

Abstract
Plasmid DNA (pDNA) has been widely used for non-viral gene delivery. After pDNA molecules enter a mammalian cell, they may be trapped in subcellular structures or degraded by nucleases. Only a fraction of them can function as templates for transcription in the nucleus. Thus, an important question is, what is the minimal amount of pDNA molecules that need to be delivered into a cell for transgene expression? At present, it is technically a challenge to experimentally answer the question. To this end, we developed a statistical framework to establish the relationship between two experimentally quantifiable factors — average copy number of pDNA per cell among a group of cells after transfection and percent of the cells with transgene expression. The framework was applied to the analysis of electrotransfection under different experimental conditions in vitro. We experimentally varied the average copy number per cell and the electrotransfection efficiency through changes in extracellular pDNA dose, electric field strength, and pulse number. The experimental data could be explained or predicted quantitatively by the statistical framework. Based on the data and the framework, we could predict that the minimal number of pDNA molecules in the nucleus for transgene expression was on the order of 10. Although the prediction was dependent on the cell and experimental conditions used in the study, the framework may be generally applied to analysis of non-viral gene delivery.
Keywords: non-viral gene delivery, copy number of DNA, electrotransfection, electroporation, mathematical modeling
1. Introduction
Electrotransfection can be used to deliver nucleic acids into cells both in vitro and in vivo [1-5]. For gene delivery, the electrotransfection efficiency, measured by the extent of transgene expression, depends on the rates of intracellular transport, DNA degradation by nucleases, transcription, and translation as well as the amount of DNA being delivered into cells. As a result, the efficiency can be influenced by a large number of factors in cells and experimental conditions [6-12]. To optimize electrotransfection, it is critical to understand how each factor affects the efficiency. At present, it is possible to establish empirical relationships between the efficiency and the key factors if other factors can be controlled experimentally. Furthermore, enabling techniques have been developed to measure DNA concentrations in the cytoplasm and the nucleus, providing quantitative information on rate limiting steps in intracellular gene delivery.
Transport of plasmid DNA (pDNA) in cells has been investigated in previous studies of non-viral gene delivery facilitated by synthetic materials, such as lipids and polymers [13-18], or pulsed electric field [1, 3, 19-22]. One important question in non-viral gene delivery is, what is the minimal number of pDNA molecules that need to be delivered into a cell for transgene expression? Different approaches may be used to answer the question. One is to directly quantify the initial pDNA copy number and the transgene expression in the same cell. Since the copy number has to be measured within a period post transfection to avoid pDNA degradation by intracellular nucleases, which is generally much shorter than the period required for transgene protein production, a technical challenge to this approach is whether the copy number measurement can be achieved without destruction of cells. Alternatively, different cells can be used to separately quantify the copy number and the transgene expression. However, these quantities distribute heterogeneously among different cells within the same sample. As a result, the experimental data cannot be directly used to establish the relationship between the initial pDNA copy number and the transgene expression in a cell. To circumvent this problem, we developed a statistical framework for determination of the relationship between two experimentally quantifiable factors — the average copy number of pDNA per cell among a group of cells after transfection and the percent of the cells with the transgene expression. To demonstrate the framework, we applied it to the analysis of electrotransfection. Based on the experimental data and the framework, we could predict the minimal number of pDNA molecules in a cell required for transgene expression.
2. Materials and Methods
2.1. Cell culture and DNA preparation
HCT116 cells (human colorectal carcinoma, ATCC, Manassas, VA, USA) were incubated in McCoy's 5a Medium (GIBCO) with 10% bovine calf serum and 1% penicillin-streptomycin (GIBCO) at 37°C in 5% CO2 and 95% air, and passaged every 2-3 days. pDNA encoding enhanced green fluorescence protein (EGFP) (pEGFP-N1) was purchased from Clontech (Mountain View, CA, USA), amplified using DH5α E.Coli, and purified from single colonies using Maxi DNA purification kits (QIAGEN) according to the manufacturer’s instruction. In some experiments, pDNA was labeled with rhodamine (Rho-pDNA), a fluorescent marker, using Label IT® CX-Rhodamine Labeling kit (Mirus Bio) according to the manufacturer’s instruction.
2.2. Degradation of extracellular DNA
To remove extracellular pDNA after electrotransfection, all samples were treated with a DNA removal product (DRP) (iQ-Check Free DNA Removal Solution, Bio-Rad). To determine its optimal concentration, the mixture was added to 1 ml of a pDNA solution prepared with the complete culture medium (1 μg/ml) at the volume ratios of 1:40, 1:80, 1:160, 1:320, and 1:640, respectively. In control groups, the pDNA solution was prepared with either pure water or complete culture medium without the DRP. After the samples were treated for 30 min, 10 μl of each sample was mixed with 2 μl loading dye containing 10x SYBR™ green I (ThermoFisher Scientific, Waltham, MA, USA), and loaded onto the 1% agarose gel prepared with 1x Tris-Acetate-EDTA buffer for electrophoresis of DNA. Thereafter, bands of DNA in the gel were analyzed for determining the optimal dilution factor for the DRP.
2.3. Electrotransfection
The cultured cells were collected by trypsinization, and 106 cells were counted and re-suspended in 100 μl of the pulsing buffer M1 (5 mM KCl, 15 mM MgCl2, 120 mM sodium phosphate, 50 mM mannitol, pH7.2), containing 1 μg pDNA unless indicated in Results section. The sample was loaded into disposable 2-mm gap aluminum cuvette (Bio-Rad), and treated with either one electric pulse (650 V and 400 μs) or two to three pulses (650 V, 400 μs, and 2 s interval) (BTX ECM830 Square Wave Electroporation System, Harvard Apparatus), unless indicated in Results section. In the 2- and 3-pulse groups, the buffer was supplemented with 0.2% type B gelatin (Sigma) that could significantly increase the cell viability by reducing electrotransfection-induced plasma membrane damage [23]. To verify the effect of gelatin on cell viability in the current study, we electrotransfected 1 μg pDNA into cells with 1 or 3 pulses, and measured the viability with the Trypan Blue Exclusion assay. Our data showed that the supplementation increased the viability (mean±SD) from 81.0%±0.8% to 90.8%±0.6% (n = 3) in the 3-pulse group, but had little effects on the viability in the 1-pulse group (91.8%±0.7% vs 92.4%±1.4%, n = 3), presumably because the latter was already high without the supplementation.
After pulsing, 1 ml of fresh complete medium pre-warmed at 37°C was added into the cuvette; and the cells were centrifuged to remove free pDNA in the medium, resuspended in 1 ml of the pre-warmed medium, and incubated at 37°C for 30 min. To further remove extracellular pDNA, 1 ml sample was treated with 10 μl solution of the DRP (1:100 dilution) for 30 min at 37°C. In control groups, the samples were treated with fresh medium without the DRP. After treatment, the medium was removed by centrifugation; and the cells were resuspended in 1 ml fresh medium for subsequent analysis.
