

Abstract
Purpose:
We report results from a phase II study assessing the efficacy of the WEE1 inhibitor adavosertib with cisplatin in metastatic triple-negative breast cancer (mTNBC).
Experimental Design:
Patients with mTNBC treated with 0–1 prior lines of chemotherapy received cisplatin 75 mg/m2 IV followed 21 days later by cisplatin plus adavosertib 200 mg oral twice daily for 5 doses every 21 days. The study had 90% power to detect the difference between null (20%) and alternative (40%) objective response rates (ORRs) with a one-sided type I error of 0.1: an ORR >30% was predefined as making the regimen worthy of further study. RNA sequencing and multiplex cyclic immunofluorescence on pre- and post-adavosertib tumor biopsies, as well as targeted next-generation sequencing on archival tissue, were correlated with clinical benefit, defined as stable disease ≥6 months or complete or partial response.
Results:
34 patients initiated protocol therapy; median age was 56 years, 2 patients (6%) had BRCA2 mutations, and 14 (41%) had one prior chemotherapy. ORR was 26% (95%CI 13–44), and median progression-free survival was 4.9 months (95%CI 2.3–5.7). Treatment-related grade 3–5 adverse events occurred in 53% of patients, most commonly diarrhea in 21%. One death occurred due to sepsis, possibly related to study therapy. Tumors from patients with clinical benefit demonstrated enriched immune gene expression and T cell infiltration.
Conclusions:
Among patients with mTNBC treated with 0–1 prior lines, adavosertib combined with cisplatin missed the prespecified ORR cutoff of >30%. The finding of immune-infiltrated tumors in patients with clinical benefit warrants validation.
Keywords: breast cancer, targeted therapy, gene expression, immune pathways, antigen presentation, T cell infiltration
Statement of Translational Relevance
This phase II clinical trial evaluated whether the WEE1 inhibitor adavosertib combined with cisplatin improved clinical outcomes for patients with metastatic triple-negative breast cancer (mTNBC) and investigated molecular correlates of response to this therapy. A total of 34 patients were treated with adavosertib and cisplatin as first or second-line therapy for mTNBC. The endpoint was not met but objective response rate was 26% and median progression-free survival was 4.9 months. Transcriptomic and immunofluorescence analyses revealed that adavosertib treatment and clinical benefit may correlate with intratumoral immune gene expression and T cell infiltration. These findings suggest that adavosertib may augment antitumor immunity and warrant validation in future studies, including an ongoing trial of adavosertib with the PD-L1 inhibitor durvalumab.
Introduction
Patients with metastatic triple-negative breast cancer (mTNBC) have few treatment options, and a median overall survival (OS) of 1–1.5 years after diagnosis.1 In the TNT and TBCRC009 trials, median progression-free survival (PFS) was 3 months on first or second-line platinum agents.2,3 Although median PFS increased to around 7 months in the atezolizumab and nab-paclitaxel arm of the IMpassion130 study,4 this regimen is only approved for patients with PD-L1-positive mTNBC, a small subset of patients with mTNBC,5 and most patients experience progression after a few months.5 Therefore, more effective treatment strategies are needed.
One potential therapeutic target is WEE1, a tyrosine kinase that arrests the cell cycle at the G2 checkpoint by inhibiting cyclin-dependent kinase 1 (CDK1) and preventing entry into mitosis, thus providing an opportunity for DNA damage repair.6 WEE1 may be particularly important in p53-deficient cancers, such as mTNBC, in which TP53 mutations occur in 80% of tumors,7 since loss-of-function mutations in TP53 leave cells completely dependent on the ATR-CHK1-WEE1 axis for G2 checkpoint control and also prevent cell cycle arrest at the G1 checkpoint, allowing entry into the S phase of DNA synthesis prior to repair of damaged DNA.6 WEE1 also slows cell cycle progression through the S phase by inhibiting CDK2, stabilizing replication forks and preventing DNA double-strand breaks.6, 8 WEE1 inhibition may cause replication stress in a p53-independent manner8 and sensitize tumors with p53 deficiency or high genomic instability to DNA-damaging therapies.9, 10
Adavosertib (AZD1775) is a potent and selective WEE1 inhibitor.6, 11 Preclinical studies have demonstrated that adavosertib sensitizes cells to DNA-damaging chemotherapies independent of TP53 status.12, 13 A phase I dose escalation study of adavosertib in combination with gemcitabine, cisplatin, or carboplatin showed safety and efficacy with a numerical but not significant improvement in response rate among patients with advanced solid tumors with versus without TP53 mutations.6 Three phase II studies of adavosertib with chemotherapy demonstrated encouraging antitumor efficacy in patients with TP53-mutated or TP53-unselected ovarian cancer who were sensitive or refractory/resistant to platinum-based therapy,14–16 including significant improvements in PFS and OS as compared to chemotherapy alone.15 Subsequent trials of adavosertib in combination with docetaxel and cisplatin, gemcitabine and radiation, or irinotecan showed favorable clinical activity in patients with TP53-unselected locally advanced head/neck squamous cell carcinoma,8 pancreatic cancer,17 and relapsed refractory pediatric solid tumors,18 respectively. Single-agent adavosertib also demonstrated high activity in recurrent uterine serous carcinoma, which has frequent TP53 mutations and increased replication stress.19
Here we report the results of a phase II study assessing the objective response rate (ORR) of adavosertib and cisplatin as first- or second-line therapy for mTNBC. We also evaluted molecular correlates of response, including genomic, transcriptomic, and tumor microenvironment characteristics.
Methods
Study Design and Participants
We conducted a single-arm, two-stage phase II study of adavosertib, a WEE1 inhibitor, with cisplatin for patients with mTNBC (estrogen receptor <1%, progesterone receptor <1%, and HER2 negative by the American Society of Clinical Oncology/College of American Pathologists guidelines) treated with 0–1 prior lines of chemotherapy in the metastatic setting. Participants could not have received prior platinum chemotherapy or a prior WEE1 inhibitor. Eligible patients included those with a history of treated brain metastases, defined as no ongoing requirement for corticosteroids and no evidence of progression for ≥1 month after treatment as ascertained by clinical examination and imaging. Patients were included regardless of tumor TP53 status based on the range of mechanisms by which adavosertib may induce cytotoxicity, coupled with the promising preclinical and clinical data in TP53-unselected cancers.12, 13, 17, 18
Treatment consisted of one cycle of cisplatin monotherapy (75 mg/m2 IV) followed 21 days later by adavosertib 200 mg oral twice daily for 5 doses over 2.5 days from days 1–3 in combination with cisplatin 75 mg/m2 IV every 21 days. The dosing schedule for adavosertib was selected based on the relatively short half-life observed in the phase I clinical trial,6 as well as preclinical data suggesting that multiple doses increase combinatorial efficacy with chemotherapy without affecting tolerability.20 AstraZeneca provided funding for the study and provided adavosertib. All patients with biopsy-accessible disease underwent paired site-matched research biopsies 5–48 hours after the monotherapy cisplatin dose on cycle 1 day 1 and 5–8 hours +/− 24 hours after the last dose of adavosertib on cycle 2 day 3.
