

Abstract
Background:
Antagonism of peripheral opioid receptors by methylnaltrexone (MNTX) was recently proposed as a potential mechanism to attenuate the development of opioid analgesic tolerance based on experiments conducted in mice. However, reports indicate that MNTX is demethylated to naltrexone (NTX) in mice, and NTX may subsequently cross the blood-brain barrier to antagonize centrally-mediated opioid effects. The goal of this study was to determine whether MNTX alters centrally-mediated behaviors elicited by the opioid analgesics, morphine and oxycodone, and to quantify concentrations of MNTX and NTX in blood and brain following their administration in mice.
Methods:
Combinations of MNTX and morphine were tested under acute and chronic conditions in thermal nociceptive assays. Effects of MNTX and NTX pretreatment were assessed in an oxycodone discrimination operant procedure. Blood and brain concentrations of these antagonists were quantified after their administration using liquid chromatography-mass spectrometry.
Results:
MNTX dose-dependently attenuated acute and chronic morphine antinociception. MNTX and NTX dose-dependently antagonized the discriminative stimulus effects of oxycodone. MNTX and NTX were detected in both blood and brain after administration of MNTX, confirming its demethylation and demonstrating that MNTX itself can cross the blood-brain barrier.
Conclusions:
These results provide converging behavioral and analytical evidence that MNTX administration in mice attenuates centrally-mediated effects produced by opioid analgesics and results in functional concentrations of MNTX and NTX in blood and brain. Collectively, these findings indicate that MNTX cannot be administered systemically in mice for making inferences that its effects are peripherally restricted.
Keywords: methylnaltrexone, morphine, oxycodone, antinociception, tolerance, mouse
1. Introduction
Methylnaltrexone (MNTX) is a quaternary amine derivative of the opioid receptor antagonist naltrexone (NTX). MNTX features an added methyl group to the amine ring of the parent compound, resulting in increased polarity and decreased lipophilicity. Consequentially, MNTX does not cross the blood-brain barrier in humans at clinically effective doses, and does not interfere with opioid analgesia (Pergolizzi et al., 2020). MNTX is clinically indicated for the reversal of opioid-induced constipation, a common consequence of chronic opioid use that includes pain, reflux, fecal impaction, and infection (Webster, 2015).
Preclinical researchers have used MNTX as a tool to antagonize opioid receptors while assuming a lack of central activity (for review, see Brown and Goldberg, 1985); however, some have reported results incompatible with that assumption. Ramabadran (1982) reported that MNTX produced dose- and time-dependent hyperalgesia in mice in a hot plate assay, speculating that the drug “might be converted to an active metabolite”. Also, antinociceptive effects of morphine in the hot plate assay can be blocked by MNTX in rats (Bianchi et al., 1982) and in mice (Russell et al., 1982). Illuminating the tenuousness of this assumption especially when disregarding species differences, a study of metabolic differences across species conducted by Kotake et al. (1989) indicated that MNTX was demethylated in mice and more so than in dogs or humans. Thus, it is possible that NTX, as a metabolite of MNTX demethylation, could explain some effects of MNTX on centrally-mediated responses in mice, while compromising experimental interpretations presuming MNTX to be peripherally restricted in mice. MNTX may even act centrally in humans in limited situations. For example, miosis is elicited by 0.45 mg/kg MNTX administered subcutaneously (Zacny et al., 2015), and an oral dose of 19.2 mg/kg MNTX reduces morphine-induced alterations in subjective ratings including “stimulated” and “nauseous” (Yuan et al., 1998). This latter finding correlated with plasma MNTX concentrations. In general, however, the scientific consensus is that clinically-utilized MNTX doses (i.e., up to 0.3 mg/kg s.c.) do not serve as antagonists on centrally located opioid receptors (Webster et al., 2015; Webster and Israel, 2018; Yuan et al., 2000; Yuan et al., 2002).
A highly cited study (111 citations, Web of Science, 20201023) using mice concluded that peripheral nociceptors had a key role in the development of antinociceptive tolerance and opioid-induced hyperalgesia to morphine (Corder et al., 2017). Requisite for this conclusion was the assumption that the blood-brain barrier was impermeable to MNTX, and its administration would produce peripherally restricted antagonism of morphine’s effects. MNTX was found to prevent the development of opioid antinociceptive tolerance in several nociceptive assays and models of perioperative and chronic pain without disrupting morphine antinociception. The ability to potentially repurpose a clinically approved medication to decrease the development of opioid analgesic tolerance without affecting analgesia would represent a breakthrough transformation in chronic pain management. However, this study was conducted in mice which metabolize MNTX into NTX as described above.
The objective of the present study was to assess the peripheral restrictedness of MNTX in mice using behavioral and analytical assays. Combinations of MNTX and morphine were administered acutely or repeatedly to mice and tested in the warm-water tail-withdrawal antinociception assay. Other groups of mice were implanted with morphine pellets and assessed for daily tail-withdrawal or hot-plate latencies. These nociceptive assays were selected because they involve differential CNS recruitment with the tail-withdrawal response from a noxious thermal stimulus being a spinally-mediated reflex, whereas escape behaviors in the hot plate assay recruit supraspinal processes (Barrot, 2012). Also determined was whether MNTX could attenuate the discriminative stimulus properties of oxycodone, a centrally-mediated model of a drug’s subjective effects (Porter et al., 2018). Lastly, the dose-response and time-course relationships of MNTX and NTX concentrations in blood and whole brain were evaluated to corroborate the findings from behavioral studies. The results indicated that MNTX could antagonize opioid effects of CNS-mediated behaviors in mice and its administration results in functionally meaningful concentrations of MNTX and NTX in the brain.
2. Material and Methods
2.1. Subjects
Adult male Swiss Webster mice (Envigo, Dublin, VA) were used in acute warm-water tail withdrawal and in morphine pellet experiments in which warm-water tail withdrawal and hot plate responses were tested. Adult male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were used in the chronic, peripherally administered morphine warm-water tail-withdrawal, oxycodone discrimination, and mass spectrometry experiments. A subset of mice from a previously reported study (Walentiny et al., 2019) were used in the oxycodone discrimination experiments; all other mice were drug naive prior to testing. Subjects were housed in temperature-controlled (20-22°C) AAALAC-accredited facilities and maintained on a 12 h/12 h light-dark cycle (lights on 0600-1800 h) with behavioral testing occurring during the light cycle. Food and water were available ad libitum in the home cage, except for oxycodone discrimination subjects that were maintained at 85-90% of their free-feeding body weights by controlled post-session feeding to promote food-reinforced responding. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (2011), and approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
2.2. Apparatus
Nociception was assessed using water baths (Thermo Electron Corp, Marietta, OH; Grant Instruments Ltd., Cambridge, UK) and a hot plate (Life Science Inc, Woodland Hills, CA). Latencies to elicit nociceptive behaviors were measured with a digital stopwatch. Drug discrimination experiments were conducted with commercially obtained operant conditioning chambers (ENV-307W-CT; Med Associates Inc., Fairfax, VT) housed in sound- and light-attenuating cubicles (SAC-022-SA, Med Associates Inc.). The front panel of each chamber was equipped with two retractable levers. A recessed food receptacle connected to a pellet hopper was centered between the levers. A house light on the rear panel was illuminated during sessions and fans provided ventilation and masking noise. A computer operating with Med-PC IV software (Med Associates Inc.) was used to control session parameters and record data.
2.3. Procedure
2.3.1. Effects of acute administration of MNTX and morphine in the warm-water tail-withdrawal assay
Baseline tail-withdrawal latencies to a noxious warm-water bath set to 56°C were initially determined by gently restraining mice with a cloth and submerging the distal third of the tail in the water. Next, mice were injected with a dose MNTX (0.5-25 mg/kg) or vehicle 20 min prior to administration of 10 mg/kg morphine. Thirty min after morphine administration, tail-withdrawal latencies were re-determined. A maximum exposure of 10 s was imposed to minimize tissue damage.
2.3.2. Effects of MNTX on nociceptive behaviors in a TNBS-induced model of inflammatory bowel disease
To evaluate MNTX’s effectiveness in antagonizing centrally-mediated morphine antinociception in the presence or absence of peripheral inflammation, we utilized a model of 2,4,6-trinitrobenzenesulfonic acid (TNBS) to model symptoms of inflammatory bowel disease (Silva et al., 2019). Mice were administered TNBS or its vehicle, and were subsequently administered MNTX or vehicle daily to evaluate MNTX’s effectiveness in antagonizing morphine antinociception in the presence or absence of chronic inflammatory pain. On Day 0, Swiss Webster mice were implanted subcutaneously with 75 mg morphine pellets in the dorsum under anesthesia (2.5% isoflurane). A small cut was made at the base of the neck with scissors after the hair was shaved and the area was thoroughly cleansed with 10% povidone iodine (General Medical Corp., Walnut, CA) and rinsed with 70% ethanol. The pellet was inserted into the small opening and the skin was stapled together with Clay Adams Brand, MikRon AutoClip 9-mm Wound Clips (Becton Dickinson, Franklin Lakes, NJ). The pelleted mice were then injected with 25 mg/kg MNTX s.c. under anesthesia followed by intra-rectal administration of 100 μL 2,4,6-trinitrobenzenesulfonic acid (TNBS) solution containing a 1:1 dilution mixture of picrylsulfonic acid solution (5% w/v in H2O; Sigma-Aldrich, St. Louis, MO) and 50% ethanol. The control group received 100 μL of a 1:1 dilution mixture of saline and 50% ethanol. Approximately 24 h after morphine pellet implantation, separate groups of mice were assessed daily for tail flick (56°C; previously described methodology) or hot plate latencies 20 min after administration of 25 mg/kg MNTX. For the hot plate assay, mice were placed inside a cylindrical tube on the hot plate (56°C) to prevent escaping and the latency to lick the hind paw was recorded. A 30 s maximum exposure time was imposed to minimize tissue damage. On Day 3, after the daily MNTX injection and nociceptive testing, mice with sub-maximal baseline recordings (i.e., < 10 s in tail withdrawal; < 30 s in hot plate) were challenged with 10 mg/kg morphine (s.c.) and then tested again 30 min later to determine if MNTX’s effects on morphine-induced antinociception were surmountable with a booster dose of morphine.
