

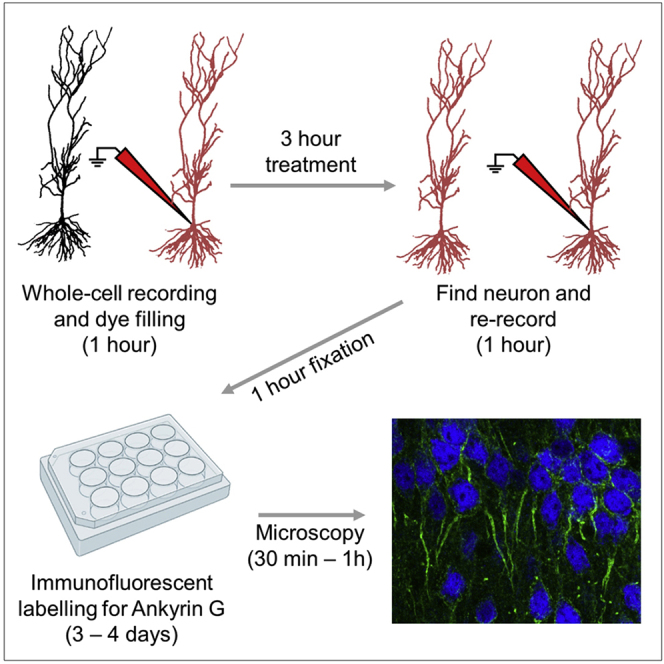
Summary
This protocol allows repeated whole-cell patch-clamp recordings from individual rodent CA1 hippocampal neurons, followed by immunohistological labeling of the axon initial segment. This overcomes the need to maintain whole-cell recordings over the timescales required for homeostatic modification to cellular excitability, allowing for correlative analysis of the structure and function of neurons. Moreover, this protocol allows for paired analysis of physiological properties assessed before and after pharmacological treatment, thus providing increased statistical power, despite the relatively low-throughput nature of the recordings.
For complete details on the use and execution of this protocol, please refer to Booker et al. (2020a).
Subject areas: Single cell, Microscopy, Neuroscience
Graphical abstract
Highlights
-
•
Protocol allows repeated whole-cell patch-clamp recording in ex vivo hippocampal neurons
-
•
Produces within neuron controls, increasing statistical power through paired analysis
-
•
Allows for axon initial segment labeling following whole-cell patch-clamp recordings
-
•
Facilitates comparison of neuron structure and function following long-term pharmacology
This protocol allows repeated whole-cell patch-clamp recordings from individual rodent CA1 hippocampal neurons, followed by immunohistological labeling of the axon initial segment. This overcomes the need to maintain whole-cell recordings over the timescales required for homeostatic modification to cellular excitability, allowing for correlative analysis of the structure and function of neurons. Moreover, this protocol allows for paired analysis of physiological properties assessed before and after pharmacological treatment, thus providing increased statistical power, despite the relatively low-throughput nature of the recordings.
Before you begin
Acute hippocampal brain slice preparation
Timing: 30 min
Full protocols for the preparation of acute brain slices in our research group have been previously described (Booker et al., 2014, Booker et al., 2017, Booker et al., 2020a, Booker, 2020b). This protocol is not in specific chronological order, but rather with respect to each experimental aspect, in their respective order. It is more efficient to perform a number of days of electrophysiological recording, followed by subsequent batch processing of tissue and images. Briefly:
-
1.Prepare holding chamber:
-
a.Obtain plastic inserts to form 2 compartments in the chamber, one for holding slices and one to place carbogen bubbler:
-
i.A cylindrical plastic insert of 2.5 cm diameter and 2 cm height can be obtained by cutting a 50 mL syringe.
-
ii.From a 20 mL syringe, remove the plunger and the cut off top portion of the syringe body. Cut off the syringe tip and make small holes in the bottom of the syringe (Figure 1B).Note: A heated scalpel blade can be used to more easily cut through the plastic.
-
i.
-
b.Build holding compartment by stretching mesh over the smaller cylinder and applying superglue (Figure 1C).
-
c.Once superglue has dried, cut excess mesh to match the diameter of the plastic insert (Figure 1D).
- d.
-
a.
-
2.Prepare high-sucrose artificial cerebrospinal fluid (ACSF) for slicing:
-
a.Place ∼200 mL sucrose-ACSF into a glass flask and put into a freezer at −20°C for ∼30 min prior to preparation of acute slices. This will allow partial freezing of sucrose-ACSF to form a slushy consistency.
-
b.Fill a submerged slice holding chamber with sucrose-ACSF in a water bath at 35°C and aspirate with carbogen (95% O2/5% CO2).
-
a.
-
3.Prepare the workspace:
-
a.Add crushed ice to slicing chamber (Figure 2A).
-
b.Layout tools used for brain dissection (Figure 2B).
-
c.Agitate slushed sucrose-ACSF to ensure no large lumps of ice. Place flask of sucrose-ACSF on ice and then aspirate with carbogen for at least 5–10 min prior to dissection of the brain.
