

Abstract
A defining feature of protracted sepsis is development of immunosuppression that is thought to be a major driving force in the morbidity and mortality associated with the syndrome. The immunosuppression that occurs in sepsis is characterized by profound apoptosis-induced depletion of CD4 and CD8 T cells and severely impaired T cell function. OX40, a member of the tumor necrosis factor receptor superfamily, is a positive co-stimulatory molecule expressed on activated T cells. When engaged by OX40 ligand, OX40 stimulates T cell proliferation and shifts the cellular immune phenotype toward TH1 with increased production of cytokines that are essential for control of invading pathogens. The purpose of the present study was to determine if administration of agonistic antibody to OX40 could reverse sepsis-induced immunosuppression, restore T cell function, and improve survival in a clinically relevant animal model of sepsis.
The present study demonstrates that OX40 agonistic antibody reversed sepsis-induced impairment of T cell function, increased T cell IFN-γ production, increased the number of immune effector cells, and improved survival in the mouse cecal ligation and puncture model of sepsis. Importantly, OX40 agonistic antibody was not only effective in murine sepsis but also improved T effector cell function in peripheral blood mononuclear cells from patients with sepsis. The present results provide support for the use of immune adjuvants that target T cell depletion and T cell dysfunction in the therapy of sepsis-induced immunosuppression. In addition to the checkpoint inhibitors anti-PD-1 and anti-PD-L1, OX40 agonistic antibody may be a new therapeutic approach to the treatment of this highly lethal disorder.
Keywords: sepsis, immunosuppression, lymphocytes, programmed cell death, OX40
Summary:
Sepsis induces T cell dysfunction and T cell depletion which is prevented by treatment with agonistic antibody to OX40
Graphical Abstract
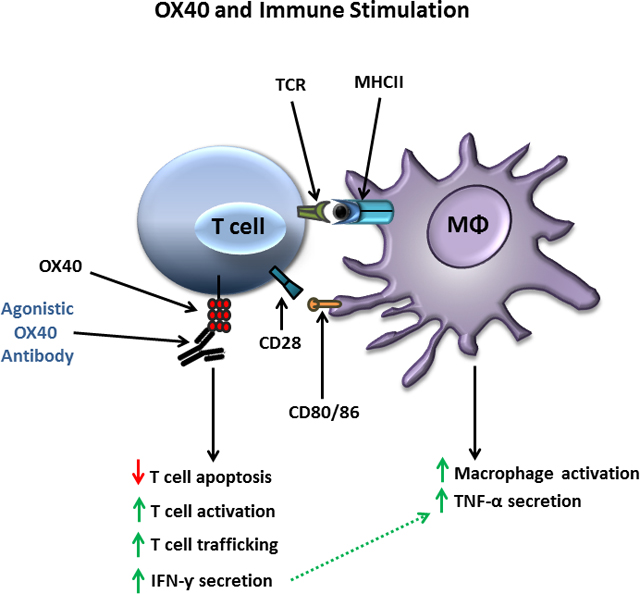
Introduction
Sepsis is defined as life threatening organ dysfunction due to a dysregulated host immune response to infection [1]. Sepsis is a major health care problem and is the most common cause of death in most intensive care units [2,3]. The dysregulated host response that occurs in sepsis is frequently typified by an initial hyper-inflammatory phase characterized by fever, shock, and respiratory failure. Following the initial exaggerated inflammatory response, patients often develop a more protracted phase of immunosuppression [4]. The immunosuppression phase of sepsis is manifested by an inability of the patient to clear the initial inciting infection and acquisition of new secondary infections. Additional hallmarks of the immunosuppressive phase of sepsis are reactivation of latent viruses, e.g., cytomegalovirus, Epstein Barr Virus, Herpes Simplex-1, and infections with relatively weakly virulent pathogens including, for example, fungal organisms, vancomycin resistant enterococci, stenotrophomonas, and acinetobacter [5–7].
The realization that immunosuppression is a key pathophysiologic abnormality in sepsis has led to investigation of therapies to boost the host immune system. The remarkable success of immune therapies in oncology have further stimulated interest in immune therapies in sepsis as these two seemingly disparate disorders share many of the same immunosuppressive mechanisms [8]. A variety of immune adjuvants have been tested in sepsis including leukocyte growth factors, immunostimulatory cytokines, and inhibitors of negative cellular co-stimulatory pathways, e.g., anti-programmed death one (anti-PD-1) and its ligand (anti-PD-L1) [9–12]. Testing of these agents in animal models of sepsis has demonstrated their ability to reverse immunosuppression and improve survival [13–16]. Importantly, IL-7 and the checkpoint inhibitors anti-PD-1/PD-L1 have shown efficacy in case reports in patients with life threatening JC virus associated progressive multi-focal leukoencephalopathy and hepatitis C, thereby providing support for their potential efficacy against bacterial pathogens [17–19]. Given the success of these immune agents in animal models of sepsis as well as their efficacy in patients with severe viral infections, phase1/2 clinical trials in sepsis have been recently conducted [10–12]. In general, the immune-based therapies have been well tolerated and, although the trials have contained relatively small numbers of patients, there has been no evidence of precipitation of a cytokine storm or detection of major adverse autoimmune effects in patients with sepsis [10–12]. Importantly, there was evidence that these agents enhance immunity of the septic patient [10–12].
Another promising immune adjuvant that has shown activity in both animal infectious models and in clinical trials in oncology, is agonistic targeting of OX40, a member of the tumor necrosis factor (TNF) receptor superfamily [20]. Activation of OX40 via OX40 ligand (OX40L) stimulates lymphocyte proliferation and skews CD4 T cell differentiation toward a T helper type 1 (TH1) phenotype [21]. In addition, OX40 agonism augments a variety of lymphocyte functions including production of IFN-γ, a cytokine that is essential for macrophage activation and host defense in sepsis [22]. An additional action of OX40 that may be particularly beneficial in sepsis is its effect to suppress the generation and/or function of T regulatory cells that act to impair immunity. Importantly, investigators have reported that OX40 agonists enhance host immunity and improve survival in animal models of malaria and in hepatitis B viral infections, which highlight the potential benefit of OX40 based immunotherapy in infectious disease and sepsis [23,24].
The purpose of this investigation was to determine the potential efficacy of OX40 agonists in reversing sepsis-induced immune suppression and improving survival in a clinically relevant animal model of sepsis. The effect of OX40 agonistic antibodies on immune effector cells from septic and critically ill non-septic patients was also examined to extend potential efficacy to humans.