Patients underwent restaging scans every 6 weeks, and response was evaluated by RECIST 1.1. All patients were enrolled at Dana-Farber Cancer Institute (DFCI). The DFCI institutional review board approved the study, and participants provided written informed consent before study entry. The study was monitored by the Data Safety Monitoring Committee at Dana-Farber/Harvard Cancer Center and conducted in accordance with the ethical guidelines outlined by the Belmont Report.
Molecular Studies
Exploratory analyses evaluated the association of response with genomic, transcriptomic, and immunofluorescence profiles of on-treatment tumors (eMethods 1). Targeted panel sequencing was available on archival tumor specimens from 30 enrolled patients (81%).21, 22 To enrich for functional genomic alterations, only non-synonymous mutations and significant copy number alterations were included. Whole transcriptome sequencing was performed on 45 on-study tumor biopsies from 26 treated patients on cisplatin monotherapy and/or on cisplatin plus WEE1 inhibitor combination therapy (76%; eMethods 2). Gene pathway expression was evaluated with gene set enrichment analysis (GSEA) using the hallmark gene sets from the Molecular Signatures Database (MSigDB),23 and tumor immune cell composition was assessed with CIBERSORTx.24 For these analyses, RECIST responses were categorized by the clinical benefit rate (CBR), defined as the proportion of patients achieving stable disease ≥6 months or complete or partial response. The clinical benefit group included one patient with an unconfirmed partial response, who had new lesions on repeat imaging.
To investigate the tumor immune microenvironment, 6 paired site-matched pre- and post-WEE1 inhibitor biopsies were evaluated with cyclic immunofluorescence (CyCIF).25–27 CyCIF was also used to measure WEE1 inhibitor-induced changes in cell cycle state and DNA damage related markers, including decreased phopho-CDK1/2 as an indication of target engagement.6, 28 Formal statistical tests for significance were not performed in the CyCIF cohort given the power limitations of the small sample size (n=6).
Statistical Analysis
The primary endpoint of the study was ORR, defined as the proportion of patients with complete or partial responses per RECIST 1.1. The study used a Simon optimal two-stage design that had 90% power to detect the difference between null (20%) and alternative (40%) ORRs with a one-sided type I error of 0.1. The null hypothesis was based on the ORR of 20% previously observed with first- or second-line platinum therapy in mTNBC without BRCA1/2 mutations.3 In the first stage, 17 patients were enrolled. Since there were at least 4 responses, the study continued to stage 2, where another 20 patients were enrolled. An ORR >30% would identify the regimen as worthy of further study. The analysis population consisted of all patients who initiated protocol therapy. Secondary endpoints included PFS, defined as the time from study enrollment until disease progression or death, whichever occurred first, and OS. Patients alive and without disease progression were censored at date of last disease evaluation. PFS was assessed by the Kaplan-Meier method.
Continuous molecular variables were compared between response groups (clinical benefit versus none) with the non-parametric Wilcoxon rank-sum test, and proportion of patients with somatic alterations were compared with two-sided Fisher’s exact tests. Kaplan-Meier analyses evaluated the association of molecular correlates with survival. Significance testing for differences in PFS and OS were calculated using the log-rank test. All comparisons were two-sided with an alpha level of 0.05. The false discovery rate (FDR) for GSEA was controlled with the Benjamini-Hochberg method with a threshold of q <0.05. All statistical analyses were performed in SAS 9.4 and R 3.5.1.
Results
Patient Characteristics
From January 26, 2017 to December 11, 2018, 37 patients enrolled in this study; 3 withdrew consent prior to therapy initiation, and 34 initiated study treatment (Supplementary Fig. S1). The median follow-up at the time of data cutoff was 32 months, and no patients remained on study therapy. Baseline characteristics are shown in Table 1. The median age was 56 years (range, 22–69 years), and only 2 patients (6%) had known BRCA1/2 mutations. Most patients (59%) had received no prior therapy for mTNBC, and the remainder had received one prior line of chemotherapy. In the early-stage setting or as first-line therapy for mTNBC, 91% of patients had been treated with taxanes and cyclophosphamide, and 79% had been treated with anthracyclines. Only 11% of patients had received prior immune checkpoint inhibitors. The median time from adjuvant therapy completion to metastatic cancer diagnosis was 12.8 months, and 50% (15/30) of the participants without de novo metastatic disease had a disease-free interval < 12 months. Most patients had visceral metastases, including liver (53%), lung (53%), and both liver and lung (32%). Among the 30 patients with targeted genomic sequencing, TP53 and ROS1 were the most frequently mutated genes with nonsynonymous mutations in 28 (93%) and 5 (17%) patients, respectively (Supplementary Fig. S2). Without matched normal samples, ROS1 variants may represent germline variants, which have previously been reported at a similar frequency in breast cancer.29
Table 1.
Baseline Patient Characteristics
| Characteristic | All (%) n = 34 |
CRs (%) n = 3 |
|---|---|---|
| Median age (range), y | 56 (22–69) | 55 (47–60) |
| ECOG performance status | ||
| 0 | 30 (88) | 3 (100) |
| 1 | 4 (12) | |
| BRCA1/2 carriers | ||
| Yes | 2 (6) | |
| No | 27 (79) | 3 (100) |
| Unknown | 5 (15) | |
| Patients with de novo metastatic disease | 4 (12) | 1 (33) |
| Prior lines of metastatic chemotherapy | ||
| 0 | 20 (59) | 2 (67) |
| 1 | 14 (41) | 1 (33) |
| Prior breast cancer therapy | ||
| Taxanes | 31 (91) | 2 (67) |
| Cytoxan | 31 (91) | 2 (67) |
| Anthracyclines | 27 (79) | 2 (67) |
| Capecitabine | 8 (23) | |
| Eribulin | 4 (12) | 1 (33) |
| Immune checkpoint inhibitors | 4 (12) | |
| PARP inhibitors | 3 (9) | |
| Disease free interval < 12 monthsa | 15 (50) | 1 (33) |
| Metastatic sites | ||
| Liver | 18 (53) | 3 (100) |
| Lung | 18 (53) | 2 (67) |
| Bone | 13 (38) | 1 (33) |
| Brain | 1 (3) |
Percent with disease free interval < 12 months was calculated from the 30 participants without de novo metastatic disease.
