but did not change the amount of total Tau (Figure 4C and 4D), increased the ratio of Tau-PS202/Tau-PT205 to total Tau (Figure 4E), and decreased PSD-95 amount (Figure 4F and 4G) in the hippocampi of mice compared to control plus vehicle or control plus yohimbine plus clonidine.
Figure 4. Yohimbine inhibits the protective effect of clonidine on attenuating sevoflurane-induced Tau phosphorylation and sevoflurane-induced reduction of PSD-95 in the hippocampi of young mice.
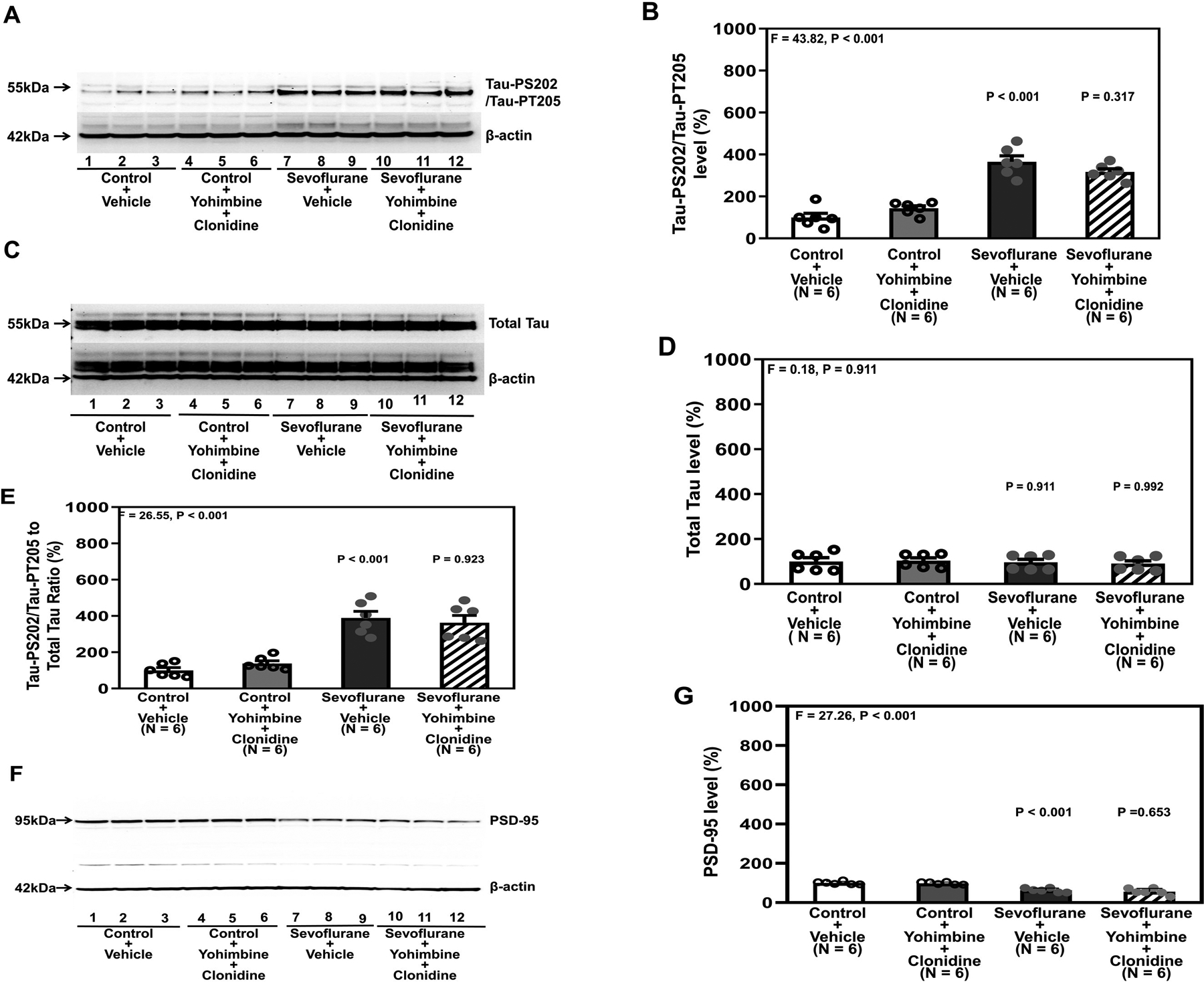
A. Tau-PS202/Tau-PT205 in the hippocampi of young mice after various treatments. B. Quantification of the western blot in A. There were no significant changes in total Tau in the different conditions (C and D). E. Quantification of the western blots in A and C presented as the ratio of Tau-PS202/Tau-PT205 to total Tau in the hippocampi of young mice. F. PSD-95 in the hippocampi of young mice under various treatment conditions. G. Quantification of the western blot in F. There were no significant differences in the β-Actin amounts in the hippocampi of the young mice among the different conditions. P, postnatal; PSD, postsynaptic density; Tau-PS202, Tau phosphorylated at serine-202; Tau-PT205, Tau phosphorylated at threonine 205. A one-way ANOVA with Bonferroni’s multiple comparison test was used to analyze the data. N = 6 in each group.