MATERIALS AND METHODS
Mice
Animal studies were performed at Washington University School of Medicine (WUSM). Male C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Outbred male CD1 mice were purchased at Charles River Laboratory (Kingston, NY). All animal studies were approved by the Animal Studies Committee.
Sepsis model: cecal ligation and puncture
The cecal ligation and puncture (CLP) model was used to induce intra-abdominal peritonitis, as described previously. Mice were anesthetized with isoflurane, and a transverse abdominal incision was performed. The cecum was identified, ligated, and punctured as previously described [25]. Following cecal ligation and puncture, the abdomen was closed in two layers and 0.03 mg Buprenex in 1ml of 0.9% saline was administered subcutaneous for pain medication and to compensate for third space fluid losses that occur following surgery and during sepsis.
C57BL/6J mice were used for survival studies and for examining the effect of OX40 agonistic antibody on splenocyte numbers and function. For survival studies, the ligated cecum was punctured twice with a 25 gauge needle. In studies involving spleen harvest and immune testing of isolated cells, a 27 gauge needle was used for cecal puncture in order to have a sufficient number of surviving animals. CD1 mice were employed for studies testing the delayed type hypersensitivity (DTH) immune response with an injury level of a 23 gauge with 2 punctures. Previous work from our group showed that this strain of mice had a more robust immune response to recall antigenic challenge (13). For all studies, mice received 1 dose of 200μg of anti-OX40 or isotype-control intra-peritoneal at 6 hours and 48 hours post CLP. Investigators were blinded to mouse group identification and mice were randomized in a blinded fashion to receive either OX40 agonistic antibody or isotype control antibody.
Flow cytometric analysis of splenocytes
Splenocytes were prepared from septic and naive mice 2– 5 days post surgery as described [13]. Red cells were lysed using RBC lysis buffer (Biolegend, cat.# 420301). Total cell counts per spleen were determined via Vi-Cell counter (Beckman Coulter, Fullerton, CA). The percentage of the individual cell phenotypes (e.g., CD4, CD8, B cell, and macrophages) in the splenocyte population was determined via flow cytometric analysis as described below. Splenocytes were stained for CD4/CD3/CD8/B220. All living cells in the forward and sideways scatter were selected and plotted for CD3/CD4 double positive for CD4 T cells, CD3/CD8 double positive for CD8 T cells and B220+ CD3− for B cells. In the staining combination CD11c/B220/CD11b/Ly6G, DCs were determined as CD11chigh, CD11b+, Ly6G−, B220−. Macrophages were defined as CD11cneg-low, CD11b+, Ly6G−, B220−. Granulocytes were determined as CD11b+, Ly6Ghigh, B220−. Samples were acquired in a FACScan (BD Biosciences) with a 5-color upgrade (CyTek ), and analysis was performed in CellQuest Pro (BD Biosciences). The absolute cell counts for each splenic population were calculated, as previously described [13]. The gating strategy for determination of cell types and OX40 positivity is shown in Figure 1. Briefly, after the lymphocyte cell population was identified, based upon forward x side scatter plot gating, cells were investigated for CD3 and CD4, or CD3 and CD8 double positivity. The double positive populations were then investigated for OX40 expression.
Figure 1: OX40 expression on CD4 T cells increases during sepsis.

A.) Splenocytes of healthy and septic mice (2days and 5 days after the onset of sepsis) were harvested for the determination of OX40 expression on CD4 and CD8 T cells. OX40 expression was significantly increased on septic CD4 T cells compared to naïve healthy control cells but not on CD8 T cells. B.) CD3 and CD28 stimulation overnight induced a general significant increase of OX40 in both healthy and septic T cells versus unstimulated T cells. C.) The gating strategy to measure OX40 on lymphocytes is displayed. A rough lymphocyte gate was drawn on a Forward Scatter (FSC) by Side Scatter (SSC) plot. Cells from within the lymphocyte gate were then examined for CD4 and CD3 double positivity or CD8 and CD3 positivity. CD4+ and CD8+ T cells were then analyzed for OX40 expression. A representative plot from a septic animal is displayed. Data in panel A and B were analyzed by one-way ANOVA with multiple comparison post-tests, CD4 T cell results were significant (overall p<0.0001), Displayed are the p-values correspond to post tests.
Quantitation of IFN-γ and TNF-α production via ELISA
To determine the impact of OX40 agonistic antibody treatment on the amount of cytokines produced by T cells and monocytes upon stimulation, ELISAs were performed. 5×106 splenocytes were plated in 24 wells and stimulated with αCD3 (1μg/ml) and αCD28 (5μg/ml) for IFN-γ expression and with LPS (2μg/ml) for TNF-α expression in 1 ml of complete RPMI1640 overnight. The supernatants were obtained and IFN-γ and TNF-α were quantified using ELISA MAX Standard Sets from Biolegend (cat.# 430801 for IFN-ƴ, cat.#430901 for TNF-α) respectively, employing the μQuant Scanning Microplate Spectrophotometer (Bio-Tek Instruments, Winooski, VT) as described [26,27].
Quantitation of IFN-γ and TNF-α via ELISpot
In addition to measuring the total amount of cytokine produced by splenocytes via ELISA, ELISpot assays were performed to examine the frequency of responsive T cells and monocytes. 5×104 splenic PBMC (IFN-γ) or 5×103 cells (TNF-α) were plated per 96 well in 200μl of complete RPMI. Cells were stimulated overnight with CD3/CD28 (500ng/ml and 5μg/ml) for IFN-γ production and with LPS (2.5ng/ml) for the production of TNF-α [27].
R&D’s Elispot Development Kit (mouse IFN-γ, cat. # SEL485: mouse TNF-α, cat. # SEL410) was used following manufacturer instruction. Following development, images were captured and analyzed on Cellular Technologies Ltd (Cleveland, OH) ImmunoSpot 7.0 plate reader and software [27].
Determination of delayed type hypersensitivity (DTH) response
CD1 mice underwent CLP surgery as previously described [27]. One cohort of mice received 200μg of agonistic antibody to OX40 via intra-peritoneal injection 6 and 48 hours post CLP. A second cohort of control mice were treated identically except for receiving isotype control instead of active antibody. At 3 days post CLP, a time point of maximum immune suppression, all mice were immunized with 100μl TNBS (10mM) subcutaneously. At day 7, all mice had an antigenic challenge with 50μl of 10mM TNBS into the right footpad. PBS was injected into the left footpad as an internal control. The immunologic recall response was measured by determining the footpad swelling (in micrometers) 24 hours post challenge. Numbers represent the difference between the antigenic challenge and the saline control. Measurements of footpad thickness were performed by an investigator blinded to mouse group identification.
