

Abstract
Neurodegenerative diseases represent some of the most devastating neurological disorders, characterized by progressive loss of the structure and function of neurons. Current therapy for neurodegenerative disorders is limited to symptomatic treatment rather than disease modifying interventions, emphasizing the desperate need for improved approaches. Abundant evidence indicates that impaired mitochondrial function plays a crucial role in pathogenesis of many neurodegenerative diseases and so biochemical factors in mitochondria are considered promising targets for pharmacological-based therapies. Peroxisome proliferator-activated receptors-γ (PPARγ) are ligand-inducible transcription factors involved in regulating various genes including peroxisome proliferator-activated receptor gamma co-activator-1 alpha (PGC1α). This review summarizes the evidence supporting the ability of PPARγ-PGC1α to coordinately up-regulate the expression of genes required for mitochondrial biogenesis in neurons and provide directions for future work to explore the potential benefit of targeting mitochondrial biogenesis in neurodegenerative disorders. We have highlighted key roles of NRF2, uncoupling protein-2 (UCP2), and paraoxonase-2 (PON2) signaling in mediating PGC1α-induced mitochondrial biogenesis. In addition, the status of PPARγ modulators being used in clinical trials for Parkinson’s disease (PD), Alzheimer’s disease (AD) and Huntington’s disease (HD) has been compiled. The overall purpose of this review is to update and critique our understanding of the role of PPARγ-PGC1α-NRF2 in the induction of mitochondrial biogenesis together with suggestions for strategies to target PPARγ-PGC1α-NRF2 signaling in order to combat mitochondrial dysfunction in neurodegenerative disorders.
Keywords: Neurodegenerative disorders, Mitochondrial biogenesis, PPARγ, PGC1α, NRF2, UCP2, PON2
1. Introduction
Neurodegeneration typically refers to steady demise of nerve cells and damage to brain tissue. Neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and others are common neurological disorders that currently affect approximately a hundred million people worldwide (Reddy & Reddy, 2011; Golpich et al., 2017; Magalingam, Radhakrishnan, Ping, & Haleagrahara, 2018). Despite numerous advances in the areas of molecular biology, genetics, and pharmaceutical sciences, there are still no effective drugs to halt or retard neurodegeneration. Due to their high energy requirement, neurons are particularly dependent on mitochondrial function (Cenini, Lloret, & Cascella, 2019; Grimm & Eckert, 2017) and mitochondrial dysfunction has long been recognized as a prominent pathological feature of several neurodegenerative diseases (Chen, Turnbull, & Reeve, 2019; Gao et al., 2017; Golpich et al., 2017; Wu, Chen, & Jiang, 2019). Under physiological conditions, mitochondria produce ATP through oxidative phosphorylation, preserve calcium homeostasis, and control antioxidant defense signaling (Bhatti, Bhatti, & Reddy, 2017; Cenini et al., 2019; Tarasov, Griffiths, & Rutter, 2012) whereas under pathological stress, mitochondria generate excessive reactive oxygen species (ROS) and receive a massive influx of calcium, initiating the formation of the mitochondrial permeability transition pore complex which can lead to cell death (Gao et al., 2017; Tarasov et al., 2012; Wu et al., 2019).
Peroxisome proliferator-activated receptor (PPARs) are ligand-activated transcription factors that belong to the superfamily of nuclear receptors and are involved in regulating various genes and metabolic processes, such as regulation of redox balance and mitochondrial function (Corona & Duchen, 2015, 2016). Recent studies suggest that activation of PPARγ-peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC1α) stimulates mitochondrial biogenesis and induces beneficial effects on mitochondrial function in animal models of neurodegenerative disorders including PD, AD, HD (Aguiar Jr, Bristot, Alves, Cardoso, & Scheffer, 2019; Corona & Duchen, 2015, 2016; Gureev, Shaforostova, & Popov, 2019; Komen & Thorburn, 2014). Therefore, stimulation of mitochondrial biogenesis is considered an innovative approach for the treatment of neurodegenerative disorders.
Mitochondrial biogenesis is a process by which new mitochondria are produced from existing mitochondria (Dominy & Puigserver, 2013; Gureev et al., 2019; Shiota, Traven, & Lithgow, 2015). It is a complex process requiring the coordinated expression and assembly of over 1100 proteins encoded by both the nuclear and mitochondrial genomes (Brown, Murphy, Jornayvaz, & Shulman, 2010; Calvo, Clauser, & Mootha, 2016; Cuperfain, Zhang, Kennedy, & Gonçalves, 2018; Safdar et al., 2018). The mitochondrial genome encodes just 37 proteins, thus the vast majority of mitochondrial protein synthesis is under the control of the nuclear genome (Cuperfain et al., 2018; Gureev et al., 2019). PPARγ co-activator PGC1α is deemed a master regulator of mitochondrial biogenesis by virtue of its ability to augment the expression as well as activity of several critical transcription factors (Aguiar Jr et al., 2019; Gureev et al., 2019; Ye et al., 2017). Notably, PGC1α activates nuclear respiratory factor 2 (NRF2), and subsequently mitochondrial transcription factor A (TFAM) (Gureev et al., 2019; Piantadosi & Suliman, 2012), with activation of this PGC-1α-NRF2-TFAM pathway leading to synthesis of mitochondrial DNA and proteins and eventually generation of new mitochondria (Aguiar Jr et al., 2019; Gureev et al., 2019; Piantadosi & Suliman, 2012). PPARγ-PGC1α expression also mediates mitochondrial uncoupling through the induction of proteins such as uncoupling protein-2 (UCP2). UCP2 is an inner mitochondrial membrane protein that dissipates the mitochondrial membrane potential to uncouple electron transport from ATP synthesis and consequently reduces reactive oxygen species (ROS) production (Aguiar Jr et al., 2019; Hermes et al., 2016; Mehta & Li, 2009; Morrow, Roth, Redmond Jr, Diano, & Elsworth, 2012). UCP2 has also been shown to reduce pro-inflammatory cytokines production, mitochondrial calcium overload and potential apoptotic events (Aguiar Jr et al., 2019; Mehta & Li, 2009). Experimentally-induced reduction in UCP2 expression is associated with mitochondrial dysfunction, ROS accumulation, and elevated cell death; therefore, UCP2 may contribute to the pathogenesis of several diseases including neurodegenerative disorders (Andrews et al., 2005; Andrews, Diano, & Horvath, 2005; Cenini et al., 2019; Ho et al., 2012).
Studies have revealed that activation of the PPARγ-PGC1α axis also induces expression of another protein with potent antioxidant and neuroprotective properties, paraoxonase-2 (PON2) (Camps et al., 2012; Costa, Garrick, Roque, & Pellacani, 2016; Parsanejad et al., 2014). PON2 is highly expressed in brain and is predominately localized to mitochondrial and endoplasmic reticulum membranes (Costa et al., 2014). In fact, evidence indicates that PON2 is critical for properly functioning mitochondria, playing an important role in mitigating oxidative stress (Devarajan et al., 2018; Garrick et al., 2016). This review updates the current status of our knowledge and understanding of the mechanism of PPARγ-PGC1α induced mitochondrial biogenesis in neurodegenerative diseases, with emphasis on AD, PD, HD, and the importance of mitochondrial biogenesis as a potential novel therapeutic target for their treatment. Likewise, we present a concise overview of the key roles of several factors, including NRF2, UCP2, PON2, in regulating PPARγ-PGC1α induced mitochondrial biogenesis (Fig. 1). In addition, we summarize the status of PPARγ modulators in recent clinical trials for neurodegenerative disorders.
Fig. 1.
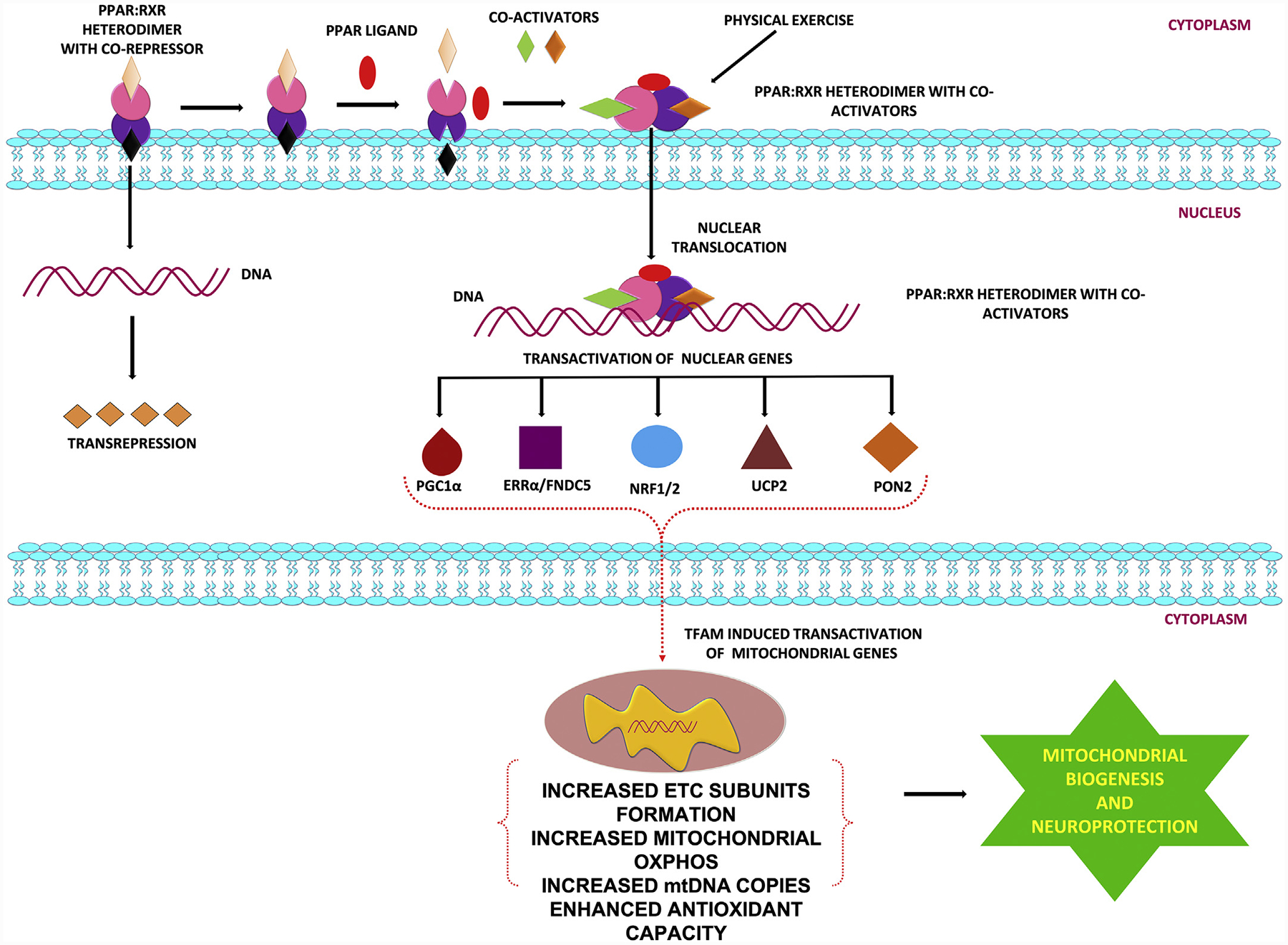
PPARγ signaling and neuroprotection: Role of PGC1α, NRF2, ERR-α, UCP2 and PON-2.
2. PPAR γ: family, expression, activation and modulators
PPARs belong to the superfamily of the nuclear receptors, which regulate gene expression using various ligand-dependent and independent molecular processes (Bordet, Gelé, Duriez, & Fruchart, 2006; Tyagi, Gupta, Saini, Kaushal, & Sharma, 2011; Warden et al., 2016). PPARs play an important role in energy homeostasis, for example by acting as lipid sensors, regulating metabolism in response to dietary lipid intake by directing metabolism and storage of lipids (Tyagi et al., 2011; Varga, Czimmerer, & Nagy, 2011). Three different isoforms of the PPARs exist, which are encoded by separate genes: PPARα (NR1C3), PPARβ/δ (NR1C1), and PPARγ (NR1C2) (Brunmeir & Xu, 2018; Tyagi et al., 2011; Varga et al., 2011). PPARα receptor is expressed in brain, liver, heart and brown adipose tissue, where its main function is to stimulate the breakdown of fatty acids and cholesterol, driving gluconeogenesis and reducing triglyceride levels (Tyagi et al., 2011; Varga et al., 2011). The PPARβ/δ receptor is expressed in heart, brain, liver and skeletal muscles, where it binds and responds to very low density lipoprotein-derived fatty acids, eicosanoids and is involved in fatty acid oxidation (Tyagi et al., 2011; Varga et al., 2011). There are 2 isoforms of PPARγ, PPARγ1 and PPARγ2, which result from alternative splicing (Chang & Ha, 2018; Yun, Han, & Park, 2018). Differences in activity and locations distinguish them; PPARγ2 has stronger transcriptional activity compared with PPARγ1 with expression limited to adipose tissue, whereas PPARγ1 is expressed in many cell types (Tyagi et al., 2011; Varga et al., 2011). Within the CNS, PPARγ is present in several cell types including neurons, astrocytes, oligodendrocytes and microglia (Bernardo & Minghetti, 2008; Corona & Duchen, 2015, 2016; Warden et al., 2016). Particularly high levels of PPARγ have been found in cortex, hippocampus, basal ganglia and in areas expressing dopamine receptors (Corona & Duchen, 2015, 2016). Structurally, PPARγ consists of a ligand-independent transcriptional activation domain, a DNA binding domain, a hinge region for co-factor docking, and a ligand-binding domain (Wang, Xu, Yu, Yang, & Han, 2006; Yun et al., 2018). PPARs can regulate gene transcription either by transactivation that is DNA-dependent, which involves binding to PPAR response elements of target genes, or by transrepression, a mechanism in which PPARs interferes with other transcription factor pathways in a DNA-independent manner (Ricote & Glass, 2007; Varga et al., 2011). PPARγ is activated by small, lipophilic compounds and regulates gene expression by forming a heterodimer with retinoid-X-receptors (RXR) (Tyagi et al., 2011) and binding to the peroxisome proliferator response element (PPRE) located in the promoter region of its target genes (Tyagi et al., 2011; Wang et al., 2006; Yun et al., 2018). Gene transcription also requires recruitment of co-activators, such as PGC1α, to induce conformational changes and release co-repressor molecules that PPARγ is otherwise bound to (Govindarajulu et al., 2018; Muralikumar, Vetrivel, Narayanasamy, & Das, 2017; Ricote & Glass, 2007; Tyagi et al., 2011; Varga et al., 2011; Viswakarma et al., 2010). Thus, the involvement of co-activators and co-repressors makes PPAR-regulated transcriptional activation quite complex. Natural ligands for PPARγ are unsaturated fatty acids, eicosanoids, oxidized phospholipids and nitroalkenes. The prostaglandin, 15-deoxy-delta-12,14-prostaglandin J2 (15d-PGJ2), is the most potent and most commonly used naturally occurring ligand for PPARγ (Grygiel-Górniak, 2014). Synthetic PPARγ agonists include pioglitazone and rosiglitazone, which are currently in clinical use as insulin-sensitizing agents for the treatment of type 2 diabetes (Grygiel-Górniak, 2014).
3. PPARγ and mitochondrial dynamics: fission, fusion and biogenesis
Mitochondria are subcellular organelles containing DNA that are responsible for energy production through oxidative phosphorylation (Roger, Muñoz-Gómez, & Kamikawa, 2017). Each mitochondrion has two membranes and inside the inner compartment (matrix) there are multiple copies of mitochondrial DNA (mtDNA), which are maternally inherited and packed in higher-order nucleoprotein structures called nucleoids (Kasashima, Nagao, & Endo, 2014; Kaufman et al., 2007). Although nucleoids are distributed throughout the mitochondrial matrix, they are preferentially located in proximity of the inner membrane, the location of the oxidative phosphorylation process (Kasashima et al., 2014). Mitochondria respond to changing cellular conditions, with biogenesis (mitogenesis), altered dynamics (fission and fusion), and autophagy (mitophagy), which are all important for proper mitochondrial function, distribution, structure, and movement (Fu, Liu, & Yin, 2019; Seo, Yoon, & Do, 2018). Mitochondrial fusion and fission play significant roles in maintaining mitochondrial function and morphology when cells experience stress (Walczak, Partyka, Duszyński, & Szczepanowska, 2017; Youle & Van Der Bliek, 2012). Fusion helps mitigate stress by mixing the contents of partially damaged mitochondria as a form of complementation (Youle & Van Der Bliek, 2012). Fission is needed to create new mitochondria, but it also contributes to quality control by enabling the removal of damaged mitochondria and can facilitate apoptosis during high levels of cellular stress (Youle & Van Der Bliek, 2012). Disruptions of these processes has been implicated in neurodegenerative diseases, including PD, AD and HD (Bertholet et al., 2016). Mitochondrial fission and fusion processes are both mediated by large guanosine triphosphatases (GTPases) in the dynamin family (Youle & Van Der Bliek, 2012). Mitochondrial fusion results in an elongated and interconnected mitochondrial network by the coordinated merging of the outer and inner mitochondrial membranes under the control of three conserved transmembrane GTPases proteins, mitofusin1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy 1 protein (OPA1) (Fig. 2) (Bertholet et al., 2016; Youle & Van Der Bliek, 2012). Fusion allows exchange of matrix contents and mtDNA molecules between mitochondria, which favors optimal mitochondrial physiology by diluting mutated mtDNA and rescuing damaged mitochondria by the acquisition of key components from healthy mitochondria (Youle & Van Der Bliek, 2012; Zorzano & Claret, 2015). In contrast, fission of preexisting mitochondria generates new mitochondria without mtDNA replication under the control of the master mediator, dynamin related protein Drp1, whose activity is regulated through post-translational modifications and interactions with specific receptor proteins, such as the Fission 1 protein (Youle & Van Der Bliek, 2012; Zorzano & Claret, 2015). Using PC12 cell lines, it has been shown that PGC1α (a key regulator of mitochondrial dynamics) participates in mitochondrial fusion and mitochondrial fission through regulation of Drp1 and Mfn2 proteins (Fig. 2) (Peng et al., 2017).
