

Abstract
Background
Immunotherapy in prostate cancer (PCa) lags behind the progresses obtained in other cancer types partially because of its limited immune infiltration. Tumor-resident immune cells have been detected in the prostate, but the regulatory mechanisms that govern tumor infiltration are still poorly understood. To address this gap, we investigated the role of Wolf-Hirschhorn syndrome candidate 1 (WHSC1), a histone methyltransferase enzyme that targets dimethyl and trimethyl H3K36. WHSC1 is known to promote malignant growth and progression in multiple tumors, but its role in the interface between PCa and immune system is unknown.
Methods
RNA Sequencing (RNASeq) data from patients with PCa from The Cancer Genome Atlas (TCGA) were collected and divided into top/bottom 30% based on the expression of WHSC1 and disease-free survival was calculated. Publicly available chromatin immunoprecipitation (ChIPSeq) data were obtained from Cistrome and integrated with the available RNASeq data. RNASeq, ATACSeq and methylomic were analyzed using R Bioconductor packages comparing C42 cells with or without stable knockdown on WHSC1. Flow cytometry was used to measure Major Histocompatibility complex (MHC) levels, MHC-bound ovalbumin and tumor infiltration. C57B6 and NOD scid gamma (NSG) mice were subcutaneously grafted with TRansgenic Adenocarcinoma of the Mouse Prostate (TRAMP) C2 cells and treated with MCTP39 (10 mg/kg); tumor size was monitored over time and curves were compared using permutation analyses. All analyses used a significance threshold of 0.05.
Results
Leveraging TCGA data, we demonstrated that elevated WHSC1 levels positively correlate with the presence of an immunosuppressive microenvironment. We validated those results in vitro, demonstrating that genetic and pharmacological inhibition of WHSC1 restores antigen presentation. This occurs via an elegant epigenetic regulation of gene expression at the chromatin and DNA methylation levels. In vivo studies in immunocompetent mice also show an increased frequency of CD8+ T cells in tumors from mice treated with WHSC1 inhibitor, supporting the hypothesis that the antitumor effect following WHSC1 inhibition requires a fully functional immune system.
Conclusions
This study demonstrates a novel role for WHSC1 in defining immune infiltration in PCa, with significant future implications for the use of immunotherapies in prostate malignancies.
Keywords: T-lymphocytes, immunotherapy, gene expression profiling, tumor escape, tumor microenvironment
Introduction
Prostate cancer (PCa) growth is intrinsically driven by the androgen receptor (AR), and androgen deprivation therapy (ADT) is a common therapeutic intervention aimed to reduced tumor size by blocking the androgen signaling. ADT also favors the establishment of an immunopermissive tumor microenvironment by promoting tumor homing of CD4+ and CD8+ T lymphocytes, natural killer (NK) cells and macrophage.1–3 Furthermore, parallel inhibition of the androgen pathway and FOXP3+ T regulatory (Treg) cells increases the response to anti-CTLA4, an effect that was not observed in the absence of ADT.4 These results support the concept of a tumor-driven immunosuppressive mechanism that reduces the ability of the immune system to recognize tumor cells. Although promising, these results are mainly descriptive and the actual mechanism by which PCa defines infiltrating moieties is still poorly understood; thus, identifying the determinants of immune cell infiltration into the prostate could shed light on the regulatory interface between PCa and the immune system.
The Wolf-Hirschhorn syndrome candidate 1 (WHSC1) protein gene encodes for a SET-domain-containing histone methyltransferase that targets H3K36me2/me3.5–8 Elevated WHSC1 expression in the tumors correlates with worse prognosis, promoting resistance to chemotherapy and metastatic phenotype in PCa and other tumors.9–13 While the role of WHSC1 in tumor progression is well described, its role as epigenetic modifier in the crosstalk between PCa and the immune system remains mainly unexplored. In fact, the breadth of studies that investigate the regulatory role of WHSC1 on specific components of tumor-resident immune pathways is limited. Elevated WHSC1 reduces Human Leukocyte Antigen (HLA) levels in malignant peripheral sheath tumors in a mechanism antagonistic to PRC214 and mediates interferon-induced transcription through interaction with SPT6.15 Lastly, studies in cervical cancer revealed that WHSC1 promotes tumor progression and metastases by inducing transforming growth factor-beta (TGF-β)-mediated immunosuppression.16 Taken together, these results suggest that tumor-resident WHSC1 might act as the culprit for the poor antitumor immune response in PCa, which led us to investigate WHSC1’s role in defining the pathways regulating immune infiltration in prostate tumors.
In this paper, we examined the role of WHSC1 in regulating tumor-resident immune pathways and we demonstrate that the anti-tumor effect of WHSC1 inhibition is mediated by increased recruitment of immune cells to the tumor site.
Methods
Cell lines
TRAMP C2 cells were a kind gift from Dr Barbara Foster17; they were maintained in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) supplemented with 1 nM Dihydrotestosterone (DHT), 0.008 mg/mL insulin and penstrep. C42 cells were maintained in RPMI1640 with 10% FBS and P/S. C42 cells were validated via microsatellite PCR at the Roswell Genomics core. Both cell lines were mycoplasma negative.
WHSC1 knockdown
Short hairpin RNA (shRNA) knockdown of WHSC1 in C42 cells was prepared by the Roswell Park Gene Editing shared resource. Briefly, stable knockdown cells were generated using lentiviral-based shRNA (containing shRNA-pGIPZ plasmid or pGIPZ non-silencing control plasmid) produced in HEK293T cells as per manufacturer instructions. These lentiviral-based shRNAs express green fluorescent protein (GFP) and have a puromycin selectable marker. For stable transduction of target cells, media from 70% confluent C42 cells were removed, and virus supernatant plus 4 μg/mL polybrene (total 1 mL for six-well plate) was directly added on top of the cells and the plate was sealed and centrifuged at 1800 rpm for 45 min at room temperature (RT). Next, the plate was incubated for 3–6 hours at 37°C/5% CO2 and the media were replaced with normal target cell media and incubated again overnight at 37°C/5% CO2. The next day, cells were split 1:5 and were allowed to attach for 1 hour, and the media were replaced with puromycin selection media every 3–4 days (normal target cell media+puromycin (2 μg/mL). The resistant transduced C42 cells cultured for at least four to five passages in puromycin selection media and GFP expression were observed by microscopy before being using for further experiments.
Transient knockdown experiments in TRAMP C2 cells were made using SiWHSC1 (4390771, ThermoFischer Scientific) or SiCTR (4390843, ThermoFischer Scientific), and those in DU145 cells using siWHSC1 (SR305101) or siCTR (SR30004) from Origene.
For siRNA knockdown, 200×103 C42 cells or TRAMP C2 cells in 200 µL optiMEM were plated in a 24-well plate overnight and the next day were transfected with or without siWHSC1 (Invitrogen) using lipofectamine RNAimax at 37C, 5% CO2. After 6 hours, media were gently aspirated and replenished with fresh RPMI1640 or DMEM supplemented with DHT and further incubated at 37°C, 5% CO2 for 48 hours. Cells were harvested after 48 hours for RNA isolation using TRIzol (Invitrogen, 15596026) for quantitative PCR (qPCR) or for protein isolation by radioimmunoprecipitation assay (RIPA) buffer for western blot. The concentration of RNA was measured by nanospectrometer, and for RT-qPCR, 2 ng/µL of RNA was used for cDNA synthesis using iscript cDNA synthesis kit (Biorad, 170–8891). SYBR Green/Rox qPCR master mix (ThermoFischer Scientific, K0221) was used to analyze the expression of WHSC1, PDL-1, DNMT1 and GAPDH. The primer sequence is for these genes is provided in online supplemental table S2.
jitc-2020-001374supp007.pdf (44.4KB, pdf)
OVA overexpression
Soluble OVA gene, which was amplified from pCI-neo-sOVA (a gift from Maria Castro, Addgene plasmid 25 09818), and monomeric enhanced green fluorescent protein (mEGFP) gene, which was amplified from mEGFP-N1 (a kind gift from Michael Davidson, Addgene plasmid 54767), were genetically fused via P2A translational skipping sequence and were cloned in the Sleeping Beauty transposon plasmid with the human elongation factor 1α promoter.19 This plasmid, pT2-EF-OVA-mEGFP, was electroporated together with the Sleeping Beauty Transposase plasmid, pCMV(CAT)T7-SB100 (a gift from Zsuzsanna Izsvak; Addgene plasmid #34 87920), into TRAMP-C2 by Nucleofector 4D instrument. The electroporated cells were kept in maintenance medium (DMEM supplemented with 10% FBS, 0.005 mg/mL bovine insulin, 1 nM DHT and cell sorted based on mEGFP expression using FACS Aria I cell sorter. The expression of OVA on sorted cells were confirmed by western blotting using rabbit polyclonal OVA antibody (ab186717) at 1:4000 dilution and flow cytometry using PE anti-mouse H-2Kb bound to SIINFEKL (BioLegend) before incubating with or without MCTP-39 for 48 hours. The expression of OVA was analyzed after 48 hours by flow cytometry using BDLSRIIA cytometer, and data were analyzed by FCS Express V.7 Research Edition.