Dexmedetomidine and clonidine attenuate sevoflurane-induced cognitive impairment in young mice.
There was no significant interaction of treatment (vehicle versus dexmedetomidine) and time (day 31 to day 37), and dexmedetomidine did not significantly alter the escape latency of the young mice in the MWM test compared to vehicle (Figure 5A). Moreover, dexmedetomidine did not significantly change the platform crossing number of the young mice in the MWM test compared to vehicle (Figure 5B).
Figure 5. Dexmedetomidine mitigates sevoflurane-induced cognitive impairment in young mice.
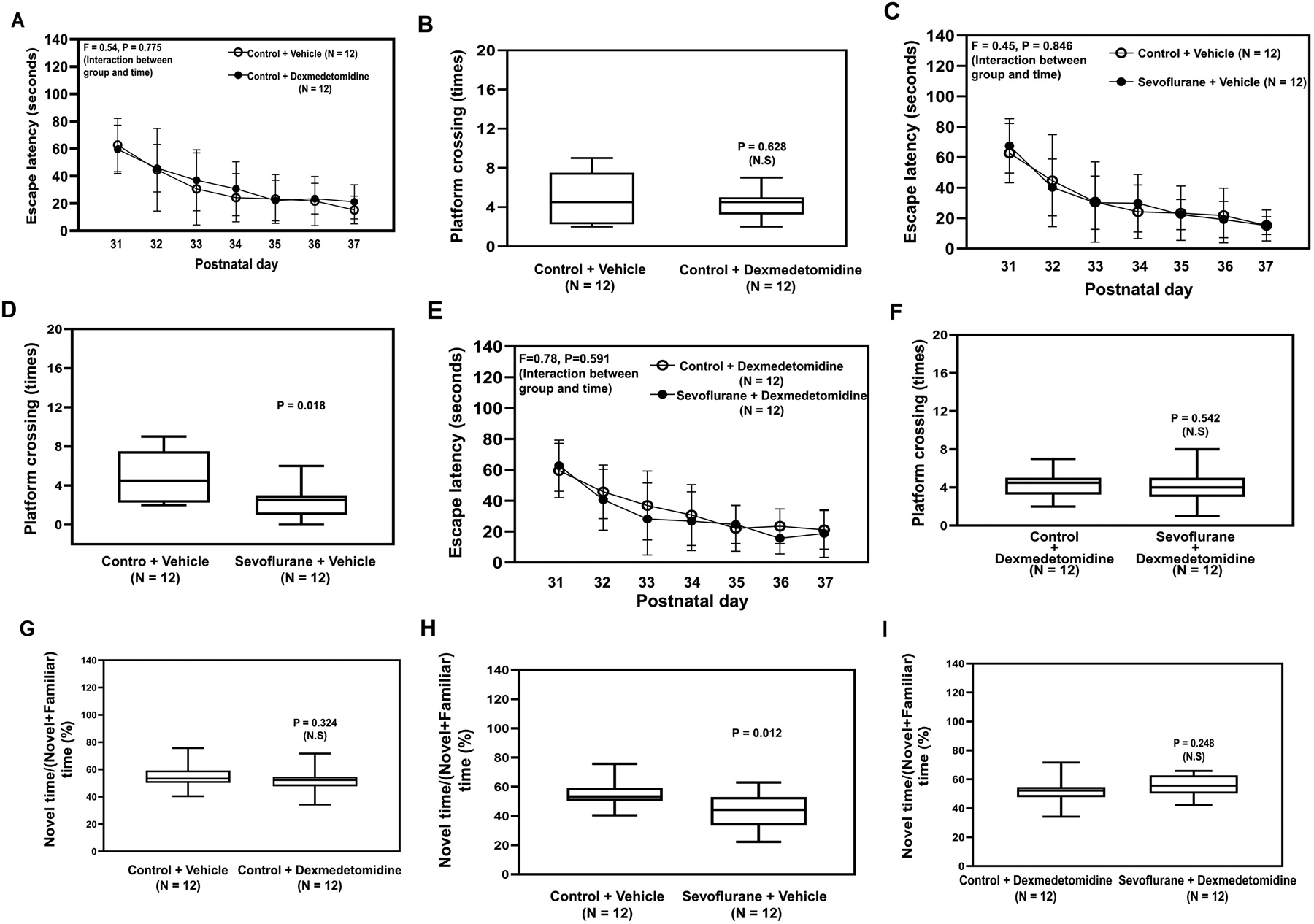
Escape latency (A) and platform crossing number (B) of the young mice in the MWM test after dexmedetomidine or vehicle. Escape latency (C) and platform crossing number (D) in the MWM test after sevoflurane or vehicle. Escape latency (E) and platform crossing number (F) in the MWM after pretreatment with dexmedetomidine followed by sevoflurane G. Ratio of the exploring novel object time to the total object time of the NOR test in young mice treated with dexmedetomidine or control. H. Ratio of exploring novel object time to the total object time in NOR test in mice treated with sevoflurane compared to control condition. I. Ratio of exploring novel object time to the total object time in the NOR test compared to