

Despite advanced treatments for heart failure (HF), morbidity and mortality rates remain high1. Understanding the gene regulatory networks and signaling pathways involved in different cardiomyopathy phenotypes leading to HF may provide potential mechanistic insights for developing therapeutic strategies.
We expanded our RNA-seq analysis using the dataset obtained from the Myocardial Applied Genomics Network consortium, as previously described2. The data included whole-transcriptome of left ventricle (LV) tissues curated from patients who experienced end-stage HF due to dilated cardiomyopathy (DCM, n=162), hypertrophic cardiomyopathy (HCM, n=28), and from unmatched non-failing hearts from organ donors as controls (NF, n=162). Based on a threshold of 1TPM (Transcripts Per Kilo-base Million), we identified 9,511, 9,771, and 9,236 genes expressed in DCM, HCM, and NF, respectively (Figure A). The three groups shared 8,773 expressed genes in the LV. One hundred ninety-four genes were only expressed in NF but not in HF. There were 895 (9% of differentially expressed genes, DEGs) and 1,282 (13.2%) DEGs (fold changes [FC] ≥ 1.5) in DCM and HCM, respectively, in comparison to the NF. Among these genes, 805 (8.1%) and 1,140 (11.7%) genes were upregulated in DCM and HCM, respectively, whereas 90 (0.9%) or 142 (1.5%) genes were downregulated in DCM or HCM significantly.
Figure.
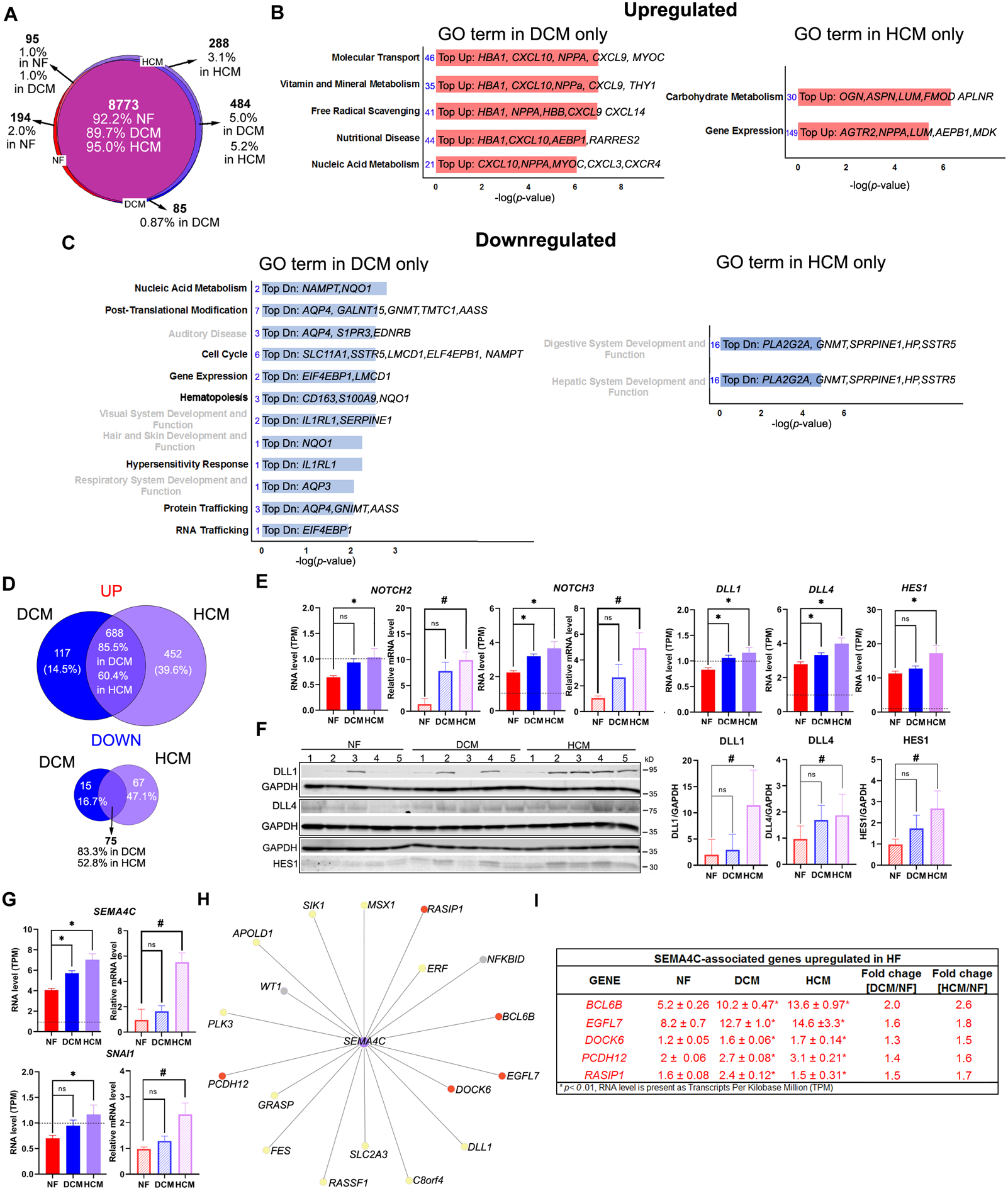
Whole-transcriptome analysis reveals differential transcriptome and gene regulatory networks in non-failing and failing human hearts. (A) Venn diagram showing number and percentage of expressed genes of non-failing (NF), dilated cardiomyopathy (DCM) and left ventricular hypertrophy (HCM) (B) Ingenuity Pathway analysis (IPA) for all differentially expressed genes (DEGs) upregulated in DCM or HCM revealing the different gene ontology (GO) functions presented only in DCM (left) or HCM (right). The number of genes (number in blue) and the top upregulated (Top-Up) genes in each GO function were listed (C) IPA for all DEGs downregulated in DCM or HCM revealing the different gene ontology (GO) functions presented only in DCM (left) or HCM (right). The number of genes (number in blue) and the top downregulated (Top-Dn) genes in each GO function was listed. The GO terms related to cardiovascular disease and function were highlighted in bold black font. (D) Venn diagram showing the number and percentage of differential expressed genes of DCM and HCM that were upregulated (left) and down-regulated in both groups. (E) Bar graphs showing the RNA levels in NF (n=162), DCM (n=162), HCM (n=28) determined using RNA-seq for NOTCH2, NOTCH3, HES1 DLL1, and DLL4 and using quantitative RT-PCR for NOTCH2 and NOTCH3 (n=5 in each group). Relative mRNA levels were calculated using the ΔΔCt method, with 18s levels as a reference and normalized to the value obtained from NF control. (H) Western blot analysis and quantification of NF, DCM and HCM (n=5 in each group) left ventricle lysates for HES1(Abcam, ab119776), DLL1 (Abcam, ab10554) and