Western blotting
Protein concentration was measured using bicinchoninic acid (BCA) kit and 30 µg of protein was loaded into sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gel from either transfected or untransfected C42 or TRAMP C2 cells. The protein from gel was transferred into PVDF and further incubated with human (anti-WHSC1, Abcam, ab225625) or mouse (anti-WHSC1, Abcam, ab75359) primary antibody to WHSC1 using 1:2000 dilution for C42 lysate and 1:1000 for TRAMP C2 protein lysate. The primary antibody was further detected using either goat anti-rabbit (Abcam, dilution 1:10,000) or goat anti-mouse (Abcam, dilution 1:5000), while the house keeping gene, GAPDH, in both C42 and TRAMP C2 protein lysate was detected using anti-GAPDH antibody at 1:50 000 dilution (Abcam, ab181602).
Cell proliferation
Knockdown of WHSC1 in C42 and TRAMP C2 cells is discussed in previous sections. Cells were counted at 48, 96 and 144 hours. For pharmacological inhibition, cells either C42 or TRAMP C2 were seeded overnight at 4×103 cells per 100 µL media in a 96-well plate; the following day, cells were treated with vehicle control or different concentrations of MCTP-39 (0–10 µM) for 48 hours. After 48 hours, C42 or TRAMP C2 cells were either counted or used for staining with MHCI/II antibodies. Briefly, cells were detached using trypsin and washed with fluorescence-activated cell sorting (FACS) buffer for 5 min at 300 g. The pellet was resuspended in FACS buffer and stained with MHC-I/II antibodies for 20 min at 4°C. After incubation, cells were washed with FACS buffer two times and resuspended in 200 µL of FACS buffer before acquiring the data on a flow cytometer.
Mice and in vivo experiments
Male C57B/6J mice (6–8 weeks of age) and NSG mice (10–12 weeks of age) were obtained from the Roswell Park’s Center For Immunotherapy breeding colonies. All in vivo experiments were made following institutional and Institutional Animal Care and Use Committee (IACUC) regulations. In vivo experiments were not blinded. TRAMP C2 cells (1 × 106 cells/100 µL) were injected subcutaneously into the right flank of male C57BL/6J mice (n=6/group) or NSG mice (n=5/group) using a 27 G needle. Sample size was chosen based on pilot studies and chosen to include the potential loss of mice prior treatment. Tumor volume was monitored weekly with an electronic caliper and calculated as V=(W2×L)/2, where V is tumor volume, W is tumor width and L is tumor length. When tumors reached 100 mm3, mice were randomized prior to treatment with either MCTP-39 (10 mg/kg 5×/week/4 weeks) or vehicle control. Mice were euthanized either when tumors reached 2000 mm in any dimension, as per institutional IACUC regulations, when mice showed signs of advanced disease or after 4 weeks of treatment. Tumors were harvested and weighted; single cell suspension was prepared using the tumor dissociation kit (Miltenyi Biotech) as per manufacturing instructions prior to flow cytometry analysis.
Flow cytometry and antibodies
CD45-positive and CD45-negative fraction from single cell suspension was stained with the antibodies listed in online supplemental table S3. Gating strategies are described in online supplemental figures S4–S6). For C42 cells and TRAMP C2, cells were trypsinized and washed with FACS buffer two times for 5 min at 300 g. The pellet of C42 or TRAMP C2 cells were resuspended in FACS buffer and C42 cells were surface stained with HLA-B7, HLA-F, HLA-E, and HLA-DQ, while TRAMP C2 cells were stained with a cocktail of I-A/I-E and H-2Kb antibodies for 20 min at 4°C. To detect HLA-DM, C42 cells were first surface stained followed by fixation and permeabilization followed by staining with anti HLA-DM antibody for 30 min at RT. Unstained C42 cells or TRAMP C2 cells were used as negative control. After incubation, cells were washed with FACS buffer two times and resuspended in 200 μL of FACS buffer before acquiring data on BDLSRIIA cytometer. For mice, single cell suspension from tumors were incubated with anti-mouse CD16/32 antibody (FcR blocker) for 10 min at RT and subsequently stained with Zombie aqua for 15 min at RT followed by staining with anti-CD45, CD8a, CD3 and H-2Kb for 20 min at 4°C. After staining, cells were washed and fixed with fixation buffer for 15 min at 4°C followed by washing two times with FACS buffer. Cells were resuspended in 200 μL of FACS buffer before acquiring data on flow cytometer. For compensation, the Ultracomp beads was used to stain for individual fluorochrome-conjugated antibodies as single-color compensation controls, and each drop of bead contained a positive population that captures fluorochrome-conjugated antibody and a negative population that does not react with antibody. This bimodal distribution was used a single-color compensation control in multiparameter analyses. For the live/dead cell compensation, live cells were divided into two aliquots and saturating volume of methanol was added to one of the aliquots, vortexed and incubated at 4°C for 5 min. After 5 min, cells were washed three times with FACS buffer and mixed with original aliquot of live cells to get the heterogenous population of live/dead cells that was used for compensation in flow cytometry. Data were analyzed using FCS express V.7 Research Edition.
jitc-2020-001374supp008.pdf (44.1KB, pdf)
jitc-2020-001374supp009.pdf (133.9KB, pdf)
jitc-2020-001374supp010.pdf (156.8KB, pdf)
jitc-2020-001374supp011.pdf (143.1KB, pdf)
Analysis of published datasets
Publicly available PCa datasets were retrieved from cBioportal (https://www.cbioportal.org/). Detailed statistical methods are explained further in the appropriate section. GSEA analysis on TCGA data was done using the package DOSE,21 clusterprofiler22 and enrichplot (https://github.com/GuangchuangYu/enrichplot) using Reactome gene signature downloaded from the GSEA website (https://www.gsea-msigdb.org/gsea/index.jsp). To create the antigen processing and presentation signature, the following gene sets were used: REACTOME ANTIGEN PROCESSING CROSS PRESENTATION, REACTOME ANTIGEN PROCESSING UBIQUITINATION PROTEASOME DEGRADATION, REACTOME MHC CLASS II ANTIGEN PRESENTATION, REACTOME ANTIGEN PRESENTATION FOLDING ASSEMBLY AND PEPTIDE LOADING OF CLASS I MHC, REACTOME CLASS I MHC MEDIATED ANTIGEN PROCESSING PRESENTATION. ChIPSeq data were downloaded from Cistrome (http://cistrome.org/) with the following accession numbers: GSM1527830, GSM1679107, GSM353624, GSM353627, GSM875813, GSM875814, GSM1679108, GSM2252903, ENCSR849APH_1, and ENCSR849APH_2. Predicted target genes from Cistrome that scored 0 were removed, leaving a total of 13 878 unique genes (online supplemental file S2). Differential gene expression analysis from TCGA data comparing patients with highversus low WHSC1 transcript levels was performed using the limma R package at a significance level of FDR<0.05 and LogFC 0.25.23 Precomputed TCGA immune infiltration data were downloaded from xCell.24
jitc-2020-001374supp012.txt (88.7KB, txt)
RNASeq
RNA extraction and library preparation were performed by the Roswell Park Genomics Shared Resources. RNA libraries were constructed using the KAPA mRNA HyperPrep Kit (Roche Sequencing Solutions), and the libraries were sequenced on the Illumina NextSeq 500 sequencer with 2×75 cycle sequencing. Raw reads were compiled into fastq files, mapped onto the human hg38 reference genome using STAR25 and quantified at the gene level using the tximport R package.26 Genes differentially expressed between conditions were identified using limma.23 GSEA analysis was performed as described previously. APM signature was generated using HLA genes and genes involved in the antigen processing and presentation.
