

Abstract
Delays in achieving the global eradication of wild poliovirus transmission continue to postpone subsequent cessation of all oral poliovirus vaccine (OPV) use. Countries must stop OPV use to end all cases of poliomyelitis, including vaccine-associated paralytic polio (VAPP) and cases caused by vaccine-derived polioviruses (VDPVs). The Global Polio Eradication Initiative (GPEI) coordinated global cessation of all type 2 OPV (OPV2) use in routine immunization in 2016 but did not successfully end the transmission of type 2 VDPVs (VDPV2s), and consequently continues to use type 2 OPV (OPV2) for outbreak response activities. Using an updated global poliovirus transmission and OPV evolution model, we characterize outbreak response options for 2019–2029 related to responding to VDPV2 outbreaks with a genetically stabilized novel OPV (nOPV2) strain or with the currently licensed monovalent OPV2 (mOPV2). Given uncertainties about the properties of nOPV2, we model different assumptions that appear consistent with the evidence on nOPV2 to date. Using nOPV2 to respond to detected cases may reduce the expected VDPV and VAPP cases and the risk of needing to restart OPV2 use in routine immunization compared to mOPV2 use for outbreak response. The actual properties, availability, and use of nOPV2 will determine its effects on type 2 poliovirus transmission in populations. Even with optimal nOPV2 performance, countries and the GPEI would still likely need to restart OPV2 use in routine immunization in OPV-using countries if operational improvements in outbreak response to stop the transmission of cVDPV2s are not implemented effectively.
Keywords: polio, dynamic modeling, outbreak response
1. INTRODUCTION
As efforts to eradicate polio continue, countries continue to use oral poliovirus vaccine (OPV) and inactivated poliovirus vaccine (IPV) in their immunization programs. Using a global polio model (Duintjer Tebbens, Pallansch, Wassalik, Cochi, & Thompson, 2015) that included OPV evolution (Duintjer Tebbens, Pallansch, Kalkowska, et al., 2013), we recommended outbreak response strategies (Duintjer Tebbens, Pallansch, Wassilak, Cochi, & Thompson, 2016) to support the Global Polio Eradication Initiative (GPEI) and its 2013–2018 Strategic Plan (World Health Organization Global Polio Eradication Initiative, 2013). Recognizing the importance of the availability of vaccine for outbreak response, we also used the model to explore vaccine demands and the dynamics of stockpiles of monovalent OPV (mOPV) (Duintjer Tebbens, Pallansch, et al., 2016). The model includes consideration of all recognized risks associated with the use of OPV, including vaccine-associated paralytic polio (VAPP) and vaccine-derived polioviruses (VDPVs). To manage VAPP and VDPV risks, in 2008 the World Health Assembly resolved to eventually end all OPV use in routine immunization (RI) following successful eradication of wild polioviruses (World Health Assembly, 2008). Modelers recommended that OPV-using countries conduct sufficient supplementary immunization activities (SIAs) using OPV2-containing vaccines before OPV2 cessation to raise population immunity to transmission enough such that, after OPV2 cessation, the OPV2-related viruses would die out instead of evolving to become circulating VDPVs (cVDPVs) (Thompson & Duintjer Tebbens, 2014).
In late April-early May 2016, the GPEI globally coordinated the cessation of type 2-containing OPV (OPV2) (Hampton et al., 2016), which implied ending all use of trivalent OPV (tOPV, which contains all three OPV types) and replacing it with bivalent OPV (bOPV, which contains only types 1 and 3 OPV). This led to the interruption of most OPV2-related virus transmission (Diop et al., 2017) and ended all type 2 VAPP (VAPP2) associated with RI. Unfortunately, not all countries achieved sufficiently high levels of population immunity in all geographies prior to OPV2 cessation to prevent transmission, and this led to the identification of some type 2 cVDPV (cVDPV2) outbreaks shortly after OPV2 cessation (Duintjer Tebbens & Thompson, 2018) (mostly, but not all, due to undetected VDPV2 circulating prior to the cessation of OPV2). Transmission of cVDPV2s continues to date in increasing numbers of geographies (Blake et al., 2018; Kalkowska, Pallansch, et al., 2020; Macklin et al., 2020), some of which appear to be associated with use of mOPV2 for outbreak response leading to the emergence of new cVDPV2s. We recently updated our global model to account for recent programmatic and epidemiological experience (i.e., delays in achieving global eradication and ceasing OPV2 use) and new scientific evidence available as of the end of 2019 including figures showing the model estimates of cases compared to reported cases up through 2018 (Alleman et al., 2020; Kalkowska, Wassilak, Cochi, Pallansch, & Thompson, 2020). In addition, we recently identified the need to consider the unexpected detection of transmission of OPV2-related viruses in areas where they should not be present (Blake et al., 2018; Kalkowska, Pallansch, et al., 2020; Macklin et al., 2020). Thus, we provided an updated estimate of the likely need to restart OPV2 use for routine immunization in OPV-using countries and showed the expected value of modeled cases compared to reported cases for 2019–2020 for serotype 2 (Kalkowska, Pallansch, et al., 2020).
Despite prior recommendations for aggressive outbreak response to any cVDPVs and clear GPEI standard operating procedures for responses to cVDPV2 outbreaks (Duintjer Tebbens, Pallansch, et al., 2016), most countries with outbreaks did not conduct prompt, high-quality, and in some cases sufficiently large, outbreak response SIAs (oSIAs) to stop transmission. In addition, despite model results characterizing the use of IPV for oSIAs when used instead of or in addition to IPV as not effective at stopping transmission and not cost-effective (i.e., not a good value for money) (Duintjer Tebbens & Thompson, 2017a), some countries used IPV for oSIAs. As of the beginning of 2020, the GPEI does not appear on track with respect to stopping transmission of cVDPV2s or wild poliovirus type 1 (WPV1) (Kalkowska, Wassilak, et al., 2020). Characterizing the transmission dynamics of cVDPV2s in 2020 and the potential role of recently developed, genetically stabilized novel OPV2 (nOPV2) offers the potential to support future GPEI strategies and to anticipate OPV2 needs (Thompson & Kalkowska, 2019). The serious health and socioeconomic consequences of the Coronavirus Disease 2019 (COVID-19) pandemic beginning in early 2020, including disruption of polio immunization activities, will likely further exacerbate the situation with cVDPV2 outbreaks and increase the likelihood of OPV2 restart, which we leave to future studies. We previously estimated the probabilities of needing to restart OPV2 in RI and SIAs using the available type 2 mOPV (mOPV2) vaccine (Kalkowska, Pallansch, et al., 2020; Kalkowska, Wassilak, et al., 2020), but did not include the potential use of nOPV2. Given the prominent role of nOPV2 in the recent GPEI strategy to manage cVDPV2s (World Health Organization Global Polio Eradication Initiative, 2020b), and GPEI policy to limit OPV2 use to emergency use in outbreak populations only (World Health Organization Global Polio Eradication Initiative, 2018, 2020a), we explore the impacts of using nOPV2 for outbreak response. Given the complex nature of the biological issues involved in disease transmission, vaccine development and modeling, Section 2 provides a review of polioviruses and OPV, and Section 3 discusses the development of nOPV strains and the lead nOPV2 candidate for use in oSIAs. Sections 4–6 present the methods, results, and discussion.
2. POLIOVIRUSES AND OPV
Each of the three types (1, 2, and 3) of the small (i.e., virus particle size of 28 nm) and non-enveloped polioviruses contain a single-stranded, positive-sense ribonucleic acid (RNA) genome, and a genomic organization common to all 110+ enterovirus (EV) types (Coyne, Oberste, & Pallansch, 2017; Oh, Pathak, Goodfellow, Arnold, & Cameron, 2009; Adi Stern et al., 2017). Traditionally, laboratories identified all enteroviruses by their antigenic properties, which defined the serotypes. With advances in technology, molecular typing based on partial genome sequencing supplanted antigenic typing, so virologists now often simply refer to “type” instead of “serotype.” The ~7500 nucleotide (nt) genome includes a single open reading frame (ORF) that encodes a polyprotein that is subsequently processed by viral proteases (i.e., 2A and 3C) into all the structural and functional viral proteins (Fig. 1(a)) (Adi Stern et al., 2017). The ORF for a poliovirus encodes four viral capsid proteins (VP1, VP2, VP3, and VP4) that provide the outer structure of the virus particle, and also encodes multiple non-structural proteins that play key roles in virus replication. Key non-structural proteins for EVs include the 2A and 3C proteases, the RNA-dependent RNA polymerase (RdRp) (i.e., 3Dpol for all EVs), and viral genome-linked protein (VPg) (3B) covalently linked to the 5’-end of the genomic RNA in the virus particle. These and other proteins collectively control key viral and cellular functions and structures to facilitate viral protein synthesis and viral RNA replication in the cytoplasm of infected host cells. Specifically, the 3C protease cleaves several host cell proteins, thus turning off those cellular functions (e.g., protein translation) to make the cellular resources available to support viral RNA replication and the production of viral proteins. Untranslated regions (UTRs, interchangeably called non-translated regions or NTRs) flank each end of the coding region, designated as the 5’-UTR (beginning) and 3’-UTR (ending). The 5’-UTR includes highly conserved sequences that fold into three-dimensional structures and form critical functional units for viral replication. These include a cloverleaf structure (CL), essential for initiating RNA synthesis, and an internal ribosome entry site (IRES) that facilitates preferential translation of the viral RNA to produce the viral proteins. Finally, a cis replication element (cre) located in the 2C region of the genome functions as a template for uridylylation of the VPg protein. The uridylated VPg, termed VPg-pUpU, then functions as a primer to initiate positive-sense viral RNA synthesis at the 3’-end of the negative-sense genome RNA.
Fig. 1.
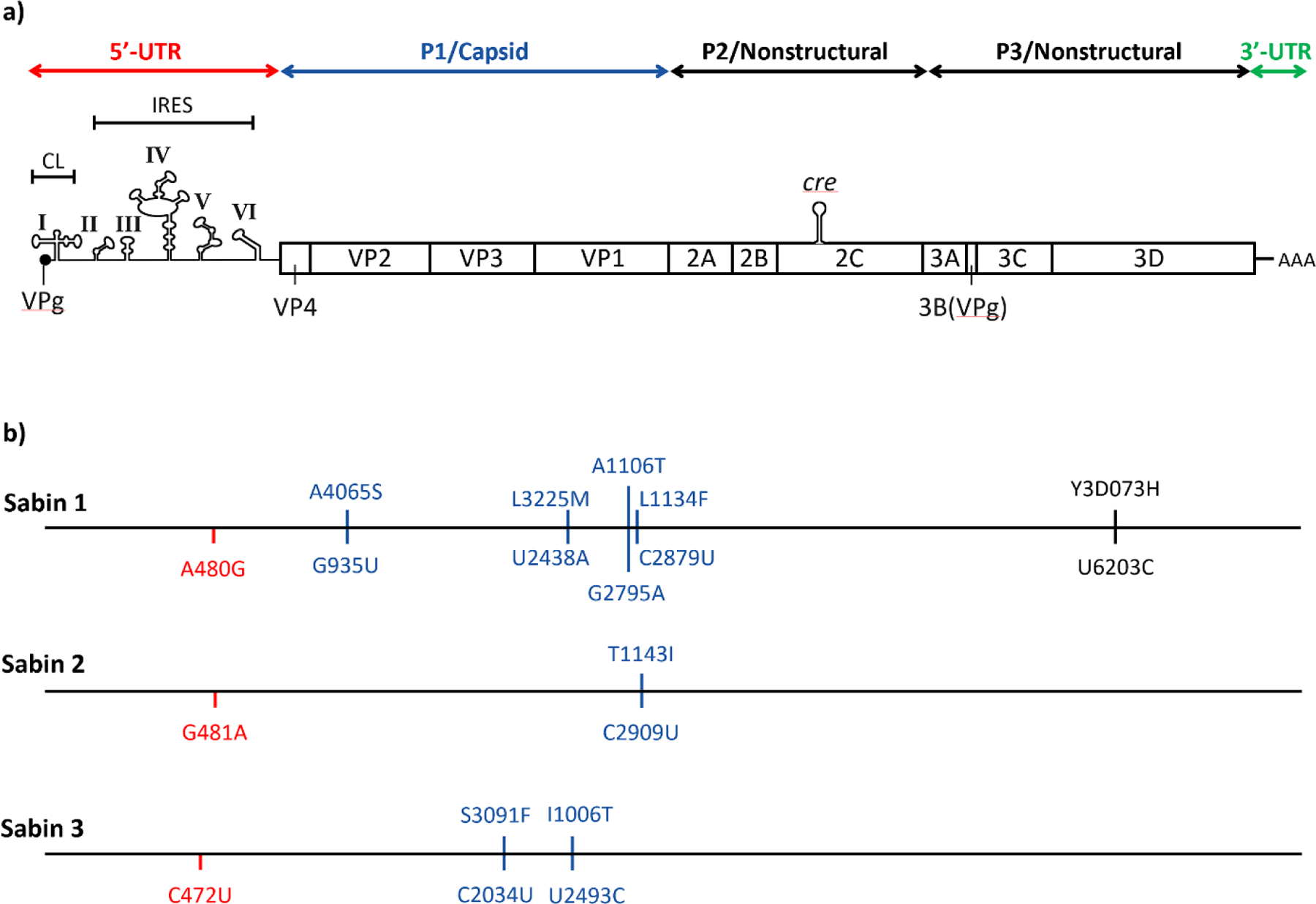
Poliovirus genome: (a) Genomic organization and primary translational open reading frame and processed proteins for polioviruses and (b) Key attenuating mutations for each OPV type (nucleotide changes shown below the line and amino acid changes shown above the line).