DLL4 (Cell Signaling, 2589S) GAPDH (proteintech,60004) was used for normalization. HES1 (fold change increased 1.7 ± 0.3 in DCM/NF and 2.7 ± 0.4 fold in HCM/NF), DLL1 (fold change increased 2.9 ± 3.0 in DCM/NF and 11.4 ± 3.1 fold in HCM/NF), and DLL4 (fold change increased 1.6 ± 0.3 in DCM/NF and 1.8 ± 0.4 fold in HCM/NF) (I) Bar graphs showing the RNA levels in NF (n=162), DCM (n=162), HCM (n=28) determined using RNA-seq (left) and quantitative RT-PCR (right) for SEMA4C and SANI1. Relative mRNA levels were calculated using the ΔΔCt method, with 18s levels as a reference and normalized to the value obtained from NF control. Data are shown as mean± SE. *, p<0.0001; #, p<0.05, ONE-way ANOVA in G-I. (H) Co-expressed gene network analysis reveals SEMA4C-associated genes in human LV tissues. Red dots indicate genes whose RNA level is significant changes ≥ 1.5 folds in HCM. Yellow dots indicate genes are normally expressed in three groups. Gray dots indicate genes RNA level is less than 1 TPM. (I) Showing the RNA level determined by RNA-seq for SEMA4C-associated genes whose RNA level is significant changes ≥ 1.5 folds in HCM.
Only ~10% of the DEGs were present between NF and failing human hearts. These genes likely contributed significantly to HF as they were associated with the pathological processes of HF such as inflammatory response (p-value range: [DCM:1.54E-06–1.43E-46]; [HCM:5.43E-04–6.4E-43]) and cardiovascular diseases ([DCM:1.1E-02–2.33E-09]; [HCM:2.81E-04–4.54E-16]). The gene ontology (GO) analysis for all DEGs also revealed different geneterms between DCM and HCM. For upregulated DEGs, the GO terms found in DCM specifically were involved in molecular transport, nutritional diseases, and metabolism, whereas carbohydrate metabolism and gene expression were specially linked to HCM (Figure B). For downregulated DEGs, nucleic acid metabolism, post-translation modification, protein, and RNA trafficking were linked to DCM only, whereas no specific disease GO terms were linked to HCM specifically (Figure C). More than 50% of these DEGs were overlapping between DCM and HCM (Figure D), suggesting DCM and HCM shared the same regulatory mechanisms in HF. Nevertheless, we found that 46% (651 genes) of DEGs (1,414 genes) were uniquely expressed in DCM or HCM (Figure D). Patients with HCM have a higher expression of genes associated with Notch signaling in their LVs than DCM patients. These include two members of the Notch family of receptors, NOTCH2 and NOTCH3 [FC = +1.6 in HCM/NF; +1.4 in DCM/NF] for both genes) and a well-known Notch signaling target gene, hairy and enhancer of split-1, (HES1, [FC = +1.5 in HCM/NF; +1.1 in DCM/NF]) (Figure E).To a lesser extent, two well-known Notch ligands, DLL4 and DLL1 (Delta-like ligand 4 &1) whose RNA levels were increased > 1.4 fold in the HCM, were only upregulated ~1.3 fold in the DCM comparing to the NF (Figure E). The data were validated by qPCR (NOTCH2 and NOTCH3) or by Western analysis (HES1, DLL1, and DLL4)(Figure F), which were compatible with the RNA-seq findings.