2.3.3. Effects of MNTX and chronic morphine in the warm-water tail-withdrawal assay
We next examined the dose-response relationship of MNTX by repeated systemic co-administration of MNTX (0.5-10 mg/kg) with 10 mg/kg morphine once daily. Morphine cumulative-dose response curves were determined on Day 1 and Day 7. First, each mouse was placed in a restraint cloth, and the distal 3 cm of its tail was submerged in a water bath maintained at 52.5°C to determine its baseline withdrawal latency. Immediately thereafter, mice received consecutive injections of saline (i.e., morphine’s vehicle) and their randomly-assigned antagonist condition (i.e., 0.1, 0.5, 1, or 10 mg/kg MNTX, 1 mg/kg NTX, or saline vehicle), and were returned to their home cage. This design permitted detection of antagonist-induced changes to the morphine dose-response curve relative to saline-treated controls. After a 30 min pretreatment period had elapsed, tail-flick latencies were re-determined, and mice were immediately injected with the lowest dose of morphine (1 mg/kg). Following the 30 min pretreatment period, tail-flick latencies were re-determined, and the next highest dose of morphine was administered (i.e., acutely 2.2 mg/kg resulting in a cumulative dose of 3.2 mg/kg). This process was repeated with cumulative doses of 10 and 32 mg/kg morphine.
On non-test days (i.e., Days 2-6), mice received two consecutive injections. The first injection was either 10 mg/kg morphine or saline and the second injection was the respective antagonist condition administered on Day 1. Approximately 24 h after receiving the last set of injections on Day 6, mice were re-assessed in the warm-water tail-withdrawal procedure with cumulative morphine dosing identical to that conducted on Day 1 to assess for potential rightward shifts in the morphine dose-response curve indicative of tolerance. A 10 s maximum exposure time was imposed across all assessments to minimize tissue damage.
2.3.4. Effects of MNTX on the Discriminative Stimulus Effects of Oxycodone
Oxycodone discrimination training and testing were conducted as described by Walentiny et al. (2019). Briefly, mice were trained to discriminate between 1.3 mg/kg oxycodone (i.e., “training dose”) and vehicle administered 15 min prior to the start of experimental sessions in a two lever, food-reinforced procedure. During training, responding on one lever following oxycodone administration resulted in delivery of a 20 mg food pellet (Bio-Serv, Flemington, NJ; Catalog #F0071) according to a fixed ratio 10 (FR10) schedule (i.e., 10 consecutive responses on the designated lever were required for reinforcer delivery). Responding on the opposite lever was reinforced in an identical manner following vehicle administration. Once subjects demonstrated accurate discrimination between the two conditions (i.e., emitted the first ten consecutive responses on the injection-appropriate lever and ≥ 80% of all responses on this lever as well) for 8 out of 10 consecutive sessions, testing commenced. A test session was conducted typically on Tuesdays and Fridays to permit a minimum 72 h drug washout and only if the mice had accurately discriminated during their most recent oxycodone and vehicle training sessions; otherwise, a training session was conducted. During test sessions, presses of either lever were reinforced according to an FR10 schedule, but the response contingency was reset if a mouse switched responding on one lever to the other prior to completing the FR10 requirement.
After generating an oxycodone dose-response curve, initial antagonism tests were conducted with NTX (0.01, 0.1, 1 mg/kg) and MNTX (0.1, 0.5, 1, 10 mg/kg) administered 25 or 45 min, respectively, prior to the test session. During these initial tests, MNTX demonstrated antagonistic efficacy and the temporal parameters of these effects were subsequently evaluated. During these time-course tests, 10 mg/kg MNTX was administered 15, 30, 45, 60, 120, or 240 min prior to session start to assess the onset and duration of MNTX antagonism. During each antagonism test, the oxycodone training dose was administered 15 min prior to session start and the test session lasted 15 min. Prior to each dose-response or time-course determination, control tests were conducted with the oxycodone training dose and vehicle to ensure that subjects were accurately discriminating between the two conditions and to serve as comparators for statistical analysis.
2.3.5. Quantification of MNTX and NTX in Mouse Blood and Brain using Liquid Chromatography Mass Spectrometry
To corroborate the behavioral results that suggested centrally mediated actions of MNTX, concentrations of MNTX and NTX were determined in blood and brain in mice using identical antagonist dosing parameters as used during the oxycodone discrimination experiments. Dose-response evaluations were conducted with MNTX (0.1, 0.5, 1, 10 mg/kg, 45 min pretreatment) and NTX (0.01, 0.1, 1 mg/kg, 25 min pretreatment). A time-course evaluation (15, 30, 60, 120, 240 min) also was conducted with 10 mg/kg MNTX. Groups of mice administered saline (vehicle for MNTX and NTX) were sacrificed at 45, 25, or 15 min post-treatment to serve as controls for these respective experiments. Following the designated pretreatment time, mice were sacrificed via cervical dislocation and decapitated. Whole brain samples were collected and stored individually, and trunk blood of mice from a given treatment condition was aggregated in a single vial to yield a sufficient sample volume for analytical quantification. Samples were stored at −80°C until analyzed.
The identification and quantification of MNTX and NTX in mouse blood and brain was performed using a modification of previously described methods (Schwienteck et al., 2019). MNTX and NTX were extracted from both blood and brain using a liquid/liquid extraction. Prior to extraction the brain tissue specimens were homogenized 1-part tissue to 3-parts ddH2O. Matrix matched seven-point calibration curves (1- 100 ng/mL or ng/g), drug free control, a negative control without internal standard (ISTD), and quality control specimens (3, 30 and 75 ng/mL or ng/g) were prepared and analyzed with each batch of samples. NTX-d3, the ISTD, was added at 10 ng/mL to aliquots of either 100 μL for blood or 400 μL for brain homogenate to each calibrator, control or specimen except the negative control. To these samples 0.5 mL of saturated carbonate/bicarbonate buffer (1:1, pH 9.5) and 1.0 mL of chloroform:2-propanol (9:1) was added. The samples were then mixed and centrifuged. The top aqueous layer was aspirated, and the organic layer was transferred to a clean test tube and evaporated to dryness under nitrogen. The samples were then reconstituted in methanol and transferred to autosampler vials for analysis.
Chromatographic separation of MNTX and NTX was achieved using a Shimadzu Nexera X2 liquid chromatography system with a Zorbax XDB-C18 4.6 x 75 mm, 3.5-micron column (Agilent Technologies, Santa Clara, CA). The mobile phase consisted of water/methanol (80:20, v/v) with 1 g/L ammonium formate and 0.1 % fomic acid with a flow rate was 1 mL/min. The systems detector was a Sciex 6500 QTRAP system with an IonDrive Turbo V source for TurbolonSpray® (Sciex, Ontario, Canada) that had the curtain gas flow rate set at 30 mL/min and the ion source gases 1 and 2 at 60 mL/min. The source temperature was set at 650°C with an ionspray voltage was 5500 V. The declustering potential and collection energy was 86 eV and 39 eV, respectively. The following quantification and qualifying transition ions were monitored in positive multiple reaction monitoring (MRM) mode: MNTX 356> 340 & 358 > 254; NTX 342> 267 & 342 > 282; NTX-d3 345> 270 & 345 > 285. The total run time was 6 mins with MNTX and NTX eluting at 1.9 and 3.3 mins respectively. Calibration curves were constructed using peak area of ratios of the quantification transition ions and the ISTD transition ion. These curves were then used to determine the concentrations of the blood or brain samples.
2.4. Drugs
Morphine sulfate for systemic injections and morphine base pellets for subcutaneous implantation were generously provided by the National Institute on Drug Abuse (NIDA; Bethesda, MD) drug supply program. MNTX bromide used in the acute morphine and morphine pellet experiments (Swiss Webster mice) was synthesized by Dr. Yan Zhang at Virginia Commonwealth University and dissolved in ddH2O. MNTX bromide used in the chronic, systemically administered morphine warm-water tail withdrawal, oxycodone discrimination, and quantification experiments (C57BL/6J mice) was provided by NIDA and dissolved in sterile saline. Oxycodone HCl (Mallinckrodt Inc., St. Louis, MO) and NTX HCl (Sigma-Aldrich, St. Louis, MO; Catalog #N3136) were obtained commercially and dissolved in sterile saline. Pretreatment times for each experiment are described in the preceding methodology section. All solutions were administered s.c. in a volume equivalent to 10 mL/kg body weight.
2.5. Data Analysis
For nociceptive assays, data were expressed as percent maximum possible effect (%MPE) using the following formula: [(test latency – baseline latency) / (maximum possible latency – baseline latency)] × 100. Maximum possible latencies for the warm-water tail withdrawal and hot plate assays were 10 s and 30 s, respectively. For the acute morphine tail-withdrawal experiment (Figure 1), data were analyzed using one-way between-subjects ANOVA followed by Dunnett’s post-hoc test to identify differences in %MPE relative to vehicle control. For experiments involving morphine-pelleted mice (Figure 2), daily baseline latencies (top panels) and morphine challenge in the tail withdrawal assay (bottom right panel) were analyzed using two-way ANOVA. Dunnett’s post-hoc test was used to 1) compare latencies between-groups relative to the non-TNBS control mice administered vehicle (designated as vehicle + vehicle group), and 2) compare latencies within-group over the course of the experiment relative to Day 1 to assess for tolerance development. To determine whether morphine could differentially surmount MNTX antagonism as a function of TNBS injury, mice displaying sub-maximal antinociception on Day 3 were administered a booster dose of 10 mg/kg morphine and results were analyzed using a two-way ANOVA followed by Bonferroni’s post-hoc test to compare within-group latencies pre- and post-morphine bolus.
