

Summary
Interferon (IFN)-induced activation of the signal transducer and activator of transcription (STAT) family is an important event in antiviral immunity. Here, we show that the nonreceptor kinases c-Abl and Arg directly interact with STAT1 and potentiate the phosphorylation of STAT1 on Y701. c-Abl/Arg could mediate STAT1 phosphorylation independent of Janus kinases in the absence of IFNγ and potentiate IFNγ-mediated STAT1 phosphorylation. Moreover, STAT1 dimerization, nuclear translocation, and downstream gene transcription are regulated by c-Abl/Arg. c-Abl/Arg (abl1/abl2) deficiency significantly suppresses antiviral responses in vesicular stomatitis virus-infected cells. Compared to vehicle, administration of the c-Abl/Arg selective inhibitor AMN107 resulted in significantly increased mortality in mice infected with human influenza virus. Our study demonstrates that c-Abl plays an essential role in the STAT1 activation signaling pathway and provides an important approach for antiviral immunity regulation.
Subject areas: Biological Sciences, Virology, Viral Microbiology
Graphical abstract
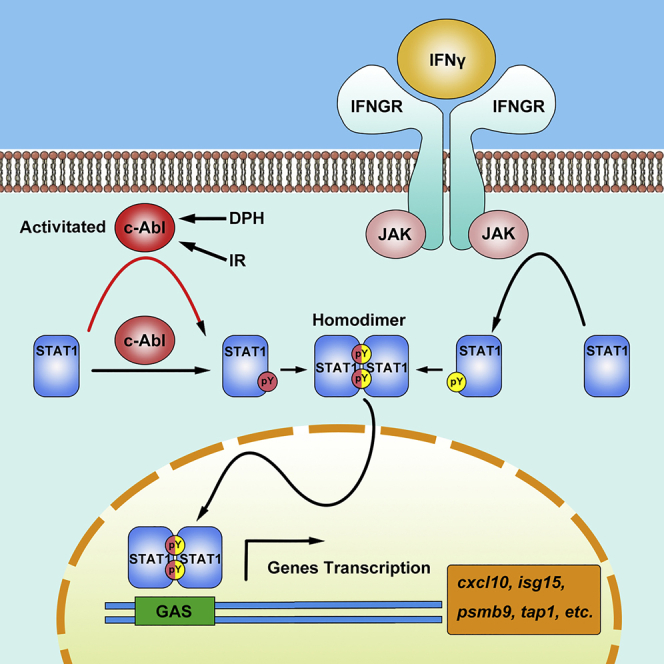
Highlights
-
•
c-Abl/Arg mediates a JAK2-independent STAT1 phosphorylation
-
•
Abl kinase potentiates IFNγ-induced STAT1 phosphorylation and activation
-
•
c-Abl-mediated STAT1 phosphorylation contributes to IFN-induced antiviral effects
Biological Sciences; Virology; Viral Microbiology
Introduction
Interferons (IFNs) play key roles in innate immunity against viral infection and in immune surveillance by regulating the activation of Janus kinases (JAKs) and signal transducer and activator of transcription (STAT) and by reprogramming cellular gene expression (Stark and Darnell, 2012). The binding of IFNs to their respective receptors results in the cross-phosphorylation of three associated JAKs (JAK1, JAK2, and TYK2) and the recruitment of STATs, allowing single tyrosine residues near the C-terminal ends of these STATs to be phosphorylated by JAKs (Platanias, 2005). The activated STATs then dimerize, migrate to the nucleus, and bind to the promoters of IFN-responsive genes to induce their transcription.
The tyrosine phosphorylation of STATs is an essential step in the JAK-STAT pathway of IFN signaling and is involved in STAT dimerization, nuclear translocation, and DNA binding (Stark and Darnell, 2012). Thus, tyrosine phosphorylation functions as an STAT activation switch. Evidence obtained through targeted disruption of JAK genes in mice suggests that JAKs are the most important STAT tyrosine kinases (Villarino et al., 2017). However, tyrosine residues in STAT1, STAT3, and STAT5 have also been shown to be phosphorylated in JAK-deficient cells by epidermal growth factor and platelet-derived growth factor receptors with intrinsic tyrosine kinase activity (Leaman et al., 1996; Vignais et al., 1996). In addition, constitutive activation of STATs has been observed in transformed cells expressing oncogenic tyrosine kinases such as v-Src, v-Abl, and BCR-Abl, but whether STATs are the direct substrates of these kinases remains to be determined (Danial and Rothman, 2000; Reddy et al., 2000).
The mammalian Abelson kinase c-Abl encoded by the c-abl (abl1) gene and Abl-related gene protein Arg encoded by the arg (abl2) gene are members of a family of intracellular nonreceptor tyrosine kinases that are required for multiple cellular processes, including proliferation, apoptosis, adhesion, cell migration, and stress responses (Bradley and Koleske, 2009; Colicelli, 2010; Greuber et al., 2013; Pendergast, 2002). The kinase activity of c-Abl is tightly regulated under normal physiological conditions. Retroviral transduction and chromosomal translocation events give rise to the expression of v-Abl or BCR-Abl, an oncogenic form of Abl, respectively (Advani and Pendergast, 2002). In addition, targeted deletion of the c-abl gene in mice results in pleiotropic phenotypes associated with immune dysfunction, including high perinatal lethality, splenic and thymic atrophy, lymphopenia, and increased susceptibility to infection (Schwartzberg et al., 1991). Moreover, treatment of BCR-Abl-positive chronic myeloid leukemia (CML) with the Abl inhibitors STI571 (imatinib) and AMN107 (nilotinib) is associated with immunosuppression in a subset of patients (Dietz et al., 2004; Hochhaus et al., 2016; Mattiuzzi et al., 2003). Previous studies have demonstrated that c-Abl plays a role in the regulation of TCR (T cell receptor)- and BCR (B cell receptor)-mediated signal transduction, development, proliferation, and cytokine production (Gu et al., 2009; Pendergast, 2002; Silberman et al., 2008). Abl-deficient mice also exhibit reduced numbers of T/B cells (Gu et al., 2009; Liberatore and Goff, 2009; Schwartzberg et al., 1991). However, the mechanisms underlying the impaired immunity that arise in the absence or inhibition of c-Abl remain elusive.