Human Studies
In order to relate the findings regarding OX40 in murine sepsis to clinical sepsis, OX40 agonistic antibody was also evaluated in clinical samples from septic and critically-ill non-septic patients.
Patient selection
Septic patients:
Patients at Barnes Jewish Hospital who were older than 18 years of age and who fulfilled a consensus panel definition of sepsis [11,12] were included in the study (Table 1). Sepsis was defined as the presence of systemic inflammatory response syndrome (SIRS) and a known or suspected source of infection as previously described [11]. Patients with HIV infection, viral hepatitis, or who were receiving immunosuppressive medications (except corticosteroids at a dose of < 10 mg of prednisone or the pharmacological equivalent per day) were excluded. Consent for study entry and blood draws was obtained from the patient or a legally authorized representative.
Table 1.
Patient Clinical Characteristics
| Septic patients (n = 22) | Critically Ill Non-Septic (CINS) patients (n = 11) | |
|---|---|---|
| Age, mean (range) | 51 (18–89) | 47 (23–61) |
| Male/Female | 12/10 | 8/3 |
| Comorbidities: | ||
| Cancer | 1 (4.5%) | 2 (18.2%) |
| Cardiovascular disease | 4 (18.2%) | 4 (36.4%) |
| Diabetes | 6 (27.3%) | 3 (27.3%) |
| GI disease | 2 (9.1%) | 0 (0%) |
| Hematologic disease | 1 (4.5%) | 1 (9.1%) |
| Hepatic disease | 2 (9.1%) | 0 (0%) |
| Neurologic disease | 3 (13.6%) | 2 (18.2%) |
| No significant PMH | 8 (36.4%) | 4 (36.4%) |
| Renal disease | 2 (9.1%) | 1 (9.1%) |
| Respiratory disease | 4 (18.2%) | 1 (9.1%) |
| Substance abuse | 3 (13.6%) | 0 (0%) |
| Primary Diagnosis | Gastrointestinal disease (n=6, 27.3%) | Cardiovascular disease (n=3, 27.3%) |
| Neurologic disease (n=3, 13.6%) | Respiratory disease (n=1, 9.1%) | |
| Respiratory disease (n=4, 18.2%) | Spinal complications (n=2, 18.2%) | |
| Sepsis/Septic shock (n=1, 4.5%) | Trauma (n=5, 45.5%) | |
| Skin/Soft Tissue Infection (n=3, 13.6%) | ||
| Trauma (n=5, 22.7%) | ||
| 90 day readmission (total patients) | 3 (13.6%) | 3 (27.3%) |
| 90 day readmission (total admissions) | 4 | 3 |
| 30 day mortality | 2 (9.1%) | 0 (0%) |
| 31–90 day mortality | 2 (9.1%) | 1 (9.1%) |
| Total 90 day mortality | 4 (18.2%) | 1 (9.1%) |
| SOFA score at time of blood draw, mean (range) | 7 (0–18) | N/A |
| APACHE II score at time of blood draw, mean (range) | 18 (7–27) | N/A |
Abbreviations: PMH: past medical history; SOFA: sequential organ failure assessment; APACHE: Acute Physiology and Chronic Health Evaluation; GI; gastrointestinal
Critically-ill non-septic patients:
Control subjects consisted of critically-ill non-septic patients admitted to the ICU for care following major surgery, trauma, or myocardial ischemia (Table 1). Exclusion criteria were identical to that for patients with sepsis – see above. Consent for study entry and blood draws was obtained from the patient or a legally authorized representative. All protocols were approved by the Washington University Institutional Review Board.
Determination of T cell IFN-γ and monocyte TNF-α production by human PBMCs
To confirm that the mouse results were consistent with human septic patients, the ELISPOT assay was also performed on patient samples. Patient PBMCs were harvested from whole blood via Ficoll-Paque™, counted with a Vi-Cell™ counter from Beckman Coulter (Brea, CA, USA), plated at a standardized density, (2.5 × 104 cells per well for IFN-γ and 2.5×103 cells per well for TNF-α), and incubated overnight with RPMI media containing anti-CD3/anti-CD28 (500ng/ml and 5μg/ml) or LPS (2.5ng/ml) for stimulation. Each well had a volume of 200 μl. rhOX40 ligand was obtained from R&D Systems (cat.# 1054-OX). Final concentration: 100ng/ml of rhOX40 ligand. IFN-γ and TNF-α were detected using a colorimetric reagent kit (CTL, Shaker Height, OH, cat. # hIFNgp-2M/5 and # hTNFap-2M/5). ELISPOT plates were made by Millipore (Hampton, NH; catalog number M8IPS4510). Assays were performed per manufacturer’s instructions. Following development, images were captured and analyzed on Cellular Technologies Ltd (Cleveland, OH) ImmunoSpot 7.0 plate reader and software.
Antibodies and reagents:
The following antibodies were purchased from Biolegend (San Diego, CA): CD4-FITC (cat. # 100510), CD3-Pe (cat. # 100308), CD8-PerCP/Cy5.5 (cat. # 100734), B220-APC (cat. # 103212), CD11b-PerCP/Cy5.5 (cat. # 101227), CD11c-FITC (cat. # 117305), Ly-6G-Pe (cat. # 127607), anti-mouse CD3 (cat. # 100202), anti-mouse CD28 (cat. # 10112).
An agonistic antibody that activates the mouse OX40 receptor (mOX40.23-mg1 clone 6E10) was provided by Bristol Myers Squibb [28]. A control non-binding mouse IgG1 antibody from BioXcel was also provided by Bristol Myers Squibb
Recombinant OX40-ligand that activates the human OX40 receptor was purchased from R&D Systems (cat. 1054-OX).
Statistical analysis
Data were analysed with the statistical software Prism (GraphPad, San Diego, CA, USA). Survival analysis was tested with Gehan-Breslow-Wilcoxon. Data are reported as the mean ± SEM. For comparison of two groups, the Student’s t-test was employed. A paired t-test was used when comparing samples from the same patient which were treated identically except for incubation with either OX40 agonistic antibody or isotype control antibody. One-way ANOVA with Tukey’s multiple comparison tests was used to analyse data in which there were more than two groups. Two-way ANOVA was used to analyse differential effects of CD3/CD28 treatment on OX40 surface expression between naïve animals and OX40 antibody treated animals. Significance was reported at p <0.05.