ATACSeq
ATACSeq was run on C42 cells with stable knockdown of WHSC1 via shRNA and control. The ATACSeq libraries were sequenced using NextSeq 500 sequencer at 2×75 cycle sequencing. Raw data were processed with MACS227 28 and further processed using ChIPSeeker29 to annotate identify the genes within the genomic regions within ATAC peaks. To calculate the fold changes between the two conditions, we created a consensus list of genomic regions covered by both conditions (shCTR and shWHSC1) using the soGGi R Bioconductor package.30 Reads spanning over these regions were then quantified using Rsubread,31 summarized at the gene level using peaks that are within 1000 bp to the nearest TSS and used to calculate the log2FC between WHSC1 knockdown and control. Results were merged with the RNASeq DEGs to identify genes that positively correlate with RNASeq data, hence higher ATAC signal, higher gene expression. The ATACSeq log2FC was then used to rank genes used as input for GSEA analysis using the GO signature. GSEA analysis was run in R using the clusterprofiler package and a p value cut-off of 0.05.
Methylation analysis
Methylation analysis was run on C42 cells with stable knockdown of WHSC1 via shRNA and control using the Illumina Infinium MethylationEPIC BeadChip Kit (Illumina). Raw files were processed using the Champ Bioconductor R package32 using default parameters. Methylation probes that coincided with known SNPs were removed. Probe IDs from the differentially methylated probe (DMP) list were merged with RNASeq DEG data, aggregated using mean intensity values at the gene level and correlated with RNASeq log2FC results to identify genes with reduced methylation and increased gene expression, or vice versa, on WHSC1 knockdown.
Statistical methods
Survival analysis was performed dividing patients based on the upper/lower 25% of the expression levels of WHSC1; significance, HR and CIs were calculated using Cox proportional hazard model available in the R package survival.33 The AUC analysis to evaluate WHSC1 as predictor for biochemical recurrence was done using the R package survivalROC,34 censoring for biochemical recurrence. Since TCGA does not offer the history of PSA testing per each patient over time, we used a threshold of PSA>0.4 ng/mL as described in Brockman et al.35 Significance for the AUC analysis was calculated by simulating 10 000 AUCs using randomly selected genes in the RNASeq dataset. The empirical p value was calculated by dividing the number of expected/simulated AUCs higher than our observed value by 10 000 (number of simulations).36 Significance when comparing two groups was calculated via two-tailed Student’s t-test at a significance threshold of 0.05. When more than two groups were compared, one-way analysis of variance with Tukey’s post hoc correction was used at a significance threshold of 0.05. In both cases, bar plots indicated the mean and SE of at least three biological replicates unless specified otherwise. The correlation between WHSC1 and AR gene expression was calculated using Spearman correlation using the log2 expression values from TCGA RNASeq data. Growth curves in mice were compared using permutation test with 10 000 simulations via the statmod R package using the compareGrowthCurves function.37 38
Results
WHSC1 levels positively correlate with the presence of an immunosuppressive microenvironment in PCa
We first evaluated WHSC1 expression in 489 PCa samples from The Cancer Genome Atlas (TCGA) collected following radical prostatectomy39 (online supplemental table S1) and its association with patients disease-free survival (DFS). This analysis revealed that patients with elevated WHSC1 expression (top 25%, online supplemental file S3) have significantly shorter DFS than patients with low WHSC1 levels (bottom 25%) (figure 1A and online supplemental figure S1A) (Cox p=0.000121, Cox HR=3.2221, 95% CI 1.801 to 6.127). Raising prostate-specific antigen (PSA) concentration post-therapy in PCa are often indicative of disease recurrence,35 and we found that WHSC1 gene expression levels are a modest, but significant, predictor for biochemical recurrence with an area under the curve (AUC) of 0.742 (online supplemental figure S1B, C; empirical p=0.0046). Moreover, PCa is an androgen-driven disease, and we found a positive correlation between WHSC1 and AR expression (online supplemental figure S1D) (Spearman’s coefficient=0.397, p<2.2e-16), corroborating previously published data on WHSC1 function as a transcriptional regulator of AR.13 We then investigated whether WHSC1 expression levels correlate with the presence of specific immune populations and found that patients with elevated WHSC1 have a highly immunosuppressive tumor microenvironment composed of high Th2 and Treg and low Th1 cells, NKT cells and M1 macrophages (figure 1B) (p value<0.001, Student’s t-test). Next, we interrogated TCGA RNA Sequencing (RNASeq) data to evaluate whether different WHSC1 levels correlate with altered HLA expression in the tumor, which would undermine the tumor’s ability to present tumor antigens to the immune system. Indeed, patients with elevated WHSC1 were found with consistently low levels of HLA class I and class II transcripts (figure 1C). We then investigated which transcriptional pathways were altered in patients with high versus low expression of WHSC1. Gene set enrichment analysis (GSEA) revealed that patients with elevated WHSC1 have increased expression of genes involved with cell proliferation pathways such as cell division, DNA replication and DNA repair (figure 1D), corroborating the results from previous studies.9 10 13 However, we also identified a downregulation of pathways involved with response to interferon gamma (IFN-γ), antigen processing and presentation and cytokine secretion (figure 1E), suggesting that high levels of WHSC1 negatively correlate with the status of both tumor-resident immune pathways and the antigen processing and presentation machinery (APM), which we previously demonstrated as a determinant of T cells’ ability to recognize neoantigens.40
Figure 1.

Exploratory analysis of TCGA data focused on WHSC1. (A) Kaplan-Meier plot showing disease free survival comparing patients with high versus low expression levels of WHSC1 using the TCGA PCa cohort. Patients were divided based on the top/bottom 25% gene expression, Cox HR, p value and median survival were calculated for the two groups and are shown on the plot. (B) Predicted infiltration levels of immune cells in TCGA PCa data using precomputed data from xCell. High and low (red and blue) refer to patients with the top/bottom 25% of WHSC1 expression. P value shown in figure, calculated with two-tailed Student’s t-test, n=125/group. (C) Heatmap showing HLA expression in the TCGA PCa cohorts. Red and black cells indicate upregulated and downregulated genes, respectively. Side annotation indicates the patients’ groups based on WHSC1 expression levels. (D, E) GSEA analysis comparing patients with high versus low WHSC1 levels highlighting upregulated (C) and downregulated (D) pathways in patients with elevated WHSC1 expression levels. GSEA, gene set enrichment analysis; NES, normalized enrichment score; IFN-γ, interferon gamma; NA, not applicable; PCa, prostate cancer; TCGA, The Cancer Genome Atlas; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
jitc-2020-001374supp001.pdf (61.8KB, pdf)
jitc-2020-001374supp002.txt (4.2KB, txt)
jitc-2020-001374supp003.pdf (1MB, pdf)
Immune and APM genes are transcriptionally regulated by WHSC1 in PCa
We sought to pinpoint potential mechanistic events that allow WHSC1 to regulate the expression of APM genes. To this end, we downloaded publicly available ChIPSeq data for PCa cell lines from the Cistrome database41 42 and generated a consensus list of genes residing in H3K36me2 and H3K36me3 loci as proxy for WHSC1 targets, exploiting the fact that WHSC1 can deposit both H3K36me2 and H3K36me3 marks.7 We ran differential gene expression analysis for this signature in the TCGA PCa dataset comparing patients with high versus low WHSC1 levels. A total of 5788 genes were differentially expressed, including ubiquitinases, proteases and HLAs, involved with protein and antigen processing, DNA methylatransferases and immune genes (figure 2). Notably, patients with low WHSC1 tend to present with high expression of HLA-C and HLA-F, low CD276/B7H3 and CD274/PDL1, and low DNMT1, DNMT3A and DNMT3B levels (figure 2). To validate our computational observations, we stably knocked down WHSC1 in C42 cells and noted a significant reduction in cell proliferation (online supplemental figure S2A), also observed on transient knockdown of WHSC1 in DU145 cells (online supplemental figure S2B). On WHSC1 knockdown, 3783 genes were differentially expressed (1834 down and 1949 up, false discovery rate (FDR)<0.05) (figure 3A). We first confirmed the downregulation of AR and its downstream target KLK2, indicating a transcriptional suppression of the androgen signaling (figure 3B). GSEA analysis revealed a downregulation of TGF-β signaling and upregulation of IFN-γ and tumor necrosis factor signaling (figure 3C). The APM signature, curated from the Reactome database, was also upregulated on WHSC1 knockdown (figure 3D). At the gene level, we noticed an increase in HLAs, proteinases and ubiquitinase genes, parallel to a downregulation of DNMT1 and CD276 (figure 3E), consistent with the initial computational observations. A number of differentially expressed genes (DEGs) were common with those identified integrating ChIPSeq and TCGA data, including UBL5, UBXN1, UBA52, PSME1, UBE2N, PSMC5, UBL7, UBE2E3, UBR7, UBR3 and UBE4A, suggesting that protein processing and degradation could be directly regulated by WHSC1 via methylation of the H3K36 histone mark. The degree of dissimilarity at the gene level between the predicted H3K36me2-H3K36me3 Cistrome and the DEGs following WHSC1 knockdown is likely caused by the different biological sources from which data were originated (Cistrome/cell lines vs TCGA/patient biopsies) and by the fact that multiple enzymes can methylate H3K36.43–48 These results support a mechanistic role for WHSC1 in regulating the expression of APM genes in PCa.