It has been widely reported that the JAK-STAT signaling pathway is essential for BCR-Abl-induced transformation (Carlesso et al., 1996; Danial et al., 1998; de Groot et al., 1999). Therefore, it is worth investigating whether JAK-STAT can also contribute to c-Abl-involved innate immunity. Here, we report that STAT1 interacts with and is phosphorylated by c-Abl in a JAK-independent manner, by which the dimerization, nuclear localization, and downstream gene transcription of STAT1 are regulated. These findings revealed that, in addition to JAK kinase, c-Abl is indispensable for the full activation of STAT1 and the subsequent IFN-mediated antiviral responses.
Results
The transcription of interferon gamma-activated sequence downstream genes is regulated by Abl kinase
Our previous study suggested that c-Abl widely participates in the regulation of gene transcription (Dong et al., 2017). To illustrate the role of c-Abl in gene expression, the transcription of approximately 22,000 genes in wild-type (WT) and c-abl−/−arg−/− MEFs (mouse embryonic fibroblasts) was detected using Affymetrix GeneChips (Mouse Genome 430 2.0) (GEO accession: GSE154568). A total of 1,744 differentially expressed genes (DEGs) with fold changes >4 were identified in c-abl/arg double-knockout (DKO) and WT MEFs, of which 960 were upregulated and 784 were downregulated (Figure 1A, left). To further investigate the potential cellular functions and associated pathways of these DEGs, we performed Gene Ontology (GO) analysis (for terms in the biological process, molecular function, and cellular component) (Figure S1A). The genes downregulated by c-Abl and Arg were mainly associated with virus defense responses (GO: 0009615, GO: 0051607, and GO: 0045071) and immune responses (GO: 0002376, GO: 0045087). Further analysis showed that IFN-specific genes (Liu et al., 2012) were enriched among the downregulated genes (Figures 1A, right, and S1B).
Figure 1.

c-Abl regulates the promoters of IFN-responsive genes via GAS elements
(A) The differentially expressed genes were identified by RNA sequencing in c-abl/arg double-knockout (DKO) and WT MEFs. Left, the volcano plot of the c-Abl-regulated differentially expressed genes with >4-fold change. Right, the downregulation of IFN-related genes in DKO MEFs compared with that in WT MEFs. c-abl/arg double-knockout efficiency was detected in Figure S3A.
(B) The transcription levels of the indicated genes were determined via quantitative RT-PCR. The data were normalized to the value of psma5 to exclude nonspecific regulation due to adenoviral infection.
(C) A schematic diagram shows that the psmb9 and tap1 genes are transcribed from a shared bidirectional promoter and regulated by IFNγ.
(D) The transcription of tap1 and psmb9 genes was detected by luciferase reporter constructs in RAW264.7 cells with or without AMN107 treatment. All quantitative data are shown as the mean ± SD of three independent experiments (unpaired Student's t-test). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To validate the differential expression profiles, the transcript levels of antiviral genes, including cxcl10, psmb9, tap1, and isg15, were determined via quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) assays and normalized to the levels of psma5, which is not highly regulated by c-Abl. Compared with WT MEFs, c-abl−/−arg−/− MEFs exhibited significantly lower transcript levels of these genes, but the DKO-induced reductions were completely reversed by c-Abl rescue in a dose-dependent manner (Figures 1B and S1C). These data suggested that a panel of IFN-induced genes were regulated by c-Abl and Arg.
To investigate the key transcriptional elements regulated by Abl kinases in response to IFN, a luciferase reporter system was constructed based on the tap1/psmb9 shared bidirectional promoter regulated by IFNγ (Figure 1C). As expected, the c-Abl/Arg-selective inhibitor AMN107 significantly inhibited the transcriptional activity of the tap1 promoter (red) (Figure 1D). Notably, unlike deletion of NFκB-acting (blue) or IFN consensus sequence (yellow) elements, AMN107 treatment had little if any effect on tap1 promoter activity when the gamma-activated sequence (GAS) element (green) was deleted (Figure 1D). These findings indicate that the STAT1-targeted GAS cis-element is involved in c-Abl-regulated tap1 transcription, which strongly suggests that STAT1 is responsible for c-Abl-mediated regulation of IFN-responsive gene expression.
c-Abl directly interacts with STAT1
IFN-induced TAP1 expression is regulated by c-Abl, possibly through STAT1, suggesting that STAT1 may be associated with c-Abl. To confirm this hypothesis, lysates of MCF-7 cells were subjected to anti-c-Abl immunoprecipitation followed by immunoblotting with an anti-STAT1 antibody. STAT1 was present in anti-c-Abl but not IgG immunoprecipitates (Figure 2A). Next, 293T cells were cotransfected with Myc-c-Abl and Flag-STAT1 or Flag-Vector as a control. The presence of Myc-c-Abl in anti-Flag immunoprecipitates prepared from cells coexpressing Flag-STAT1 but not Flag-Vector showed an association between ectopically expressed Myc-c-Abl and Flag-STAT1 (Figure 2B, left). A similar result was also obtained in a reciprocal experiment (Figure 2B, right). Moreover, the other member of the Abl family, Arg (Abl2), which is highly homologous to c-Abl in its N-terminal domain (NTD), also interacted with STAT1 (Figure S2A).
Figure 2.

c-Abl interacts with STAT1
(A) Lysates from MCF-7 cells were subjected to immunoprecipitation with anti-c-Abl or normal rabbit IgG as a control and subsequently analyzed by immunoblotting with the indicated antibodies.
(B) 293T cells were transfected with the indicated plasmids. Anti-Flag or IgG immunoprecipitates were analyzed by immunoblotting with the indicated antibodies.
(C) Lysates from 293T cells transfected with the Flag-c-Abl expression plasmid were incubated with GST or GST-STAT1 fusion protein-conjugated agarose beads. The adsorbates were analyzed by immunoblotting and Coomassie blue staining.
(D) Anti-Flag or IgG immunoprecipitates prepared from 293T cells transfected with the indicated plasmids were subjected to SDS-PAGE and transferred onto a PVDF membrane. The PVDF membrane was incubated with soluble GST-STAT1 or GST only and then detected with an anti-GST antibody.