RESULTS
Sepsis increases OX40 expression on murine CD4 but not CD8 T cells
OX40 is reported to be expressed at low levels on resting naïve T cells and is inducible upon activation of CD4 and CD8 T cells [20]. We investigated if T cell OX40 expression increases during sepsis. Mice were made septic via cecal ligation and puncture (CLP) and splenocytes were harvested two and five days post CLP surgery. OX40 expression on CD4 and CD8 T cells was compared to the OX40 expression of naïve healthy animals.
Two days after CLP, the percentage of CD4 T cells expressing OX40 was significantly increased compared to naive controls (7.5 ± 0.39% vs. 12.8 ± 1.16% on naïve and CLP CD4 T cells respectively (Fig. 1A). This increase persisted, but did not increase further by day 5. In contrast to the effect of sepsis to increase OX40 expression on CD4 T cells, there was no significant increase in OX40 expression on CD8 T cells at either 2 or 5 days post sepsis onset compared to CD8 T cells of naïve mice.
In addition to examining the effect of sepsis on OX40 expression in CD4 and CD8 T cells, we also examined whether overnight stimulation of CD4 and CD8 T cells with anti-CD3/anti-CD28 agonistic antibodies would lead to additional increases in OX40 expression. Splenocyte CD3/CD28 stimulation led to increased OX40 expression in both CD4 and CD8 T cells from both naïve and day 5 CLP animals (p<0.0001 for treatment effect for both CD4 and CD8). The percentage of OX40 positive CD4 T cells increased to 73.6% in splenocytes from naïve animals and 86.3% in splenocytes from septic animals 5 days post CLP This increase was, to a lesser extent, found to also be present on stimulated CD8 T cells (4.2% and 6.6% respectively, Fig. 1B. The flow cytometry gating strategy is shown in Figure 1C.)
Agonistic OX40 improves the delayed type hypersensitivity (DTH) response in CLP
The DTH response requires a coordinated interaction of cells of the innate and adaptive immune system and is therefore a reflection of the overall functional status of host immunity [30]. In our model of delayed-type hypersensitivity, the ability to respond to a newly introduced antigen (TNBS, 10mM) is tested by immunizing septic animals on day 3 after CLP, a previously proven time point of significant immune suppression (13). Four days after immunization an antigen recall is tested by injecting TNBS into the footpad. The amount of footpad swelling 24 hours later indicates the immune response at the time point of immunization. Healthy animals that served as a positive control were subjected to immunization and recall antigen challenge whereas healthy control mice that did not receive the primary antigen immunization but only the recall antigen injection into the footpad served as negative immune controls.
Mice with sepsis failed to mount a significant immune response to the antigenic challenge compared to the positive control, consistent with a profound sepsis-induced defect in immune response (Fig. 2B). Importantly, septic mice that received isotype control (CLP control) were not statistically different from the negative control group but were statistically different from the positive control group (p=0.0001, Fig. 2B). In contrast, septic mice that were treated with agonistic-OX40 antibody showed a benefit of the immune therapy as evidenced by the fact that their DTH response was significantly greater than the negative control group (p=0.0066) and not statistically different from the positive control group. However, OX40 agonistic antibody treated mice were not different from CLP mice that were treated with the isotype control antibody (p=0.19). A representative image of the swollen mouse paw following recall antigen challenge is depicted in Fig. 2C.
Figure 2: Sepsis induces a marked decrease in the DTH response that is partially restored by OX40 agonistic antibody.

A.) Mice underwent CLP surgery and were divided into 2 treatment groups. One group received 200μg OX40 agonistic antibody 6 hours after CLP and again at 48 hours. The control group received isotype control. 3 days post surgery mice were sensitized with trinitrophenyl (TNP) subcutaneously and challenged into the footpad with TNP four days post sensitization. B.) The degree of footpad swelling was quantified 24 hours post challenge and compared to the swelling of the saline injected control foot (internal control). C.) Example of a recall reaction after sensitization and challenge. A dial gauge in micrometer is used to measure the immune response. Data in Panel B was analyzed by one-way ANOVA (Krusk-Wallace) with multiple comparison post-tests, results were significant (overall p<0.0001), displayed p values correspond to the results of the multiple comparison post-tests.
OX40 agonist therapy decreases circulating lymphocyte counts
Previous studies have shown that activation of OX40 leads to trafficking of lymphocytes from the bloodstream to sites of inflammation which is likely related to upregulation of lymphocyte adhesion molecules by OX40 agonist[31]. This trafficking from the blood to the sites of inflammation or infection is often reflected by a decrease in circulating lymphocytes. To determine if peripheral white blood cell numbers and distributions were altered by sepsis and OX40 agonist therapy, mice underwent CLP surgery. One cohort of mice was treated with OX40 agonistic antibody at 6 and 48 hours after CLP while the second cohort was injected with isotype control antibody. Blood was obtained at 5 days post-CLP. Total white blood cell (WBC) and differential cell counts were analyzed using a Hemavet cell counter. Naïve mice that had no surgery were used to provide the normal blood cell counts in mice.
Although there was no difference in total WBC count in naïve vs. CLP control mice, there was a statistically significant decrease in the WBC count in CLP mice that were treated with OX40 agonist vs naïve mice; p<0.0003; (Fig. 3). The number of circulating lymphocytes was decreased in both CLP controls and in CLP mice treated with OX40 agonist versus naïve mice (Fig. 3). Furthermore, the circulating lymphocytes in CLP mice treated with OX40 agonist was less than 50% of that in the CLP mice treated with isotype control antibody; p=0.0016. The number of blood neutrophils was increased approximately two-fold in CLP control mice compared to naïve mice, p= 0.003; (Fig. 3). Although there was a trend toward increased numbers of neutrophils in CLP mice treated with OX40 agonist antibody, it did not achieve statistical significance.
Figure 3: OX40 agonist decreases white blood cell (WBC) counts in peripheral blood.

Peripheral blood was harvested 5 days post-surgery by cardiac puncture and the different blood cell subsets were analyzed by Hemavet. White blood cells (WBCs), lymphocytes, and neutrophils were different between groups (p=0.0004, p<0.0001, and p=0.0005 respectively). Total WBC and lymphocytes were significantly decreased in blood following OX40 treatment relative to naïve and control CLP animals. OX40 agonistic antibody treatment had little effect the numbers of neutrophils and monocytes. Results were analyzed by one-way ANOVA, and multiple comparison post-tests were then performed. Displayed p-values correspond to the results of the multiple comparisons.