CR, complete responder; ECOG, Eastern Cooperative Oncology Group; PARP, poly (ADP-ribose) polymerase
Efficacy and Toxicity
The ORR was 26% (9/34, 95% CI 13–44) overall (Figure 1A), 30% (6/20, 95% CI 12–54) in the first-line cohort, and 21% (3/14, 95% CI 5–51) in the second-line cohort (Table 2). All responses were confirmed with subsequent repeat imaging scans. Of the three complete responders, two had lymph nodes as target lesions, which are considered normal when the short axis measures < 10 mm and so do not require a 100% reduction in lesion size to be classified as a complete response.30 Two of the complete responders had no prior chemotherapy for metastatic disease, and the third had de novo metastatic disease (Table 1). None were BRCA1/2 carriers. Of the two complete responders with targeted panel sequencing results, notable DNA repair alterations include both having TP53 mutations and one having an ATM mutation (Figure 2A).
Figure 1. Clinical efficacy of adavosertib with cisplatin in metastatic TNBC.
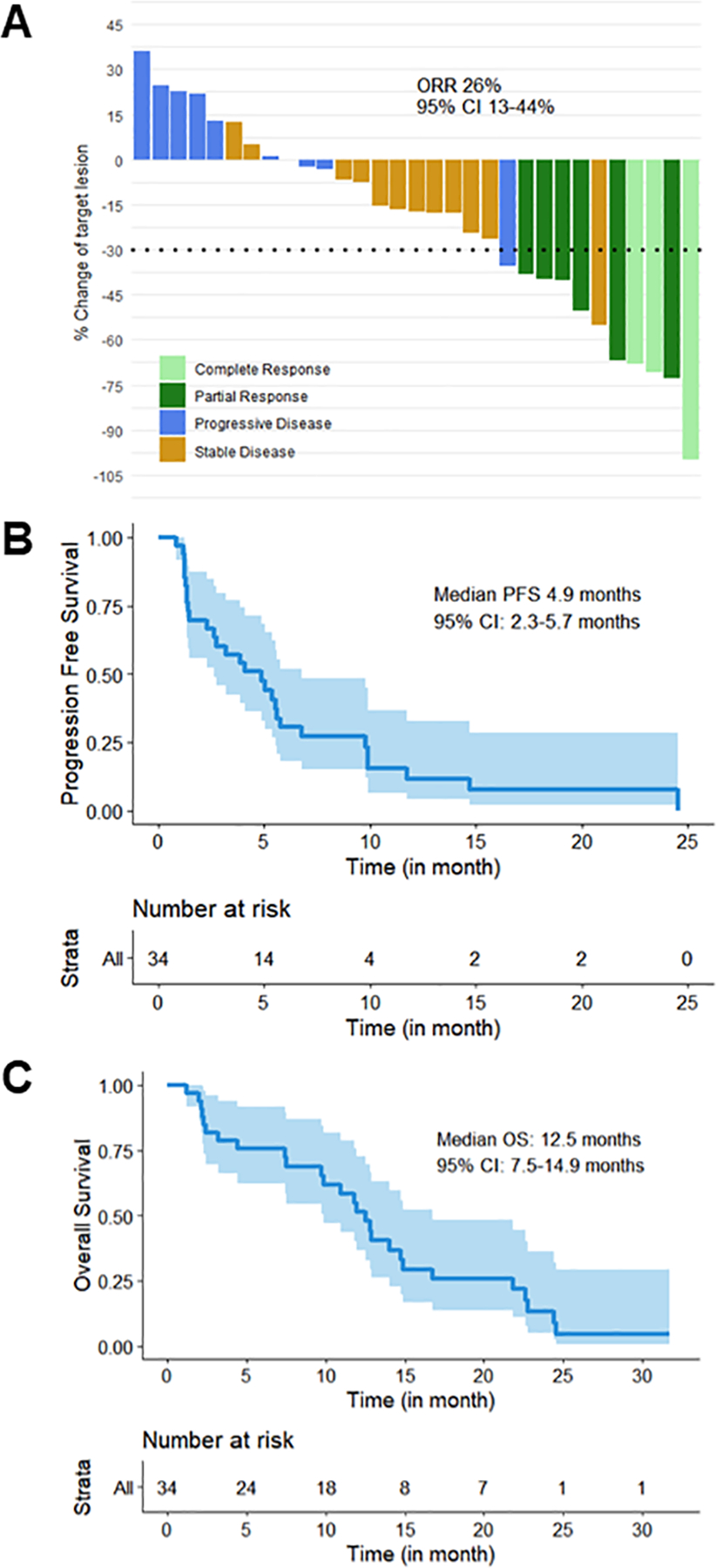
A) Waterfall plot of best tumor size change and overall response rate (ORR): two patients with > 30% reduction in tumor size had new lesions on initial scan or on a subsequent scan after initial scans showed stable disease and then unconfirmed partial response. In addition, two of the three complete responders had lymph nodes as target lesions, which are considered normal when the short axis measures < 10 mm and so do not require a 100% reduction in lesion size to be classified as a complete response.30 B, C) Kaplan-Meier curves for progression-free survival (PFS, B) and overall survival (OS, C).
Table 2.
Best Clinical Response
| Response Type | Overall (n = 34) | 1st Line (n = 20) | 2nd Line (n = 14) |
|---|---|---|---|
| Complete response (CR), n (%) | 3 (9) | 2 (10) | 1 (7) |
| Partial response (PR), n (%) | 6(18) | 4 (20) | 2 (14) |
| Stable disease (SD), n (%) | 13(38) | 7 (35) | 6 (43) |
| Progressive disease, n (%) | 10 (29) | 7 (35) | 3 (21) |
| Not evaluable, n (%) | 2 (6) | 0 (0) | 2 (14) |
| Overall response rate, % (95% CI) | 26 (13–44) | 30 (12–54) | 21 (5–51) |
| Clinical benefit rate, % (95% CI) | 32 (17–51) | 30 (12–54) | 36 (13–65) |
| Median PFS, months (95% CI) | 4.9 (2.3–5.7) | 3.8 (1.3–5.6) | 5.0 (1.3–6.8) |
| Median OS, months (95% CI) | 12.5 (7.5–14.9) | 12.5 (9.7–14.0) | 16.7 (2.3–24.4) |
CI, confidence interval; Clinical benefit rate = CR + PR + SD ≥ 6 months; CR, clinical response; PR, partial response; SD, stable disease; PFS, progression-free survival; OS, overall survival
Figure 2. Genomic analyses of adavosertib with cisplatin in metastatic TNBC demonstrated immune pathway enrichment in patients with clinical benefit versus without (CB).