(E) Upper panel, schematic diagram of the STAT1 domain. Lower panel, 293T cells expressing Flag-c-Abl were incubated with the indicated GST-STAT1 truncated mutants or GST-conjugated agarose beads. The adsorbates were analyzed by anti-Flag immunoblotting.
(F) The interaction between Flag-STAT1 and GST-Abl SH3, GST-Abl SH2, and GST only was detected by far-Western blotting as described in (D).
(G) MEFs were stimulated with IFNγ at different concentrations for 3 hr. The complexes of endogenous c-Abl and STAT1 were detected by in situ PLA using anti-c-Abl and anti-STAT1 antibodies. PLA signals were shown in red, and nuclei were stained with DAPI (blue). Scale bars, 10 μm.
Next, lysates prepared from 293T cells expressing Flag-c-Abl were incubated with agarose-conjugated GST-STAT1 or GST alone, and Flag-c-Abl was detected in the GST-STAT1 but not the GST adsorbates (Figure 2C). To rule out indirect binding mediated by other components in the cell lysates, anti-Flag immunoprecipitates prepared from 293T cells expressing Flag-c-Abl were subjected to SDS-PAGE (sodium dodecyl sulphate–polyacrylamide gel electrophoresis), and the proteins were transferred to a PVDF (polyvinylidene fluoride) membrane and then blotted with soluble GST-STAT1 or GST (as a control). The results showed that GST-STAT1, but not GST, bound c-Abl directly in vitro (Figure 2D).
To define the c-Abl-binding domain, agarose-conjugated and GST-fused full length or truncated STAT1 (Figure 2E, upper panel) were incubated with the lysates of 293T cells expressing Flag-c-Abl. Analysis of the adsorbates by immunoblotting with an anti-Flag antibody demonstrated that amino acids 577–750 of STAT1, which compose a region that contains the SH2 domain and transactivation domain, were responsible for the c-Abl interaction (Figure 2E, lower panel). Similarly, Flag-STAT1 immunoprecipitated by anti-Flag antibody was fractioned by SDS-PAGE and was transferred onto a PVDF membrane. Then, the membrane was blotted with a soluble GST-c-Abl SH3 or GST-c-Abl SH2 and GST only. The result showed that c-Abl SH3 domain was the major domain responsible for STAT1 association (Figure 2F).
Furthermore, the in situ interaction of endogenous c-Abl with STAT1 was assessed by Duolink proximity ligation assay (PLA). STAT1:c-Abl complexes were observed mainly in the cytoplasm, and the formation of these complexes was substantially potentiated by IFNγ stimulation (Figure 2G). Collectively, these findings demonstrate a direct association between c-Abl and STAT1 both in vitro and in vivo.
c-Abl mediates JAK-independent STAT1 phosphorylation
To investigate whether STAT1 is a substrate of c-Abl, purified His-tagged STAT1 was incubated with recombinant c-Abl (containing the catalytic domain at amino acids 237–643) in the presence of ATP for an in vitro kinase assay. Immunoblotting of the reaction product with anti-phospho-tyrosine and anti-phospho-STAT1 Y701 demonstrated that STAT1 was directly phosphorylated by c-Abl at tyrosine residue(s), including Y701, in vitro (Figure 3A). Next, we found that Flag-STAT1 expressed in 293T cells was tyrosine phosphorylated, especially on residue Y701, by Myc-c-Abl but not the kinase-inactive mutant Myc-c-Abl (K290R). Furthermore, upon treatment with the c-Abl/Arg-selective inhibitor AMN107, c-Abl-mediated STAT1 phosphorylation was nearly eliminated, which indicates that the tyrosine phosphorylation of STAT1 is dependent on c-Abl kinase activity (Figure 3B). c-Abl-mediated STAT1 phosphorylation was substantially impaired when STAT1 Y701 was substituted with phenylalanine, indicating that Y701 is the major phosphosite of Abl (Figure 3C). Moreover, STAT1 was also phosphorylated by Arg (Figure S2B).
Figure 3.

STAT1 is phosphorylated by c-Abl
(A) The immunoprecipitates prepared from 293T cells expressing His-STAT1 or His only were incubated with 10 ng of human Abl protein (Merck, 14–529) in the presence of 0.2 mM ATP. The reaction products were analyzed with the indicated antibodies.
(B and C) Anti-Flag or IgG immunoprecipitates prepared from 293T cells transfected with the indicated plasmids were analyzed by immunoblotting.
(D and E) Lysates of MCF-7 cells subjected to the indicated treatment were analyzed by immunoblotting.
(F) Lysates from JAK2-deficient or wild-type MCF-7 cells expressing the indicated plasmids were analyzed by immunoblotting.
(G) Wild-type or c-abl/arg-knockout MEFs were stimulated with IFNγ at 40 ng/mL for the indicated time (left) or with IFNγ at the indicated concentrations for 120 min (right). Lysates were analyzed by immunoblotting.
To further delineate the specific STAT1 residues phosphorylated by c-Abl, Flag-STAT1 coexpressed with Myc-c-Abl was subjected to liquid chromatography coupled with tandem mass spectrometry analysis. In addition to the well-defined and functionally important STAT1 phosphosite Y701 (Schindler et al., 1992; Shuai et al., 1993), two other phosphosites, Y106 and Y665, were also identified (Figure S4A). Compared with WT STAT1, both Y106F and Y701F mutants showed compromised tyrosine phosphorylation (Figure 3C). However, the phosphorylation of STAT1 Y701 was not considerably affected by the phosphorylation of Y106 and Y665, indicating that Y106 and Y665 may play distinct roles (Figure S4B). Together, these findings indicate that tyrosine residues of STAT1, including the previously reported residue Y701, can be phosphorylated by Abl family kinases.