Virologists classify EVs that infect humans into four species: Enterovirus A, B, C, or D (Simmonds et al., 2020). Polioviruses comprise 3 of the 23 types within Enterovirus C (EV-C). While all EVs share the same genome structure and replication strategy, the four EV species can be differentiated from one another based on shared nucleotide and protein sequence similarities in their coding regions. The 3’-UTR sequences correlate with the four coding region groups (i.e., species), but the 5’-UTR sequences fall into only two distinct groups, with species C and D in group I and species A and B in group II. During replication, the viral RdRp can “fall off” its template molecule and reinitiate synthesis on another viral RNA molecule (termed “template-switching”), often at the same location along the genome. As a result, two EVs of different type but of the same species that happen to simultaneously infect a single host cell can exchange genetic material (Oberste, 2008). Similarly, two virus types from the same 5’-UTR group can also exchange sequences in the 5’-UTR, even if their coding regions come from separate EV species. In RNA viruses, virologists use the term “recombination” to describe this phenomenon, although the biochemical details of the process differ from genetic recombination that occurs in organisms with a DNA genome (e.g., bacteria or higher life forms).
Modeling the risks associated with OPV-related viruses, including the risks of VAPP, chronic infection in individuals with immune deficiencies, and the emergence of cVDPVs, requires the representation of the key inherent properties of OPV as a live, attenuated virus (Duintjer Tebbens & Thompson, 2017b). OPV offers the favorable properties of providing superior mucosal immunity and the potential for secondary vaccination through shedding from vaccine recipients to contacts. As an attenuated virus, OPV exhibits significantly reduced neurovirulence compared to its parent wild virus, largely because of reduced replicative fitness upon infecting the human host. In the 1950s, Albert Sabin developed the attenuated OPV strains empirically through a series of passages in alternative hosts (Dowdle, de Gourville, Kew, Pallansch, & Wood, 2003; Sabin, 1985; Sutter, Kew, Cochi, & Aylward, 2017). Further research many years later revealed the genetic nature of the attenuations for the three types of OPV to include only simple changes at a small number of sites (e.g., two sites for type 2 (Macadam et al., 1993)), most of which differ by type (Duintjer Tebbens, Pallansch, Kim, et al., 2013; Minor, Macadam, Stone, & Almond, 1993). Fig. 1(b) highlights the genome sequence differences of OPV strains compared to the parent wild poliovirus that represent the locations of known key attenuating mutations. The one common analogous attenuating mutation of all OPV types occurs in domain V (domV) of the 5’-UTR (i.e., nucleotides 480, 481, and 472 for types 1, 2, and 3, respectively). The EV RdRp (3Dpol) lacks a “proofreading” function, which makes it error-prone during RNA replication (Ward, Stokes, & Flanegan, 1988). Thus, multiple rounds of replication in one or more host cells leads to a pool of viral RNAs that constitute a “quasispecies” of RNA molecules, which differ from one another at only a few different sites across the genome. Therefore, when OPV and OPV-related viruses replicate, they can potentially mutate to reverse the attenuation sites. The resulting reverted virus increases in fitness, which correlates with both a replicative and transmission advantage and an increase in neurovirulence (compared to the attenuated OPV vaccine strain) (Sutter et al., 2017).
Reversion of attenuation can also occur through recombination in a co-infected cell with a different poliovirus strain without the attenuating mutations or through recombination with an EV-C or 5’-UTR Group I EV. Numerous studies demonstrate that these recombination events can occur among the three OPV strains in a single vaccine recipient (Cammack, Phillips, Dunn, Patel, & Minor, 1988; Furione et al., 1993; Georgescu et al., 1994; Lipskaya et al., 1991). The survival of these reverted virus progeny, however, depends on their successful infection of other cells in the same host and subsequent infection of a new host to establish a new infection. These types of “bottleneck” events affect the transmission dynamics at a cellular, individual, and population level (Escarmis, Lazaro, & Manrubia, 2006; Novella, Quer, Domingo, & Holland, 1999).
As described previously, the construct of our differential equation-based transmission and OPV evolution model (Duintjer Tebbens, Pallansch, Kalkowska, et al., 2013; Kalkowska, Wassilak, et al., 2020) uses a relatively simple 20-stage process to abstractly model the complex dynamic genomic changes that occur as polioviruses spread in populations and vary by type (Duintjer Tebbens, Pallansch, Kim, et al., 2013). Fig. 2 shows the assumed progression of reversion for (a) the relative basic reproductive number (R0) and (b) paralysis-to-infection ratio (PIR) of OPV-related viruses by type through the 20-stage process (Duintjer Tebbens, Pallansch, Kalkowska, et al., 2013; Kalkowska, Wassilak, et al., 2020). This approach does not map directly to the stochastic loss of key attenuating mutations, but instead seeks to simulate the evolution of quasispecies in the population. Thus, while loss of attenuation for any given OPV (e.g., a simple two-step process for type 2 OPV) in an individual may occur, this does not instantly change the composition of the poliovirus quasispecies in the population. The model and processes assumed in Fig. 2 seek to match the overall collective dynamics of the evolution of the different OPV types as they progress to their homologous cVDPVs in populations (Duintjer Tebbens, Pallansch, Kim, et al., 2013).
Fig. 2:
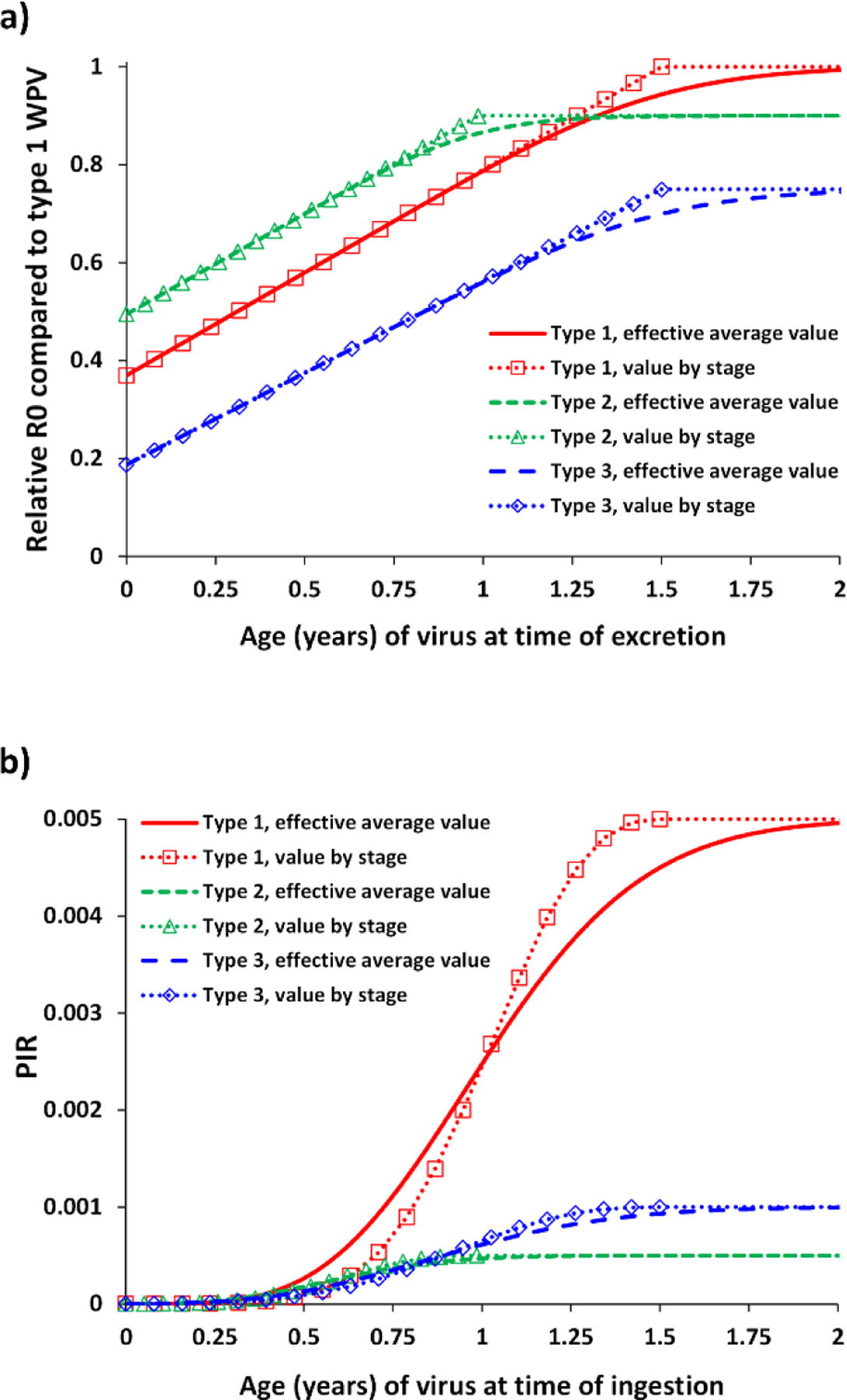
Assumed effect of reversion of OPV-related viruses by stage for the (a) relative basic reproductive number (R0) and (b) paralysis-to-infection ratio (PIR)
3. NOVEL OPV STRAINS
The recognized risks and consequences of continued use of OPV, combined with the potential longer-term necessity of its use to achieve polio eradication objectives, prompted researchers to explore the development of improved live attenuated polio vaccines that retain all of the advantages of the current OPV, but reduce the risks of VAPP and VDPVs (Macadam et al., 2006). A growing body of knowledge describing the biology of key attenuation components of OPV supports these development efforts. Multiple lines of research to better understand specific mechanisms of both attenuation and reversion provided insights and several research groups continue to apply that knowledge to improve and stabilize poliovirus attenuation. These lines of work included: (i) direct genetic stabilization of the primary attenuating mutation in the 5’-UTR common to all three types (Knowlson et al., 2015), (ii) modifications to the 3Dpol to increase replication fidelity to lower the mutation rate and suppress recombination (Pfeiffer & Kirkegaard, 2003; Runckel, Westesson, Andino, & DeRisi, 2013; Vignuzzi, Wendt, & Andino, 2008), (iii) multiple site attenuation through codon deoptimization in the capsid region to modulate replicative fitness (Burns et al., 2009; Burns et al., 2006), (iv) construction of hybrid viruses, such as the poliovirus strain with a rhinovirus 5’-UTR, which has proven useful for treating glioblastoma (Brown et al., 2017; Gromeier, Alexander, & Wimmer, 1996), (v) sequence rearrangement of critical genetic elements, such as movement of the cis-replicative-element (cre) in 2CATPase to the 5’ UTR (Yeh et al., 2020) and (vi) combinations of these. Table I(a) summarizes these different OPV stabilization strategies.