Fig. 2.
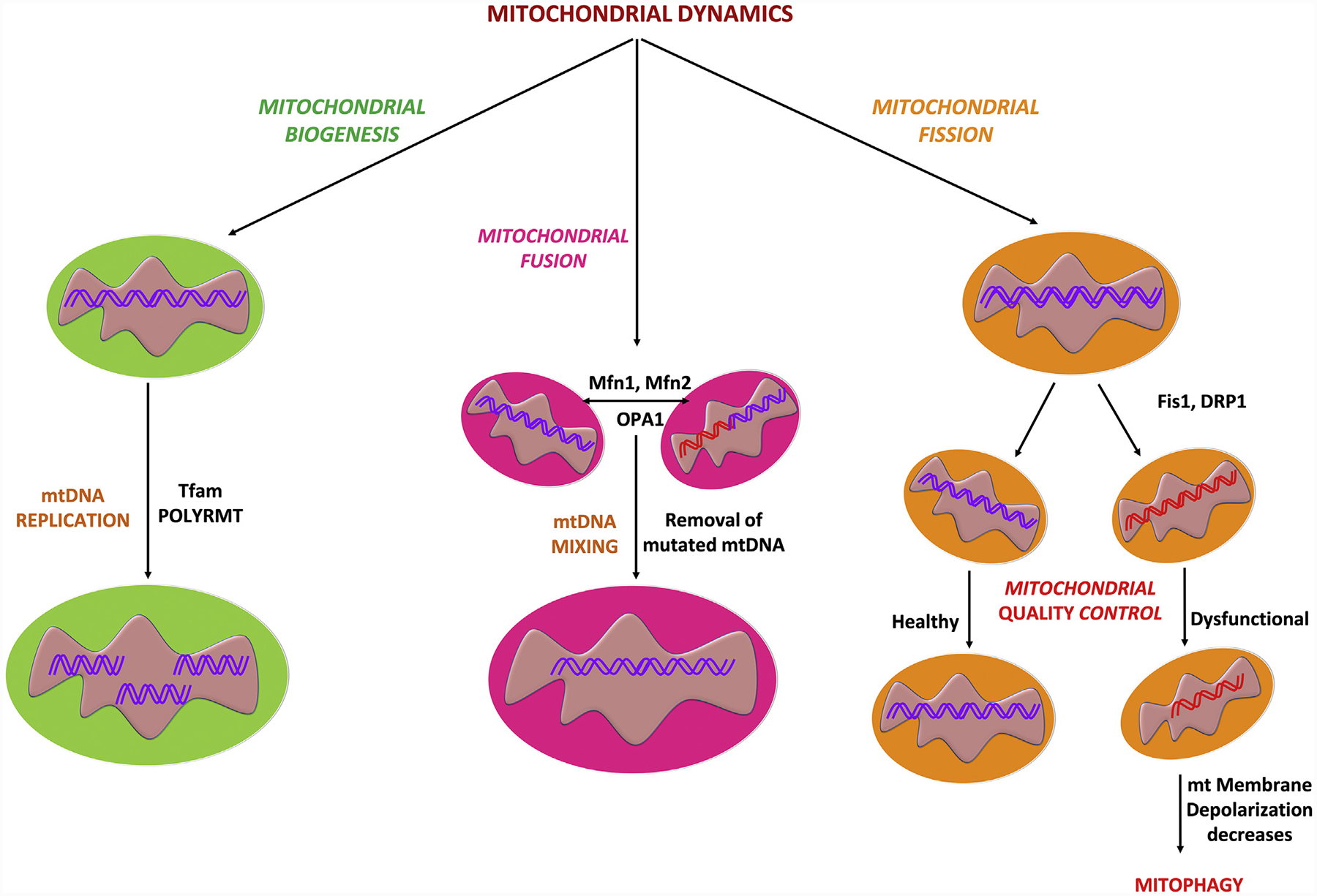
The contribution of biogenesis, fusion, fission and quality control processes to overall mitochondrial dynamics.
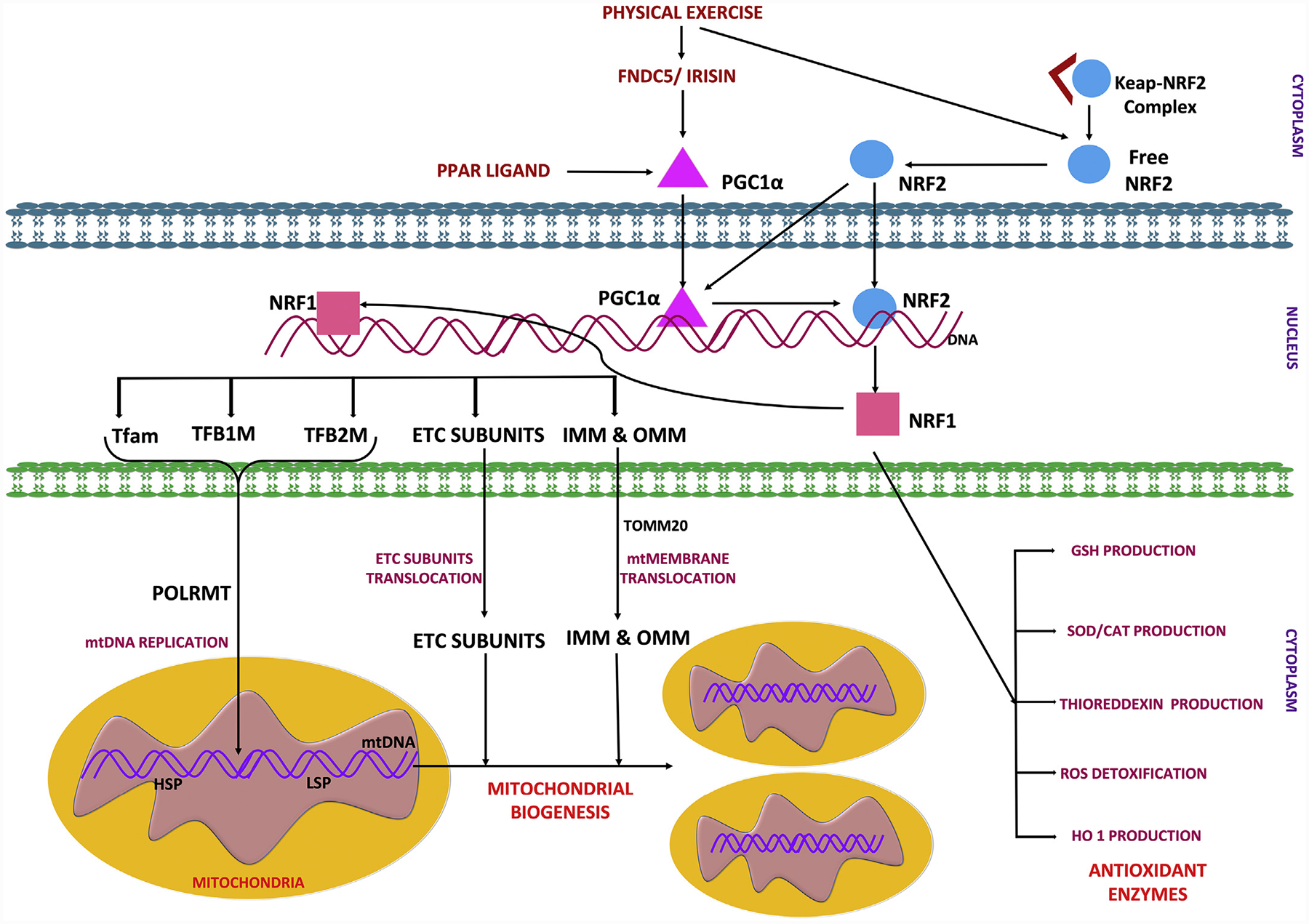
A mitochondrial quality control system is essential to maintain a functional mitochondrial population in neurons and is dependent on ubiquitin proteasome system (UPS) and mitophagy to clear aged and dysfunctional mitochondria (Evans & Holzbaur, 2020; Pickles, Vigié, & Youle, 2018). Mitochondrial biogenesis is a complex process which involves several distinct processes: 1) synthesis of inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM) 2) synthesis of mitochondrial-encoded proteins 3) synthesis and import of nuclear-encoded mitochondrial proteins; and 4) replication of mtDNA (Dhar, Ongwijitwat, & Wong-Riley, 2008; Uittenbogaard & Chiaramello, 2014). Mitochondrial biogenesis requires a coordinated regulation of both the nuclear and mitochondrial genomes, as most mitochondrial proteins are encoded by nuclear genes (Ryan & Hoogenraad, 2007; Sharma & Sampath, 2019). PGC1α stimulates the expression of nuclear-encoded subunits of the mitochondrial electron transport chain via the NRF2 pathway, thereby directly stimulating the mitochondrial bioenergetic output (Scarpulla, 2011). NRF2 helps to maintain cellular redox homeostasis by regulating a number of genes with antioxidant properties including, but not limited to, glutathione, thioredoxin, hemeoxygenase (HO1), and NAD(P)H dehydrogenase (NQO1) (Tonelli, Chio, & Tuveson, 2018; Vomund, Schäfer, Parnham, Brüne, & Von Knethen, 2017). NRF2 is known to positively regulate NRF1 by binding to the four-antioxidant response element promoter sequences of NRF1, leading to activation of the NRF1-mediated mitochondrial biogenesis pathway (Dinkova-Kostova & Abramov, 2015; Gureev et al., 2019; Holmström, Kostov, & Dinkova-Kostova, 2016). Further, NRF1 and NRF2 activate TFAM and bind to promoter regions of nuclear and mitochondrial DNA encoding subunits of mitochondrial respiratory complexes as well as regulating genes involved in heme biosynthesis, the import of nuclear encoded mitochondrial proteins, and mtDNA replication and transcription (Dhar et al., 2008; Dinkova-Kostova & Abramov, 2015; Gureev et al., 2019; Holmström et al., 2016). mtDNA is transcribed by the mitochondrial RNA polymerase (POLRMT) (Shokolenko & Alexeyev, 2017) and key enhancer protein for this process is TFAM, which ensures appropriate mtRNA unwinding and flexing required for the POLRMT binding to the mtDNA promoters (Gureev et al., 2019; Kühl et al., 2016). TFAM binds to mitochondrial promoter sequences, the two heavy chain specific promoters (HSP1 and HSP2) and the light chain-specific promoter (LSP) (Ngo, Kaiser, & Chan, 2018) in a sequence-specific manner to initiate transcription together with the two basal mitochondrial transcription factors, TFB1M & TFB2M, and mitochondrial POLRMT (Fig. 3) (Gureev et al., 2019; Kühl et al., 2016; Ngo et al., 2018).
Fig. 3.
Activation of PPARγ- PGC1α signaling leading to mitochondrial biogenesis.
Several cell-signaling pathways tightly regulate mitochondrial biogenesis. The AMP-activated kinase (AMPK)-PGC1α axis and the sirtuin 1 (SIRT1)-PGC1α axis are two major pathways that regulate mitochondrial biogenesis. AMPK can either directly phosphorylate PGC1α or activate Sirt1 by increasing NAD1 levels (Cantó & Auwerx, 2009; Li, Hou, & Hao, 2017). Upon phosphorylation, PGC1α translocates from the cytoplasm into the nucleus to trigger mitochondrial biogenesis (Dominy & Puigserver, 2013). Various physiological and pharmacological factors can promote mitochondrial biogenesis. For example, caloric restriction, and exercise both stimulate mitochondrial biogenesis (Naoi et al., 2019) by activating AMPK-Sirt1 signaling (Dominy & Puigserver, 2013; Li et al., 2017). In summary, mitochondrial biogenesis plays a critical role in maintaining cell function and promoting cellular functional recovery after injuries. Activating mitochondrial biogenesis is a promising treatment strategy for neurodegenerative disorders.
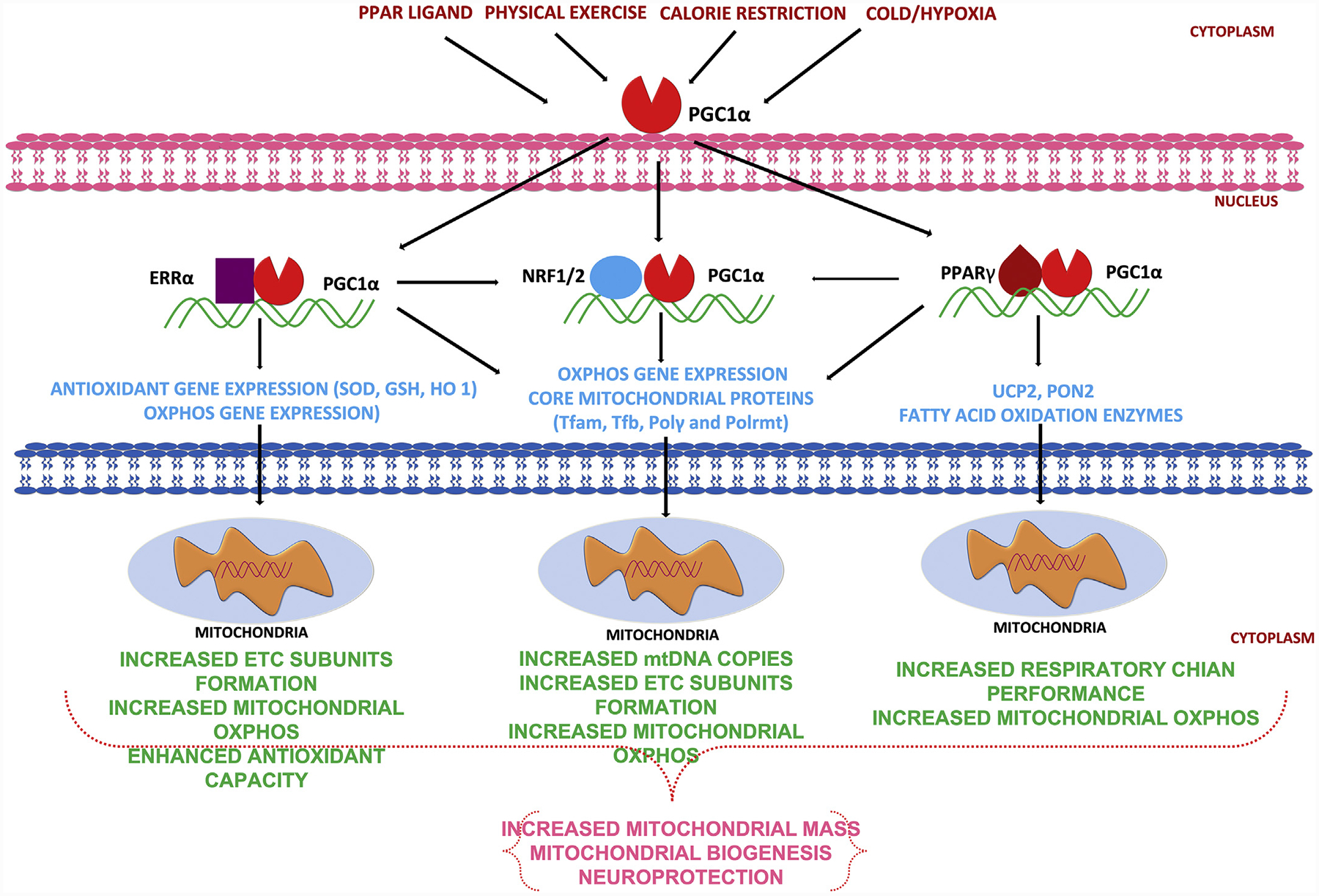
4. PPAR γ and PGC1α signaling: role of NRF2, estrogen related receptor and irisin
As mentioned above, PGC1α is classified as a transcription coactivator and is considered a principal controller of mitochondrial biogenesis that plays a vital role in the regulation of cellular energy metabolism (Austin & St-Pierre, 2012; Brown et al., 2010; Gureev et al., 2019; Schreiber, Knutti, Brogli, Uhlmann, & Kralli, 2003). A transcription co-activator is a protein that raises the possibility of a gene being transcribed by interacting with transcription factors without binding to DNA in a sequence-specific manner (Gureev et al., 2019). The PGC1 family consists of three members, namely PGC1α, PGC1β and the PGC-related co-activator (Austin & St-Pierre, 2012; Gureev et al., 2019). The PGC1 family members have related modular structure and are similarly potent at increasing mitochondrial function when overexpressed. The N-terminal half of PGC1α interacts with many transcription factors, whereas the C-terminal end of PGC1α interacts with the TRAP/DRIP/Mediator complex (Yun et al., 2018). PGC1α is highly expressed in tissues where mitochondria are plentiful and oxidative metabolism is active, such as adipose tissue, heart, skeletal muscle and brain (Cheng, Ku, & Lin, 2018; Gill & La Merrill, 2017). The expression of PGC1α is highly inducible by physiological cues, including exercise, cold temperature and fasting (Fig. 4); however, to date such increases in PGC1α have been reported only in humans and few rodent models (rats and mice), so their generality across other species awaits confirmation (Gureev et al., 2019; Shoag & Arany, 2010; Teng, Li, Stockton, & Foley, 2011; Villena, 2015). Exercise has been reported to have beneficial effects in neurological diseases like PD, AD and HD (Yau, Gil-Mohapel, Christie, & So, 2014). Recent studies have implicated an “exercise-hormone”, FNDC5, and its active or secretory form “irisin” in the neuroprotective effects of exercise (Islam, Young, & Wrann, 2017; Wrann, 2015). FNDC5 is highly expressed in skeletal muscles, heart and nervous tissue. In mammals, FNDC5, the precursor of irisin, is secreted during exercise and promotes thermogenesis (Arhire, Mihalache, & Covasa, 2019; Cao, Zheng, Redfearn, & Yang, 2019). Irisin consists of 112 amino acid peptide that is cleaved from FNDC5 and released into the bloodstream in a PGC1α-dependent manner through a muscle contraction-mediated transcription mechanism (Aguiar Jr et al., 2019; Cao et al., 2019). Although skeletal muscles are the main source of irisin production, it remains unclear whether neuronal irisin is derived from muscles or is produced in neurons (Delezie & Handschin, 2018). In the CNS, FNDC5-irisin regulates central mechanisms that mediate adaptive responses by refining neuronal mitochondrial uncoupling and enhancing the expression of neurotrophins and neuroprotective proteins, such as BDNF, neuronal PAS domain protein 4, cFOS, activity-regulated cytoskeleton-associated protein, and zinc finger protein 268 (ZIF268) (Aguiar Jr et al., 2019). In neurons, PGC1α interacts with estrogen-related receptor alpha (ERRα) to regulate the expression of FNDC5 (Aguiar Jr et al., 2019). Moreover, increased expression of FNDC5 stimulates neuronal development and differentiation in cell cultures as well as in vivo (Aguiar Jr et al., 2019; Wrann, 2015). FNDC5 is widely distributed in brain, being expressed in neurons, astrocytes and microglia (Aguiar Jr et al., 2019; Delezie & Handschin, 2018). Reduced irisin levels have been linked with mood impairment and increased concentrations of irisin in blood have been shown to produce antidepressant-like effects in mice (Aguiar Jr et al., 2019; Korta, Pocheć, & Mazur-Biały, 2019). Brain derived neurotrophic factor (BDNF) is involved in learning and memory processes, and in neuronal differentiation, survival and maintenance (Kowiański et al., 2018) and it is activated by PGC1α. Therefore, activation of the PGC1α-BDNF pathway by irisin may be a key biochemical component underlying the antidepressant-like effects of exercise (Aguiar Jr et al., 2019; Wrann, 2015). In conclusion, FNDC5-irisin can modulate synaptic plasticity as well as mitochondrial biogenesis and represents a highly promising pharmacological target for neurodegenerative and some psychiatric disorders.
