

Abstract
The rates and patterns by which cells acquire mutations profoundly shape their evolutionary trajectories and phenotypic potential. Conventional models maintain that mutations are independently acquired over many successive generations. Yet, recent evidence suggests that cells can also experience mutagenic processes that drive rapid genome evolution. One such process manifests as punctuated bursts of genomic instability, in which multiple new mutations are acquired simultaneously during transient episodes of genomic instability. This mutational mode is reminiscent of the theory of punctuated equilibrium, proposed by Stephen Jay Gould and Niles Eldredge in 1972 to explain the burst-like appearance of new species in the fossil record. In this review, we survey the dominant and emerging theories of eukaryotic genome evolution with a particular focus on the growing body of work that substantiates the existence and importance of punctuated bursts of genomic instability. In addition, we summarize and discuss two recent studies from our own group, the results of which indicate that punctuated bursts systemic genomic instability (SGI) can rapidly reconfigure the structure of the diploid genome of Saccharomyces cerevisiae.
Keywords: Genomic Instability, Aneuploidy, LOH, Mitotic recombination, Punctuated evolution, Genome evolution
Paradigms of evolution in the genomics era
Genomic mutations are essential for the evolution of biological systems. In the Origin of Species, Charles Darwin wrote: ‘As natural selection acts solely by accumulating slight, successive, favorable variations, it can produce no great or sudden modification; it can act only by very short and slow steps’ (Darwin 1859). Drawing from the concept of ‘Natura non facit saltum’ (i.e., ‘Nature makes no leaps’), Darwin portrayed evolution as an iterative machine powered by rare, independently acquired mutations and directed by the pressures of natural selection. Gradualism has become a central tenet of modern biology and remains a dominant lens through which biologists from diverse fields interpret genotypic and phenotypic change. Yet, gradualism fails to explain some evolutionary processes, particularly those that occur at seemly accelerated tempos. Perhaps the best example of this apparent ‘accelerated’ evolution is seen in the development of cancer. By the time they are detected, many cancer cells harbor remarkably complex patterns of chromosomal and karyotypic structural alterations (Alexandrov, et al. 2013, Garraway and Lander 2013, Kandoth, et al. 2013, Lawrence, et al. 2013, Vogelstein, et al. 2013, Zhang and Pellman 2015). Given that the known rates of these classes of mutation are low, it has remained unclear how cancer cells accumulate so many mutations so rapidly by a gradual mode of evolution alone. One alternative model, known as the mutator or hypermutation phenotype, suggests that some cancer cells can proliferate in a constant state of genomic instability, and as a result, acquire new mutations at elevated rates (Fig. 1). The hypermutation phenotype has been observed in cells harboring deleterious mutations which lower the fidelity of DNA polymerases or the activity of DNA repair proteins (Loeb 2016, Loeb, et al. 1974). This accelerated tempo also manifests as the phenotype known as chromosomal instability (CIN), a condition defined by constitutively increased rates of whole chromosome mis-segregation and loss-of-heterozygosity (LOH)(Weaverand Cleveland 2008). Another emerging model, which we refer to as ‘saltational bursts’ or ‘punctuated bursts’, proposes that cancer cells may acquire multiple mutations simultaneously during short-lived episodes of genomic instability (Fig. 1). This model derives from the theory of punctuated equilibrium, originally proposed by Niles Eldredge and Stephen Jay Gould in 1972 (Eldredge and Gould 1972), and describes a biphasic mode of evolution consisting of long periods of genome stability (i.e., stasis) punctuated by short bursts of destabilization during which many genomic changes are rapidly acquired.
Fig. 1.