JAKs are primarily responsible for IFN-induced STAT1 Y701 phosphorylation (Villarino et al., 2017). As expected, STAT1 Y701 phosphorylation was induced by IFNγ and was significantly inhibited by ruxolitinib, a bispecific inhibitor of JAK1 and JAK2 (Figure 3D). Notably, a compromised Y701 phosphorylation was also observed upon Abl inhibition (Figure 3D, lane 4), indicating that c-Abl partially contributes to IFNγ-induced STAT1 activation. Further, STAT1 Y701 phosphorylation was also induced by DPH, a cell-permeable c-Abl activator, to a lesser extent than by IFNγ, which could be completely reversed by AMN107 (Figure 3E). To confirm DPH-induced Abl activation, c-Abl Y412 phosphorylation, an indicator of Abl activation, was also detected (Figure 3E). It has been demonstrated that JAK2 activation occurs first and is needed for the subsequent activation of JAK1 (Briscoe et al., 1996). To exclude the effect of JAK2 on STAT1 activation, we established a jak2-KO MCF-7 cell line via CRISPR. Decreased but detectable STAT1 Y701 phosphorylation was observed in jak2-KO MCF-7 cells compared to that in parental MCF-7 cells, which could be similarly potentiated in the presence of Myc-c-Abl but not Myc-c-Abl (K290R) (Figure 3F) and was further diminished after AMN107 treatment. In c-abl−/−arg−/− cells, IFNγ-induced phosphorylation of STAT1 at Y701 was greatly compromised in a time- and dose-dependent manner (Figure 3G). All these observations demonstrate that c-Abl kinase could directly phosphorylate and activate STAT1 independent of the IFNγ-JAK signaling pathway and that, importantly, these two kinases may have synergistic effects on STAT1 phosphorylation at Y701.
c-Abl-mediated phosphorylation regulates STAT1 transactivation activity
The tyrosine phosphorylation of STATs is an essential step in the JAK-STAT pathway of IFN signaling and is involved in STAT dimerization, nuclear translocation, and DNA binding. To investigate if c-Abl-mediated phosphorylation regulates STAT dimerization, GFP-STAT1 and Flag-STAT1 were coexpressed in 293T cells in the presence or absence of Myc-c-Abl. The levels of GFP-STAT1 in anti-Flag immunoprecipitates were examined to indicate STAT1 dimerization. As shown in Figure 4A, coexpression of c-Abl potentiated the formation of STAT1-STAT1 homodimers. Immunofluorescence microscopy of MCF-7 cells further demonstrated that ablation of c-abl/arg or treatment with AMN107 significantly impaired IFNγ-induced STAT1 nuclear translocation (Figures 4B and 4C).
Figure 4.

c-Abl promotes STAT1 dimer formation and nuclear import
(A) Anti-Flag or IgG immunoprecipitates prepared from 293T cells transfected with the indicated plasmids were analyzed by immunoblotting.
(B) In situ cellular localization of endogenous STAT1 (green) in wild-type and c-abl/arg-knockout MCF-7 cells was detected by anti-STAT1 immunofluorescence microscopy. Nuclei were stained with DAPI (blue). c-abl/arg double-knockout efficiency was detected in Figure S3B.
(C) The nuclear level of STAT1 in Figure 4B was calculated by the ratio of fluorescence intensity in the nucleus to that in the cytoplasm in the same cell. At least 15 cells were calculated, and the results are expressed as the mean ± SD.
(D) Nuclear extracts isolated from 293T cells transfected with the indicated plasmids were incubated with a biotin-tagged nucleic acid probe containing the IRF1 promoter sequence, resolved via native PAGE, and analyzed with ECL. A 1000-fold molar excess of unlabeled oligonucleotide was used as a DNA-binding competitor. The efficiency of nuclear extract isolation was shown in Figure S5B. All quantitative data are shown as the mean ± SD of three independent experiments (unpaired Student's t-test). ∗∗p < 0.01.
Furthermore, electromobility shift assays were employed to analyze the promoter binding activity of STAT1. A biotin-tagged IRF1 promoter region containing the STAT1 binding consensus sequence was used as the detection probe (Aaronson and Horvath, 2002). This probe was incubated with nuclear extracts from 293T cells exogenously expressing STAT1 with or without c-Abl, which was balanced basing on Figure S5A. As shown in Figure 4D, numerous IRF1 probe-bound complexes containing STAT1 were observed in nuclear extracts (Figure 4D, lane 1). Complex formation increased slightly with IFNγ treatment (Figure 4D, lane 2) and increased significantly in cells ectopically expressing c-Abl (Figure 4D, lane 3). However, compared with WT STAT1, c-Abl showed little if any effect on promoter binding of STAT1 harboring Y701F (Figure 4D, lane 7 and 8), while Y106F and Y665F mutations showed nearly no difference with WT STAT1. The specificity of the complexes containing STAT1 and the detection probe was confirmed upon the addition of an untagged competitor probe (Figure 4D, lane 9). Moreover, the addition of an anti-STAT1 antibody resulted in the formation of a supershifted band (Figure 4D, lane 11). When the tagged probes were incubated with nuclear extracts of 293T cells as a control, no complexes were observed (Figure 4D, lane 10). These results demonstrate that c-Abl-mediated STAT1 Y701 phosphorylation potentiates the binding of STAT1 with its target promoters.
Then, IFNγ-induced transactivation of major STAT1-regulated genes, including ccl5, cxcl10, cxcl11, gbp2, ido1, ifi35, irf1, isg15, oasl, psmb9, and tap1, was assessed in WT or c-abl/arg DKO MCF-7 cells via RT-PCR. As expected, gene transcription was significantly impaired by c-abl/arg depletion (Figure 5A). Moreover, genes typically induced by IFNγ and IFNα failed to be stimulated by IFNs in the presence of AMN107 (Figures 5B and 5C). These findings demonstrate that the transactivation activity of STAT1 is positively regulated by c-Abl.
Figure 5.

c-Abl regulates IFN-related gene expression
The mRNA transcript level of IFN-induced genes was analyzed by RT-PCR. IFN-induced gene expression was measured by the ratio of specific gene transcription with IFN treatment to that without IFN treatment.