As mentioned above, the electrotransfection was achieved with one to three pulses. For samples treated with one pulse, the analysis was performed immediately. For samples treated with two to three pulses, the cells were cultured prior to the analysis for 3 hours in 6-well plates at 37°C to allow cell recovery from pulsing-induced damages. The analysis included visual examination of samples under a light microscope, and quantitative measurement of cell viability and pDNA copy numbers in cells. The electrotransfection efficiency in all groups was quantified at 24 hours post pulsing.
2.4. Plasmid and genomic DNA extraction
DNA extraction from cells was performed for measurement of their copy numbers. In the experiment, cell suspension prepared after electrotransfection was washed twice with cold PBS. Intracellular pDNA and genomic DNA were extracted using the E.Z.N.A. tissue DNA kit (Omega Bio-Tek, Norcross, GA, USA) according to the manufacturer’s instruction for cultured cells. Briefly, BL buffer and proteinase K were added to cell suspension. After heated at 70°C for 10 min, the sample was mixed with pure ethanol (1:1, v/v), and loaded into the HiBind® DNA Mini column. After washing the column with different buffers, DNA molecules bound to the column were eluted twice using an elution buffer. For PCR analysis, the DNA solution might be diluted with the EB buffer if necessary.
To quantify the percent loss of DNA due the processes above, 105 untreated cells were lysed using the E.Z.N.A. tissue DNA kit, and mixed with 1 μl pDNA solution (1 μg/μl) before the sample was loaded into the purification column for DNA extraction. DNA sample eluted from the column was used to quantify GAPDH (for genomic DNA) and EGFP (for pDNA) copy numbers, using the qPCR technique. Data were compared with the known copy number of DNA in the sample for calculation of the percent loss of DNA.
2.5. Quantitative real-time PCR (qPCR)
It was performed with the CFX Connect™ Real-Time PCR Detection System (Bio-Rad), and the DNA amplification was determined, based on the SYBR® Green I detection chemistry. To quantify the copy number of the pDNA, the forward and the reverse primer sequences for EGFP were TGAACCGTCAGATCCGCTAG and GCTGAACTTGTGGCCGTTTA, respectively. To quantify the genomic DNA, we measured the copy number of a housekeeping gene, GAPDH. Its primer sequences were ACATCATCCCTGCCTCTAC and TCAAAGGTGGAGGAGTGG, respectively. All primers were purchased from Integrated DNA Technologies. In each PCR reaction, 18 μl of a solution containing 1x SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) and the two primers at 300 nM for each type, was mixed with 2 μl of the sample in a PCR tube. The sequences for EGFP and GAPDH were amplified in different tubes. The thermal cycle protocol was 3 min at 98°C followed by 45 cycles for amplification (98°C for 10 s, 60°C for 30 s). After the PCR reaction, the cycle threshold was converted to the copy number of pDNA or the number of cells, based on the standard curves (see below), for calculation of the copy number per cell. To minimize system errors, the melting curve analysis was performed for all samples (65-95°C at 0.5°C increment); the sample PCR data and the standard curves for both pDNA and genomic DNA were generated on the same PCR plate, and the PCR efficiency was determined for each plate with the standard curve data. If the efficiency was outside the range between 91% and 96%, all sample data for the plate would be discarded. Each reported PCR data was the mean of triplicates.
2.6. Standard curves of DNA
Mass concentration of DNA in the stock solution was determined with NanoDrop spectrophotometer (ThermoFisher Scientific). The stock solutions of pEGFP-N1 and human genomic DNA (EMD Millipore, Burlington, MA, USA) were diluted serially with pure water on the day of measurement, and loaded into different PCR tubes. After the PCR reaction (see above), the mass concentration of DNA was plotted as a function of the cycle threshold to generate the standard curves. The mass concentration was converted to the copy number of pDNA per unit volume based on its molecular weight, or to the number of cells per unit volume assuming that DNA loss during the extraction was negligible, and that each genome contained two copies of GAPDH.
2.7. Fluorescence microscopy and image analysis
Cells were maintained on ice after electrotransfection, and preloaded with CellTracker™ Green CMFDA Dye (ThermoFisher Scientific) before imaging. Fluorescence images of cells were acquired using a Revolution XD spinning disk confocal microscope (Andor Technology, Concord, MA) equipped with a 60x/NA1.2W corr UPlanApo objective. Super resolution images were obtained, using the super-resolution radial fluctuations (SRRF) method implemented in a Dragonfly spinning disk confocal microscope (Andor Technology) equipped with a 63X/1.47 TIRF HC PL APO corr oil objective. To count the number of Rho-pDNA molecules per cell, we converted the 3-D image stack to a 2-D image through maximum intensity projection, and segmented the cells in the image using CellProfiler. A manual threshold was set on the rhodamine channel (red) and the total number of pixels above this threshold was measured. Red objects in the image were identified as any group of pixels above this threshold with a diameter of 2-15 pixels (set to encompass the smallest and the largest punctate structures observed).
2.8. Electrotransfection efficiency measured with flow cytometry
Flow cytometry analysis was performed as previously described [3]. In brief, cells were trypsinized and resuspended in 350 μl of medium containing 5 μg/ml propidium iodide (PI). The sample was load into a flow cytometer (BD FACSCanto II, Becton Dickinson, Franklin Lakes, NJ). For each sample, 10,000 events were collected. Data analysis was performed using FlowJo. Electrotransfection efficiency was characterized by three parameters. One was electrotransfection effectiveness (eTE) defined as the percent of viable (PI−) cells that are EGFP+; the second parameter was average EGFP expression level, or the geometric mean (GM) of the fluorescence intensity (arbitrary unit) among EGFP+ cells; and third parameter was the apparent expression level per viable cell calculated as eTE*GM.
2.9. Mathematical model of non-viral gene delivery
The number of pDNA molecules transfected into individual cells is non-uniform. Some cells have more pDNA molecules than others, due to variations in different factors, such as cell and pDNA locations in the transfection buffer and activities of subcellular structures. To model the non-uniformity, we assume that the pDNA copy number per cell has a log-normal distribution within a short period post transfection when pDNA degradation is negligible,
| (1) |
where f is the probability density function, σ and μ are the shape and the location parameters, respectively, Dc is the number of pDNA molecules in a cell. We had also tested other functions (e.g., Poisson and half-normal) that satisfied the requirement — Dc ≥ 0 since the number of pDNA molecules in a cell cannot be negative, and observed that the log-normal function was the best choice under the current experimental conditions. After pDNA molecules are internalized by cells, a fraction of them will be degraded by intracellular nucleases. Therefore, only a small fraction of pDNA molecules can reach the nucleus for transgene expression, suggesting that Dc has to be greater than a threshold level, D0, to achieve successful transfection. Integration of Equation (1) from 0 to D0 yields the cumulative distribution function, F,
| (2) |
where erf is the error function. Since F is also the probability that the pDNA copy number in a cell is less than or equal to D0, the eTE can be calculated by,
| (3) |
and the average copy number per cells, E[Dc], can be calculated by,
| (4) |
To determine copy numbers of pDNA in different subcellular compartments, we developed a mathematical model of intracellular pDNA transport based on those reported in previous studies [17, 18] (see Fig. 1). The model assumes that cellular uptake and intracellular transport of pDNA are mediated by endocytosis, which is a common mechanism involved in most non-viral gene delivery approaches including electrotransfection [1, 3, 11, 22].