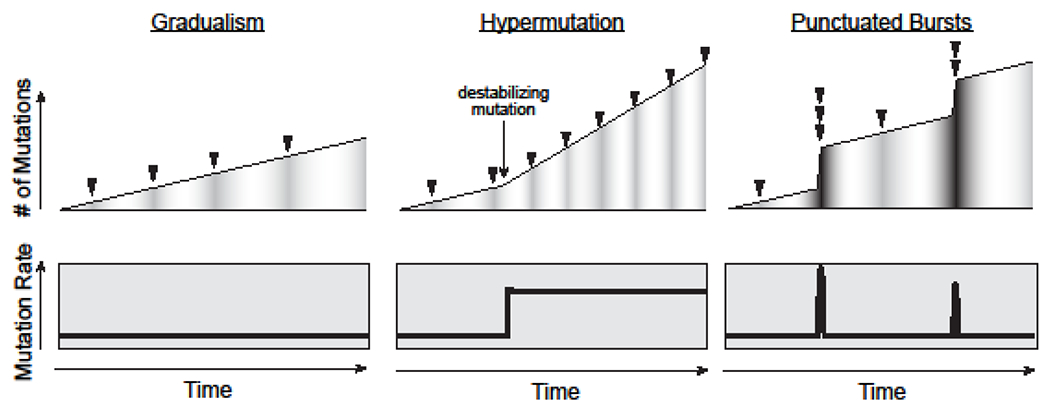
Darwinian gradualism (left) predicts that genomes acquire new mutations (arrowheads) sequentially and independently of one another (top) at linear and constant rates (bottom). The hypermutation model (center) predicts that following acquisition of a destabilizing “mutator” mutation, a genome may become chronically unstable and acquire new mutations independently at an elevated rate. The punctuated bursts model (right) predicts that genomes can experience discrete and transient episodes of systemic genomic instability (SGI) and acquire multiple new mutations rapidly but will resume stable genome propagation for extended periods.
The advent of modern whole genome sequencing (WGS) technologies and bioinformatic analysis has enabled researchers to begin investigating the extent to which these mutational modes actually contribute to the evolution of genomes. Since then, numerous studies have provided compelling data suggesting that genomes can become dramatically restructured by bursts of mutation. Such bursts have been postulated to underlie types of genome evolution ranging from the expansion of gene duplications (Jiang, et al. 2007), to the activity of transposable elements (Oliver and Greene 2009). Additionally, a growing body of work supports the idea that punctuated genome evolution drives clonal expansion of human cancers (Cross, et al. 2016, Markowetz 2016). Indeed, numerous independent studies that analyzed the mutational landscape of breast, melanoma, and colorectal tumors using WGS analysis discovered that these tumors were composed of several sub-clonal populations that likely arose rapidly early in tumor initiation and stably expanded as the tumor grew (Casasent, et al. 2018, Cross, et al. 2018, Field, et al. 2018, Gao, et al. 2016, Gerstung, et al. 2020, Sottoriva, et al. 2015, Stepanenko, et al. 2015). Such population structures indicate that these tumors experienced a punctuated burst of genomic instability and were not driven by a hypermutation phenotype, which would result in a tumor consisting of a highly heterogeneous population of genetically distinct cells. These studies, together with broader surveys of cancer genome evolution indicate that bursts could be a feature common to the development of many cancer types (for an excellent review on the topic, we recommend (Cross, et al. 2016)). Despite these advances, deducing the mutational modes at play in cancer genome evolution remains complicated by the fact that the genome of a tumor sample is typically characterized long after neoplastic initiation. As such, it is difficult to rule out the degrees to which modes like bursts may have contributed to the current mutational landscape of the genome. Thus, although punctuated bursts appear to significantly contribute to the evolution of diverse biological systems, critical outstanding questions remain unanswered: What types of mutations arise in bursts? On what time scale do bursts occur? What molecular and cellular events cause these bursts of genomic instability? How do these events contribute to the phenotypic plasticity, adaptive potential, and long-term evolution of the cell?
Using budding yeast to study the tempos of structural genome evolution
This year, our group published two parallel studies in which we used diploid Saccharomyces cerevisiae cells to define the tempos with which two classes of structural alterations arise in the genome (Heasley, et al. 2020, Sampaio, et al. 2020). In Sampaio et al., 2020 we investigated the patterns by which cells acquire LOH resulting from mitotic recombination, and in Heasley et al., 2020, we investigated the patterns by which cells acquire whole chromosome copy number alterations (CCNAs)(e.g., aneuploidies). Our results, summarized and discussed below, suggested that these large-scale alterations often arise in a burst-like pattern, and that these bursts can rapidly and dramatically alter the structure and content of the diploid genome.
