

Abstract
Purpose:
Immunomonitoring of chimeric antigen receptor (CAR) T cells relies primarily on their quantification in the peripheral blood, which inadequately quantifies their biodistribution and activation status in the tissues. Non-invasive molecular imaging of CAR T cells by positron emission tomography (PET) is a promising approach with the ability to provide spatial, temporal and functional information. Reported strategies rely on the incorporation of reporter transgenes or ex vivo biolabeling, significantly limiting the application of CAR T cell molecular imaging. In the present study, we assessed the ability of antibody-based PET (immunoPET) to non-invasively visualize CAR T cells.
Experimental Design:
After analyzing human CAR T cells in vitro and ex vivo from patient samples to identify candidate targets for immunoPET, we employed a syngeneic, orthotopic murine tumor model of lymphoma to assess the feasibility of in vivo tracking of CAR T cells by immunoPET using the 89Zr-DFO-anti-ICOS tracer we previously reported.
Results:
Analysis of human CD19-CAR T cells during activation identified the Inducible T-cell COStimulator (ICOS) as a potential target for immunoPET. In a preclinical tumor model, 89Zr-DFO-ICOS mAb PET-CT imaging detected significantly higher signal in specific bone marrow-containing skeletal sites of CAR T cell treated mice compared with controls. Importantly, administration of ICOS-targeting antibodies at tracer doses did not interfere with CAR T cell persistence and function.
Conclusions:
This study highlights the potential of ICOS-immunoPET imaging for monitoring of CAR T cell therapy, a strategy readily applicable to both commercially available and investigational CAR T cells.
Introduction
Chimeric antigen receptor (CAR) T cell therapy has shown impressive results in B-cell hematologic malignancies leading to the clinical approval of the first engineered cellular therapy for cancer (1,2). In contrast to conventional pharmacological approaches, CAR T cells are living drugs that after administration actively migrate to target tumor-infiltrated tissues, expand and persist in order to exert their function efficiently. The development of new CAR T cell-based therapies and the improvement of existing ones are greatly influenced by our ability to monitor their in vivo dynamics to gain greater insights into the success or failure of this treatment approach. The measurement of circulating CAR T cells in the peripheral blood by flow cytometry and/or polymerase chain reaction (PCR) is the only immuno-monitoring modality currently available. Some studies have reported an association between the peak and the duration of circulating CAR T cell levels and the clinical outcome (3–7) while other studies have failed to confirm these results (8,9) suggesting that such a measure might not accurately reflect the actual biodistribution and persistence of the cells in the body. Such a discrepancy can be particularly relevant when CAR T cells are employed for the treatment of solid tumors for which lack of efficient migration to the tumor site is considered one of the main barriers limiting the efficacy of CAR T cell therapy (2,10).
Molecular imaging is an attractive strategy for non-invasive and longitudinal monitoring of CAR T cell distribution in vivo. To date, several studies using positron emission tomography (PET) imaging have been conducted, due to the high sensitivity and quantitative capabilities of this clinically relevant imaging modality. The majority of the preclinical and clinical pilot studies published so far involves the use of reporter genes inserted into the CAR construct for the detection of CAR T cells (11–15). Despite the enormous potential of this indirect labeling approach, major limitations of this strategy are the requirement for the generation and approval of entirely new CAR T cells specifically designed to incorporate the imaging functionality and a significant risk of immunogenicity. To circumvent these limitations, an alternative approach tested has been the ex vivo labeling of CAR T cells with radioactive tracers prior to administration (16–18). This approach has the advantage of being applicable to any CAR T cells including the currently commercially available ones, but has several limitations, principally limited temporal resolution, a function of the radioactive decay of the label employed. Even if radioisotopes with long half-lives are employed, imaging is limited to a relatively short time frame after administration and is further compounded by signal dilution as CAR T cells proliferate in vivo.
ImmunoPET is a rapidly expanding area of molecular imaging that employs monoclonal antibodies and antibody fragments radiolabeled with PET isotopes, thereby combining the ultra-high specificity and affinity of antibodies for cell surface markers, with the superior sensitivity of PET (19). Advantageously, the radiolabeled antibodies can be injected at different time points post administration of the cells and even repeatedly to allow longitudinal and serial assessment of CAR T cell persistence. Target molecules evaluated to date to study T cell immune responses principally include lineage defining molecules such as CD3 (20,21), CD4 (22,23), CD7 (24) and/or CD8 (25,26) and T-cell surface activation markers such as OX40 (27), HLA-DR (28) and ICOS (29). Our group and others have evaluated such approaches for monitoring different classes of cancer immunotherapies in preclinical models, including tumor vaccination (27,29), immune checkpoint blockade(29) and allogeneic hematopoietic stem cell transplantation (28,30).
In the present work, we identified Inducible T-cell COStimulator (ICOS or CD278), a costimulatory molecule upregulated during T cell activation (31), as a candidate target for CAR T cell monitoring using immunoPET. Moreover, we assessed the utility of ICOS-targeted immunoPET to monitor CD19-specific CAR T cell activation, expansion and homing to target tumor-infiltrated tissues in a murine model of B-cell lymphoma.
Methods
Analysis of RNA sequencing data obtained from human CAR T cells during in vitro activation
We analyzed an RNA sequencing dataset we recently published (32) and deposited in NCBI Gene Expression Omnibus (GEO), series accession number: GSE136891. RNAseq data were obtained at serial time points from CD19.CD28z CAR T cells generated from naive (CD45RA+, CD45RO−, CD62L+, CCR7+, CD95−, and CD122−) CD4+ or CD8+ T cells as detailed in (32).
Mass cytometry analysis of human CAR T cells
We retrospectively analyzed ex vivo ICOS expression on human CD19–28z CAR T cells in prospectively collected data obtained from 31 patients receiving commercial axicabtagene ciloleucel (Axi-cel) at Stanford for relapsed or refractory diffuse large B-cell lymphoma (r/r DLBCL). Mass cytometry analysis was performed as previously described (33). The mass cytometry panel assessed expression of 33 surface or intracellular proteins relevant to T cell function in blood collected on day 7 (peak expansion), and on day 21 (late expansion) post-CAR T cell infusion (33). ICOS expression was evaluated on live CAR-expressing T cells identified by anti-idiotype antibody provided by Dr. Laurence Cooper (34). Written informed consent was provided by all patients enrolled, and the study was approved by the Stanford Institutional Review Board. Experimental procedures were carried out in accordance with the ethical principles of the declaration of Helsinki.
Animals
BALB/cJ mice were purchased from the Jackson Laboratory (Sacramento, CA). Firefly Luciferase (Luc)+ transgenic BALB/c mice have been previously reported (35) and were bred in our animal facility at Stanford University. All procedures were performed on sex-matched animals between 8 and 12 weeks of age and approved by Stanford University’s Administrative Panel for Laboratory Animal Care (APLAC)/Institutional Animal Care and Use Committee (IACUC) in compliance with the guidelines of humane care of laboratory animals.