(A) Wild-type or c-abl/arg-knockout MCF-7 cells were treated with 40 ng/mL IFNγ for 6 hr
(B and C) MCF-7 cells were treated with or without 5 μM AMN107 for 24 hr and then subjected to 40 ng/mL IFNγ (B) or 1000 U/ml IFNα (C) stimulation for 6 hr. All quantitative data are shown as the mean ± SD of three technical experiments (unpaired Student's t-test). ∗∗p < 0.01.
c-Abl promotes IFN-dependent antiviral effects
TAP1 and PSMB9 are involved in the production and presentation of peptides in the MHC class I antigen-processing pathway (Ghannam et al., 2014; Vitale et al., 1998). In accordance with previous finding, the antigen presentation of ovalbumin by JAWS II cells to B3Z cells was significantly suppressed by AMN107 because of the reduced IL2 production (Figure 6A). To further assess the antiviral biological consequences of STAT1 regulation by c-Abl, WT and c-abl−/−arg−/− MEFs pretreated with or without serially diluted IFNγ were infected with vesicular stomatitis virus (VSV). VSV infection caused more severe cell death in c-abl−/−arg−/− MEFs and WT cells treated with AMN107 than in WT MEFs treated with vehicles (Figure 6B). IFNγ treatment notably decreased cell death induced by viral infection, but it had a far less pronounced effect in the presence of AMN107. Consistent with this finding, c-abl/arg ablation in MEFs resulted in insensitivity to IFN stimulation, but this effect was notably reversed by c-Abl rescue (Figure 6B). Next, A549 cells treated with or without AMN107 were infected with Newcastle disease virus (NDV) expressing GFP, and the results revealed that viral replication was also potentiated by AMN107 treatment but suppressed by c-Abl expression (Figure 6C). In accordance with the finding that DNA transfection leads to activation of the endogenous IFN response (Park et al., 2003), transfection with the pcDNA vector inhibited viral proliferation. Notably, transfection with Flag-c-Abl resulted in a more pronounced inhibitory effect on viral proliferation than transfection with an empty vector (Figure 6C). Furthermore, IFNγ treatment had little, if any, effect on viral infection in the presence of the c-Abl-selective inhibitor AMN107 (Figure 6D). These data suggest that c-Abl plays important roles in STAT1-mediated antiviral effects.
Figure 6.

c-Abl promotes IFN-dependent antiviral effects
(A) Antigen presentation by JAWS II dendritic cells was analyzed by B3Z-secreted IL2 levels.
(B) Wild-type and c-abl/arg double-knockout MEFs infected with Ad-Vector or Ad-Abl or treated with or without 5 μM AMN107 were infected with VSV (MOI = 10) for 48 hr. Cell survival was then analyzed by MTS assay.
(C) A549 cells were treated with 5 μM AMN107 for 18 hr or transfected with Flag-c-Abl for 24 hr and samples treated with DMSO or empty vector served as controls. Then, the cells were infected with NDV-GFP (MOI = 1) for 24 hr, and GFP fluorescence was quantified by flow cytometry analysis (left) and is shown as the geometric mean ± SD of three independent measurements (right).
(D) AMN107 or ruxolitinib-treated A549 cells were stimulated with IFNγ for 24 hr. Then, the cells were infected with NDV-GFP for 24 hr, and GFP fluorescence was analyzed by FACS.
(E) Survival analysis of mice following H1N1 infection. 8- to 10-week-old WT mice and c-abl-cKO mice (10 in each group) intravenously treated with AMN107 or saline for 12 days were intranasally infected with H1N1 at 0.1 LD50, and mortality was monitored. All quantitative data are shown as the mean ± SD of three independent experiments (unpaired Student's t-test or log rank test). ∗∗p < 0.01.
We next investigated the roles of c-Abl in viral infection in c-ablfl/fl,Lck-Cre (c-abl-conditional knockout, c-abl-cKO) mice, in which c-abl was specifically knocked out in thymocytes (Figure S7A), since germline knockout of c-abl gene results in runting and death within the first two weeks after birth (Koleske et al., 1998; Schwartzberg et al., 1991).
Then, WT and c-abl-cKO mice were intranasally infected with influenza A virus (IAV). A statistically nonsignificant higher mortality was observed among c-abl-cKO mice than among WT mice. WT mice continuously administered AMN107, which systematically suppresses c-Abl/Arg kinases in all tissues, demonstrated significantly increased sensitivity to IAV (Figure 6E). Collectively, these findings indicate that c-Abl is required for antiviral immunity in animals.
Discussion
IFNγ is one of the most important immune-modulating cytokines and plays vital roles in innate immunity against viral infection. In canonical IFNγ signaling, upon IFNγ stimulation, JAK1 and JAK2 are recruited to the cytoplasmic tails of aggregated IFNγ receptors (IFNGRs) and activated through sequential autophosphorylation and transphosphorylation events (Stark, 2007; Villarino et al., 2017). Then, STAT1 docks to IFNGR by the association of its SH2 domain with a recognition sequence in IFNGR (Y440DKPH444), within which Y440 is phosphorylated by JAKs (Greenlund et al., 1995). Following its phosphorylation at Y701 by JAKs, STAT1 dimerizes and translocates into the nucleus, where it promotes the transcription of an array of IFNγ-responsive genes (Aaronson and Horvath, 2002; Stark and Darnell, 2012). Stat1 ablation in mice results in an absence of transcriptional responses to IFN and a lack of IFN-induced antimicrobial and antiviral activities in cells (Meraz et al., 1996). Stat1−/− mice also demonstrate increased susceptibility to the development of spontaneous and chemical (3-methylcholanthrene)-induced tumors (Kaplan et al., 1998) and hypersensitivity to certain inflammatory pathologies (Bettelli et al., 2004; Villarino et al., 2010). Moreover, STAT1 signaling shields T cells from natural killer cell-mediated cytotoxicity (Kang et al., 2019). These findings suggest that STAT1 mediates innate immune activation and tumor immunity.