In our studies, we took advantage of several strengths of the budding yeast model system to comprehensively asses the tempos with which LOH and CCNAs arise in the diploid genome: 1) The ability to recover clones harboring specific mutations using counter-selectable selection, 2) The ability to conduct quantitatively rigorous mutational analyses using small clonal populations (≤35 generations), and 3) the ability to inexpensively sequence the genomes of numerous clones with deep coverage. Operating under the conventional Darwinian premise that genomic alterations are acquired independently and gradually over many generations, we predicted that the rate at which a cell acquires two defined mutations (e.g., rateA+B) should be the multiplicative product of the rates at which each individual mutation occurs (rateA+B = rateA x rateB). We constructed diploid strains from which clones harboring defined structural alterations at two distinct loci in the genome could be selected. With these, we grew cells in normal conditions for fewer than 35 generations and used fluctuation analysis to determine the rates at which each individual mutation occurred as well as the rate at which both mutations arose in the same cell. For our analysis of the tempos by which de novo LOH occurs, we inserted hemizygous copies of the counter-selectable markers URA3 and CAN1 at genomic loci on chromosome IV (Chr4), Chr5, and Chr13 to create a suite of strains that could be selected for individual LOH events on Chr4, Chr5, or Chr13, as well as pairs of LOH events on Chr4 and Chr5, and Chr5 and Chr13 by plating cultures to media containing 5-fluoroorotic acid (5-FOA)(Boeke, et al. 1984), canavanine (Larimer, et al. 1978), or a combination of both drugs. With the rates derived from these experiments, we compared the ‘predicted’ rates at which a cell would be expected to acquire both tracts of LOH independently to the ‘observed’ rates at which cells harboring both LOH events actually appeared in a population. Intriguingly, cells harboring two selected tracts of LOH arose at rates 15- to 150-fold higher than predicted by the conventional model of mutation acquisition.
This higher-than-predicted incidence of double mutants was even more pronounced in our parallel study which examined patterns of coincident aneuploidization (Heasley, et al. 2020). We engineered diploid strains with two copies each of either the URA3, CAN1, or TRP1 markers inserted on either side of the centromere of a specific chromosome such that we could select for different combinations of aneuploidies (loss of: Chr1 and Chr3, Chr1 and Chr5, Chr3 and Chr5, Chr9 and Chr5, Chr12 and Chr5). We found that aneuploidies of each individual chromosome occurred at rates ranging between 1.2x10−6-2.1x10−8 events/cell/generation. Per Darwinian principles, cells harboring any of the above pairs of aneuploidies would be predicted to arise at the exceedingly low rates of 10−12–10−14 events/cell/division. Remarkably, and in contrast to this prediction, we recovered cells harboring two aneuploidies at rates 600-fold to 3800-fold higher than expected. Together, results from our quantitative approach indicated that cells harboring multiple structural mutations arose more often than could be explained by a gradual model of genome evolution.
We independently validated the quantitative results of the selection assays described above by sequencing the genomes of derivative clones harboring one or two selected structural mutations. Using WGS analysis, we were able to descriptively assess the structure of these genomes, as well as to detect unselected mutations that co-occurred with the primary selected events. From this analysis, we found that unselected mutations frequently accompanied a primary selected event (Fig. 2). Whereas control clones isolated without any selection were free of structural alterations, 15.5% of clones selected for LOH possessed additional unselected tracts of LOH elsewhere in the genome and 44% of clones selected for aneuploidy harbored additional unselected aneuploidies. The read coverage depths deduced from our WGS analysis demonstrated that the majority of these unselected mutations had not accumulated after selection of the primary mutation and instead suggested that they arose during the same temporally restricted episode of genomic instability that had resulted in the acquisition of the primary selected mutation(s). Moreover, this result also indicated that, rather than showing signs of continued instability (i.e., a mutator phenotype), most clones had stably propagated these newly reconfigured genomes throughout the growth of the colony that formed on the selective media plate.
Fig. 2.