control in the young mice pretreated with dexmedetomidine and then sevoflurane. NOR, New Object Recognition Test; MWM; Morris Water Maze; P, postnatal. A two-way ANOVA and Mann-Whitney U test were used to analyze the data. N = 12 in each group.
Sevoflurane did not significantly alter the escape latency of mice in the MWM (Figure 5C), but sevoflurane decreased the platform crossing number of the young mice in the MWM test compared to control condition (P = 0.018, Mann–Whitney U test, Figure 5D). However, sevoflurane anesthesia did not cause cognitive impairment in the mice that received pre-treatment with dexmedetomidine (Figure 5E and 5F). Consistently, sevoflurane anesthesia induced cognitive impairment in the mice in the NOR as evidenced by the decreased ratio of novel time to the novel plus familiar time in young mice following sevoflurane anesthesia compared to control (Figure 5H). Whereas dexmedetomidine alone did not cause cognitive impairment in the mice (Figure 5G), dexmedetomidine attenuated the sevoflurane-induced cognitive impairment in the young mice in the NOR (Figure 5I). Similarly, clonidine attenuated the sevoflurane-induced cognitive impairment indicated by both the MWM test (Figure 6A to 6D) and the NOR (Figure 6E and 6F).
Figure 6. Clonidine mitigates sevoflurane-induced cognitive impairment in young mice.