OX40 agonist increases splenic CD4 but not CD8 T cells
A hallmark of sepsis is a profound apoptosis-induced depletion of CD4 and CD8 T cells. OX40 signaling causes activation of anti-apoptotic pathways that may inhibit apoptosis. OX40 activation also induces lymphocyte proliferation. These two actions of OX40 to block apoptosis and increase lymphocyte proliferation may blunt the sepsis-induced depletion of lymphocytes. Consequently, we examined the impact of OX40 agonistic therapy on absolute lymphocyte counts in the mouse spleens. Mice underwent CLP with one cohort being treated with OX40 agonistic antibody at 6, and 48 hours post-surgery. A control group received isotype control antibody. Healthy unoperated mice (naïve) served as controls to quantitate normal splenic cell numbers and phenotypes. Five days post-surgery splenocytes were harvested and flow cytometry was performed to identify the different splenic cell subsets. We and other investigators have shown previously that sepsis-induced lymphocyte apoptosis is detected in the spleen within 8–12 hours after sepsis onset, peaks around 24–48 hours, and that decreased blood lymphocyte counts (lymphopenia) persists for days. In contrast to the decrease in splenic lymphocytes, there is an increase in myeloid derived cells in the spleens of septic mice due to an influx of neutrophils and other myeloid derived cells as sepsis persists. Five days post CLP total splenocyte counts in the OX40 agonistic antibody treated group were significantly increased compared to naïve and CLP isotype controls where as mice in the CLP control group showed a decrease in both CD3 and CD4 T cells compared to naïve mice (Fig. 4). Conversely, CLP mice that had been treated with OX40 agonistic antibody, had an increase in CD3 and CD4 T cells compared to both CLP mice treated with isotype control antibody and naïve mice. There was no effect of OX40 agonist on CD8 T cells (Fig. 4).
Figure 4: OX40 agonist prevents the loss of splenic CD4 T cells and B cellsand increases counts of Granulocytes and Macrophages.

Splenocytes were harvested 5 days post-surgery, counted, and then stained for the diverse cell subsets and analyzed by flow cytometry. With the exception of CD8 T cells, the displayed cell subsets showed a significant increase in cell numbers following OX40 agonistic antibody treatment. Results were analyzed by One-way ANOVA, and multiple comparison post-tests were then performed. Displayed p-values correspond to the results of the multiple comparisons.
Additionally, a decrease in splenic B cells was prevented in septic mice that received therapy with OX40 agonist (Fig. 4).
OX40 increases macrophage and neutrophil cell numbers
In contrast to the effect on lymphocytes, sepsis does not induce apoptosis of mature macrophages [32]. In addition, spleens of mice that have undergone CLP typically are increased in size by day fivebecause of an influx of numerous myeloid derived cells including neutrophils and macrophages [25]. Cells of the innate arm of the immune system demonstrate a more rapid rate of repopulation after sepsis with numbers quickly surpassing cell counts of the healthy control group. Five days post CLP, both macrophages and neutrophils were significantly increased in septic versus naïve mice and this increase was further enhanced in septic mice treated with OX40 agonistic antibody (Fig. 4).
OX40 agonist increases not only cell numbers but also cell function after sepsis
A key pathophysiologic mechanism of sepsis is impaired lymphocyte IFN-γ production. IFN-γ is essential for effective activation of monocytes and macrophages that play an important role in pathogen elimination [22]. One of the critical cytokines produced by activated macrophages is TNF-α. Consequently, we investigated the effect of OX40 agonistic antibody therapy on lymphocyte IFN-γ and monocyte TNF-α production. Assays were performed by two independent methods, i.e., ELISA and ELISpot assays (Fig. 5).
Figure 5: OX40 agonist increases IFN-γ and TNF-α production.

Mice underwent CLP surgery and were either treated with OX40 agonist or isotype control 6 hours post surgery with an additional dose at 48 hours. Splenocytes were harvested at day 5 post surgery. A.) ELISA were performed OX40 agonistic antibody treatment increased the secretion of both IFN-γ and TNF-α relative to either naïve animals or control CLP animals. B.) OX40 agonistic antibody treatment significantly increased the number of IFN-ƴ producing cells compared to isotype control whereas the number of TNF-α expressing cells was unchanged. C.) Representative ELISpot images for the number of IFN-ƴ producing cells for isotype control and OX40 agonistic antibody treatment group are displayed. Data was analyzed by one way ANOVA (p<0.0001 overall for both A and B), displayed p-values correspond to multiple comparison post-tests.
The ELISA assay demonstrated that IFN-γ secretion in the naïve and CLP control mice were similar. However, OX40 agonist treatment significantly improves IFN-γ production compared to both CLP control and naïve groups (p<0.0001 for both). A similar result was observed for the TNF-α production. While TNF-α production in response to LPS stimulation is not different in naïve versus CLP control mice, there was an approximate doubling of TNF-α secretion in the OX40 agonistic antibody treated mice (p<0.0001 compared to both groups)(Fig.5A).
In contrast to the ELISA assay that provides information on the total amount of cytokine produced, the ELISpot assay detects the number of cytokine producing cells. Thus, both assays provide distinct and important information. The ELISpot assay demonstrated that mice treated with OX40 agonistic antibody had an approximate 2–3 fold increase in the number of IFN-γ producing lymphocytes compared to both naïve and CLP control mice(p<0.0001 for both comparisons Fig. 5B). In contrast, although the number of TNF-α secreting cells was increased in septic vs naïve mice, there was no statistical difference between the OX40 agonistic antibody treated versus the isotype control antibody treated CLP groups (Fig.5B).
Collectively, the results of ELISA and ELISpot demonstrate that OX40 agonistic antibody therapy caused an increase not only in the number of immune effector cells but also an increase in their function which is critical in control of invading pathogens.
OX40 agonist improves survival in sepsis
Having demonstrated that therapy with agonistic antibody to OX40 improves both DTH and the number and function of splenic CD4 T cells and other cell types, we next tested its impact on sepsis-induced mortality. C57BL/6J mice underwent CLP surgery and were injected 6 hours and 48 hours after CLP with OX40 agonist antibody or isotype control antibody. Agonistic antibody to OX40 treatment resulted in a significant improvement in survival at 7 days post surgery; p≤0.05; (Fig. 6).
Figure 6: OX40 agonist improves survival in sepsis.

C57BL/6J mice were injected with 200μg OX40 agonistic antibody 6 hours after CLP and again at 48 hours. Septic control animals received isotype control. Survival was recorded for 7 days. Mice receiving OX40 agonist had a markedly improved survival. Data was analyzed by Gehan-Breslow-Wilcoxon test.