Figure 1.

Schematic of kinetic model for intracellular pDNA transport. It includes endosomal escape, complex formation between pDNA and nuclear import protein (NIP), binding to nuclear pore complex (NPC), nuclear entry, dissociation of pDNA from NIP, protein synthesis, and degradation of pDNA and protein.
The kinetic equations of the model are as follows.
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
| (11) |
where D and DI denote the numbers of pDNA and the complex formed by pDNA and nuclear import protein (NIP), respectively; the subscripts of D and DI indicate different compartments (see Fig. 1); NPC is the abbreviation of nuclear pore complex; and P denotes transgene protein. ki (i = 1,2,…,8) are rate constants; and their baseline values are listed in Table 1. Furthermore, the model assumes that pDNA molecules are transfected into cells within a short period, within which pDNA degradation in cells is negligible. Therefore, the initial value of Dendosome is equal to the total number of pDNA molecules delivered into the cell (i.e., Dc); and there are no pDNA molecules in other subcellular compartments at time zero. With these initial conditions, Equations (5) through (11) can be solved simultaneously using MATLAB® to calculate the values of P and D in each compartment as functions of time.
Table 1.
Baseline values of rate constants
| Name | Description | Value (min−1) |
|---|---|---|
| k1 | endosomal escape | 10 −2 |
| k2 | complex formation between pDNA and nuclear import protein | 2 × 10 −3 |
| k3 | pDNA degradation | 5 × 10 −3 |
| k4 | association of pDNA complex with NPC | 103 |
| k5 | entry of pDNA complex into the nucleus | 3 × 10 −3 |
| k6 | dissociation of pDNA from nuclear import protein | 103 |
| k7 | protein production | 10 −2 |
| k8 | protein degradation | 7.7 × 10 −4 |
2.10. Statistical analysis of experimental data
Difference among different experimental groups was compared using the Kruskal-Wallis test followed by Dunn’s post-hoc analysis (GraphPad Prism). A difference was considered to be statistically significant if the p-value was < 0.05.
3. Results
3.1. Quantitative measurement of average pDNA copy number per cell
Different techniques are available for measurement of DNA copy numbers in aqueous solutions. They include Southern blot [25], qPCR [13, 15, 16, 26], and droplet digital PCR [27]. We chose to use qPCR because its measurement is accurate over at least 5 orders of magnitude. In addition to the large dynamic range, qPCR allows to simultaneously quantify both pDNA and genomic DNA in the same sample for calculation of the average pDNA copy number per cell. However, two technical issues need to be addressed first before the technique could be used reliably in the study. The first was DNA loss during sample preparation that could cause underestimation of the copy number. To determine the percent loss, we lysed 105 untreated cells and mixed the lysate with 1 μg pDNA. The copy numbers of genomic DNA and pDNA in the mixture were then measured with qPCR (see the Materials and Methods section). Our data from 3 independent samples showed that the percent loss (mean±SD) was 10.1±1.7% for genomic DNA and 4.4±3.1% for pDNA, indicating that the DNA loss during extraction from cell lysates was insignificant.
The second issue was the large amount of extracellular pDNA in experimental samples after electrotransfection that could cause a significant overestimation of pDNA copy number in cells. To remove extracellular pDNA, we washed the cells with PBS twice prior to DNA extraction. The process could remove the majority of free pDNA molecules in the medium, but the amount of pDNA left in the medium was still higher than that in cells, especially when the electrotransfection efficiency was low. Additionally, the washing step could not effectively remove pDNA aggregates in the medium and pDNA bound to cell membrane. To solve the problem, we treated the washed cells with different reagents, such as trypsin and different DNase, and found that the DRP from Bio-Rad was the most efficient reagent for elimination of both membrane-bound and extracellular pDNA.
To determine the optimal concentration of the DRP, the stock solution was diluted at different times before being used to treat pDNA, and the fragmented DNA was subjected to gel electrophoresis after treatment. The results shown in Fig. 2A indicated that untreated pDNA in the complete medium migrated slower than that in pure water, presumably due to changes in pDNA conformation or partial neutralization of pDNA charge by chemicals in the medium. In the treated groups, a non-specific band of DNA was noticed in each lane near the 5000 bp location. They were unlikely to be DNA fragments because the intensity and the size of the bands were independent of the concentration of the DRP. They were likely to be the gel loading dye. In other regions of the gel, no significant bands were visible in the lanes of 1:40 and 1:160, indicating that all DNA fragments were smaller than 100 bp after the treatment at these concentrations of the DRP. When the stock solution of the DRP was diluted by a factor of 320, an extra DNA band was observed at 200 bp (Fig. 2A), and the band at the same location were larger and darker in the lane of 1:640, indicating that the DRP at low concentrations were ineffective for complete DNA degradation. Based on the data, we chose to use the DRP at the dilution factor of 1:100 to treat all samples after electrotransfection. At this concentration, the DRP treatment for 30 min caused insignificant changes in cell viability as determined with the Trypan Blue exclusion assay (data no shown).
Figure 2.

Validation of qPCR method for intracellular pDNA copy number measurement. (A) Typical image of agarose gel showing pDNA and its degraded fragments. Gel electrophoresis was performed after pDNA solutions prepared with complete culture medium were treated with the DRP diluted by different factors (1:40, 1:160, 1:320, and 1:640). The pDNA solutions in untreated control groups were prepared with pure water or complete culture medium (Med). (B)-(E) Typical fluorescence images of pDNA (red) and cells (green) after the buffer containing Rho-pDNA and cells (stained with CellTracker™ Green CMFDA) was treated with one electric pulse (650 V and 400 μs) (Pulsed). To remove extracellular pDNA after electrotransfection, the cells were treated with the DRP (1:100 dilution). The non-treated groups served as controls. (F)-(I) Typical fluorescence images of Rho-pDNA and cells. The experimental condition was similar to that in Panels B-E, except that the pulsing buffer was supplemented with 0.2% type B gelatin, and that 3 pulses (650 V/cm, 400 μs, 2 s interval) were applied to the cells. (J)&(K) Images of Rho-pDNA and cells at a higher magnification. The samples were from the same groups as those shown in Panels G&I, respectively. These images show apoptotic cells (black arrows) and pDNA aggregates either alone or complexed with membrane debris (white arrowheads). Scale bars: 40 μm in B-I; 20 μm in J&K.