Table I.
nOPV design elements
| (a) Different strategies to stabilize OPV | ||
|---|---|---|
| Modification | Description | Ref |
| Substitution of key single nucleotides | Direct genetic stabilization of the primary attenuating mutation in the 5’-UTR common to all three types | (Knowlson et al., 2015) |
| Polymerase modification | Modifications to the 3Dpol to increase replication fidelity to lower mutation rates and suppress recombination | (Pfeiffer & Kirkegaard, 2003; Runckel et al., 2013; Vignuzzi et al., 2008) |
| Capsid codon deoptimization | Multiple site attenuation through codon deoptimization in the capsid region to modulate replicative fitness | (Burns et al., 2009; Burns et al., 2006) |
| Hybrid construction | Constructing hybrid viruses, such as the poliovirus strain with a rhinovirus 5’-UTR | (Brown et al., 2017; Gromeier et al., 1996) |
| Genome modification | Rearrangement of the genetic sequence to render certain recombinants non-viable (e.g. moving functional cre to the 5’-UTR) | (Yeh et al., 2020) |
| Combinations of the above | Use multiple of the above strategies | |
| (b) nOPV2 candidates | |||
|---|---|---|---|
| Modification | Candidate | Purpose | |
| 1 | 2 | ||
| S15 domain V | X | X | Improves genetic stability of the known 5’-UTR attenuation determinant by preventing reversion by single nucleotide change |
| cre relocation | X | Suppresses viable recombination events that might replace S15 domV attenuation determinant in 5’-UTR | |
| Polymerase (HiFi) |
X | Improves fidelity of viral replication leading to less genetic mutation and reversion | |
| Polymerase (Rec1) | X | Reduces frequency of recombination events, thereby reducing ability of population to improve replication fitness through that mechanism | |
| Capsid codon deoptimization | X | Reduces replication fitness and may enhance innate immune response in the gut, thereby reducing shedding | |
In 2011, a consortium for a new OPV initiated efforts to coordinate nOPV research with the goal of developing new candidate vaccine strains to address the inherent genetic instability of the current OPV strains (Konopka-Anstadt et al., 2020). The prioritization of nOPV2 was reinforced by the frequent emergence and circulation of VDPV2, the apparent elimination of wild poliovirus type 2 (WPV2) transmission by 1999, certification of the eradication of indigenous WPV2 in 2015 (Global Polio Eradication Initiative, 2015), and anticipation of cessation of OPV2-containing vaccine use (i.e., OPV2 cessation). Notably, immunity of WPV2 prevented the occurrence of cVDPV2s prior to WPV2 eradication in countries with poor immunization, and after WPV2 eradication the observance of cVDPV2 outbreaks occurred in areas with poor OPV2 immunization coverage. Building on combinations of strategies in Table I(a), investigators pursued multiple different nOPV2 design concepts, from which two candidates prevailed. Table I(b) summarizes the characteristics of these candidates (1 and 2) and Fig. 3 shows details about their resulting genomes.
Fig. 3.
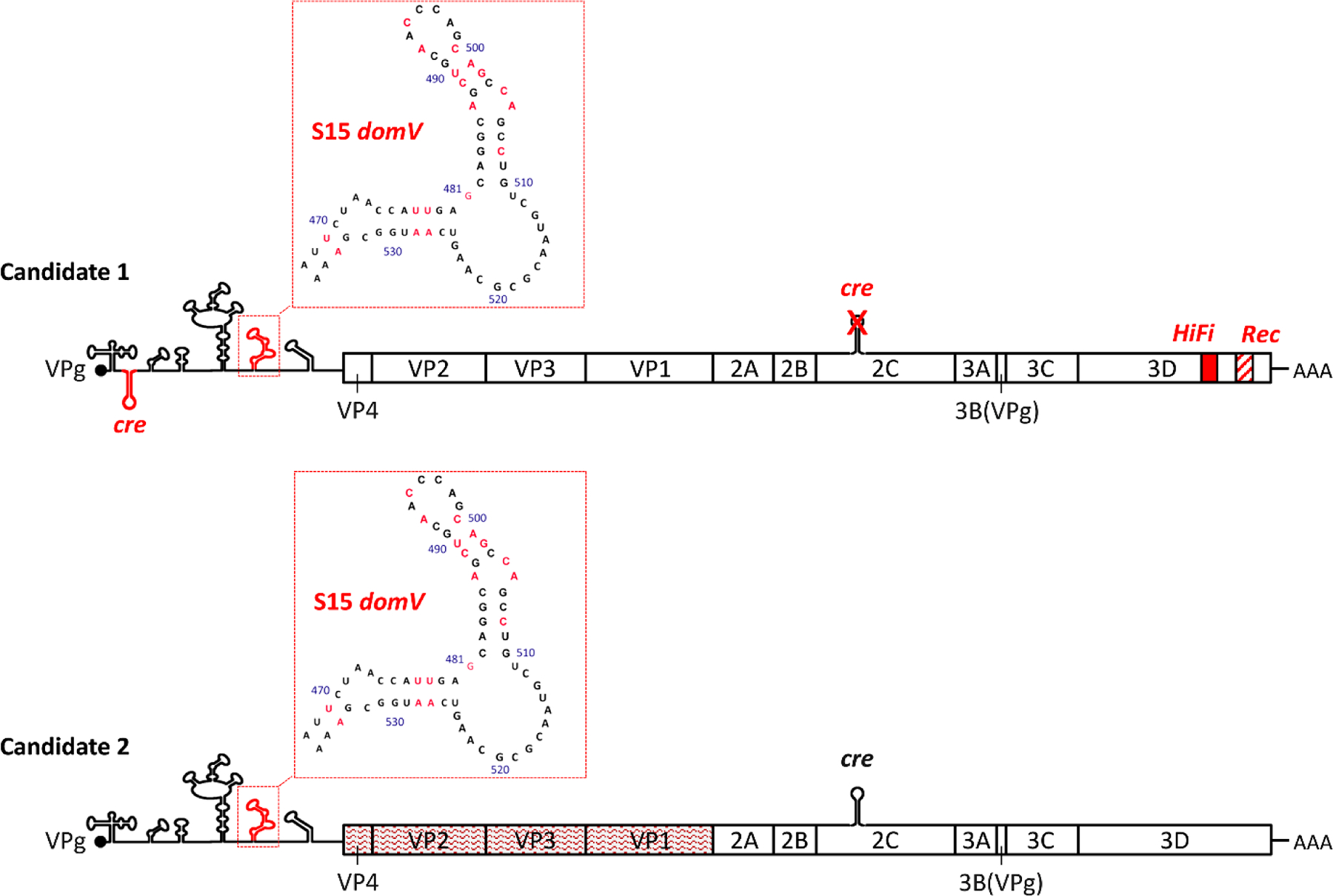
Schematic representation of the genetic changes introduced into different nOPV2 candidate strains (based on (Konopka-Anstadt et al., 2020) (Yeh et al., 2020)). All genomic changes that characterize the Candidate strain are highlighted in red color and/or font.
As shown in Fig. 3, both nOPV2 candidates build on a common OPV2 backbone and contain a modified 5’-UTR. All three Sabin OPV strains contain mutations within domV that function as key “gate-keeper” attenuation determinants. These domV attenuation determinants face constant pressure to revert during replication in the human gut, with single nucleotide reversions leading to enhanced thermostability and increased virulence. Reversion in domV often precedes all other events leading to cVDPV virulence (A. Stern et al., 2017). A specific stabilized form of domV known as S15 domV (Konopka-Anstadt et al., 2020) (Yeh et al., 2020) (Fig. 3 detail shown in inset), is included within the 5’-UTR of the nOPV2 vaccine candidates and eliminates the ability of the virus to revert by single nucleotide changes within this key determinant, thus rendering the attenuation of nOPV2 more genetically stable than OPV2.
In addition to stabilization of domV, nOPV2 candidate 1 also features relocation of the functional cre from the 2C region to the 5’-UTR (Yeh et al., 2020). The original cre is modified through several mutations (cremut) that eliminate replication functionality and make direct reversion unlikely, while preserving the amino acid coding of the 2C protein. The modified relocated cre (designated cre5) is located in the spacer sequence between CL and IRES (Fig. 3). This move makes any potential recombination in the 5’-UTR ineffective to restore the original domV, because such a recombination event would eliminate the only functional cre motif and thus render the progeny non-viable. Candidate 1 also includes two modifications to the 3Dpol viral polymerase that (1) enhance the fidelity of the viral polymerase (via the high fidelity, or “HiFi,” modification), thereby reducing the high mutation rate and probability of reversion, and (2) reduce the frequency of recombination events (via the recombination, or “Rec1,” modification) and thereby suppress the ability to improve fitness through that mechanism. The 3Dpol modifications followed in vitro screening studies designed to identify variants with the desired features (Yeh et al., 2020). The polymerase mutations minimally affect the kinetics of virus replication for candidate 1.
Candidate 2 includes the same stabilized 5’-UTR as candidate 1, as well as modifications that target the capsid region rather than non-structural regions (Fig. 3). The replacement of naturally preferred codons across the candidate 2 capsid region with “nonpreferred” synonymous codons increased the CpG dinucleotide content but introduced no changes at the protein-coding level. Codon-deoptimized polioviruses demonstrate reduced replicative fitness, modulated by the number of codon replacements made and the resulting relative dinucleotide content (Burns et al., 2009; Burns et al., 2006). Codon deoptimization leads to delivery of an increased amount of viral RNA per infectious particle for nOPV2 compared to OPV2, which may enhance the antiviral response within the gut of vaccine recipients (Konopka-Anstadt et al., 2020). This increased RNA quantity combined with reduced replicative fitness may lessen the duration and/or magnitude of viral shedding in vaccine recipients, potentially leading to less infectious circulating vaccine virus (Van Damme et al., 2019). Additionally, targeting the capsid with these modifications safeguards the attenuated phenotype even if recombination occurs, which offers particularly important stabilization due to retention of only the capsid during recombination events. While candidate 2 nOPV2 includes >80 nucleotide changes in the codon-deoptimized capsid region, the antigenic structure of the virus remains unchanged due to the absence of any amino acid changes in the engineered capsid protein region. In addition, the extensive changes increase the inherent stability of candidate 2, due to the requirement for many mutations to produce significant reversion of the phenotype.
The selection process for both nOPV2 candidate 1 and 2 strains for initial pre-clinical development included both in vitro and in vivo studies to assess growth properties (as related to potential vaccine production), antigenicity, immunogenicity, genetic stability upon serial passage in cell culture, and retention of attenuation as measured in a transgenic mouse model of neurovirulence (Van Damme et al., 2019). Results from the pre-clinical evaluation of both candidates informed the decision to initiate head-to-head clinical trial testing.
As of mid-2020, three human clinical trials of nOPV2 candidates 1 and 2 have been conducted, including studies in adults, children, and infants. A phase I trial conducted in 2017 under biological containment among adults previously immunized exclusively with IPV demonstrated the safety, immunogenicity, and genetic stability of both nOPV2 candidates (Van Damme et al., 2019). More recently, phase II trials conducted in previously immunized adults and in toddlers (OPV-IPV vaccinated) and infants (bOPV-IPV vaccinated) further support the safety and immunogenicity of both candidates compared to mOPV2 (Bandyopadhyay, 2020a, 2020b). The initial findings from ongoing clinical trials also appear to confirm the general outlines of the stability designs. To date, no studies of either candidate strain of nOPV2 included administration to immunologically naïve individuals and in particular, to naïve infants, who represent the key primary target recipients of nOPV2. Considering the urgent need for nOPV2 doses at large scale, favorable clinical data and the greater production yield for candidate 1 nOPV2, the next phase of development began for candidate 1, including vaccine production at risk on a commercial scale and application for its use under a pending WHO Emergency Use Listing (EUL) by the vaccine manufacturer.
Despite the proof of concept and favorable results from clinical trials that suggest only very limited reversion for candidate 1 nOPV2, reversion remains theoretically possible. Reversion for candidate 1 would require two serial and ordered recombination events to restore replicative fitness to the virus and make it at least as fit as mOPV2 (Fig. 4). The first recombination event would need to occur between candidate 1 nOPV2 (red) and any EV-C (light blue), including other poliovirus types and strains, which would simultaneously restore a functional cre in the 2C region and remove the HiFi and Rec1 mutations in the progeny virus. Fig. 4 (top) depicts such an event occurring at the 2B-2C junction to create progeny 1, although the recombination could occur anywhere between the VP1–2A junction and cremut in 2C. This progeny virus strain would then include two functional, but redundant, cre coding regions. This first progeny would still maintain the attenuation of the 5’-UTR intact, so in principle it would likely not benefit from a replication advantage. However, because the cre in the 2C region could fully support replication and make the progeny virus viable if the cre5 is removed, a second recombination event with another 5’-UTR Group I EV (dark blue) could substitute a non-attenuated functional 5’-UTR for the cre5 and attenuated S15 domV. Fig. 4 (bottom) depicts the second event occurring at the 5’-UTR-VP4 junction to create progeny 2, but the recombination could occur anywhere between the S15 domV and VP4. Fig. 4 conveys one possible pathway of the many potential paths from a candidate 1 nOPV2 strain to a more transmissible and neurovirulent strain. However, even though these other multiple serial recombination events could result in equivalent functional genomes to the one shown in Fig. 4, they are less likely to occur since they would require an increased number of recombination events. On a population level with millions of infections, recombination events like these occur relatively quickly; although theoretically possible, it is not known if all the required events could occur in a single individual. Depending on the donor viruses in these recombination events, the second recombination progeny could behave like mOPV2, partially reverted OPV2-related viruses, or fully reverted OPV (i.e., cVDPV2). Notably, since most recombinant cVDPV2s include already reverted sequences or other 5’-UTR Group I sequences in the 5’-UTR and EV-C sequences in the 3’-half of the genome (rather than OPV sequences), the latter possibility (i.e. behavior like fully reverted OPV) seems the most likely (Burns et al., 2013; Famulare et al., 2015; Jegouic et al., 2009; Liu et al., 2003).