Figure 1:
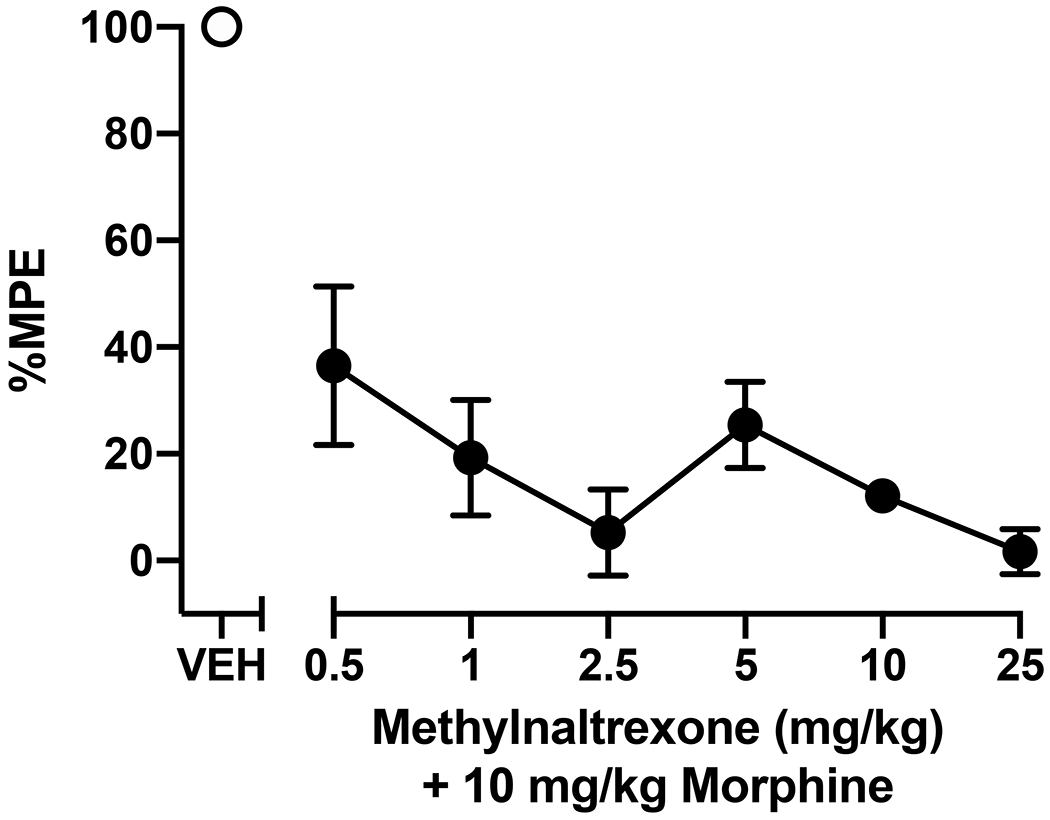
Effects of MNTX on acute morphine-induced antinociception in a warm-water tail-withdrawal assay. Mice were administered a MNTX dose (0.5, 1, 2.5, 5, 10, 25 mg/kg, s.c.) or vehicle (VEH) followed 20 min later by injection of 10 mg/kg morphine (s.c.), then tested 30 min after morphine administration. Data represent the mean ± SEM percent maximum possible effect (%MPE) of 5-6 mice per treatment condition. Significant differences in %MPE relative to vehicle-pretreated mice are denoted by filled symbols (p<0.05).
Figure 2:
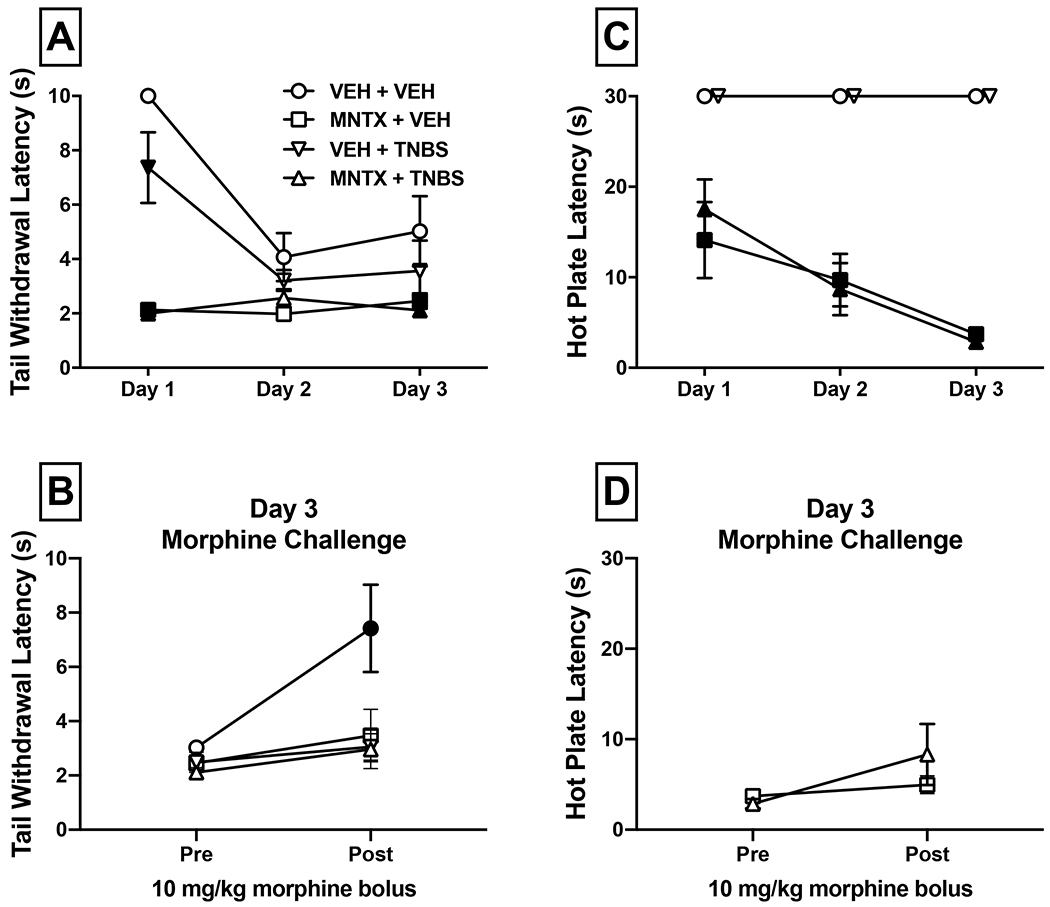
Effects of MNTX on morphine antinociception in a TNBS-induced model of inflammatory colitis. On Day 0, mice were implanted with 75 mg morphine pellets and administered a TNBS solution to induce colonic inflammation or vehicle. Separate groups of mice received once daily administration of 25 mg/kg MNTX (s.c.) or vehicle and were tested for daily nociceptive responses (s) in a warm-water tail-withdrawal (panel A) or hot plate assay (panel C). Significant between-group differences compared to the vehicle + vehicle condition at each day are denoted by filled symbols (p<0.05); graphical denotation of within-group differences are omitted for clarity but are included in text (section 3.2). After the baseline assessment on Day 3, mice with sub-maximal response latencies were challenged with 10 mg/kg morphine (s.c.) to determine if MNTX antagonism of morphine’s effects was surmountable (panels B and D). Significant within-subject differences in post-morphine response latencies compared to pre-morphine latencies are denoted by filled symbols, * p<0.05.
For the chronic morphine warm-water tail withdrawal experiments, data generated on Day 1 (Figure 3, panel A) and Day 7 (Figure 3, panel B) were analyzed separately using a mixed-design ANOVA, with antagonist treatment condition (MNTX, NTX, or vehicle) as the between-subjects factor and morphine dose as the within-subjects factor. Data were subsequently analyzed using Dunnett’s post-hoc tests comparing antagonist conditions to the saline + saline control group at each morphine dose. Additionally, two-way repeated measures ANOVA was used to analyze within-subject responses to morphine on Day 1 vs. Day 7 (Figure 4), followed by Bonferroni post-hoc tests to identify shifts in the morphine dose-response curves indicative of tolerance.
Figure 3:
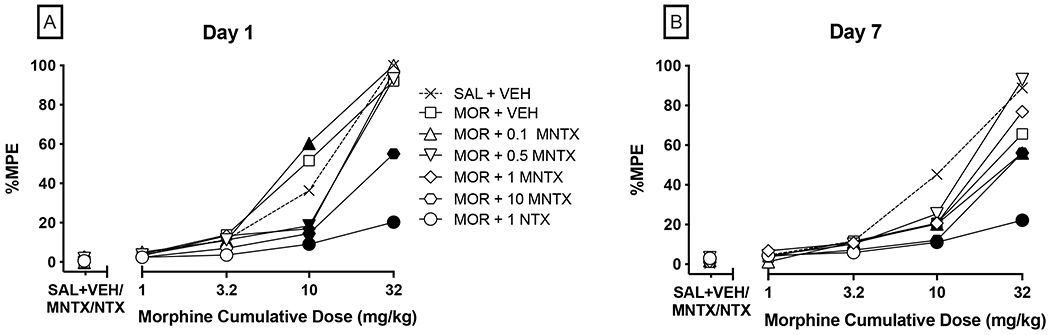
Between-subjects effects of chronic co-administration of MNTX and 10 mg/kg morphine in a warm-water tail-withdrawal assay. Panels A-B show antinociceptive response (%MPE) on Day 1 and Day 7, respectively, to cumulative doses of morphine in mice pretreated with either saline, 0.1-10 mg/kg MNTX, or 1 mg/kg NTX. SAL + VEH refers to mice assigned to receive chronic co-administration of saline (morphine vehicle) and saline (vehicle for MNTX and NTX). MOR refers to mice receiving chronic administration of 10 mg/kg morphine (s.c.) once daily along with the respective co-treatment condition on Days 2-6. Values represent mean %MPE of 6-12 mice per condition. Error bars are omitted for clarity but are included in the within-subjects presentation of this data in Figure 4. Significant differences (p<0.05) in %MPE compared to the respective saline + antagonist test condition are denoted by filled symbols.