To visualize effects of the DRP on extracellular DNA, cell suspensions were treated with the DRP for 30 min after electrotransfection of Rho-pDNA. Thereafter, the cytoplasm was labeled with CellTracker™ Green dye; and the samples were examined under a confocal microscope (Fig.2B through K). We observed that compared with the untreated controls, the DRP treatment significantly reduced signals from extracellular Rho-pDNA in the non-pulsed controls (Fig. 2B, D, F, H), and in samples treated with 1 pulse (Fig. 2C, E). Similarly, we observed that the treatment of samples with the DRP could reduce signals from extracellular Rho-pDNA in samples treated with 3 pulses (Fig. 2G, I) although the reduction was less significant compared with those in samples treated with none or 1 pulse, presumably due to pDNA aggregation caused by pulsing [28]. The aggregated DNA is more resistant to enzymatic degradation than free DNA molecules.
An increase in the pulse number also created large structures with red fluorescence (Fig 2J, K), which could be pDNA aggregates [28] or pDNA complexed with debris of cell membrane [2, 29-32]. The number of these structures was dramatically reduced after the DRP treatment (Fig. 2K). Quantitatively, we observed that the treatment could reduce the total copy number of pDNA in individual samples by 6 to 13 folds, indicating that the treatment could effectively remove extracellular and membrane bound pDNA. In subsequent experiments, all cell samples were treated with the DRP prior to DNA extraction from cells for qPCR analysis.
It is well-known that electrotransfection can induce cell apoptosis [33-35]. In the current study, the number of apoptotic cells in samples treated with 3 pulses was observed to be higher than those in the non-pulsed and 1-pulse groups. One concern was that a large amount of pDNA could accumulate in dead cells, causing overestimation of average pDNA copy number in samples. To address it, we examined the whole cell population after electrotransfection, and observed no cells (live or dead) containing a large amount of pDNA in the cytoplasm either before or after the DRP treatment (Fig. 2J, K).
3.2. Effects of pulsing conditions on average copy number of pDNA per cell
The effects were analyzed by varying DNA dose or concentration in the pulsing buffer, and electric pulse parameters (voltage and number). We observed that the average DNA copy number per cell at 1 h post electrotransfection (3250 V/cm, 400 μs, 1 pulse) increased with increasing the dose from 1 μg to 4 μg, which was equivalent to 1.96x1011 to 7.83x1011 copies of pDNA (Fig. 3A). The relationship was nearly linear, indicating that the ratio of intracellular vs extracellular copy numbers was dose-independent. Since the number of cells in each group was 106, the data indicated that only 0.1% of pDNA in the buffer was delivered into cells.
Figure 3.

Dependence of average pDNA copy number on pDNA dose and pulsing conditions. The copy number increased with increasing (A) the dose, (B) the pulse number, and (C) the pulse strength. The curves are the results of linear fitting. Error bar: SD; n = 3. *P < 0.05, the highest vs the lowest data points. (D)&(E) Super-resolution image of a sample. The experimental condition was the same as that described in Panel E of Figure 2. The signals from both red (Rho-pDNA) and green (cytosol) channels are shown in (D), and the red signal alone is shown in (E). The numbers 1 through 9 indicate the cells used for quantitative image analysis. Scale bar: 10 μm.
To quantify the dependence of pDNA copy number on pulse number and magnitude, 1 μg DNA was electrotransfected into cells with 1 to 3 pulses (at 650 V, 400 μs duration, and 2 s interval) in the buffer supplemented with 0.2% type B gelatin, and the cells were analyzed at 4 h after pulsing. The data showed that the average pDNA copy number per cell increased with increasing the pulse number (Fig. 3B). Interestingly, an increase in pulse number from 1 to 3 resulted in a similar increase in the pDNA copy number, compared with those achieved by increasing the DNA dose from 1 to 4 μg while the pulse number was fixed at unity (Fig. 3A). In a similar experiment, the pulse number was fixed at 3, but the pulse magnitude was increased from 350 to 650 V. We observed that the pDNA copy number per cell increased with increasing the pulse magnitude (Fig. 3C). To directly visualize individual pDNA molecules in cells, 1 μg of Rho-pDNA was electrotransfected into cells with one pulse; the sample was treated with the DRP, followed by fluorescence imaging with super-resolution confocal microscopy. The 3-D image stack was converted to a 2-D image through maximum intensity projection. A representative image is shown in Fig. 3D, E. Each red spot or object in the image could be a single or a small group of pDNA molecules, depending on the size of the spots. Through 2-D image analysis, the numbers of red pixel and red object per cell (mean ± SD, n = 9) were observed to be 1596 ± 835 and 60 ± 20, respectively.
3.3. Dependence of transgene expression on pDNA dose
One application of the method developed in the study was to determine the minimal pDNA copy number in a mammalian cell required for transgene expression. Viral vectors are known to be highly efficient in gene delivery. Some viral vectors can achieve transgene expression in certain mammalian cells with just one viral particle per cell [36-38]. However, the minimum copy number per cell is still unknown for electrotransfected pDNA. To estimate the number, we serially diluted pDNA solution so that its concentration in the pulsing buffer ranged from 15.625 ng to 500 ng. At 1 h post electrotransfection (650 V, 400 μs, 1 pulse), data from qPCR analysis showed that intracellular pDNA copy number increased linearly with increasing the pDNA dose (Fig. 4A). The data were consistent with those shown in Fig. 3A, where the pDNA dose was increased from 1 and 4 μg. When the DNA dose was 15.625 ng, which is equivalent to 3.06x109 copies of pDNA, the average copy number of pDNA was 3.5 per cell. Under the same experimental condition, we quantified the electrotransfection efficiency with three parameters defined in Section 2.8: (i) the eTE or the percent of viable cells that are EGFP+, (ii) the average EGFP expression level or the geometric mean (GM) of the fluorescence intensity (arbitrary unit) among EGFP+ cells; and (iii) the apparent expression level per viable cell calculated as eTE*GM. The eTE increased with increasing the dose (Fig. 4B), but the GM was insensitive to the dose (Fig. 4C). As a result, the apparent expression level increased with increasing the dose (Fig. 4D).
Figure 4.

Dependences of electrotransfection efficiency and average pDNA copy number on pDNA dose. (A) The copy number increased linearly with the pDNA dose. n = 4. (B)-(D) Effects of pDNA dose on the electrotransfection efficiency measured with three parameters: (B) eTE, (C) geometric mean of expression level, and (D) apparent expression level. n = 3. The curves are the results of linear fitting. Error bar: SD.
3.4. Numerical simulation of electrotransfection
Experimental data shown in Fig. 4 indicated that the copy number of pDNA per cell could significantly affect the eTE but had minor effects on the geometric mean of the transgene expression level. To explain the observations quantitatively, we developed a statistical framework for analyzing pDNA transfection (see the Materials and Methods section). Based on Equations (3) and (4) in the model, we performed regression analysis of the combined experimental data shown in Fig. 4A, B to determine the values of σ and D0. Result of the analysis is shown in Fig. 5A, which required σ and D0 to be equal to 2.184 and 172, respectively.
Figure 5.