-
d.Once satisfactorily carbogenated, transfer a small volume (∼50 mL) of sucrose-ACSF to a small glass beaker.
-
a.
Note: All tools and equipment required for removal of the brain and preparation for slicing can be found in Figure 2B.
Figure 1.

Assembly of a submerged slice holding chamber
(A) Material necessary to build holding chamber (in order: alcohol burner, beaker, nylon mesh, 50 mL syringe, 20 mL syringe, superglue, scalpel).
(B) Plastic inserts cut to form 2 compartments, one to hold the slices and another to place carbogen bubbler.
(C) Nylon mesh stretched over smaller insert.
(D) Finalized holding compartment.
(E) Top view of finalized chamber with 2 compartments.
(F) Side view of finalized chamber.
Figure 2.

Preparation of tools and chambers for acute brain slicing and storage/treatment of slices
(A) Preparation of the vibratome chamber.
(B) Tools and equipment for the dissection of the brain and slicing of the hippocampus.
Whole-cell patch-clamp recordings: prepare solutions
-
4.Prepare recording ACSF:
-
a.Place recording ACSF on water bath set to 40°C on the recording set up and bubble with carbogen.
-
a.
-
5.Prepare intracellular solution.Note: Intracellular solution can be prepared and stored in small aliquots (∼1 mL) at −20°C for 4–8 weeks prior to use.
-
a.On the day of recording, add Alexa Fluor hydrazide 568 (final concentration 100 μM) to aliquot of intracellular solution.Note: Other fluorescent dyes with different wavelengths can be used as appropriate to the optical pathway installed on the microscope being used for recording.Note: Keep intracellular solution on ice and protected from light throughout the experimental day.
-
a.
For whole-cell patch-clamp recordings: prepare equipment
-
6.Prepare glass electrodes:
-
a.Pull glass electrodes with tip resistance of 3–4 MΩ.
-
a.
-
7.Start-up equipment:
-
a.Switch on all equipment necessary for whole-cell patch-clamp recording:
-
i.Amplifier, digitizer, PC.
-
ii.Digital camera, infrared transmission, and epifluorescent light sources.
-
iii.Micromanipulators and microscope controllers.
-
iv.In-line heater and water bath.Note: The specific equipment used for this protocol and our previous study is listed in the key resources table, however suitable alternatives can be used according to the experimental set up available in your lab.
-
i.
-
b.Start flow of pre-warmed and carbogenated recording ACSF and switch on in-line heater. Set temperature of in-line heater to maintain a temperature of 30 ± 1°C in the recording chamber.
-
a.
-
8.Prepare holding chambers with control (15 mM NaCl) and treatment (15 mM KCl) ACSF:
-
a.Measure required ACSF to fill holding chamber (typically 100–200 mL).
-
b.Prepare sufficient ACSF with added NaCl or KCl. by adding the respective salt from 1 M stock solution. Add solution to separate, labeled holding chambers.
-
c.Keep holding chambers bubbling with carbogen in a water bath at 35°C.
-
a.
Note: The specific treatment should be kept blind to the researcher performing recordings and AIS labeling and imaging. This can be achieved by labeling solutions A and B, and the key to the treatment kept blind until all analysis is completed.
Prepare histological solutions
-
9.
Prepare 0.4 M Phosphate Buffer (PB) stock solution (see Materials and equipment).
Note: stock solution can be prepared prior to experimental day and kept at 20°C ± 2°C (room temperature) for up to 1 month.
-
10.
Prepare 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB) for tissue fixation.
Note: PFA can be made prior to experimental day, and stored in aliquots of 10–20 mL for 2–3 months at −20°C.
CRITICAL: PFA in its powdered form is highly toxic when inhaled. All PFA should be weighed and handled in a fume-hood or in a lab where appropriate personal protective equipment is in use by all lab members.
-
11.
Prepare phosphate buffered saline (PBS; 0.1 M PB with 0.9% NaCl) from tablets.
-
12.