Figure 4:
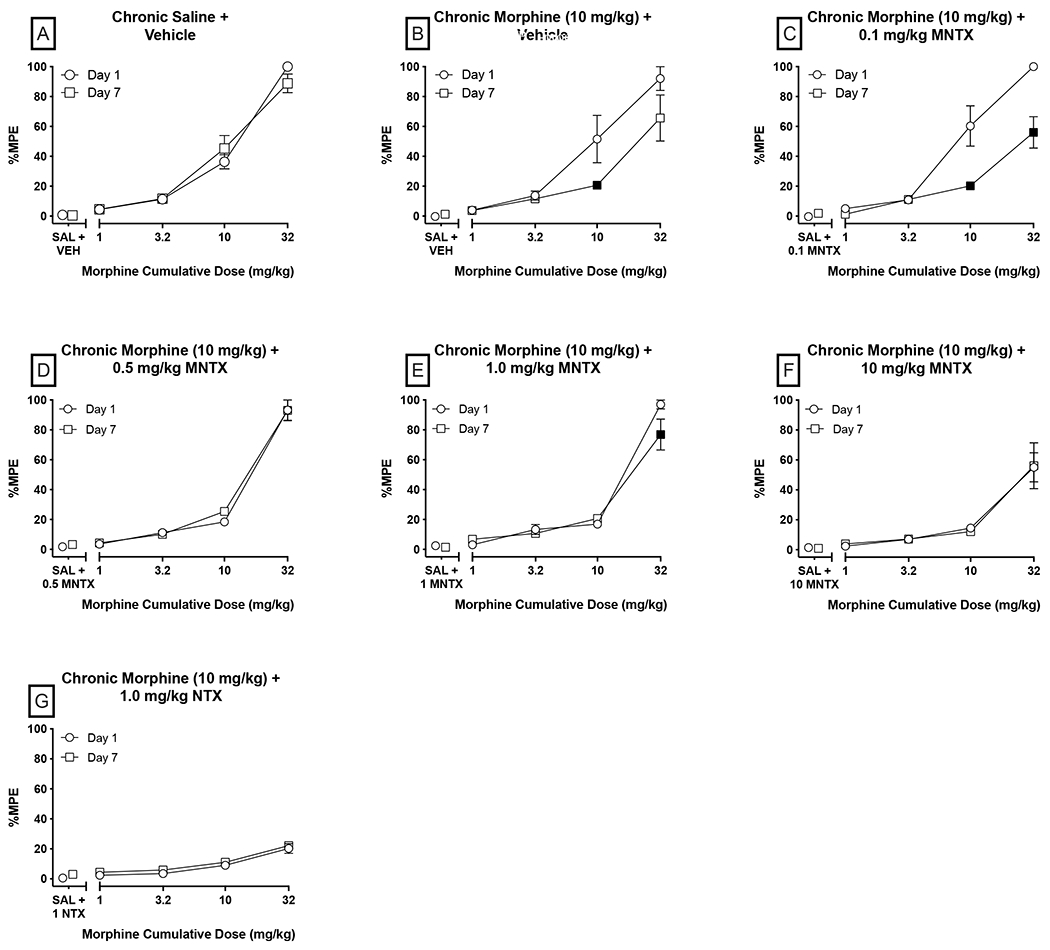
Within-subjects effects of effects of chronic coadministration of MNTX and 10 mg/kg morphine in a warm-water tail-withdrawal assay. Data are reorganized from those depicted in Figure 3 to show shifts in the morphine dose-response curve as a function of repeated co-administration of 10 mg/kg morphine and the respective antagonist condition. Significant differences (p<0.05) in %MPE at a given morphine dose relative to Day 1 are denoted by filled symbols. All other details same as Figure 3.
In the oxycodone discrimination studies (Figures 5–6), the number of responses on the oxycodone-designated lever was divided by total responses emitted on both levers, and this dividend was then multiplied by 100 to calculate percent oxycodone lever responding (%OLR). Full substitution for oxycodone was defined as ≥ 80% OLR. Response rates (total lever presses/minute) for each session were calculated for each mouse. If a mouse failed to complete 10 total responses, its data were included in calculations of response rate but excluded in calculating %OLR to prevent a disproportionate influence on %OLR by low rates of responding. Oxycodone generalization curves were analyzed using repeated-measures ANOVA following by Dunnett’s post-hoc tests comparing each dosing condition to vehicle control to determine oxycodone doses that significantly increased %OLR or altered response rates. The ED50 and 95% confidence intervals for oxycodone generalization were calculated via variable slope, curvilinear analysis. NTX and MNTX data analyses were conducted similarly, except that the oxycodone test value served as control for post-hoc analysis to determine whether the antagonist condition + oxycodone significantly decreased %OLR or altered rates. Subsequently, AD50 values and 95% confidence intervals for reducing %OLR were calculated via variable slope, curvilinear analysis for MNTX and NTX when co-administered with the 1.3 mg/kg oxycodone training dose.
Figure 5:
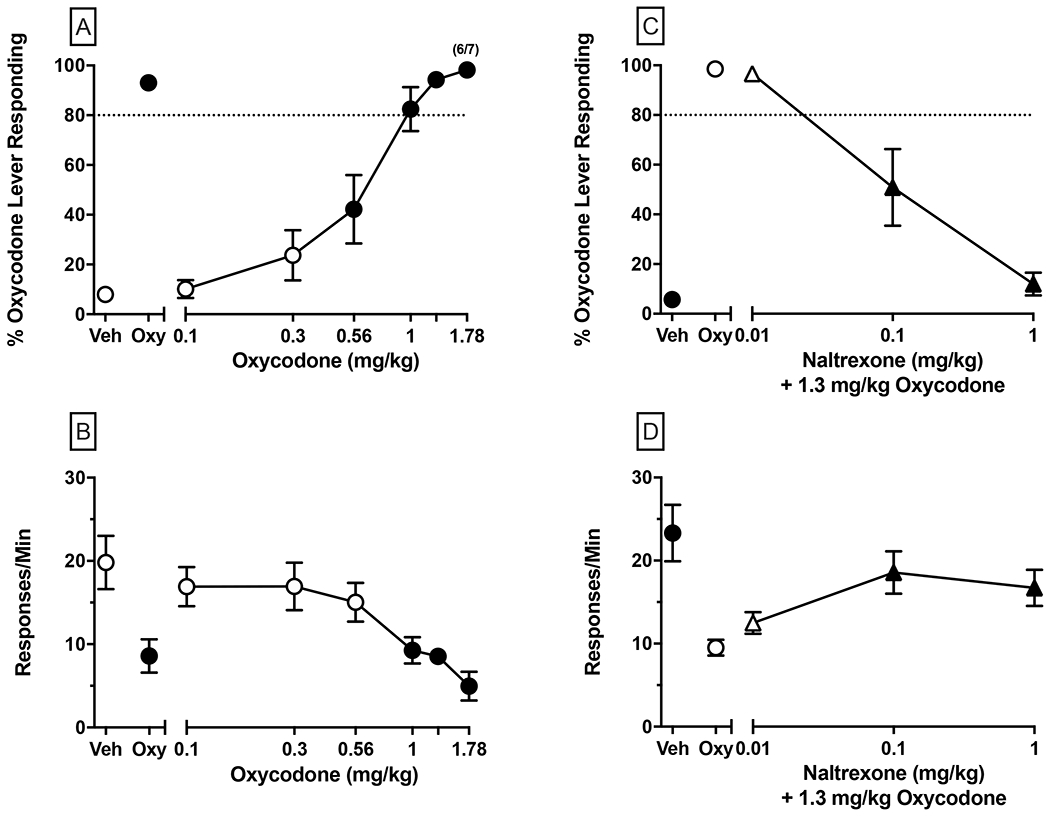
Oxycodone generalization (left panels; A-B), and dose-dependent NTX antagonism of the 1.3 mg/kg oxycodone training dose (right panels; C-D). NTX and oxycodone were administered 25 and 15 min prior to the test session, respectively. Upper panels depict percent responding on the oxycodone-associated lever (i.e., %OLR) and the dashed line represents the threshold for full substitution (i.e., 80% OLR). Bottom panels depict response rate (resp/min). Values above Veh and Oxy represent control test data conducted with saline vehicle and the oxycodone training dose, respectively. Test conditions contain values (i.e., mean ± SEM) for 7 mice. Numbers in parentheses above a %OLR data point represent the number of subjects meeting inclusion criteria (i.e., ≥ 10 total responses; N=6/7 for the 1.78 mg/kg oxycodone test); otherwise all mice met inclusion criteria for a given data point. Significant differences (p<0.05) in %OLR and response rate compared to the respective control condition (i.e., vehicle for oxycodone generalization; oxycodone for NTX testing) are denoted by filled symbols.