Schlatterer et al. reported that c-Abl, but not Arg, could induce neuronal loss by prompting STAT1 activation and interferon production. However, the exact mechanism responsible for c-Abl-dependent STAT1 activation has not been elucidated (Schlatterer et al., 2012). Here, we show that c-Abl binds and phosphorylates STAT1 and interacts with STAT2 directly in vitro (Figures 2 and 3, S8A, S9A, and S10A). STAT1 Y701 is exclusively phosphorylated by JAK kinases, which regulates STAT1 dimer formation (Schindler et al., 1992; Shuai et al., 1993) and nucleocytoplasmic shuttling during IFNγ signaling (Mao et al., 2005; Mertens et al., 2006; Zhong et al., 2005). Our study unexpectedly found that c-Abl, another kinase other than JAKs, also contributes to STAT1 Y701 phosphorylation independently (Figure 3). Although c-Abl-mediated Y701 phosphorylation is not as power as that of JAKs, it seems to be necessary for Y701 to reach maximal phosphorylation since seriously impaired Y701 phosphorylation was observed in c-abl−/−arg−/− cells upon IFNγ stimulus, and IFNγ-induced STAT1 Y701 phosphorylation was inhibited by AMN107, from 1.25 μM to 5 μM, in a dose-dependent manner (Figures 3G and S11A). However, Y701 phosphorylation is not much affected by the phosphorylation state of Y106 and Y665, the other two phosphosites of c-Abl, indicating that potentiated STAT1 Y701 phosphorylation does not result from c-Abl-mediated multiple-site phosphorylation of STAT1 but is possibly attributed to enhanced JAK activity. Previous studies have shown that constitutive activation of JAK1 and variable activation of JAK2 have been observed in BCR-Abl-expressing cells (Chai et al., 1997; Henderson et al., 1997; Shuai et al., 1996). Moreover, BCR-Abl has been shown to modestly activate STAT1 and STAT2 through the tyrosine phosphorylation of JAK1, JAK2, and JAK3 (Henderson et al., 1997). Current evidence supports that Abl and JAKs may have pronounced synergistic effects on STAT1 phosphorylation at Y701 (Figure 3E).
In addition to Y701, the phosphorylation of Y106 and Y665 was identified simultaneously (Figures 3C and S4A). These phosphosites were not involved in STAT1 Y701 phosphorylation or STAT1 homodimerization (Figures S4B and S4C). However, Murphy and colleagues found that the interaction between the NTDs (N-terminal protein interaction domain, amino acids 1–136) of monomers is necessary for the dimerization of nonphosphorylated full-length STAT molecules (Ota et al., 2004). STAT1 mutants (F77A and/or L78A) fail to form nonphosphorylated dimers, and these mutants exhibit phenotype-persistent phosphorylation in IFN-treated cells and resistance to TC45-mediated dephosphorylation in vitro (Mertens et al., 2006). Y106 in the NTD, which is near F77 and L78, may be responsible for maintaining STAT1 in a phosphorylated dimer state and for maintaining STAT1 in the nucleus under ionizing radiation (Figures S6A and S6B). Moreover, the SH2 domain interacts with the phosphorylated tyrosine-containing domain when phosphorylated homodimer formation is induced by IFN (Shuai et al., 1994). Y665, which lies in the SH2 domain, may affect dimerization. The functions of c-Abl-mediated phosphorylation sites (Y106 and Y665) require further investigation.
Similar to JAKs, c-Abl-mediated phosphorylation regulates STAT1 dimer formation (including STAT1-STAT1 homodimer and STAT1-STAT2 heterodimer formation (Figures 4A, S12A, and S12B)), nuclear translocation, DNA binding, and transcription of IFN-stimulated genes. Abl disruption contributed to potentiated viral proliferation and increased virus-induced cell death (Figures 6B, 6C, and 6D). Moreover, we found that ionizing radiation (IR) induced STAT1 nuclear translocation in the absence of the c-Abl inhibitor (Figures S6A and S6B). IR activates c-Abl directly (Pendergast, 2002) and subsequently STAT1-dependent IFNγ signaling, which may provide an underlying mechanism by which radiation therapy initiates an IFN-cascading innate and adaptive immune attack on the tumor (Burnette et al., 2011) and offers a stronger innate immune status in response to IR stimuli. Compared to WT littermates, mice in which c-abl was conditionally knocked out in thymocytes showed higher but not statistically significant mortality. Notably, mice systematically administered the c-Abl/Arg inhibitor AMN107 had higher mortality (Figure 6E), which indicates that c-Abl expression in a variety of tissues contributed to antiviral immunity in animals. From these data, we suggest that endogenous Abl offers latent basal resistance to viral infection and that activated Abl stimulated by ionizing radiation shields cells.
Abl kinases are constitutively activated in most patients with CML. The Abl kinase inhibitors STI571 and AMN107 represent the frontline treatment for CML therapy. However, upper respiratory tract infection and immunosuppression are commonly observed, which may be caused by the repression of the STAT-mediated immune regulatory pathway (Hochhaus et al., 2016; Mattiuzzi et al., 2003). Our findings added a theoretical foundation to optimize the therapy.
In summary, the tyrosine kinase c-Abl was found to associate with STAT1 in vivo and in vitro, to promote STAT1 phosphorylation at Y701 independent of JAKs, to potentiate transactivation, and to mediate innate immune responses against infection, particularly in the context of c-Abl-related stress. Our finding provides a supplementary approach for STAT1 activation, which contributes to the growing body of knowledge regarding the regulation of IFN downstream signaling pathway.
Limitations of the study
In this study, we indicated that c-Abl-mediated STAT1 phosphorylation contributed to IFN-induced antiviral effects. However, the mechanism of IFN-induced Abl activation, which was not reported previously, and the cross-talk between Abl and JAK2 under IFN simulation still need to be addressed more clearly. Although Abl kinase activity is obviously involved in STAT1-mediated antiviral effects in cytologic analysis as shown in Figure 6, the effect of JAK2 in this process could not be completely excluded. The similar situation should also be considered when understanding the result of animal experiments. In addition, the exact role of Abl-STAT1 axis in antiviral innate immunity should also be evaluated more specifically to exclude the involvement of other regulator that might be similarly targeted by Abl activation, considering the wide range of Abl substrates. Besides STAT1 Y701, the other phosphosites, such as Y106 and Y665, were also identified in this study, the potential function of which is worthy to be further investigated.
Resource availability
Lead contact
Xuan Liu, PhD, Beijing Institute of Biotechnology, Beijing 100850, China; email: liux931932@163.com.
Material availability
This study did not generate any new unique reagents and/or materials.