Fig. 4.
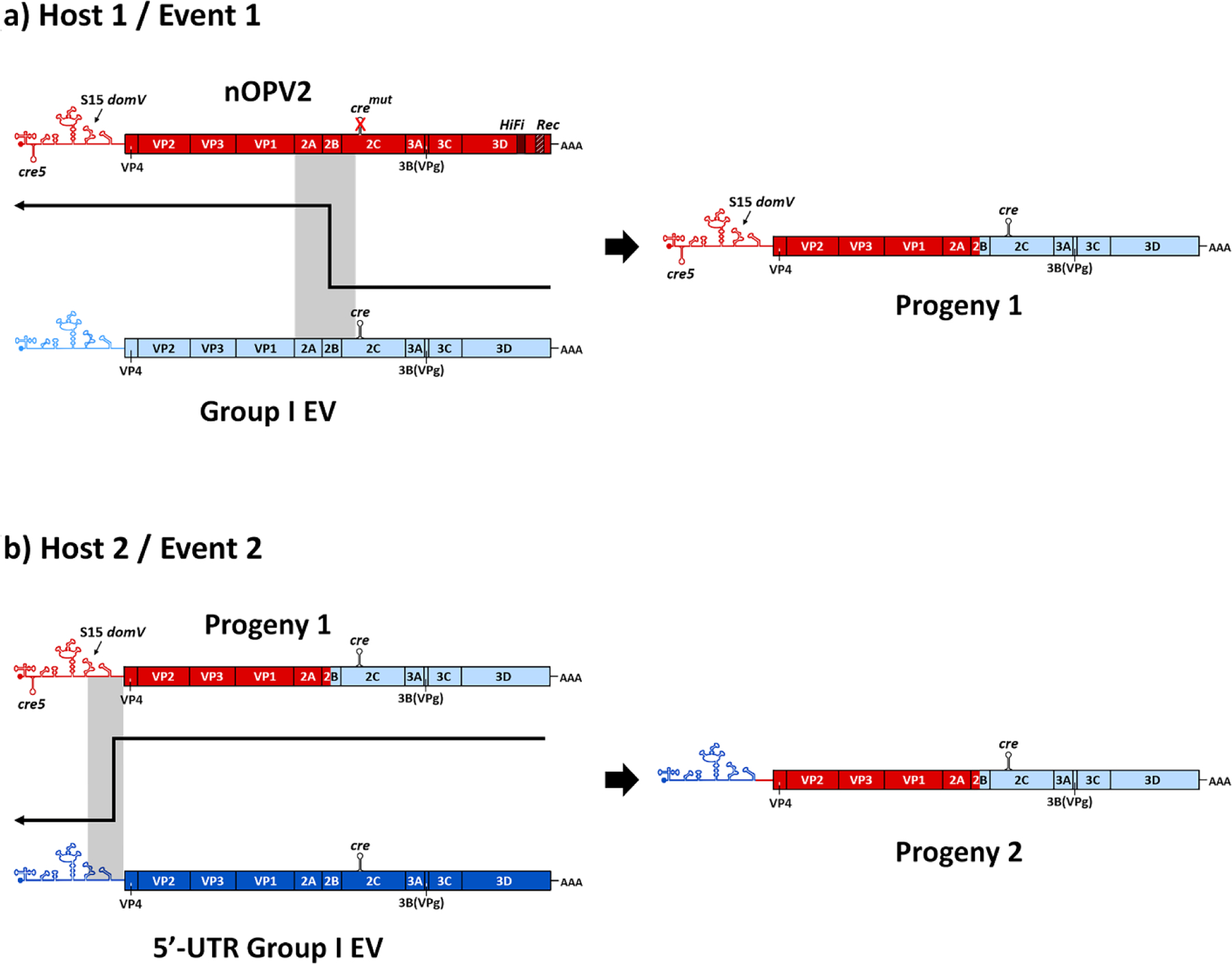
Theoretical pathway for loss of engineered changes and reversion of attenuation for nOPV2 candidate 1 by serial recombination events during replication and transmission. The resulting species (Progeny 2) is a strain that has lost all attenuating mutations compared with nOPV2.
In contrast to the decades of experience and evidence related to OPV and reversion of its attenuating mutations, the evidence related to nOPV2 in humans remains limited to observations from early clinical trials, which do not include sufficient power to observe relatively rare events associated with OPV (e.g., VAPP, VDPVs). Both VAPP and cVDPVs were only observed following the use of OPV at scale (millions of doses) and more commonly in the absence of homotypic WPVs. For modeling nOPV2 use, we consider the potential similarities and differences between mOPV2 (>105 CCID50) as licensed and used in historic control trials and SIAs, and nOPV2 data from preclinical studies and clinical trials. We focus on the nOPV2 candidate selected for production (i.e., candidate 1, formulated with a “low dose” titer of >105 CCID50). We note that the initial clinical trials only included a “high dose” titer of >106 CCID50 (Bandyopadhyay, 2020a, 2020b), but that an ongoing clinical trial among infants includes arms for both the “low dose” and “high dose” titers.
Although limited, some observations support the development of bounding assumptions for mathematical modeling. First, from the observations in the clinical trials, shedding of candidate 1 nOPV2 given at the high dose to previously immunized adults appears similar to mOPV2 shedding by a similar population (Van Damme et al., 2019), but uncertainty remains about shedding for the low dose for candidate 1 nOPV2 compared to mOPV2 and when given to immunologically naïve individuals, particularly infants. Based on the available data, we might expect significant secondary transmission with nOPV2 (similar to that of mOPV2), which not only increases secondary immunization but also increases the opportunities for recombination and reversion. However, in contrast to secondary spread of mOPV2, the effective reproductive number (Reff) of nOPV2 in the population may remain below 1, because the benefit of increased fitness that comes with the reversion of mOPV2 at the primary attenuation site would not occur (at all or to the same extent) with nOPV2. Thus, the nOPV2 progeny would likely die out in most circumstances. In addition, because phenotypic reversion by direct point mutation does not appear to occur at any significant rate for nOPV2, we would not expect a significant risk of VAPP with nOPV2 use even with limited transmission. Data related to this point is currently limited by the small number of individuals in the clinical trials, and changes during transmission can only be inferred in the absence of direct observation.
Second, significant uncertainty exists about how nOPV2 may revert in individuals through recombination events and potentially evolve as a result of any secondary transmission that occurs in the population. For modeling purposes, this means characterizing the distinctly different processes by which nOPV2 may achieve phenotypic reversion primarily through recombination (compared to mOPV2). In general, while evidence and experience demonstrate that recombination of live polioviruses with each other and with other EVs occurs, the rates of serial recombination events remain poorly characterized. Such events depend on the population and vary by season and year, which makes any characterization approximate. Specifically, the context related to the prevalence of other circulating viruses that could recombine due to co-infection with nOPV2 (e.g., bOPV used in RI, bOPV or mOPV2 used in SIAs, cVDPVs, EV-C) could affect the probability of co-infection and therefore the probability of the required recombination events in unpredictable ways. Multiple studies of WPV and cVDPV circulation suggest relatively common recombination of these polioviruses with EV-C (Burns et al., 2013; Famulare et al., 2015; Jegouic et al., 2009; Liu et al., 2003). However, only limited information exists to support the characterization of rates of recombination in OPV recipients or contacts with other OPV strains (Cuervo et al., 2001) or with EV-C. We can expect that two ordered, serial recombination events with nOPV2 would occur with much lower probability than the common point mutation reversions seen with OPV2. Consistent with this expectation, the current clinical trials that provide limited available evidence to date do not report the observation of any recombination (although the studies lack sufficient power to observe these low-probability events). However, after generation of the first recombinant with a replacement in the 3’-half of the genome, the virus might become more transmissible and therefore available for the second recombination event in the 5’-UTR. Because we cannot expect the limited number of individuals in the studies to adequately inform the nOPV2 behavior when widely administered to a general population of children under 5 years of age, modeling should recognize the uncertain potential for nOPV2 evolution and reversion when used widely in real populations. Given the expected use of nOPV2 for outbreak response SIAs, we seek to model its potential benefits and possible limitations.
4. METHODS
We explore a range of options for outbreak response using an updated global poliovirus transmission and OPV evolution model (Kalkowska, Pallansch, et al., 2020; Kalkowska, Wassilak, et al., 2020) with different scenarios and including the potential use of nOPV2 modeled assuming a range of different properties for low-dose nOPV2 candidate 1. The model divides the global population into 72 blocks of 10 subpopulations each (10.7 million people per subpopulation in 2019 (Population Division of the Department of Economic and Social Affairs of the United Nations Secretariat, 2019)), grouped into 9 preferential mixing areas (PMAs) of different sizes, which represent larger geographical regions (e.g., Africa, Australasia, Europe) (Kalkowska, Wassilak, et al., 2020). The 72 blocks represent a simplified global population structure that reflects the global population of approximately 7.2 billion people. The simplified structure does not aim to explicitly identify individual countries or populations due to their variable sizes, limited existing information to characterize all countries and heterogeneity within them, and the costs of computational resources. The subpopulations mix homogenously among the people but heterogeneously by age, and export preferentially to other subpopulations based on the PMAs in which the blocks belong. The model accounts for global variability in conditions, costs, and preferences by further classifying the blocks by World Bank Income Level (World Bank, 2019). We also characterize current vaccine use into multiple RI schedules (World Health Organization, 2019): OPV+IPV (former OPV-only, with one added IPV dose simultaneously with the third bOPV dose), IPV/OPV (sequential schedules that give IPV first followed by bOPV), and IPV-only (Kalkowska, Wassilak, et al., 2020). The epidemiological, demographic, and transmission assumptions of the model at the beginning of the analytical time horizon (T0 = January 1, 2019) represent conditions that existed in the world as of the end of 2018 (Kalkowska, Wassilak, et al., 2020), with updated cVDPV2-related inputs for 2019 (Kalkowska, Pallansch, et al., 2020).
The model characterizes poliovirus transmission and OPV evolution for each poliovirus type and considers both fecal–oral and oral-oral transmission. For each subpopulation, the model divides the population into seven age groups, eight immunity states, and it includes a five-stage immunity waning process, a six-stage infection process (i.e., two latent and four infectious stages), and a 20-stage OPV evolution process. We use generic model inputs (Kalkowska, Wassilak, et al., 2020) based on an extensive expert review (Duintjer Tebbens, Pallansch, et al., 2013a) and elicitation process (Duintjer Tebbens, Pallansch, et al., 2013b) and a fitting process to a cross section of situations (Duintjer Tebbens, Pallansch, Kalkowska, et al., 2013; Kalkowska et al., 2015). Only individuals fully susceptible to the type can become paralyzed upon first infection with an LPV of that type (i.e., IPV and prior homotypic live poliovirus infection provide life-long protection from paralysis and thus from presenting with acute flaccid paralysis (AFP) caused by that type) (Kalkowska, Wassilak, et al., 2020). The model accounts for the differences in the nature of immunological protection provided by each type and dose of vaccine, and tracks infections, which include infections in individuals with no immunity, IPV-only induced immunity, and waned immunity despite prior LPV infection (Kalkowska, Wassilak, et al., 2020). The model includes subpopulation-specific and time-varying threshold-based AFP surveillance and stochastic environmental surveillance (ES) to detect poliovirus transmission (Kalkowska, Wassilak, et al., 2020). The model triggers oSIAs when the cumulative incidence of polio cases per 10 million people exceeds subpopulation-specific AFP detection thresholds or the environmental detections exceed the subpopulation-threshold for subpopulations that include ES (Kalkowska, Wassilak, et al., 2020) based on population specific surveillance sensitivity. Once an outbreak occurs in any subpopulation in a block, the model automatically ensures faster detection in another subpopulation in the same block (lower detection threshold) and leads to an oSIA starting 30 days after detection (15 days sooner compared to first detection in that block), which mimics a situation of heightened awareness in the area (Kalkowska, Wassilak, et al., 2020). For this analysis, we focused on modeling oSIAs consistent with actual GPEI oSIA performance (Kalkowska, Wassilak, et al., 2020), instead of assuming aggressive, large, and high-quality oSIAs that we assumed in earlier modeling (Duintjer Tebbens et al., 2015; Duintjer Tebbens, Pallansch, et al., 2016; Duintjer Tebbens & Thompson, 2018). Prior to type-specific OPV cessation, the model accumulates the incidence from any ongoing transmission (e.g., cVDPV2s), effective importations, and indigenous cVDPV emergences. After type-specific OPV cessation, the model accounts for any homotypic incidence, including cases caused by all live polioviruses (i.e., OPV, OPV-related, VDPVs and WPVs) including failures in containment (Kalkowska, Wassilak, et al., 2020). The model clears the cumulative incidence relevant to the oSIA trigger after each completed outbreak response or every 6 months without any oSIAs (Kalkowska, Wassilak, et al., 2020). During the analytical time horizon, oSIAs target children <5 years old or all new birth cohorts starting 5 years since the type-specific OPV cessation. For oSIAs, we assume the use of 2 rounds separately by 30 days, followed by 2 additional rounds in the event of breakthrough transmission. The scope of the oSIA includes the outbreak subpopulation only when R0 < 10, or the outbreak subpopulation and its 4 worst-performing neighboring subpopulations within the same block when R0 ≥ 10 (Kalkowska, Wassilak, et al., 2020). The vaccine choice for the oSIA depends on timing, vaccine availability, and policy decisions (Kalkowska, Wassilak, et al., 2020). The updated reference case used in prior studies (RC2) (Kalkowska, Pallansch, et al., 2020) assumes the use of mOPV of types 1, 2 and 3 (mOPV1, mOPV2, and mOPV3) for 5, 8, and 5 years, respectively, post homotypic OPV cessation. After that period, the model uses only IPV for oSIAs and assumes that the IPV oSIAs are conducted with lower intensity (i.e., only 2 rounds independent of breakthrough and with assumed lower coverage compared to OPV oSIAs due to the inability to go house-to-house) because of the costs and challenges associated with delivery of an injected vaccine (Kalkowska, Wassilak, et al., 2020). For this analysis, we also added a reference case (RC3) that assumes the use of mOPV2 for outbreak response throughout the model time horizon to facilitate comparison with the alternative scenarios that assume nOPV2 use throughout the time horizon.