Prepare 0.1 M PB from stock buffer on day of tissue processing.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-AnkyrinG (1 μg/mL, 1:1,000) | NeuroMAB | N106/36 |
| Rabbit polyclonal anti-AnkyrinG (0.4 μg/mL, 1:500) | Santa Cruz | sc-28561 |
| Rabbit polyclonal anti-NeuN (1 μg/mL, 1:1,000) | Millipore EMD | ABN78 |
| Mouse monoclonal anti-NeuN (1 μg/mL, 1:1,000) | Millipore EMD | MAB377 |
| Alexa 488 goat anti-mouse (4 μg/mL, 1:500) | Thermo Fisher Scientific | A-11001 |
| Alexa 568 goat anti-mouse (4 μg/mL, 1:500) | Thermo Fisher Scientific | A-11004 |
| Alexa 568 goat anti-rabbit (4 μg/mL, 1:500) | Thermo Fisher Scientific | A-11011 |
| Alexa 633 goat anti-rabbit (4 μg/mL, 1:500) | Thermo Fisher Scientific | A-21071 |
| Streptavidin-Alexa Fluor 568 conjugate (4 μg/mL, 1:500) | Thermo Fisher Scientific | S11226 |
| Chemicals, peptides, and recombinant proteins | ||
| Isofane (100% isoflurane) | Henry Schein | 988-3245 |
| K-gluconate | Sigma-Aldrich | P1847 |
| HEPES | Sigma-Aldrich | H3375 |
| Na2ATP | Sigma-Aldrich | A2383 |
| Na2Phosphocreatine | Sigma-Aldrich | P7936 |
| EGTA | Sigma-Aldrich | E3889 |
| Na2GTP | Sigma-Aldrich | G8877 |
| Biocytin | Sigma-Aldrich | B4261 |
| MgCl2 (1 M stock) | Honeywell Fluka | 63020 |
| Ca2Cl2 (1 M stock) | Honeywell Fluka | 21114 |
| Alexa Fluor 568 hydrazide | Thermo Fisher Scientific | A10437 |
| PBS tablets | Sigma-Aldrich | P4417 |
| Triton X-100 | Sigma-Aldrich | X100 |
| Sodium azide | Sigma-Aldrich | S2002 |
| Normal goat serum | Vector Laboratories | S-1000-20 |
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6J (P28–35 males) | Jackson Laboratory | 000664 |
| Rat: Long-Evans hooded (Crl:LE, P28–35 males) | Charles River | 006 |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com |
| R/R Studio | R Core Team, 2013 | https://www.r-project.org; https://rstudio.com |
| pClamp | Molecular devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite#gref |
| Stimfit | Guzman et al., 2014 | https://github.com/neurodroid/stimfit |
| ImageJ | Schneider et al., 2012 | https://imagej.net/Fiji/Downloads |
| Imaris | Oxford Instruments | Imaris for Neuroscientists package |
| Huygen’s Deconvolution Software | Scientific Volume Imaging | Huygens Essential |
| Other | ||
| Vibratome | Leica | VT1200S |
| Horizontal pipette puller | Sutter Instruments | P-97 |
| Capillary glass (1.5/0.75 mm outer/inner diameter) | Warner Instruments | GC150F-7.5 |
| Headstage amplifier | Molecular Devices | CV-7B |
| Amplifier | Molecular Devices | Multiclamp 700B |
| Digitizer | Molecular Devices | Digidata 1440 |
| Micromanipulators | Luigs and Neumann | SM7 |
| Camera | Hamamatsu | Orca 2 |
| Microscope | Olympus | BX61WI |
| Epifluorescent light source | Prior Scientific | Lumen200 |
| Sharp dissection scissors | Fine Science Tools | 14501-14 |
| Fine dissection scissors | Fine Science Tools | 14106-09 |
| Curved tip forceps | Fine Science Tools | 91117-10 |
| Vectashield mounting medium | Vector Laboratories | H-1400 |
| Microscope slides | Thermo Scientific | 12362098 |
Materials and equipment
Solution compositions for slice preparation and recording:
| Reagent | Concentration (mM) | Mass/volume |
|---|---|---|
| Sucrose-ACSF | ||
| NaCl | 86 | 5.03 g |
| KCL | 2.5 | 0.19 g |
| D-glucose | 25 | 4.51 g |
| Sucrose | 75 | 25.67 g |
| NaH2PO4 | 1.2 | 0.19 g |
| NaHCO3 | 25 | 2.10 g |
| CaCl2 | 0.5 | 0.5 mL |
| MgCl2 | 7 | 7 mL |
| ddH2O | - | adjust to 1 L total volume |
| Recording ACSF | ||
| NaCl | 124 | 14.49 g |
| KCl | 2.5 | 0.37 g |
| D-glucose | 20 | 7.21 g |
| NaH2PO4 | 1.2 | 0.37 g |
| NaHCO3 | 25 | 4.21 g |
| Ca2Cl2 (1 M stock) | 2 | 4 mL |
| MgCl2 (1 M stock) | 1 | 2 mL |
| ddH2O | - | adjust to 2 L total volume |
Store sucrose-ACSF at 4°C for up to 5 days. Prepare fresh recording ACSF on the day of recording. Following bubbling with carbogen, the pH of ACSF should be 7.3–7.4. Osmolarity should be 310–320 mOsm/L for recording ACSF and 320–330 mOsm/L for sucrose-ACSF.