Data and code availability
The profile of gene transcription in WT and abl/arg knockout MEFs (GEO accession numbers: GSE154568) can be accessed on GEO (Gene Expression Omnibus).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This investigation was supported by grant 31170854 awarded by the Natural Science Foundation of China. We thank George Stark (Cleveland, OH) for the parental 2C4 and γ2A cell lines; Dr. Yuwei Gao (Institute of Military Veterinary) for NDV-GFP, the mouse-adapted UI182 virus derived from A/Changchun/01/2009 (H1N1) and the B3Z hybridoma cell line; Dr. Xiao Yang (Beijing Proteome Research Center) for the Lck-Cre mice; and Dr. Shang Gao (Department of Pharmacology, University of Illinois at Chicago) for making the bioinformatics charts.
Author contributions
H.L. and Y.C. performed the experiment and data analysis. H.L., Y.C., C.C., and X.L. designed the experiments. Y.B., Y.F., T.G., G.W., L.Z., Q.D., S.Z., Y.Y., C.S., and X.N. assisted in some experiments. Y.J. and P.L. developed protocols and provided reagents. H.L. wrote the manuscript; C.C. and X.L. revised the manuscript. All authors read and approved the final submission.
Declaration of interests
The authors declare no competing interests.
Published: February 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102078.
Contributor Information
Cheng Cao, Email: caoc@nic.bmi.ac.cn.
Xuan Liu, Email: liux931932@163.com.
Supplemental information
References
- Aaronson D.S., Horvath C.M. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Advani A.S., Pendergast A.M. Bcr-Abl variants: biological and clinical aspects. Leuk. Res. 2002;26:713–720. doi: 10.1016/s0145-2126(01)00197-7. [DOI] [PubMed] [Google Scholar]
- Bettelli E., Sullivan B., Szabo S.J., Sobel R.A., Glimcher L.H., Kuchroo V.K. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley W.D., Koleske A.J. Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J. Cell Sci. 2009;122:3441–3454. doi: 10.1242/jcs.039859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J., Rogers N.C., Witthuhn B.A., Watling D., Harpur A.G., Wilks A.F., Stark G.R., Ihle J.N., Kerr I.M. Kinase-negative mutants of JAK1 can sustain interferon-gamma-inducible gene expression but not an antiviral state. EMBO J. 1996;15:799–809. [PMC free article] [PubMed] [Google Scholar]
- Burnette B.C., Liang H., Lee Y., Chlewicki L., Khodarev N.N., Weichselbaum R.R., Fu Y.X., Auh S.L. The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 2011;71:2488–2496. doi: 10.1158/0008-5472.CAN-10-2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlesso N., Frank D.A., Griffin J.D. Tyrosyl phosphorylation and DNA binding activity of signal transducers and activators of transcription (STAT) proteins in hematopoietic cell lines transformed by Bcr/Abl. J. Exp. Med. 1996;183:811–820. doi: 10.1084/jem.183.3.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai S.K., Nichols G.L., Rothman P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 1997;159:4720–4728. [PubMed] [Google Scholar]
- Colicelli J. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci. Signal. 2010;3:re6. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial N.N., Losman J.A., Lu T., Yip N., Krishnan K., Krolewski J., Goff S.P., Wang J.Y., Rothman P.B. Direct interaction of Jak1 and v-Abl is required for v-Abl-induced activation of STATs and proliferation. Mol. Cell Biol. 1998;18:6795–6804. doi: 10.1128/mcb.18.11.6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial N.N., Rothman P. JAK-STAT signaling activated by Abl oncogenes. Oncogene. 2000;19:2523–2531. doi: 10.1038/sj.onc.1203484. [DOI] [PubMed] [Google Scholar]
- de Groot R.P., Raaijmakers J.A., Lammers J.W., Jove R., Koenderman L. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood. 1999;94:1108–1112. [PubMed] [Google Scholar]
- Dietz A.B., Souan L., Knutson G.J., Bulur P.A., Litzow M.R., Vuk-Pavlovic S. Imatinib mesylate inhibits T-cell proliferation in vitro and delayed-type hypersensitivity in vivo. Blood. 2004;104:1094–1099. doi: 10.1182/blood-2003-12-4266. [DOI] [PubMed] [Google Scholar]
- Dong Q., Li C., Qu X., Cao C., Liu X. Global regulation of differential gene expression by c-abl/arg oncogenic kinases. Med. Sci. Monit. 2017;23:2625–2635. doi: 10.12659/MSM.904888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghannam K., Martinez-Gamboa L., Spengler L., Krause S., Smiljanovic B., Bonin M., Bhattarai S., Grutzkau A., Burmester G.R., Haupl T. Upregulation of immunoproteasome subunits in myositis indicates active inflammation with involvement of antigen presenting cells, CD8 T-cells and IFNGamma. PLoS one. 2014;9:e104048. doi: 10.1371/journal.pone.0104048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlund A.C., Morales M.O., Viviano B.L., Yan H., Krolewski J., Schreiber R.D. Stat recruitment by tyrosine-phosphorylated cytokine receptors: an ordered reversible affinity-driven process. Immunity. 1995;2:677–687. doi: 10.1016/1074-7613(95)90012-8. [DOI] [PubMed] [Google Scholar]
- Greuber E.K., Smith-Pearson P., Wang J., Pendergast A.M. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat. Rev. Cancer. 2013;13:559–571. doi: 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J.J., Ryu J.R., Pendergast A.M. Abl tyrosine kinases in T-cell signaling. Immunol. Rev. 2009;228:170–183. doi: 10.1111/j.1600-065X.2008.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson Y.C., Guo X.Y., Greenberger J., Deisseroth A.B. Potential role of bcr-abl in the activation of JAK1 kinase. Clin. Cancer Res. 1997;3:145–149. [PubMed] [Google Scholar]
- Hochhaus A., Saglio G., Hughes T.P., Larson R.A., Kim D.W., Issaragrisil S., le Coutre P.D., Etienne G., Dorlhiac-Llacer P.E., Clark R.E. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30:1044–1054. doi: 10.1038/leu.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.H., Biswas A., Field M., Snapper S.B. STAT1 signaling shields T cells from NK cell-mediated cytotoxicity. Nat. Commun. 2019;10:912. doi: 10.1038/s41467-019-08743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan D.H., Shankaran V., Dighe A.S., Stockert E., Aguet M., Old L.J., Schreiber R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. U S A. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koleske A.J., Gifford A.M., Scott M.L., Nee M., Bronson R.T., Miczek K.A., Baltimore D. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron. 1998;21:1259–1272. doi: 10.1016/s0896-6273(00)80646-7. [DOI] [PubMed] [Google Scholar]