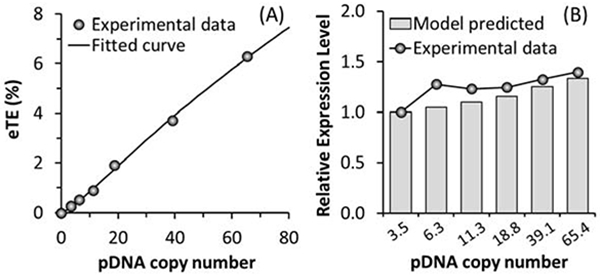
Numerical simulations of electrotransfection efficiency and its dependence on average pDNA copy number per cell. (A) Regression analysis of experimental data for determination of model constants (σ and D0). The symbols are experimental data of the eTE and the curve is the result of fitting the model to the data. (B) Model prediction of relative transgene expression level. The predicted values are compared with the experimental data described in Figure 4C after the pDNA dose was converted to the pDNA copy number using the data shown in Figure 4A.
The transgene expression level or the geometric mean of fluorescence intensity per cell (GM) was simulated by solving the kinetic equations described in the Materials and Methods section. The model predicted that the EGFP concentration at 24 hours post electrotransfection was a linear function of the amount of pDNA delivered into a cell (data not shown). Assuming the fluorescence intensity to be proportional to the EGFP concentration, the relative GM was calculated based on the simulated data (Fig. 5B). The model predictions were slightly lower than the normalized experimental data shown in Fig. 4C although both results demonstrated that the GM was less sensitive to changes in the pDNA copy number than the eTE. Furthermore, the kinetic model predicted that the copy number of pDNA in the nucleus was time-dependent (Fig. 6A) and increased linearly with increasing the number of pDNA molecules (Dc) delivered into cells; and that the value of Dc had minor effects on the time at which the peak level was reached, they were all close to 9.1 hours post electrotransfection. The peak copy number was approximately equal to 11 if Dc = D0, the threshold level above which the transgene expression becomes detectable experimentally.
Figure 6.

Numerical simulations of time-dependent pDNA copy numbers in the nucleus. (A) Baseline profiles of the simulated copy number at different initial conditions (Dc). The values of Dc for different curves (from bottom to top) are D0/3, D0/2, D0, 2D0, 3D0, 4D0, and 5D0, respectively; and D0 = 172. The peak level of the profile increases monotonically with increasing the value of Dc. (B) Profiles of the copy number when the rate constant for pDNA degradation is reduced from the baseline by 50%.
We discovered in a previous study that pretreatment of cells with a non-degradable sugar (e.g., sucrose) could inhibit pDNA degradation in cells [39]. To simulate the inhibition, we reduced the value of k3 by 50%, and re-calculated the profiles of pDNA copy number in the nucleus. Results shown in Fig. 6B indicated that the reduction in k3 increased the peak time from 9.1 to 12.4 hours although the shape of the profiles was still similar to that shown in Fig.6A. The inhibition of pDNA degradation would also increase the copy number in the nucleus and decrease the threshold, D0, thereby increasing the electrotransfection efficiency. For example, Equations (2)-(4) predicted that a decrease in D0 by 50% (i.e., from 172 to 86) could lead to 79% increase in the eTE if the average copy number per cells, E[Dc], was 65.4, which could be achieved experimentally at the pDNA dose of 500 ng (Fig.4A)
4. Discussion
We developed a statistical framework to explain the observations in electrotransfection experiments. The experimental data demonstrated that increasing pDNA dose, pulse strength, and pulse number could increase the average copy number of electrotransfected pDNA per cell. Under the experimental conditions used in the study, the eTE was approximately a linear function of the dose varying from 15.6 ng to 4 μg, , but the ratio in pDNA copy numbers between intracellular and extracellular spaces ( ≈ 0.001) was nearly independent of the dose. The ratio could be increased significantly by increasing the pulse magnitude or the pulse number. Based on the experimental data and the statistical framework, we could predict the minimal number of pDNA molecules required to be electrotransfected into a cell for transgene expression.
4.1. Error analysis in quantitative measurement of pDNA copy numbers
qPCR technique is currently the most sensitive approach to measurement of pDNA copy numbers, but it is also prone to overestimation when used to measure the intracellular copy number, due to the contamination by extracellular and cell membrane bound pDNA. Meanwhile, the copy number can be underestimated because the efficiency of pDNA extraction from cells is less than 100%. Therefore, the qPCR data need to be validated with different control experiments. In the current study, the first control experiment was to determine the efficiency of DNA extraction from cell samples. We lysed 105 cells and mixed the lysate with 1 μg pDNA, followed by DNA extraction from the mixture. Our data indicated that the extraction efficiency was 89.9% for genomic DNA and 95.6% for pDNA, indicating that the relative error in the pDNA copy number per cell, caused by incomplete extraction, was 5.7%.
The second control experiment was to determine if extracellular pDNA, including those bound to cell membrane, could be removed post electrotransfection. The majority of extracellular pDNA could be removed by simply washing the cells with PBS, but the amount of pDNA removed was still inadequate because only ~0.1% pDNA molecules were internalized by cells. To further remove the extracellular pDNA, we treated the washed cells with the DRP prior to cell lysing. The treatment could reduce the amount of pDNA in samples by 6 to 13 folds, presumably due to the degradation of extracellular DNA since the DRP could not enter the cells. To confirm the reduction, we examined the samples under a fluorescence microscope after electrotransfection of Rho-pDNA. It was observed that the fluorescence signal was significantly reduced after the DRP treatment. For samples where the electrotransfection was achieved with a single electric pulse, no red fluorescence was detectable in the extracellular medium (see Fig. 2E). To our knowledge, this is the first time that extracellular pDNA could be effectively removed for quantitative analysis of intracellular copy number of electrotransfected pDNA. However, the red fluorescent aggregates were still detectable in the medium after the DRP treatment if the electrotransfection was performed with 3 pulses, indicating that the DRP was less effective to degrade pDNA aggregates [28] or pDNA complexed with membrane debris from dead cells [30-32]. The observation suggested that the reported pDNA copy number per cell could be overestimated in experimental groups where 2 to 3 pulses were used for electrotransfection (see Fig. 3B). New techniques are needed in future studies to completely remove extracellular and membrane bound pDNA after multiple pulses are applied to samples for electrotransfection.
Advances in fluorescence microscopy have allowed for direct visualization of pDNA in live cells. In the third experiment for the validation of qPCR data, we used a state-of-the-art super resolution microscope to image Rho-pDNA molecules in live cells. The microscope with a 63x objective can resolve objects ~100 nm apart, which is adequate for detection of pDNA at the single molecule level. In the current study, we compared the pixel number, the object number, and the pDNA copy number in cells after electrotransfection with 1 pulse. They were approximately 1600, 60, and 180, respectively, which were consistent with the expectation. Generally, the number of pixels with red fluorescence should be larger than the pDNA copy number, presumably due to light scattering in samples; whereas the number of fluorescent objects in the image should be smaller than the copy number because each fluorescent object could represent a single or a group of pDNA molecules accumulated in an intracellular vesicle (e.g., endosome) as observed previously [1, 3, 39, 40].