Figure 6:
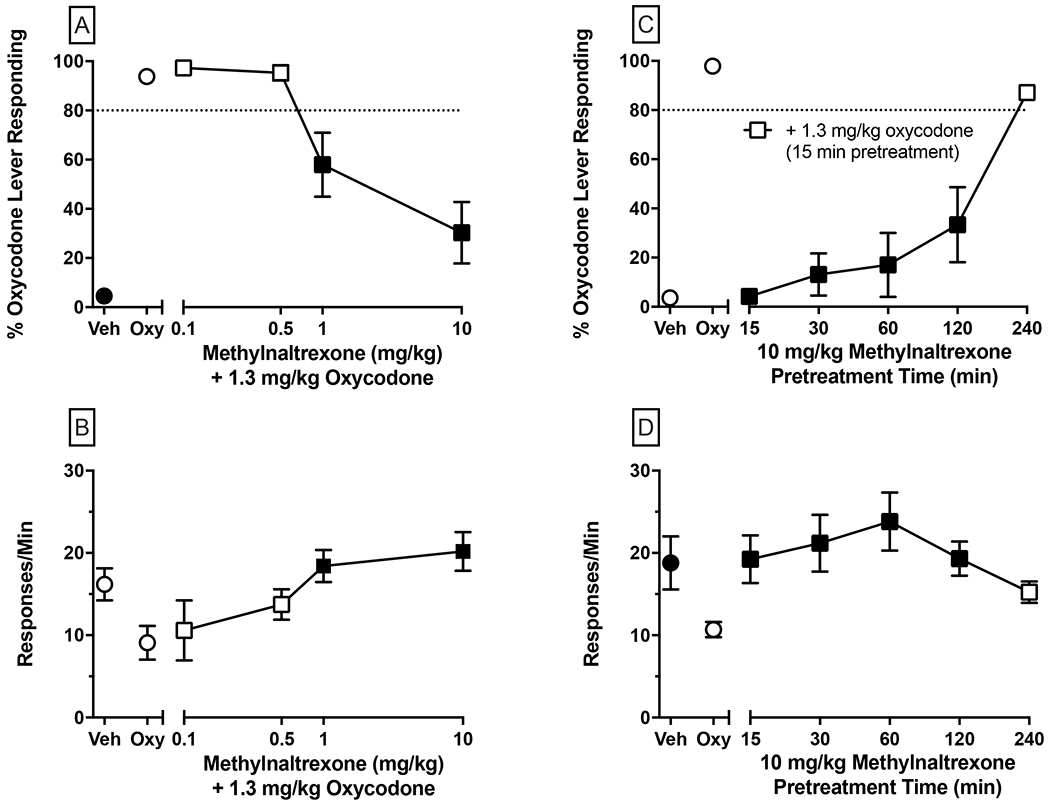
Dose-dependent antagonism by MNTX (left panels; A-B) using a 45 min MNTX pretreatment time and time-course determination of 10 mg/kg MNTX’s antagonism of the oxycodone training dose (right panels; C-D). Oxycodone was administered 15 min prior to each test session. Values represent the mean ± SEM of 6-7 mice. Significant differences (p<0.05) in %OLR and response rate compared to oxycodone control are denoted by filled symbols. All other details same as Figure 5.
For analytical studies (Figure 7), brain concentrations of MNTX and NTX were expressed as a function of tissue weight (ng/g) and were analyzed using one-way ANOVA followed by Dunnett’s post-hoc test to identify differences compared to vehicle. Because blood samples from all subjects administered a given manipulation were aggregated into a single sample, antagonist concentrations were reported as a function of blood volume (ng/mL) but were not analyzed. All statistical analyses were conducted using GraphPad Prism 8 (GraphPad Software, LLC, La Jolla, CA) and statistical significance was inferred at a level of p<0.05.
Figure 7:
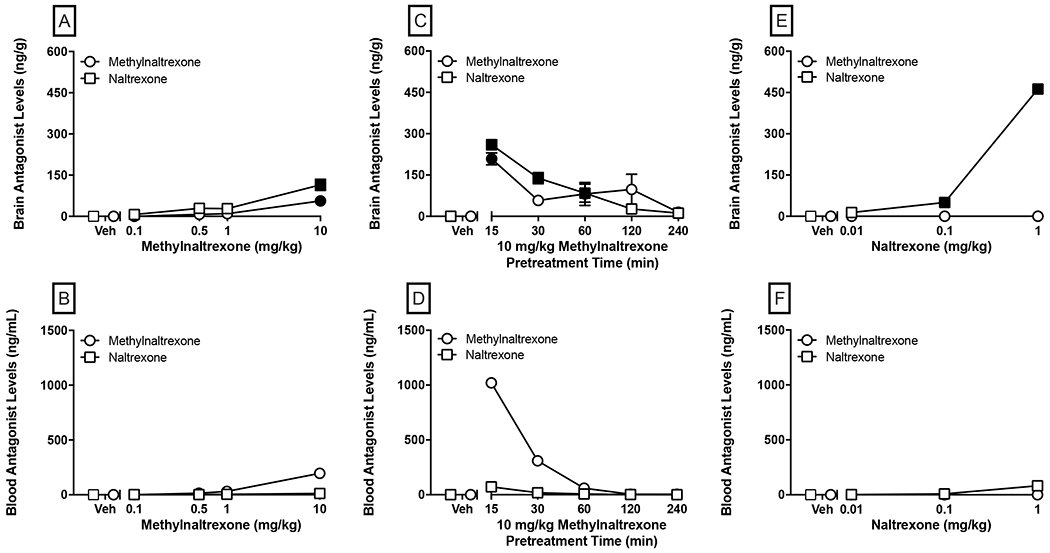
Brain (top panels) and blood (bottom panels) concentrations of MNTX and NTX in mice administered 0.1-10 mg/kg MNTX 45 min prior to sacrifice (panels A-B), 10 mg/kg MNTX administered at varying pretreatment times (0-240 min prior to sacrifice; panels C-D), or 0.01-1 mg/kg NTX administered 25 min prior to sacrifice (panels E-F). Values represent the mean ± SEM of brain concentration (ng/g) or calculated blood concentration (ng/mL) from a pooled blood sample from each given condition. Significant differences (p<0.05) in brain antagonist level compared to the respective vehicle condition are denoted by filled symbols.
3. Results
3.1. Effects of MNTX on acute morphine-induced antinociception
We evaluated the effect of MNTX on morphine-induced antinociception by injecting mice with a range of MNTX doses (0.5-25 mg/kg) 20 min prior to morphine (10 mg/kg) administration in the warm-water tail-withdrawal assay. When mice were pretreated with MNTX vehicle, morphine evoked characteristic antinociception with all mice leaving their tail exposed in the warm water for the maximum 10-s trial period (i.e., 100% MPE). As shown in Figure 1, all doses of MNTX reduced %MPE from vehicle control levels. The ANOVA indicated there was a significant main effect of MNTX treatment [F(6, 31)=20.21, p<0.0001], and post-hoc analysis indicated that all doses of MNTX significantly (p<0.0001) decreased %MPE indicative of effective antagonism of morphine’s antinociceptive properties.
3.2. Effects of MNTX on chronic morphine-induced antinociception
We next sought to determine if MNTX would differentially alter development of centrally mediated antinociceptive tolerance to morphine in the presence or absence of a peripheral inflammatory pain state. TNBS or vehicle-treated mice were pelleted with 75 mg morphine pellets on Day 0, injected daily with 25 mg/kg MNTX or vehicle, and assessed for response latencies in the warm-water tail-withdrawal assay (Figure 2, Panels A and B). Analysis indicated there were main effects of treatment condition [F(3, 23)=17.26, p<0.0001] and day [F(2, 46)=15.67, p<0.0001], and an interaction [F(6, 46)=6.763, p<0.0001]. Within-subjects post-hoc analysis indicated that tolerance to morphine’s antinociceptive effects only developed in groups not administered MNTX, as tail-withdrawal latencies were significantly faster on Days 2 and 3 compared to Day 1 in both the vehicle + vehicle control and vehicle + TNBS groups. Between-subjects analysis indicated that tail-withdrawal latencies were significantly decreased in all other treatment groups relative to control mice on Day 1, suggesting that TNBS inflammation, MNTX, or a combination of both initially reduced morphine antinociception. No between-subjects differences emerged on Day 2, but on Day 3 both MNTX-treated groups had significantly lower latencies compared to the control group.
Mice with sub-maximal tail-withdrawal latencies were challenged with 10 mg/kg morphine on Day 3 (panel B). There were significant main effects of chronic treatment condition [F(3, 20)=3.859, p=0.0250], and morphine [F(1, 20)=13.79, p=0.0014], as well as a significant interaction [F(3, 20)=3.480, p=0.0351). Post-hoc analysis indicated that the supplemental morphine injection significantly increased tail withdrawal latencies in vehicle + vehicle mice (p=0.0010), but not in any other group. These data indicate that this morphine dose was insufficient to reverse decreased tail-withdrawal latencies produced by MNTX, TNBS-induced inflammation, or a combination thereof in mice implanted with morphine pellets.
A differential pattern was observed in the hot plate assay, whereby maximal antinociception was sustained over the three-day period in control mice or TNBS mice in the absence of MNTX treatment (Figure 2, Panel C); therefore, morphine challenge tests were not conducted in these groups on Day 3. There were significant main effects of treatment condition [F(3, 16)=49.84, p<0.0001] and day [F(2, 32)=25.86, p<0.0001], and an interaction [F(6, 32)=9.304, p<0.0001]. In both MNTX-treated groups, response latencies were attenuated to a similar degree compared to control mice and decreased further on Days 2 and 3. Between-group analysis indicated that both MNTX groups had significantly lower hot plate latencies across all 3 test days compared to the control group.
When a supplemental 10 mg/kg morphine dose was administered to MNTX-treated mice (Figure 2, Panel D), there was no main effect of chronic treatment condition [F(1, 8)=0.3727, p=0.5585] or morphine [F(1, 8)=4.859, p=0.0586], nor an interaction [F(1, 8)=1.887, p=0.2068]. Moreover, planned comparisons indicated there was no difference in hot plate latencies post-morphine in either TNBS (p=0.0705) or vehicle mice (p>0.9999) administered MNTX. These data demonstrate that MNTX antagonizes the antinociceptive properties of morphine similarly in mice with or without TNBS-induced inflammation. Although these experiments were not explicitly designed to examine dependence, signs of opioid withdrawal-related behavior such as jumping, paw tremors, or diarrhea were not noted following MNTX administration.
Next, we sought to extend the findings that MNTX antagonizes the antinociceptive and tolerance-related effects of acute and chronic morphine, respectively, by employing a lower MNTX dose range and extending the duration of treatment. Pretreatment with MNTX or NTX was associated with decreases in morphine antinociception relative to saline-treated controls on Day 1 (Figure 3, Panel A). There were main effects of antagonist condition [F(6, 41)=21.87, p<0.0001], morphine [F(4, 164)=402.5, p<0.0001], and an interaction [F(24, 164)=11.30, p<0.0001]. Post-hoc analysis indicated that relative to the group assigned to the chronic saline condition, antinociception following the 10 mg/kg cumulative morphine dose was reduced by pretreatment with MNTX at 0.5 (p=0.0450), 1 (p=0.0178), and 10 mg/kg (p=0.0036) doses, and by 1 mg/kg NTX (p<0.0001). Additionally, 10 mg/kg MNTX and 1 mg/kg NTX attenuated antinociception at the 32 mg/kg cumulative morphine dose (p<0.0001) that otherwise produced near-maximal antinociception in all other treatment groups. These findings in C57BL/6J mice (an inbred strain) extend our results obtained in outbred Swiss Webster mice demonstrating that these MNTX doses antagonize acute morphine-induced antinociception under a range of testing conditions. Compared to vehicle pretreated mice, 0.1 mg/kg MNTX pretreatment significantly increased %MPE at the 10 mg/kg morphine cumulative dose (p=0.0008).