Murine CAR T cells generation
CAR T cells specifically recognizing the murine CD19 molecule and including a CD28 costimulatory domain were generated as previously reported (36,37). Murine CD19 (mCD19) CAR stable producer cell line (37) was kindly provided by Dr. Terry J. Fry. T cells were enriched from BALB/c mouse spleen single-cell suspensions using the mouse Pan T Cell Isolation Kit II (Miltenyi Biotec) according to the manufacturer’s protocol. T cells were activated for 24 hours with Dynabeads® Mouse T-Activator CD3/CD28 (Life Technologies, Grand Island, NY) in the presence of human IL-2 (30 U/ml) and murine IL-7 (10 ng/ml; PeproTech) in RPMI 1640 media supplemented with 10% heat-inactivated FBS, 1 mmol/L sodium pyruvate, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C with 5% CO2. Activated T cells were then transduced with mCD19 CAR by culturing them for 48 hours in retronectin coated plates loaded with supernatant harvested from the stable producer line 48 hours after culture. Dynabeads were then removed and after washing cells were rested 24 hours in fresh medium containing IL-2 and IL-7 before use. Transduction efficiency was measured by flow cytometry after protein L staining (38). Cell numbers were adjusted based on transduction efficacy (50% on average) before in vitro or in vivo use.
In vitro cytotoxic assay
Murine CD19.28z CAR T cells were co-cultured with luciferase-transduced A20 cells (A20luc (39)) at different ratios adjusted based on transduction efficiency in culture medium consisting of RPMI 1640, supplemented with L-glutamine (2 mM), penicillin (100 U/mL), streptomycin (0.1 mg/mL), 2-mercaptoethanol (5× 10e-5 M), and 10% fetal bovine serum. After 24 hours of culture, D-luciferin (PerkinElmer) was added at 5 μg/ml and incubated for 5 min at room temperature before imaging using an IVIS Spectrum imaging system (Perkin Elmer). For ICOS expression analysis, CAR T cells cultured with A20Luc+ at 1:1 ratio or unstimulated controls were harvested and analyzed by flow cytometry.
In vivo murine tumor model
We employed a systemic B-cell lymphoma mouse model we previously reported in which tumor cells infiltrate secondary lymphoid organs and bone marrow (39). Briefly, CD19-expressing A20 cells were purchased from ATCC (Rockville, MD) in 2017, expanded for three passages and cryopreserved until use. A20 cells were thawed, cultured for maximum a week, resuspended in PBS, and injected (2.5 × 10e5 cells/mouse) by tail vein intravenously (i.v.) into sub-lethally (4.4 Gy) irradiated Thy1.2+ BALB/c recipient mice. Seven days after tumor injection, Luc+ CAR T cells (1×10e6 transduced cells/mouse adjusted on transduction efficiency) or equivalent numbers of in vitro expanded untransduced Luc+ T cells (UT) were administered by retro-orbital intravenous injection. In tumor homing experiments, A20Luc+ cells were employed and mice left untreated.
Flow cytometry analysis
Single-cell suspensions were prepared from spleen and bone marrow in phosphate-buffered saline (PBS) containing 2% fetal bovine serum. Extracellular staining was preceded by incubation with purified FC blocking reagent (Miltenyi Biotech). Cells were stained with: FITC anti-CD45.1 (clone A20); BV785 anti-ICOS (clone C398.4A) or appropriate isotype control (clone HTK888); APC anti-Thy1.1 (clone OX-7); APC/Fire750 anti-CD19 (clone 6D5); BV421 anti-CD4 (clone GK1.5); BV605 anti-CD3 (clone 17A2); BV650 anti-CD8 (clone 53–6.7). Dead cells were excluded using Fixable Viability Dye eFluor® 506 (eBioscience). All antibodies were purchased from Biolegend. Samples were acquired on a BD LSR II flow cytometer (BD Biosciences), and analysis was performed with FlowJo 10.5.0 software (Tree Star).
In vivo bioluminescence imaging
For in vivo bioluminescence imaging (BLI), mice were injected with D-luciferin (10 mg/kg; intraperitoneally) and anesthetized with 2% isoflurane in oxygen. Imaging was conducted using an IVIS Spectrum imaging system (Perkin Elmer) and data were analyzed with Living Image software version 4.1 (Perkin Elmer) or using an Ami imaging system (Spectral Instruments Imaging, Tucson, AZ) and data analyzed with Aura Software (Spectral Instruments Imaging).
Bioconjugation and radiolabeling of ICOS-targeted monoclonal antibody (ICOS mAb)
ICOS mAb (clone:7E.17G9, Bio-X-cell) was modified with the bifunctional chelator deferoxamine (DFO/p-SCN-Bn-Deferoxamine) (Macrocyclics). Briefly, unconjugated mAb was prepared at 1 mg/ml in PBS (pH=7.4), and buffer exchanged with PBS solution adjusted to pH 8.8–9.0 using 1M Na2CO3. Following this, 10-fold molar excess DFO was added to the ICOS mAb solution and the conjugation was allowed to proceed for 1-hour incubation at 37°C. The mixture was subsequently thoroughly buffer exchanged using PBS (pH=7.4) to remove unreacted DFO using a 2 ml vivaspin centrifugal concentrator with a 50K cutoff (Sartorius). The concentration of the final DFO-ICOS mAb conjugate was determined by Thermo Scientific™ NanoDrop™ One Microvolume UV-Vis Spectrophotometer.
For 89Zr labeling, 37 MBq (~1 mCi) of 89Zr-oxalate (3D imaging) was diluted in 0.5 mL of HEPES buffer (0.5 M) to ensure a pH range of 7.0–7.5 followed by the addition of ~166 μg of DFO-ICOSmAb. Radiolabeling was carried out for 1h at 37°C with shaking, after which the radiolabeled antibody 89Zr-DFO-ICOS mAb was purified using a 7K MW cut-off zeba spin desalting column (Thermofisher) centrifuged for 1 minute at 1000g. The radiochemical yield and purity were determined via instant thin-layer chromatography (iTLC). Two μL samples of the radiolabeling reaction and purified radiolabeled antibody were spotted into silica-impregnated radio iTLC plates, run with 50 mM EDTA (pH = 4.5), and developed in a phosphor-plate reader (Perkin Elmer). Radiolabeled antibody remained at the origin (Rf = 0) while free 89Zr moved with the solvent front (Rf = 1). A final radiochemical purity of 99% was achieved, along with a final specific activity of 6 μCi/μg/ml. The final formulation was prepared in PBS.
Small animal PET/CT and ex vivo biodistribution studies
Mice were anesthetized using isoflurane (2.0–2.5% for induction and 1.5–2.0% for maintenance) delivered by 100 % oxygen. 89Zr-DFO-ICOSmAb (45 μCi ± 3.6, 7.5 μg± 0.6) was administered i.v. in untreated and Luc+ untransduced T cell or CAR T cell treated A20 tumor-bearing mice 5 days post-CAR T cell injection. Radiotracer administration corresponded to day 5 post-CAR T cell injection. At 24 and 48 hours post 89Zr-DFO-ICOS mAb tracer administration, mice underwent imaging with static 20 minute PET scans followed by 10 minute transmission scans for PET attenuation correction on the Inveon dedicated PET scanner (DPET, Siemens), and CT scans on the GNEXT (Sofie Biosciences) for anatomical co-registration of the PET data. PET image reconstruction and image analysis were conducted as previously described (27).