Potassium-gluconate based intracellular solution composition (pH 7.3, 290–300 mOsm/L):
| Reagent | Concentration (mM) | Mass/volume |
|---|---|---|
| K-gluconate | 142 | 1.6628 g |
| HEPES | 10 | 0.1191 g |
| KCl | 4 | 0.0149 g |
| MgCl2 (1 M stock) | 2 | 100 μL |
| Na2ATP | 2 | 0.0551 g |
| Na2Phosphocreatine | 1 | 0.0128 g |
| EGTA | 0.5 | 0.0095 g |
| Na2GTP | 0.3 | 0.0078 g |
| Biocytin | 2.7 | 0.0500 g |
| ddH2O | - | adjust to 50 mL total volume |
Intracellular solution can be prepared and stored in small aliquots (∼1 mL) at −20°C for 4–8 weeks prior to use.
Alternatives: Internal solution can be made without biocytin, and biocytin can be added to each individual aliquot of internal solution on the day before starting recordings.
Phosphate buffer 0.4 M stock solution composition (pH 7.4):
| Reagent | Concentration (mM) | Weight (g) |
|---|---|---|
| NaH2PO4 | 300 | 6.2 g |
| Na2HPO4 | 100 | 22 g |
| ddH2O | - | adjust final volume to 500 mL |
Stock solution can be kept at 20°C ± 2°C for up to a month.
Phosphate buffered saline (PBS), pH 7.4
Prepare from Sigma-Aldrich PBS tablets. Add 5 tablets to 1 L of ddH2O. Prepare PBS on the day of use and keep at 4°C for up to 2 weeks.
Alternatives: prepare PBS from 0.4 M PB stock solution: 0.1 M PB with 0.9% NaCl.
Step-by-step method details
Acute slice preparation
This step describes the preparation of acute hippocampal slices to be used for electrophysiological recordings and subsequent histological processing.
Note: All work outlined in this protocol has been performed in line with UK Home Office guidelines (ASPA 2013) and The University of Edinburgh Ethical Board.
-
1.Dissect brain:
-
a.Anesthetize mouse or rat by placing in an anesthetic chamber containing suitable concentration of a volatile anesthetic (e.g., isoflurane).Note: We use ∼1 mL of isoflurane in a 5 L anesthetic chamber, however the exact method and anesthetic agent used should be in line with local guidelines.
-
b.Confirm deep anesthesia, reduction of breathing, and absence of reflexes.
-
c.Decapitate rodent using a pair of sharp scissors or guillotine.
-
d.Rapidly remove skull, using a pair of fine scissors.
-
e.Using curved, blunt-tip forceps to remove the skull from between bregma and the zygomatic arch.
-
f.Using a small spatula, gently separate the brain from the skull, taking care to sever the cranial nerves. Roll brain into small beaker of sucrose-ASCF (Before you begin step 2c.)Note: All tools and equipment required for removal of the brain and preparation for slicing can be found in Figure 2.CRITICAL: The dissection of the brain following decapitation should ideally take <30 s to ensure high quality brain slices.
-
a.
-
2.Prepare horizontal brain slices of the hippocampus:
-
a.Transfer brain from small beaker to glass Petri dish, lined with disc of filter paper.
-
b.Using a scalpel, remove cerebellum and frontal third of brain.
-
c.Apply a small amount of cyanoacrylate glue (Superglue) to slicing stage of vibratome. Use probe to spread glue to form a thin film.
-
d.Place brain, ventral surface down onto the glue.
-
e.Using a wide-mouth Pasteur pipette, apply a few drops of sucrose-ACSF to the brain to wash excess glue away from brain block.
-
f.Place slicing stage in vibratome and flood slicing chamber with remaining ice-cold sucrose-ACSF. Take care to position the brain so that the entorhinal cortices are positioned closest to the vibratome blade. Place a carbogen line in the slicing chamber (Figures 3A and 3B).
-
g.Move blade to brain surface, and then cut 400 μm thick horizontal slices containing the hippocampus (Figure 3C).Note: Slices tend to have better preservation of neuronal structure if they are cut slowly. Ideally cut slices at 0.05–0.1 mm/s forward blade movement, with a horizontal blade oscillation of 1–1.5 mm.Note: A scalpel blade or a bent hypodermic needle can be used to trim the slices and separate the slices from each hemisphere before transferring into the holding chamber.Note: From a P28–35 mice it should be possible to obtain 8 slices (4 per hemisphere) containing the ventromedial portion of the hippocampus. For an equivalent age of rat, approximately 10–12 slices of the ventromedial hippocampus can be obtained.
-
h.Transfer slices to a submerged holding chamber filled with carbogenated sucrose-ACSF pre-warmed to 35°C and recover for 30 min (Figure 3D).
-
i.Following recovery, transfer the holding chamber to the lab bench at 20°C ± 2°C (room temperature) until the slices are required for recording.CRITICAL: Keep solutions bubbled with 95% O2/5% CO2 at all times to allow a sufficient partial pressure of O2 to build up in solution to supply brain tissue and preserve cell viability.
-
a.
Figure 3.

Obtaining acute hippocampal slices
(A) Brain positioned on the vibratome stage.
(B) Vibratome chamber filled with carbogenated sucrose-ACSF.
(C) Cutting horizontal slices containing the hippocampus.
(D) Trimmed slices in the holding chamber.