Fig. 4.
Induction of mitochondrial biogenesis and neuroprotection in response to PPAR ligands, physical exercise, calorie restriction, cold/hypoxic conditions.
PGC1α needs to interact with a transcription factor to induce neuronal FNDC5 gene expression (Cheng et al., 2018; Wrann et al., 2013) and several clues indicate that the relevant PGC1α binding partner is orphan nuclear estrogen-related receptor alpha (ERRα). ERRs contain DNA-binding domains comprising two highly conserved zinc finger motifs that target the receptor to a specific DNA sequence (TCAAGGTCA) called the estrogen-related response element (ERRE) (Cheng et al., 2018; Wrann et al., 2013). ERRs bind to ERRE as a monomer or a homodimer or as a heterodimer with co-activators (Mohideen-Abdul et al., 2017; Saito & Cui, 2018). Several transcriptional co-activators interacting with ERRs have been identified, which include PGC1α and PGC1β (Saito & Cui, 2018) and the transcriptional activities driven by these interactions have been shown to be essential for mitochondrial biogenesis and cellular energy metabolism (Saito & Cui, 2018). A central function of PGC1α that is intimately linked to mitochondrial biogenesis is the detoxification of ROS (Austin & St-Pierre, 2012; Bhatti et al., 2017). ROS are inevitably generated during mitochondrial respiration, and PGC1α has emerged as a key player controlling their removal by regulating the expression of numerous ROS detoxifying enzymes (Austin & St-Pierre, 2012; Baldelli, Aquilano, & Ciriolo, 2014; Bhatti et al., 2017). Overall, past studies have revealed that PGC1α increases mitochondrial biogenesis in parallel with enhancing cellular ROS-detoxifying capacity, such that cells benefit from increased mitochondrial respiration and ATP production without any oxidative damage (Austin & St-Pierre, 2012; Ježek, Cooper, & Strich, 2018). For these reasons, PGC1α has been suggested to coordinate a ‘clean energy program’, in which the generation of mitochondrial high-energy metabolites (ATP) and removal of toxic derivatives (ROS) are coordinately regulated (Bhatti et al., 2017). In addition, PGC1α also acts as a co-activator for NRF2 and TFAM (Gureev et al., 2019) and this is relevant as NRF2 is a central player in the regulation of cellular defense mechanisms through its anti-inflammatory, antioxidant, detoxification, autophagy, and proteasome actions (Gureev et al., 2019). Furthermore, PGC1α-NRF2 regulates the expression of some ROS detoxifying enzymes, such as SOD1 and 2, catalase and glutathione peroxidase-1 (Corona & Duchen, 2015). In addition, PGC1α-NRF2 induces the expression of nucleus-encoded mitochondrial genes, including TFAM and oxidative phosphorylation (OXPHOS) proteins (Baldelli et al., 2014) (Fig. 4). Therefore, it can be concluded that PPARγ-PGC1α expression improves mitochondrial function, decreases oxidative damage, neuroinflammation, and apoptotic events through the induction of NRF2, FNDC5, Irisin and EREs.
4.1. PPARγ and UCP2
It has long been known that respiration and mitochondrial ATP synthesis are coupled. More recently research has revealed interesting properties exhibited by a class of mitochondrial transporters, known as “uncoupling proteins (UCPs)”, which are located in the inner membrane and control the level of respiration coupling. UCPs belong to the super family of mitochondrial transporter proteins, which share structural and functional similarities (Pierelli et al., 2017; Robbins & Zhao, 2011). A common feature is a tripartite structure, with three repeats of ~100 amino acids, each containing two hydrophobic stretches that correspond to transmembrane alpha helices. Thus, UCPs consist of six alpha-helical regions that span the lipid bilayer. The two transmembrane helices in each repeat are linked by a long hydrophilic loop, which is orientated towards the matrix side of the membrane, and the amino and carboxyl termini extend into the intermembrane space (Pierelli et al., 2017). Although the five members of the UCP family (UCP1–5) share structural similarities, they demonstrate differences in regional expression and function. UCP1 and UCP3 are expressed in peripheral locations: UCP1 (also known as thermogenin/real UCP) is limited to brown adipose tissue, whereas UCP3 is located in heart and skeletal muscle. UCP2 mRNA and protein are found throughout the CNS and has been recognized for its potential to protect mitochondrial function in neurological disorders, which is discussed below (Andrews, Diano, & Horvath, 2005; Andrews, Horvath, et al., 2005; Mehan, Kaur, Dudi, Rajput, & Kalra, 2017; Pierelli et al., 2017). UCP4 and UCP5 have received less attention, but they are expressed at relatively high levels in specific regions of the CNS, such as hypothalamus, hippocampus and cortex (Andrews, Diano, & Horvath, 2005; Andrews, Horvath, et al., 2005; Bouillaud, Alves-Guerra, & Ricquier, 2016; Pierelli et al., 2017). Mechanistically, UCPs function to uncouple mitochondrial oxygen consumption (respiration) from adenosine triphosphate (ATP) synthesis by facilitating proton flux through the inner mitochondrial membrane, which provides an alternative route to partially dissipate the mitochondrial membrane potential that drives ATP synthesis. (Aguiar Jr et al., 2019; Baffy, Derdak, & Robson, 2011). The consequence of this is to reduce ROS, mitochondrial membrane potential and ATP/ADP ratio, and for UCP1 this chemical energy is dissipated in the form of heat (Pierelli et al., 2017; Robbins & Zhao, 2011). While acute mitochondrial decoupling reduces mitochondrial ATP production, prolonged mitochondrial uncoupling promotes an increase in the number of mitochondria and so an overall increased level of ATP production can be achieved (Demine, Renard, & Arnould, 2019). Therefore, UCP activity can enhance mitochondrial function and diminish damaging processes associated with ROS. Indeed, there is evidence to demonstrate that UCP2 serves to protect nervous tissue from multiple sources of acute damage (Hass & Barnstable, 2019; Ho et al., 2012). In brain, the roles of UCP2 in hypothalamus and substantia nigra have received most attention. In hypothalamus it has been established that UCP2 participates in the regulation of mitochondrial fission and glucose homeostasis (Toda et al., 2016). In neurons of the substantia nigra pars compacta (SNpc) activation of UCP2 induces mitochondrial uncoupling and protection of dopaminergic cells from damage inflicted by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a protoxin that induces PD-like pathophysiology (Andrews et al., 2009; Andrews, Diano, & Horvath, 2005; Andrews, Horvath, et al., 2005). An explanation for UCP2’s neuroprotective properties in response to pharmacological and physical insults appears to rely on its important role in preserving mitochondrial function by reducing ROS production, with consequent relief of oxidative stress without compromising the production of ATP (Aguiar Jr et al., 2019; Andrews, Diano, & Horvath, 2005; Andrews, Horvath, et al., 2005; Cadenas, 2018; Kukat et al., 2014). Interestingly, there are reports to indicate that the stimulation of mitochondrial biogenesis and reduction of ROS induced by PPARγ-PGC1α is dependent on induction of UCP2 (de Oliveira Bristot, de Bem Alves, Cardoso, da Luz Scheffer, & Aguiar Jr, 2019; Demine et al., 2019). Furthermore, UCP2 regulation of overall mitochondrial function appears necessary for the beneficial bioenergetic adaptations in response to physical activity (Aguiar Jr et al., 2019; Dietrich, Andrews, & Horvath, 2008; Sreedhar & Zhao, 2017). Therefore, it can be concluded that under certain conditions UCP2-induced mitochondrial uncoupling can be neuroprotective and a target for preventing neurodegeneration.
4.2. PPARγ and PON2
The paraoxonase family has three members: paraoxonase 1 (PON1), paraoxonase 2 (PON2) and paraoxonase 3 (PON3). The name paraoxonase was derived from PON1’s ability to hydrolyze paraoxon, the toxic metabolite of the insecticide parathion (Furlong, Marsillach, Jarvik, & Costa, 2016). PON2 and PON3 do not share this ability to hydrolyze paraoxon; however, all three PONs hydrolyze microbial N-acyl homoserine lactone quorum-sensing factors and can modulate oxidative stress and inflammation (Costa et al., 2014; Costa, Cole, Garrick, Marsillach, & Furlong, 2017; Draganov et al., 2005; Furlong et al., 2016; Levy, Reichert, & Bydlowski, 2019). PON1 and PON3 are predominately expressed in liver, where they are secreted into blood and subsequently associate with high-density lipoproteins. PON2, on the other hand, has been detected at the mRNA and protein level in several tissues such as brain, muscles, lungs, heart and liver. As PON2 has antioxidant properties and is the only paraoxonase found in brain, it has received much attention as target for neuroprotection (Costa et al., 2014; Giordano, Cole, Furlong, & Costa, 2011). Subcellular studies have associated PON2 with intracellular membranes, primarily the inner mitochondrial membrane and endoplasmic reticulum (Levy et al., 2019). In mitochondria, PON2 enhances the function of coenzyme Q, which associates with mitochondrial complex III in the electron transport chain, and subsequently reduces the production of ROS that can lead to oxidative stress. As mitochondria are chief source of ROS, PON2’s location in the mitochondria is consistent with its critical role in protecting cells from oxidative damage (Costa et al., 2014, 2016) and this is emphasized by the finding that PON2 deficiency induces mitochondrial dysfunction (Devarajan et al., 2018; Parsanejad et al., 2014).
In male and female mice, PON2 levels are higher in astrocytes than in neurons, but overall expression is higher in both brain and peripheral tissues of females (Giordano et al., 2011; Giordano et al., 2013). Within mouse brain, PON2 protein is most highly expressed in substantia nigra, striatum, and nucleus accumbens, which are dopamine-rich regions, while lower levels occur in cerebral cortex, cerebellum, hippocampus and brainstem (Giordano et al., 2011; Giordano et al., 2013). As dopaminergic neurons are particularly susceptible to oxidative stress and are preferentially lost in PD (Pacelli et al., 2015), it is possible that the elevated level of PON2 expression in these regions is a coping mechanism to offset the impact of ROS. Indeed, neurons lacking PON2 are hypersensitive to damage inflicted by MPP+, the toxic metabolite of MPTP (Parsanejad et al., 2014). It is not known whether the patterns of preferential cellular and regional distribution observed in the mouse are present in primate brain. However, PON2 expression is highest in the infant brain, in both mice and nonhuman primates (Garrick et al., 2016; Giordano et al., 2011). The higher expression of PON2 in brain early in life in nonhuman primates parallels the resistance at that developmental stage to the parkinsonian protoxin MPTP or methamphetamine at doses that cause extensive damage to dopamine neurons in adult animals (Morrow et al., 2012). As PON2 expression is consistently observed to be higher in females than in males (Giordano et al., 2011; Giordano et al., 2013), the relatively low PON2 levels in dopaminergic neurons in males may provide less defense against oxidative stress and conceivably could be linked with the higher incidence of PD in males as compared to females (Giordano et al., 2013). In cell cultures, it has been demonstrated that rosiglitazone, a synthetic activator of PPARγ, up-regulated the mRNA and protein expression of PON2, while activation of PPARα did not induce PON2 expression (Viladomiu, Hontecillas, Lu, & Bassaganya-Riera, 2013). This finding has been extended recently by the demonstration that pioglitazone administration to mice upregulates PON2 expression in mouse striatum (Blackburn, Curry, Thomsen, Roth, & Elsworth, 2020). Strategies aimed at increasing PON2 expression in brain may therefore be useful in combatting neurodegenerative disorders linked with oxidative stress and neuroinflammation. So far though, relatively little research has been carried out to test this conjecture. Thus, dietary or pharmacological modulation of PON2 may provide new avenues for neuroprotection and indeed PON2 activation may be revealed as a contributor to the neuroprotective properties of established drugs. In summary, PON2 has emerged as a potentially important intracellular defense mechanism against oxidative stress and the neuroprotective benefits linked with PPARγ activators may depend on PON2 expression and stimulation of mitochondrial biogenesis.
5. PPARγ /PGC1α/NRF2 signaling in neurodegenerative disorders
5.1. PPARγ and PD
PD is characterized by the progressive neurodegeneration of dopaminergic neurons of the SNpc, causing tremor, bradykinesia, rigidity and contributing to a deterioration of cognitive function (Maiti, Manna, & Dunbar, 2017). There are extensive data pointing to impaired mitochondrial function and oxidative stress as key mechanisms in the pathophysiology of the disease, most notably implicating dysfunction of mitochondrial complex I (Moon & Paek, 2015; Park, Davis, & Sue, 2018). The role of PGC1α has been strongly implicated in pathogenesis of idiopathic PD (Chen et al., 2019; Poljsak, 2011; Steinbacher & Eckl, 2015; Zheng et al., 2010). Furthermore, there is a link between PGC1α and two genes that are mutated in autosomal recessive Parkinsonism, PINK1 and Parkin. The proteins appear to function in the same pathway with PINK1 acting upstream of Parkin. A defect in the PINK1/Parkin pathway leads to impairment of mitophagy, which is critical for mitochondrial quality control (Ashrafi & Schwarz, 2013; Truban, Hou, Caulfield, Fiesel, & Springer, 2017). Furthermore, both parkin and PINK1 have been implicated in regulating mitochondrial biogenesis (Pickrell & Youle, 2015; Truban et al., 2017). The link between Parkin and PGC1α arises because of the regulation of PGC1α by Parkin-Interacting Substrate (PARIS, also known as zinc finger protein 746, ZNF746). In the absence of Parkin, PARIS is bound to the promoter of PGC1α gene and suppresses its expression, but in the presence of parkin, proteasomal degradation of PARIS is promoted, leading to expression of PGC1α-dependent genes and biogenesis (Castillo-Quan, 2011; Stevens et al., 2015; Zheng et al., 2017). PARIS is highly expressed in the substantia nigra (Zheng et al., 2017) and consistent with the interactions described above, provisional knockout of Parkin in rodents leads to progressive degeneration of dopaminergic neurons that is contingent on PARIS expression (Jiang et al., 2016; Lee et al., 2017; Stevens et al., 2015). Additionally, overexpression of PARIS results in a decrease in PGC1α expression and selective death of dopaminergic neurons in the SNpc, which can be prevented by co-expression of either PARKIN or PGC1α (Dickey & La Spada, 2016; Shin et al., 2011; Zheng et al., 2017). Other studies have shown that transgenic overexpression of PGC1α or activation of PGC1α by resveratrol protects dopaminergic neurons in the MPTP mouse model of PD (Ur Rasheed, Tripathi, Mishra, Shukla, & Singh, 2016). An increased vulnerability to MPTP-induced degeneration of nigral dopaminergic neurons is observed in PGC1α knockout mice, suggesting a critical role of PGC1α in neuroprotection (Jiang et al., 2016). Consistent with this, activation of PGC1α augments the expression of nuclear-encoded subunits of the mitochondrial ETC and rescues the loss of dopaminergic neurons induced by mutant α-synuclein or rotenone administration in animal models (Zhang et al., 2017; Zheng et al., 2010). However, contrary to expectations, an initial study of adenoviral delivery of PGC1α to the nigrostriatal system revealed increased dopaminergic neuronal death in MPTP-treated mice (Clark et al., 2012), demonstrating that viral vector-mediated overexpression of full-length PGC1α in the SNpc does not necessarily protect against neuronal loss and DA depletion. A similar result was observed in another study where AAV2/6 viral vector was used to induce a 400-fold increase in PGC1α mRNA in rat SNpc, which culminated in degeneration of DA neurons (Ciron, Lengacher, Dusonchet, Aebischer, & Schneider, 2012). These negative effects appear to be the result of chronic overexpression of supraphysiological levels of PGC1α, resulting in adverse adaptions such as mitochondrial hyperactivity, excessive proliferation of mitochondria and increased production of ROS. Overall, studies on PGC1α activation or overexpression in PD models have provided inconsistent data. Nevertheless, there is sufficient evidence to suggest a key role of PGC1α and its downstream effectors in PD and offer targets for pharmacological treatment of the disease.