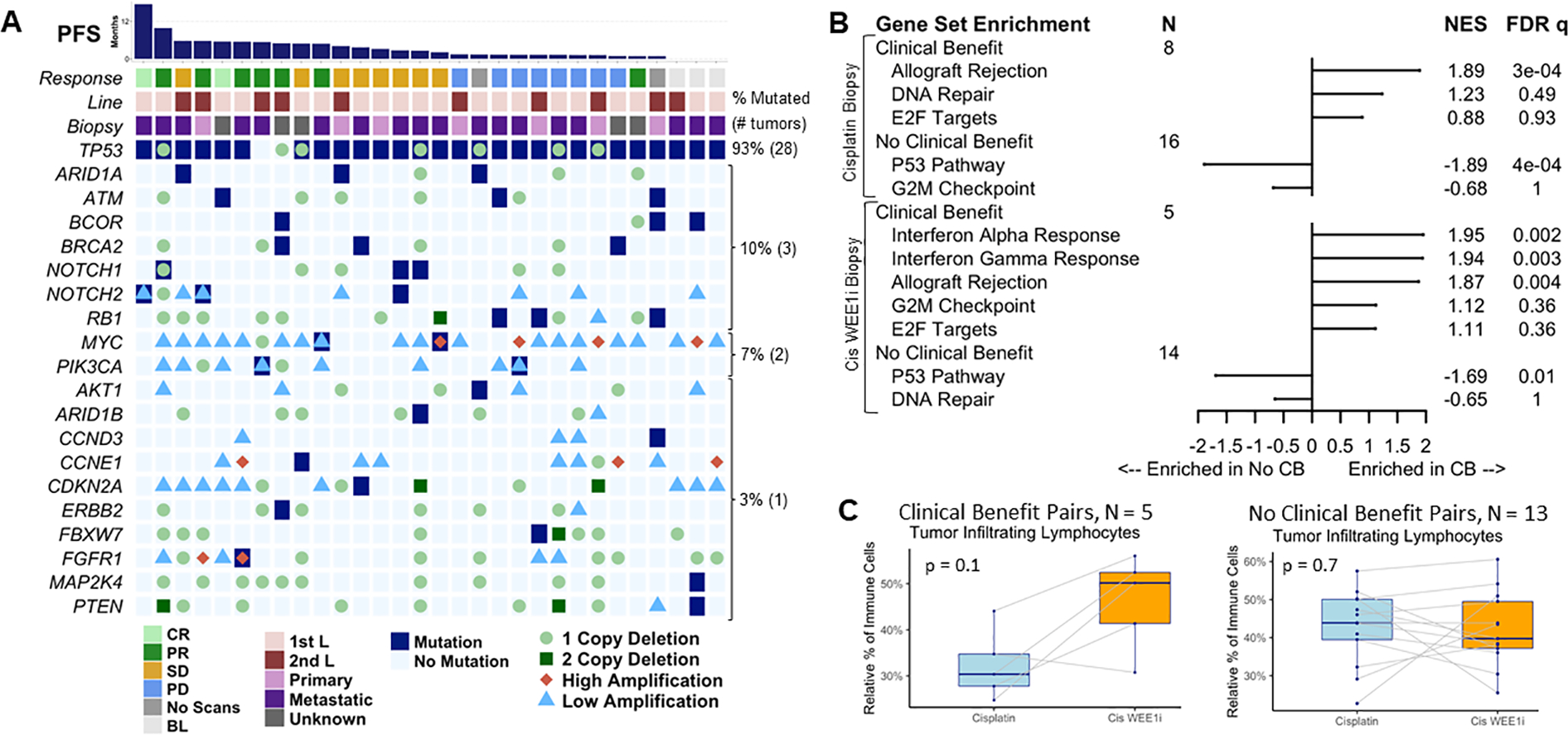
A) Breast cancer gene alterations in archival primary or metastatic tumors from 30 patients with targeted panel sequencing showed no clear association with response: each column represents a patient ordered from longest to shortest progression-free survival (PFS). B) Molecular Signature Database hallmark gene set enrichment analysis revealed immune gene set enrichment in patients with versus without CB at both biopsy (bx) timepoints on cisplatin monotherapy and on cisplatin WEE1 inhibitor (Cis WEE1i) combination therapy. C) Paired site-matched biopsy analysis of tumor infiltrating lymphocytes (TILs) measured by RNA sequencing indicated a nonsignificant increase in TILs in patients with but not without CB using paired Wilcoxon p values.
BL, baseline scans only as withdrew prior to treatment; CB, clinical benefit; Cis WEE1i, cisplatin WEE1 inhibitor; CR, complete response; FDR, false discovery rate corrected; L, line; NES, normalized enrichment score; PD, progressive disease; PR, partial response; SD, stable disease
Median PFS was 4.9 months (95% CI 2.3–5.7) overall (Figure 1B), 3.8 months (95% CI 1.3–5.6) in the first-line cohort, and 5.0 months (95% CI 1.3–6.8) in the second-line cohort (Table 2). The CBR was 32% (95% CI 17–51) overall, 30% (95% CI 12–54) in the first-line cohort, and 36% (95% CI 13–65) in the second-line cohort (Table 2). At the time of data cutoff, 29% of patients were still in survival follow-up. With the available survival data, median OS was 12.5 months (95% CI 7.5–14.9) overall (Figure 1C), 12.5 months (95% CI 9.7–14.0) in the first-line cohort, and 16.7 months (95% CI 2.3–24.4) in the second-line cohort.
All patients who initiated protocol therapy (n=34) were evaluated for safety outcomes. Adverse events (AEs) related to study therapy occurred in 31 (91%) patients, while grade 3–5 AEs occurred in 18 (53%) patients. The most common treatment-related AEs regardless of grade were nausea (50%), diarrhea (35%), anemia (29%), and neutropenia (29%) (Table 3). The most common grade 3–4 treatment-related AEs were diarrhea (21%), neutropenia (18%), and anemia (12%) (Table 3). One death occurred from culture-negative sepsis, possibly related to study therapy. This patient was hospitalized for abdominal pain, duodenitis, and lactic acidosis with mild transient neutropenia to absolute neutrophil count of 700, responsive to filgrastim. She subsequently developed atrial fibrillation with rapid ventricular rate complicated by persistent hypotension requiring vasopressors and respiratory failure requiring intubation.
Table 3.
Treatment-Related Adverse Events (> 20% of patients)
| Adverse Event | All Grades | Grade 3–5* |
|---|---|---|
| n (%) | n (%) | |
| Any | 31 (91) | 18 (53) |
| Nausea | 17 (50) | 2 (6) |
| Diarrhea | 12 (35) | 7 (21) |
| Anemia | 10 (29) | 4 (12) |
| Neutropenia | 10 (29) | 6 (18) |
| Fatigue | 9 (26) | 0 (0) |
| Vomiting | 7 (21) | 0 (0) |
| Tinnitus | 7 (21) | 0 (0) |
1 death occurred from sepsis possibly related to study therapy.
Molecular Correlates of Response
Targeted panel sequencing of archival tumor biopsies from 30 enrolled patients showed no clear association of response with frequently mutated breast cancer driver genes31 (Figure 2A): no single gene nonsynonymous mutation or deleterious copy number change was significantly associated with clinical benefit (eResults; Supplementary Fig. S3), including TP53 and BRCA1/2 alterations (Supplementary Figs. S4–5B). This was also true of MSigDB hallmark DNA repair, E2F, and G2M pathway alterations:23 none were associated with PFS or OS by the Kaplan-Meier method (Supplementary Figs. S5C–6). However, tumors with MSigDB G2M pathway alterations had slightly worse PFS in unadjusted analyses (p=0.04, Supplementary Fig. S6C).