Figure 2.
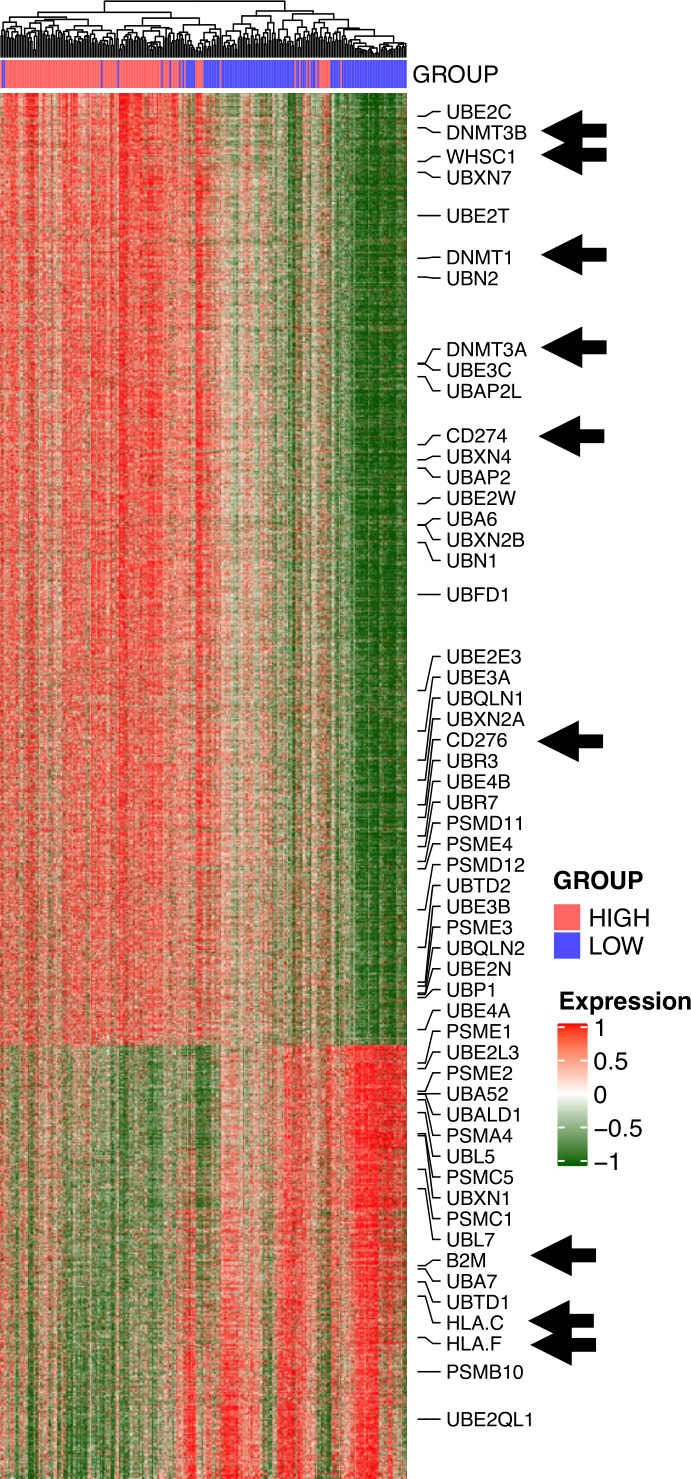
Differential gene expression analysis in TCGA data. (A) Heatmap of degS within H3K36me3 and H3K36me2 loci identified using ChIPSeq data from Cistrome and RNAseq data from TCGA. Red and green indicate high and low expression levels, respectively. Blue and red annotations indicate patients with low and high WHSC1 gene expressions, respectively. Columns are clustered using Pearson correlation, and rows are ranked based on log2FC comparing tumors with low versus high WHSC1. Black arrows indicate selected genes (DNMTs, CD274, B2M, HLAs and WHSC1). RNASeq, RNA Sequencing; TCGA, The Cancer Genome Atlas; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
Figure 3.

Transcriptional analysis of WHSC1 knockdown in C42 cells. (A) RNAseq analysis in C42 cells following shRNA knockdown of WHSC1. Red dots indicate genes with FDR<0.05 (B) Box plots showing the expression levels of Ar and KLK2 following WHSC1 knockdown, p value on figure calculated with limma, n=3/group. (C) GSEA indicating pathways downregulated and upregulated following knockdown of WHSC1. (D) GSEA analysis using a custom APM-MHC gene signature (E) Heatmap showing expression of HLAs, DNA repair genes and DNMT1 in C42 comparing knockdown versus control. Red and cyan cells indicate high and low expression levels, respectively. Black arrows highlight HLAs, DNMT1, CD276 and WHSC1 transcripts. APM, antigen processing and presentation machinery; AR, androgen receptor; GSEA, gene set enrichment analysis; IFN-γ, interferon gamma; MHC, Major Histocompatibility complex; shRNA, short hairpin RNA; TGF-β, transforming growth factor-beta; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
jitc-2020-001374supp004.pdf (1.1MB, pdf)
WHSC1 regulates protein degradation and immune components via DNA methylation
Our computational and transcriptional analyses indicate that one of the mechanisms by which elevated WHSC1 regulates target gene expression is by upregulating DNMT1, corroborating previous studies indicating a link between H3K26me2 and DNA methylation.49 To evaluate the link between DNA methylation and the expression of genes in immune pathways or APM, we performed methylomic analyses in C42 cells following WHSC1 knockdown (figure 4A, B).
Figure 4.