Repeated whole-cell patch-clamp recordings with dye filling
This step allows for assessment of cellular physiology of hippocampal neurons both under control conditions and following a prolonged depolarization or osmotic controls within the same cell. Specific aspects of performing the whole-cell patch-clamp technique have been described previously (Booker et al., 2014, Neher and Sakmann, 1992).
-
3.Transfer hippocampal slice to the recording chamber:
-
a.Turn off inflow and outflow of recording ACSF.
-
b.Using a wide-mouth Pasteur pipette transfer a brain slice from the holding chamber to the slice chamber.
-
c.Take care to ensure that the dorsal surface is kept upwards.
-
d.Place slice weight over slice, ensuring that anchor wires do not overlay the CA1 region of the hippocampus.
-
e.Restart recording ACSF flow. ACSF flow rate should be approx. 4–6 mL/min and should not be recirculated.
-
a.
Note: It may be helpful to modify the slice weight to give a large gap between anchor wires to prevent obfuscation of the CA1 region.
-
4.
Under low magnification (10× objective) identify str. pyramidale of CA1 (Figure 4A).
-
5.Exchange objective lens to high magnification (40× objective) and identify healthy region of pyramidal cells (Figure 4B).
-
a.Healthy CA1 pyramidal cells should have an ovoid cell body that is lightly contrasted with a halo of diffracted light around it. Any cells that are shriveled or shrunken are likely damaged or undergoing cell death.
-
b.Typically identify a healthy looking cluster of cells that are located 50–100 μm below the slice surface.
-
a.
-
6.
Fill a pre-prepared patch pipette with intracellular solution and place on electrode holder.
Note: Ensure that Ag wire of headstage amplifier is suitably coated with AgCl prior to any recording. Any chips in AgCl coating will lead to undue capacitive currents and offset errors during recording.
-
7.
Apply positive pressure to patch pipette via pneumatic tube attached to electrode holder.
-
8.
Move patch pipette through recording ACSF and “park” the pipette just above the slice so that the cell to be recorded is located within the proximal region of the pipette tip (Figure 4B).
-
9.
Obtain a whole-cell patch-clamp recording from the first identified CA1 pyramidal cell, as previously described in detail (Booker et al., 2014) (Figures 4C and 4D).
-
10.Perform intrinsic physiology analysis in current-clamp, including:
-
a.30× −10 pA steps from the zero-current level (500 ms duration). This will assess resting membrane potential, input resistance, and membrane time constant.
-
b.From a membrane potential of −70 mV, 3× families of hyper-to depolarizing current steps (−100 to +400 pA in 25 pA steps, 500 ms duration).
-
a.
-
11.
Confirm spontaneous synaptic inputs by recording between 1 and 5 min of continuous recording at −70 mV voltage-clamp.
Optional: Specific requirements of the recording may differ from this basic protocol. Depending on the hypothesis being tested, alternative whole-cell protocols may be required.
-
12.Once the recording is complete, confirm dye filling under epifluorescent imaging (typically 5–10 min of recording is sufficient to dye fill the somatodendritic regions) and perform an outside-out patch:
-
a.Close IR-DIC light source and increase exposure time of digital camera (100–200 ms works well).
-
b.Move filter turret to correct filter-set for fluorescent dye used.
-
c.Move focus to identify filled cell (Figure 4D).
-
d.Form outside-out patch by slowly retracting the patch electrode under visual guidance.
-
e.When access resistance begins to increase, slowly increase holding potential toward ∼−40 mV.Note: in performing outside out seals, access resistance is observed to smoothly increase from 5–30 MΩ to the 1–10 GΩ range. Increasing the holding potential, facilitates rupture of the retracted membrane.
-
f.Continue retracting pipette until well clear of resealed cell.
-
g.Confirm continued presence of filled cell both under IR-DIC and fluorescence.
-
a.
-
13.Repeat procedure for a further 2–4 cells:
-
a.Make note of the order of which cells are recorded. This can be aided by drawing a map or taking images of the fluorescently labeled cells.
-
a.
-
14.Once all recordings are complete and viability of resealing confirmed, place slice into appropriate treatment:
-
a.Stop inflow and outflow of recording ACSF.
-
b.Remove slice weight.
-
c.Using the wide-mouth Pasteur pipette remove slice from the recording chamber and place in treatment chamber.
-
d.Note time slice treatment was started.
-
e.Rinse transfer pipette with distilled water.
-
a.
Note: To account for time since slicing affecting the recording conditions for individual slices, recorded slices should be treated with different KCl or NaCl in a counterbalanced manner, i.e., alternating treatment within day and rotating of which treatment is used first between days.
Optional: This protocol is based on our recent study (Booker et al., 2020a) in which we employed prolonged depolarization of neurons by application of 15 mM KCl to examine cell excitability. Many other methods of modifying cell excitability could be employed (e.g., 4-aminopyridine, tetraethylammonium, tetrodotoxin, AP-5, CNQX, or bicuculline). The only requirement is that the agent used is easily and rapidly washed out of the tissue to allow comparison of physiological states under identical conditions.