On Day 7, morphine cumulative dose testing in the tail-withdrawal procedure was repeated after mice had received daily co-administration of 10 mg/kg morphine and a dose of MNTX, NTX, or saline (Figure 3, Panel B). There were main effects of antagonist condition [F(6, 41)=6.498, p<0.0001], morphine [F(4, 164)=155.0, p<0.0001], and an interaction [F(24, 164)=4.239, p<0.0001]. Post-hoc analysis indicated that, following chronic morphine treatment, %MPE values at the 10 mg/kg morphine cumulative dose were decreased in mice pretreated with vehicle (p=0.0004), 0.1 (p=0.0004), and 10 mg/kg MNTX (p=0.0440), or 1 mg/kg NTX (p=0.0264) compared to mice chronically administered saline and vehicle. A similar attenuation at 32 mg/kg morphine was observed in the 0.1 (p=0.0004), and 10 mg/kg MNTX (p=0.0005), and 1 mg/kg NTX (p<0.0001) pretreatment groups. Vehicle-pretreated mice showed a similar trend that did not reach statistical significance (p=0.0585).
To facilitate evaluation of tolerance development, within-subjects dose-response data for all conditions are presented in Figure 4. Significant main effects of morphine treatment were observed for all groups (p<0.0001). There were no significant differences at any morphine dose between Day 1 and Day 7 in the chronic saline + saline group (Panel A), indicating the stability of antinociceptive response to morphine over time. In contrast, mice chronically administered morphine and vehicle had a decreased antinociceptive response to 10 mg/kg morphine on Day 7 (p=0.0002), indicating development of tolerance (Panel B). Similarly, 0.1 mg/kg MNTX pretreatment, which did not alter antinociception on Day 1, failed to alter tolerance development as indicated by lower %MPE when tested with 10 and 32 mg/kg morphine on Day 7 (p<0.0001; Panel C). There was no main effect of repeated treatment in mice administered MNTX doses of 1 mg/kg or higher or the NTX group. Similar morphine dose effect curves were obtained on Days 1 and 7 in the chronic 0.5, 1.0, and 10 mg/kg MNTX and 1 mg/kg NTX groups (with the sole exception of 1 mg/kg MNTX at 32 mg/kg morphine) indicating these doses of MNTX and NTX were able to prevent the development of tolerance (Panels D, E, F, and G, respectively); however, each of these treatment conditions significantly attenuated morphine’s antinociceptive effects on Day 1 relative to vehicle-pretreated controls at one or more morphine doses (Figure 3, Panel A). These findings do not support the hypothesis that co-administering MNTX prevents or attenuates the development of morphine antinociceptive tolerance in mice at doses that do not also acutely antagonize morphine-induced antinociception. Anecdotal observation of somatic opioid withdrawal signs was not noted following administration of any MNTX dose or NTX.
3.3. Effects of MNTX on the discriminative stimulus properties of oxycodone
Results of dose-response tests with oxycodone in mice trained to discriminate 1.3 mg/kg oxycodone are presented in Figure 5, Panels A-B. Increasing the oxycodone dose (0.1, 0.3, 0.56, 1.0, 1.3, 1.78 mg/kg) resulted in dose-dependent increases in the percentage of responses made on the oxycodone-paired lever with doses of 0.56 mg/kg and higher significantly increasing %OLR compared to vehicle, and doses of 1 mg/kg and above fully substituted (i.e., ≥80% OLR) for the 1.3 mg/kg oxycodone training stimulus (Figure 5, Panel A). The ED50 of oxycodone dose for generalization was 0.5600 mg/kg (95% CI: 0.4583-0.6617 mg/kg). Oxycodone dose-dependently decreased response rates [F(7, 42)=9.493, p<0.0001] and post-hoc analysis indicated that the oxycodone control test, and 1.0, 1.3, and 1.78 mg/kg oxycodone doses reduced response rates relative to vehicle (Figure 5, Panel B).
NTX (0.01, 0.1, 1 mg/kg) administered 10 min prior to the 1.3 mg/kg oxycodone training dose (Figure 5, Panels C-D), which was tested as a positive control for MOR antagonism, resulted in dose-dependent decreases in %OLR with a calculated AD50 of 0.1114 mg/kg (95% CI: 0.04683-0.1761 mg/kg). Relative to oxycodone control, 0.1 and 1 mg/kg NTX significantly reduced %OLR levels (p<0.001 and p<0.0001, respectively) and significantly reversed the suppression of response rate produced by the 1.3 mg/kg oxycodone control test (p<0.01 and p<0.05, respectively).
Results of tests with MNTX (0.01, 0.5, 1, 10 mg/kg) administered 30 min prior to the 1.3 mg/kg oxycodone training dose (i.e., 45 min pre-session) are shown in Figure 6, Panels A-B. MNTX dose-dependently decreased %OLR (Figure 6, Panel A) with a calculated AD50 of 0.9647 mg/kg (95% CI: 0.4085-1.521 mg/kg). MNTX doses of 1 and 10 mg/kg significantly reduced levels of %OLR (p<0.01 and 0.0001, respectively) and significantly reversed the suppression of response rates (p<0.05 and 0.01, respectively) of 1.3 mg/kg oxycodone.
To determine whether this incomplete antagonism of 10 mg/kg MNTX (30.33% OLR) relative to the more robust antagonism produced by 1 mg/kg NTX (12.00 %OLR) of oxycodone’s discriminative stimulus effects was attributable to MNTX dose or temporal factors, the pretreatment time of 10 mg/kg MNTX was systematically varied (15, 30, 60, 120, 240 min pre-session). As shown in Figure 6, Panel C, when mice were co-administered 10 mg/kg MNTX and oxycodone (i.e., 15 min time point), mean %OLR was 5.06%, suggesting that MNTX was rapidly demethylated to NTX. As MNTX pretreatment time increased, so too did %OLR values culminating with a mean of 87.16% OLR obtained with a 240 min MNTX pretreatment time. Oxycodone-appropriate responding was significantly (p<0.0001) decreased throughout the 15-120 min time point assessments. A similar profile was observed with response rates (Figure 6, Panel D), as post-hoc analysis indicated that rates were significantly elevated relative to oxycodone at all time points except at 240 min post 10 mg/kg MNTX. Overall, these findings extend results demonstrating the ability of MNTX to suppress CNS-mediated effects of opioid agonists and elucidate the pharmacokinetic properties of MNTX that contribute to this antagonism.
3.4. Quantification of MNTX and NTX concentrations in mouse whole brain and blood
Given these findings in the discrimination procedure, we sought to quantify concentrations of MNTX and NTX using otherwise identical treatment parameters but without oxycodone co-administration. Concentrations of MNTX and NTX from mouse whole brain and blood are depicted in Figure 7. Results from the MNTX dose-response experiment indicated significant effects of MNTX treatment on brain concentrations of MNTX [F(4, 25)=22.08, p<0.0001] and NTX [F(4, 25)=14.13, p<0.0001], and post-hoc analysis indicated 10 mg/kg MNTX produced significant increases in concentrations of MNTX and NTX (p<0.0001 for both; Panel A). Blood concentrations are shown in Panel B and demonstrate a dose-dependent increase in MNTX, peaking at 196 ng/mL after administration of 10 mg/kg MNTX. NTX concentrations showed a similar, but less robust increase peaking at 12.1 ng/mL for the same dose. These findings importantly demonstrate that MNTX crosses the blood-brain barrier in mice and its peripheral administration results in blood and brain concentrations of NTX. Time course data are presented in Panels C and D and corroborate results of the oxycodone discrimination experiment demonstrating rapid demethylation of MNTX to NTX that peaked at the earliest time point assessed (15 min). There were significant effects of pretreatment time on brain concentrations of MNTX [F(5, 30)=6.368, p=0.0004] and NTX [F(5, 30)=32.20, p<0.0001]. A significant increase in MNTX was only observed at the 15 min time point, whereas significant elevation of NTX concentrations were sustained through the 60 min time point. Similar to the dose-response blood data, peak MNTX concentrations were markedly higher than corresponding NTX concentrations, with both peaking at the 15 min time point (MNTX: 1020 ng/mL; NTX: 71.7 ng/mL). Results from NTX dose-response tests are shown in Panels E-F. There was a main effect of treatment on NTX brain concentrations [F(3, 20)=1422.0, p<0.0001], with significant increases observed at 0.1 and 1 mg/kg, doses that partially and completed antagonized the discriminative stimulus effects of oxycodone, respectively. MNTX was not detected in any of the samples in brain or blood from NTX-treated mice.
4. Discussion
The present study evaluated whether peripheral administration of MNTX antagonizes the antinociceptive effects of morphine or the discriminative stimulus effects of oxycodone in mice. We chose to examine these effects because they involve a range of differential levels of CNS mediation from antinociceptive reflexive tail withdrawal in the warm-water tail-withdrawal procedure, to antinociceptive escape behavior in the hot plate assay, to a procedure thought to be correlated with the subjective effects of drugs in the oxycodone discrimination procedure. MNTX was effective at blocking or reversing each of these CNS-mediated effects in a dose-dependent manner across assays. In addition, analysis for MNTX and NTX in blood and brain confirmed the presence of both in these bodily compartments following MNTX treatment in mice. These data provide compelling evidence that peripherally administering MNTX in the mouse does not restrict it nor its metabolite, NTX, to the periphery, and is the first quantification of these antagonists concentrations in mouse blood and brain following MNTX administration.