Dysregulation of PPARγ has been linked to the development of neurodegenerative diseases with an inflammatory component such as PD (Golpich et al., 2017). The neuroprotective effect of PPARγ activation has been studied in a number of PD models. Using cultured human neuroblastoma cells, rosiglitazone was protective against mitochondrial dysfunction produced by the complex I inhibitor MPTP by increasing mitochondrial membrane potential, expression of SOD and catalase, as well as expression of Bcl2 and Bax, thus promoting both antioxidant defense and limiting apoptosis (Jung et al., 2007). In another study, pioglitazone preserved the mitochondrial function of oligodendrocyte progenitors from the inhibitory impact of rotenone and tumor necrosis factor α (TNFα), by maintaining mitochondrial membrane potential, reducing mitochondrial ROS production and by increasing the expression of PGC1α, UCP2 and cytochrome oxidase subunit COX1 (De Nuccio et al., 2015). In differentiated neuroblastoma SH-SY5Y cells exposed to rotenone to elicit chronic partial inhibition of complex I, rosiglitazone increased mitochondrial biogenesis, oxygen consumption, mitochondrial mass, mitochondrial membrane potential and mtDNA copy number along with decreased autophagy and free radical generation (Corona, de Souza, & Duchen, 2014). In vivo studies also support a neuroprotective action of PPARγ in PD models. For example, elevated indices of oxidative stress in brains of MPTP-treated rats were attenuated by pioglitazone treatment (Kumar, Kaundal, More, & Sharma, 2009). In addition, in mice treated with MPTP plus probenecid, rosiglitazone was effective in protecting against neuroinflammation, partial degeneration of the SN as well as decline of striatal dopamine (Carta et al., 2011). A novel non-TZD partial PPARγ agonist, LSN862 protects against MPTP-induced neurodegeneration in mice, an effect that was associated with modulation of PPARγ and PGC1α expression, downregulation of neuroinflammation and decreased oxidative stress (Swanson, Du, Johnson, Johnson, & Emborg, 2013). In an important nonhuman primate study, pioglitazone achieved neuroprotection in rhesus monkeys when administered soon after MPTP, as demonstrated by significant behavioral recovery along with preservation of dopaminergic markers and reduced infiltration of the nigrostriatal area by CD68-positive macrophages (Swanson et al., 2011). Interestingly, after administration of pioglitazone to nonhuman primates, dopaminergic neurons that express PPARγ were found more likely to survive MPTP-induced damage (Swanson & Emborg, 2014). PPARγ agonists have been shown to diminish inflammatory cascades in a mouse model of PD by inducing decreases in both microgliosis and astrogliosis (Pisanu et al., 2014). Interestingly, while repeated oral administration of pioglitazone protected against deterioration of motor function and neuronal loss in the acute MPTP mouse model, it failed to protect in a rat model using 6-hydroxydopamine (6-OHDA) as a dopaminergic selective toxin delivered bilaterally to the median forebrain bundle, and this apparent discrepancy was thought to reflect the greater severity of damage caused by 6-OHDA (Laloux, Petrault, Lecointe, Devos, & Bordet, 2012). However, in another study short-term treatment in the unilateral intranigral 6-OHDA rat model, intraperitoneal administration of rosiglitazone did significantly attenuate reduction in TH+ neurons (Lee, Lee, Park, Shin, & Koh, 2012). Thus, there is solid evidence that the beneficial effects of PPARγ agonists in combatting inflammation and promoting mitochondrial biogenesis may be beneficial for treating brain disorders such as PD.
5.2. PPARγ and AD
AD is the most common neurodegenerative disorder, marked by progressive loss of memory and characterized neuropathologically by the increased presence of extraneuronal amyloid plaques derived from aggregation of amyloid beta peptide (Aβ) and intraneuronal neurofibrillary tangles comprising aggregates of hyperphosphorylated tau protein in the brain (Mokhtar, Bakhuraysah, Cram, & Petratos, 2013; Singh, Srivastav, Yadav, Srikrishna, & Perry, 2016). Recent evidence has led many in the field to regard hyperphosphorylated forms of tau as a primary driver of neurodegeneration in AD (Long, Maloney, Rogers, & Lahiri, 2019). However, mitochondrial dysfunction is also an early feature of AD, so it is possible that intervention at the mitochondrial level could improve dysfunction in AD (Golpich et al., 2017; Lejri et al., 2019). This is supported by evidence of Aβ accumulation inside brain mitochondria of patients with AD (Pavlov, Petersen, Glaser, & Ankarcrona, 2009; Swerdlow, 2018). In fact, the level of mitochondrial Aβ in AD appears related to the extent of mitochondrial dysfunction in different regions of brain and to the degree of cognitive impairment (Swerdlow, 2018). Additionally, defective mitochondrial oxidative phosphorylation has been reported in the brain tissue of patients with AD (Desler, Lillenes, Tønjum, & Rasmussen, 2018; Kawamata & Manfredi, 2017). Multiple studies have concluded that mitochondrial dysfunction, particularly deficiencies in mitochondrial respiratory chain complex I, may be a potential unifying factor in pathogenesis of the several neurodegenerative disorders including AD (Johri & Beal, 2012; Kausar, Wang, & Cui, 2018; Lin & Beal, 2006). Notably, hyperphosphorylated forms of tau can selectively impair complex I, leading to increased ROS levels, and resulting in reduced levels of ATP (Cheng & Bai, 2018; Pérez, Jara, & Quintanilla, 2018). Furthermore, reduced activity of mitochondrial enzymes, such as complex III and complex IV activity has been documented in patients with AD and such changes have been linked with excitotoxic cell death (Golpich et al., 2017; Kilbride, Gluchowska, Telford, O’Sullivan, & Davey, 2011). Moreover, altered expression genes encoding proteins necessary for mitochondrial biogenesis such as PGC1α, NRF1, NRF2, and TFAM have been described in some neurodegenerative diseases such as AD (Golpich et al., 2017; Gureev et al., 2019; Qin et al., 2009). Interestingly, several researchers have shown in vitro and in mouse models that expression of PGC1α decreases Aβ generation by reducing β-secretase (BACE1) gene transcription (Wang et al., 2013; Katsouri et al., 2011). However, PGC1α overexpression has been noted to exacerbate Aβ and hyperphosphorylated tau deposition in a mouse model of AD (Dumont et al., 2014), so as in PD models, the level of PGC1α expression and conditions under which it is delivered probably have a substantial impact on the outcome. Aβ oligomers are now generally thought to be the toxic species that initiates downstream AD pathology (Cline, Bicca, Viola, & Klein, 2018), although the pathway linking them to tau protein hyperphosphorylation and NFTs formation is not fully known. Provocation of oxidative stress and mitochondria dysfunction in AD has been emphasized by some investigators and advocated as targets for treatment (Liu et al., 2015; Tönnies & Trushina, 2017). Corona & Duchen, 2016; Golpich et al., 2017; Gureev et al., 2019). Induction of mitochondrial biogenesis by activation of the AMPK-SIRT1 pathway in brain protects against learning impairments and hippocampal degeneration in mouse models of AD; the proposed mechanism for this action is SIRT1-mediated deacetylation of PGC1α, resulting in PGC1α activation with a concomitant reduction in amyloid pathology (Golpich et al., 2017; Wang & Chen, 2016; Xu, Jackson, Khoury, Escobar, & Perez-Pinzon, 2018). Based on the data reviewed, impaired mitochondrial biogenesis has been suggested to be an underlying factor in mitochondrial dysfunction observed in AD and so increasing mitochondrial biogenesis represents a novel pharmacological strategy for AD treatment.
PPARγ has emerged as a drug target for the management of neurological disorders like AD (Cheng et al., 2019; Khan et al., 2019; Omeragic, Hoque, Choi, & Bendayan, 2017). The potential value of a PPARγ agonist in the treatment of AD has been illustrated by several studies employing mouse models of the disease. An early study showed that short-term treatment of APPV717I transgenic mice with pioglitazone or ibuprofen significantly decreased the number of activated microglia and astrocytes, reduced the expression of the proinflammatory enzymes COX2 and iNOS as well as reduced Aβ deposits in the hippocampus and cortex (Heneka et al., 2005). Also, in aged APPV717I transgenic mice, repeated pioglitazone treatment significantly attenuated astroglial activation, reduced signs of oxidative stress, normalized the cerebral blood flow and restored glucose uptake in response to increased neuronal activity, but failed to improve spatial memory (Nicolakakis et al., 2008). In Apolipoprotein E (APOE) knockout mice, rosiglitazone increased mitochondrial biogenesis and improved glucose utilization (Strum et al., 2007). Treatment with rosiglitazone improved cognition in the Tg2576 mice, another transgenic mouse model of AD that presents with accumulation of Aβ, neuronal loss, and cognitive deficits (Rodriguez-Rivera, Denner, & Dineley, 2011). In another study with Tg2576 mice, rosiglitazone treatment enhanced cognitive function and normalized dentate granule cell presynaptic function (Nenov et al., 2014). Using a different model of AD, APPswe/PS1Δe9 mice, treatment with pioglitazone reduced brain levels of soluble and insoluble Aβ levels, which correlated with the loss of both diffuse and dense-core plaques within the cortex (Mandrekar-Colucci, Karlo, & Landreth, 2012). Also, in APPswe/PS1Δe9 mice, the novel PPARγ modulator, DSP-8658, and pioglitazone, improved spatial memory and augmented microglial Aβ phagocytosis, which was followed by a reduction in cortical and hippocampal Aβ levels (Yamanaka et al., 2012). Activation of Wnt signaling by rosiglitazone was posited to be responsible for the drug’s ability to reduce spatial memory impairment and neurodegeneration in brains of APPswe/PS1de9 mice, along with preventing changes in presynaptic and postsynaptic marker proteins (Toledo & Inestrosa, 2010). The mechanism of action of PPARγ agonists relevant to AD has been explored in cell culture studies. Activation by troglitazone and rosiglitazone protected rat hippocampal neurons in culture against toxicity elicited by exposure to synthetic Aβ1–40 peptide, as a result of modulating Wnt signaling components, including an increase of cytoplasmic and nuclear β-catenin levels and inhibition of GSK3β; the latter effect is especially relevant as GSK3β is a major kinase that phosphorylates tau (Inestrosa, Godoy, Quintanilla, Koenig, & Bronfman, 2005). Furthermore, in cultured hippocampal neurons, rosiglitazone protected against mitochondrial damage, oxidative stress and apoptosis induced by synthetic Aβ1–40 peptide, through a mechanism involving elevated expression of Bcl2 (Fuenzalida et al., 2007). Therefore, preclinical research strongly indicates that administration of PPARγ agonists may be beneficial to patients with AD.
5.3. PPARγ and HD
HD is an autosomal dominant neurodegenerative disorder characterized by psychiatric disturbances, cognitive deterioration, and motor impairment (Francelle, Lotz, Outeiro, Brouillet, & Merienne, 2017; Jamwal & Kumar, 2015, 2016). HD is caused by the expansion of cytosine–adenine–guanine (CAG, translated into glutamine) triplet repeats in the huntingtin (Htt) gene and characterized by accumulation of insoluble polyglutamine-containing Htt protein aggregates in affected neurons (Francelle et al., 2017; Jamwal & Kumar, 2015). Mitochondrial dysfunction is strongly associated with the pathogenesis of HD (Golpich et al., 2017; Taherzadeh-Fard et al., 2011). Notably, PGC1α is downregulated in patients with HD (Wenz, 2011; Zheng, Winderickx, Franssens, & Liu, 2018) and in mouse models genetic repression of PGC1α by mutant huntingtin (mHtt) increases HD-like striatal neurodegeneration and motor coordination (Cui et al., 2006; McMeekin et al., 2018), findings that pinpoint a vital contribution of PGC1α-dependent pathways in HD. Thus, deficiency in PGC1α and its downstream genes may lead to mitochondrial dysfunction in HD (Chaturvedi et al., 2009, 2010; Zheng et al., 2018). Likewise, several downstream targets of PGC1α such as NRF1 and TFAM could contribute to the HD mitochondrial dysfunction (Taherzadeh-Fard et al., 2011). Also, several studies revealed that mHtt impairs mitochondrial functions in mHtt-expressing striatal cells (Intihar, Martinez, & Gomez-Pastor, 2019; Jin & Johnson, 2010). Consistent with this, mHtt expression suppresses PPARγ transcriptional activity, which is important for mitochondrial stabilization (Corona & Dunchen, 2016; Golpich et al., 2017). Additionally, there is evidence that mitochondrial ATP levels and ETC activity are reduced in patients with HD (Farshbaf & Ghaedi, 2017; Johri & Beal, 2012). In striatal neurons of patients with late-stage HD, decreased activity of mitochondrial complexes II, III, and IV has been identified (Golpich et al., 2017; Intihar et al., 2019; Jamwal & Kumar, 2015; Reddy & Reddy, 2011). Further, in research using knock-in and HD transgenic mice as well as experimental rodent models of HD, reductions complexes I, II, III, and IV activities were detected in brain tissues, further suggesting that mitochondria deficiency contributes to HD pathogenesis (Golpich et al., 2017; Jamwal & Kumar, 2015, 2016).
There are several publications that discuss the role of PGC1α in HD-related mitochondrial impairment and its potential as a therapeutic target to treat HD. Particularly compelling is the evidence that polymorphisms at the PGC1α gene modify the age of onset of HD symptoms (Weydt, Soyal, Landwehrmeyer, & Patsch, W. European Huntington Disease N., 2014) and also that PGC1α expression is lower in striatal neurons of HD patients and in mouse models (Intihar et al., 2019; Johri, Chandra, & Beal, 2013). Genetic elimination of PGC1α in a mouse model of HD enhances progression, as evaluated by measures of neuronal neurodegeneration and motor coordination, further indicating a likely crucial contribution of PGC1α in HD (Golpich et al., 2017; Intihar et al., 2019; Tsunemi et al., 2012). A generalized loss of PGC1α in mice results in HD-like motor deficits in addition to vacuolation in the cortex and striatum (Lucas et al., 2012). Interestingly though, a recent study suggested that PGC1α loss specifically in the spiny striatal neuron population alone does not fully replicate this HD-like pheno-type, indicating important contributions of PGC1α dysregulation in extra-striatal regions to the signs observed with the unrestricted loss of PGC1α (McMeekin et al., 2018). In moderate-to-severe HD patients a grade-dependent decrease in mitochondria number in striatal spiny neurons has been reported and is associated with decreases in TFAM and PGC1α (Golpich et al., 2017). Moreover, significant decreases in TFAM and PGC1α have been observed in both myoblast cultures and muscle biopsies from patients with HD. Together, these findings robustly pinpoint a decreased expression of PGC1α in HD pathogenesis (Golpich et al., 2017; McMeekin et al., 2018). Interestingly, PGC1α has been demonstrated to promote removal of protein aggregates and Htt turnover by activating transcription factor EB (TFEB) (Tsunemi et al., 2012). TFEB is a major controller of the autophagy–lysosome pathway and alone it can diminish Htt aggregation as well as toxicity, revealing that it is downstream from PGC1α (Tsunemi et al., 2012). Thus, PGC1α and TFEB emerge as viable therapeutic targets in HD and possibly other neurodegenerative disorders that result from protein misfolding (Golpich et al., 2017; Tsunemi et al., 2012).