-
15.
Repeat procedure for the remaining slices and alternate treatment conditions.
Pause point: If break is required, following treatment of slice a pause can be taken.
-
16.Following treatment, remove slice from treatment chamber and return to recording chamber:
-
a.Ensure that the same side of slice is positioned up, to allow direct access to dye filled cells.
-
b.Place slice weight on slice, avoiding CA1 region of the slice.
-
c.Allow 3–5 min for ACSF to fully wash out treatment ACSF.
-
a.
Note: If recirculation of ACSF is unavoidable, ensure that it is not recirculated while any residual treatment solution is washed through the perfusion tubing to prevent effected on cell excitability.
-
17.
Using fluorescence microscopy, identify the previously filled cells, comparing to the map/images taken previously (Figure 4E).
-
18.
Perform whole-cell patch-clamp recordings on dye filled cells, starting with first filled cell and moving through the selection that have survived treatment (Figure 4F).
-
19.
For all cells, once recording has been completed, reseal the patch of membrane with outside-out patch formation as in step 12.
Note: For consistency, the faster that these recording can be performed the more likely they reflect the state of the slice immediately after treatment. We aim to re-record all cells within ∼20 min of slice returning to normal ACSF. This may dictate how many cells are filled in the first phase of recording and the number of recording protocols used. Using a very brief recording protocol, and experienced electrophysiologist can re-record 3–4 cells within this time frame.
-
20.Once all filled cells have been recorded and resealed, slices should be fixed within 1 min of last recording. Once removed from the perfusing ACSF, the slice should be fixed immediately (less than 1 min):
-
a.Start by turning off all inflow and outflow pumps.
-
b.Remove the slice weight.
-
c.Carefully extract the slice using the slice transfer pipette.
-
d.Place the slice into a fixation vessel (e.g., a multi-well plate or bijou bottle).
-
a.
-
21.Fix slices using 4% PFA in 0.1 M PB:
-
a.Extract excess recording ACSF using a Pasteur pipette.
-
b.Apply 2–3 mL of fresh 4% PFA in PB to the slice.
-
c.Swirl or agitate the slice slightly to ensure that it is free-floating and not adhered to the wall of the vessel.
-
d.Keep slices in 4% PFA at 20°C ± 2°C (room temperature) for 1 h.
-
e.Transfer slices into PBS and store at 4°C until used for immunohistochemical labeling.
-
a.
Figure 4.

Placement of slice in chamber, recording and dye filling of CA1 pyramidal cells
(A and B) (A) low power overview of the hippocampus under infrared differential inference contrast (IR-DIC) microscopy, with recording site of (B) indicated (black box). Hippocampal subregions (white text), layers (black text), and the hippocampal fissure (dashed gray line) are indicated for reference. Scale bar: 200 μm. (B) High-power (40×) IR-DIC image of a CA1 pyramidal cell with a patch pipette placed on the cell body. Scale bar: 20 μm.
(C) Epifluorescent image of the same neuron, before breaking in to whole-cell configuration. Note the absence of labeling in the cell body. Scale bar: 20 μm.
(D) The same cell, but now in whole-cell configuration and having had filled for 5 min. Scale bar: 20 μm.
(E) Fluorescent identification of the same cell after 3 h incubation in 15 mM KCl. Scale bar: 20 μm.
(F) Whole-cell patch-clamp recording from the same cell under IR-DIC optics. Scale bar: 20 μm.
Figure 5.

Immunolabeling for the AIS from acute slices
(A) AnkyrinG immunolabeling (AnkG, green pseudocolor) shown in comparison to NeuN labeling (blue pseudocolor) allows the visualization of AISs in a field of view including CA1 of the hippocampus. Scale bar: 20 μm.
(B) Higher magnification view of a single AIS. Scale bar: 10 μm.
(C) Poor quality AnkyrinG labeling in over-fixed tissue. Scale bar: 20 μm.
(D) Visualization and measurement of the AIS (AnkyrinG, green, top) in a biocytin labeled neuron (Bioc, grayscale, middle) and merged (lower). Scale bar: 20 μm.
Note: At this point, it can be helpful to transfer fixed slices to a multi-well plate for storage and future immunolabeling. Each experimental slice should occupy its own individual well and be labeled with a unique experimental identifier (i.e., date and slice number)
Immunohistochemistry of fixed acute brain slices
This step allows for recovery of the recorded cells to assess their morphology and concomitant visualization and measurement of axon initial segments (AIS) in the same slices (Figures 5A and 5B). This process is highly reliable and allows for accurate detection of AIS structure, as well as measurement of somatodendritic structure. Inclusion of anti-NeuN primary antibodies labels neuronal somata and can help visualize the hippocampal formation.
-
22.Wash slices thoroughly in PBS:
-
a.Remove excess storage PBS from well with plastic Pasteur pipette.