OX40 agonist increases IFN-γ in T cells from septic patients
To determine if the beneficial effects of OX40 stimulation on lymphocyte IFN-γ production observed in murine sepsis occurs also in cells of septic patients, ELISpot assays were performed on blood samples from septic and critically-ill, non-septic patients. Peripheral blood mononuclear cells from both patient groups were stimulated overnight with CD3/CD28 plus either recombinant human OX40-agonistic antibody or isotype control antibodies. Approximately 22 hours after incubation, the number of IFN-γ producing lymphocytes was determined as described previously [27]. Treatment with the OX40-ligand increased the number of IFN-γ secreting cells by an average of 49.2%, p<0.0001, (Fig. 7). As there was no clear breaking point in the dispersal of OX40 stimulation mediated increase in spot numbers, we considered an increase of 20% or greater in IFN-γ secreting cells a positive response to the stimulation as previously described [26]. 82% of septic blood samples displayed a positive response to OX40 agonism (18 of 22 patient samples).
Figure 7: OX40-ligand increases IFN-γ production in PBMCs from septic and critically ill non-septic patients.

PBMCs isolated from patient blood were plated on 96 well filter plates and stimulated with CD3 and CD28 overnight with and without OX40-ligand. 24 hours later the number of IFN-γ producing cells was determined by ELISpot and expressed as percent increase in IFN-γ production in CD3/CD28 and OX40 agonistic antibody stimulated culture over cell culture stimulated by CD3 and CD28 alone. Comparing samples with and without OX40 treatment showed that OX40 agonistic antibody treatment significantly increased the number of IFN-ƴ producing cells (p<0.0001).
OX40-ligand treatment led to a very similar result in CINS blood samples, as expected from prior studies showing OX40 signaling activates receptor-expressing cells (21). Treatment with the OX40-ligand increased the number of IFN-γ secreting cells by an average of 57%, with 72.7% (8 of 11 patients) of patients showing a positive response.
DISCUSSION
The present study adds to the growing body of research indicating that OX40 agonistic agents may be efficacious in improving outcomes in infectious diseases [23,24]. Previous studies of OX40 in a mouse malaria model of infection demonstrated that OX40 agonistic antibody lead to a 3- to 4-fold expansion of parasite-specific CD4 T cells [23]. Significantly, this increase in parasite specific CD4 T cells was associated with a decrease in the number of circulating blood parasites consistent with an improvement in the ability of the host to eradicate the pathogens [23]. OX40 agonistic agents also improved host immunity against hepatitis B infections resulting in enhanced T cell responses, decreased hepatic injury, and clearance of the virus [24]. The current results show that OX40 agonistic antibody therapy is beneficial in a polymicrobial bacterial model of sepsis and improves effector T cell function in patients with sepsis. Collectively, these studies underscore the potential utility of the OX40 therapy in infectious diseases.
A second important implication of the present study is the finding that therapeutic approaches in sepsis that are targeted to enhance lymphocyte function and adaptive immunity may be beneficial in reducing morbidity and mortality. Most previous therapies of sepsis have been directed at blocking a presumed hyper-inflammatory, cytokine mediated host response. Over 100 clinical trials of anti-inflammatory and or anti-cytokine agents have failed to show benefit in sepsis [2,3]. An increasing appreciation of the profound immunosuppressive effects of sustained sepsis has led to new therapeutic approaches that act to restore and enhance host immunity. In that regard, IL-7 and the checkpoint inhibitors anti-PD-1 and anti-PD-L1 have been efficacious not only in animal models of sepsis but also in patients with life threatening JC virus induced progressive multi-focal leukoencephalopathy[13,14,18,19]. Although there are several important differences in the immunologic effects of IL-7, anti-PD-1/PD-L1, and OX40 agonistic antibody, all three agents act to ameliorate sepsis-induced apoptotic death of lymphocytes and restore T cell production of the key cytokine IFN-γ [13–16,28]. Collectively, the studies of these three immune adjuvants, i.e., IL-7, anti-PD-1/PD-L1, and OX40 stimulation, make a strong case for a new therapeutic approach targeting the depressed adaptive immunity in sepsis.
The present results showing beneficial effects of OX40 agonistic antibody in sepsis stand in contrast to the findings of Karulf et al who reported that mice that had genetic deletion of OX40 ligand (OX40 ligand null mice) or mice that had treatment with antagonistic antibody to OX40 ligand had improved survival in sepsis [33]. We speculate that the reason for the different results regarding the effect of the OX40 pathway in sepsis relates to differences in the severity of the sepsis models employed by Karulf et al and the present model. Karulf and associates used a much larger gauge needle (#19 gauge) to puncture the cecum compared to the #25 gauge needle employed for tests of survival in the present study. The larger gauge needle used by Karulf et al results in the release of an increased bacterial inoculum as well as augmented release of endotoxins from the cecum thereby inducing more cytokine-mediated inflammation. This more intense initial inflammation results in a high early mortality as reflected by the fact that in two of the sepsis models employed by Karulf and colleagues, the control mice had over 50% mortality within the first 24 hours after CLP. In contrast, in our less fulminant CLP model, only 10% of control mice had died by 24 hours. The increased severity of the sepsis model employed by Karulf et al. is also evidenced by data showing that the control mice had 100% mortality by the end of the study while in the present study, 40% of the control mice were still alive at day 7 (Fig. 6). Thus, it is therefore likely that the mice dying in the study by Karulf and associates were dying of a hyper-inflammatory cytokine-mediated death. Therefore, blocking the OX40 pathway with antagonistic antibody to OX40 ligand in this hyper-inflammatory model would improve survival. It is important to note that with better treatment algorithms, the large majority of deaths in septic patients now occur after the initial hyper-inflammatory phase of sepsis and during the immunosuppressive phase of the disorder, which represents the greatest opportunity for life-saving therapies [34]. Therefore, treatment to enhance host immunity is the current focus of many sepsis investigators [3,7,9].