We consider 4 alternative outbreak response scenarios compared to RC2 (and RC3) assuming the use of low-dose candidate 1 nOPV2 instead of mOPV2 for all oSIAs for cVDPV2s starting in January 1, 2021 and assuming continued use throughout the remainder of the model time horizon, while bOPV remains the primary choice for RI and pSIAs in OPV-using blocks. We focus on exploring the bounds of the potential benefits of the current GPEI plans to use nOPV2 exclusively in oSIAs (World Health Organization Global Polio Eradication Initiative, 2020b) and on comparing the potential dynamics of nOPV2 with mOPV2. We assume the same efficacy of nOPV2 as mOPV2 (i.e., we assume that licensure and acceptance of nOPV2 would require its non-inferiority with respect to safety and efficacy compared to mOPV2). For this analysis, we name the scenarios: (i) “No reversion, no VAPP,” which assumes the same shedding and infectious dose as mOPV2 leading to the same take rates and transmissibility (R0) for nOPV2 candidate 1 as mOPV2, but with no reversion or paralysis despite transmissibility (i.e., no VAPP and no VDPVs) (Duintjer Tebbens & Thompson, 2016); (ii) “No reversion, same VAPP” which assumes the same effectiveness, no reversion despite transmissibility, but paralysis occurs at the mOPV2 VAPP rates (i.e., the same conditions as the “No reversion, no VAPP” scenario except with VAPP); (iii) “Some reversion, same VAPP”, and (iv) “Some reversion, lower VAPP”, which assume that nOPV2 candidate 1 follows the reversion characteristics shown in Fig. 5(a) for R0 and 5(b) for PIR for the 20-stage process in the model compared to mOPV2 and may cause VAPP at (iii) the same rate as mOPV2 or (iv) a rate of 1/250th the mOPV2 VAPP rate (Yeh et al., 2020) (difference not visible on the scale in Fig. 5(b) given values so many orders of magnitude below 1). As shown in Fig. 5, we essentially make the impacts of the reduced reversion rate reflected in slower and non-linear changes in phenotypic properties while progressing to higher model stages (i.e., unchanged implied “fitness” and potential for transmission (relative R0 compared to the R0 for the subpopulation for WPV1) for stages 0–9 and retain the attenuated properties related to neurovirulence during these stages). The net effect is to make nOPV2 less likely to increase transmissibility and neurovirulence compared to mOPV2 at equivalent stages of reversion. We use the same reversion structure to model both mOPV2 and nOPV2, and consequently we apply a stepwise delay process starting at the time of nOVP2 introduction to phase in the implementation of the nOPV2 reversion characteristics (shown in Fig. 5). This allows the mOPV2-related viruses transmitting at the time of nOPV2 introduction to progress to higher reversion stages normally, while making any nOPV2 introduced behave according to appropriate nOPV2 characteristics. The progression implements time delays, each equal to the average time per reversion stage, and applies the nOPV2 characteristics progressively to each stage for stages 0–9 (i.e., after a time delay since the introduction of nOPV2 at stage 0, apply the nOPV2 characteristics to stage 1, etc.). The model assumes a global threshold of 5,000 cVDPV and/or WPV cases (i.e., OPV reversion stage 19 cases or WPV cases if any stochastic reintroductions of WPVs occur) to trigger the need to restart OPV2 for RI in OPV-using countries (Kalkowska, Wassilak, et al., 2020). For OPV2 restarts implemented during the time horizon, the model uses mOPV2 for the RC2 and RC3 scenarios and nOPV2 for the 4 alternative outbreak response scenarios.
Fig. 5.
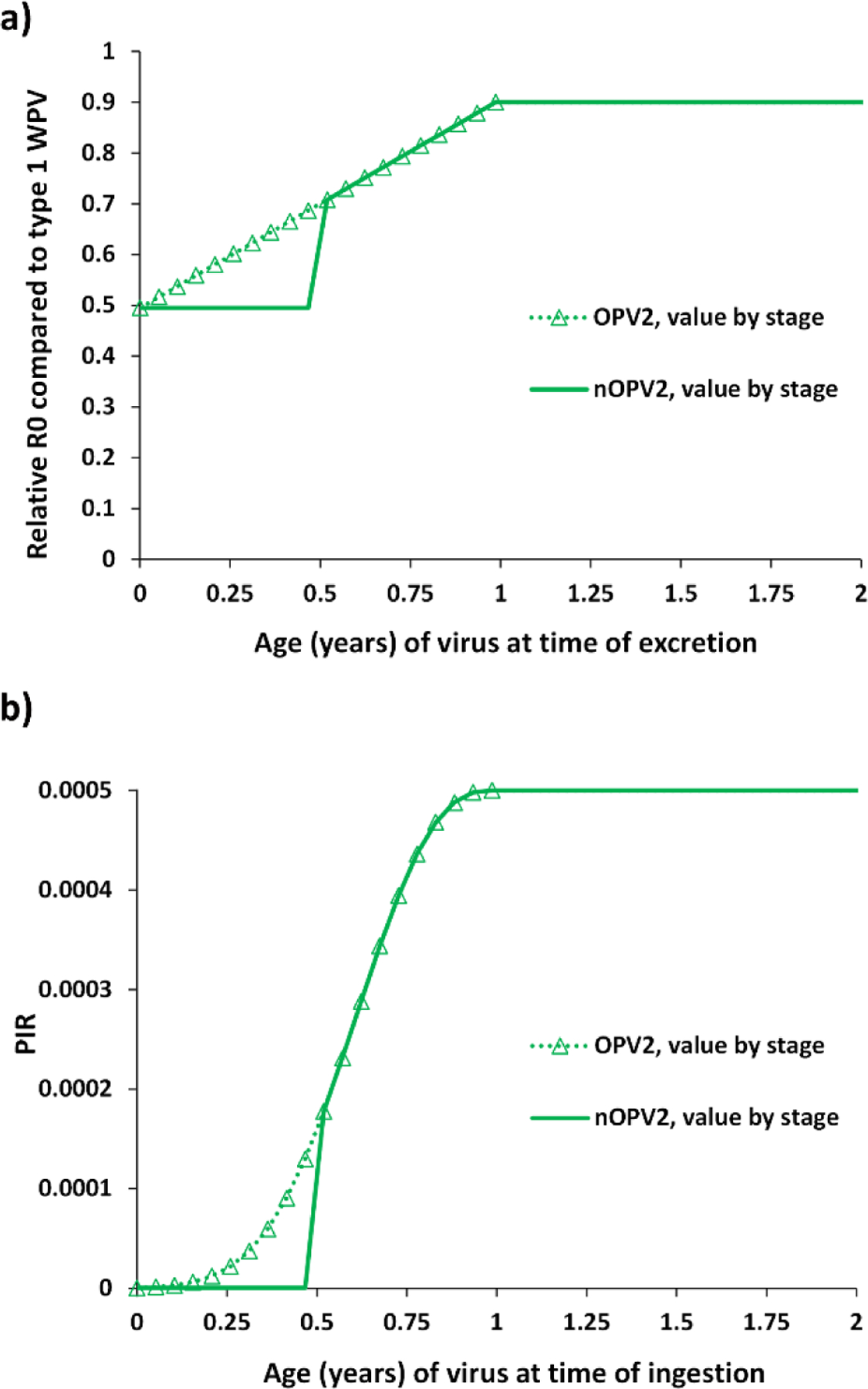
Characterization of model inputs assumed for nOPV2 candidate 1 compared to mOPV2 for the (a) relative basic reproductive number (R0) and (b) paralysis-to-infection ratio (PIR)
We run the updated model with the assumption of no constraints on vaccine availability to respond to outbreaks (i.e., unlimited stockpiles), which allows us to determine potential vaccine supply needs (Kalkowska, Wassilak, et al., 2020). We code the model using the general-purpose programming language JAVA™ and the integrated development environment Eclipse™, and we simulate 100 stochastic iterations on the Amazon Elastic Compute Cloud (Amazon EC2). We use the same random number seeds and initial conditions for the 100 iterations for each scenario to focus on direct comparisons between the scenarios.
5. RESULTS
As reported in Table II, using nOPV2 for oSIAs exclusively does not lead to a high probability of eradication of existing cVDPV2s by 2029 (range of 39% to 57%), although its use represents a substantial improvement from the 7% probability of eradication that occurs for RC2. However, compared to extended use of mOPV2 in RC3 (43% probability of eradication), that improvement becomes less pronounced and only advantageous under the “No reversion” scenarios. In general, we find that nOPV2 use will lead to expected prolonged, but low-level transmission throughout the time horizon (2019–2029) in nearly half of the iterations of existing cVDPV2s and OPV2-related viruses from mOPV2 use prior to 2021, even if nOPV2 does not revert and causes no VAPP or low VAPP (i.e., the “No reversion, no VAPP” or “No reversion, same VAPP” scenarios). The introduction of nOPV2 use for oSIAs for the “Some reversion” scenarios leads to probabilities of eradication between these other bounding cases.
Table II.