Following the completion of the scan 48 hours after injection of the tracer, mice were euthanized and ex vivo biodistribution studies were performed to measure tissue-specific radioactivity, and further corroborate PET findings. Briefly, we collected blood (~100 μl) via cardiac puncture as well as the following tissues: heart, spleen, kidney, liver, muscle, femur, tibia, iliac bone and lumbar spine. Harvested hearts were rinsed in clean water after an incision was made to remove residual blood and then gently dabbed to remove excess moisture. Tissues were placed in a tube, weighed and radioactivity measured using an automated gamma counter (Hidex AMG Automatic Gamma Counter). Tissue-associated radioactivity was normalized to tissue weight and amount of radioactivity administered to each mouse, decay-corrected to the time of radiotracer injection. Data were expressed as percentage of injected dose per gram of tissue (%ID/g) values.
Assessment of the impact of ICOSmAb administration at imaging doses on CAR T cell homeostasis and function
Murine specific ICOS monoclonal antibody (ICOSmAb, clone 7E.17G9; BioXcell) or isotype control (clone: LTF-2; BioXcell) were administered as a single dose intravenously on day 5 post CAR T administration (10 μg in 100 μL of PBS). The dose was determined based on the upper limit of antibody administered during PET imaging studies.
Statistical Analysis
Student’s 2-tailed t test, the Mann–Whitney U test or the Wilcoxon signed-rank test were used as appropriate to determine statistical significance. BLI data (photons/s) are displayed as mean and standard deviation for each group over time and analyzed by two-way ANOVA with Bonferroni correction. Survival curves were represented with the Kaplan-Meier method and compared by log-rank test. Statistical analyses were performed using Prism 8 (GraphPad Software, La Jolla, CA) and R version 3.5.1 (Comprehensive R Archive Network (CRAN) project (http://cran.us.r-project.org) with R studio version 1.1.453.
Results
ICOS expression is upregulated and sustained during in vitro and in vivo activation of human CD19.28z CAR T cells
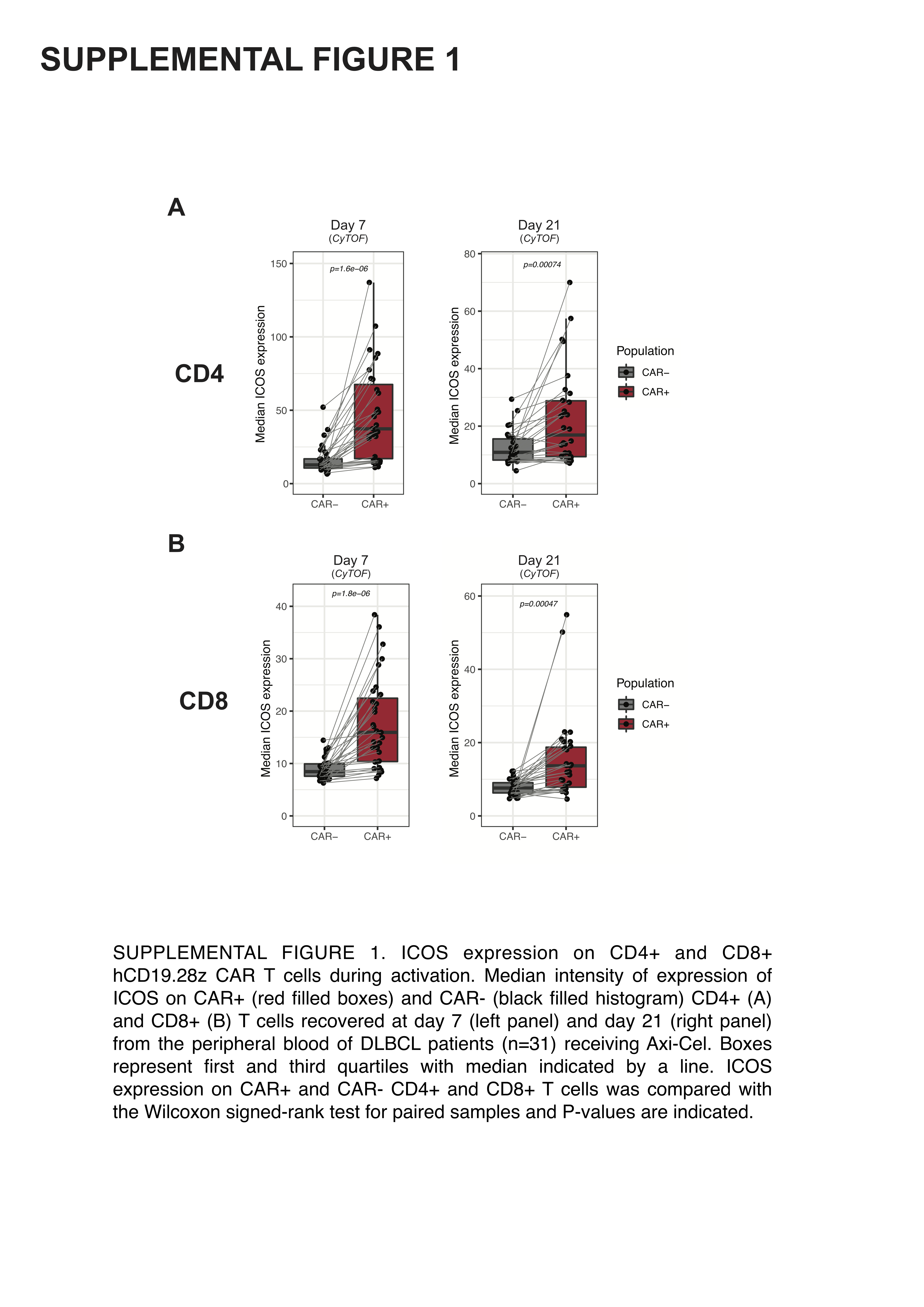
In an effort to identify surface activation markers as candidate targets for immunoPET, we analyzed recently published RNA sequencing data obtained from human CD4+ and CD8+ CD19.28z CAR T cells cultured in vitro (32). The analysis was restricted to a selected list of markers known to be upregulated during activation on the T cell surface and previously employed as targets for PET imaging (27,29,40,41). As shown in Figure 1A, transcripts of most of the selected markers were expressed at high levels early after activation (day 7) and decreased at later time points (day 10 and day 14). ICOS transcription exhibited a different trend, progressively increasing and being sustained at later time points in both CD4+ and CD8+ CAR T cells (Figure 1A). To further validate ICOS as a candidate target for immunoPET, we next assessed its ex vivo expression on CD19.28z CAR T cells isolated from 31 patients receiving commercial axicabtagene ciloleucel (KTE-C19, Axi-cel) for relapsed/refractory B-cell lymphoma, at day 7 and 21 post therapy administration. CAR-expressing T cells identified using specific anti-idiotype staining were compared to circulating T cells not expressing the CAR. ICOS was expressed at significantly higher levels on CAR-expressing (CAR+) T cells compared with CAR-nonexpressing (CAR-) T cells (Figure 1B). This difference was observed at day 7, corresponding to the peak of CAR T cell expansion (median intensity ±SEM: CAR+: 26.6±4.7; CAR−: 9.6±0.9; p=0.00002), and further maintained at day 21 (CAR+: 16.3±3.2; CAR−: 9.9±1.0; p=0.00068; Figure 1C). In accordance with the RNA sequencing data (Figure 1A), we detected higher ICOS expression on both CD4+ and CD8+ CAR T cells (Supplemental figure 1). Collectively, these results identify ICOS as a promising target for CAR T cell monitoring by immunoPET.