-
b.Fill well containing slice with fresh PBS (approx. 3–5 mL).
-
c.Repeat a further 2–3 times to fully remove any residual buffer and PFA. Washes should last 5–10 min per repetition.
-
a.
-
23.Block non-specific antigenicity in slices, to improve signal to noise of immunolabeling:
-
a.Prepare blocking solution containing 10% normal goat serum (NGS), 0.3% Triton X-100 (to allow membrane permeabilization and antibody penetration) and 0.05% NaN3 (to limit microbial growth), in PBS.
-
b.Add 0.5 mL of blocking solution to each well of a 24-well plate labeled with the unique slice identifiers.
-
c.Transfer each slice to their according well with a soft, fine paintbrush.Note: Care should be taken to not damage the slice. Slices are best lifted out of the well by sliding the brush under the slice and lifting out of the buffer in an upwards motion.
-
d.Block slices for 1 h at 20°C ± 2°C (room temperature) in the blocking solution.
-
a.
-
24.Incubate slices with primary antibodies:
-
a.Prepare fresh antibody dilution solution, containing: 5% NGS, 0.3% Triton X-100, 0.05% NaN3 all diluted in PBS.
-
b.Add primary antibodies:
- Mouse anti-AnkyrinG (1 μg/mL or 1:1,000 of neat antisera)
- Rabbit anti-NeuN (1 μg/mL or 1:1,000 of neat antisera)Note: Antibodies should be stored according to the manufacturer’s guidelines. For most primary antibodies, we prepare aliquots of neat antisera (5–20 μL) and store at −20°C until used. Repeated freeze-thaw cycles of antibodies severely impair their antigenicity.
-
c.Add 0.5 mL of prepared antibody solution to each well of a fresh, clean 24-well plate.
-
d.Transfer slices to their according well with a paintbrush, rinsing the brush in clean PBS between slices.
-
e.Cover the 24-well plate with lab film (i.e., Parafilm) to prevent evaporation of buffer.
-
f.Place 24-well plate on orbital shaker in cool room or fridge.
-
g.Incubate slices for at least 18 to 24 h (overnight) at 4°C.Note: For thick slices, longer incubation can improve the penetration of antibodies deeper into the slice. As such, we often incubate for 2–3 days rather than just 1 overnight period.
-
a.
-
25.Wash slices thoroughly in PBS:
-
a.Fill clean multi-well plates with PBS buffer.
-
b.Transfer slice from primary antibody plate to PBS buffer using a fine paintbrush.
-
c.Remove excess storage PBS from well with plastic Pasteur pipette.
-
d.Fill well containing slice with fresh PBS (approx. 3–5 mL).
-
e.Repeat a further 2–3 times to fully remove any residual buffer. Washes should last 5–10 min per repetition.
-
a.
-
26.Apply secondary antibodies:
-
a.Prepare secondary antibody buffer: 3% NGS, 0.3% Triton X-100, 0.05% NaN3 in PBS.
-
b.Add secondary antibodies:
- Goat anti-Mouse Alexa Fluor 488 (4 μg/mL or 1:500 of neat sera)
- Goat anti-Rabbit Alexa Fluor 633 (4 μg/mL or 1:500 of neat sera)
- Streptavidin-Alexa Fluor 568 conjugate (4 μg/mL or 1:500 of neat sera)Note: Use streptavidin conjugated fluorophore with same spectral properties as the Alexa Fluor dye used to fill neurons during the recording. (i.e., Streptavidin-AlexaFluor568 conjugate is used with Alexa 568 hydrazide dye) to avoid potential contamination of signals with residual fluorescence from filling.Note: Secondary antibody combinations can be tailored to your specific confocal microscope configuration. Given the relative size and labeling strength of AnkyrinG, shorter wavelengths are optimal. As NeuN is less important for reconstruction of neurons, we often use this in the far-red channel.
-
c.Add 0.5 mL of prepared antibody solution to each well of a fresh, clean 24-well plate.
-
d.Transfer slices to their according well with a paintbrush, rinsing the brush in clean PBS between slices.
-
e.Cover the 24-well plate with lab film (i.e., Parafilm) to prevent evaporation of buffer.
-
f.Place 24-well plate on orbital shaker in cool room or fridge.
-
g.Incubate for 18 to 24 h at 4°C.
-
a.
-
27.Wash slices thoroughly in PBS:
-
a.Fill clean multi-well plates with PBS buffer.
-
b.Transfer slice from primary antibody plate to PBS buffer using a fine paintbrush.
-
c.Remove excess storage PBS from well with plastic Pasteur pipette.
-
d.Fill well containing slice with fresh PBS (approx. 3–5 mL).
-
e.Repeat a further 2–3 times to fully remove any residual buffer. Washes should last 5–10 min per repetition.
-
a.
-
28.Desalt slices:
-
a.Fill clean multi-well plates with 0.1 M PB buffer (3–5 mL/well).