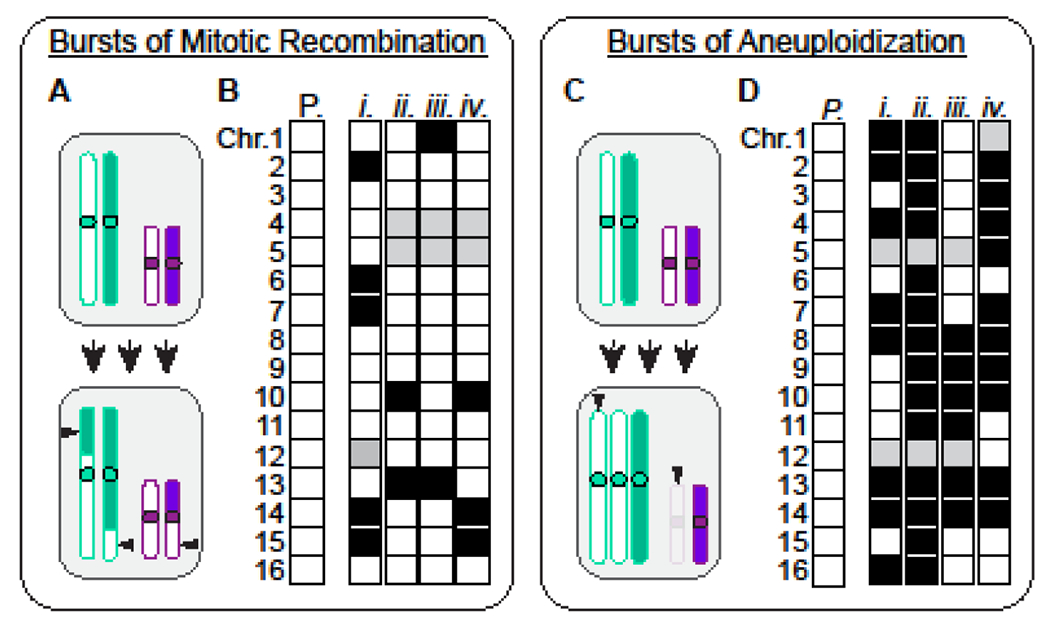
Yeast cells can acquire multiple mutations simultaneously. A) a cartoon illustrating a burst of mitotic recombination leading to multiple new LOH events. Shown are two pairs of homologous chromosomes (upper cell) and a derivative clone harboring numerous tracts of LOH (lower cell, arrowheads). B) Representative schematics of the parental diploid yeast genome and four selected clones that acquired multiple unselected tracts of LOH (Sampaio, et al. 2020). Each square represents both homologs of the denoted chromosome. White squares represent chromosomes that maintained the parental configuration. Grey squares represent the chromosome harboring the selected mutation. Black squares represent chromosomes that had concomitantly acquired an unselected tract of LOH. C) A cartoon illustrating a how a burst of aneuploidization can produce a clone harboring multiple aneuploidies. D) Representative schematics of the parental yeast genome and four selected clones that had acquired multiple unselected aneuploidies(Heasley, et al. 2020). White squares represent chromosomes that maintained the parental configuration (i.e., one copy of each homolog). Grey squares represent the selected chromosome that was lost. Black squares represent chromosome pairs affected by unselected aneuploidy.
Tempos and modes of genome evolution reconsidered
Together with the work of others (Forche, et al. 2011, Forche, et al. 2018, Hickman, et al. 2015), our studies demonstrate that cells can gain multiple mutations non-independently during very short-lived episodes of systemic genomic instability (SGI). In contrast with the principles of gradualism, it appears that sometimes, nature does make leaps. How do we reconcile this finding with established models of mutagenesis and genomic instability? Reports documenting the coincident acquisition of disparate mutations in the yeast genome date back nearly 60 years (Fogel and Hurst 1963, Freeman and Hoffmann 2007, Golin and Esposito 1984, Golin and Tampe 1988, Wood 1982). We suspect that the same fundamental mode of punctuated SGI was responsible for those observations, but could not be fully appreciated and characterized without the benefit of modern whole-genome analysis. Selectable assays, such as the those used in our studies, have been a primary methodology with which to define various metrics of genome stability (e.g., mutation rates)(Klein, et al. 2019, Putnam and Kolodner 2017). Yet, the historic and preferential use of highly homozygous isogenic laboratory strains for such assays has limited our ability to determine how the structure and composition of the entire genome changes when a specific selectable mutation is acquired. Because isogenic strains lack genetic markers throughout in the genome, the selectable mutation was likely the only genomic change detectable in past assays, resulting in the common assumption that it was the only mutation acquired in the genome of the selected cell (Heasley, et al. 2021). In our studies, we used highly heterozygous diploid strains so as to be able to interrogate greater than 25,000 loci distributed across the genome by WGS analysis. In doing so, we were able to detect other unselected and unrelated mutations that arose elsewhere in the genome coincident with a selected mutation.