Estimated probability of cVDPV2 eradication, restarts triggered (and implemented), expected value (median) and [range] of cases and millions of doses of vaccine used for outbreak response in 100 stochastic iterations for 2019–2029 for the scenarios modeled (see main text for descriptions)
| Scenario | Probability of cVDPV2 eradication | Restarts triggered (imple-mented) | Expected cVDPV2 cases (median) [range] |
Expected VAPP2 cases (median) [range] |
Expected total cases* (median) [range] |
Expected millions of mOPV2 doses used during years when mOPV2 use is allowed (median) [range] |
Expected millions of IPV doses used (median) [range] |
Expected millions of nOPV2 doses used (median) [range] |
|---|---|---|---|---|---|---|---|---|
| RC2 | 7% | 89 (34) | 26,226 (18,706) [721 – 111,354] |
24 (9) [5 – 124] |
32,788 (27,142) [878 – 111,637] |
518 (427) [177 – 1,258] |
615 (517) [0 – 2,241] |
NA |
| RC3 | 43% | 47 (19) | 6,765 (4,603) [697 – 51,852] |
26 (13) [5 – 110] |
12,321 (7,216) [878 – 52,557] |
821 (679) [189 – 2,488] |
0.4 (0) [0 – 8] |
NA |
| No reversion, no VAPP | 57% | 22 (3) | 4,116 (2,689) [485 – 42,915] |
4 (4) [3 – 6]** |
4,398 (2,899) [666 – 43,099] |
100 (95) [81 – 160] |
0.3 (0) [0 – 8] |
414 (370) [54 – 1,509] |
| No reversion, same VAPP | 57% | 22 (3) | 4,116 (2,689) [485 – 42,915] |
13 (10) [5 – 117] |
4,407 (2,908) [669 – 43,210] |
100 (95) [81 – 160] |
0.3 (0) [0 – 8] |
414 (379) [54 – 1,509] |
| Some reversion, same VAPP | 39% | 67 (17) | 9,895 (7,640) [846 – 41,074] |
28 (16) [5 – 124] |
14,562 (9,638) [1,362 – 50,895] |
100 (95) [81 – 160] |
0.8 (0) [0 – 26] |
842 (693) [66 – 2,379 () |
| Some reversion, lower VAPP | 40% | 65 (18) | 9,742 (7640) [846 – 41,077] |
4 (4) [3 – 6] |
14,571 (9,617) [1,360 – 50,772] |
100 (95) [81 – 160] |
0.8 (0) [0 – 26] |
841 (680) [66 – 2,379] |
Abbreviations: cVDPV2, circulating vaccine-derived poliovirus of type 2; IPV, inactivated poliovirus vaccine; mOPV, type 2 monovalent oral poliovirus vaccine; NA, not applicable; nOPV2, candidate 1 nOPV2; RC2, control reference case with outbreak response using mOPV2 through 2024 then IPV; RC3, control reference case with outbreak response using mOPV2 for the full time horizon; VAPP, vaccine-associated paralytic polio; VDPV, vaccine-derived poliovirus
Note:
includes all type 2 cases (i.e., totals from all infections with live polioviruses, including WPV, VDPVs, and VAPP, which sums to more than the prior two columns due to cases associated with OPV-related viruses and cases associated with rare, but non-zero stochastic risks such as containment breaches)
VAPP2 cases occurring in 2019–2020 (before nOPV2 candidate 1 use begins)
Table II shows an 89% probability of OPV2 restart for RC2, with 34% of iterations leading to more than 5,000 cases and triggering a restart of OPV2 use in RI within the model time horizon. Using mOPV2 throughout the time horizon (RC3) dramatically decreases the probability of OPV2 restart to 47% triggered and 19% implemented within the time horizon. The use of nOPV2 under the “No reversion, no VAPP” and “No reversion, same VAPP” scenarios leads to fewer OPV2 restarts during the time horizon (22% triggered, 3% implemented), but does not drop the OPV2 restarts to 0 due to the extent of cVDPV2 transmission already seeded prior to the start of nOPV2 use. The scenarios that include the possibility of “Some reversion” reduce the probability of OPV2 restart compared to RC2 from 89% to 67% for “same VAPP” and 65% for “lower VAPP”, with 17% for “same VAPP” and 18% for “lower VAPP” of iterations implementing OPV2 use in RI within the model time horizon. The difference in the number of iterations triggering and implementing OPV2 restart in the time horizon for these scenarios depends on the VAPP assumptions for stage 0 and occurs due to the change in outbreak response dynamics. The model tracks the transmission of virus regardless of reversion stage until the accumulated number of infections results in an AFP case or ES detection. The model also incorporates a delay in the outbreak response that approximates programmatic delays and surveillance and epidemiologic uncertainties in the actual response process. Specifically, while the detection of AFP cases after type-specific OPV cessation triggers an outbreak response, lower paralytic incidence from stages 0–9 in the “Some reversion, lower VAPP” scenario (compared to the “Some reversion, same VAPP” scenario) changes the dynamics of the accumulation of paralytic cases. This change shifts the timing of oSIAs later during the period 2026–27, which consequently leads to more transmission and higher incidence in 2028–29 and earlier implementation of restarting OPV2 (either mOPV2 or tOPV) use in RI in some iterations. The use of nOPV2 compared to the extended use of mOPV2 in RC3 offers an improvement for the “No reversion” scenarios (i.e., 25 fewer OPV2 restarts triggered for the “No reversion” scenarios compared to RC3). In contrast, the use of nOPV2 compared to extended use of mOPV2 (RC3) does not show an improvement for the “Some reversion” scenarios (i.e., 18–20 more OPV2 restarts triggered for the “Some reversion” scenarios compared to RC3). These results highlight the importance of resolving the uncertainty about the actual ability of nOPV2 to revert.
Table II also summarizes the expected numbers of cVDPV2 and VAPP cases and the doses of vaccine used for each scenario. The iterations that include the implementation of OPV2 restart within the time horizon typically lead to large-scale OPV2-containing vaccine use starting as early as 2025, which leads to notable increases in VAPP cases for those iterations and wide ranges for the VAPP results summarized in Table II. We include IPV doses because RC2 assumes no mOPV2 use after 2024 and a shift in 2025 to IPV only for oSIAs. For the RC3 or nOPV2 scenarios, which allow the use of mOPV2 or nOPV2, respectively, throughout the time horizon, the iterations include a few exportations of cVDPV2 viruses into high-income countries that perform oSIAs with IPV, which leads to IPV doses shown for these scenarios in Table II. Comparisons of the expected values, medians, and upper values of the ranges suggest that some of the iterations yield much higher values than others and imply right-skewed distributions. Specifically, some of the upper range of cVDPV2 reflect low-probability but high-consequence stochastic iterations (e.g., 1–3 iterations out of the 100 with much higher numbers). The behavior in these small number of iterations occurs due to a mix of the model structure (i.e., exportation to higher R0 homogenously mixing subpopulations in which virus can spread rapidly), timing (exportation occurring in or after 2025 when population immunity to transmission is very low), and the nature of outbreak response (exportation to badly performing subpopulations). We emphasize the importance of focusing on the high-level insights that emerge from consistent comparison across the scenarios and not on any specific point estimates.
Fig. 6 shows the expected values of the 100 iterations of each scenario for: (a) cVDPV2 and (b) VAPP2 annual paralytic cases over the time horizon that correspond to the totals summarized in Table II. The “No reversion, no VAPP” and “No reversion, same VAPP” scenarios in Fig. 6(a) show overlapping and substantially lower expected cVDPV2 cases than expected with mOPV2 use in RC2, with divergence starting in 2022 due to oSIAs starting in 2021 using nOPV2 not leading to the seeding of new cVDPV2s. However, as noted above, the nOPV2 use in these scenarios does not immediately stop the cVDPV2s already seeded due to its use for outbreak response at the same coverage level as mOPV2 oSIAs. Notably, the “No reversion, no VAPP” and “No reversion, same VAPP” scenarios include nOPV2 use for oSIAs throughout the time horizon as the detection of cases leads to recognition of transmission, which then leads to an oSIA and use of nOPV2. For these scenarios, our assumption about the same R0 for nOPV2 as mOPV2 at stage 0 (see Fig. 5(a)) implies the same initial extent of secondary spread and associated benefits in increasing population immunity, but lower (or no) risks of seeding new cVDPVs due to nOPV2 use, and thus fewer new cVDPV outbreaks later in the time horizon compared to mOPV2 use. While counterintuitive, in 2021–24, the results of the “Some reversion” scenarios show slightly greater numbers of expected cVDPV2 cases than RC2, because of the relative disadvantage of nOPV2 compared to mOPV2 with respect to population immunity from decreased relative R0 for stages 0–9 (see Fig. 5(a). However, the use of nOPV2 does not seed new cVDPV2s, which leads to fewer cVDPV2 cases after 2024. Additionally, the slower accumulation of cases attributable to transmission of the nOPV2-related viruses relative to mOPV2 leads to later detection of outbreaks and later outbreak response. Fig. 6(b) shows that if nOPV2 use implies any risk of VAPP, then we should expect a very low incidence of VAPP cases associated with nOPV2 use over the time horizon. The number of VAPP cases depends on the number of nOPV2 doses given to immunologically naïve children, including cases associated with OPV2 restarts in the iterations in which they occur, and the actual VAPP rate for nOPV2. Fig. 6 also shows that compared to the extended use of mOPV2 in RC3, the “No reversion” scenarios perform only moderately better, while the diminished secondary spread in “Some reversion” scenarios leads to more expected cVDPV2 cases than in RC3. Overall, the results show the inability to completely contain cVDPV2 outbreaks even with non-reverting nOPV2, which highlight the underlying problems that arise from low-quality outbreak response and the inability of using nOPV2 (or mOPV2) for oSIAs only to confidently stop all transmission of OPV2-related viruses and achieve successful OPV2 cessation. Notably, circulation of LPV2s will continue in populations with sufficiently low population immunity to transmission, with all populations becoming increasingly vulnerable to sustaining transmission of an imported LPV2 as a function of time since the last use of OPV2 in the population (Duintjer Tebbens, Hampton, & Thompson, 2016a, 2016b).
Fig. 6.
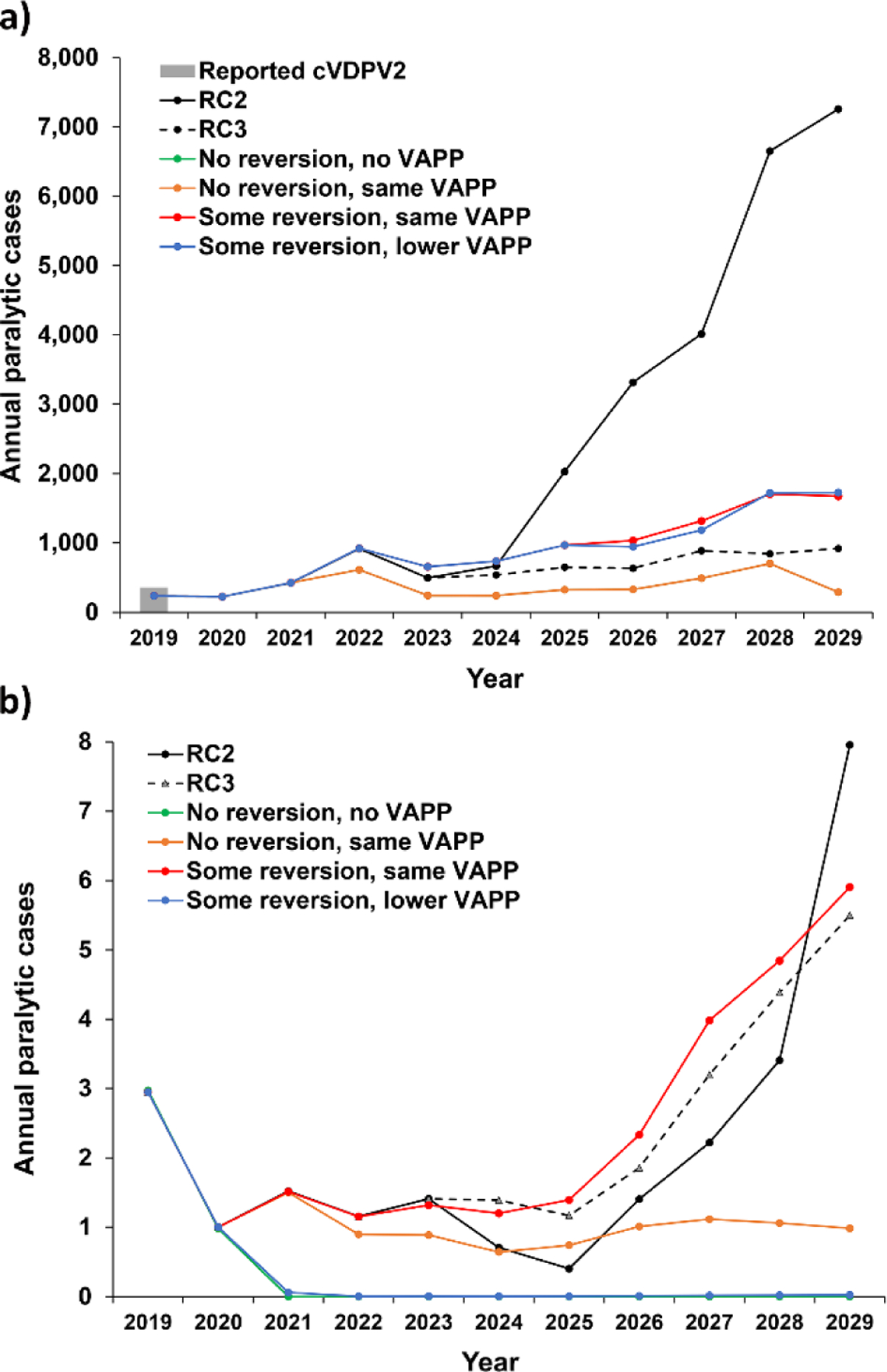
Expected value of annual serotype 2 paralytic cases for 2019–2029 based on 100 iterations for modeled scenarios that use different vaccine options to respond to outbreaks: mOPV2 through 2024 then IPV (RC2), mOPV2 for outbreak response for the full time horizon (RC3), or using nOPV2 starting in 2021 with different characteristics: (a) Incidence of cVDPV2s (“No reversion, no VAPP” and “No Reversion, same VAPP” lines overlap) (b) Incidence of type 2 VAPP (“No reversion,no VAPP” and “Some reversion, lower VAPP” lines overlap)
6. DISCUSSION
Now 4 years after the 2016 tOPV-bOPV switch, continued circulation of existing cVDPV2s and seeding of new emergences by using mOPV2 for outbreak response both contribute to the projected failure to complete successful OPV2 cessation (Kalkowska, Pallansch, et al., 2020). The impacts of nOPV2 use will depend on its properties and the nature of its use. This analysis suggests that switching to using nOPV2 instead of mOPV2 in oSIAs in 2021 by itself will likely not solve the current cVDPV2 problem, even if the introduction of nOPV2 may somewhat improve the possibility of success compared to RC2. This limited impact is due to the population dynamics of existing cVDPV2 transmission and current operational challenges in improving the quality of outbreak response (Kalkowska, Pallansch, et al., 2020). The success of the current endgame strategy of OPV cessation requires stopping existing cVDPV2 transmission (Thompson & Duintjer Tebbens, 2012). The impacts from the disruption of RI, WPV1 eradication efforts, and cVDPV outbreak response due to COVID-19 in 2020 will likely become more evident in the future and will complicate prior expectations and prospects for OPV cessation as a polio endgame strategy within the current 2019–2023 strategic plan (World Health Organization Global Polio Eradication Initiative, 2019, 2020b).