Perhaps the most compelling behavioral evidence of MNTX acting centrally involved results from oxycodone discrimination tests. It is well-established that the discriminative stimulus properties of drugs are mediated by the CNS (Colpaert, 1999). In the present study, MNTX antagonized oxycodone’s discriminative stimulus effects in a dose- and time-dependent manner. A time-course evaluation of 10 mg/kg MNTX showed a rapid onset of antagonist activity, as mice co-treated with MNTX and oxycodone 15 min prior to the test session emitted only 4.21% of their responses on the oxycodone-associated lever. There was a systematic increase in oxycodone-like responding as MNTX pretreatment time increased, and by 4 h subjects responded predominantly on the oxycodone-paired lever. The ability of MNTX to significantly antagonize oxycodone’s discriminative stimulus effects for up to 120 min post-administration was consistent with effective pretreatment times in our antinociceptive experiments (e.g., 50 min pretreatment time in Figure 1; 120-150 min pretreatment time in Figures 3–4 for morphine cumulative doses of 10 and 32 mg/kg). Minimally, the present results indicate that systemically administered MNTX rapidly results in the availability of MNTX and NTX for binding to brain opioid receptors. This onset of action is consistent with absorption pharmacokinetics of s.c. administered MNTX in humans (Osinski et al., 2002; Oswald et al., 2011), but the present research suggests differences in distribution pharmacokinetics.
The analytical findings support and extend previous research examining MNTX metabolism in different species (Chandrasekaran et al., 2010; Kotake et al., 1989). MNTX has been shown to bind to MOR receptors in vitro (Beattie et al., 2007) albeit with approximately 30-fold lower affinity than NTX (Cassel et al., 2005). Individual contributions by MNTX and NTX for antagonizing the behavioral effects observed are difficult to separate. For instance, in the oxycodone discrimination procedure, higher concentrations of NTX than MNTX were detected in brain under MNTX dosing conditions that produced the greatest levels antagonism. However, in the chronic warm-water tail-withdrawal tests, behaviorally active doses were administered approximately 120 min prior to testing 10 mg/kg morphine. At this time point, blood levels of MNTX and NTX were both low (4.75 and 2.02 ng/mL, respectively), but MNTX levels in the brain were nearly 4-fold higher than NTX (97.33 vs 26.62 ng/g).
A recent preclinical study that has gained much attention reported that peripherally administered MNTX prevented the development of antinociceptive tolerance to morphine in mice, and the subsequent speculation that peripherally-restricted opioid receptor antagonists could be clinically relevant to treat development of opioid analgesic tolerance (Corder et al., 2017). One desired property of a candidate medication to prevent analgesic opioid tolerance would be that it does not interfere with the opioid analgesia. Using an outbred mouse strain, we observed that acute MNTX doses as low as 0.5 mg/kg attenuated antinociception elicited by the same 10 mg/kg morphine dose utilized by Corder et al. (2017). Chronic morphine antinociception was also antagonized by MNTX in outbred mice (Figure 2), serving as further proof of concept that MNTX had central actions in mice. In subsequent behavioral and analytical experiments (Figures 3–7), the same mouse strain (C57BL/6J) and identical MNTX doses as Corder et al. (2017) were tested to control for these key variables. Chronic morphine delivered via daily peripheral injections resulted in antinociceptive tolerance in the absence of MNTX treatment. In contrast, doses of 0.5 mg/kg MNTX and higher (as well as 1 mg/kg NTX) significantly decreased morphine antinociception on the initial test day and the morphine dose-response curves were similar for these conditions on the initial and final test day. These findings demonstrate that any potential effects of MNTX in the mouse on antinociceptive tolerance development arise under conditions that would enable the antagonism of acute morphine antinociception.
It should be noted that the present studies were not an exact replication of methodologies reported by Corder et al. (2017). Some methodological differences that could have contributed to the differing results include differences in the dosing parameters, pain modalities, and noxious stimuli used as well as the dependent measures reported. For example, while both studies utilized the hot plate assay, Corder et al. (2017) reported additional dependent measures across a fixed, inescapable 45-s observational period including “affective-motivational” responses of jumping, as well as flinching, guarding, and attending to an exposed paw, as opposed to the latency to initial paw licking over a maximum 30-s exposure period measured. Thus, the summation of different “affective-motivational” and reflexive behaviors as opposed to the initial reflexive behavior latency, recruitment of different neuronal processes underlying these behaviors, along with possible differences in the degree of tissue insult with escapable vs. non-escapable noxious stimulus exposure could have played a role in the different results. The pattern and delivery of chronic morphine also differed between studies, as Corder et al. (2017) administered 10 mg/kg morphine once daily, whereas the present chronic studies employed either morphine pellets or utilized a cumulative dosing procedure for assessing tolerance. Nonetheless, MNTX’s ability to antagonize morphine’s antinociceptive effects in the present studies was robust in nature, consistent across several commonly utilized preclinical nociceptive assays, in both inbred and outbred mouse strains, and following acute and chronic treatment. Lastly, our analytical pharmacokinetic results (also obtained in C57BL/6J mice using identical MNTX doses) supported our behavioral results and refute the notion that MNTX is peripherally restricted in mice. Overall, our results provide evidence that it is erroneous to assume peripheral seclusion of MNTX for its ability to attenuate morphine antinociceptive tolerance in mice. This observation impairs if not invalidates speculation of using peripheral antagonists clinically for abating opioid tolerance based solely upon their effects in mice.
The present findings and past reports of MNTX acting centrally force reassessment of conclusions of recent reports in which MNTX was used as a pharmacological tool in mice to infer peripherally-mediated actions. For instance, co-treatment of 1 mg/kg MNTX i.p. with an escalating morphine dosing regimen in mice resulted in prevention of morphine antinociceptive tolerance (Ruhela et al., 2020), but likely as a function of MNTX antagonism of MORs in the CNS. NTX and MNTX have also been reported to reduce pruritis induced by serotonin administration in mice, with the latter finding used to suggest a role of peripheral opioid receptors in mediating this response (Haddadi et al., 2018). Other hot plate studies in mice support our general conclusions but required a higher dose of 30 mg/kg MNTX to induce hyperalgesia (Ramabadran, 1982) or reverse morphine antinociception in the hot plate assay (Russell et al., 1982).
Generalization of these findings to rats, another commonly utilized rodent species in preclinical research, would be tenuous. Most germane to the present work is a recent study which found that NTX, but not MNTX, prevented the development of morphine tolerance in multiple nociceptive assays, including those used in our studies (Blomqvist et al., 2020). Albeit lower MNTX doses were utilized (0.5-2 mg/kg) in the rat study, the authors reported that repeated co-administration of 2 mg/kg MNTX and escalating doses of morphine resulted in the presence of a small concentration of MNTX in brain (~5 ng/g) and slight, but significantly higher concentrations in spinal cord and blood plasma. In contrast, our acute dosing results showed nearly two-fold higher (9.95 ng/g) higher brain concentrations following administration of a lower dose (1 mg/kg MNTX) that was behaviorally active in antinociceptive testing. Moreover, we report peak MNTX brain concentrations of 208.83 ng/g following administration of 10 mg/kg MNTX using a 15 min pretreatment time. Earlier studies also showed a separation in effects as a function of MNTX route of administration, suggesting that the blood-brain barrier permeability of MNTX and metabolic conversion to NTX in mice does not occur to a similar extent to rats. For instance, Brown and Holtzman (1981) demonstrated that i.c.v., but not peripheral administration of MNTX, decreases water intake in water-deprived rats. Similarly, morphine-induced catalepsy was only partially attenuated by peripheral administration of 56 mg/kg MNTX but completely reversed when administered i.c.v. (Brown et al., 1983).
We have previously shown accelerated antinociceptive tolerance to morphine in the TNBS model (Komla et al., 2019) and therefore, another objective of our studies was to determine whether inhibition of peripheral opioid receptors could mitigate the development of tolerance in this inflammatory model. The TNBS model mimics key clinical characteristics of inflammatory bowel disease, such as Crohn’s disease, and opioids are prescribed for its treatment. Opioid use carries a high risk for mortality and enhanced infection from those suffering from Crohn’s disease (Cross et al., 2005; Lichtenstein et al., 2012). Furthermore, inflammatory bowel disease patients taking opioids are three times more likely to become immoderate opioid users than their matched controls (Targownik et al., 2014), making this a clinically-relevant model to investigate pharmacotherapies to blunt opioid tolerance development. In general, the only notable difference produced by TNBS administration was a decreased tail-withdrawal latency on the initial test day, consistent with our previous findings under otherwise identical morphine dosing and testing conditions (Komla et al., 2019). Tolerance did not develop in the hot plate assay over the 3-day period in the morphine pelleted mice not administered MNTX, reflecting differential rates of tolerance development between the nociceptive assays tested. Interestingly, hot plate latencies continued to decrease over time in MNTX-treated mice irrespective of TNBS condition. Analytical data suggest this outcome is not likely attributable to accumulation of MNTX or NTX in blood or brain over time as these concentrations were at or near zero 4 h post-MNTX administration. Withdrawal-related hyperalgesia is one possible explanation for this progressive decrease in latencies, as the morphine pellet dose used is sufficient to induce physical dependence (Bhargava, 1978), and hyperalgesia is one consequence of morphine withdrawal (Schmidt and Way, 1980). In both assays antinociceptive latencies were generally similar as a function of MNTX condition with negligible effects of TNBS manipulation. Furthermore, an additional 10 mg/kg morphine dose on the final test day did not surmount MNTX antagonism. These observations suggest that the addition of a colonic inflammatory pain state does not alter the robust capacity of MNTX to antagonize morphine’s central effects.
The primary objective of the present study was to evaluate whether behavioral effects of opioids that are presumably CNS-mediated are immune to the effects of MNTX when it is administered peripherally as has been assumed in some other reports when tested under otherwise comparable dosing conditions. To this end, the methodologies employed in the present study are utilized extensively in preclinical studies investigating opioid sites of action and their underlying mechanisms. Collectively, the results of this study demonstrate that MNTX is not an appropriate pharmacological tool to study peripherally-isolated biological processes in mice. Additional studies, such as determining conditions under which MNTX reverses opioid-induced gastric transit inhibition in mice, would be valuable to determine whether the MNTX doses utilized herein are similar to those producing clinically-relevant outcomes.