The protective effects of PPARγ agonists have been studied in several different models of HD. In cultured striatal cells expressing mutant huntingtin, STHdh (Q111), rosiglitazone increased mitochondrial biogenesis and protected against thapsigargin-induced calcium-dependent mitochondrial dysfunction (Quintanilla, Jin, Fuenzalida, Bronfman, & Johnson, 2008). Similarly, in cultured rat primary cortical neurons expressing mutant huntingtin STHdh (Q111), PPARγ activation by rosiglitazone significantly attenuated thapsigargin-induced cell death, concomitant with reduced caspase activation, a delay in the loss of mitochondrial membrane potential, and a reduction of ROS generation (Jin, Hwang, Jo, & Johnson, 2012). In addition, the PPARγ agonist, rosiglitazone rescued the reduction in mitochondrial mass associated with the expression of mutant huntingtin in N2A cells (Chiang, Chern, & Huang, 2012). Rosiglitazone also significantly increased survival in N2A cells expressing mHtt, while also upregulating the expression of PPARγ, PGC1α, NRF1, TFAM and improving mitochondrial function in mutant cells (Chiang et al., 2015). Furthermore, rosiglitazone treatment normalized endoplasmic reticulum stress sensors, Bip, CHOP and ASK1, and reduced mHtt aggregates, which included ubiquitin and heat shock factor 1, and increased the levels of the functional ubiquitin-proteasome system and heat shock protein 70 (Chiang et al., 2015). In a 3-nitropropionic acid (3-NP)-induced experimental model of HD, PPARγ activation by pioglitazone protected striatal cells from mitochondrial dysfunction and oxidative stress and the beneficial effects on mitochondrial dysfunction were attributed to interference with the NFκB signaling pathway, which has been implicated in the pathogenesis of HD (Napolitano et al., 2011). In the R6/2 mouse model of HD, treatment with rosiglitazone rescued the progressive weight loss, motor deterioration, formation of mHtt, as well as expression of BDNF and Bcl2 (Chiang et al., 2010). Also, in the R6/2 mouse model, bezafibrate, improved survival while increasing mitochondrial biogenesis and reducing pathology in brain, muscle and brown adipose tissue (Johri et al., 2011). Treatment with rosiglitazone also reduced huntingtin aggregates and normalized the expression of PGC1α, NRF2, and TFAM in the cortex of transgenic mice R6/2 (Chiang et al., 2012). Likewise, in N171–82Q HD mice, prolonged administration of rosiglitazone considerably improved motor function, increased BDNF levels in cerebral cortex, prevented degeneration of orexin-A-immunopositive neurons in the hypothalamus, averted PGC1α reduction and increased Sirt6 protein levels (Jin et al., 2013). Taken together, these results indicate that activation of pathways promoting mitochondria biogenesis, such as with administration of PPARγ agonists, is a potential valuable strategy for advancing HD treatment.
5.4. PPARγ/PGC1α and other neurodegenerative and neuropsychiatric disorders
While the therapeutic focus of this review is directed towards PD, AD and HD, it is interesting to briefly consider the potential of PPAR activation in other neurodegenerative disorders. Several studies have reported that mitochondrial deficiency plays vital role in ALS, incriminating altered structure, localization and function in patients (Da Cruz et al., 2012a, 2012b; Golpich et al., 2017; Jiang, Wang, Perry, Zhu, & Wang, 2015; Khalil & Liévens, 2017). In ALS, mitochondrial dysfunction in skeletal muscle cells has been linked with a decrease in mRNA and protein content of PGC1α and NRF2 (Da Cruz et al., 2012a, 2012b; Nierenberg et al., 2018; Qi et al., 2015; Russell et al., 2013; Uittenbogaard & Chiaramello, 2014). In a mouse model of ALS, activation of PGC1α has promoted several beneficial effects on muscle (Da Cruz et al., 2012a, 2012b; Nijssen, Comley, & Hedlund, 2017). A practical approach to stimulate PGC1α pathways and improve mitochondrial function is through PPAR activators. Indeed, pioglitazone has been reported to produce neuroprotective effect in the transgenic mouse models of ALS (Kiaei, Kipiani, Chen, Calingasan, & Beal, 2005; Schütz et al., 2005; Shibata et al., 2008). Together, these observations strongly suggest that fine-tuning PPARγ transcriptional activity within the CNS may represent a novel approach to limit the progression of ALS.
Numerous studies have linked Down’s syndrome with deficient mitochondrial function (Arbuzova, Hutchin, & Cuckle, 2002; Izzo et al., 2014, 2018). Notably, the expression of mitochondrial complexes I, III and V are decreased in the brain of DS subjects and, in addition, numerous mtDNA mutations are found in DS brain tissue (Izzo et al., 2018). PGC1α has been suspected as the culprit behind altered mitochondrial mechanisms in DS as it is drastically downregulated in affected subjects (Izzo et al., 2017; Izzo et al., 2017; Izzo et al., 2018). It is now becoming evident that counteracting mitochondrial dysfunction in DS may be possible by targeting the PPARγ-PGC1α axis. Recent in vitro and in vivo studies using models of DS suggest that treatment with drugs such as metformin, resveratrol and epigallocatechin-3-gallate exerts beneficial effects on mitochondrial function (Izzo et al., 2018; Izzo, Nitti, et al., 2017; Valenti et al., 2016). It will be valuable to test other pharmacological activators of PGC1α pathway, including thiazolidinediones, glitazones, and bezafibrates, which selectively stimulate PPARs, for their ability to normalize altered mitochondrial function in DS.
The neuronal cell bodies in both primary and secondary progressive MS display a spectrum of molecular changes such as altered mitochondrial function, transport and bioenergetics (Correale, Gaitán, Ysrraelit, & Fiol, 2016; Correale, Marrodan, & Ysrraelit, 2019; Friese, Schattling, & Fugger, 2014). Molecular changes within neurons in progressive MS have been shown to impact the mitochondrial ETC complexes and consequently impair the ability of the neuron to produce ATP by oxidative phosphorylation (Barcelos, Troxell, & Graves, 2019; Campbell & Mahad, 2018; Fischer et al., 2012; Gonzalo et al., 2019; Mahad et al., 2009). In MS, a significant decrease in PGC1α has been detected at the mRNA and protein levels (Witte et al., 2013) and also a decreased PPARγ expression has been reported in blood mononuclear cells that inversely correlated with disease activity (Ferret-Sena et al., 2016). Therefore, it is reasonable to speculate that PPARγ agonists may have beneficial effects in MS.
In addition to the potential of PPARγ agonists as a treatment for the neurodegenerative disorders reviewed above, research with animal models has predicted that this class of drugs provides a promising therapeutic approach to several neuropsychiatric disorders (e.g., depression, anxiety, mood disorders) and for certain brain injuries (e.g., stroke, ischemia and traumatic brain injury). Mechanisms thought to be responsible for these beneficial actions include their ability to protect mitochondria and exert antioxidant and anti-apoptotic activities. Several investigators using neuropsychiatric models have emphasized that a disease-modifying effect of PPARγ agonists appears to depend on their anti-inflammatory properties (Rolland et al., 2013; Tufano & Pinna, 2020). Furthermore, clinical trials to treat depressive symptoms by targeting PGC1α through administration of PPARγ agonists have produced some encouraging outcomes, such as the use of pioglitazone or rosiglitazone in the treatment of patients with major depressive disorder or bipolar disorder (Tufano & Pinna, 2020). Traditional therapies have typically been designed to target a single pathogenic mechanism and, this may have contributed to their limited effectiveness. A PPARγ agonist can act as a powerful agent for inducing several neuroprotective processes relevant to clinical therapies for neuropsychiatric disorders and brain injuries; this provides a strong rationale for further research on the use of PPARγ agonists as a novel treatment for such conditions.
6. PPARγ modulators and neurodegenerative disorders: Recent update on clinical trials
There is ample preclinical evidence to indicate that PPARγ agonists have the potential to treat mitochondrial dysfunction and energy metabolism defects associated with the pathogenesis of neurodegenerative and neuropsychiatric disorders. PPARγ agonists have been clinically tested in a wide range of health conditions with varying degrees of success. It should be noted that different drugs targeted at PPARs do not always have similar efficacy, safety profiles and clinical outcomes, which sometimes makes it difficult to generalize about their therapeutic value. PPARγ agonists and other experimental pharmacological treatments for neurodegenerative conditions are often tested in the advanced/late stage of disease when interventions are less likely to be effective. PPARγ agonists in clinical trials for neurological and neurodegenerative diseases have yielded mixed results. The conflicting results are indicative of knowledge gaps in our understanding of PPARγ actions in brain together with the role of PPARγ in these diseases. In order to appreciate the momentum in the field, we have summarized all the clinical trials of PPARγ in various neurodegenerative disorders to date (Table 1).
Table 1.
Summary of the clinical trials of PPARγ agonists in various neurodegenerative disorders (AD, PD, ALS and DS)
| Name of Disease | Title of Study | Drug | Clinical Phase (Sample Size) | Findings | Status | Clinical Trial Identifier |
|---|---|---|---|---|---|---|
| AD | Pioglitazone in Alzheimer Disease Progression | Pioglitazone | Phase II (25) | No result was posted for this study. | Completed | NCT00982202 |
| AD | Effects of Pioglitazone or Exercise in Older Adults with Mild Cognitive Impairment and Insulin Resistance: A Pilot Study | Pioglitazone | Phase II (66) | Pioglitazone improved insulin resistance but not cognitive performance in older adults with MCI and insulin resistance. | Completed | NCT00736996 |
| AD | A Double Blind, Randomized, Placebo Controlled, Parallel Group Study to Simultaneously Qualify a Biomarker Algorithm for Prognosis of Risk of Developing Mild Cognitive Impairment Due to Alzheimer’s Disease (MCI Due to AD) and to Test the Safety and Efficacy ofPioglitazone (AD-4833 SR 0.8 mg QD) to Delay the Onset of MCI Due to AD in Cognitively Normal Subjects | Pioglitazone | Phase III (3494) | Terminated (Lack of efficacy of the drug; no safety concern) | Terminated | NCT01931566 |
| AD | A Blinded Long-term Extension Study to Evaluate the Safety and Efficacy of Pioglitazone (AD-4833 Sustained Release 0.8 mg Daily) to Slow the Progression of Cognitive Decline in Subjects Who Have Completed the AD-4833/TOMM40_301 Study with Diagnosis of Mild Cognitive Impairment Due to Alzheimer Disease | Pioglitazone | Phase III (40) | Terminated (Lack of efficacy of the drug; no safety concern) | Terminated | NCT02284906 |
| AD | Effect of Activation of the Receptor PPAR-γ/RXR as a Possible Treatment for Alzheimer’s Disease. Role of Genistein. | Genistein | Interventional Clinical Trial (50) | Changes in Amyloid beta concentration in cerebrospinal fluid (CSF) | On-going | NCT01982578 |
| AD | An Open-label Extension to Study 49653/461, to Assess the Long-term Safety of Rosiglitazone (Extended Release Tablets) in Subjects with Mild to Moderate Alzheimer’s Disease | Rosiglitazone | Phase II (33) | Number of Participants showed Adverse Events (AE’s) leading to permanent discontinuation of study drug or withdrawal were reported. | Completed | NCT00381238 |
| AD | An Open Label Single Oral Dose Study in Patients with Mild Alzheimer’s Disease to Assess the Pharmacokinetics of Extended Release Formulation of Rosiglitazone (RSG XR) in This Population | Rosiglitazone (Extended Release) | Phase I (14) | No Result Posted for this study | Completed | NCT00688207 |
| AD | A Double-blind, Randomized, Placebo-controlled, Parallel-group Study to Investigate the Effects of Rosiglitazone (Extended Release Tablets) on Cerebral Glucose Utilization and Cognition in Subjects with Mild to Moderate Alzheimer’s Disease (AD) | Rosiglitazone | Phase II (80) | Result Not yet Available | Completed | NCT00265148 |
| AD | A 24-week, Double-blind, Double-dummy, Randomized, Parallel-group Study to Investigate the Effects of Rosiglitazone (Extended Release Tablets), Donepezil, and Placebo as Monotherapy on Cognition and Overall Clinical Response in APOE ε4-stratified Subjects with Mild to Moderate Alzheimer’s Disease. (REFLECT-1) | Rosiglitazone (Extended Release) | Phase III (862) | Rosiglitazone (Extended Release Tablets) as Monotherapy in Subjects with Mild to Moderate Alzheimer’s Disease | Completed | NCT00428090 |
| AD | A 54-week, Double-blind, Randomized, Placebo-controlled, Parallel-group Study to Investigate the Effects of Rosiglitazone (Extended Release Tablets) as Adjunctive Therapy to Donepezil on Cognition and Overall Clinical Response in APOE ε4-stratified Subjects with Mild to Moderate Alzheimer’s Disease. | Rosiglitazone (Extended Release) | Interventional Clinical Trial (1496) | Rosiglitazone (Extended Release Tablets) as Adjunctive Therapy for Subjects with Mild to Moderate Alzheimer’s Disease (REFLECT-2) | Completed | NCT00348309 |
| AD | A 54 Week, Double-blind, Randomized, Placebo-controlled, Parallel Group Study to Investigate the Effects of Rosiglitazone (Extended Release Tablets) as Adjunctive Therapy to Acetylcholinesterase Inhibitors on Cognition and Overall Clinical Response in APOE4-stratified Subjects with Mild to Moderate Alzheimer’s Disease | Rosiglitazone (Extended Release) | Phase III (1468) | Rosiglitazone (Extended Release Tablets) As Adjunctive Therapy in Subjects with Mild to Moderate Alzheimer’s Disease (REFLECT-3) | Completed | NCT00348140 |
| AD | A Double-blind, Randomized, Placebo-controlled, Parallel-group Study to Investigate the Effects of Rosiglitazone (Extended Release Tablets) on Cerebral Glucose Utilization and Cognition in Subjects with Mild to Moderate Alzheimer’s Disease | Rosiglitazone (Extended Release) | Phase II (12) | Study Terminated (Slow recruitment) | Terminated | NCT00334568 |
| AD | An Open-label Extension to Study AVA105640, to Assess the Long-term Safety and Efficacy of Rosiglitazone (Extended Release Tablets) on Cognition in Subjects with Mild to Moderate Alzheimer’s Disease | Rosiglitazone (Extended Release) | Phase III (331) | Number of Participants with Serious AEs and Deaths Study Terminated (Based on preliminary parent study results) |
Terminated | NCT00550420 |
| AD | An Open-label Extension to Study AVA102670 and AVA102672, to Assess the Long-term Safety and Efficacy of Rosiglitazone (Extended Release Tablets) as Adjunctive Therapy on Cognition in Subjects with Mild to Moderate Alzheimer’s Disease. | Rosiglitazone (Extended Release) | Phase III (1461) | Number Participants with Serious Adverse Events (SAEs) and Deaths Study Terminated (Based on preliminary parent study results) |
Terminated | NCT00490568 |
| AD | An Open-Label, Randomized, Crossover Study to the Dose Proportionality of RSG XR in Healthy Volunteers in Fasting Conditions | Rosiglitazone (Extended Release) | Phase I (60) | No result posted for this study | Completed | NCT00733785 |
| AD | An Open-Label, Randomized, Two-Period Crossover Study to Demonstrate the Bioequivalence of a Tablet Formulation of Rosiglitazone XR (BRL-049653) 8mg Manufactured at Two Different Sites in Healthy Volunteers in Fasting Conditions | Rosiglitazone (Extended Release) | Phase I (50) | No result posted for this study | Completed | NCT00468897 |
| AD | Insulin, Neurogenetics and Memory in Alzheimer’s Disease: A Novel Therapeutic Approach’ | Rosiglitazone | Phase II | No result posted for this study | Completed | NCT00018382 |
| AD | A Study to Evaluate the Effect of Extended Release Rosiglitazone (RSG XR) on Cardiac Conduction as Compared to Placebo and a Single Oral Dose of Moxifloxacin | Rosiglitazone | Phase I (0) | No result posted for this study | Withdrawn | NCT00884533 |
| AD | A Phase I, Double Blind, Randomized, Two-Way Cross Over, Single- Centre Study in Healthy CYP2D6 Extensive Metabolizers and Poor Metabolizers to Investigate the Potential of AZD3480 to Inhibit Cytochrome P450 1A2, 2C19, 3A4, 2C8, 2B6 and UGT1A1 Activity | AZD3480, Caffeine, Bupropion, Rosiglitazone, Omeprazole, Midazolam, Bilirubin | Phase I (18) | No result posted for this study | Completed | NCT00692510 |
| AD | The Effects of Rosiglitazone on Cognition in Patients With MCI | Rosiglitazone | Phase II (120) | No result posted for this study | Unknown | NCT00242593 |
| PD | A Multi-Center, Double-Blind, Placebo-Controlled Phase II Study of Pioglitazone in Early Parkinson’s Disease | Pioglitazone | Phase II (210) | Pioglitazone at the doses studied here is unlikely to modify progression in early Parkinson’s disease. Further study of pioglitazone in a larger trial in patients with Parkinson’s disease is not recommended | Completed | NCT01280123 |
| ALS | Efficacy, Safety and Tolerability Study of 45 mg Pioglitazone in Patients with Amyotrophic Lateral Sclerosis (ALS) Receiving Standard Therapy (Riluzole) | Pioglitazone | Phase II (219) | No result posted for this study | Terminated | NCT00690118 |
| ALS | Phase IIA Trial: Tretinoin and Pioglitazone HCL Combination Therapy in Amyotrophic Lateral Sclerosis | Pioglitazone and Tretinoin Pioglitazone HCl and Tretinoin |
Phase I & II (28) | No result posted for this study | Completed | NCT00919555 |
| MS | Pilot Test of ACTOS in Multiple Sclerosis: Safety and Tolerability | Pioglitazone | Phase I (30) | No result posted for this study | Completed | NCT00242177 |
| MS | Targeting Residual Activity by Precision, Biomarker-Guided Combination Therapies of Multiple Sclerosis (TRAP-MS) | Pioglitazone Montelukast Losartan Hydroxy-chloroquine |
Phase I & II (250) | On-going study; Still no result posted for this study | Recruiting | NCT03109288 |
7. Knowledge gaps and future directions
Within the scaffold of this article, we have assessed studies on the involvement of mitochondria in neurodegenerative disorders published in the last 2–3 decades. These discoveries constitute a large body of evidence supporting the role of mitochondrial dysfunction in the pathophysiology of various neurodegenerative disorders. Current research suggests that this role is complex and multifaceted, with an interplay of elements involving the mitochondrial respiratory chain, mitochondrial genome, mitophagy/mitochondrial homeostasis and mitochondrial biogenesis. The next challenge is to better characterize these molecular interactions as they are emerging as potential clues for treatment of neurodegenerative disease. Targeting mitochondrial defects may open new avenues for the development of therapies and this concept is supported by numerous preclinical studies. The lack of success in some clinical trials can be attributed to an incomplete understanding of the molecular pathways and targets, a patient population that is too heterogeneous or skewed towards late-stage disease, and the lack of adequate biomarkers to monitor drug efficacy. Therapy should be developed to target pathways more specifically such as mitophagy, mitochondrial biogenesis, fusion/fission and trafficking. Improvements in clinical trial design would be to include more uniform sample populations and especially those with earlier stages of disease. Highly specific and sensitive disease biomarkers would allow detection of even subtle improvements arising from novel drug therapies. New research is emerging at a rapid pace, providing further insights into mitochondrial dysfunction in neurodegenerative disorders, yet despite this progress, there is still much to learn about the precise disease mechanisms involved. Mitochondrial dysfunction clearly plays a fundamental role in neurodegenerative disorders and should be a key theme for future research.