Modeling the performance of a new vaccine remains a challenge for a large and evolving global program, and several limitations of our analysis warrant mention. First, as with all models, the structure and assumptions of the model limit its ability to provide useful insights (see extensive discussion elsewhere of limitations of the global model (Kalkowska, Wassilak, et al., 2020)) and our use of an existing model implies all of the limitations of that model. Second, this analysis does not consider an alternative path that would achieve WPV1 eradication by 2023 (e.g., (Kalkowska & Thompson, 2020)) or the reality that as part of considering OPV2 restart, the GPEI and countries will need to also consider the potential impacts of the COVID-19 pandemic on transmission dynamics and the risk of an OPV2 restart. Such analysis falls beyond the scope of this work, but the outcomes of disruption of GPEI activities should become more apparent by the end of 2020. Third, uncertainty remains about the actual nOPV2 properties and the nature and extent of its transmission in populations. Although we considered the current evidence from published clinical trials and preliminary insights from phase II trials in previously immunized adults, young children, and infants, more evidence from these studies may lead to more accurate assumptions in future modeling. Actual nOPV2 use in large populations will provide the ultimate evidence of VAPP and VDPV risks. Fourth, the model shows conceptual insights, but does not and cannot predict the future, because future outcomes depend on the current and future actions taken by the GPEI and countries (Thompson & Kalkowska, 2020) and the actual performance of the vaccine and field operation effectiveness in SIAs. We emphasize that our model seeks to provide high-level behavioral insights about what might occur in the context of different possible scenarios and not specific estimates of what will occur in the future. Specifically, by exploring many possible futures, we hope that this work highlights the importance of considering all possibilities and not just hoping for the “best” case. Fifth, our assumptions of unlimited supplies of all vaccines for this analysis lead to better possible futures than could occur, because limited supplies will constrain the timing and scope of oSIA responses and lead to more extensive transmission prior to effective oSIAs. We intended for this analysis to highlight the important potential differences between mOPV2 and nOPV2, not to characterize any transition between mOPV2 and nOPV2 use or to consider any mixed use or rationing that could occur during the time of limited availability of both mOPV2 and nOPV2.
Favorable performance of nOPV2 in controlling cVDPV2 outbreaks (i.e., low or no VAPP, no or minimal reversion, high immunogenicity and effectiveness) will likely influence future development and evaluation of nOPV1 and nOPV3 candidates. However, the failure of OPV2 cessation as a successful strategy could lead to a pre-switch strategy of polio control through RI and preventive SIAs and oSIAs for cVDPV2 outbreaks. In this context some OPV-using countries may consider using tOPV instead of bOPV and IPV, since IPV costs considerably more to purchase and deliver. As efforts to eliminate polio continue, the development of nOPVs may create other possible long-term poliovirus vaccine options. The availability of nOPV1 and nOPV3 would increase the prospects for successful Sabin OPV1 and OPV3 cessation, and development of nOPVs for all types could theoretically lead to a trivalent nOPV formulation. The prospect of Sabin OPV2 restart in RI increases the urgency for a consideration of the options for all nOPV use and timeframes for inclusion of potential alternative type 2 vaccines. However, the cost and time required for development, licensure, production, and procurement of alternative OPV2 containing vaccines would need to be balanced with the potential benefits as the GPEI and countries evaluate future strategies. With initial use of nOPV2 expected in the latter half of 2020 and large-scale use in early 2021, the properties of nOPV2 in actual use will influence vaccine demands, stockpile needs, and program outcomes.
The insights from this modeling suggest the need for expansion of the GPEI strategy of using nOPV2 only for oSIAs to include considerations for RI or larger, preventive SIAs (World Health Organization Global Polio Eradication Initiative, 2019, 2020b), which may include more aggressive oSIAs as suggested in earlier modeling (Duintjer Tebbens et al., 2015; Duintjer Tebbens, Pallansch, et al., 2016; Duintjer Tebbens & Thompson, 2018). The development of nOPVs represents a substantial investment that may prove essential for a successful polio endgame. Although uncertainty exists about actual nOPV2 properties and the future of nOPV2 use, this analysis provides an example of how modeling can inform decisions and policy based on the potential benefits of nOPV2. Future studies should consider other potential uses for nOPVs, including their possible use in RI in the event of OPV restart and the potential use of nOPVs for oSIAs after homotypic OPV cessation for types 1 and 3 instead of mOPVs.
ACKNOWLEDGMENTS
The first and last two authors acknowledge support for this publication under Cooperative Agreement Number 5NU2RGH001913-04-00 funded by the Centers for Disease Control and Prevention. We thank Abhijeet Anand and Stephen Cochi for helpful comments and the sponsors of ongoing nOPV clinical trials, including the University of Antwerp and Fighting Infectious Diseases in Emerging Countries for sharing information. The views expressed are solely those of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention or Department of Health and Human Services.
REFERENCES
- Alleman MM, Jorba J, Greene SA, Diop OM, Iber J, Tallis G, … Burns CC (2020). Update on Vaccine-Derived Poliovirus Outbreaks - Worldwide, July 2019–February 2020. MMWR. Morbidity and mortality weekly report, 69(16), 489–495. 10.15585/mmwr.mm6916a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay AS (2020a). Clinical data from novel type-2 oral polio vaccine trials and plan for emergency use listing. Meeting of the Strategic Advisory Group of Experts (SAGE) on Immunization, March – April 2020 Retrieved from https://www.who.int/immunization/sage/meetings/2019/october/Bandyopadhyay_polio_sage_october_2019.pdf?ua=1 [Google Scholar]
- Bandyopadhyay AS (2020b). Summary of nOPV2 clinical data. Meeting of the Strategic Advisory Group of Experts (SAGE) on Immunization, March – April 2020 Retrieved from https://www.who.int/immunization/sage/meetings/2020/april/SAGE_Slidedeck_Mar2020-Web.pdf?ua=1 [Google Scholar]
- Blake IM, Pons-Salort M, Molodecky NA, Diop OM, Chenoweth P, Bandyopadhyay AS, … Grassly NC (2018). Type 2 Poliovirus Detection after Global Withdrawal of Trivalent Oral Vaccine. N Engl J Med, 379(9), 834–845. 10.1056/NEJMoa1716677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, … Nair SK (2017). Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen–specific CTLs. Sci Transl Med, 9(408), eaan4220 10.1126/scitranslmed.aan4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CC, Campagnoli R, Shaw J, Vincent A, Jorba J, & Kew O (2009). Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and UpA dinucleotides within and across synonymous capsid region codons. J Virol, 83(19), 9957–9969. 10.1128/jvi.00508-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CC, Shaw J, Campagnoli R, Jorba J, Vincent A, Quay J, & Kew O (2006). Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J Virol, 80(7), 3259–3272. 10.1128/jvi.80.7.3259-3272.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CC, Shaw J, Jorba J, Bukbuk D, Adu F, Gumede N, … Kew O (2013). Multiple independent emergences of type 2 vaccine-derived polioviruses during a large outbreak in northern Nigeria. J Virol, 87(9), 4907–4922. 10.1128/jvi.02954-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammack N, Phillips A, Dunn G, Patel V, & Minor PD (1988). Intertypic genomic rearrangements of poliovirus strains in vaccinees. Virology, 167(2), 507–514. [PubMed] [Google Scholar]
- Coyne CB, Oberste MS, & Pallansch MA (2017). Enteroviruses: Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruse In Howley PM & Knipe DM (Eds.), Fields Virology: Emerging Viruses (pp. 86–128). Philadelphia, PA: Wolthers Kluwer, Inc.. [Google Scholar]
- Cuervo NS, Guillot S, Romanenkova N, Combiescu M, Aubert-Combiescu A, Seghier M, … Delpeyroux F (2001). Genomic features of intertypic recombinant sabin poliovirus strains excreted by primary vaccinees. Journal of Virology, 75(13), 5740–5751. 10.1128/JVI.75.13.5740-5751.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diop OM, Asghar H, Gavrilin E, Moeletsi NG, Benito GR, Paladin F, … Quddus A (2017). Virologic Monitoring of Poliovirus Type 2 after Oral Poliovirus Vaccine Type 2 Withdrawal in April 2016 - Worldwide, 2016–2017. Morbidity and Mortality Weekly Report, 66(20), 538–542. 10.15585/mmwr.mm6620a4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle WR, de Gourville E, Kew OM, Pallansch MA, & Wood DJ (2003). Polio eradication: the OPV paradox. Reviews in Medical Virology, 13(5), 277–291. [DOI] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Hampton LM, & Thompson KM (2016a). Implementation of coordinated global serotype 2 oral poliovirus vaccine cessation: Risks of inadvertent trivalent oral poliovirus vaccine use. BMC Infectious Diseases, 16, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Hampton LM, & Thompson KM (2016b). Implementation of coordinated global serotype 2 oral poliovirus vaccine cessation: Risks of potential non-synchronous cessation. BMC Infectious Diseases, 16, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Pallansch MA, Chumakov KM, Halsey NA, Hovi T, Minor PD, … Thompson KM (2013a). Expert review on poliovirus immunity and transmission. Risk Analysis, 33(4), 544–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Pallansch MA, Chumakov KM, Halsey NA, Hovi T, Minor PD, … Thompson KM (2013b). Review and assessment of poliovirus immunity and transmission: Synthesis of knowledge gaps and identification of research needs. Risk Analysis, 33(4), 606–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Pallansch MA, Kalkowska DA, Wassilak SGF, Cochi SL, & Thompson KM (2013). Characterizing poliovirus transmission and evolution: Insights from modeling experiences with wild and vaccine-related polioviruses. Risk Analysis, 23(4), 703–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Pallansch MA, Kim J-H, Burns CC, Kew OM, Oberste MS, … Thompson KM (2013). Review: Oral poliovirus vaccine evolution and insights relevant to modeling the risks of circulating vaccine-derived polioviruses (cVDPVs). Risk Analysis, 23(4), 680–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Pallansch MA, Wassalik SGF, Cochi SL, & Thompson KM (2015). An economic analysis of poliovirus risk management policy options for 2013–2052. BMC Infectious Diseases, 15(389), 10.1186/s12879-12015-11112-12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, Pallansch MA, Wassilak SGF, Cochi SL, & Thompson KM (2016). Characterization of outbreak response strategies and potential vaccine stockpile needs for the polio endgame. BMC Infectious Diseases, 16, 137 10.1186/s12879-016-1465-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, & Thompson KM (2016). The potential benefits of a new poliovirus vaccine for long-term poliovirus risk management. Future Microbiology, 11(12), 1549–1561. [DOI] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, & Thompson KM (2017a). Costs and benefits of including inactivated in addition to oral poliovirus vaccine in outbreak response after cessation of oral poliovirus vaccine use. Med Decis Making Policy & Practice, 2(1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, & Thompson KM (2017b). Poliovirus vaccination during the endgame: Insights from integrated modeling. Expert Review of Vaccines, 16(6), 577–586. 10.1080/14760584.2017.1322514 [DOI] [PubMed] [Google Scholar]