Methylomic data analysis following WHSC1 knockdown in C42 cells. (A) Distribution of the beta values in our methylomic analysis. (B) PCa analysis of the normalized methylation data; values were scaled and centred prior to PCa analysis. Blue and red dots indicate controls (shCTR) and knockdown (shWHSC1) samples, respectively. (C) Scatterplot showing the relationship between the changes in percentage methylation and gene expression following WHSC1 knockdown. Red dots highlight genes with negative correlation (high methylation, low expression and vice versa) that were selected (D) Heatmap visualization of the methylation intensity versus expression values, highlighting genes that belong to immune and APM pathways. For methylation data (left), orange and blue indicate high and low intensity/beta values, respectively; for RNAseq data, red and cyan indicate high and low gene expression, respectively. APM, antigen processing and presentation machinery; CTR, control; PCa, prostate cancer; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
A total of 2209 DEGs contained differentially methylated probes, and in 651 genes, changes in transcript levels negatively correlated with changes in DNA methylation on WHSC1 knockdown (figure 4C, D). In accordance with the data presented earlier, the results includes six genes involved in peptide proteosomal degradation (UBE2E1, UBE2E6, UBE2L6, UBE4A, RNF135 and PSMD8); RUNX1, which is involved in promoting class I Major Histocompatibility complex (MHC) expression50; SMAD7, which negatively regulates the immunosuppressive TGF-β signaling (reviewed in Stolfi et al51); a number of membrane trafficking proteins (SRL, RIMS1, STXBP6, and NAPB); interleukin (IL)-6R, increased during PCa development52 and in breast cancer53; INHBB, which belongs to the TGF-β family and is increased in PCa54; IL-5, which was shown to promote cancer metastases modulating the TME55 56; PARP10, which limits cell proliferation and metastases57; and CD276, which is elevated in several cancers and is a potential immunotherapeutic target.58–60 Lastly, we noticed that probes associated with the DNMT1 gene had increased methylation with a parallel reduction in DNMT1 gene expression (figure 4D). These results suggest that WHSC1 can regulate tumor-resident immune pathways and APM by altering the DNA methylation landscape in PCa.
WHSC1 epigenetically regulates genes in the APM by changing chromatin status
To further narrow the mechanistic role of WHSC1 in modulating APM genes, we performed ATACSeq analysis of C42 cells following knockdown of WHSC1. We first created a consensus peak list, compared the peak intensity between the two conditions and noticed that the biggest differences were, as expected, in the peaks for mononucleosomal or dinucleosomal regions with no differences in larger peaks (figure 5A, B). After annotating genes to the peaks, we kept those with open chromatin and increased gene expression, or vice versa, as indicated by a positive correlation between the log fold changes in ATACSeq and RNASeq (figure 5C), and performed GSEA analysis. Within the results (online supplemental file S1), we identified upregulation of genes in immune signaling and protein ubiquitination pathways (figure 5D, E). Next, we evaluated the presence of peaks at the gene level for the core enrichment of the upregulated pathways and demonstrated that increased peak magnitude at the transcriptional start site (TSS) is associated with increased gene expression (figure 5F). Interestingly, while the results between the H3K36 Cistrome, ATACSeq and DNA methylation have a modest overlap, genes consistently belong to immune and antigen processing pathways. This suggests that WHSC1 plays complementary direct and indirect roles in regulating cellular behavior by modifying chromatin accessibility and DNA methylation in cancer.
Figure 5.

ATACSeq analysis in C42 cells with stable knockdown of WHSC1. (A) ATAC seq log2FC obtained by calculating the difference in read abundance from common loci in control and knockdown cells. (B) Summary of the log2FC versus fragment length, with the red lines indicating the periodicity of the nucleosomes. (C) Correlation between log2FC in ATACSeq and RNAseq data; each dot is a gene; highlighted in purple are genes showing positive correlation between the two datasets. (D) GSEA analysis using the aforementioned data. (E) Box plots indicating gene expression in the genes involved in the upregulated pathways (n=3/group, FDR values on figure, calculated with limma) (F) ATACSeq reads for representative genes confirming increased peaks in red boxes following knockdown of WHSC1. GSEA, gene set enrichment analysis; PCa, prostate cancer; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
jitc-2020-001374supp005.txt (13.5KB, txt)
WHSC1 and DNMT1 expression reflect tumor phenotype in vivo
Following the results from computational and in vitro experiments, we sought to evaluate in vivo whether the pattern of expression of WHSC1 and companion markers was conserved. Prostate samples were isolated from TRAMP mice61 with tumors at 20/25 weeks of age, with aggressive palpable tumors (~35 weeks) and healthy wild type (WT) mice. The protein levels of WHSC1, DNMT1, H3K36me2 and CD274/PD-L1 were evaluated via western immunoblotting. CD274 was included since it is elevated in patients from TCGA with high WHSC1 levels (figure 2) and is a downstream target of IFN-γ,62 63 which is elevated on WHSC1 knockdown (figure 3C). Furthermore, CD274 has potential translational relevance as target for checkpoint inhibitor therapy. Results show that healthy WT prostates have no detectable expression of any of the aforementioned proteins while higher levels of DNMT1, WHSC1, CD274/PD-L1 and H3K36me2 are detected in the tumor samples via western blot (figure 6A). We then tested whether the effect of WHSC1 knockdown in human C42 cells was reproducible in the murine TRAMP C2 cells,17 as proxy for potential in vivo effects. Knockdown of Whsc1 via siRNA led to a significant reduction in cell proliferation (online supplemental figure S3A, B) and increased transcript levels of DNMT1 and CD274 (figure 6B).
Figure 6.

Expression of WHSC1 in murine samples. (A) Protein data from prostates isolated from WT and TRAMP mice at different stages of PCA development testing the protein levels for WHSC1, DNMT1, CD274 and H3K36me2. (B) qPCR validation for DNMT1 and CD274 following WHSC1 knockdown (n=3/group, two-tailed Student’s t-test). *P<0.05. qPCR, quantitative PCR; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
jitc-2020-001374supp006.pdf (951KB, pdf)
Pharmacological inhibition of WHSC1 upregulates MHC molecules and increases immune infiltration in vivo
Since we detected increased MHC transcript levels following WHSC1 knockdown, we investigated whether this observation can be replicated at the protein level on pharmacological inhibition of WHSC1 (with MCTP-399) in both human (C42) and murine (TRAMP C2) cell lines. The combined treatment with MCTP-39 and IFN-γ had an additive effect on upregulating HLA-B7, HLA-F, HLA-E, HLA-DQ, HLA-DM (figure 7A) and murine H2Kb I-A/I-E (figure 7B), as measured via flow cytometry analysis. We then tested whether antigen-bound MHC was also elevated on WHSC1 inhibition using ovalbumin (OVA)-overexpressing TRAMP C2 cells, and demonstrated that treatement with MCTP39 increased the OVA-bound H2Kb fraction (figure 7C, D). These results suggest that pharmacolocycal inhibition of WHSC1 increases PCa cells’ ability to present processed antigens on the cell’s surface via upregulation of MHC molecules.
Figure 7.

Effect of WHSC1 inhibition on antigen presentation. (A, B) Flow cytometry analysis of MHC expression levels on pharmacological WHSC1 inhibition and IFN-γ treatment in human C42 (A) and murine C2 (B) cells (n=3/group, one-way analysis of variance with post hoc Tukey correction). *P<0.05, **P<0.01, ***P<0.001. (C) Flow plot showing increased H2Kb-bound to OVA following treatment with MCTP39 and its quantification (right bar plot). IFN-γ, interferon gamma; OVA, ovalbumin; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
Functional immune system is needed to mediate anti-WHSC1 tumor growth
Hypothesizing that higher tumor antigen presentation would affect tumor growth and the levels of infiltrating immune cells in the tumor, we grafted C57B/6 mice with TRAMP C2 cells, administered MCTP39 (10 mg/kg) for 4 weeks intraperitoneally and evaluated tumor size weekly and T-cell infiltration at endpoint. Tumor growth was significantly reduced on treatment with MCTP39 (figure 8A) (p=0.0023), while this inhibitory effect was not observed when we grafted the same number of TRAMP C2 cells in immunocompromised NSG mice (figure 8B). Moreover, the reduction in tumor growth in C57B/6 mice was accompanied by increased H2Kb expression, increased CD8+ T-cell infiltration and reduced tumor weight (figure 8C). These results indicate that that the antitumor effect observed following Whsc1 inhibition requires the presence of a functional immune system for optimal tumor control.
Figure 8.