GSEA comparing biopsies before and after WEE1 inhibitor therapy (n=26 patients) revealed that DNA repair and cell cycle pathways, specifically E2F and G2M,23 were enriched in 25 cisplatin biopsies compared to 20 cisplatin plus WEE1 inhibitor biopsies (q values <0.001; normalized enrichment scores [NES] 2.22, 2.64, and 1.81, for DNA repair, E2F and G2M, respectively), suggesting that adavosertib downregulates DNA repair and cell cycle genes. Upregulation of the interferon-α response pathway following WEE1 inhibitor therapy (NES −1.72, q <0.01) was also observed. Conversely, GSEA comparing biopsies from patients with clinical benefit versus without demonstrated no differences in DNA repair or E2F/G2M cell cycle pathways (Figure 2B). An evaluation of previously reported WEE1 inhibitor gene expression correlates,6, 32, 33 including CCNE1,34 showed that only EGR1 exhibited differential expression (p=0.04) involving higher expression in patients with no clinical benefit prior to WEE1 inhibitor therapy (Supplementary Fig. S7).
However, GSEA demonstrated upregulation of immune-related pathways and downregulation of the p53 pathway in patients with clinical benefit versus without (Figure 2B). The allograft rejection pathway was the most enriched gene set in cisplatin tumor biopsies from patients with (n=8) versus without (n=16) clinical benefit, while the interferon-α response, interferon-γ response, and allograft rejection pathways were the first, second, and fourth most enriched gene sets in tumors from patients with (n=5) versus without (n=14) clinical benefit after WEE1 inhibitor therapy, all with NES >1.8 and FDR q values <0.01 (Figure 2B). A comparison of genes within the allograft rejection pathway revealed that antigen presentation genes, including HLA-DQA1, HLA-DRA, CD74 (HLA-DR), HLA-DOA, HLA-DMA, and HCLS1, had higher expression in responders than non-responders in cisplatin biopsies (Mann-Whitney p <0.05 without FDR correction, Supplementary Fig. S8A), while T cell infiltration genes, including CD8A, CD4, CXCL9, and CSK, had higher expression in responders than non-responders in cisplatin WEE1 inhibitor biopsies (Mann-Whitney p <0.05 without FDR correction, Supplementary Fig. S8B).
Tumor infiltrating lymphocytes (TILs) proportions inferred from RNA sequencing data did not differ by clinical benefit group at either biopsy timepoint (Supplementary Fig. S9). While TILs increased after WEE1 inhibitor treatment in 4/5 patients with clinical benefit, this difference was not significant (paired Wilcoxon p=0.1; Figure 2C). Memory CD4-positive T cells were significantly increased after WEE1 inhibitor treatment in 18 patients with paired biopsies (paired Wilcoxon p=0.01) with no difference by clinical benefit (Supplementary Fig. S10). Anti-tumor M1 macrophages trended towards being higher in post-WEE1 inhibitor biopsies from patients with clinical benefit (Wilcoxon p=0.09, Supplementary Fig. S11).
Tumor immunostaining studies were undertaken to explore the effects of WEE1 inhibitor therapy on immune infiltration and DNA damage markers. Stromal TILs measured using the International Immuno-Oncology Biomarker Working Group criteria35 in 6 available paired biopsies demonstrated a non-significant increase from a median of 1% to 7.5% following WEE1 inhibitor treatment (paired Wilcoxon p=0.1; Supplementary Fig. S12). Immunohistochemistry for DNA damage markers showed positive RAD51 and negative phospho-histone H3 (pHH3) staining in biopsies following cisplatin that switched to negative RAD51 and positive pHH3 staining in some biopsies after cisplatin WEE1 inhibitor treatment, as well as positive geminin and γ-H2AX staining in both biopsies (Supplementary Fig. S13, Supplementary Table S1). While reduced RAD51 suggested suppressed homologous recombination, the small number of paired biopsy samples with high quality immunohistochemistry data cannot establish whether or not RAD51 is a predictive biomarker.
Using CyCIF, we stained tumor sections with 24 different tumor, stromal, and immune cell markers to characterize 6 of the paired site-matched pre- and post-WEE1 inhibitor biopsies (Supplementary Table S2). Mean marker fluorescence intensity in single cells across all samples revealed changes between monotherapy and combination therapy (Figure 3A). CyCIF also demonstrated the expected changes in cell cycle and DNA damage markers with WEE1 inhibitor therapy. In tumor cells, defined as pan-cytokeratin-positive (panCK+) cells, CyCIF showed that phospho-CDK1/2/3/5, the target for WEE1, decreased on average by 56% in combination therapy versus monotherapy biopsies (Figure 3B, Supplementary Fig. S14, Supplementary Table S3). The number of positive pHH3 tumor cells increased by an average of 28-fold from monotherapy to combination therapy biopsies (Figure 3B, Supplementary Fig. S14, Supplementary Table S3), suggesting increased entrance into mitosis following WEE1 inhibitor therapy. These data serve as pharmacodynamics evidence of WEE1 engagement by adavosertib.6, 28
Figure 3. Cyclic immunofluorescence (CyCIF) analyses of adavosertib with cisplatin in metastatic TNBC corroborated immune enrichment in responders.