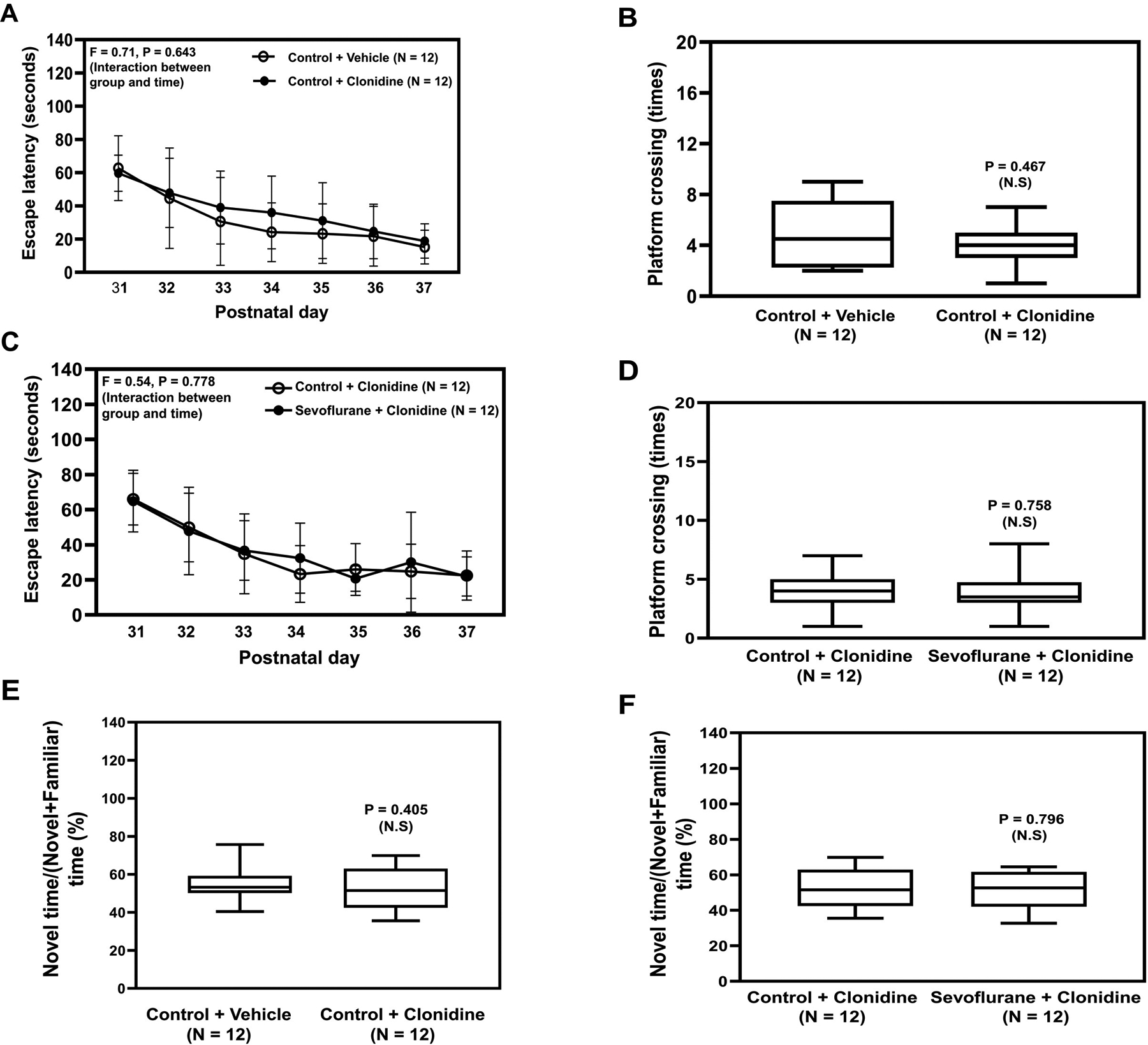
Escape latency (A) and platform crossing number (B) of young mice on the MWM test compared to vehicle after clonidine treatment. Escape latency (C) and platform crossing number (D) on the MWM test after pretreatment with clonidine followed by sevoflurane treatment. E. Ratio of exploring novel object time to the total object time on the NOR test compared to vehicle after clonidine treatment. F. Ratio of exploring novel object time to the total object time on the NOR test compared to control in young mice pretreated with clonidine followed by sevoflurane. NOR, New Object Recognition Test; MWM; Morris Water Maze; P, postnatal. A two-way ANOVA and Mann-Whitney U test were used to analyze the data. N = 12 in each group.
Yohimbine inhibits the protective effects of dexmedetomidine or clonidine on sevoflurane-induced cognitive impairment in young mice.
There was no interaction of treatment (control plus vehicle versus control plus yohimbine plus dexmedetomidine) and time (P31 to P37) on the escape latency of mice in the MWM test (Supplemental Figure 1A), and there was no significant difference in the platform crossing number of the young mice following the treatment of control plus vehicle or control plus yohimbine plus dexmedetomidine (Supplemental Figure 1B). Sevoflurane did not significantly change the escape latency of MWM of the young mice that received yohimbine plus dexmedetomidine (Supplemental Figure 1C). However, mice that received yohimbine plus dexmedetomidine as pretreatment experienced cognitive impairment following sevoflurane anesthesia as evidenced by decreased platform crossing number (P = 0.027, Mann–Whitney U test, Supplemental Figure 1D). Yohimbine plus dexmedetomidine did not significantly change the ratio of novel time to novel plus familiar time compared to vehicle (Supplemental Figure 1E). On the other hand, sevoflurane anesthesia caused cognitive impairment as evidenced by the decreased ratio of novel time to novel plus familiar time compared to control (Supplemental Figure 1F). Consistently, yohimbine reversed the effects of clonidine on attenuating the sevoflurane-induced cognitive impairment indicated in both the MWM test (Supplemental Figure 2A to 2D) and NOR (Supplemental Figure 2E and 2F).