In vivo effect of WHSC1 inhibition on tumor growth and T-cell infiltration. (A) Growth curve of TRAMP C2 cells grafted in C57B/6 mice treated with MCTP39 for 4 weeks. Blue and red lines indicate growth in control and MCTP39-treated mice, respectively. n=6/group, p=0.0023, permutation test. (B) Growth curve of TRAMP C2 cells in NSG mice following 4 weeks treatment with MCTP39. Black and gray lines indicate growth in control and MCTP39-treated mice, respectively. P=0.1461, permutation test. (C) Quantification of flow cytometry data in tumors at endpoint evaluating, from left to right, CD8+ T-cell infiltration, H2Kb expression on the tumor and tumor weight in gram in C56B/6 mice. n=6/group, two-tailed Student’s t-test. *P<0.05, **P<0.01. NS, not significant; WHSC 1, Wolf-Hirschhorn syndrome candidate 1.
Discussion
The use of immunotherapy in PCa has been pioneered by the use of vaccines targeting the PSA (Sipuleucel T/Provenge)64 or the prostate acid phosphatases (Prostvac VF)65 that showed significant clinical benefits for patients with metastatic PCa. However, there is still an incomplete understanding of the regulatory interface that mediates the infiltration of T cells into the prostate. The relevance of filling this gap in knowledge is highlighted by negative results or minimal benefits recorded in subsequent clinical trials testing immunotherapeutic approaches in patients with PCa.66–72 Therefore, a thorough understanding of the mechanism by which PCa evades the immune system and limits T-cell infiltration could have significant translational consequences.
Here we presented an epigenetic tumor-driven mechanism by which prostate tumors remain relatively cold due to increased levels of the epigenetic enzyme WHSC1. While the role of tumor WHSC1 in PCa has been investigated by other groups,9 13 73 74 we offer a novel complementary evidence of its role in promoting immune evasion by rendering tumor cells less visible to the immune system. This is achieved by an elegant and coordinated downregulation of MHC molecules and APM genes through alteration of the chromatin status and by modifying the methylation of APM genes.
We first used bioinformatics approaches to investigate the relationship between the transcript levels of WHSC1 and genes in immune-related pathways mining publicly available data. Using RNASeq data from TCGA, we demonstrated that patients with PCa with increased WHSC1 transcript levels have shorter time to recurrence and lower expression of HLAs, and tend to have an immunosuppressive tumor microenvironment. By integrating TCGA RNASeq data with H3K36 methylation data from different PCa cell lines, as proxy for WHSC1 activity, we identified HLAs, DNMTs, ubiquitinases and proteosomal and immune genes as candidate targets of WHSC1. Following knockdown of WHSC1 in vitro, we confirmed a pattern in which HLAs; ubiquitinases and proteosomal genes were mostly upregulated, while DNMT1 and CD276 were downregulated. This pattern of expression is remarkably consistent with the TCGA/Cistrome results, suggesting that WHSC1 regulates protein degradation, antigen processing and presentation, while potentially altering DNA methylation via DNMT1. Our RNASeq results also confirmed a downregulation of AR and KLK2, suggesting that inhibition of WHSC1 in C42 cells limits AR signaling, corroborating previous studies showing increased immune infiltration in the tumor following ADT.1–3 While complementary to our study, previous studies did neither investigate nor identify the tumor-resident immune pathways that define the interface between prostate tumors and immune system. To this end, we corroborated the RNASeq results via flow cytometry analysis, demonstrating that silencing of WHSC1 increased MHC expression on PCa cells, augmenting the levels of MHC-bound antigen on the cell’s surface. Consequentially, we observed increased T-cell infiltration in grafted tumors following pharmacological inhibition of WHSC1 with MCTP39, which correlated with reduced tumor growth. Previous studies using DU145 xenografts also showed reduced tumor weight on MCTP39 treatment,9 but the use of immunocompromised mice did not allow capturing of any effect on the immune system. The TRAMP C2 cells were shown to be a reliable subcutaneous in vivo model for studying the behavior of the immune system in PCa in response to therapy.75–78 In our study, we used immunocompetent C57B/6 mice grafted with syngeneic TRAMP C2 cells and found reduced tumor weight, increased T-cell infiltration and increased MHC expression in treated tumors. The observed changes in T-cell infiltration and MHC expression are consistent with the induction of an antitumor immune response. Furthermore, we tested whether a similar magnitude in tumor reduction was observed in immunocompromised mice and found only a limited and non-significant effect of MCTP39 in delaying tumor growth. While this suggests that the immune system plays a certain role in mediating the observed antitumor effect, more studies are needed to pinpoint the exact immune populations responsible for this effect. Since NSG mice lack of T, B and NK and have a mutation in the C5 component of the complement pathway, impairing complement-dependent cytolysis, it is possible that the reduction in tumor growth observed in immunocompetent mice is the result of the coordinated action of multiple immune cell types. To this point, we demonstrate that, following WHSC1 inhibition, there is higher antigen presentation in vitro and higher T-cell infiltration in vivo. This suggests that T cells are the most likely driver of the antitumor response that we observed.
Mechanistically, we demonstrate a close relationship between WHSC1 and DNMT1 expression, suggesting a role for WHSC1 in maintaining the DNA methylation status in PCa. A similar relationship was shown between H3K36me2 and DNA methylation, where ablation of Nsd1 and Nsd2 in mouse changes the genomic localization of DNMT3A, which is redeployed intragenically, leading to reduced intergenic methylation levels.49 This happens because of changes in the methylation status of H3K36me2, which is required for efficient DNMT3A binding. While our results do not directly indicate which DNA methyltransferase enzyme drives the changes we observed, RNASeq data point to DNMT1, which is located within H3K36me2- H3K36me3 loci and is downregulated following WHSC1 knockdown. Lastly, due to the effect of WHSC1 on cellular proliferation, we acknowledge that a degree of the observed changes in the epigenome might be secondary to the reduced proliferative potential following WHSC1 inhibition.
WHSC1 casts a broad regulatory net that affects a heterogeneous panel of genes and cellular pathways. First, it appears to have complementary, yet separate, mechanisms of action that mold the epigenetic landscape of PCa cells by altering chromatin accessibility and DNA methylation. At the gene level, WHSC1 alters ubiquitinases and proteosomal genes, which mediate protein degradation and generate potentially antigenic peptides. The upregulation of immune and MHC genes following WHSC1 inhibition suggests that processed peptides can then reach the cell’s surface for recognition by the immune system. WHSC1 also controls the expression of CD274/PD-L1 and CD276. Although CD274/PD-L1 is regulated by IFN-γ following immune activation,62 63 it is also indicative of T-cell exhaustion and is overexpressed in numerous tumors favoring an immunosuppressive microenvironment,79 80 similarly to CD276.58–60 This suggests that WHSC1 has the potential to limit the antitumor immune response to the tumor microenvironment by different, yet complementary, mechanisms.
In conclusion, here we offer a novel function for WHSC1/NSD2 as key regulator of tumor-resident immune pathways, and WHSC1 pharmacological inhibition has the potential to act as a potent adjuvant for combination immunotherapy in PCa.
Footnotes
Twitter: @thethornelab
Contributors: SB conceived the project and performed the bioinformatics analyses. MYW and TT developed the ovalbumin vector and prepared transfected cells. PKS performed the ATACSeq experiment in vitro. MYW and JLT performed in vitro viability experiments. MYW and JM performed the immunological assays. MYW, EK and BG performed the in vivo experiments. SB, MYW, AL, and ECG worked on the data analysis. SB, RCK and KO developed the experimental design and wrote the paper. All authors reviewed and approved the final version of the manuscript prior to submission.
Funding: SB Roswell Park Alliance Foundation and Roswell Park Startup Funds. This work was also supported by Roswell Park Comprehensive Cancer Center and National Cancer Institute (grant P30CA016056) involving the use of the Genomic Shared Resources, Immune Analysis Shared Resources and the Gene Modulation Shared Resources.
Competing interests: None declared.
Patient consent for publication: Not required.