The remarkably altered genomes characterized in our studies bring to mind the ‘hopeful monsters’ described in Richard Goldschmidt’s theory of macromutation in which he proposed that large-scale mutations (e.g., chromosomal rearrangements) were likely to drive the large-scale evolutionary events that could not be explained by Darwinian models of gradualism (Dietrich 2000, Goldschmidt 1940, Wright 1941). As he wrote in The Material Basis of Evolution ‘A single mutational step affecting the right process at the right moment can accomplish everything…’(Goldschmidt 1940). He used the term ‘hopeful monsters’ to refer to individuals that had acquired such alterations because while most would likely suffer tremendous fitness deficits, a rare ‘monster’ with dramatic genotypic and phenotypic changes might survive and define a new species. Indeed punctuated bursts of SGI appear to be capable of producing a spectrum of ‘hopeful monsters’, a number of which do display new phenotypic variations. While Goldschmidt’s application of this premise to the complex and variable processes underlying speciation was in some ways flawed (Wright 1941), it remains tempting to speculate that bursts of SGI could produce novel phenotypes that would be unlikely to appear by gradualism alone. Moving forward, we are broadening our investigations of punctuated bursts to define how these events impact the phenotypic variation and adaptive potential of the cells which experience them.
What cellular events might cause these punctuated bursts of mutation? While numerous possibilities exist, we speculate that these events may occur when the activity of cellular processes such as DNA damage repair (Craven, et al. 2002), replication (Wilhelm, et al. 2019), sister chromatid cohesion (Covo, et al. 2014, Daum, et al. 2011), spindle assembly (Maiato and Logarinho 2014, Mattiuzzo, et al. 2011), and mitotic checkpoint activity (Musacchio 2015) become transiently perturbed (Ninio 1991, Rosenberg, et al. 1998). For example, because the mitotic checkpoint is a global surveillance system that monitors the attachment of all chromosomes to the mitotic spindle, any stochastic defect in mitotic checkpoint activity renders every chromosome vulnerable to erroneous segregation at anaphase. A logical extension of this systemic vulnerability may be that during the infrequent instances when the mitotic checkpoint does fail, multiple chromosomes can be mis-segregated during a single aberrant division to give rise to the complex karyotypes observed in our study of aneuploidization (Heasley, et al. 2020).
Decades of research have revealed how cellular processes such as those listed above work together to safeguard the integrity of the genome (Putnam and Kolodner 2017). Indeed, it is because of the efficacy of these pathways that cells maintain high-fidelity genome transmission for many generations without acquiring new mutations. However, our results indicate that on the rare occasions when such safeguards briefly falter, cells may experience an episode of SGI and acquire numerous mutations throughout the genome. Does this pattern represent punctuated equilibrium at the most fundamental level? Are the punctuated bursts we observed in our studies simply the result of stochastic failures in the very pathways that maintain prolonged genome stability (i.e., stasis)? Additional work will be required to comprehensively investigate this concept, but if true, then perhaps punctuated equilibrium represents a mutational mode integral to eukaryotic genome evolution.