FIGURE 1. ICOS is upregulated on human CD19.28z CAR T cells in vitro and in vivo.

(A) Row scaled heatmap visualization of RNA expression of the surface activation markers CD38, CD69, IL2RA, OX40 and ICOS in CD4+ (left panel) and CD8+ (right panel) CAR T cells on day 7, 10 and 14 of in vitro culture. (B) Representative mass cytometry histogram showing ICOS expression at the surface of CAR+ (red filled histogram) and CAR- (black filled histogram) T cells recovered at day 7 from the peripheral blood of a DLBCL patient receiving Axi-Cel. (C) Median intensity of expression of ICOS on CAR+ (red-filled boxplot) and CAR- (gray-filled boxplot) T cells recovered at day 7 (left panel) and day 21 (right panel) from the peripheral blood of DLBCL patients (n=31) receiving Axi-Cel. Boxes represent first and third quartiles with median indicated by a line. ICOS expression on CAR+ and CAR- T cells was compared with the Wilcoxon signed-rank test for paired samples and P-values are indicated.
ICOS is upregulated during in vitro and in vivo activation of murine CD19.28z CAR T cells
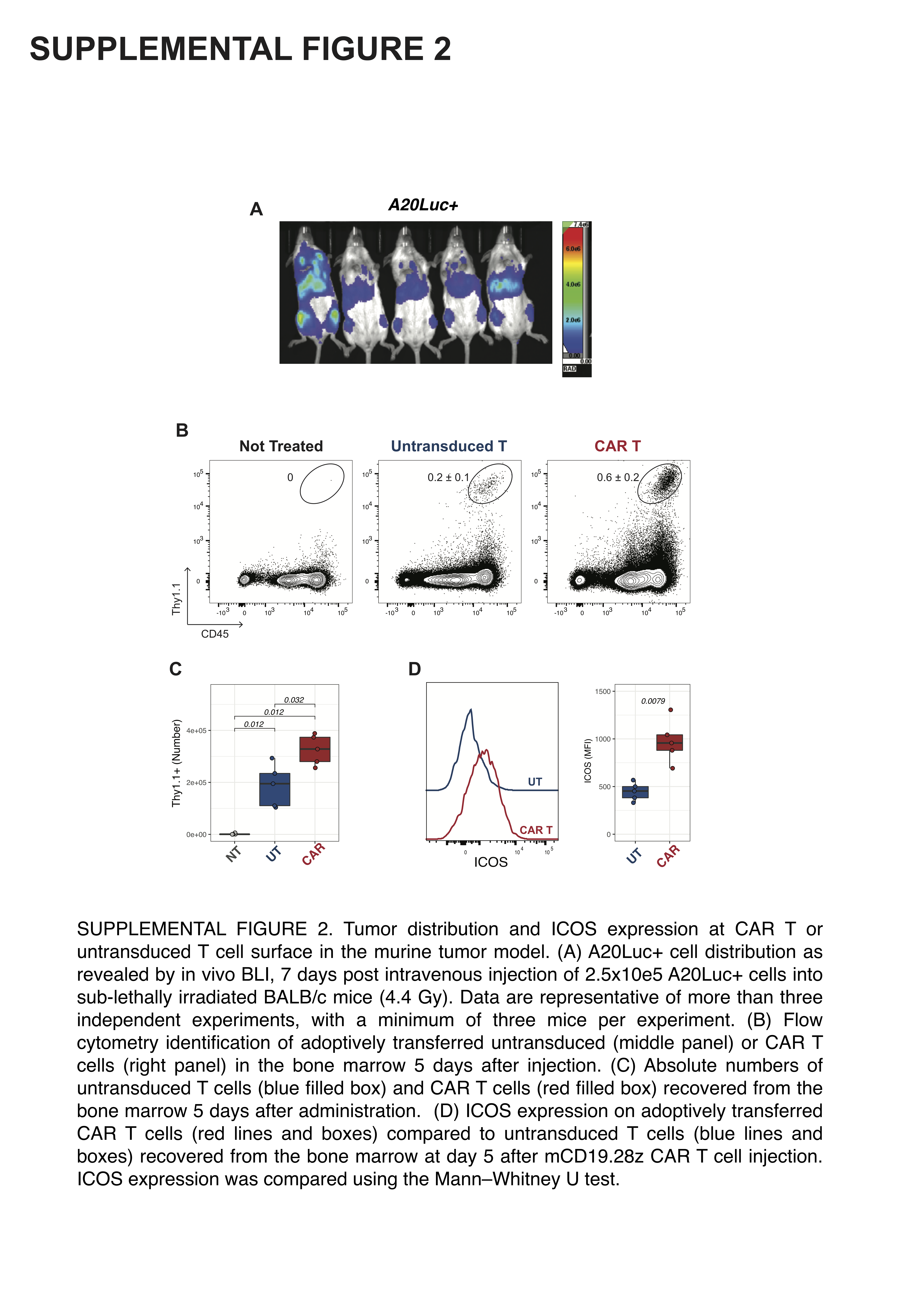
In order to test the utility of ICOS-immunoPET for in vivo monitoring of CAR T cells in a preclinical mouse model, we went on to assess ICOS expression during murine CD19.28z (mCD19.28z) CAR T cell activation. First, we analyzed ICOS expression during a 24-hour cytotoxic assay incubating mCD19–28z CAR T cells together with the Luc+ CD19-expressing murine B-cell lymphoma cell line A20. As expected, mCD19–28z CAR T exerted a dose-dependent cytotoxic effect against A20 cells (Figure 2A). ICOS expression, as assessed by flow cytometry, was significantly upregulated on murine CAR T cells exposed to A20 target cells (1:1 CAR to target ratio) compared to CAR T cells alone (Figure 2B). We next assessed ICOS expression on mCD19.28z CAR T cells activated in vivo. To better mimic the clinical scenario, we employed a systemic tumor model in which A20-lymphoma cells were injected intravenously (Figure 2C). In agreement with what we previously reported (39), by the time of CAR T cell administration, 7 days after tumor inoculation, A20Luc+ cells were infiltrating the liver, the lymphoid organs and the bone marrow (BM; Supplemental figure 2). Flow cytometry analysis was performed at day 5 post administration of Thy1.1+ CAR T cells into Thy1.2+ recipients, a time point when CAR T cells had expanded and were detectable within the spleen and the bone marrow as confirmed by in vivo bioluminescence (Figure 2D). ICOS expression was significantly higher at the surface of administered Thy1.1+ infused cells when compared to endogenous Thy1.1− cells both in the spleen (Figure 2E) and in the bone marrow (Figure 2F). To further assess the specificity of ICOS upregulation for CAR-mediated activation of CAR T cells and distinguish it from activation resulting from the lymphopenic context, we used ex vivo expanded untransduced T cells (UT) as an additional control. As shown in Supplemental Figure 2B–C, lower percentages and absolute numbers of untransduced T cells were detected within the bone marrow, suggesting preferential homing and/or expansion of CAR T cells in the tumor-infiltrated bone marrow. Phenotypically, bone-marrow infiltrating CAR T cells expressed significantly higher levels of ICOS compared with untransduced T cells (Supplemental Figure 2C) confirming the specificity of ICOS expression for activated CAR T cells. Collectively, these results confirm that ICOS is specifically expressed on murine CAR T cell surface during ex vivo and in vivo activation, supporting its potential utility as a biomarker for CAR T monitoring by immunoPET.