Ethics approval: Patients' data used in this manuscript are publicly available and may be obtained from The Cancer Genome Atlas, which offers deidentified data from participating patients.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. All sequencing data generated from the studies in this paper were submitted to Gene Expression Omnibus (GEO) and are available under the GSE153336 accession number.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
References
- 1.Gannon PO, Poisson AO, Delvoye N, et al. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J Immunol Methods 2009;348:9–17. 10.1016/j.jim.2009.06.004 [DOI] [PubMed] [Google Scholar]
- 2.Mercader M, Bodner BK, Moser MT, et al. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc Natl Acad Sci U S A 2001;98:14565–70. 10.1073/pnas.251140998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen Y-C, Ghasemzadeh A, Kochel CM, et al. Combining intratumoral Treg depletion with androgen deprivation therapy (ADT): preclinical activity in the Myc-CaP model. Prostate Cancer Prostatic Dis 2018;21:113–25. 10.1038/s41391-017-0013-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flammiger A, Weisbach L, Huland H, et al. High tissue density of Foxp3+ T cells is associated with clinical outcome in prostate cancer. Eur J Cancer 2013;49:1273–9. 10.1016/j.ejca.2012.11.035 [DOI] [PubMed] [Google Scholar]
- 5.Hu M, Sun X-J, Zhang Y-L, et al. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc Natl Acad Sci U S A 2010;107:2956–61. 10.1073/pnas.0915033107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 2012;13:115–26. 10.1038/nrm3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nimura K, Ura K, Shiratori H, et al. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature 2009;460::287-–91. 10.1038/nature08086 [DOI] [PubMed] [Google Scholar]
- 8.Li N, Xue W, Yuan H, et al. AKT-mediated stabilization of histone methyltransferase Whsc1 promotes prostate cancer metastasis. J Clin Invest 2017;127:1284–302. 10.1172/JCI91144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aytes A, Giacobbe A, Mitrofanova A, et al. Nsd2 is a conserved driver of metastatic prostate cancer progression. Nat Commun 2018;9:5201. 10.1038/s41467-018-07511-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah MY, Martinez-Garcia E, Phillip JM, et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016;35:5905–15. 10.1038/onc.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ezponda T, Popovic R, Shah MY, et al. The histone methyltransferase MMSET/WHSC1 activates Twist1 to promote an epithelial-mesenchymal transition and invasive properties of prostate cancer. Oncogene 2013;32:2882–90. 10.1038/onc.2012.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang P, Guo L, Duan ZJ, et al. Histone methyltransferase NSD2/MMSET mediates constitutive NF-κB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol Cell Biol 2012;32:3121–31. 10.1128/MCB.00204-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang H-B, Choi Y, Lee JM, et al. The histone methyltransferase, NSD2, enhances androgen receptor-mediated transcription. FEBS Lett 2009;583:1880–6. 10.1016/j.febslet.2009.05.038 [DOI] [PubMed] [Google Scholar]
- 14.Wojcik JB, Marchione DM, Sidoli S, et al. Epigenomic reordering induced by polycomb loss drives oncogenesis but leads to therapeutic vulnerabilities in malignant peripheral nerve sheath tumors. Cancer Res 2019;79:3205–19. 10.1158/0008-5472.CAN-18-3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouda R, Sarai N, Nehru V, et al. Spt6 interacts with NSD2 and facilitates interferon-induced transcription. FEBS Lett 2018;592:1681–92. 10.1002/1873-3468.13069 [DOI] [PubMed] [Google Scholar]
- 16.Zhu L, Yu C-L, Zheng Y. Nsd2 inhibition suppresses metastasis in cervical cancer by promoting TGF-β/TGF-βRI/SMADs signaling. Biochem Biophys Res Commun 2019;519:489–96. 10.1016/j.bbrc.2019.08.020 [DOI] [PubMed] [Google Scholar]
- 17.Foster BA, Gingrich JR, Kwon ED, et al. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res 1997;57:3325–30. [PubMed] [Google Scholar]
- 18.Yang J, Sanderson NSR, Wawrowsky K, et al. Kupfer-type immunological synapse characteristics do not predict anti-brain tumor cytolytic T-cell function in vivo. Proc Natl Acad Sci U S A 2010;107:4716–21. 10.1073/pnas.0911587107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsuji T, Yoneda A, Matsuzaki J, et al. Rapid construction of antitumor T-cell receptor vectors from frozen tumors for engineered T-cell therapy. Cancer Immunol Res 2018;6:594–604. 10.1158/2326-6066.CIR-17-0434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mátés L, Chuah MKL, Belay E, et al. Molecular evolution of a novel hyperactive sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 2009;41:753–61. 10.1038/ng.343 [DOI] [PubMed] [Google Scholar]
- 21.Yu G, Wang L-G, Yan G-R, et al. Dose: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics 2015;31:608–9. 10.1093/bioinformatics/btu684 [DOI] [PubMed] [Google Scholar]
- 22.Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. 10.1186/s13059-017-1349-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dobin A, Davis CA, Schlesinger F, et al. Star: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-Seq: transcript-level estimates improve gene-level inferences. F1000Res 2015;4:1521. 10.12688/f1000research.7563.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu T Use model-based analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol 2014;1150:81–95. 10.1007/978-1-4939-0512-6_4 [DOI] [PubMed] [Google Scholar]
- 29.Yu G, Wang L-G, He Q-Y. ChIPseeker: an R/Bioconductor package for CHIP peak annotation, comparison and visualization. Bioinformatics 2015;31:2382–3. 10.1093/bioinformatics/btv145 [DOI] [PubMed] [Google Scholar]
- 30.Dharmalingam GBD, Carroll T. soGGi: Visualise ChIP-Seq, MNase-seq and motif occurrence as aggregate plots summarised over grouped genomic intervals R package version 1180, 2019. [Google Scholar]
- 31.Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 2019;47:e47. 10.1093/nar/gkz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris TJ, Butcher LM, Feber A, et al. Champ: 450k CHIP analysis methylation pipeline. Bioinformatics 2014;30:428–30. 10.1093/bioinformatics/btt684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.TMTaPM G Modeling survival data: extending the COX model, 2000. [Google Scholar]
- 34.Heagerty PJ, Lumley T, Pepe MS. Time-Dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000;56:337–44. 10.1111/j.0006-341X.2000.00337.x [DOI] [PubMed] [Google Scholar]
- 35.Brockman JA, Alanee S, Vickers AJ, et al. Nomogram predicting prostate cancer-specific mortality for men with biochemical recurrence after radical prostatectomy. Eur Urol 2015;67:1160–7. 10.1016/j.eururo.2014.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.SJaG M Areas beneath the relative operating characteristics (ROC) and relative operating levels (rol) curves: statistical significance and interpretation. QJR Meteorol Soc 2006;128:2145–66. 10.1256/003590002320603584 [DOI] [Google Scholar]
- 37.Giner G, Smyth Gordon, K.. statmod: probability calculations for the inverse Gaussian distribution. R J 2016;8:339–51. 10.32614/RJ-2016-024 [DOI] [Google Scholar]
- 38.Phipson B, Smyth GK. Permutation P-values should never be zero: calculating exact P-values when permutations are randomly drawn. Stat Appl Genet Mol Biol 2010;9:39. 10.2202/1544-6115.1585 [DOI] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Research Network The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–25. 10.1016/j.cell.2015.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S, Matsuzaki J, Wei L, et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J Immunother Cancer 2019;7:156. 10.1186/s40425-019-0629-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mei S, Qin Q, Wu Q, et al. Cistrome data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res 2017;45:D658–62. 10.1093/nar/gkw983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu T, Ortiz JA, Taing L, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol 2011;12:R83. 10.1186/gb-2011-12-8-r83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rayasam GV, Wendling O, Angrand P-O, et al. Nsd1 is essential for early post-implantation development and has a catalytically active set domain. Embo J 2003;22:3153–63. 10.1093/emboj/cdg288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Trojer P, Xu C-F, et al. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem 2009;284:34283–95. 10.1074/jbc.M109.034462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka Y, Katagiri Z-I, Kawahashi K, et al. Trithorax-Group protein ASH1 methylates histone H3 lysine 36. Gene 2007;397:161–8. 10.1016/j.gene.2007.04.027 [DOI] [PubMed] [Google Scholar]