- Leaman D.W., Pisharody S., Flickinger T.W., Commane M.A., Schlessinger J., Kerr I.M., Levy D.E., Stark G.R. Roles of JAKs in activation of STATs and stimulation of c-fos gene expression by epidermal growth factor. Mol. Cell Biol. 1996;16:369–375. doi: 10.1128/mcb.16.1.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberatore R.A., Goff S.P. c-Abl-deficient mice exhibit reduced numbers of peritoneal B-1 cells and defects in BCR-induced B cell activation. Int. Immunol. 2009;21:403–414. doi: 10.1093/intimm/dxp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.Y., Sanchez D.J., Aliyari R., Lu S., Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. U S A. 2012;109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X., Ren Z., Parker G.N., Sondermann H., Pastorello M.A., Wang W., McMurray J.S., Demeler B., Darnell J.E., Jr., Chen X. Structural bases of unphosphorylated STAT1 association and receptor binding. Mol. Cell. 2005;17:761–771. doi: 10.1016/j.molcel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Mattiuzzi G.N., Cortes J.E., Talpaz M., Reuben J., Rios M.B., Shan J., Kontoyiannis D., Giles F.J., Raad I., Verstovsek S. Development of Varicella-Zoster virus infection in patients with chronic myelogenous leukemia treated with imatinib mesylate. Clin. Cancer Res. 2003;9:976–980. [PubMed] [Google Scholar]
- Meraz M.A., White J.M., Sheehan K.C., Bach E.A., Rodig S.J., Dighe A.S., Kaplan D.H., Riley J.K., Greenlund A.C., Campbell D. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Mertens C., Zhong M., Krishnaraj R., Zou W., Chen X., Darnell J.E., Jr. Dephosphorylation of phosphotyrosine on STAT1 dimers requires extensive spatial reorientation of the monomers facilitated by the N-terminal domain. Genes Dev. 2006;20:3372–3381. doi: 10.1101/gad.1485406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota N., Brett T.J., Murphy T.L., Fremont D.H., Murphy K.M. N-domain-dependent nonphosphorylated STAT4 dimers required for cytokine-driven activation. Nat. Immunol. 2004;5:208–215. doi: 10.1038/ni1032. [DOI] [PubMed] [Google Scholar]
- Park M.S., Shaw M.L., Munoz-Jordan J., Cros J.F., Nakaya T., Bouvier N., Palese P., Garcia-Sastre A., Basler C.F. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 2003;77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergast A.M. The Abl family kinases: mechanisms of regulation and signaling. Adv. Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- Platanias L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Reddy E.P., Korapati A., Chaturvedi P., Rane S. IL-3 signaling and the role of Src kinases, JAKs and STATs: a covert liaison unveiled. Oncogene. 2000;19:2532–2547. doi: 10.1038/sj.onc.1203594. [DOI] [PubMed] [Google Scholar]
- Schindler C., Shuai K., Prezioso V.R., Darnell J.E., Jr. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science. 1992;257:809–813. doi: 10.1126/science.1496401. [DOI] [PubMed] [Google Scholar]
- Schlatterer S.D., Suh H.S., Conejero-Goldberg C., Chen S., Acker C.M., Lee S.C., Davies P. Neuronal c-Abl activation leads to induction of cell cycle and interferon signaling pathways. J. Neuroinflammation. 2012;9:208. doi: 10.1186/1742-2094-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzberg P.L., Stall A.M., Hardin J.D., Bowdish K.S., Humaran T., Boast S., Harbison M.L., Robertson E.J., Goff S.P. Mice homozygous for the ablm1 mutation show poor viability and depletion of selected B and T cell populations. Cell. 1991;65:1165–1175. doi: 10.1016/0092-8674(91)90012-n. [DOI] [PubMed] [Google Scholar]
- Shuai K., Halpern J., ten Hoeve J., Rao X., Sawyers C.L. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996;13:247–254. [PubMed] [Google Scholar]
- Shuai K., Horvath C.M., Huang L.H., Qureshi S.A., Cowburn D., Darnell J.E., Jr. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994;76:821–828. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- Shuai K., Stark G.R., Kerr I.M., Darnell J.E., Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- Silberman I., Sionov R.V., Zuckerman V., Haupt S., Goldberg Z., Strasser A., Ben-Sasson Z.S., Baniyash M., Koleske A.J., Haupt Y. T cell survival and function requires the c-Abl tyrosine kinase. Cell Cycle. 2008;7:3847–3857. doi: 10.4161/cc.7.24.7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G.R. How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev. 2007;18:419–423. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark G.R., Darnell J.E., Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignais M.L., Sadowski H.B., Watling D., Rogers N.C., Gilman M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol. Cell Biol. 1996;16:1759–1769. doi: 10.1128/mcb.16.4.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino A.V., Gallo E., Abbas A.K. STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J. Immunol. 2010;185:6461–6471. doi: 10.4049/jimmunol.1001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino A.V., Kanno Y., O'Shea J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017;18:374–384. doi: 10.1038/ni.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale M., Rezzani R., Rodella L., Zauli G., Grigolato P., Cadei M., Hicklin D.J., Ferrone S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998;58:737–742. [PubMed] [Google Scholar]
- Zhong M., Henriksen M.A., Takeuchi K., Schaefer O., Liu B., ten Hoeve J., Ren Z., Mao X., Chen X., Shuai K. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc. Natl. Acad. Sci. U S A. 2005;102:3966–3971. doi: 10.1073/pnas.0501063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The profile of gene transcription in WT and abl/arg knockout MEFs (GEO accession numbers: GSE154568) can be accessed on GEO (Gene Expression Omnibus).