Discussion
In this proof of concept study, we determined that the alpha-2 adrenergic receptor contributes to the development of anesthesia-induced Tau phosphorylation in young mice and that activation of alpha-2 adrenergic receptor can attenuate sevoflurane-induced Tau phosphorylation and neurocognitive impairment in young mice. These findings have established a system and revealed that alpha-2 adrenergic receptor could be one of the up-stream mechanisms by which sevoflurane induces Tau phosphorylation and cognitive impairment in young mice. Moreover, anesthetic dexmedetomidine may mitigate the sevoflurane-induced Tau phosphorylation and cognitive impairment in young mice, pending more confirmative studies.
Specifically, the alpha-2 adrenergic receptor agonists dexmedetomidine (Figure 1) and clonidine (Figure 2) attenuated the sevoflurane-induced Tau phosphorylation and sevoflurane-induced reduction in PSD-95 amount in the hippocampi of young mice at the end of three sevoflurane anesthesia exposure periods on postnatal day 12 or on postnatal day 37, respectively. Importantly, the alpha-2 adrenergic receptor antagonist yohimbine mitigated the protective effects of dexmedetomidine (Figure 3) and clonidine (Figure 4) on sevoflurane-induced Tau phosphorylation and reduction in PSD-95. Moreover, dexmedetomidine (Figure 5) and clonidine (Figure 6) attenuated the sevoflurane-induced cognitive impairment in the young mice tested at postnatal days 29 to 30 (NOR) and postnatal days 31 to 37 (MWM) while yohimbine reversed the protective effects of dexmedetomidine (Supplemental Figure 1) and clonidine (Supplemental Figure 2). These data indicate that activation of the alpha-2 adrenergic receptor mitigated the sevoflurane-induced Tau phosphorylation and cognitive impairment in young mice. Furthermore, anesthetic dexmedetomidine attenuated the development of anesthesia-induced Tau phosphorylation and cognitive impairment.
Whittington et al. reported that a single administration (300 ug/kg) of dexmedetomidine induced Tau phosphorylation in the hippocampi of 8 to 10 week old wild type mice and in 3 month old transgenic mice overexpressing human Tau on a murine Tau knockout background.10 This treatment also caused cognitive impairment in mice along with the inhibition of ERK, JNK, CaMKII, and GSK-3β in the brain.10 However, in the present study, treatment with 10 μg/kg dexmedetomidine did not induce Tau phosphorylation. Moreover, dexmedetomidine treatment attenuated the sevoflurane-induced Tau phosphorylation, synaptic loss and cognitive impairment observed in the young mice. Important differences between the two studies are: (1) the age of mice (8 to 10 weeks versus postnatal day 6 to 12); (2) single treatment with 300 ug/kg of dexmedetomidine versus three treatments of dexmedetomidine with 10 ug/kg; and (3) measurement of Tau-PS202/Tau-PT205 at 30 minutes and 6 hours after the administration of dexmedetomidine versus measurement of Tau-PS202/Tau-PT205 at the end of the last anesthesia on postnatal day 12, which was 6 days after the first administration of dexmedetomidine. Note that escalating the cumulative dose of dexmedetomidine, e.g., 125 and 250 ug, but not 50 ug, induced apoptosis22.
Interestingly, Whittington et al. also indicated that the alpha-2 adrenergic receptor antagonist atipamezole blocked dexmedetomidine-induced Tau phosphorylation in the brain tissues of mice. In the present study, we found that the alpha-2 adrenergic receptor antagonist yohimbine reversed the effects of dexmedetomidine and clonidine on mitigating sevoflurane-induced Tau phosphorylation and synaptic loss in the hippocampi of young mice, along with cognitive impairment. These findings demonstrate that different doses of dexmedetomidine in different ages of mice could have different effects on Tau phosphorylation and cognitive function. Moreover, the effects of dexmedetomidine on Tau phosphorylation, either attenuation or induction, were likely through its action on the alpha-2 adrenergic receptor. Future studies should systematically assess whether different dosages of dexmedetomidine might have different effects on Tau phosphorylation, synaptic loss and other effects in rodents of varying ages.