Methylnaltrexone is a peripherally restricted opioid receptor antagonist
In mice, methylnaltrexone antagonized centrally-mediated morphine antinociception
Methylnaltrexone reversed the discriminative stimulus effects of oxycodone
Methylnaltrexone was detected in mouse brain after its administration
Methylnaltrexone is not suitable to study peripherally-mediated processes in mice
Acknowledgements
The authors wish to thank Molly Creighton and Alejandro Andrade for their excellent technical assistance and to Dr. Yan Zhang for his generous supply of methylnaltrexone. Research support was provided by the National Institute on Drug Abuse (P30DA033934; R01DA036975; R01DA039942; N01DA-17-8932). The authors wish to thank Drs. D. White and J. Acri, NIDA project officers (N01DA-17-8932), for their helpful intellectual contributions to study design and analysis for a subset of antinociceptive tests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrot M, 2012. Tests and models of nociception and pain in rodents. Neuroscience 211, 39–50. [DOI] [PubMed] [Google Scholar]
- Beattie DT, Cheruvu M, Mai N, O’Keefe M, Johnson-Rabidoux S, Peterson C, Kaufman E, Vickery R, 2007. The in vitro pharmacology of the peripherally restricted opioid receptor antagonists, alvimopan, ADL 08-0011 and methylnaltrexone. Naunyn Schmiedebergs Arch Pharmacol 375, 205–220. [DOI] [PubMed] [Google Scholar]
- Bhargava HN, 1978. The effects of naltrexone on the development of physical dependence on morphine. Eur J Pharmacol 50, 193–202. [DOI] [PubMed] [Google Scholar]
- Bianchi G, Fiocchi R, Tavani A, Manara L, 1982. Quaternary narcotic antagonists’ relative ability to prevent antinociception and gastrointestinal transit inhibition in morphine-treated rats as an index of peripheral selectivity. Life Sci 30, 1875–1883. [DOI] [PubMed] [Google Scholar]
- Blomqvist KJ, Dudek KA, Viisanen H, Matlik K, Ahlstrom FHG, Laitila J, Kalso EA, Rauhala PV, Lilius TO, 2020. Antagonism of peripheral opioid receptors by methylnaltrexone does not prevent morphine tolerance in rats. Journal of Neuroscience Research. [DOI] [PubMed] [Google Scholar]
- Brown DR, Goldberg LI, 1985. The use of quaternary narcotic antagonists in opiate research. Neuropharmacology 24, 181–191. [DOI] [PubMed] [Google Scholar]
- Brown DR, Holtzman SG, 1981. Opiate antagonists: central sites of action in suppressing water intake of the rat. Brain Res 221, 432–436. [DOI] [PubMed] [Google Scholar]
- Brown DR, Robertson MJ, Goldberg LI, 1983. Reversal of morphine-induced catalepsy in the rat by narcotic antagonists and their quaternary derivatives. Neuropharmacology 22, 317–321. [DOI] [PubMed] [Google Scholar]
- Cassel JA, Daubert JD, DeHaven RN, 2005. [(3)H]Alvimopan binding to the μ opioid receptor: comparative binding kinetics of opioid antagonists. Eur J Pharmacol 520, 29–36. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran A, Tong Z, Li H, Erve JC, DeMaio W, Goljer I, McConnell O, Rotshteyn Y, Hultin T, Talaat R, Scatina J, 2010. Metabolism of intravenous methylnaltrexone in mice, rats, dogs, and humans. Drug Metab Dispos 38, 606–616. [DOI] [PubMed] [Google Scholar]
- Colpaert FC, 1999. Drug discrimination in neurobiology. Pharmacol Biochem Behav 64, 337–345. [DOI] [PubMed] [Google Scholar]
- Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, Sotoudeh C, Clark JD, Barres BA, Bohlen CJ, Scherrer G, 2017. Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat Med 23, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross RK, Wilson KT, Binion DG, 2005. Narcotic use in patients with Crohn’s disease. American Journal of Gastroenterology 100, 2225–2229. [DOI] [PubMed] [Google Scholar]
- Haddadi NS, Foroutan A, Shakiba S, Afshari K, Ostadhadi S, Daneshpazhooh M, Dehpour AR, 2018. Attenuation of serotonin-induced itch by sumatriptan: possible involvement of endogenous opioids. Arch Dermatol Res 310, 165–172. [DOI] [PubMed] [Google Scholar]
- Komla E, Stevens DL, Zheng Y, Zhang Y, Dewey WL, Akbarali HI, 2019. Experimental Colitis Enhances the Rate of Antinociceptive Tolerance to Morphine via Peripheral Opioid Receptors. Journal of Pharmacology and Experimental Therapeutics 370, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotake AN, Kuwahara SK, Burton E, McCoy CE, Goldberg LI, 1989. Variations in demethylation of N-methylnaltrexone in mice, rats, dogs, and humans. Xenobiotica 19, 1247–1254. [DOI] [PubMed] [Google Scholar]
- Lichtenstein GR, Feagan BG, Cohen RD, Salzberg BA, Diamond RH, Price S, Langholff W, Londhe A, Sandborn WJ, 2012. Serious Infection and Mortality in Patients With Crohn’s Disease: More Than 5 Years of Follow-Up in the TREAT (TM) Registry. American Journal of Gastroenterology 107, 1409–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osinski J, Wang A, Wu JA, Foss JF, Yuan CS, 2002. Determination of methylnaltrexone in clinical samples by solid-phase extraction and high-performance liquid chromatography for a pharmacokinetics study. J Chromatogr B Analyt Technol Biomed Life Sci 780, 251–259. [DOI] [PubMed] [Google Scholar]
- Oswald S, Schumacher G, Siegmund W, 2011. Quantitative determination of methylnaltrexone in human serum using liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal 56, 1079–1084. [DOI] [PubMed] [Google Scholar]
- Pergolizzi JV Jr., Christo PJ, LeQuang JA, Magnusson P, 2020. The Use of Peripheral mu-Opioid Receptor Antagonists (PAMORA) in the Management of Opioid-Induced Constipation: An Update on Their Efficacy and Safety. Drug Des Devel Ther 14, 1009–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JH, Prus AJ, Overton DA, 2018. Drug Discrimination: Historical Origins, Important Concepts, and Principles. Curr Top Behav Neurosci 39, 3–26. [DOI] [PubMed] [Google Scholar]
- Ramabadran K, 1982. Effects of N-methylnaloxone and N-methylnaltrexone on nociception and precipitated abstinence in mice. Life Sci 31, 1253–1256. [DOI] [PubMed] [Google Scholar]
- Ruhela D, Bhopale VM, Yang M, Yu K, Weintraub E, Greenblatt A, Thom SR, 2020. Blood-borne and brain-derived microparticles in morphine-induced anti-nociceptive tolerance. Brain Behav Immun. [DOI] [PubMed] [Google Scholar]
- Russell J, Bass P, Goldberg LI, Schuster CR, Merz H, 1982. Antagonism of gut, but not central effects of morphine with quaternary narcotic antagonists. Eur J Pharmacol 78, 255–261. [DOI] [PubMed] [Google Scholar]
- Schmidt WK, Way EL, 1980. Hyperalgesic effects of divalent cations and antinociceptive effects of a calcium chelator in naive and morphine-dependent mice. J Pharmacol Exp Ther 212, 22–27. [PubMed] [Google Scholar]
- Schwienteck KL, Blake S, Bremer PT, Poklis JL, Townsend EA, Negus SS, Banks ML, 2019. Effectiveness and selectivity of a heroin conjugate vaccine to attenuate heroin, 6-acetylmorphine, and morphine antinociception in rats: Comparison with naltrexone. Drug Alcohol Depend 204, 107501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva I, Pinto R, Mateus V, 2019. Preclinical Study in Vivo for New Pharmacological Approaches in Inflammatory Bowel Disease: A Systematic Review of Chronic Model of TNBS-Induced Colitis. J Clin Med 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targownik LE, Nugent Z, Singh H, Bugden S, Bernstein CN, 2014. The Prevalence and Predictors of Opioid Use in Inflammatory Bowel Disease: A Population-Based Analysis. American Journal of Gastroenterology 109, 1613–1620. [DOI] [PubMed] [Google Scholar]
- Walentiny DM, Moisa LT, Beardsley PM, 2019. Oxycodone-like discriminative stimulus effects of fentanyl-related emerging drugs of abuse in mice. Neuropharmacology 150, 210–216. [DOI] [PubMed] [Google Scholar]
- Webster LR, 2015. Opioid-Induced Constipation. Pain Med 16 Suppl 1, S16–21. [DOI] [PubMed] [Google Scholar]
- Webster LR, Brenner DM, Barrett AC, Paterson C, Bortey E, Forbes WP, 2015. Analysis of opioid-mediated analgesia in Phase III studies of methylnaltrexone for opioid-induced constipation in patients with chronic noncancer pain. Journal of Pain Research 8, 771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster LR, Israel RJ, 2018. Oral methylnaltrexone does not negatively impact analgesia in patients with opioid-induced constipation and chronic noncancer pain. J Pain Res 11, 1503–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan CS, Foss JF, O’Connor M, Osinski J, Karrison T, Moss J, Roizen MF, 2000. Methylnaltrexone for reversal of constipation due to chronic methadone use - A randomized controlled trial. Jama-Journal of the American Medical Association 283, 367–372. [DOI] [PubMed] [Google Scholar]
- Yuan CS, Foss JF, O’Connor M, Osinski J, Roizen MF, Moss J, 1998. Efficacy of orally administered methylnaltrexone in decreasing subjective effects after intravenous morphine. Drug Alcohol Depend 52, 161–165. [DOI] [PubMed] [Google Scholar]
- Yuan CS, Wei G, Foss JF, O’Connor M, Karrison T, Osinski J, 2002. Effects of subcutaneous methylnaltrexone on morphine-induced peripherally mediated side effects: a double-blind randomized placebo-controlled trial. J Pharmacol Exp Ther 300, 118–123. [DOI] [PubMed] [Google Scholar]
- Zacny JP, Wroblewski K, Coalson DW, 2015. Methylnaltrexone: its pharmacological effects alone and effects on morphine in healthy volunteers. Psychopharmacology (Berl) 232, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]