8. Concluding remarks
Mitochondria have crucial roles in cell survival as well as cell death and are able to alter their morphology, number and function in response to stressors and physiological conditions. Altered mitochondrial dynamics as well as mtDNA mutations, gene mutations and impaired transcription contribute to mitochondrial dysfunction that results in or contributes to the pathogenesis of several neurodegenerative disorders. Mitochondria should be considered as a potential target for pharmacological-based therapies as it has become evident that counteracting mitochondrial dysfunction in neurodegenerative disorders may be possible by targeting the PPARγ-PGC1α axis, which includes proteins such as UCP2, PON2 and NRF2. It is somewhat surprising that although there are a range of drugs capable of modulating the activity of PPARγ-PGC1α axis and/or the downstream PPAR proteins, relatively few therapeutic approaches have been so far undertaken to attempt to correct the overall mitochondrial dysfunction in various neurodegenerative disorders using this approach. This review has distilled recent literature relevant to the PPARγ-PGC1α axis control of mitochondrial biogenesis. The key conclusion is that PGC1α activity is mainly controlled by PPARs, AMPKs and SIRT1 and can be stimulated pharmacologically or by non-pharmacological therapies like physical exercise, calorie restriction and acute stress (cold and hypoxia) (Fig. 5). PPARγ agonists exert multiple positive effects on mitochondrial activity in animal models of various neurodegenerative disorders but their effect on mitochondrial biogenesis has not been reported yet. Other pharmacological activators of mitochondrial biogenesis i.e. PGC1α activators, which selectively stimulate PPARγ, should be tested to restore mitochondrial function in neurodegenerative disorders. This will open new therapeutic perspectives to improve the mitochondrial function in neurodegenerative disorders, namely AD, PD, HD and other neurodegenerative disorders.
Fig. 5.
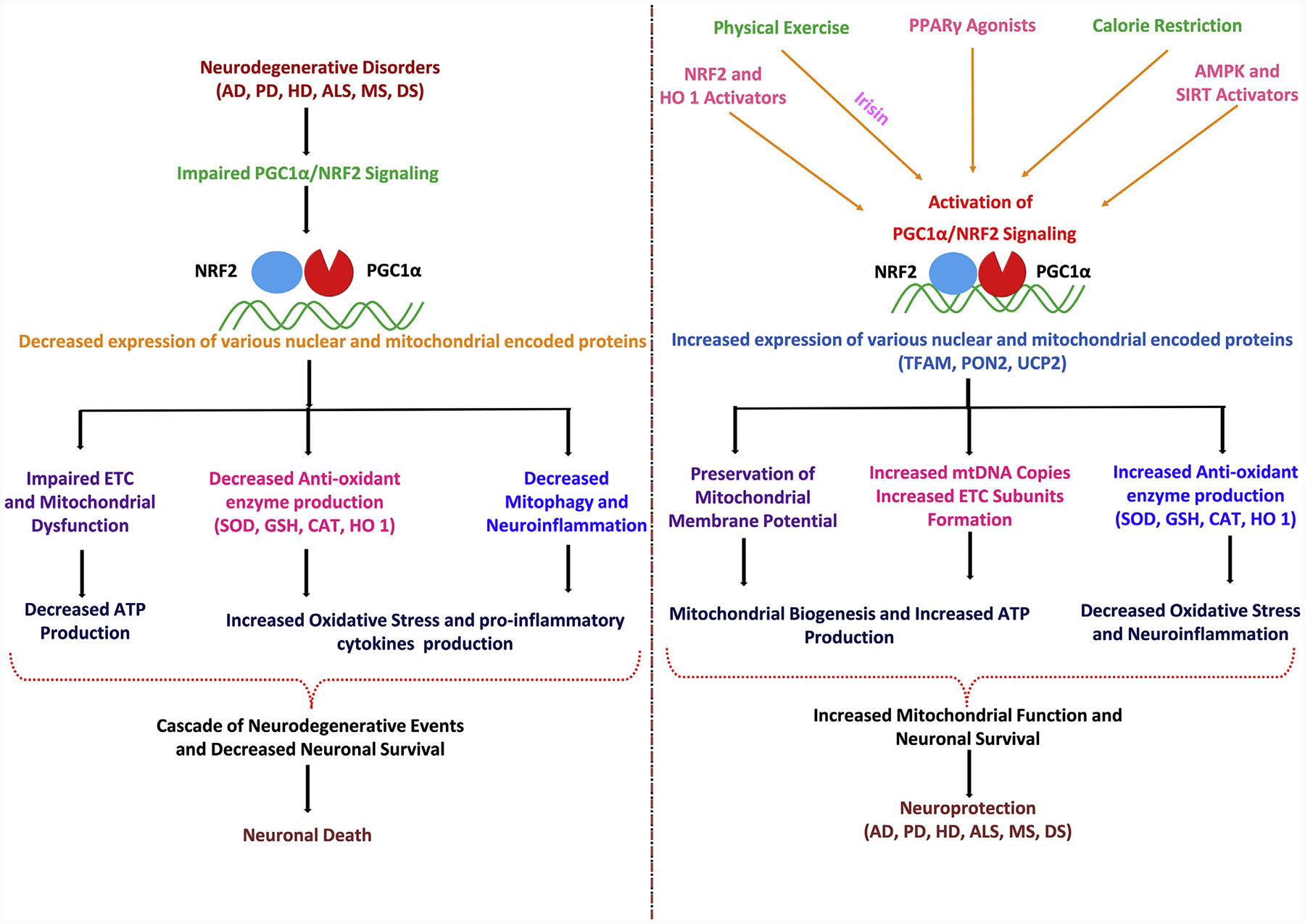
PGC1α-NRF2 pathways involved in neurodegeneration and neuroprotection.
Funding information
Supported by NIH grant AG048918.
Abbreviations:
- 3-NP
3-nitropropionic acid
- 6-OHDA
6-hydroxydopamine
- AD
Alzheimer’s disease
- ALS
Amyotrophic lateral sclerosis
- Aβ
amyloid beta
- AMPK
AMP-activated kinase
- BDNF
brain derived neurotrophic factor
- CNS
central nervous system
- DS
Down syndrome
- ERRα
estrogen-related receptor alpha
- ERRE
estrogen-related response element
- ETC
electron transport chain
- GTPases
guanosine triphosphatases
- HO1
heme oxygenase-1
- HSP1
heavy chain specific promoters
- UCP
uncoupling protein
- HD
Huntington’s disease
- IMM
inner mitochondrial membrane
- LBD
ligand binding domain
- LSP1
light chain-specific promoter
- MS
Multiple sclerosis
- mHtt
mutant huntingtin
- MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MPP+
1-Methyl-4-phenylpyridinium ion
- Mfn1
mitofusin1
- Mfn2
mitofusin 2
- mtDNA
mitochondrial DNA
- NEMGs
nuclear encoded mitochondrial genes
- NQO1 NAD(P)H
quinone oxidoreductase 1
- NRF1
nuclear respiratory factor-1
- NRF2
nuclear respiratory factor-2
- OPA1
optic atrophy 1 protein
- OMM
outer mitochondrial membrane
- PON
paraoxonase
- PD
Parkinson’s disease
- PPARγ
peroxisome proliferator-activated receptor gamma
- PPRE
peroxisome proliferator response element
- PGC1α
peroxisome proliferator-activated receptor gamma coactivator-1 alpha
- PINK1
PTEN-induced kinase 1
- PARKIN
parkin-interacting substrate
- POLRMT
mitochondrial RNA polymerase
- RXR
retinoid-X-receptor
- ROS
reactive oxygen species
- SNpc
substantia nigra pars compacta
- SOD
superoxide dismutase
- SIRT1
sirtuins 1
- TFAM
mitochondrial transcription factor A
- UPS
ubiquitin proteasome system
Footnotes
Declaration of Competing Interest
The authors declare that there are no conflicts of interest.
References
- Aguiar AS Jr., Bristot V, Alves A, Cardoso L, & Scheffer D (2019). The role of PGC1α/UCP2 signaling in the beneficial effects of physical exercise on the brain. Frontiers in Neuroscience 13, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Diano S, & Horvath TL (2005). Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nature Reviews Neuroscience 6, 829. [DOI] [PubMed] [Google Scholar]
- Andrews ZB, Erion D, Beiler R, Liu ZW, Abizaid A, Zigman J, & Roth RH (2009). Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. Journal of Neuroscience 29, 14057–14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Horvath B, Barnstable CJ, Elseworth J, Yang L, Beal MF, … Horvath TL (2005). Uncoupling protein-2 is critical for nigral dopamine cell survival in a mouse model of Parkinson’s disease. Journal of Neuroscience 25, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuzova S, Hutchin T, & Cuckle H (2002). Mitochondrial dysfunction and Down’s syndrome. Bioessays 24, 681–684. [DOI] [PubMed] [Google Scholar]
- Arhire LI, Mihalache L, & Covasa M (2019). Irisin: a hope in understanding and managing obesity and metabolic syndrome. Frontiers in Endocrinology 10, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, & Schwarz TL (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death and Differentiation 20, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin S, & St-Pierre J (2012). PGC1α and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. Journal of Cell Science 125, 4963–4971. [DOI] [PubMed] [Google Scholar]
- Baffy G, Derdak Z, & Robson SC (2011). Mitochondrial recoupling: a novel therapeutic strategy for cancer? British Journal of Cancer 105, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldelli S, Aquilano K, & Ciriolo MR (2014). PGC1α buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death & Disease 5, e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelos IPD, Troxell RM, & Graves JS (2019). Mitochondrial dysfunction and multiple sclerosis. Biology 8, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo A, & Minghetti L (2008). Regulation of Glial Cell Functions by PPAR-γ Natural and Synthetic Agonists. PPAR Research 2008, 864140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertholet AM, Delerue T, Millet AM, Moulis MF, David C, Daloyau M, … Rojo M (2016). Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiology of Disease 90, 3–19. [DOI] [PubMed] [Google Scholar]
- Bhatti JS, Bhatti GK, & Reddy PH (2017). Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochemica et Biophysica Acta (BBA)-Molecular Basis of Disease 1863, 1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn J, Curry D, Thomsen A, Roth R, & Elsworth J (2020). Pioglitazone activates paraoxonase-2 in the brain: a novel neuroprotective mechanism. Experimental Neurology 327, 113324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordet R, Gelé P, Duriez P, & Fruchart JC (2006). PPARs: a new target for neuroprotection. Journal of Neurology, Neurosurgery and Psychiatry 77, 285–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillaud F, Alves-Guerra MC, & Ricquier D (2016). UCPs, at the interface between bioenergetics and metabolism. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1863, 2443–2456. [DOI] [PubMed] [Google Scholar]
- Brown GC, Murphy MP, Jornayvaz FR, & Shulman GI (2010). Regulation of mitochondrial biogenesis. Essays in Biochemistry 47, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmeir R, & Xu F (2018). Functional regulation of PPARs through post-translational modifications. International Journal of Molecular Sciences 19, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas S (2018). Mitochondrial uncoupling, ROS generation and cardioprotection. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1859, 940–950. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, & Mootha VK (2016). MitoCarta2. 0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Research 44, D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell G, & Mahad D (2018). Neurodegeneration in progressive multiple sclerosis. Cold Spring Harbor Perspectives in Medicine 8, a028985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camps J, García-Heredia A, Rull A, Alonso-Villaverde C, Aragones G, Beltrán-Debón R, … Joven J (2012). PPARs in regulation of paraoxonases: control of oxidative stress and inflammation pathways. PPAR Research 2012, 616371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, & Auwerx J (2009). PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current Opinion in Lipidology 20, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao RY, Zheng H, Redfearn D, & Yang J (2019). FNDC5: a novel player in metabolism and metabolic syndrome. Biochimie 158, 111–116. [DOI] [PubMed] [Google Scholar]
- Carta AR, Frau L, Pisanu A, Wardas J, Spiga S, & Carboni E (2011). Rosiglitazone decreases peroxisome proliferator receptor-gamma levels in microglia and inhibits TNF-alpha production: new evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 194, 250–261. [DOI] [PubMed] [Google Scholar]
- Castillo-Quan JI (2011). Parkin’ control: regulation of PGC1α through PARIS in Parkinson’s disease. Disease Models & Mechanisms 4, 427–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenini G, Lloret A, & Cascella R (2019). Oxidative stress in neurodegenerative diseases: from a mitochondrial point of view. Oxidative Medicine and Cellular Longevity 2019, 2105607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JS, & Ha K (2018). A truncated PPAR gamma 2 localizes to mitochondria and regulates mitochondrial respiration in brown adipocytes. PLoS One 13, e0195007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L, … Ciammola A (2009). Impaired PGC1α function in muscle in Huntington’s disease. Human Molecular Genetics 18, 3048–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi RK, Calingasan NY, Yang L, Hennessey T, Johri A, & Beal MF (2010). Impairment of PGC-1alpha expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington’s disease following chronic energy deprivation. Human Molecular Genetics 19, 3190–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Turnbull DM, & Reeve AK (2019). Mitochondrial dysfunction in parkinson’s disease—cause or consequence? Biology 8, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CF, Ku HC, & Lin H (2018). PGC1α as a pivotal factor in lipid and metabolic regulation. International Journal of Molecular Sciences 19, 3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HS, Tan WR, Low ZS, Marvalim C, Lee JYH, & Tan NS (2019). Exploration and development of PPAR modulators in health and disease: an update of clinical evidence. International Journal of Molecular Sciences 20, 5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, & Bai F (2018). The association of tau with mitochondrial dysfunction in alzheimer’s disease. Frontiers in Neuroscience 12, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MC, Chen CM, Lee MR, Chen HW, Chen HM, Wu YS, … Wu YR (2010). Modulation of energy deficiency in Huntington’s disease via activation of the peroxisome proliferator-activated receptor gamma. Human Molecular Genetics 19, 4043–4058. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Cheng YC, Nicol CJ, Lin KH, Yen CH, Chen SJ, & Huang RN (2015). Rosiglitazone activation of PPARγ-dependent signaling is neuroprotective in mutant huntingtin expressing cells. Experimental Cell Research 338, 183–193. [DOI] [PubMed] [Google Scholar]
- Chiang MC, Chern Y, & Huang RN (2012). PPAR gamma rescue of the mitochondrial dysfunction in Huntington’s disease. Neurobiology of Disease 45, 322–328. [DOI] [PubMed] [Google Scholar]
- Ciron C, Lengacher S, Dusonchet J, Aebischer P, & Schneider BL (2012). Sustained expression of PGC1α in the rat nigrostriatal system selectively impairs dopaminergic function. Human Molecular Genetics 21, 1861–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Silvaggi JM, Kiselak T, Zheng K, Clore EL, Dai Y, … Simon DK (2012). PGC1α overexpression downregulates Pitx3 and increases susceptibility to MPTP toxicity associated with decreased BDNF. PLoS One 7, e48925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline EN, Bicca MA, Viola KL, & Klein WL (2018). The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. Journal of Alzheimers Disease 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona JC, de Souza SC, & Duchen MR (2014). PPARγ activation rescues mitochondrial function from inhibition of complex I and loss of PINK1. Experimental Neurology 253, 16–27. [DOI] [PubMed] [Google Scholar]
- Corona JC, & Duchen MR (2015). PPARγ and PGC1α as therapeutic targets in Parkinson’s. Neurochemical Research 40, 308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona JC, & Duchen MR (2016). PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radical Biology and Medicine 100, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correale J, Gaitán MI, Ysrraelit MC, & Fiol MP (2016). Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 140, 527–546. [DOI] [PubMed] [Google Scholar]
- Correale J, Marrodan M, & Ysrraelit MC (2019). Mechanisms of neurodegeneration and axonal dysfunction in progressive multiple sclerosis. Biomedicines 7, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LG, Cole TB, Garrick JM, Marsillach J, & Furlong CE (2017). Metals and paraoxonases In Aschner M, & Costa L (Eds.), Neurotoxicity of Metals. Advances in Neurobiology 18 (pp. 85–111). Switzerland: Springer, Cham. [DOI] [PubMed] [Google Scholar]
- Costa LG, de Laat R, Dao K, Pellacani C, Cole TB, & Furlong CE (2014). Paraoxonase-2 (PON2) in brain and its potential role in neuroprotection. Neurotoxicology 43, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LG, Garrick J, Roque PJ, & Pellacani C (2016). Nutraceuticals in CNS diseases: potential mechanisms of neuroprotection In Gupta RC (Ed.), Nutraceuticals - Efficacy, Safety and Toxicity (pp. 3–13). Cambridge: Academic Press, Elsevier. [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, & Krainc D (2006). Transcriptional repression of PGC1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. [DOI] [PubMed] [Google Scholar]
- Cuperfain AB, Zhang ZL, Kennedy JL, & Gonçalves VF (2018). The complex interaction of mitochondrial genetics and mitochondrial pathways in psychiatric disease. Molecular Neuropsychiatry 4, 52–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, … Williams DS (2012a). Elevated PGC1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metabolism 15, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, … Williams DS (2012b). Elevated PGC1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metabolism 15, 778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nuccio C, Bernardo A, Cruciani C, De Simone R, Visentin S, & Minghetti L (2015). Peroxisome proliferator activated receptor-γ agonists protect oligodendrocyte progenitors against tumor necrosis factor-alpha-induced damage: Effects on mitochondrial functions and differentiation. Experimental Neurology 271, 506–514. [DOI] [PubMed] [Google Scholar]
- Delezie J, & Handschin C (2018). Endocrine crosstalk between skeletal muscle and the brain. Frontiers in Neurology 9, 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demine S, Renard P, & Arnould T (2019). Mitochondrial uncoupling: a key controller of biological processes in physiology and diseases. Cells 8, 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desler C, Lillenes MS, Tønjum T, & Rasmussen LJ (2018). The role of mitochondrial dysfunction in the progression of Alzheimer’s disease. Current Medicinal Chemistry 25, 5578–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devarajan A, Su F, Grijalva V, Yalamanchi M, Yalamanchi A, Gao F, … Srinivasa TR (2018). Paraoxonase 2 overexpression inhibits tumor development in a mouse model of ovarian cancer. Cell Death & Disease 9, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar SS, Ongwijitwat S, & Wong-Riley MT (2008). Nuclear respiratory factor 1 regulates all ten nuclear-encoded subunits of cytochrome c oxidase in neurons. Journal of Biological Chemistry 283, 3120–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey AS, & La Spada AR (2016). Transcription modulation of mitochondrial function and related pathways as a therapeutic opportunity in parkinson’s disease In Buhlman L (Ed.), Mitochondrial mechanisms of degeneration and repair in Parkinson’s disease (pp. 231–253). Cham: Springer. [Google Scholar]
- Dietrich MO, Andrews ZB, & Horvath TL (2008). Exercise-induced synaptogenesis in the hippocampus is dependent on UCP2-regulated mitochondrial adaptation. Journal of Neuroscience 28, 10766–10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, & Abramov AY (2015). The emerging role of Nrf2 in mitochondrial function. Free Radical Biology and Medicine 88, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy JE, & Puigserver P (2013). Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harbor Perspectives in Biology 5, a015008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draganov DI, Teiber JF, Speelman A, Osawa Y, Sunahara R, & La Du BN (2005). Human paraoxonases (PON1, PON2, and PON3) are lactonases with overlapping and distinct substrate specificities. Journal of Lipid Research 46, 1239–1247. [DOI] [PubMed] [Google Scholar]
- Dumont M, Stack C, Elipenahli C, Jainuddin S, Launay N, Gerges M, … Pujol A (2014). PGC-1α: overexpression exacerbates β-amyloid and tau deposition in a transgenic mouse model of Alzheimer’s disease. The FASEB Journal 28, 1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CS, & Holzbaur EL (2020). Quality Control in Neurons: Mitophagy and Other Selective Autophagy Mechanisms. Journal of Molecular Biology 432, 240–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshbaf MJ, & Ghaedi K (2017). Huntington’s disease and mitochondria. Neurotoxicity Research 32, 518–529. [DOI] [PubMed] [Google Scholar]