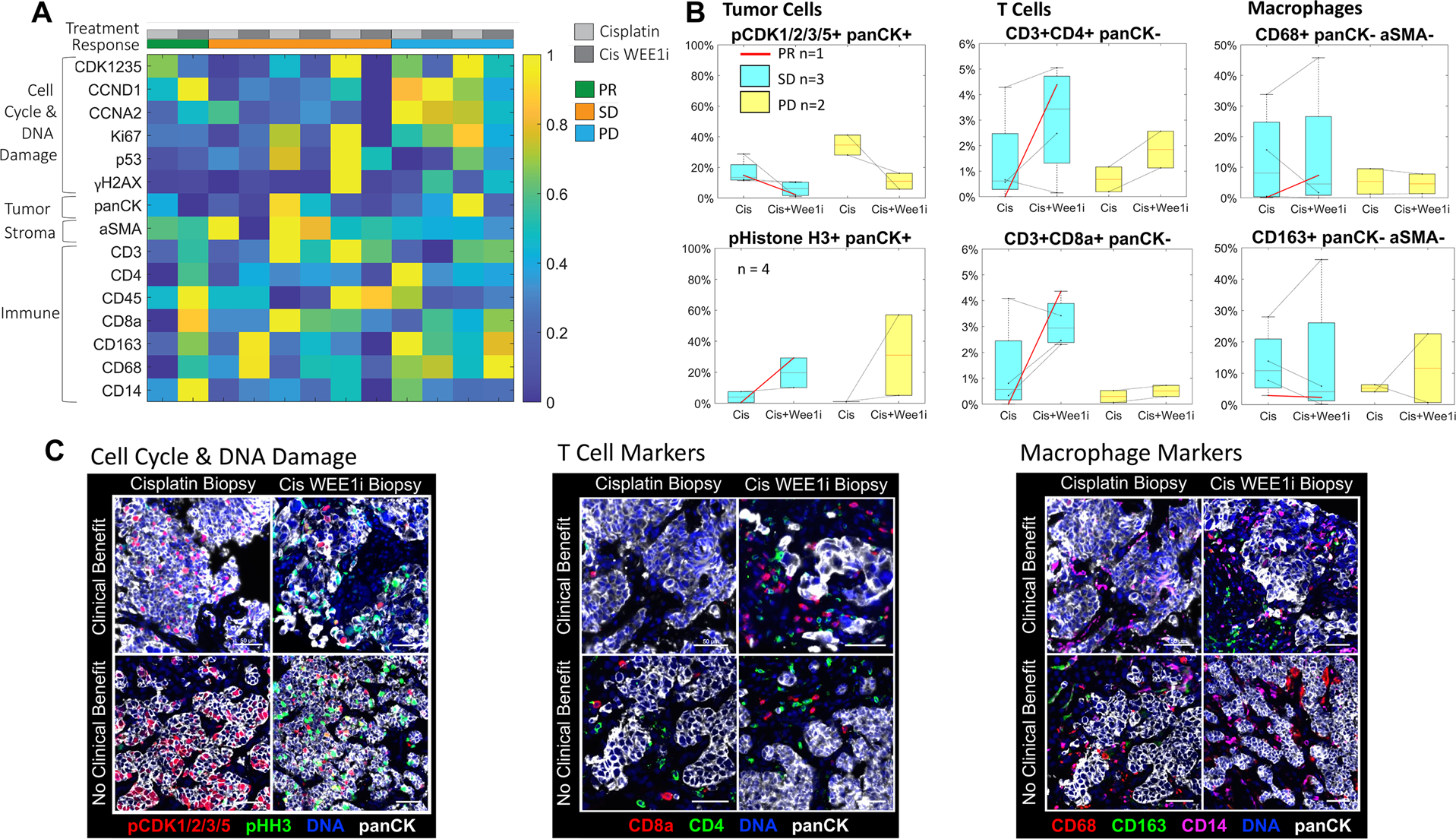
A) Heatmap of mean marker fluorescence intensity of single cells across 6 patients with paired site-matched tumor biopsies on cisplatin (Cis) and on cisplatin WEE1 inhibitor (Cis WEE1i) therapy. B) The mean percentage of positive single cells stratified by response (red corresponds to partial response [PR], blue to stable disease [SD], yellow to progressive disease [PD]). Tumor cells were identified using panCK and showed decreased phospho-CDK1/2/3/5 expression and increased pHistoneH3 (pHH3) expression in Cis WEE1i versus Cis biopsies. Immune cells were identified using the panCK-negative cell population and demonstrated a greater increase in CD3+CD4+ and CD3+CD8+ T cells in patients with PR and SD versus PD. Values on the y-axis are mean percentages. C) Merged CyCIF images from tumor biopsies of patients with clinical benefit versus without demonstrated decreased phospho-CDK1/2/3/5 in Cis WEE1i versus Cis biopsies and greater increases in CD3+CD4+ and CD3+CD8+ T cells in patients with clinical benefit versus without.
Cis, cisplatin; Cis WEE1i, cisplatin WEE1 inhibitor; PD, progressive disease; pHH3, pHistoneH3; PR, partial response; SD, stable disease.
CyCIF analysis of immune cells demonstrated increased intratumoral T cell infiltration following WEE1 inhibitor therapy in patients having more favorable responses. The percent of panCK-negative cells positive for T cell markers (CD3+CD4+ or CD3+CD8+) increased from monotherapy to combination therapy biopsies in 5/6 patients (Figure 3B). In addition, CD3+CD4+ and CD3+CD8+ T cell numbers exhibited larger increases in patients with partial response or stable disease versus progressive disease: the number of CD3+ CD4+ and CD3+CD8+ cells increased by an average fold-change of 12.4 and 13.6 versus 4.0 and 3.0, respectively (Supplementary Table S3). Conversely, there was no clear change in the expression of macrophage markers CD68 and CD163 across biopsy timepoints or response groups (Figure 3B–C, Supplementary Table S3).
Discussion
This phase II study combining adavosertib and cisplatin in patients with mTNBC, the first to our knowledge, demonstrated an ORR of 26%, which missed the prespecified ORR cutoff of 30%, and a median PFS of 4.9 months. This median PFS compares favorably to prior trials of first-line platinum chemotherapy in this patient population, such as the TNT and TBCRC009 trials that showed a median PFS of 3 months for first- or second-line platinum therapy.2,3 Notably, our cohort included many patients with early relapse, so the median disease-free interval of 13 months in the present trial was shorter than the median disease-free interval of 21 months in the historical control TBCRC009 trial by which our study was powered,3 raising the question of whether adavosertib had benefit beyond cisplatin in our more treatment-refractory population. In contrast to some prior studies,6, 12 we did not observe differential response rates by TP53 alteration status perhaps due to our use of combination therapy or the small number of tumors without a TP53 alteration. Despite a previous report of responses to adavosertib monotherapy in some patients with germline BRCA mutations,28 genomic alterations in DNA repair and cell cycle pathways, including BRCA1/2 alterations, did not correlate with response, consistent with results from the randomized NCI mpact trial.36 Future studies of WEE1 inhibitors with more comprehensive genomic assays will likely elucidate the role of genomic instability in response to WEE1 inhibition in clinical mTNBC cohorts.
CyCIF on paired site-matched pre- and post-WEE1 inhibitor biopsies demonstrated decreased phosphorylation of CDK1/2/3/5 in patients treated with combination therapy, as well as increased staining for the mitotic marker pHH3. These anticipated changes in cell cycle state provide pharmacodynamics evidence of successful target engagement. Likewise, gene expression analyses of the pre- and post-WEE1 inhibitor biopsies found that DNA repair and cell cycle pathways were downregulated following WEE1 inhibitor treatment, consistent with the mechanism of action of adavosertib.
The gene expression results also showed enriched p53 gene pathway expression in patients with no clinical benefit but did not reveal an association of DNA repair or other cell cycle genes with response. In line with prior reports demonstrating that EGR1 expression decreases with higher doses of WEE1 inhibitor therapy,6, 32 EGR1 was found to have higher pre-WEE1 inhibitor expression in patients with no clinical benefit, suggesting that adavosertib may not have adequately suppressed EGR1 levels in these non-responding tumors. Our results did not detect a role for other previously reported WEE1 inhibitor response correlates, such as CCNE1 and G1/S regulatory genes,33, 34 perhaps due to the greater heterogeneity of clinical samples compared to the preclinical experimental conditions of these prior reports.