A dramatic effect of OX40 agonistic antibody therapy was to decrease the number of circulating lymphocytes in sepsis (Fig. 3). While mice with sepsis had significantly less circulating lymphocytes compared to naïve mice, septic mice that were treated with OX40 agonistic antibody had an even more dramatic reduction. The number of circulating lymphocytes in septic mice which were treated with agonistic antibody to OX40 was less than approximately 50% of the circulating lymphocytes in septic mice which received the isotype control antibody. This effect of OX40 therapy to decrease circulating lymphocytes seems paradoxical given OX40’s effect to increase splenic CD3 and CD4 T cell counts (Fig. 4). A likely explanation for OX40 agonistic antibody to decrease circulating lymphocytes is its well-known effect to increase lymphocyte activation and expression of lymphocyte adhesion molecules [30, 31]. Activated lymphocytes with increased adhesion molecules traffic to sites of infection and interact with their cognate receptors expressed on endothelial and organ parenchymal cells. Subsequently, these lymphocytes leave the circulation and assist in modulating the local immune response that is essential to control the invading pathogens. Indeed, a similar acute drop in circulating lymphocytes is observed after administration of IL-7 that also induces T cell activation and upregulation of cell adhesion molecules [10]. We speculate that this drop in circulating lymphocytes following administration of OX40 agonistic antibody reflects an important therapeutic action of the drug in sepsis.
Remarkably, the immunosuppression that occurs in sepsis and cancer share many common mechanisms [8,36]. Therefore, the fact that clinical trials of OX40 agonistic antibodies are showing activity in patients with cancer provides support for their potential applicability in patients with sepsis-induced immune suppression [20]. OX40 agonistic antibody is currently being tested in multiple clinical trials including in patients with head and neck malignancies (NCT02274155) and in colon cancer (NCT02559224). As is often the case when immunotherapeutics are being used, there is concern that OX40 agonistic antibodies may induce autoimmune reactions in patients [37]. Such autoimmune effects have been reported to occur in patients treated with the checkpoint inhibitors anti-PD-1 and anti-PD-L1 [18]. However, OX40’s mechanism of action to directly stimulate T cells by co-stimulation is distinct from the effect of anti-PD-1/anti-PD-L1 antibodies that act to block co-inhibitory T cell receptors. Stated another way, OX40 agonistic antibodies “step on the accelerator” while checkpoint inhibitors “release the brakes” on the T cell. Although definitive comparative studies have not been conducted, these different molecular mechanisms of action may result in a different likelihood of inducing autoimmunity [38]. Both anti-PD-1 and anti-PD-L1 antibodies have been tested in phase 1 clinical trials in patients with sepsis [11,12]. Although it is important to emphasize that the number of septic patients included in these trials was small, the treatments appeared to have a reasonable safety profile and did not induce a hyper-inflammatory state. If anti-PD-1 and anti-PDL1 antibodies are ultimately shown to be effective in patients with sepsis and to have an acceptable safety profile, it can be argued that an OX40 agonistic antibody may also be beneficial.
In conclusion, OX40 agonistic antibody reversed sepsis-induced impairment of T cell function, increased the number of immune effector cells, and improved survival in a clinically-relevant animal model of sepsis. OX40 agonistic antibody was not only effective in murine sepsis but also improved T effector cell function in peripheral blood mononuclear cells from patients with sepsis. The present results provide additional support for the use of immune adjuvants in the therapy of sepsis.
Acknowledgments
This work was supported by the National Institutes of Health Grants GM1269128 to RSH, grant K23GM129660 to AMD, and funding from Bristol Myers Squibb to RSH.
Abbreviations
- ANOVA
Analysis of variance
- APACHE II
Acute Physiology and Chronic Health Evaluation II
- CARS
Compensatory anti-inflammatory response syndrome
- CINS
Critically ill non-septic
- CLP
Cecal ligation puncture
- DTH
Delayed-type hypersensitivity
- ELISA
Enzyme linked immunosorbent assay
- ELIspot
Enzyme-linked immune absorbent spot
- FACS
Fluorescence activated cell sorter
- FSC
forward scatter
- HIV
Immunodeficiency virus
- IFN
Interferon
- IL-7
Interleukin-7
- ICU
Intensive care unit
- LPS
Lipopolysaccharide
- mAb
monoclonal antibody
- MFI
Mean fluorescence intensity
- PBS
Phosphate-buffered saline
- PD-1
Programmed Death receptor 1
- PBMCs
Peripheral blood mononuclear cells
- PD-L1
Programmed Death Ligand 1
- PMN
Polymorphonuclear cells
- RBCs
Red blood cells
- SSC
side scatter
- SIRS
Systemic inflammatory response syndrome
- SOFA
Sequential organ failure assessment
- TNBS
2,4,6-Trinitrobenzenesulfonic acid solution
- TNF
Tumor necrosis factor
- WBC
White blood cell
Footnotes
The authors from Bristol Myers Squibb (Dr. Michael Quigley and Dr. Dan Tenney) confirm that they are not involved in the development of OX40 agonist antibody for treatment of sepsis. Dr. Quigley was involved in the development of an OX40 agonist antibody (BMS −986178) for the treatment of cancer.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. 2016. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 315:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angus DC, and van der Poll T. 2013. Severe sepsis and septic shock. N Engl J Med 369:840–851. [DOI] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, and Vincent JL 2016. Sepsis and septic shock. Nat Rev Dis Primers 2:16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hotchkiss RS, and Karl IE 2003. The pathophysiology and treatment of sepsis. N Engl J Med 348:138–150. [DOI] [PubMed] [Google Scholar]
- 5.Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, Pachot A, Brooks TL, Deych E, Shannon WD, et al. 2014. Reactivation of multiple viruses in patients with sepsis. PLoS One 9:e98819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Efron PA, Mohr AM, Bihorac A, Horiguchi H, Hollen MK, Segal MS, Baker HV, Leeuwenburgh C, Moldawer LL, Moore FA, et al. 2018. Persistent inflammation, immunosuppression, and catabolism and the development of chronic critical illness after surgery. Surgery 164:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venet F, Rimmele T, and Monneret G. 2018. Management of Sepsis-Induced Immunosuppression. Crit Care Clin 34:97–106. [DOI] [PubMed] [Google Scholar]
- 8.Hotchkiss RS, and Moldawer LL 2014. Parallels between cancer and infectious disease. N Engl J Med 371:380–383. [DOI] [PubMed] [Google Scholar]