- 46.Eom GH, Kim K-B, Kim JH, et al. Histone methyltransferase SETD3 regulates muscle differentiation. J Biol Chem 2011;286:34733–42. 10.1074/jbc.M110.203307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S-H, Oshige M, Durant ST, et al. The set domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc Natl Acad Sci U S A 2005;102:18075–80. 10.1073/pnas.0503676102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown MA, Sims RJ, Gottlieb PD, et al. Identification and characterization of SMYD2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer 2006;5:26. 10.1186/1476-4598-5-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinberg DN, Papillon-Cavanagh S, Chen H, et al. The histone mark H3K36me2 recruits Dnmt3a and shapes the intergenic DNA methylation landscape. Nature 2019;573:281–6. 10.1038/s41586-019-1534-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Howcroft TK, Weissman JD, Gegonne A, et al. A T lymphocyte-specific transcription complex containing Runx1 activates MHC class I expression. J Immunol 2005;174:2106–15. 10.4049/jimmunol.174.4.2106 [DOI] [PubMed] [Google Scholar]
- 51.Stolfi C, Marafini I, De Simone V, et al. The dual role of Smad7 in the control of cancer growth and metastasis. Int J Mol Sci 2013;14:23774–90. 10.3390/ijms141223774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Azevedo A, Cunha V, Teixeira AL, et al. Il-6/Il-6R as a potential key signaling pathway in prostate cancer development. World J Clin Oncol 2011;2:384–96. 10.5306/wjco.v2.i12.384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weng Y-S, Tseng H-Y, Chen Y-A, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer 2019;18:42. 10.1186/s12943-019-0988-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hofland J, van Weerden WM, Steenbergen J, et al. Activin A stimulates AKR1C3 expression and growth in human prostate cancer. Endocrinology 2012;153:5726–34. 10.1210/en.2011-2065 [DOI] [PubMed] [Google Scholar]
- 55.Zaynagetdinov R, Sherrill TP, Gleaves LA, et al. Interleukin-5 facilitates lung metastasis by modulating the immune microenvironment. Cancer Res 2015;75:1624–34. 10.1158/0008-5472.CAN-14-2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simson L, Ellyard JI, Dent LA, et al. Regulation of carcinogenesis by IL-5 and CCL11: a potential role for eosinophils in tumor immune surveillance. J Immunol 2007;178:4222–9. 10.4049/jimmunol.178.7.4222 [DOI] [PubMed] [Google Scholar]
- 57.Zhao Y, Hu X, Wei L, et al. Parp10 suppresses tumor metastasis through regulation of Aurora A activity. Oncogene 2018;37:2921–35. 10.1038/s41388-018-0168-5 [DOI] [PubMed] [Google Scholar]
- 58.Seaman S, Zhu Z, Saha S, et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-Positive tumor cells and tumor vasculature. Cancer Cell 2017;31:501–15. 10.1016/j.ccell.2017.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Inamura K, Takazawa Y, Inoue Y, et al. Tumor B7-H3 (CD276) expression and survival in pancreatic cancer. J Clin Med 2018;7. 10.3390/jcm7070172. [Epub ahead of print: 10 Jul 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai D, Li J, Liu D, et al. Tumor-expressed B7-H3 mediates the inhibition of antitumor T-cell functions in ovarian cancer insensitive to PD-1 blockade therapy. Cell Mol Immunol 2020;17:227–36. 10.1038/s41423-019-0305-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hurwitz AA, Foster BA, Allison JP, et al. The TRAMP mouse as a model for prostate cancer. Curr Protoc Immunol 2001;Chapter 20:Unit 20.5. 10.1002/0471142735.im2005s45 [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 2019;29:3766. 10.1016/j.celrep.2019.11.113 [DOI] [PubMed] [Google Scholar]
- 63.Garcia-Diaz A, Shin DS, Moreno BH, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 2017;19:1189–201. 10.1016/j.celrep.2017.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411–22. 10.1056/NEJMoa1001294 [DOI] [PubMed] [Google Scholar]
- 65.Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol 2010;28:1099–105. 10.1200/JCO.2009.25.0597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gulley JL, Borre M, Vogelzang NJ, et al. Phase III trial of PROSTVAC in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J Clin Oncol 2019;37:1051–61. 10.1200/JCO.18.02031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beer TM, Kwon ED, Drake CG, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic Chemotherapy-Naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40–7. 10.1200/JCO.2016.69.1584 [DOI] [PubMed] [Google Scholar]
- 68.Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 2014;15:700–12. 10.1016/S1470-2045(14)70189-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Slovin SF, Higano CS, Hamid O, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol 2013;24:1813–21. 10.1093/annonc/mdt107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McNeel DG, Chen Y-H, Gulley JL, et al. Randomized phase II trial of docetaxel with or without PSA-TRICOM vaccine in patients with castrate-resistant metastatic prostate cancer: a trial of the ECOG-ACRIN cancer research Group (E1809). Hum Vaccin Immunother 2015;11:2469–74. 10.1080/21645515.2015.1062190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Twardowski P, Wong JYC, Pal SK, et al. Randomized phase II trial of sipuleucel-T immunotherapy preceded by sensitizing radiation therapy and sipuleucel-T alone in patients with metastatic castrate resistant prostate cancer. Cancer Treat Res Commun 2019;19:100116. 10.1016/j.ctarc.2018.100116 [DOI] [PubMed] [Google Scholar]
- 72.Rini BI, Weinberg V, Fong L, et al. Combination immunotherapy with prostatic acid phosphatase pulsed antigen-presenting cells (Provenge) plus bevacizumab in patients with serologic progression of prostate cancer after definitive local therapy. Cancer 2006;107:67–74. 10.1002/cncr.21956 [DOI] [PubMed] [Google Scholar]
- 73.García-Carpizo V, Sarmentero J, Han B, et al. Nsd2 contributes to oncogenic RAS-driven transcription in lung cancer cells through long-range epigenetic activation. Sci Rep 2016;6:32952. 10.1038/srep32952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuo AJ, Cheung P, Chen K, et al. Nsd2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol Cell 2011;44:609–20. 10.1016/j.molcel.2011.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poggio M, Hu T, Pai C-C, et al. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 2019;177:414–27. 10.1016/j.cell.2019.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu P, Steel JC, Zhang M, et al. Simultaneous inhibition of two regulatory T-cell subsets enhanced interleukin-15 efficacy in a prostate tumor model. Proc Natl Acad Sci U S A 2012;109:6187–92. 10.1073/pnas.1203479109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mikyšková R, Štěpánek I, Indrová M, et al. Dendritic cells pulsed with tumor cells killed by high hydrostatic pressure induce strong immune responses and display therapeutic effects both in murine TC-1 and TRAMP-C2 tumors when combined with docetaxel chemotherapy. Int J Oncol 2016;48:953–64. 10.3892/ijo.2015.3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pai C-CS, Huang JT, Lu X, et al. Clonal deletion of tumor-specific T cells by interferon-γ confers therapeutic resistance to combination immune checkpoint blockade. Immunity 2019;50:477–92. 10.1016/j.immuni.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khosravi N, Mokhtarzadeh A, Baghbanzadeh A, et al. Immune checkpoints in tumor microenvironment and their relevance to the development of cancer stem cells. Life Sci 2020;256:118005. 10.1016/j.lfs.2020.118005 [DOI] [PubMed] [Google Scholar]
- 80.Toor SM, Sasidharan Nair V, Decock J, et al. Immune checkpoints in the tumor microenvironment. Semin Cancer Biol 2020;65:1–12. 10.1016/j.semcancer.2019.06.021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2020-001374supp007.pdf (44.4KB, pdf)
jitc-2020-001374supp008.pdf (44.1KB, pdf)
jitc-2020-001374supp009.pdf (133.9KB, pdf)
jitc-2020-001374supp010.pdf (156.8KB, pdf)
jitc-2020-001374supp011.pdf (143.1KB, pdf)
jitc-2020-001374supp012.txt (88.7KB, txt)
jitc-2020-001374supp001.pdf (61.8KB, pdf)
jitc-2020-001374supp002.txt (4.2KB, txt)
jitc-2020-001374supp003.pdf (1MB, pdf)
jitc-2020-001374supp004.pdf (1.1MB, pdf)
jitc-2020-001374supp005.txt (13.5KB, txt)
jitc-2020-001374supp006.pdf (951KB, pdf)