However, our gene expression data showed upregulation of immune response pathways in patients with clinical benefit both before and after WEE1 inhibitor treatment. This was corroborated by the multiplex CyCIF finding of greater T cell infiltration following WEE1 inhibitor therapy in tumors with more favorable responses. Further studies will be needed to elucidate the mechanism of increased T cell infiltration following WEE1 inhibition. Based on our results with pHH3 immunofluorescence and γ-H2AX immunohistochemistry, WEE1 inhibition facilitated entry into mitosis with ongoing evidence of DNA damage. These biological changes may be associated with micronucleation and ultimate activation of the cGAS/STING pathway, which is expected to result in a Type I interferon response and the production of cytokines that drive cytotoxic T cell infiltration and activation.37, 38 These results also raise the question of whether the favorable response of tumors with enriched immune pathway gene expression is specific to adavosertib and cisplatin, more generalizable to a number of different therapies, or indicative of better overall prognosis. Beyond requiring validation in future larger data sets of WEE1 inhibitor-treated tumors, these findings highlight the need to evaluate the role of immune pathways in response to other non-immune therapies. If confirmed in larger prospective studies, the additional finding of interferon-α gene expression enrichment following WEE1 inhibitor therapy raises the questions of whether PD-L1 expression may be induced in adaptation and whether WEE1 inhibitors may synergize with immunotherapy. A clinical trial evaluating the combination of adavosertib with the PD-L1 inhibitor durvalumab is ongoing (NCT02617277).
The AEs observed in this study were similar to those observed in previous studies combining adavosertib with chemotherapy, consisting most commonly of bone marrow and gastrointestinal toxicity.6, 8, 14, 17, 18 The frequency of serious AEs ≥ grade 3 was also similar in the prior combination studies, occurring in about half of all participants,6, 8, 14, 17, 18 although a prior possible treatment-related death had not previously been reported. The first selective WEE1 kinase degrader, generated by linking adavosertib to the cereblon-binding ligand, pomalidomide, recently demonstrated G2/M accumulation at lower doses than adavosertib in cancer cell lines,39 raising the question of whether such novel WEE1 kinase degraders will show lower dose-limiting AEs and thereby greater efficacy in clinical studies.
A strength of this study is its deep multi-modal molecular characterization of paired site-matched biopsies pre- and post-WEE1 inhibitor. However, the study is limited by its small sample size that curtails the power of correlative analyses to find significant differences between response and biopsy groups. Furthermore, its non-randomized design may have introduced biases, such as a healthier enrolled study population than historical control studies of platinum monotherapy used to estimate the null hypothesis and response rate cutoff. In addition, the use of archival biopsies for the targeted next generation sequencing assessments raises the possibility that our lack of genomic correlates of response, including TP53 alterations, may be due to an incomplete assessment of the mutation status of the tumors at the time of protocol therapy rather than a true absence of an association. Finally, our ability to isolate the effect of adavosertib in our paired biopsy comparisons may have been affected by the slightly different timing of the biopsies in relation to cisplatin, and differential gene expression between these two timepoints may partially be due to different exposure periods to cisplatin.
In conclusion, among patients with mTNBC treated with 0–1 lines of prior therapy, the combination of adavosertib and cisplatin did not meet the prespecified efficacy cutoff of ORR >30% required to show a statistically significant improvement in response rate compared to cisplatin monotherapy as defined by this study. This result may be due to small sample size, nonrandomized study design, or true lack of benefit. Similar to prior combination studies of WEE1 inhibitors and chemotherapy, about half of patients experienced serious AEs.6, 8, 14, 17, 18 Ultimately, the recent approval of immune checkpoint inhibitors for patients with treatment-naïve PD-L1-positive mTNBC makes WEE1 inhibitor combinations most relevant in PD-L1-negative populations and second-line treatment settings. Alternatively, the combination of WEE1 inhibition with chemotherapy and PD-1/L1 inhibitors in the first-line mTNBC setting may be important if future studies confirm our finding of an augmented immune response in WEE1 inhibitor-treated tumor biopsies and show that the association of enriched immune signaling in responders is specific to WEE1 inhibition rather than general treatment response.
Supplementary Material
Acknowledgments:
This work was supported by AstraZeneca, the Mehlman Family Fund, the Harvard Ludwig Center, NCI grant U54-CA225088, and the NCI Specialized Program of Research Excellence (SPORE) Grant 1P50CA168504. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health/NCI. TEK acknowledges grant support from T32CA009172. TV acknowledges grant support from the Finnish Medical Foundation, Relander Foundation, Turku University Foundation, Maud Kuistila Memorial Foundation, the Finnish Society of Oncology, and the Cancer Society of Southwest Finland. EAM acknowledges the Rob and Karen Hale Distinguished Chair in Surgical Oncology for support. We kindly thank Kate Bifolck for her editorial support to this work. The authors would also like to acknowledge the DFCI Oncology Data Retrieval System (OncDRS) for the aggregation, management, and delivery of the genomic research data used in this project.
Footnotes
Data availability: The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest: JLG is a consultant for Glaxo-Smith Kline (GSK), Array BioPharma, Codagenix, Verseau, and Kymera and receives sponsored research support from GSK, Eli Lilly and Array BioPharma. PKS serves on the SAB or BOD of Glencoe Software, Applied Biomath and RareCyte Inc. and has equity in these companies; he is a member of the NanoString SAB. EPW receives consulting fees from Carrick Therapeutics, G1 Therapeutics, Genentech/Roche, Genomic Health/Exact Sciences, GSK, Jounce, Lilly, Novartis, Seattle Genetics, Syros and Leap; and institutional research funding from Genentech/Roche. EAM reports personal financial interests: research support for lab from Glaxo Smithkline; honoraria from Physician Education Resource; compensated service on Scientific Advisory Boards for Astra-Zeneca, Exact Sciences (formerly Genomic Health), Merck, Peregrine Pharmaceuticals, Roche/Genentech, Sellas Lifesciences, TapImmune Inc; uncompensated service on Steering Committees for BMS, Lilly, Roche/Genentech. EAM reports institutional financial interests from MD Anderson: clinical trial funding from AstraZeneca, EMD Serono, Galena Biopharma, Roche/Genentech; and institutional financical interests from DFCI: clinical trial funding from Roche/Genentech (via SU2C grant). EMV serves as a consultant/advisor to Tango Therapeutics, Invitae, Genome Medical, Dynamo, Foresite Capital, and Illumina; holds research support from Novartis and Bristol-Myers Squibb; and holds equity in Synapse, Genome Medical, Tango, and Microsoft Corp. G.I.S. has received research funding from Eli Lilly, Merck KGaA/EMD-Serono, Merck, and Sierra Oncology. He has served on advisory boards for Pfizer, Eli Lilly, G1 Therapeutics, Roche, Merck KGaA/EMD-Serono, Sierra Oncology, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, Daiichi Sankyo, Seattle Genetics, Boehringer Ingelheim, ImmunoMet, Asana, Artios, Atrin, Concarlo Holdings, Syros and Zentalis. In addition, he holds a patent entitled, “Dosage regimen for sapacitabine and seliciclib,” also issued to Cyclacel Pharmaceuticals, and a pending patent, entitled, “Compositions and Methods for Predicting Response and Resistance to CDK4/6 Inhibition,” together with Liam Cornell. SMT receives institutional research funding from AstraZeneca, Lilly, Merck, Nektar, Novartis, Pfizer, Genentech/Roche, Immunomedics, Exelixis, Bristol-Myers Squibb, Eisai, Nanostring, Cyclacel, Odonate, and Seattle Genetics; has served as an advisor/consultant to AstraZeneca, Lilly, Merck, Nektar, Novartis, Pfizer, Genentech/Roche, Immunomedics, Bristol-Myers Squibb, Eisai, Nanostring, Puma, Sanofi, Celldex, Paxman, Puma, Silverback Therapeutics, G1 Therapeutics, AbbVie, Anthenex, OncoPep, Outcomes4Me, Kyowa Kirin Pharmaceuticals, Daiichi-Sankyo, and Samsung Bioepsis Inc.