Another study by Hu et al. indicated that a single treatment with 50 ug/kg dexmedetomidine attenuated neuroinflammation and cognitive impairment in adult mice (12 to 14 weeks old) induced by HMGB1 protein and surgery23. These data further indicate that a low dose of dexmedetomidine could have neuroprotective effects. This is supported by our finding that 10 ug/kg dexmedetomidine attenuated the sevoflurane-induced Tau phosphorylation, synaptic loss and cognitive impairment in young mice. Hu et al. demonstrated that dexmedetomidine inhibited HMGB1-induced inflammation through an imidazoline- and α7-nicotinic acetylcholine receptor–dependent mechanism, but not an alpha-2 adrenergic receptor-dependent mechanism.23 Therefore, future studies should determine whether dexmedetomidine can act on α−7 nicotinic acetylcholine receptor to attenuate sevoflurane-induced Tau phosphorylation, synaptic loss and cognitive impairment.
Our study has several limitations. First, we determined the effects of sevoflurane, dexmedetomidine, clonidine, and yohimbine on a group of mixed sex mice, so it is unknown whether these effects are different between females and males. Second, we used Tau-PS202/Tau-PT205 to determine the role of the alpha-2 adrenergic receptor on sevoflurane-induced Tau phosphorylation based on our previous studies.11 It is possible that dexmedetomidine and clonidine could have different effects on sevoflurane-induced Tau phosphorylation at different sites, e.g., Tau-PS262. Further study of the interaction between dexmedetomidine, clonidine, yohimbine, and sevoflurane is necessary to determine their effect on different sites of Tau phosphorylation.
In conclusion, the alpha-2 adrenergic receptor agonists dexmedetomidine and clonidine attenuated the sevoflurane-induced Tau phosphorylation and synaptic loss in the hippocampi of young mice along with sevoflurane-induced cognitive impairment. Moreover, the alpha-2 adrenergic receptor antagonist yohimbine reversed the protective effects of dexmedetomidine and clonidine. These findings indicate that the alpha-2 adrenergic receptor may contribute to the development of sevoflurane-induced Tau phosphorylation and synaptic loss. Dexmedetomidine could be used to prevent or treat the anesthesia-induced cognitive impairment in young mice. These findings support a need to determine the clinical relevance of these pre-clinical findings.
Supplementary Material
Key points summary.
Question: What are the interventions of anesthesia-induced cognitive impairment in young mice ?
Findings: Alpha-2 adrenergic receptor agonist dexmedetomidine and clonidine mitigated the sevoflurane-induced Tau phosphorylation, synaptic loss and cognitive impairment in young mice.
Meaning: These findings suggest that alpha-2 adrenergic receptor may contribute, at least partially, to the sevoflurane-induced Tau phosphorylation and cognitive impairment in young mice, and alpha-2 adrenergic receptor agonist dexmedetomidine and clonidine may mitigate the effects of sevoflurane in young mice.
Funding:
This research was supported by the National Institutes of Health, Bethesda, Maryland to Zhongcong Xie (HD086977). These studies were performed in Massachusetts General Hospital and Harvard Medical School and are attributed to the Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital and Harvard Medical School.
Glossary
- HMGB1
High molecular group box 1 protein
- MWM
Morris Water Maze
- NOR
New Object Recognition Test
- P
postnatal day
- PSD
Postsynaptic density
- Tau-PS202
Tau phosphorylated at serine-202
- Tau-PT205
Tau phosphorylated at threonine 205
Footnotes
Conflict of interest: Dr. Zhongcong Xie provides consulting service to Shanghai 9th and 10th hospitals, Baxter (as an invited speaker), and Novartis.