FIGURE 2. ICOS is selectively expressed on activated murine CD19.28z CAR T cells in vitro and in vivo.

(A) Representative in vitro bioluminescence (BLI) image of in vitro cytotoxic activity of mCD19.28z CAR T cells cocultured for 24 hours with A20Luc+ cells at the indicated ratios of Effector: Target (E:T). (B) ICOS expression on mCD19.28z CAR T cells cultured for 24 hours in the presence (solid red line and red filled box) or in the absence (dashed red line and empty box) of A20Luc+ cells (E:T = 1:1). Data were pooled from two independent experiments performed in triplicate. (C) Schematic representation of the in vivo lymphoma tumor model employed as detailed in the text. (D) Representative in vivo bioluminescence (BLI) image of the biodistribution of Luc+ mCD19.28z CAR T cells in the lymphoma model at day 5 post injection (i.v.). Data are representative of more than five independent experiments, using a minimum of three mice per experiment. (E-F) ICOS expression on adoptively transferred Thy1.1+ cells (red lines and boxes) compared to Thy1.1- cells (gray lines and boxes) recovered from the spleen (E) or bone marrow (F) at day 5 after mCD19.28z CAR T cell injection.
ICOS-immunoPET enables visualization of activated CAR T cells in the bone marrow
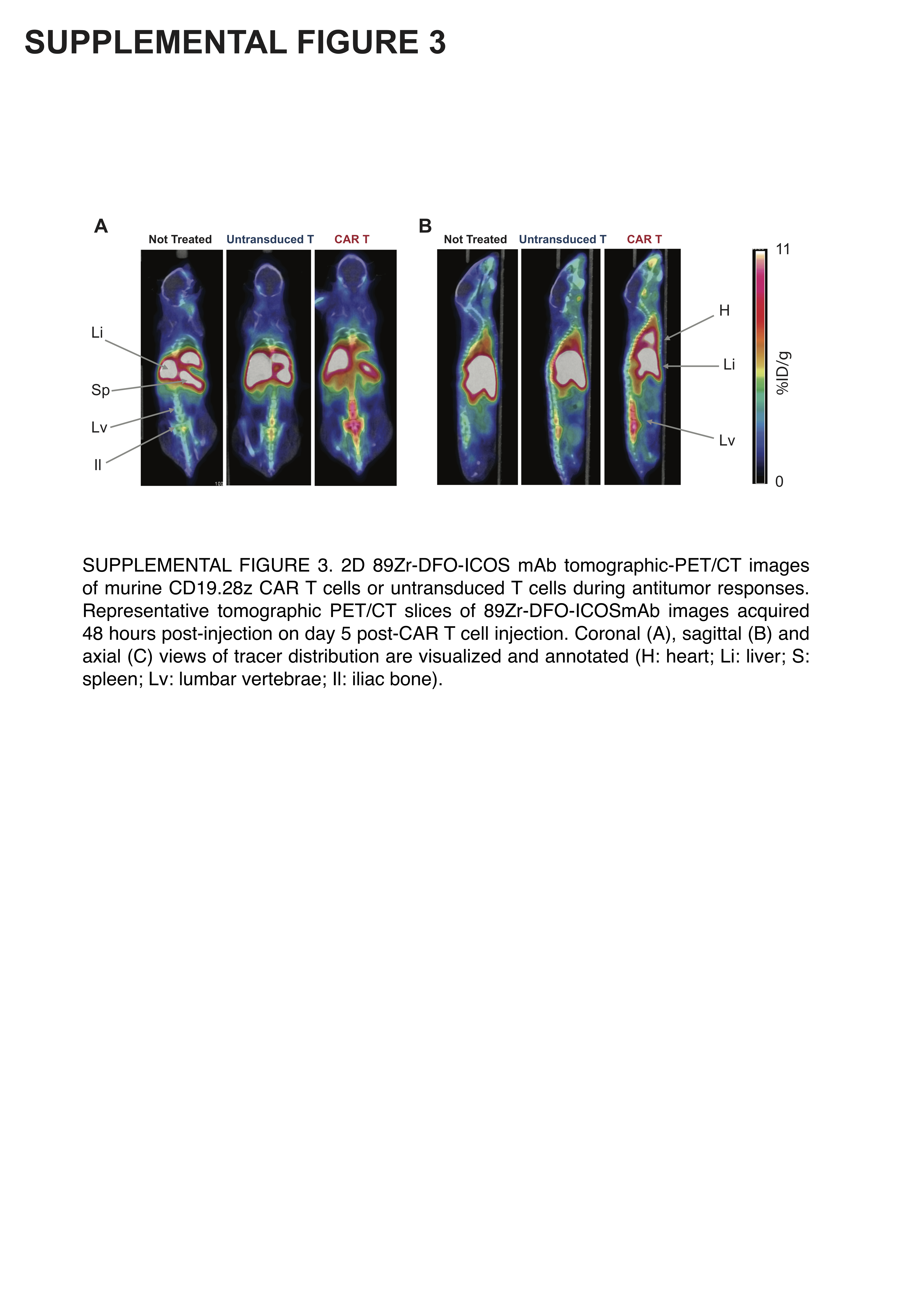
We next tested the ability of ICOS-targeted immunoPET to visualize CAR T cell migration, activation, migration and expansion during an antitumor response using 89Zr-DFO-ICOS mAb in vivo. Employing the aforementioned murine tumor model, 89Zr-DFO-ICOS mAb was intravenously injected at day 5 post-T cell administration and PET/CT images were acquired at 24 and 48 hours post-tracer injection. The 48-hour time point was selected as the one providing superior signal-to-noise ratio. Figure 3A and B show representative volume-rendered technique (VRT) PET/CT images of tumor-bearing mice that either received no treatment (Not Treated, left panels), untransduced T cells (middle panel) or mCD19.28z CAR T cells (CAR T, right panels). PET/CT images of untreated mice showed that 89Zr-DFO-ICOS mAb primarily accumulated in the heart, spleen and liver, but not in bones, consistent with the biodistribution and clearance of intact antibodies (Figure 3A and B, left panels; Supplemental Figure 3). Mice receiving untransduced T cells as well as CAR T cells displayed a tracer biodistribution in highly vascularized organs (heart, liver and spleen) similar to mice that were not treated. Importantly, we detected significantly higher 89Zr-DFO-ICOS mAb-PET signals in the bones of CAR T cell treated mice compared with both untreated mice and mice treated with untransduced T cells (Figure 3A and B, right panels; and two dimensional maximum intensity projection (MIP) images in Supplemental Figure 3). The PET signal was particularly prominent in the lumbar spine, iliac bones, femur, tibia and humeral heads (Figure 3A and B, right panels; MIP images in Supplemental Figure 3).
FIGURE 3. 89Zr-DFO-ICOS mAb PET/CT imaging visualize murine CD19.28z CAR T cells during antitumor responses.