- 9.Hall MW, Knatz NL, Vetterly C, Tomarello S, Wewers MD, Volk HD, and Carcillo JA 2011. Immunoparalysis and nosocomial infection in children with multiple organ dysfunction syndrome. Intensive Care Med 37:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, Monneret G, Rimmele T, Blood T, Morre M, et al. 2018. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hotchkiss RS, Colston E, Yende S, Angus DC, Moldawer LL, Crouser ED, Martin GS, Coopersmith CM, Brakenridge S, Mayr FB, et al. 2019. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized, Placebo-Controlled, Single Ascending Dose Study of Antiprogrammed Cell Death-Ligand 1 Antibody (BMS-936559). Crit Care Med 47:632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hotchkiss RS, Colston E, Yende S, Crouser ED, Martin GS, Albertson T, Bartz RR, Brakenridge SC, Delano MJ, Park PK, et al. 2019. Immune checkpoint inhibition in sepsis: a Phase 1b randomized study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of nivolumab. Intensive Care Med 45:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Unsinger J, Burnham CA, McDonough J, Morre M, Prakash PS, Caldwell CC, Dunne WA, Hotchkiss RS 2012. Interleukin-7 Ameliorates Immune Dysfunction and Improves Survival in a 2-Hit Model of Fungal Sepsis. J. Infect. Dis. 206: 606–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS, et al. 2009. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A 106:6303–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, and Hotchkiss RS 2010. Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol 88:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, Unsinger J, Korman AJ, Green JM, and Hotchkiss RS 2013. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care 17:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel A, Patel J, and Ikwuagwu J. 2010. A case of progressive multifocal leukoencephalopathy and idiopathic CD4+ lymphocytopenia. J Antimicrob Chemother 65:2697–2698. [DOI] [PubMed] [Google Scholar]
- 18.Cortese I, Muranski P, Enose-Akahata Y, Ha SK, Smith B, Monaco M, Ryschkewitsch C, Major EO, Ohayon J, Schindler MK, et al. 2019. Pembrolizumab Treatment for Progressive Multifocal Leukoencephalopathy. N Engl J Med 380:1597–1605. [DOI] [PubMed] [Google Scholar]
- 19.Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, Chang K, Sulkowski M, O’Marro S, et al. .2013. A Randomized, Double-Blind, Placebo-Controlled Assessment of BMS-936558, a Fully Human Monoclonal Antibody to Programmed death-1, in Patients with Chronic Hepatitis C Virus Infection. Plos One 8:e63818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchan SL, Rogel A, and Al-Shamkhani A. 2018. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 131:39–48. [DOI] [PubMed] [Google Scholar]
- 21.Willoughby J, Griffiths J, Tews I, and Cragg MS 2017. OX40: Structure and function - What questions remain? Molecular Immunology 83:13–22. [DOI] [PubMed] [Google Scholar]
- 22.Hotchkiss RS, Chang KC, Grayson MH, Tinsley KW, Dunne BS, Davis CG, Osborne DF, and Karl IE 2003. Adoptive transfer of apoptotic splenocytes worsens survival, whereas adoptive transfer of necrotic splenocytes improves survival in sepsis. Proc Natl Acad Sci U S A 100:6724–6729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zander RA, Obeng-Adjei N, Guthmiller JJ, Kulu DI, Li J, Ongoiba A, Traore B, Crompton PD, and Butler NS 2015. PD-1 Co-inhibitory and OX40 Co-stimulatory Crosstalk Regulates Helper T Cell Differentiation and Anti-Plasmodium Humoral Immunity. Cell Host & Microbe 17:628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Publicover J, Gaggar A, Jespersen JM, Halac U, Johnson AJ, Goodsell A, Avanesyan L, Nishimura SL, Holdorf M, Mansfield KG, et al. 2018. An OX40/OX40L interaction directs successful immunity to hepatitis B virus. Science Translational Medicine 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, Hotchkiss RS 2010. Characterization and modulation of the immunosuppressive phase of sepsis. Infect. Immun. 78:1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thampy LK, Remy KE, Walton AH, Hong Z, Liu K, Liu R, Yi V, Burnham CD, and Hotchkiss RS 2018. Restoration of T Cell function in multi-drug resistant bacterial sepsis after interleukin-7, anti-PD-L1, and OX-40 administration. PLoS One 13:e0199497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazer M, Unsinger J, Drewry A, Walton A, Osborne D, Blood T, Hotchkiss R, and Remy KE 2019. IL-10 Has Differential Effects on the Innate and Adaptive Immune Systems of Septic Patients. Journal of Immunology 203:2088–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.al-Shamkhani K, Birkeland ML, Puklavec M, Brown MH, Barclay AN, 1996. OX40 is Differentially Expressed on Activated Rat and Mouse T cells and is the Sole Receptor for OX40 Ligand. Eur. J. Immunol. 26:1695–9. [DOI] [PubMed] [Google Scholar]
- 29.Wang R, Gao C, Dito G, Kabbabe D, Shao X, Hilt E, Sun Y, Pak I, Gutierrez M, Melero I, et al. 2019. An Integrative Approach to Inform Optimal Administration of OX40 Agonist Antibodies in Patients with Advanced Solid Tumors. Clinical Cancer Res. 25:6709–6720. [DOI] [PubMed] [Google Scholar]
- 30.Rosenstreich DL 1993. Evaluation of delayed hypersensitivity: from PPD to poison ivy. Allergy Proceedings 14:395–400. [DOI] [PubMed] [Google Scholar]
- 31.Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, Imamura S, and Uchiyama T. 1996. The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. Journal of Experimental Medicine 183:2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hotchkiss RS, Tinsley KW, Swanson PE, Grayson MH, Osborne DF, Wagner TH, Cobb JP, Coopersmith C, and Karl IE 2002. Depletion of dendritic cells, but not macrophages, in patients with sepsis. Journal of Immunology 168:2493–2500. [DOI] [PubMed] [Google Scholar]
- 33.Karulf M, Kelly A, Weinberg AD, and Gold JA 2010. OX40 ligand regulates inflammation and mortality in the innate immune response to sepsis. Journal of Immunology 185:4856–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD 2nd, Kreisel D, Krupnick., et al. 2011. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306:2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christou NV, Meakins JL, Gordon J, Yee J, Hassan-Zahraee M, Nohr CW, Shizgal HM, and MacLean LD 1995. The delayed hypersensitivity response and host resistance in surgical patients. 20 years later. Annals of Surgery 222:534–546; discussion 546–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moldawer LL, and Hotchkiss R. 2018. Immunotherapy: It is not just for cancer anymore. J Leukoc Biol 103:9–11. [DOI] [PubMed] [Google Scholar]
- 37.Ueno H, and Blanco P. 2015. OX40/OX40L axis: not a friend in autoimmunity. Oncotarget 6:21779–21780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linch Stefanie N., McNamara Michael J. and Redmond William L. et al. 2015. OX40 agonists and combination immunotherapy: putting the pedal to the metal Frontiers in Oncology; Vol. 5; 1–14: 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]