The relationship between the transgene expression and the pDNA copy number in cells has been investigated in previous studies of lipoplex and polyplex mediated gene delivery [13-16]. For example, the copy number in the nucleus was observed to be 75 to 50,000, depending the pDNA dose [13, 14], indicating that the average pDNA copy numbers per cell observed in these studies are much higher than the data reported in the current study. It is unclear if the discrepancy is caused by differences in gene delivery methods or contamination of the samples with pDNA outside the nucleus. Interestingly, the previous studies have also observed that the transgene expression level is a sigmodal function of the pDNA copy number in the nucleus. The expression level is saturated if the copy number is higher than a few thousands, but undetectable when the copy number is lower than a threshold level. The same threshold phenomenon had also been observed in the current study but at a much lower level. In another study, pDNA encoding Herpes simplex virus thymidine kinase (TK) was microinjected into the nucleus of a TK deficient mouse cell line for determination of the minimum number of pDNA molecules per nucleus required for transgene expression [41]. The investigator showed that as few as 5 copies of pDNA was needed for detectable TK activity in the cells, and that microinjection of the same amount of pDNA into the cytoplasm could not lead to transgene expression. A similar result was observed in an in vivo study where the authors used the hydrodynamics-based injection technique to introduce pDNA into mouse liver cells [42]. They showed that on average, only 1 to 2 pDNA molecules were needed in the nucleus for transgene expression. Considering that pDNA was delivered to only a small fraction of the cells in the liver, the true copy number per cell should be several folds higher than the estimate. Together, the data reported in the literature [41, 42] were on the same order of magnitude as that predicted by the current study (see Fig. 6).
4.2. Mathematical modeling of non-viral gene delivery
Numerous mathematical models have been developed to study non-viral gene delivery in cells [17-19, 43, 44]. Most of them are focused on the kinetics of nanoparticle-mediated delivery in cells via endocytic pathways [17, 18], which may involve binding of nanoparticles to cell membrane, internalization by vesicles, vesicular transport, endosomal escape, release of DNA from nanoparticles, and nuclear entry of DNA. One of the limitations in these studies is that they have only investigated the average behavior of gene delivery, and ignored the difference in the rate of DNA uptake by different cells. To circumvent the problem, we proposed a statistical framework to consider the heterogeneity in gene delivery. It was a combination of kinetic analysis of DNA transport in single cells and statistical modeling of the DNA uptake by a group of cells. The new framework had been used successfully to explain experimental data of electrotransfection observed in the current study, and predict the minimum number of pDNA molecules that have to be electrotransfected into a cell for transgene expression. The prediction is consistent with experimental data in the current and other studies in the literature [45-51]. In addition to eTE, our model could predict the dose dependence of the geometric mean of EGFP expression. The prediction might be considered as an evidence for validating the model. We expect that the framework can be utilized generally to investigate relationships among experimentally observed quantities: (i) average pDNA copy number per nucleus or per cell, (ii) fraction of cells expressing transgene, and (iii) geometric mean of transgene expression level in non-viral gene delivery studies.
Although the statistical framework could fit the current experimental data well, the assumptions in the kinetic model and the choice of probability density function may not be the best for other studies with different experimental conditions and cell types. However, we would like to argue that without the assumptions, the framework is still valid qualitatively for analysis of electrotransfection in different studies because of the three observations. First, the number of pDNA molecules is heterogeneously distributed among different cells after application of electric pulses. It is high in some cells; and low or equal to zero in others. Second, a fraction of pDNA molecules internalized by cells will inevitably be degraded by nucleases in cytoplasm or nucleus [29, 51]. Finally, it is unlikely for all electrotransfected pDNA molecules to enter the nucleus even without the degradation, because they can be sequestered by subcellular structures (e.g., endosome) and their transport in cells can be hindered by physiological barriers (e.g., nuclear envelope). These observations indicate that transgene expression can happen only in a fraction of the cells, measured as the eTE in the literature [51], in which the number of pDNA molecules in a cell has to be larger than the threshold (D0); and that the pDNA copy number is non-uniformly distributed among different cells. The non-uniformity can be modeled by a probability density function (e.g., log-normal function); and the area under the curve for Dc ≥ D0 is the eTE. Within a cell, the numbers of pDNA molecules in subcellular compartments and the transgene protein can be predicted by using a kinetic model (e.g., the one used in the current study). Together, these models could be used generally for quantitative analysis of electrotransfection in vitro.
Increasing pDNA dose can improve transfection efficiency, but may also induce stronger innate immune responses to exogenous DNA [9, 52, 53], preventing long-term transgene expression. Therefore, a fundamental question in non-viral gene delivery is, what is the minimum amount of plasmid that have to be delivered into a cell for transgene expression? To answer the question, we analyzed the experimental data and performed numerical simulations with the statistical framework. We could predict that under the current experimental conditions, the minimal pDNA copy number required for transgene expression (Do) was 172; and the corresponding copy number of pDNA in the nucleus varied with time (Fig.6). The peak level was 11, which was consistent with the data reported in the literature [41, 42]. The observation suggests that only a few copies of pDNA need to be delivered into the nucleus for transgene expression in a cell. Furthermore, the model predicted that inhibition of pDNA degradation could reduce the threshold level (Do), thus increase the eTE.
In conclusion, we developed a statistical framework for analysis of non-viral gene delivery. It can be used to establish the relationship between the experimental data of average pDNA copy number per cell and transfection efficiency, and to predict the minimum amount of pDNA required to be delivered into a cell for transgene expression. The framework will be useful for understanding mechanisms of pDNA transport and development of new strategies to improve of non-viral gene delivery.
Highlights.
A framework was developed for quantitative analysis of non-viral gene delivery
The framework was applied to analysis of electrotransfection data
Average copy number of plasmid per cell was quantified experimentally under different conditions
The study predicted the copy numbers of plasmid in different subcellular compartments
The copy number in a cell had to be higher than a threshold to achieve transgene expression
Acknowledgments
This research was funded partly by a grant from National Institutes of Health (GM130830).