(A-B) Representative 3D volume rendered technique (VRT) PET/CT images acquired 48 hours post-tracer administration on day 5 after untransduced T cells or mCD19.278z CAR T cells administration (i.v.). Coronal-ventral views (A) and sagittal views (B) are depicted. Location of key clearance and target tissues is indicated (H: heart; Li: liver; S: spleen; Lv: lumbar vertebrae; Il: iliac bone; F: femur and T: tibia). Images are representative of two independent experiments with 8–9 mice per group. (C) Quantitative region of interest (ROI) PET image analysis of heart, spleen, liver, muscle, lumbar spine, iliac bone, femur and tibia in mCD19.278z CAR T cell treated (CAR, red filled boxes), untransduced T cell treated (UT, blue filled boxes) and untreated controls (NT, grey filled boxes). Tracer uptake in CAR T (n=9), UT (n=9) and not treated (n=8) groups were compared using the Mann–Whitney U test.
To quantify radiotracer accumulation in specific tissues and corroborate the trends observed in the PET images, we conducted a region of interest (ROI) analysis on multiple tissues guided by CT. We detected a slight, but statistically significant increase in radiotracer accumulation in the heart of CAR T cell-treated mice compared with mice that were not treated (mean±SEM: 11.4±0.45 vs. 9.1±0.9%ID/g p<0.05, Figure 3C) while no significant differences were detected when compared with mice receiving untransduced T cells (Figure 3C). No significant differences between CAR T cell treated mice and untreated mice were detected in spleen and liver (Figure 3C). ROI quantification of PET/CT images confirmed markedly increased radiotracer uptake in bones rich in bone marrow from CAR T treated mice compared with those of both untreated mice (lumbar vertebrae p=0.014; iliac bones p=0.0012; femur p=0.0078; tibia p=0.0016; n=8–9 per group, Figure 3C) and mice receiving untransduced T cells (lumbar spine vertebrae p=0.001; iliac bones p=0.0012; femur p=0.0078; tibia p0.0056; n=9 per group, Figure 3C). As expected, there was no significant signal difference in the muscle, considered background, among the groups (Figure 3C).
To corroborate the PET results, we performed a biodistribution analysis (BioD) using ex vivo gamma counting of the different tissues following the 48 hours PET/CT acquisition. BioD analysis confirmed higher radiotracer levels in the heart of CAR T recipients compared with untreated mice (p=0.026), but not with mice receiving untransduced T cells (Figure 4). No difference among the groups was detected in blood, liver, and kidney (Figure 4). In corroboration with the higher ICOS levels detected by FACS, BioD revealed significantly higher tracer uptake in spleens from CAR T treated mice compared with the untreated group (p=0.0033) and the UT group (p=0.024; Figure 4). When analyzing the bones rich in bone marrow, we confirmed significantly higher tracer uptake in bones from CAR T treated mice compared to mice not treated (lumbar vertebrae p=0.0015; iliac bones p=0.019; femur p=0.00063; tibia p=0.00062) or treated with untransduced T cells (lumbar spine vertebrae p=0.0036; iliac bones p=0.007; femur p=0.00016; tibia p=0.00049; Figure 4). No difference was observed in muscle. Collectively, these results demonstrate the ability to specifically track the presence of CAR T cells at target tissues during antitumor responses in vivo using 89Zr-DFO-ICOSmAb immunoPET.
FIGURE 4. Quantitative 89Zr-DFO-ICOS mAb tracer biodistribution during anti-tumor responses.

Quantification of ICOS-immunoPET signal (%ID/g) from ex vivo biodistribution analysis of whole blood, heart, spleen, liver, kidney, muscle, lumbar spine, iliac bone, femur, and tibia 48 hours after tracer administration (on day 5 post CAR T therapy). Tracer uptakes in CAR T (n=9), UT (n=9) and untreated (NT, n=8) groups were compared using the Mann–Whitney U test. Results are pooled from two independent experiments with a total of 8–9 mice per group.
Administration of ICOSmAb at tracer doses does not impact the in vivo persistence and antitumor effect of mCD19.28z CAR T cells
The use of costimulatory molecules as a target for immunoPET bears the theoretical risk of impacting CAR T cell expansion, persistence and ultimately in vivo antitumor activity, especially considering that the clone we employed (7E.17G9) blocks the interaction between ICOS and its ligand, ICOSL. To assess the impact of the administration of anti-ICOS monoclonal antibodies on CAR T cell homeostasis and function, we injected doses comparable to the maximum PET dose of anti-ICOS (clone 7E.17G9) or of the appropriate isotype control at day 5 post CAR T cell administration. The BLI signals derived from expanding Luc+ CAR T cells was not significantly different between the anti-ICOS (Figure 5A, upper panels) and the isotype control groups (Figure 5A, lower panels; Figure 5B). Similarly, the contraction curve of CAR T cells at later time points (days 10–14) did not differ in mice treated with anti-ICOS compared to mice receiving isotype control (Figure 5A–B). Using overall survival as a measure of CAR T cell efficacy, we did not observe any significant impact of anti-ICOS administration on the survival of untreated and CAR T cell treated mice when compared to mice receiving isotype control (Figure 5C). Collectively, these results indicate that the administration of anti-ICOSmAb at tracer doses does not affect CAR T cell homeostasis or antitumor activity.
FIGURE 5. Impact of anti-ICOS-mAb administered at tracer dose on in vivo persistence and anti-tumor activity of mCD19.28z CAR T cells.

(A-B) Expansion of Luc+ mCD19.28z CAR T cells in tumor bearing mice treated on day 5 with ICOS-mAb (A, upper panels; B, continuous lines) or appropriate isotype control (A, lower panels; B, dashed lines), administered at a dose similar to that employed for PET/CT studies (10 μg/mouse, representing upper limit of tracer dose). Data shown are from one experiment representative of two independent experiments with 4–5 mice per group in each experiment. (C) Overall survival of mCD19.28z CAR T cell treated (CAR, red lines) or not treated (NT, black lines) mice. At day 5, mice were randomized to receive intravenous administration of ICOS-mAb (continuous lines) or appropriate isotype control (dashed lines) at a dose similar to the ones employed for PET/CT studies (10 μg/mouse, representing the upper limit of tracer dose). Results are pooled from two independent experiments with a total of 9–10 mice per group. Survival curves were plotted using the Kaplan-Meier method and compared by log-rank test.
Discussion
In this study, we identified ICOS-immunoPET as a promising strategy to non-invasively monitor CAR T cells in vivo during an antitumor response.
In vivo monitoring of CAR T cell therapy using molecular imaging is an area of intensive investigation. Several groups, including ours, have explored the utility of reporter genes (11–15) or ex vivo radiolabeling approaches (16–18) to allow tracking of CAR T cell therapies with PET. The use of immunoPET has several advantages over these approaches. First, this strategy can be applied to any CAR T cell product, bypassing the need to insert a dedicated imaging reporter gene. The feasibility of this approach is a significant advantage considering the time and efforts required for approval of new cellular products. Second, the administration of radiolabeled antibodies at different time points post CAR T cell administration, allows for serial imaging of CAR T cell dynamics in vivo. This latter aspect might be essential in the clinical setting, where detection of CAR T cell presence and activation by immunoPET might be employed to guide therapeutic decisions.