Acknowledgments
This work was supported by NIH/NIGMS awards 1K99GM13419301 to LRH and R35GM11978801 to JLA.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdes-Mas R, van Buuren MM, van’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR (2013) Signatures of mutational processes in human cancer. Nature 500: 415–421 doi: papers3://publication/doi/10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke JD, LaCroute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Molecular \& general genetics: MGG 197: 345–346 doi: papers3://publication/uuid/042E2D03-B3E9-43FB-BD01-932C628D66CD [DOI] [PubMed] [Google Scholar]
- Casasent AK, Schalck A, Gao R, Sei E, Long A, Pangburn W, Casasent T, Meric-Bernstam F, Edgerton ME, Navin NE (2018) Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 172: 205–217.e212 doi: papers3://publication/doi/10.1016/j.cell.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covo S, Puccia CM, Argueso JL, Gordenin DA, Resnick MA (2014) The sister chromatid cohesion pathway suppresses multiple chromosome gain and chromosome amplification. Genetics 196: 373–384 doi: 10.1534/genetics.113.159202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven RJ, Greenwell PW, Dominska M, Petes TD (2002) Regulation of genome stability by TEL1 and MEC1, yeast homologs of the mammalian ATM and ATR genes. Genetics 161: 493–507 doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross W, Kovac M, Mustonen V, Temko D, Davis H, Baker AM, Biswas S, Arnold R, Chegwidden L, Gatenbee C, Anderson AR, Koelzer VH, Martinez P, Jiang X, Domingo E, Woodcock DJ, Feng Y, Kovacova M, Maughan T, Jansen M, Rodriguez-Justo M, Ashraf S, Guy R, Cunningham C, East JE, Wedge DC, Wang LM, Palles C, Heinimann K, Sottoriva A, Leedham SJ, Graham TA, Tomlinson IPM, Consortium SC (2018) The evolutionary landscape of colorectal tumorigenesis. Nat Ecol Evol 2: 1661–1672 doi: 10.1038/s41559-018-0642-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross WCh, Graham TA, Wright NA (2016) New paradigms in clonal evolution: punctuated equilibrium in cancer. J Pathol 240: 126–136 doi: 10.1002/path.4757 [DOI] [PubMed] [Google Scholar]
- Darwin C 1859 On the origin of species by means of natural selection, or preservation of favoured races in the struggle for life London: : John Murray, 1859, pp. Pages. [Google Scholar]
- Daum JR, Potapova TA, Sivakumar S, Daniel JJ, Flynn JN, Rankin S, Gorbsky GJ (2011) Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Current biology: CB 21: 1018–1024 doi: papers3://publication/doi/10.1016/j.cub.2011.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich MR (2000) From Hopeful Monsters to Homeotic Effects: Richard Goldschmidt’s Integration of Development, Evolution, and Genetics. American Zoologist 40: 738–747 doi: https://doi.ora/10.1093/icb/40.5.738 [Google Scholar]
- Eldredge N, Gould SJ (1972) Punctuated Equilibria: An Alternative to Phyletic Gradualism. In: Schopf TJM (ed) Models of Paleobiology. Freeman, Cooper and Co., San Francisco, San Francisco, pp. 82–115. [Google Scholar]
- Field MG, Durante MA, Anbunathan H, Cai LZ, Decatur CL, Bowcock AM, Kurtenbach S, Harbour JW (2018) Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nature Communications 9: 116 doi: papers3://publication/doi/10.1038/s41467-017-02428-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel S, Hurst DD (1963) Coincidence relations between gene conversion and mitotic recombination in Saccharomyces. Genetics 48: 321–328 doi: papers3://publication/uuid/56DA2E3C-9B69-483D-B96D-DE1E7D408049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Abbey D, Pisithkul T, Weinzierl MA, Ringstrom T, Bruck D, Petersen K, Berman J (2011) Stress alters rates and types of loss of heterozygosity in Candida albicans. mBio 210.1128/mBio.00129-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Cromie G, Gerstein AC, Solis NV, Pisithkul T, Srifa W, Jeffery E, Abbey D, Filler SG, Dudley AM, Berman J (2018) Rapid Phenotypic and Genotypic Diversification After Exposure to the Oral Host Niche in. Genetics 209: 725–741 doi: 10.1534/genetics.118.301019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman KM, Hoffmann GR (2007) Frequencies of mutagen-induced coincident mitotic recombination at unlinked loci in Saccharomyces cerevisiae. Mutation research 616: 119–132 doi: 10.1016/j.mrfmmm.2006.11.014 [DOI] [PubMed] [Google Scholar]
- Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y, Tsai P-C, Casasent A, Waters J, Zhang H, Meric-Bernstam F, Michor F, Navin NE (2016) Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nature Genetics 48: 1119–1130 doi: 10.1038/ng.3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES (2013) Lessons from the cancer genome. Cell 153: 17–37 doi: papers3://publication/doi/10.1016/j.cell.2013.03.002 [DOI] [PubMed] [Google Scholar]
- Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, Mitchell TJ, Rubanova Y, Anur P, Yu K, Tarabichi M, Deshwar A, Wintersinger J, Kleinheinz K, Vázquez-García I, Haase K, Jerman L, Sengupta S, Macintyre G, Malikic S, Donmez N, Livitz DG, Cmero M, Demeulemeester J, Schumacher S, Fan Y, Yao X, Lee J, Schlesner M, Boutros PC, Bowtell DD, Zhu H, Getz G, Imielinski M, Beroukhim R, Sahinalp SC, Ji Y, Peifer M, Markowetz F, Mustonen V, Yuan K, Wang W, Morris QD, Spellman PT, Wedge DC, Van Loo P, Group PEHW, Consortium P (2020) The evolutionary history of 2,658 cancers. Nature 578: 122–128 doi: 10.1038/s41586-019-1907-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt RB 1940. The Material Basis of Evolution Yale University Press, New Haven, pp. Pages. [Google Scholar]
- Golin JE, Esposito MS (1984) Coincident gene conversion during mitosis in saccharomyces. Genetics 107: 355–365 doi: papers3://publication/uuid/709E8FCA-C3CC-4DC8-900E-27C4A9E315A5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golin JE, Tampe H (1988) Coincident recombination during mitosis in saccharomyces: distance-dependent and -independent components. Genetics 119: 541–547 doi: papers3://publication/uuid/3A4C096A-C24B-4D5C-A3EB-95C744C7493F [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasley LR, Sampaio NMV, Argueso JL (2021) Genome-Wide Analysis of Mitotic Recombination in Budding Yeast. Methods Mol Biol 2153: 201–219 doi: 10.1007/978-1-0716-0644-5_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasley LR, Watson RA, Argueso JL (2020) Punctuated Aneuploidization of the Budding Yeast Genome. Genetics 216: 43–50 doi: 10.1534/genetics.120.303536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman MA, Paulson C, Dudley A, Berman J (2015) Parasexual Ploidy Reduction Drives Population Heterogeneity Through Random and Transient Aneuploidy in Candida albicans. Genetics 200: 781–794 doi: 10.1534/genetics.115.178020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE (2007) Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet 39: 1361–1368 doi: 10.1038/ng.2007.9 [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, Wilson RK, Raphael BJ, Ding L (2013) Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339 doi: papers3://publication/doi/10.1038/nature12634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein HL, Bačinskaja G, Che J, Cheblal A, Elango R, Epshtein A, Fitzgerald DM, Gómez-González B, Khan SR, Kumar S, Leland BA, Marie L, Mei Q, Miné-Hattab J, Piotrowska A, Polleys EJ, Putnam CD, Radchenko EA, Saada AA, Sakofsky CJ, Shim EY, Stracy M, Xia J, Yan Z, Yin Y, Aguilera A, Argueso JL, Freudenreich CH, Gasser SM, Gordenin DA, Haber JE, Ira G, Jinks-Robertson S, King MC, Kolodner RD, Kuzminov A, Lambert SA, Lee SE, Miller KM, Mirkin SM, Petes TD, Rosenberg SM, Rothstein R, Symington LS, Zawadzki P, Kim N, Lisby M, Maikova A (2019) Guidelines for DNA recombination and repair studies: Cellular assays of DNA repair pathways. Microb Cell 6: 1–64 doi: 10.15698/mic2019.01.664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer FW, Ramey DW, Lijinsky W, Epler JL (1978) Mutagenicity of methylated N-nitrosopiperidines in Saccharomyces cerevisiae. Mutat Res 57: 155–161 doi: [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortes ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau D-A, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499: 214–218 doi: papers3://publication/doi/10.1038/nature12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA (2016) Human Cancers Express a Mutator Phenotype: Hypothesis, Origin, and Consequences. Cancer research 76: 2057–2059 doi: 10.1158/0008-5472.CAN-16-0794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Springgate CF, Battula N (1974) Errors in DNA replication as a basis of malignant changes. Cancer research 34: 2311–2321 doi: papers3://publication/uuid/162AE0AE-D154-4F95-BB3C-1AC72DD738F2 [PubMed] [Google Scholar]