Footnotes
Declaration of Competing Interest
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Cervia LD, Chang CC, Wang L, Yuan F, Distinct effects of endosomal escape and inhibition of endosomal trafficking on gene delivery via electrotransfection, PLoS One, 12 (2017) e0171699, 10.1371/journal.pone.0171699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chang CC, Wu M, Yuan F, Role of specific endocytic pathways in electrotransfection of cells, Mol Ther Methods Clin Dev, 1 (2014) 14058, 10.1038/mtm.2014.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mao M, Wang L, Chang CC, Rothenberg KE, Huang J, Wang Y, Hoffman BD, Liton PB, Yuan F, Involvement of a Rac1-Dependent Macropinocytosis Pathway in Plasmid DNA Delivery by Electrotransfection, Mol Ther, 25 (2017) 803–815, 10.1016/j.ymthe.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wu M, Yuan F, Membrane binding of plasmid DNA and endocytic pathways are involved in electrotransfection of mammalian cells, PLoS One, 6 (2011) e20923, 10.1371/journal.pone.0020923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Neumann E, Schaefer-Ridder M, Wang Y, Hofschneider PH, Gene transfer into mouse lyoma cells by electroporation in high electric fields, EMBO J, 1 (1982) 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kotnik T, Pucihar G, Rebersek M, Miklavcic D, Mir LM, Role of pulse shape in cell membrane electropermeabilization, Biochim Biophys Acta, 1614 (2003) 193–200, 10.1016/s0005-2736(03)00173-1. [DOI] [PubMed] [Google Scholar]
- [7].Agarwal A, Zudans I, Weber EA, Olofsson J, Orwar O, Weber SG, Effect of cell size and shape on single-cell electroporation, Anal Chem, 79 (2007) 3589–3596, 10.1021/ac062049e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sherba JJ, Hogquist S, Lin H, Shan JW, Shreiber DI, Zahn JD, The effects of electroporation buffer composition on cell viability and electro-transfection efficiency, Sci Rep, 10 (2020) 3053, 10.1038/s41598-020-59790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bosnjak M, Jesenko T, Kamensek U, Sersa G, Lavrencak J, Heller L, Cemazar M, Electrotransfer of Different Control Plasmids Elicits Different Antitumor Effectiveness in B16.F10 Melanoma, Cancers (Basel), 10 (2018), 10.3390/cancers10020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guo S, Jackson DL, Burcus NI, Chen YJ, Xiao S, Heller R, Gene electrotransfer enhanced by nanosecond pulsed electric fields, Mol Ther Methods Clin Dev, 1 (2014) 14043, 10.1038/mtm.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Midoux P, Pichon C, Yaouanc JJ, Jaffres PA, Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers, Br J Pharmacol, 157 (2009) 166–178, 10.1111/j.1476-5381.2009.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stewart MP, Langer R, Jensen KF, Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts, Chem Rev, 118 (2018) 7409–7531, 10.1021/acs.chemrev.7b00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cohen RN, van der Aa MA, Macaraeg N, Lee AP, Szoka FC Jr., Quantification of plasmid DNA copies in the nucleus after lipoplex and polyplex transfection, J Control Release, 135 (2009) 166–174, 10.1016/j.jconrel.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Glover DJ, Leyton DL, Moseley GW, Jans DA, The efficiency of nuclear plasmid DNA delivery is a critical determinant of transgene expression at the single cell level, J Gene Med, 12 (2010) 77–85, 10.1002/jgm.1406. [DOI] [PubMed] [Google Scholar]
- [15].Moriguchi R, Kogure K, Iwasa A, Akita H, Harashima H, Non-linear pharmacodynamics in a non-viral gene delivery system: positive non-linear relationship between dose and transfection efficiency, J Control Release, 110 (2006) 605–609, 10.1016/j.jconrel.2005.10.021. [DOI] [PubMed] [Google Scholar]
- [16].Tachibana R, Harashima H, Ide N, Ukitsu S, Ohta Y, Suzuki N, Kikuchi H, Shinohara Y, Kiwada H, Quantitative analysis of correlation between number of nuclear plasmids and gene expression activity after transfection with cationic liposomes, Pharm Res, 19 (2002) 377–381, 10.1023/a:1015162722295. [DOI] [PubMed] [Google Scholar]
- [17].Varga CM, Hong K, Lauffenburger DA, Quantitative analysis of synthetic gene delivery vector design properties, Mol Ther, 4 (2001) 438–446, 10.1006/mthe.2001.0475. [DOI] [PubMed] [Google Scholar]
- [18].Banks GA, Roselli RJ, Chen R, Giorgio TD, A model for the analysis of nonviral gene therapy, Gene Ther, 10 (2003) 1766–1775, 10.1038/sj.gt.3302076. [DOI] [PubMed] [Google Scholar]
- [19].Li J, Lin H, Numerical simulation of molecular uptake via electroporation, Bioelectrochemistry, 82 (2011) 10–21, 10.1016/j.bioelechem.2011.04.006. [DOI] [PubMed] [Google Scholar]
- [20].Zaharoff DA, Henshaw JW, Mossop B, Yuan F, Mechanistic analysis of electroporation-induced cellular uptake of macromolecules, Exp Biol Med (Maywood), 233 (2008) 94–105, https://doi.org/233/1/94 [pii] 10.3181/0704-RM-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Escoffre JM, Portet T, Wasungu L, Teissie J, Dean D, Rols MP, What is (still not) known of the mechanism by which electroporation mediates gene transfer and expression in cells and tissues, Mol Biotechnol, 41 (2009) 286–295, 10.1007/s12033-008-9121-0. [DOI] [PubMed] [Google Scholar]
- [22].Rosazza C, Deschout H, Buntz A, Braeckmans K, Rols MP, Zumbusch A, Endocytosis and Endosomal Trafficking of DNA After Gene Electrotransfer In Vitro, Mol Ther Nucleic Acids, 5 (2016) e286, 10.1038/mtna.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang Y, Chang CC, Wang L, Yuan F, Enhancing cell viability and efficiency of plasmid dna electrotransfer through reducing plasma membrane permeabilization, Bioelectricity, 2 (2020) 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Danhier P, Krishnamachary B, Bharti S, Kakkad S, Mironchik Y, Bhujwalla ZM, Combining optical reporter proteins with different half-lives to detect temporal evolution of hypoxia and reoxygenation in tumors, Neoplasia, 17 (2015) 871–881, 10.1016/j.neo.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Carapuca E, Azzoni AR, Prazeres DM, Monteiro GA, Mergulhao FJ, Time-course determination of plasmid content in eukaryotic and prokaryotic cells using real-time PCR, Mol Biotechnol, 37 (2007) 120–126, 10.1007/s12033-007-0007-3. [DOI] [PubMed] [Google Scholar]