A major challenge in immunoPET imaging of adoptively transferred cells comes from the difficulties in identifying appropriate candidate targets. An ideal target should be selectively expressed on the transferred population with minimal expression on endogenous cells. In this regard, targeting surface activation markers (27–29) displays a major advantage over targeting lineage-defining markers (20–26). In the present report, using an unbiased approach we first identified ICOS as a potential immunoPET target on human CAR T cells before transitioning to a murine model for the preclinical assessment of our strategy. Notably, ICOS-immunoPET enabled in vivo tracking of murine CAR T cells injected into immunocompetent mice bearing systemic tumors. We believe that such a model more accurately reflects the clinical scenario compared to widely employed xenogeneic tumor models, in which human CAR T cells are injected into genetically lymphopenic mice. Firstly, it allows the evaluation of the tracer in the presence of an endogenous resident population of T cells. Second, in xenogeneic models the injected cells are the only targets of antibodies recognizing human molecules in a murine system, thus artificially reducing the background noise coming from the endogenous population.
Another critical aspect that needs to be taken into account when developing new immuno-imaging approaches is the risk of interfering with CAR T cell biology. The addition of reporter genes encoding foreign proteins carries a significant risk of immunogenicity (42) that can potentially limit the therapeutic potential of the CAR T cells, a risk that cannot efficiently be evaluated in preclinical xenogeneic mouse models. Moreover, the addition of extra genes to the construct might significantly interfere with CAR expression and/or function. In the case of immunoPET, antibodies may exert biological effects after ligation of their target and, depending on the blocking, agonist or antagonist nature of the clone employed, they might interfere with CAR T cell homeostasis and function. In our animal model we show that the administration of tracer-doses of a blocking anti-ICOS does not interfere with CAR T cell persistence and antitumor effect, suggesting the safety of our approach. Although we cannot exclude any minor transient effects, overall there was no detectable impact on CAR T cell expansion (as measured by BLI), or function (as assessed by the outcome analysis). Such results will need to be confirmed on human CAR T cells during clinical translation using a fully human/humanized version of the ICOS mAb.
Our study demonstrated the feasibility of utilizing immunoPET with anti-ICOS to track CAR T cell migration into tumor-bearing mice. The early time point studied demonstrates the ability of the CAR T cells to migrate to their target tissue, the bone marrow bearing the CAR-targeted CD19 antigen on infiltrating B-cell lymphoma cells as well as on endogenous B-cell precursors. This approach is directly applicable to the clinic yet has several limitations. First, ICOS-immunoPET was affected by a substantial non-specific background coming from highly vascularized organs containing high amounts of blood such as heart, liver and spleen. Such background limited the sensitivity of the assays in tumor-containing organs such as the spleen, where a significant difference between CAR T cell treated mice and untreated mice was detectable only in ex vivo biodistribution studies but not by in vivo PET imaging. Such limitation could be circumvented by the use of alternative vector formats that are smaller in size and exhibit rapid pharmacokinetics that are more suitable for imaging, for example antibody fragments, including minibodies, diabodies, single-chain variable region fragments, and nanobodies, or engineered protein scaffolds (reviewed in (43,44)). Second, the intensity of ICOS expression on human CAR T cells was highly heterogeneous (Figure 1C), suggesting that the CAR T cell activation status and therefore the sensitivity of our approach might vary between individuals. Such heterogeneity might actually confer additional power to the ICOS-immunoPET strategy, allowing the stratification of patients based on a signal resulting from both CAR T cell expansion and in vivo activation. Moreover, our analysis of human CAR T cell phenotype was only performed on circulating CAR T cells recovered from the peripheral blood and might not reflect ICOS expression on CAR T cells at the tumor site. This may be a critical factor especially for CAR T cell imaging in solid tumors, where the tumor microenvironment might interfere with cell activation. Finally, our preclinical study was not designed to assess the predictive potential of CAR T cell monitoring by ICOS-immunoPET on animal survival given the high rates of tumor control in our mouse tumor model and the fact that all mice received the exact same number of CAR T cells. Clinical studies would be more appropriate to assess the relationship of site-specific tracer uptake with patient outcomes.
In conclusion, we describe for the first time, in vivo monitoring of CAR T cell dynamics using immunoPET targeting an endogenous biomarker, a molecular imaging approach that does not require the addition of reporter genes or ex vivo labelling and that is therefore potentially applicable to the clinical setting for the study of any commercially available and investigational CAR T cell products.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Significance.
ICOS-immunoPET enables in vivo imaging of activated CAR T cells at the tumor site without the need for incorporation of reporter transgenes or ex vivo biolabeling.
Acknowledgments
The authors would like to acknowledge the Stanford Center for Innovation in In-Vivo Imaging (SCi3) and in particular, Dr Frezghi Habte for supporting the preclinical imaging experiments. We are grateful to Rowaid Kellow and Drs. Bin Shen, Gayatri Gowrishankar and Surya Murty for supporting radiochemistry and in vivo imaging studies. We also thank the Stanford shared FACS facility for their support. We would like to dedicate this work to the memory and legacy of Professor Sanjiv Sam Gambhir.
This work was supported in part from funding from the Ben & Catherine Ivy Foundation (SSG), the Canary Foundation (SSG), NCI R01 1 CA201719-02 (SSG), R01 CA23158201 (RSN), P01 CA49605 (SSG and RSN), P5P30CA124435 (CLM) the Parker Institute for Cancer Immunotherapy (SSG, RSN, CLM and ZG), the Geneva University Hospitals Fellowship to FS, the Swiss Cancer League (BIL KLS 3806-02-2016 to FS), the Fondation de Bienfaisance Valeria Rossi di Montelera (Eugenio Litta Fellowship to FS), the American Society for Blood and Marrow Transplantation (New Investigator Award 2018 to FS), the Dubois-Ferrière-Dinu-Lipatti Foundation to FS. The Virginia and D.K. Ludwig Fund for Cancer Research (CLM) and a St Baldrick’s/Stand Up 2 Cancer Pediatric Dream Team Translational Cancer Research Grant (CLM). Stand Up 2 Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. CLM is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program.
Footnotes
Conflict-of-interest disclosures
SSG is the founder and equity holder of CellSight Inc. that develops and translates strategies for imaging cell trafficking/transplantation. CLM holds several patent applications in the area of CAR T cell immunotherapy, is a founder of, holds equity in, and receives consulting fees from Lyell Immunopharma, has received consulting fees from NeoImmune Tech, Nektar Therapeutics and Apricity Health and royalties from Juno Therapeutics for the CD22-CAR. All other authors have declared that no conflict of interest exists.