- Duintjer Tebbens RJ, & Thompson KM (2018). Polio endgame risks and the possibility of restarting the use of oral poliovirus vaccine. Expert Review of Vaccines, 17(8), 739–751. 10.1080/14760584.2018.1506333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escarmis C, Lazaro E, & Manrubia SC (2006). Population bottlenecks in quasispecies dynamics. In Quasispecies: Concept and Implications for Virology (Vol. 299, pp. 141–170). [DOI] [PubMed] [Google Scholar]
- Famulare M, Chang S, Iber J, Zhao K, Adeniji JA, Bukbuk D, … Oberste MS (2015). Sabin Vaccine Reversion in the Field: a Comprehensive Analysis of Sabin-Like Poliovirus Isolates in Nigeria. Journal of virology, 90(1), 317–331. 10.1128/JVI.01532-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furione M, Guillot S, Otelea D, Balanant J, Candrea A, & Crainic R (1993). Polioviruses with natural recombinant genomes isolated from vaccine-associated paralytic poliomyelitis. Virology, 196(1), 199–208. doi:S0042682283714686 [pii] [DOI] [PubMed] [Google Scholar]
- Georgescu MM, Delpeyroux F, Tardy-Panit M, Balanant J, Combiescu M, Combiescu AA, … Crainic R (1994). High diversity of poliovirus strains isolated from the central nervous system from patients with vaccine-associated paralytic poliomyelitis. J Virol, 68(12), 8089–8101. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7966599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Polio Eradication Initiative. (2015, September 20, 2015). Global eradication of wild poliovirus type 2 declared. Retrieved from http://www.polioeradication.org/mediaroom/newsstories/Global-eradication-of-wild-poliovirus-type-2-declared/tabid/526/news/1289/Default.aspx
- Gromeier M, Alexander L, & Wimmer E (1996). Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A, 93(6), 2370–2375. 10.1073/pnas.93.6.2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton LM, Farrell M, Ramirez-Gonzalez A, Menning L, Shendale S, Lewis I, … Immunization Systems Management Group of the Global Polio Eradication Initiative. (2016). Cessation of trivalent oral poliovirus vaccine and introduction of inactivated poliovirus vaccine - Worldwide, 2016. Morbidity and Mortality Weekly Report, 65(35), 934–938. 10.15585/mmwr.mm6535a3 [DOI] [PubMed] [Google Scholar]
- Jegouic S, Joffret M-L, Blanchard C, Riquet FB, Perret C, Pelletier I, … Delpeyroux F (2009). Recombination between polioviruses and co-circulating Coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS pathogens, 5(5), e1000412–e1000412. 10.1371/journal.ppat.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkowska DA, Duintjer Tebbens RJ, Grotto I, Shulman LM, Anis E, Wassilak SGF, … Thompson KM (2015). Modeling options to manage type 1 wild poliovirus imported into Israel in 2013. Journal of Infectious Diseases, 211(11), 1800–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkowska DA, Pallansch MA, Cochi SL, Kovacs SD, Wassilak SGF, & Thompson KM (2020). Updated characterization of post-OPV cessation risks: Lessons from 2019 serotype 2 outbreaks and implications for the probability of OPV restart. Risk Analysis, online July 6, 10.1111/risa.13555. [DOI] [PMC free article] [PubMed]
- Kalkowska DA, & Thompson KM (2020). Insights from modeling preventive supplemental immunization activities as a strategy to eliminate wild poliovirus transmission in Pakistan and Afghanistan. Risk Analysis, online March 6, 10.1111/risa.13471. [DOI] [PMC free article] [PubMed]
- Kalkowska DA, Wassilak SGF, Cochi SL, Pallansch MA, & Thompson KM (2020). Global transmission of live polioviruses: Updated integrated dynamic modeling of the polio endgame. Risk Analysis, online January 22, 10.1111/risa.13447. [DOI] [PMC free article] [PubMed]
- Knowlson S, Burlison J, Giles E, Fox H, Macadam AJ, & Minor PD (2015). New Strains Intended for the Production of Inactivated Polio Vaccine at Low-Containment After Eradication. PLoS Pathogens, 11(12), e1005316 10.1371/journal.ppat.1005316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka-Anstadt JL, Campagnoli R, Vincent A, Shaw J, Wei L, Wynn NT, … Burns CC (2020). Development of a new oral poliovirus vaccine for the eradication end game using codon deoptimization. NPJ Vaccines, 5, 26 10.1038/s41541-020-0176-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipskaya GY, Muzychenko AR, Kutitova OK, Maslova SV, Equestre M, Drozdov SG, … Agol VI (1991). Frequent isolation of intertypic poliovirus recombinants with serotype 2 specificity from vaccine-associated polio cases. J Med Virol, 35(4), 290–296. 10.1002/jmv.1890350415 [DOI] [PubMed] [Google Scholar]
- Liu HM, Zheng DP, Zhang LB, Oberste MS, Kew OM, & Pallansch MA (2003). Serial recombination during circulation of type 1 wild-vaccine recombinant polioviruses in China. Journal of Virology, 77(20), 10994–11005. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14512548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macadam AJ, Ferguson G, Stone DM, Meredith J, Knowlson S, Auda G, … Minor PD (2006). Rational design of genetically stable, live-attenuated poliovirus vaccines of all three serotypes: relevance to poliomyelitis eradication. J Virol, 80(17), 8653–8663. 10.1128/JVI.00370-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macadam AJ, Pollard SR, Ferguson G, Skuce R, Wood D, Almond JW, & Minor PD (1993). Genetic basis of attenuation of the Sabin type 2 vaccine strain of poliovirus in primates. Virology, 192(1), 18–26. 10.1006/viro.1993.1003 [DOI] [PubMed] [Google Scholar]
- Macklin GR, O’Reilly KM, Grassly NC, Edmunds WJ, Mach O, Santhana Gopala Krishnan R, … Sutter RW (2020). Evolving epidemiology of poliovirus serotype 2 following withdrawal of the serotype 2 oral poliovirus vaccine. Science, 368(6489), 401–405. 10.1126/science.aba1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor PD, Macadam AJ, Stone DM, & Almond JW (1993). Genetic basis of attenuation of the Sabin oral poliovirus vaccines. Biologicals, 21(4), 357–363. 10.1006/biol.1993.1096 [DOI] [PubMed] [Google Scholar]
- Novella IS, Quer J, Domingo E, & Holland JJ (1999). Exponential fitness gains of RNA virus populations are limited by bottleneck effects. J Virol, 73(2), 1668–1671. Retrieved from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9882378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste MS (2008). Comparative genomics of the coxsackie B viruses and related enteroviruses. Curr Top Microbiol Immunol, 323, 33–47. 10.1007/978-3-540-75546-3_2 [DOI] [PubMed] [Google Scholar]
- Oh HS, Pathak HB, Goodfellow IG, Arnold JJ, & Cameron CE (2009). Insight into Poliovirus Genome Replication and Encapsidation Obtained from Studies of 3B-3C Cleavage Site Mutants. Journal of Virology, 83(18), 9370–9387. 10.1128/jvi.02076-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer JK, & Kirkegaard K (2003). A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proceedings of the National Academy of Sciences, 100(12), 7289–7294. 10.1073/pnas.1232294100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Population Division of the Department of Economic and Social Affairs of the United Nations Secretariat. (2019). World population prospects. The 2019 revision Retrieved from https://population.un.org/wpp/
- Runckel C, Westesson O, Andino R, & DeRisi JL (2013). Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog, 9(2), e1003164 10.1371/journal.ppat.1003164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin AB (1985). Oral poliovirus vaccine: history of its development and use and current challenge to eliminate poliomyelitis from the world. Journal of Infectious Diseases, 151(3), 420–436. [DOI] [PubMed] [Google Scholar]
- Simmonds P, Gorbalenya AE, Harvala H, Hovi T, Knowles NJ, Lindberg AM, … Zell R (2020). Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch Virol, 165(3), 793–797. 10.1007/s00705-019-04520-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Yeh MT, Zinger T, Smith M, Wright C, Ling G, … Andino R (2017). The Evolutionary Pathway to Virulence of an RNA Virus. Cell, 169(1), 35–46 10.1016/j.cell.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern A, Yeh MT, Zinger T, Smith M, Wright C, Ling G, … Andino R (2017). The Evolutionary Pathway to Virulence of an RNA Virus. Cell, 169(1), 35–46. 10.1016/j.cell.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter RW, Kew OM, Cochi SL, & Aylward RB (2017). Poliovirus vaccine -- Live In Plotkin SA, Orenstein WA, Offit PA, & Edwards KM (Eds.), Plotkin’s Vaccines (Sixth ed., pp. 866–917). Philadelphia: Saunders Elsevier. [Google Scholar]
- Thompson KM, & Duintjer Tebbens RJ (2012). Current polio global eradication and control policy options: perspectives from modeling and prerequisites for oral poliovirus vaccine cessation. Expert Review of Vaccines, 11(4), 449–459. 10.1586/erv.11.195 [DOI] [PubMed] [Google Scholar]
- Thompson KM, & Duintjer Tebbens RJ (2014). Modeling the dynamics of oral poliovirus vaccine cessation. Journal of Infectious Diseases, 210(Suppl 1), S475–S484. [DOI] [PubMed] [Google Scholar]
- Thompson KM, & Kalkowska DA (2019). Logistical challenges and assumptions for modeling the failure of global cessation of oral poliovirus vaccine (OPV). Expert Review of Vaccines, 18(7), 725–736. 10.1080/14760584.2019.1635463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KM, & Kalkowska DA (2020). Reflections on modeling poliovirus transmission and the polio eradication endgame. Risk Analysis, online April 27, 10.1111/risa.13484 [DOI] [PMC free article] [PubMed]
- Van Damme P, De Coster I, Bandyopadhyay AS, Revets H, Withanage K, De Smedt P, … Gast C (2019). The safety and immunogenicity of two novel live attenuated monovalent (serotype 2) oral poliovirus vaccines in healthy adults: a double-blind, single-centre phase 1 study. Lancet, 394(10193), 148–158. 10.1016/s0140-6736(19)31279-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Wendt E, & Andino R (2008). Engineering attenuated virus vaccines by controlling replication fidelity. Nat Med, 14(2), 154–161. 10.1038/nm1726 [DOI] [PubMed] [Google Scholar]
- Ward CD, Stokes MA, & Flanegan JB (1988). Direct measurement of the poliovirus RNA polymerase error frequency in vitro. J Virol, 62(2), 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Bank. (2019). World Bank analytical classifications - calendar year 2017 GNI per capita in US$ (Atlas methodology). Retrieved from http://databank.worldbank.org/data/download/site-content/OGHIST.xls
- World Health Assembly. (2008). Poliomyelitis: mechanism for management of potential risks to eradication (resolution 61.1). Retrieved from http://apps.who.int/gb/ebwha/pdf_files/WHA61-REC1/A61_Rec1-part2-en.pdf
- World Health Organization. (2019). World schedule as of 2018/July/11. Retrieved from http://apps.who.int/immunization_monitoring/globalsummary/schedules
- World Health Organization Global Polio Eradication Initiative. (2013). Polio eradication and endgame Strategic Plan (2013–2018). Geneva; 2013. Report No: WHO/POLIO/13.02. Retrieved from http://polioeradication.org/wp-content/uploads/2016/07/PEESP_EN_A4.pdf [Google Scholar]
- World Health Organization Global Polio Eradication Initiative. (2018). Standard operating procedures: Responding to a poliovirus event or outbreak: Part 2: Protocol for poliovirus type 2. Retrieved from http://polioeradication.org/wp-content/uploads/2018/01/pol-sop-responding-polio-event-outbreak-part2-20180117.pdf
- World Health Organization Global Polio Eradication Initiative. (2019). Polio eradication and endgame strategic plan (2019–2023). Geneva; 2019. Report No: WHO/POLIO/19.04. Retrieved from http://polioeradication.org/wp-content/uploads/2019/05/polio-endgame-strategy-2019-2023.pdf [Google Scholar]
- World Health Organization Global Polio Eradication Initiative. (2020a). Standard operating procedures: Responding to a poliovirus event or outbreak: Version 3.1, March 2020. Retrieved from http://polioeradication.org/wp-content/uploads/2020/04/POL-SOP-V3.1-20200424.pdf
- World Health Organization Global Polio Eradication Initiative. (2020b). Strategy for the response to type 2 circulating vaccine-derived poliovirus 2020–2021: Addendum to the Polio eradication and endgame strategic plan (2019–2023). >Geneva; 2020. Report No: WHO/POLIO/20.02. Retrieved from http://polioeradication.org/ [Google Scholar]
- Yeh MT, Bujaki E, Dolan PT, Smith M, Wahid R, Konz J, … Andino R (2020). Engineering the Live-Attenuated Polio Vaccine to Prevent Reversion to Virulence. Cell Host & Microbe. 10.1016/j.chom.2020.04.003 [DOI] [PMC free article] [PubMed]