Finally, a novel gene for HF, Semaphorin 4C (SEMA4C), whose RNA level was found to be significantly increased in HCM at 1.7 fold in comparison to the NF but < 1.5 fold in DCM (Figure G). Its known downstream gene target, Snail family transcriptional repressor (SNAI1) is expressed and increased > 1.5 fold in HCM but not in DCM (Figure G). Using a gene network coexpressed analysis3 (Figure H), we further identified that numerous genes associated with SEMA4C are significantly upregulated ≥ 1.5 in HCM/NFmore then in that DCM LVs (Figure I). Our data suggest that SEMA4C and its associated signaling pathways and gene regulatory network may be associated with the HCM process. Evidence suggests that SMEA4C regulates epithelial- and endothelial- to mesenchymal transition (EMT and EndMT) in human diesease4. EMT and EndMT are known for contributing to cardiac fibrosis. While a massive amount of myocardial fibrosis can be seen in the HCM patients, it is less extensive in the DCM patients5. Thus, SEMA4C and its associated regulatory network may regulate cardiac fibrosis via EMT or EndMT in HCM.
In conclusion, we compare the whole-transcriptome profiles across the spectrum of HF phenotypes to individuals with non-failing left ventricular tissues. Although genes involved in or affected by the pathological processes of HF only accounted for a subset of expressed genes in the LV transcriptomes, their effects are significant enough to be associated with HF. Notch and SEMA4C signaling pathways and their associated network are statistically preferentially enriched in HCM. As a transmembrane and secreted protein, SEMA4C may serve as a diagnostic biomarker of HF. Further study the role of SEMA4C in HF is warranted.
Data created for the study are available in GEO (GSE141910). Other supporting data are available upon reasonable request. This study was approved by the Cleveland Clinic Institutional Review Board and that all subjects gave written informed consent.
Sources of Funding:
This work is supported by funding from the National Institutes of Health (NIH, R01HL105933). Dr. Ashley is supported by grants from the NIH (U01HG010218, R01HL144843). Dr. Cappola is supported by grant from the NIH (R01HL141232). Dr. Margulies is supported by grant from the NIH (R01HL149891). Dr. Tang is supported by grants from the National Institutes of Health (NIH) and the Office of Dietary Supplements (R01HL126827).
Nonstandard Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- DEG
differential expression gene
- DLL1
delta-like ligand 1
- DLL4
delta-like ligand 4
- EMT
epithelial to mesenchymal transition
- EndMT
endothelial to mesenchymal transition
- GO
gene ontology
- HCM
hypertrophic cardiomyopathy
- HES1
hairy and enhancer of split-1
- HF
heart failure
- LV
left ventricle
- SEMA4C
Semaphorin 4C
- TPM
transcripts per kilo-base million
Footnotes
Disclosures: Dr. Tang is a consultant for Sequana Medical A.G., Owkin Inc, and Relypsa Inc, and has received honorarium from Springer Nature for authorship/editorship and American Board of Internal Medicine for exam writing committee participation, all unrelated to the contents of this paper. All other authors do not have any relevant relationships to disclose.
References:
- 1.Metra M, Teerlink JR. Heart failure. Lancet. 2017;390:1981–1995. [DOI] [PubMed] [Google Scholar]
- 2.Liu CF, Abnousi A, Bazeley P, Ni Y, Morley M, Moravec CS, Hu M, Tang WHW. Global analysis of histone modifications and long-range chromatin interactions revealed the differential cistrome changes and novel transcriptional players in human dilated cardiomyopathy. J Mol Cell Cardiol. 2020;145:30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. Networkanalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019;47:W234–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gurrapu S, Pupo E, Franzolin G, Lanzetti L, Tamagnone L. Sema4c/plexinb2 signaling controls breast cancer cell growth, hormonal dependence and tumorigenic potential. Cell Death Differ. 2018;25:1259–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eijgenraam TR, Sillje HHW, de Boer RA. Current understanding of fibrosis in genetic cardiomyopathies. Trends Cardiovasc Med. 2020;30:353–361. [DOI] [PubMed] [Google Scholar]