References
- 1.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med. 2018;379:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med. 2019;25:1341–55. [DOI] [PubMed] [Google Scholar]
- 3.Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130:2317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mueller KT, Waldron E, Grupp SA, Levine JE, Laetsch TW, Pulsipher MA, et al. Clinical Pharmacology of Tisagenlecleucel in B-cell Acute Lymphoblastic Leukemia. Clin Cancer Res. 2018;24:6175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Awasthi R, Pacaud L, Waldron E, Tam CS, Jäger U, Borchmann P, et al. Tisagenlecleucel cellular kinetics, dose, and immunogenicity in relation to clinical factors in relapsed/refractory DLBCL. Blood Adv 2020;4:560–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kochenderfer JN, Somerville RPT, Lu T, Yang JC, Sherry RM, Feldman SA, et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol Ther. 2017;25:2245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keu KV, Witney TH, Yaghoubi S, Rosenberg J, Kurien A, Magnusson R, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krebs S, Ahad A, Carter LM, Eyquem J, Brand C, Bell M, et al. Antibody with Infinite Affinity for In Vivo Tracking of Genetically Engineered Lymphocytes. J Nucl Med. 2018;59:1894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emami-Shahri N, Foster J, Kashani R, Gazinska P, Cook C, Sosabowski J, et al. Clinically compliant spatial and temporal imaging of chimeric antigen receptor T-cells. Nat Commun. 2018;9:1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minn I, Huss DJ, Ahn H-H, Chinn TM, Park A, Jones J, et al. Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci Adv. 2019;5:eaaw5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sellmyer MA, Richman SA, Lohith K, Hou C, Weng C-C, Mach RH, et al. Imaging CAR T Cell Trafficking with eDHFR as a PET Reporter Gene. Mol Ther. 2020;28:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chapelin F, Gao S, Okada H, Weber TG, Messer K, Ahrens ET. Fluorine-19 nuclear magnetic resonance of chimeric antigen receptor T cell biodistribution in murine cancer model. Sci Rep. 2017;7:17748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weist MR, Starr R, Aguilar B, Chea J, Miles JK, Poku E, et al. PET of Adoptively Transferred Chimeric Antigen Receptor T Cells with 89Zr-Oxine. J Nucl Med. 2018;59:1531–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee SH, Soh H, Chung JH, Cho EH, Lee SJ, Ju J-M, et al. Feasibility of real-time in vivo 89Zr-DFO-labeled CAR T-cell trafficking using PET imaging. PLoS ONE. 2020;15:e0223814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei W, Rosenkrans ZT, Liu J, Huang G, Luo Q-Y, Cai W. ImmunoPET: Concept, Design, and Applications. Chem Rev American Chemical Society; 2020;120:3787–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beckford Vera DR, Smith CC, Bixby LM, Glatt DM, Dunn SS, Saito R, et al. Immuno-PET imaging of tumor-infiltrating lymphocytes using zirconium-89 radiolabeled anti-CD3 antibody in immune-competent mice bearing syngeneic tumors. PLoS ONE. 2018;13:e0193832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pektor S, Schlöder J, Klasen B, Bausbacher N, Wagner D-C, Schreckenberger M, et al. Using immuno-PET imaging to monitor kinetics of T cell-mediated inflammation and treatment efficiency in a humanized mouse model for GvHD. Eur J Nucl Med Mol Imaging. 2019; [DOI] [PubMed] [Google Scholar]
- 22.Freise AC, Zettlitz KA, Salazar FB, Lu X, Tavaré R, Wu AM. ImmunoPET Imaging of Murine CD4+ T Cells Using Anti-CD4 Cys-Diabody: Effects of Protein Dose on T Cell Function and Imaging. Mol Imaging Biol. 2017;19:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freise AC, Zettlitz KA, Salazar FB, Tavaré R, Tsai W-TK, Hadjioannou A, et al. ImmunoPET in inflammatory bowel disease: Imaging CD4+ T cells in a murine model of colitis. J Nucl Med. Society of Nuclear Medicine; 2018;jnumed.117.199075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer KE, Mall S, Yusufi N, Gosmann D, Steiger K, Russelli L, et al. T-cell functionality testing is highly relevant to developing novel immuno-tracers monitoring T cells in the context of immunotherapies and revealed CD7 as an attractive target. Theranostics. 2018;8:6070–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tavaré R, Escuin-Ordinas H, Mok S, McCracken MN, Zettlitz KA, Salazar FB, et al. An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy. Cancer Res. 2016;76:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo JW, Tavaré R, Mahakian LM, Silvestrini MT, Tam S, Ingham ES, et al. CD8+ T-Cell Density Imaging with 64Cu-Labeled Cys-Diabody Informs Immunotherapy Protocols. Clin Cancer Res. 2018;24:4976–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alam IS, Mayer AT, Sagiv-Barfi I, Wang K, Vermesh O, Czerwinski DK, et al. Imaging activated T cells predicts response to cancer vaccines. J Clin Invest. 2018;128:2569–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Elssen CHMJ, Rashidian M, Vrbanac V, Wucherpfennig KW, Habre ZE, Sticht J, et al. Noninvasive Imaging of Human Immune Responses in a Human Xenograft Model of Graft-Versus-Host Disease. J Nucl Med. 2017;58:1003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao Z, Mayer AT, Nobashi TW, Gambhir SS. ICOS is an indicator of T cell-mediated response to cancer immunotherapy. Cancer Res. 2020; [DOI] [PubMed] [Google Scholar]
- 30.Alam IS, Simonetta F, Scheller L, Mayer AT, Murty S, Vermesh O, et al. Visualization of activated T cells by OX40-immunoPET as a strategy for diagnosis of acute Graft-versus-Host-Disease. Cancer Res. 2020; canres.1149.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. [DOI] [PubMed] [Google Scholar]
- 32.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Good Z, Spiegel JY, Sahaf B, Malipatlolla MB, Frank MJ, Baird J, et al. Identification of Two CAR T-Cell Populations Associated with Complete Response or Progressive Disease in Adult Lymphoma Patients Treated with Axi-Cel. Blood. American Society of Hematology; 2019;134:779–779. [Google Scholar]
- 34.Jena B, Maiti S, Huls H, Singh H, Lee DA, Champlin RE, et al. Chimeric antigen receptor (CAR)-specific monoclonal antibody to detect CD19-specific T cells in clinical trials. PLoS ONE. 2013;8:e57838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, et al. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. 2005;106:1113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116:3875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin H, Ishii K, Nguyen S, Su PP, Burk CR, Kim B-H, et al. Murine pre-B-cell ALL induces T-cell dysfunction not fully reversed by introduction of a chimeric antigen receptor. Blood. 2018;132:1899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng Z, Chinnasamy N, Morgan RA. Protein L: a novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl Med 2012;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edinger M, Cao Y-A, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101:640–8. [DOI] [PubMed] [Google Scholar]
- 40.Di Gialleonardo V, Signore A, Glaudemans AWJM, Dierckx RAJO, De Vries EFJ. N-(4–18F-fluorobenzoyl)interleukin-2 for PET of human-activated T lymphocytes. J Nucl Med. 2012;53:679–86. [DOI] [PubMed] [Google Scholar]
- 41.Kang L, Jiang D, England CG, Barnhart TE, Yu B, Rosenkrans ZT, et al. ImmunoPET imaging of CD38 in murine lymphoma models using 89Zr-labeled daratumumab. Eur J Nucl Med Mol Imaging. 2018;45:1372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK–modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Freise AC, Wu AM. In vivo imaging with antibodies and engineered fragments. Mol Immunol. 2015;67:142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu R, Carroll L, Yahioglu G, Aboagye EO, Miller PW. Antibody Fragment and Affibody ImmunoPET Imaging Agents: Radiolabelling Strategies and Applications. ChemMedChem. 2018;13:2466–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.