- [26].Grandjean M, Girod PA, Calabrese D, Kostyrko K, Wicht M, Yerly F, Mazza C, Beckmann JS, Martinet D, Mermod N, High-level transgene expression by homologous recombination-mediated gene transfer, Nucleic Acids Res, 39 (2011) e104, 10.1093/nar/gkr436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Plotka M, Wozniak M, Kaczorowski T, Quantification of Plasmid Copy Number with Single Colour Droplet Digital PCR, PLoS One, 12 (2017) e0169846, 10.1371/journal.pone.0169846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chang CC, Mao M, Liu Y, Wu M, Vo-Dinh T, Yuan F, Improvement in Electrotransfection of Cells Using Carbon-Based Electrodes, Cell Mol Bioeng, 9 (2016) 538–545, 10.1007/s12195-016-0452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Henshaw JW, Yuan F, Field distribution and DNA transport in solid tumors during electric field-mediated gene delivery, J Pharm Sci, 97 (2008) 691–711, 10.1002/jps.21000. [DOI] [PubMed] [Google Scholar]
- [30].Paganin-Gioanni A, Bellard E, Escoffre JM, Rols MP, Teissie J, Golzio M, Direct visualization at the single-cell level of siRNA electrotransfer into cancer cells, Proc Natl Acad Sci U S A, 108 (2011) 10443–10447, 10.1073/pnas.1103519108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Faurie C, Rebersek M, Golzio M, Kanduser M, Escoffre JM, Pavlin M, Teissie J, Miklavcic D, Rols MP, Electro-mediated gene transfer and expression are controlled by the life-time of DNA/membrane complex formation, J Gene Med, 12 (2010) 117–125, 10.1002/jgm.1414. [DOI] [PubMed] [Google Scholar]
- [32].Sachdev S, Feijoo Moreira S, Keehnen Y, Rems L, Kreutzer MT, Boukany PE, DNA-membrane complex formation during electroporation is DNA size-dependent, Biochim Biophys Acta Biomembr, 1862 (2020) 183089, 10.1016/j.bbamem.2019.183089. [DOI] [PubMed] [Google Scholar]
- [33].Li LH, McCarthy P, Hui SW, High-efficiency electrotransfection of human primary hematopoietic stem cells, FASEB J, 15 (2001) 586–588, 10.1096/fj.00-0447fje. [DOI] [PubMed] [Google Scholar]
- [34].Wang C, Chang CC, Wang L, Yuan F, Inhibition of caspases improves non-viral T cell receptor editing, Cancers, under review (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Beebe SJ, Sain NM, Ren W, Induction of Cell Death Mechanisms and Apoptosis by Nanosecond Pulsed Electric Fields (nsPEFs), Cells, 2 (2013) 136–162, 10.3390/cells2010136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Negre D, Mangeot PE, Duisit G, Blanchard S, Vidalain PO, Leissner P, Winter AJ, Rabourdin-Combe C, Mehtali M, Moullier P, Darlix JL, Cosset FL, Characterization of novel safe lentiviral vectors derived from simian immunodeficiency virus (SIVmac251) that efficiently transduce mature human dendritic cells, Gene Ther, 7 (2000) 1613–1623, 10.1038/sj.gt.3301292. [DOI] [PubMed] [Google Scholar]
- [37].Mangeot PE, Duperrier K, Negre D, Boson B, Rigal D, Cosset FL, Darlix JL, High levels of transduction of human dendritic cells with optimized SIV vectors, Mol Ther, 5 (2002) 283–290, 10.1006/mthe.2002.0541. [DOI] [PubMed] [Google Scholar]
- [38].Wu J, Zhang S, Wu X, Dong X, Xu P, Liu X, Li C, Huang Q, Enhanced transduction and improved photoreceptor survival of retinal degeneration by the combinatorial use of rAAV2 with a lower dose of adenovirus, Vision Res, 48 (2008) 1648–1654, 10.1016/j.visres.2008.04.019. [DOI] [PubMed] [Google Scholar]
- [39].Mao M, Chang C-C, Pickar-Oliver A, Cervia LD, Wang L, Ji J, Liton PB, Gersbach CA, Yuan F, Redirecting vesicular transport to improve nonviral delivery of molecular cargo, Advanced Biosystems, 4 (2020) e2000059, 10.1002/adbi.202000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang L, Miller SE, Yuan F, Ultrastructural Analysis of Vesicular Transport in Electrotransfection, Microsc Microanal, 24 (2018) 553–563, 10.1017/S143192761801509X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Capecchi MR, High efficiency transformation by direct microinjection of DNA into cultured mammalian cells, Cell, 22 (1980) 479–488, 10.1016/0092-8674(80)90358-x. [DOI] [PubMed] [Google Scholar]
- [42].Ochiai H, Harashima H, Kamiya H, Intranuclear disposition of exogenous DNA in vivo: silencing, methylation and fragmentation, FEBS Lett, 580 (2006) 918–922, 10.1016/j.febslet.2006.01.017. [DOI] [PubMed] [Google Scholar]
- [43].Varga CM, Wickham TJ, Lauffenburger DA, Receptor-mediated targeting of gene delivery vectors: insights from molecular mechanisms for improved vehicle design, Biotechnol Bioeng, 70 (2000) 593–605, . [DOI] [PubMed] [Google Scholar]
- [44].Leguebe M, Notarangelo MG, Twarogowska M, Natalini R, Poignard C, Mathematical model for transport of DNA plasmids from the external medium up to the nucleus by electroporation, Math Biosci, 285 (2017) 1–13, 10.1016/j.mbs.2016.11.015. [DOI] [PubMed] [Google Scholar]
- [45].Gilbert R, Jaroszeski MJ, Heller L, Heller R, Electric field enhanced plasmid delivery to liver hepatocellular carcinomas, Technol Cancer Res Treat, 1 (2002) 355–364, 10.1177/153303460200100506. [DOI] [PubMed] [Google Scholar]
- [46].Atkins RM, Fawcett TJ, Gilbert R, Hoff AM, Connolly R, Brown DW, Llewellyn AJ, Jaroszeski MJ, Impedance spectroscopy as an indicator for successful in vivo electric field mediated gene delivery in a murine model, Bioelectrochemistry, 115 (2017) 33–40, 10.1016/j.bioelechem.2017.01.004. [DOI] [PubMed] [Google Scholar]
- [47].Boukany PE, Morss A, Liao WC, Henslee B, Jung H, Zhang X, Yu B, Wang X, Wu Y, Li L, Gao K, Hu X, Zhao X, Hemminger O, Lu W, Lafyatis GP, Lee LJ, Nanochannel electroporation delivers precise amounts of biomolecules into living cells, Nat Nanotechnol, 6 (2011) 747–754, 10.1038/nnano.2011.164. [DOI] [PubMed] [Google Scholar]
- [48].Heller R, Heller LC, Gene electrotransfer clinical trials, Adv Genet, 89 (2015) 235–262, 10.1016/bs.adgen.2014.10.006. [DOI] [PubMed] [Google Scholar]
- [49].Gehl J, Mir LM, Determination of optimal parameters for in vivo gene transfer by electroporation, using a rapid in vivo test for cell permeabilization, Biochem Biophys Res Commun, 261 (1999) 377–380, 10.1006/bbrc.1999.1014. [DOI] [PubMed] [Google Scholar]
- [50].May RD, Tekari A, Frauchiger DA, Krismer A, Benneker LM, Gantenbein B, Efficient Nonviral Transfection of Primary Intervertebral Disc Cells by Electroporation for Tissue Engineering Application, Tissue Eng Part C Methods, 23 (2017) 30–37, 10.1089/ten.TEC.2016.0355. [DOI] [PubMed] [Google Scholar]
- [51].Cervia LD, Yuan F, Current progress in electrotransfection as a nonviral method for gene delivery, Molecular pharmaceutics, 15 (2018) 3617–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Znidar K, Bosnjak M, Cemazar M, Heller LC, Cytosolic DNA sensor upregulation accompanies DNA electrotransfer in B16. F10 melanoma cells, Molecular Therapy-Nucleic Acids, 5 (2016) e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wu J, Chen ZJ, Innate immune sensing and signaling of cytosolic nucleic acids, Annu Rev Immunol, 32 (2014) 461–488, 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]