References
- 1.Vutskits L, Xie Z: Lasting impact of general anaesthesia on the brain: mechanisms and relevance. Nat Rev Neurosci 2016; 17: 705–717 [DOI] [PubMed] [Google Scholar]
- 2.Binder LI, Frankfurter A, Rebhun LI: The distribution of tau in the mammalian central nervous system. J Cell Biol 1985; 101: 1371–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dujardin S, Colin M, Buee L: Invited review: Animal models of tauopathies and their implications for research/translation into the clinic. Neuropathol Appl Neurobiol 2015; 41: 59–80 [DOI] [PubMed] [Google Scholar]
- 4.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM: Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 1986; 261: 6084–9 [PubMed] [Google Scholar]
- 5.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI: Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 1986; 83: 4913–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trojanowski JQ, Lee VM: Paired helical filament tau in Alzheimer’s disease. The kinase connection. Am J Pathol 1994; 144: 449–53 [PMC free article] [PubMed] [Google Scholar]
- 7.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR: Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 2000; 33: 95–130 [DOI] [PubMed] [Google Scholar]
- 8.Le Freche H, Brouillette J, Fernandez-Gomez FJ, Patin P, Caillierez R, Zommer N, Sergeant N, Buee-Scherrer V, Lebuffe G, Blum D, Buee L: Tau Phosphorylation and Sevoflurane Anesthesia: An Association to Postoperative Cognitive Impairment. Anesthesiology 2012; 116: 779–787 [DOI] [PubMed] [Google Scholar]
- 9.Whittington RA, Virag L, Marcouiller F, Papon MA, El Khoury NB, Julien C, Morin F, Emala CW, Planel E: Propofol directly increases tau phosphorylation. PLoS One 2011; 6: e16648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whittington RA, Virag L, Gratuze M, Petry FR, Noel A, Poitras I, Truchetti G, Marcouiller F, Papon MA, El Khoury N, Wong K, Bretteville A, Morin F, Planel E: Dexmedetomidine increases tau phosphorylation under normothermic conditions in vivo and in vitro. Neurobiol Aging 2015; 36: 2414–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tao G, Zhang J, Zhang L, Dong Y, Yu B, Crosby G, Culley DJ, Zhang Y, Xie Z: Sevoflurane induces tau phosphorylation and glycogen synthase kinase 3beta activation in young mice. Anesthesiology 2014; 121: 510–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu H, Liufu N, Dong Y, Xu G, Zhang Y, Shu L, Soriano SG, Zheng H, Yu B, Xie Z: Sevoflurane Acts on Ubiquitination-Proteasome Pathway to Reduce Postsynaptic Density 95 Protein Levels in Young Mice. Anesthesiology 2017; 127: 961–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu G, Lu H, Dong Y, Shapoval D, Soriano SG, Liu X, Zhang Y, Xie Z: Coenzyme Q10 reduces sevoflurane-induced cognitive deficiency in young mice. Br J Anaesth 2017; 119: 481–491 [DOI] [PubMed] [Google Scholar]
- 14.Correa-Sales C, Rabin BC, Maze M: A hypnotic response to dexmedetomidine, an alpha 2 agonist, is mediated in the locus coeruleus in rats. Anesthesiology 1992; 76: 948–52 [DOI] [PubMed] [Google Scholar]
- 15.Wunsch H, Kahn JM, Kramer AA, Wagener G, Li G, Sladen RN, Rubenfeld GD: Dexmedetomidine in the care of critically ill patients from 2001 to 2007: an observational cohort study. Anesthesiology 2010; 113: 386–94 [DOI] [PubMed] [Google Scholar]
- 16.Su X, Meng ZT, Wu XH, Cui F, Li HL, Wang DX, Zhu X, Zhu SN, Maze M, Ma D: Dexmedetomidine for prevention of delirium in elderly patients after non-cardiac surgery: a randomised, double-blind, placebo-controlled trial. Lancet 2016; 388: 1893–1902 [DOI] [PubMed] [Google Scholar]
- 17.Riker RR, Shehabi Y, Bokesch PM, Ceraso D, Wisemandle W, Koura F, Whitten P, Margolis BD, Byrne DW, Ely EW, Rocha MG, Group SS: Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 2009; 301: 489–99 [DOI] [PubMed] [Google Scholar]