References
- 1.Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov 2019;9:176–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tutt A, Tovey H, Cheang MCU, Kernaghan S, Kilburn L, Gazinska P, et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med 2018;24:628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy With Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J Clin Oncol 2015;33:1902–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. IMpassion130: updated overall survival (OS) from a global, randomized, double-blind, placebo-controlled, Phase III study of atezolizumab (atezo) + nab-paclitaxel (nP) in previously untreated locally advanced or metastatic triple-negative breast cancer (mTNBC). J Clin Oncol 2019;37:abstr 1003. [Google Scholar]
- 5.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 2018;379:2108–21. [DOI] [PubMed] [Google Scholar]
- 6.Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol 2016;34:4371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet 2019;51:1450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mendez E, Rodriguez CP, Kao MC, Raju S, Diab A, Harbison RA, et al. A Phase I Clinical Trial of AZD1775 in Combination with Neoadjuvant Weekly Docetaxel and Cisplatin before Definitive Therapy in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 2018;24:2740–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends Pharmacol Sci 2016;37:872–81. [DOI] [PubMed] [Google Scholar]
- 10.Carrassa L, Damia G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat Rev 2017;60:139–51. [DOI] [PubMed] [Google Scholar]
- 11.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, et al. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov 2012;2:524–39. [DOI] [PubMed] [Google Scholar]
- 12.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther 2009;8:2992–3000. [DOI] [PubMed] [Google Scholar]
- 13.Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, et al. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther 2013;12:2675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J Clin Oncol 2016;34:4354–61. [DOI] [PubMed] [Google Scholar]
- 15.Lheureux S, Cabanero M, Cristea MC, Mantia-Smaldone G, Olawaiye A, Ellard S, et al. A randomized double-blind placebo-controlled phase II trial comparing gemcitabine monotherapy to gemcitabine in combination with adavosertib in women with recurrent, platinum resistant epithelial ovarian cancer: A trial of the Princess Margaret, California, Chicago and Mayo Phase II Consortia. J Clin Oncol 2019;37:5518–. [Google Scholar]
- 16.Oza AM, Estevez-Diz MDP, Grischke EM, Hall M, Marme F, Provencher DM, et al. A biomarker-enriched, randomized Phase II trial of adavosertib (AZD1775) plus paclitaxel and carboplatin for women with platinum-sensitive TP53-mutant ovarian cancer. Clin Cancer Res 2020. [DOI] [PubMed] [Google Scholar]
- 17.Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J Clin Oncol 2019;37:2643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole KA, Pal S, Kudgus RA, Ijaz H, Liu X, Minard CG, et al. Phase I Clinical Trial of the Wee1 Inhibitor Adavosertib (AZD1775) with Irinotecan in Children with Relapsed Solid Tumors. A COG Phase I Consortium Report (ADVL1312). Clin Cancer Res 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu JF, Xiong N, Campos SM, Wright AA, Krasner CN, Schumer ST, et al. A phase II trial of the Wee1 inhibitor adavosertib (AZD1775) in recurrent uterine serous carcinoma. J Clin Oncol 2020;38:6009. [DOI] [PubMed] [Google Scholar]
- 20.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther 2010;9:514–22. [DOI] [PubMed] [Google Scholar]
- 21.Hanna GJ, Lizotte P, Cavanaugh M, Kuo FC, Shivdasani P, Frieden A, et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov 2017;7:818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin JR, Fallahi-Sichani M, Sorger PK. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun 2015;6:8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin JR, Fallahi-Sichani M, Chen JY, Sorger PK. Cyclic Immunofluorescence (CycIF), A Highly Multiplexed Method for Single-cell Imaging. Curr Protoc Chem Biol 2016;8:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol 2015;33:3409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein MK, Morris LK, Martin MG. Next-Generation Sequencing Identifies Novel RTK VUSs in Breast Cancer with an Emphasis on ROS1, ERBB4, ALK and NTRK3. Pathol Oncol Res 2020;26:593–5. [DOI] [PubMed] [Google Scholar]
- 30.Schwartz LH, Bogaerts J, Ford R, Shankar L, Therasse P, Gwyther S, et al. Evaluation of lymph nodes with RECIST 1.1. Eur J Cancer 2009;45:261–7. [DOI] [PubMed] [Google Scholar]
- 31.Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016;534:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizuarai S, Yamanaka K, Itadani H, Arai T, Nishibata T, Hirai H, et al. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol Cancer 2009;8:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P, et al. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc Natl Acad Sci U S A 2015;112:15160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Low KH, Alexander A, Jiang Y, Karakas C, Hess KR, et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin Cancer Res 2018;24:6594–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dieci MV, Radosevic-Robin N, Fineberg S, van den Eynden G, Ternes N, Penault-Llorca F, et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: A report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin Cancer Biol 2018;52:16–25. [DOI] [PubMed] [Google Scholar]
- 36.Kummar S, Williams M, Lih C-J, Chen AP, Rubinstein L, Antony R, et al. NCI mpact: National Cancer Institute molecular profiling-based assignment of cancer therapy. J Clin Oncol 2014;32:TPS2642-TPS. [Google Scholar]
- 37.Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 2020;10:26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548:466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Z, Pinch BJ, Olson CM, Donovan KA, Nowak RP, Mills CE, et al. Development and Characterization of a Wee1 Kinase Degrader. Cell Chem Biol 2020;27:57–65 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.