- Maiato H, Logarinho E (2014) Mitotic spindle multipolarity without centrosome amplification. Nat Cell Biol 16: 386–394 doi: 10.1038/ncb2958 [DOI] [PubMed] [Google Scholar]
- Markowetz F (2016) A saltationist theory of cancer evolution. Nature Genetics 48: 1102–1103 doi: papers3://publication/doi/10.1038/ng.3687 [DOI] [PubMed] [Google Scholar]
- Mattiuzzo M, Vargiu G, Totta P, Fiore M, Ciferri C, Musacchio A, Degrassi F (2011) Abnormal kinetochore-generated pulling forces from expressing a N-terminally modified Hec1. PLoS One 6: e16307 doi: 10.1371/journal.pone.0016307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A (2015) The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Current biology: CB 25: R1002–1018 doi: 10.1016/j.cub.2015.08.051 [DOI] [PubMed] [Google Scholar]
- Ninio J (1991) Transient mutators: a semiquantitative analysis of the influence of translation and transcription errors on mutation rates. Genetics 129: 957–962 doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KR, Greene WK (2009) Transposable elements: powerful facilitators of evolution. Bioessays 31: 703–714 doi: 10.1002/bies.200800219 [DOI] [PubMed] [Google Scholar]
- Putnam CD, Kolodner RD (2017) Pathways and Mechanisms that Prevent Genome Instability in Saccharomyces cerevisiae. Genetics 206: 1187–1225 doi: 10.1534/genetics.112.145805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SM, Thulin C, Harris RS (1998) Transient and heritable mutators in adaptive evolution in the lab and in nature. Genetics 148: 1559–1566 doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio NMV, Ajith VP, Watson RA, Heasley LR, Chakraborty P, Rodrigues-Prause A, Malc EP, Mieczkowski PA, Nishant KT, Argueso JL (2020) Characterization of Systemic Genomic Instability in Budding Yeast. Biorxiv 10.1101/2020.05.25.115535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, Marjoram P, Siegmund K, Press MF, Shibata D, Curtis C (2015) A Big Bang model of human colorectal tumor growth. Nat Genet 47: 209–216 doi: 10.1038/ng.3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanenko A, Andreieva S, Korets K, Mykytenko D, Huleyuk N, Vassetzky Y, Kavsan V (2015) Step-wise and punctuated genome evolution drive phenotype changes of tumor cells. Mutat Res 771: 56–69 doi: 10.1016/j.mrfmmm.2014.12.006 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (2013) Cancer genome landscapes. Science (New York, NY) 339: 1546–1558 doi: 10.1126/science.1235122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW (2008) The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell 14: 431–433 doi: 10.1016/j.ccr.2008.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm T, Olziersky AM, Harry D, De Sousa F, Vassal H, Eskat A, Meraldi P (2019) Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat Commun 10: 3585 doi: 10.1038/s41467-019-11584-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JS (1982) Mitotic chromosome loss induced by methyl benzimidazole-2-yl-carbamate as a rapid mapping method in Saccharomyces cerevisiae. Mol Cell Biol 2: 1080–1087 doi: 10.1128/mcb.2.9.1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S (1941) The Material Basis of Evolution. The Scientific Monthly 53: 165–170 doi: [Google Scholar]
- Zhang C-Z, Pellman D (2015) From Mutational Mechanisms in Single Cells to Mutational Patterns in Cancer Genomes. Cold Spring Harbor Symposia on Quantitative Biology 80: 117–137 doi: 10.1101/sqb.2015.80.027623 [DOI] [PubMed] [Google Scholar]