- 18.Djaiani G, Silverton N, Fedorko L, Carroll J, Styra R, Rao V, Katznelson R: Dexmedetomidine versus Propofol Sedation Reduces Delirium after Cardiac Surgery: A Randomized Controlled Trial. Anesthesiology 2016; 124: 362–8 [DOI] [PubMed] [Google Scholar]
- 19.Duan X, Coburn M, Rossaint R, Sanders RD, Waesberghe JV, Kowark A: Efficacy of perioperative dexmedetomidine on postoperative delirium: systematic review and meta-analysis with trial sequential analysis of randomised controlled trials. Br J Anaesth 2018; 121: 384–397 [DOI] [PubMed] [Google Scholar]
- 20.Deiner S, Luo X, Lin HM, Sessler DI, Saager L, Sieber FE, Lee HB, Sano M, and the Dexlirium Writing G, Jankowski C, Bergese SD, Candiotti K, Flaherty JH, Arora H, Shander A, Rock P: Intraoperative Infusion of Dexmedetomidine for Prevention of Postoperative Delirium and Cognitive Dysfunction in Elderly Patients Undergoing Major Elective Noncardiac Surgery: A Randomized Clinical Trial. JAMA Surg 2017; 152: e171505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang C, Ho YS, Ng OT, Irwin MG, Chang RC, Wong GT: Dexmedetomidine directly increases tau phosphorylation. J Alzheimers Dis 2015; 44: 839–50 [DOI] [PubMed] [Google Scholar]
- 22.Liu JR, Yuki K, Baek C, Han XH, Soriano SG: Dexmedetomidine-Induced Neuroapoptosis Is Dependent on Its Cumulative Dose. Anesth Analg 2016; 123: 1008–17 [DOI] [PubMed] [Google Scholar]
- 23.Hu J, Vacas S, Feng X, Lutrin D, Uchida Y, Lai IK, Maze M: Dexmedetomidine Prevents Cognitive Decline by Enhancing Resolution of High Mobility Group Box 1 Protein-induced Inflammation through a Vagomimetic Action in Mice. Anesthesiology 2018; 128: 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shan Y, Sun S, Yang F, Shang N, Liu H: Dexmedetomidine protects the developing rat brain against the neurotoxicity wrought by sevoflurane: role of autophagy and Drp1-Bax signaling. Drug Des Devel Ther 2018; 12: 3617–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, Wu C, Han B, Xu F, Mao M, Guo X, Wang J: Dexmedetomidine attenuates repeated propofol exposure-induced hippocampal apoptosis, PI3K/Akt/Gsk-3beta signaling disruption, and juvenile cognitive deficits in neonatal rats. Mol Med Rep 2016; 14: 769–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisenach JC, De Kock M, Klimscha W: alpha(2)-adrenergic agonists for regional anesthesia. A clinical review of clonidine (1984–1995). Anesthesiology 1996; 85: 655–74 [DOI] [PubMed] [Google Scholar]
- 27.Farrow S, Mers A, Banta G, Steigerwalt S, Lockette W: Effect of the alpha 2-adrenergic antagonist yohimbine on orthostatic tolerance. Hypertension 1990; 15: 877–80 [DOI] [PubMed] [Google Scholar]
- 28.Shen X, Dong Y, Xu Z, Wang H, Miao C, Soriano SG, Sun D, Baxter MG, Zhang Y, Xie Z: Selective Anesthesia-induced Neuroinflammation in Developing Mouse Brain and Cognitive Impairment. Anesthesiology 2013; 118: 502–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang XY, Liu ZM, Wen SH, Li YS, Li Y, Yao X, Huang WQ, Liu KX: Dexmedetomidine administration before, but not after, ischemia attenuates intestinal injury induced by intestinal ischemia-reperfusion in rats. Anesthesiology 2012; 116: 1035–46 [DOI] [PubMed] [Google Scholar]
- 30.Guo L, Yu Y, Xin N, Sun J, Chen Y, Yu M: Clonidine Protects Against Neurotoxicity Induced by Sevoflurane Through NF-kappaB Signaling Inhibition and Proinflammatory Cytokine Release in Rats. J Mol Neurosci 2018; 65: 507–513 [DOI] [PubMed] [Google Scholar]
- 31.Xie Z, Culley DJ, Dong Y, Zhang G, Zhang B, Moir RD, Frosch MP, Crosby G, Tanzi RE: The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid beta-protein level in vivo. Ann Neurol 2008; 64: 618–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.