- Ferret-Sena V, Maia Silva A, Sena A, Cavaleiro I, Vale J, Derudas B, … Staels B (2016). Natalizumab treatment modulates peroxisome proliferator-activated receptors expression in women with multiple sclerosis. PPAR Research 2016, 5716415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, … Lassmann H (2012). NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135, 886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francelle L, Lotz C, Outeiro T, Brouillet E, & Merienne K (2017). Contribution of neuroepigenetics to Huntington’s disease. Frontiers in Human Neuroscience 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese MA, Schattling B, & Fugger L (2014). Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nature Reviews Neurology 10, 225. [DOI] [PubMed] [Google Scholar]
- Fu W, Liu Y, & Yin H (2019). Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells International 2019, 9757201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, … Bronfman M (2007). Peroxisome proliferator-activated receptor γ up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. Journal of Biological Chemistry 282, 37006–37015. [DOI] [PubMed] [Google Scholar]
- Furlong CE, Marsillach J, Jarvik GP, & Costa LG (2016). Paraoxonases-1,−2 and-3: what are their functions? Chemico-Biological Interactions 259, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Wang L, Liu J, Xie F, Su B, & Wang X (2017). Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants 6, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrick JM, Dao K, de Laat R, Elsworth J, Cole TB, Marsillach J, … Costa LG (2016). Developmental expression of paraoxonase 2. Chemico-Biological Interactions 259, 168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill JA, & La Merrill MA (2017). An emerging role for epigenetic regulation of PGC1α expression in environmentally stimulated brown adipose thermogenesis. Environmental Epigenetics 3, dvx009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Cole TB, Furlong CE, & Costa LG (2011). Paraoxonase 2 (PON2) in the mouse central nervous system: a neuroprotective role? Toxicology and Applied Pharmacology 256, 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Tait L, Furlong CE, Cole TB, Kavanagh TJ, & Costa LG (2013). Gender differences in brain susceptibility to oxidative stress are mediated by levels of paraoxonase-2 expression. Free Radical Biology and Medicine 58, 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, & Ahmadiani A (2017). Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: pathogenesis and treatment. CNS Neuroscience & Therapeutics 23, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo H, Nogueras L, Gil-Sánchez A, Hervás JV, Valcheva P, González-Mingot C, … Pamplona R (2019). Impairment of mitochondrial redox status in peripheral lymphocytes of multiple sclerosis patients. Frontiers in Neuroscience 13, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajulu M, Pinky PD, Bloemer J, Ghanei N, Suppiramaniam V, & Amin R (2018). Signaling Mechanisms of Selective PPARγ Modulators in Alzheimer’s Disease. PPAR Research 2018, 2010675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm A, & Eckert A (2017). Brain aging and neurodegeneration: from a mitochondrial point of view. Journal of Neurochemistry 143, 418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grygiel-Górniak B (2014). Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications-a review. Nutrition Journal 13, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gureev AP, Shaforostova EA, & Popov VN (2019). Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC1α signaling pathways. Frontiers in Genetics 10, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass DT, & Barnstable CJ (2019). Cell Autonomous Neuroprotection by the Mitochondrial Uncoupling Protein 2 in a Mouse Model of Glaucoma. Frontiers in Neuroscience 13, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, … Landreth GE (2005). Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 128, 1442–1453. [DOI] [PubMed] [Google Scholar]
- Hermes G, Nagy D, Waterson M, Zsarnovszky A, Varela L, Hajos M, & Horvath TL (2016). Role of mitochondrial uncoupling protein-2 (UCP2) in higher brain functions, neuronal plasticity and network oscillation. Molecular Metabolism 5, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PW, Ho JW, Liu HF, So DH, Zero HM, Chan KH, … Ho SL (2012). Mitochondrial neuronal uncoupling proteins: a target for potential disease-modification in Parkinson’s disease. Translational Neurodegeneration 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmström KM, Kostov RV, & Dinkova-Kostova AT (2016). The multifaceted role of Nrf2 in mitochondrial function. Current Opinion in Toxicology 1, 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa NC, Godoy JA, Quintanilla RA, Koenig CS, & Bronfman M (2005). Peroxisome proliferator-activated receptor γ is expressed in hippocampal neurons and its activation prevents β-amyloid neurodegeneration: role of Wnt signaling. Experimental Cell Research 304, 91–104. [DOI] [PubMed] [Google Scholar]
- Intihar TA, Martinez EA, & Gomez-Pastor R (2019). Mitochondrial Dysfunction in Huntington’s Disease; Interplay Between HSF1, p53 and PGC1α Transcription Factors. Frontiers in Cellular Neuroscience 13, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MR, Young MF, & Wrann CD (2017). The role of FNDC5/Irisin in the nervous system and as a mediator for beneficial effects of exercise on the brain. hormones, metabolism and the benefits of exercise In Spiegelman B (Ed.), Source hormones, metabolism and the benefits of exercise (pp. 93). Cham: Springer. [Google Scholar]
- Izzo A, Manco R, Bonfiglio F, Calì G, De Cristofaro T, Patergnani S, … Conti A (2014). NRIP1/RIP140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in Down syndrome. Human Molecular Genetics 23, 4406–4419. [DOI] [PubMed] [Google Scholar]
- Izzo A, Manco R, de Cristofaro T, Bonfiglio F, Cicatiello R, Mollo N, et al. (2017). Over-expression of chromosome 21 miRNAs may affect mitochondrial function in the hearts of down syndrome fetuses. International Journal of Genomics 2017, 8737649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A, Mollo N, Nitti M, Paladino S, Calì G, Genesio R, … Conti A (2018). Mitochondrial dysfunction in down syndrome: molecular mechanisms and therapeutic targets. Molecular Medicine 24, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo A, Nitti M, Mollo N, Paladino S, Procaccini C, Faicchia D, et al. (2017). Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in down syndrome cells. Human Molecular Genetics 26, 1056–1069. [DOI] [PubMed] [Google Scholar]
- Jamwal S, & Kumar P (2015). Antidepressants for neuroprotection in Huntington’s disease: A review. European Journal of Pharmacology 769, 33–42. [DOI] [PubMed] [Google Scholar]
- Jamwal S, & Kumar P (2016). Spermidine ameliorates 3-nitropropionic acid (3-NP)-induced striatal toxicity: possible role of oxidative stress, neuroinflammation, and neurotransmitters. Physiology & Behavior 155, 180–187. [DOI] [PubMed] [Google Scholar]
- Ježek J, Cooper KF, & Strich R (2018). Reactive oxygen species and mitochondrial dynamics: the yin and yang of mitochondrial dysfunction and cancer progression. Antioxidants 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Kang SU, Zhang S, Karuppagounder S, Xu J, Lee YK, … Troncoso JC (2016). Adult conditional knockout of PGC1α leads to loss of dopamine neurons. eNeuro 3 (ENEURO.0183–16.2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Wang W, Perry G, Zhu X, & Wang X (2015). Mitochondrial dynamic abnormalities in amyotrophic lateral sclerosis. Translational Neurodegeneration 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Albertz J, Guo Z, Peng Q, Rudow G, Troncoso JC, … Duan W (2013). Neuroprotective effects of PPAR-γ agonist rosiglitazone in N171–82Q mouse model of Huntington’s disease. Journal of Neurochemistry 125, 410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YN, Hwang WY, Jo C, & Johnson GV (2012). Metabolic state determines sensitivity to cellular stress in Huntington disease: normalization by activation of PPARγ. PLoS One 7, e30406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YN, & Johnson GV (2010). The interrelationship between mitochondrial dysfunction and transcriptional dysregulation in Huntington disease. Journal of Bioenergetics and Biomembranes 42, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, & Beal MF (2012). Mitochondrial dysfunction in neurodegenerative diseases. Journal of Pharmacology and Experimental Therapeutics 342, 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Calingasan NY, Hennessey TM, Sharma A, Yang L, Wille E, … Beal MF (2011). Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Human Molecular Genetics 21, 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri A, Chandra A, & Beal MF (2013). PGC1α, mitochondrial dysfunction, and Huntington’s disease. Free Radical Biology and Medicine 62, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung TW, Lee JY, Shim WS, Kang ES, Kim SK, Ahn CW, … Cha BS (2007). Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. Journal of the Neurological Sciences 253, 53–60. [DOI] [PubMed] [Google Scholar]
- Kasashima K, Nagao Y, & Endo H (2014). Dynamic regulation of mitochondrial genome maintenance in germ cells. Reproductive Medicine and Biology 13, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsouri L, Parr C, Bogdanovic N, Willem M, & Sastre M (2011). PPARγ co-activator-1α (PGC1α) reduces amyloid-β generation through a PPARγ-dependent mechanism. Journal of Alzheimer’s Disease 25, 151–162. [DOI] [PubMed] [Google Scholar]
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, & Shoubridge EA (2007). The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Molecular Biology of the Cell 18, 3225–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kausar S, Wang F, & Cui H (2018). The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 7, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, & Manfredi G (2017). Proteinopathies and OXPHOS dysfunction in neurodegenerative diseases. The Journal of Cell Biology 216, 3917–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil B, & Liévens JC (2017). Mitochondrial quality control in amyotrophic lateral sclerosis: towards a common pathway? Neural Regeneration Research 12(7), 1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Alam Q, Haque A, Ashafaq M, Khan MJ, Ashraf GM, & Ahmad M (2019). Current Progress on Peroxisome Proliferator-activated Receptor Gamma Agonist as an Emerging Therapeutic Approach for the Treatment of Alzheimer’s Disease: An Update. Current Neuropharmacology 17, 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Chen J, Calingasan NY, & Beal MF (2005). Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Experimental Neurology 191, 331–336. [DOI] [PubMed] [Google Scholar]
- Kilbride SM, Gluchowska SA, Telford JE, O’Sullivan C, & Davey GP (2011). High-level inhibition of mitochondrial complexes III and IV is required to increase gluta-mate release from the nerve terminal. Molecular Neurodegeneration 6, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komen JC, & Thorburn DR (2014). Turn up the power–pharmacological activation of mitochondrial biogenesis in mouse models. British Journal of Pharmacology 171, 1818–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korta P, Pocheć E, & Mazur-Biały A (2019). Irisin as a Multifunctional Protein: Implications for Health and Certain Diseases. Medicina 55, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowiański P, Lietzau G, Czuba E, Waśkow M, Steliga A, & Moryś J (2018). BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cellular and Molecular Neurobiology 38, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühl I, Miranda M, Posse V, Milenkovic D, Mourier A, Siira SJ, … Gustafsson CM (2016). POLRMT regulates the switch between replication primer formation and gene expression of mammalian mtDNA. Science Advances 2, e1600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukat A, Dogan SA, Edgar D, Mourier A, Jacoby C, Maiti P, … Kudin AP (2014). Loss of UCP2 attenuates mitochondrial dysfunction without altering ROS production and uncoupling activity. PLoS Genetics 10, e1004385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Kaundal RK, More S, & Sharma SS (2009). Beneficial effects of pioglitazone on cognitive impairment in MPTP model of Parkinson’s disease. Behavioural Brain Research 197, 398–403. [DOI] [PubMed] [Google Scholar]
- Laloux C, Petrault M, Lecointe C, Devos D, & Bordet R (2012). Differential susceptibility to the PPARγ agonist pioglitazone in 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine and 6-hydroxydopamine rodent models of Parkinson’s disease. Pharmacological Research 65, 514–522. [DOI] [PubMed] [Google Scholar]
- Lee EY, Lee JE, Park JH, Shin IC, & Koh HC (2012). Rosiglitazone, a PPARγ agonist, protects against striatal dopaminergic neurodegeneration induced by 6-OHDA lesions in the substantia nigra of rats. Toxicology Letters 213, 332–344. [DOI] [PubMed] [Google Scholar]
- Lee Y, Stevens DA, Kang SU, Jiang H, Lee YI, Ko HS, … Kam TI (2017). PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Reports 18, 918–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejri I, Agapouda A, Grimm A, & Eckert A (2019). Mitochondria-and oxidative stress-targeting substances in cognitive decline-related disorders: from molecular mechanisms to clinical evidence. Oxidative Medicine and Cellular Longevity 2019, 9695412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Reichert CO, & Bydlowski SP (2019). Paraoxonases activities and polymorphisms in elderly and old-age diseases: an overview. Antioxidants 8, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PA, Hou X, & Hao S (2017). Mitochondrial biogenesis in neurodegeneration. Journal of Neuroscience Research 95, 2025–2029. [DOI] [PubMed] [Google Scholar]
- Lin MT, & Beal MF (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787. [DOI] [PubMed] [Google Scholar]
- Liu Z, Li T, Li P, Wei N, Zhao Z, Liang H, … Wei J (2015). The ambiguous relationship of oxidative stress, tau hyperphosphorylation, and autophagy dysfunction in Alzheimer’s disease. Oxidative Medicine and Cellular Longevity 2015, 352723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JM, Maloney B, Rogers JT, & Lahiri DK (2019). Novel upregulation of amyloid-beta precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5’-untranslated region: Implications in Alzheimer’s disease. Molecular Psychiatry 24, 345–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Trinh AT, Reid CS, & Cowell RM (2012). Developmental alterations in motor coordination and medium spiny neuron markers in mice lacking PGC1α. PLoS One 7, e42878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalingam KB, Radhakrishnan A, Ping NS, & Haleagrahara N (2018). Current concepts of neurodegenerative mechanisms in Alzheimer’s disease. BioMed Research International 2018, 3740461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, … Turnbull DM (2009). Mitochondrial changes within axons in multiple sclerosis. Brain 132, 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti P, Manna J, & Dunbar GL (2017). Current understanding of the molecular mechanisms in Parkinson’s disease: targets for potential treatments. Translational Neurodegeneration 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Karlo JC, & Landreth GE (2012). Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. Journal of Neuroscience 32, 10117–10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMeekin LJ, Li Y, Fox SN, Rowe GC, Crossman DK, Day JJ, … Cowell RM (2018). Cell-specific deletion of PGC1α from medium spiny neurons causes transcriptional alterations and age-related motor impairment. Journal of Neuroscience 38, 3273–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehan S, Kaur G, Dudi R, Rajput M, & Kalra S (2017). Restoration of mitochondrial dysfunction in 6-hydroxydopamine induced Parkinson’s disease: A complete review. Open Journal of Parkinson’s Disease and Treatment 1, 1–26. [Google Scholar]
- Mehta SL, & Li PA (2009). Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. Journal of Cerebral Blood Flow & Metabolism 29, 1069–1078. [DOI] [PubMed] [Google Scholar]
- Mohideen-Abdul K, Tazibt K, Bourguet M, Hazemann I, Lebars I, Takacs M, … Billas IM (2017). Importance of the sequence-directed DNA shape for specific binding site recognition by the estrogen-related receptor. Frontiers in Endocrinology 8, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokhtar SH, Bakhuraysah MM, Cram DS, & Petratos S (2013). The Beta-amyloid protein of Alzheimer’s disease: communication breakdown by modifying the neuronal cytoskeleton. International Journal of Alzheimer’s Disease 2013, 910502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon HE, & Paek SH (2015). Mitochondrial dysfunction in Parkinson’s disease. Experimental Neurobiology 24, 103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow BA, Roth RH, Redmond DE Jr., Diano S, & Elsworth JD (2012). Susceptibility to a parkinsonian toxin varies during primate development. Experimental Neurology 235, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralikumar S, Vetrivel U, Narayanasamy A, & Das UN (2017). Probing the intermolecular interactions of PPARγ-LBD with polyunsaturated fatty acids and their anti-inflammatory metabolites to infer most potential binding moieties. Lipids in Health and Disease 16, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano M, Costa L, Palermo R, Giovenco A, Vacca A, & Gulino A (2011). Protective effect of pioglitazone, a PPARγ ligand, in a 3 nitropropionic acid model of Huntington’s disease. Brain Research Bulletin 85, 231–237. [DOI] [PubMed] [Google Scholar]
- Nenov MN, Laezza F, Haidacher SJ, Zhao Y, Sadygov RG, Starkey JM, … Denner L (2014). Cognitive enhancing treatment with a PPARγ agonist normalizes dentate granule cell presynaptic function in Tg2576 APP mice. Journal of Neuroscience 34, 1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo HB, Kaiser JT, & Chan DC (2018). Tfam, a mitochondrial transcription and packaging factor, imposes a U–turn on mitochondrial DNA. Nature Structural & Molecular Biology 18, 1290–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, … Hamel E (2008). Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor γ agonist. Journal of Neuroscience 28, 9287–9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nierenberg AA, Ghaznavi SA, Mathias IS, Ellard KK, Janos JA, & Sylvia LG (2018). Peroxisome proliferator-activated receptor gamma coactivator-1 alpha as a novel target for bipolar disorder and other neuropsychiatric disorders. Biological Psychiatry 83, 761–769. [DOI] [PubMed] [Google Scholar]
- Nijssen J, Comley LH, & Hedlund E (2017). Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathologica 133, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Bristot VJ, de Bem Alves AC, Cardoso LR, da Luz Scheffer D, & Aguiar AS Jr. (2019). The role of PGC1α/UCP2 signaling in the beneficial effects of physical exercise on the brain. Frontiers in Neuroscience 13, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omeragic A, Hoque MT, Choi UY, & Bendayan R (2017). Peroxisome proliferator-activated receptor-gamma: potential molecular therapeutic target for HIV-1-associated brain inflammation. Journal of Neuroinflammation 14, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, & Trudeau LE (2015). Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Current Biology 25, 2349–2360. [DOI] [PubMed] [Google Scholar]
- Park JS, Davis RL, & Sue CM (2018). Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Current Neurology and Neuroscience Reports 18, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsanejad M, Bourquard N, Qu D, Zhang Y, Huang E, Rousseaux MW, … Kim RH (2014). DJ-1 interacts with and regulates paraoxonase-2, an enzyme critical for neuronal survival in response to oxidative stress. PLoS One 9, e106601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov PF, Petersen CH, Glaser E, & Ankarcrona M (2009). Mitochondrial accumulation of APP and Aβ: significance for Alzheimer disease pathogenesis. Journal of Cellular and Molecular Medicine 13, 4137–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K, Yang L, Wang J, Ye F, Dan G, Zhao Y, … Zou Z (2017). The interaction of mitochondrial biogenesis and fission/fusion mediated by PGC1α regulates rotenone-induced dopaminergic neurotoxicity. Molecular Neurobiology 54, 3783–3797. [DOI] [PubMed] [Google Scholar]
- Pérez MJ, Jara C, & Quintanilla RA (2018). Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Frontiers in Neuroscience 12, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA, & Suliman HB (2012). Redox regulation of mitochondrial biogenesis. Free Radical Biology and Medicine 53, 2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigié P, & Youle RJ (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Current Biology 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, & Youle RJ (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierelli G, Stanzione R, Forte M, Migliarino S, Perelli M, Volpe M, & Rubattu S (2017). Uncoupling protein 2: A key player and a potential therapeutic target in vascular diseases. Oxidative Medicine and Cellular Longevity 2017, 7348372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, & Carta AR (2014). Dynamic changes in pro-and anti-inflammatory cytokines in microglia after PPARγ agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiology of Disease 71, 280–291. [DOI] [PubMed] [Google Scholar]
- Poljsak B (2011). Strategies for reducing or preventing the generation of oxidative stress. Oxidative Medicine and Cellular Longevity 2011, 194586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Yin X, Wang S, Jiang H, Wang X, Ren M, … Feng H (2015). PGC1α silencing compounds the perturbation of mitochondrial function caused by mutant SOD1 in skeletal muscle of ALS mouse model. Frontiers in Aging Neuroscience 7, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, & Pasinetti GM (2009). PGC1α expression decreases in the Alzheimer disease brain as a function of dementia. Archives of Neurology 66, 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintanilla RA, Jin YN, Fuenzalida K, Bronfman M, & Johnson GV (2008). Rosiglitazone Treatment Prevents Mitochondrial Dysfunction in Mutant Huntingtin-expressing Cells: possible role of peroxisome proliferator-activated receptor-γ (pparγ) in the pathogenesis of huntington disease. Journal of Biological Chemistry 283, 25628–25637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, & Reddy TP (2011). Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Current Alzheimer Research 8, 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricote M, & Glass CK (2007). PPARs and molecular mechanisms of transrepression. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1771, 926–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins D, & Zhao Y (2011). New aspects of mitochondrial uncoupling proteins (UCPs) and their roles in tumorigenesis. International Journal of Molecular Sciences 12, 5285–5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Rivera J, Denner L, & Dineley KT (2011). Rosiglitazone reversal of Tg2576 cognitive deficits is independent of peripheral gluco-regulatory status. Behavioural Brain Research 216, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roger AJ, Muñoz-Gómez SA, & Kamikawa R (2017). The origin and diversification of mitochondria. Current Biology 27, R1177–R1192. [DOI] [PubMed] [Google Scholar]
- Rolland B, Deguil J, Jardri R, Cottencin O, Thomas P, & Bordet R (2013). Therapeutic prospects of PPARs in psychiatric disorders: a comprehensive review. Current Drug Targets 14, 724–732. [DOI] [PubMed] [Google Scholar]
- Russell AP, Wada S, Vergani L, Hock MB, Lamon S, Léger B, … Kralli A (2013). Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiology of Disease 49, 107–117. [DOI] [PubMed] [Google Scholar]
- Ryan MT, & Hoogenraad NJ (2007). Mitochondrial-nuclear communications. Annual Review of Biochemistry 76, 701–722. [DOI] [PubMed] [Google Scholar]
- Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, & Tarnopolsky MA (2018). Exercise increases mitochondrial PGC-1 α content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. The Journal of Biological Chemistry 293, 4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, & Cui H (2018). Emerging roles of estrogen-related receptors in the brain: potential interactions with estrogen signaling. International Journal of Molecular Sciences 19, 1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 1813, 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SN, Knutti D, Brogli K, Uhlmann T, & Kralli A (2003). The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor α (ERRα). Journal of Biological Chemistry 278, 9013–9018. [DOI] [PubMed] [Google Scholar]
- Schütz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schürmann B, … Heneka MT (2005). The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. Journal of Neuroscience 25, 7805–7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BJ, Yoon SH, & Do JT (2018). Mitochondrial dynamics in stem cells and differentiation. International Journal of Molecular Sciences 19, 3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, & Sampath H (2019). Mitochondrial DNA integrity: role in health and disease. Cells 8, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N, Kawaguchi-Niida M, Yamamoto T, Toi S, Hirano A, & Kobayashi M (2008). Effects of the PPARγ activator pioglitazone on p38 MAP kinase and IκBα in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology 28, 387–398. [DOI] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, … Dawson TM (2011). PARIS (ZNF746) repression of PGC1α contributes to neurodegeneration in Parkinson’s disease. Cell 144, 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota T, Traven A, & Lithgow T (2015). Mitochondrial biogenesis: cell-cycle-dependent investment in making mitochondria. Current Biology 25, R78–R80. [DOI] [PubMed] [Google Scholar]
- Shoag J, & Arany Z (2010). Regulation of hypoxia-inducible genes by PGC1α. Arteriosclerosis, Thrombosis, and Vascular Biology 30, 662–666. [DOI] [PubMed] [Google Scholar]
- Shokolenko IN, & Alexeyev MF (2017). Mitochondrial transcription in mammalian cells. Frontiers in Bioscience (Landmark edition) 22, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Srivastav S, Yadav AK, Srikrishna S, & Perry G (2016). Overview of Alzheimer’s disease and some therapeutic approaches targeting Aβ by using several synthetic and herbal compounds. Oxidative Medicine and Cellular Longevity 2016, 7361613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedhar A, & Zhao Y (2017). Uncoupling protein 2 and metabolic diseases. Mitochondrion 34, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbacher P, & Eckl P (2015). Impact of oxidative stress on exercising skeletal muscle. Biomolecules 5, 356–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, … Shin JH (2015). Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proceedings of the National Academy of Sciences 112, 11696–11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strum JC, Shehee R, Virley D, Richardson J, Mattie M, Selley P, … Roses A (2007). Rosiglitazone induces mitochondrial biogenesis in mouse brain. Journal of Alzheimer’s Disease 11, 45–51. [DOI] [PubMed] [Google Scholar]
- Swanson C, & Emborg M (2014). Expression of peroxisome proliferator-activated receptor-gamma in the substantia nigra of hemiparkinsonian nonhuman primates. Neurological Research 36, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CR, Du E, Johnson DA, Johnson JA, & Emborg ME (2013). Neuroprotective properties of a novel non-thiazoledinedione partial PPARγ agonist against MPTP. PPAR Research 2013, 582809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson CR, Joers V, Bondarenko V, Brunner K, Simmons HA, Ziegler TE, … Emborg ME (2011). The PPARγ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. Journal of Neuroinflammation 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH (2018). Mitochondria and mitochondrial cascades in Alzheimer’s disease. Journal of Alzheimer’s Disease 62, 1403–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, Saft C, Akkad DA, Wieczorek S, Haghikia A, Chan A, … Arning L (2011). PGC-1alpha downstream transcription factors NRF1 and TFAM are genetic modifiers of Huntington disease. Molecular Neurodegeneration 6, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov AI, Griffiths EJ, & Rutter GA (2012). Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 52, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng CT, Li Y, Stockton P, & Foley J (2011). Fasting induces the expression of PGC1α and ERR isoforms in the outer stripe of the outer medulla (OSOM) of the mouse kidney. PLoS One 6, e26961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu ZW, & Diano S (2016). UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell 164, 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo EM, & Inestrosa NC (2010). Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1ΔE9 mouse model of Alzheimer’s disease. Molecular Psychiatry 15, 272. [DOI] [PubMed] [Google Scholar]
- Tonelli C, Chio IIC, & Tuveson DA (2018). Transcriptional regulation by Nrf2. Antioxidants & Redox Signaling 29, 1727–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönnies E, & Trushina E (2017). Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. Journal of Alzheimer’s Disease 57, 1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truban D, Hou X, Caulfield TR, Fiesel FC, & Springer W (2017). PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? Journal of Parkinson’s Disease 7, 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RAV, … La Spada AR (2012). PGC1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Science Translational Medicine 4, 142ra97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufano M, & Pinna G (2020). Is There a Future for PPARs in the Treatment of Neuropsychiatric Disorders? Molecules 25, 1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S, Gupta P, Saini AS, Kaushal C, & Sharma S (2011). The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. Journal of Advanced Pharmaceutical Technology & Research 2, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uittenbogaard M, & Chiaramello A (2014). Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Current Pharmaceutical Design 20, 5574–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ur Rasheed MS, Tripathi MK, Mishra AK, Shukla S, & Singh MP (2016). Resveratrol protects from toxin-induced parkinsonism: plethora of proofs hitherto petty translational value. Molecular Neurobiology 53, 2751–2760. [DOI] [PubMed] [Google Scholar]
- Valenti D, de Bari L, de Rasmo D, Signorile A, Henrion-Caude A, Contestabile A, & Vacca RA (2016). The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1862, 1093–1104. [DOI] [PubMed] [Google Scholar]
- Varga T, Czimmerer Z, & Nagy L (2011). PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1812, 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladomiu M, Hontecillas R, Lu P, & Bassaganya-Riera J (2013). Preventive and prophylactic mechanisms of action of pomegranate bioactive constituents. Evidence-based Complementary and Alternative Medicine 2013, 789764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villena JA (2015). New insights into PGC-1 co-activators: redefining their role in the regulation of mitochondrial function and beyond. The FEBS Journal 282, 647–672. [DOI] [PubMed] [Google Scholar]
- Viswakarma N, Jia Y, Bai L, Vluggens A, Borensztajn J, Xu J, & Reddy JK (2010). Coactivators in PPAR-regulated gene expression. PPAR Research 2010, 250126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vomund S, Schäfer A, Parnham MJ, Brüne B, & Von Knethen A (2017). Nrf2, the master regulator of anti-oxidative responses. International Journal of Molecular Sciences 18, 2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak J, Partyka M, Duszyński J, & Szczepanowska J (2017). Implications of mitochondrial network organization in mitochondrial stress signalling in NARP cybrid and Rho0 cells. Scientific Reports 7, 14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, & Chen GJ (2016). Mitochondria as a therapeutic target in Alzheimer’s disease. Genes & Diseases 3, 220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Li JJ, Diao S, Kwak YD, Liu L, Zhi L, … Liao FF (2013). Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metabolism 17, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Xu J, Yu X, Yang R, & Han ZC (2006). Peroxisome proliferator-activated receptor γ in malignant diseases. Critical Reviews in Oncology/Hematology 58, 1–14. [DOI] [PubMed] [Google Scholar]
- Warden A, Truitt J, Merriman M, Ponomareva O, Jameson K, Ferguson LB, … Harris RA (2016). Localization of PPAR isotypes in the adult mouse and human brain. Scientific Reports 6, 27618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenz T (2011). Mitochondria and PGC1α in aging and age-associated diseases. Journal of Aging Research 2011, 810619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Soyal SM, Landwehrmeyer GB, & Patsch W European Huntington Disease N. (2014). A single nucleotide polymorphism in the coding region of PGC-1alpha is a male-specific modifier of Huntington disease age-at-onset in a large European cohort. BMC Neurology 14, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte ME, Nijland PG, Drexhage JA, Gerritsen W, Geerts D, van het Hof B, … van Horssen J (2013). Reduced expression of PGC1α partly underlies mitochondrial changes and correlates with neuronal loss in multiple sclerosis cortex. Acta Neuropathologica 125, 231–243. [DOI] [PubMed] [Google Scholar]
- Wrann CD (2015). FNDC5/Irisin–their role in the nervous system and as a mediator for beneficial effects of exercise on the brain. Brain Plasticity 1, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrann CD, White JP, Salogiannnis J, Laznik-Bogoslavski D, Wu J, Ma D, … Spiegelman BM (2013). Exercise induces hippocampal BDNF through a PGC1α/FNDC5 pathway. Cell Metabolism 18, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Chen M, & Jiang J (2019). Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 49, 35–45. [DOI] [PubMed] [Google Scholar]
- Xu J, Jackson CW, Khoury N, Escobar I, & Perez-Pinzon MA (2018). Brain SIRT1 mediates metabolic homeostasis and neuroprotection. Frontiers in Endocrinology 9, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, & Heneka MT (2012). PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. Journal of Neuroscience 32, 17321–17331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau SY, Gil-Mohapel J, Christie BR, & So KF (2014). Physical exercise-induced adult neurogenesis: a good strategy to prevent cognitive decline in neurodegenerative diseases? BioMed Research International 2014, 403120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Chen C, Si E, Cai Y, Wang J, Huang W, … Chen X (2017). Mitochondrial Effects of PGC-1alpha Silencing in MPP+ Treated Human SH-SY5Y Neuroblastoma Cells. Frontiers in Molecular Neuroscience 10, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, & Van Der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SH, Han SH, & Park JI (2018). Peroxisome proliferator-activated receptor γ and PGC1α in cancer: dual actions as tumor promoter and suppressor. PPAR Research 2018, 6727421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Du L, Zhang W, Yang Y, Zhou Q, & Du G (2017). Therapeutic effects of baicalein on rotenone-induced Parkinson’s disease through protecting mitochondrial function and biogenesis. Scientific Reports 7, 9968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, … Grünblatt E (2010). PGC1α, a potential therapeutic target for early intervention in Parkinson’s disease. Science Translational Medicine 2, 52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Winderickx J, Franssens V, & Liu B (2018). A mitochondria-associated oxidative stress perspective on Huntington’s Disease. Frontiers in Molecular Neuroscience 11, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Bernard-Marissal N, Moullan N, D’Amico D, Auwerx J, Moore DJ, … Schneider BL (2017). Parkin functionally interacts with PGC1α to preserve mitochondria and protect dopaminergic neuron s. Human Molecular Genetics 26, 582–598. [DOI] [PubMed] [Google Scholar]
- Zorzano A, & Claret M (2015). Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction. Frontiers in Aging Neuroscience 7, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]