-
b.Transfer slice from PBS buffer to PB buffer using a fine paintbrush.
-
c.Rinse paintbrush between transfers.
-
d.Stir on orbital shaker for 10–15 min.
-
e.Remove excess storage PB from well with plastic Pasteur pipette.
-
f.Fill well containing slice with fresh PB (approx. 3–5 mL).
-
g.Stir on orbital shaker for 1 h.
-
a.
-
29.Mount slices on glass slides:
-
a.Label glass microscope slide with unique slice identifier and antibodies used.
-
b.Remove slice from PB multi-well plate using paintbrush.
-
c.Blot excess PB from slice by touching brush to a piece of tissue, then gently lay slice on slide.CRITICAL: When mounting slices on glass slide, maintain the side of the slice with the recorded cell facing up. The correct side of the slice can be identified by the marks left by the threads of the slice weight used during electrophysiological recordings. Alternatively, a note of the slice orientation can be made during recording.
-
d.Blot any residual PB buffer from slice, using a cut wedge of tissue or filter paper.
-
e.Place sufficient fluorescent mounting medium (e.g., Vectashield Hardset mounting medium) on slice (2–3 drops for 22 × 22 mm coverslip).
-
f.Lay coverslip over slice.Note: To prevent bubbles forming on slice surface, it is advised to lay coverslip from one edge, then slowly allow remainder of coverslip to fall into place.
-
g.Place mounted slides in a cool, dark place for 1–2 h to allow hardset mounting medium to harden.
-
h.Seal coverslips with nail polish around the edge.
-
i.Once nail polish has cured place slides in slide box for at least 1 day before imaging.Pause point: Slides can be kept at 4°C for a few days before imaging, but it is recommended that images are taken within the first 2 weeks post-immunohistochemistry to avoid deterioration of labeling quality with time.
-
a.
Confocal imaging of the AIS and filled neurons
This step allows for recovery of morphology of the recorded cell and concomitant visualization of AISs in the slice.
-
30.
Once slides have fully equilibrated with the mounting medium (∼1 day) the refractive index of the fixed slices will have reduced to facilitate imaging.
-
31.
Place slices at 20°C ± 2°C (room temperature) ∼15 min prior to imaging to allow warming to ambient temperature.
-
32.Acquire high-resolution Z-stacks including CA1:
-
a.Place slide in confocal microscope.
-
b.Under epifluorescent imaging and a low magnification objective (×10 or ×20), identify the CA1 region by the presence of streptavidin labeling of the filled neurons.
-
c.Set appropriate light path for confocal imaging and excitation laser wavelength according to the fluorophores used.
-
d.Apply a small amount (1–2 drops) of immersion medium and exchange objective lens to a 63× oil immersion objective.
-
e.Use the volume imaging function of your confocal microscope to acquire high-resolution Z-stacks through CA1 (1,024 × 1,024 xy resolution, pixel size: 0.14 μm, 1 μm z steps).
-
a.
Note: Due to limited antibody penetration deeper in the slice, fluorescent labeling intensity is very bright in the slice surface and fainter deeper in the tissue, scan through optical sections at different depths to set the most appropriate laser intensity/detector gain that allows for high signal/noise for as much of the tissue as possible (Figure 6A). For image acquisition, we used either an Axiovert LSM 510 or LSM800 laser scanning confocal microscopes (both Zeiss, Germany), which were obtained with 63× (1.4 NA) oil or silicone immersion objective lenses.
Figure 6.

Depth profile of AnkyrinG and measurement of AIS length
(A) Multiple single plane images of AnkyrinG (AnkG, green) immunolabeling through the z plane (distance from the slice surface indicated in μm). Scale bar: 20 μm. (A, right) The orthogonal (zy) view of AnkG labeling in the zy axis. (A′) overlay of AnkyrinG labeling with measured AIS profiles (white).
(B) A single AIS followed through its z plane from start to end (white triangles) suitable for length measurement. Scale bar: 10 μm. (B′) The same AIS, but overlain with its segmented and measured extent (white overlay and text).
(C) A second AIS followed through its z plane from start (white triangle) to the surface of the slice, where it is prematurely cut (white asterisk) and thus not suitable for length measurement. (C′) Z projection of the AIS represented in (C). Scale bar: 10 μm.
Optional: This protocol is based on our recent study (Booker et al., 2020a) in which we assessed the effect of prolonged depolarization with 15 mM KCl on AIS length. Nonetheless the presence of biocytin filled neurons and the quality of AnkG labeling in these sections also allow for a deeper morphological analysis of the AIS and reconstruction of CA1 PC morphology. In that case imaging conditions must be adapted for each specific purpose as described in steps 33 and 34.
-
33.For 3-dimensional reconstruction of neurons, including the AIS, start by obtaining high-resolution images, to allow deconvolution of images and accurate surface rendering.Note: We use the Huygens deconvolution software to deconvolve images. Other software is available, however images should be collected to Nyquist sample the 3-dimensional volume.
-
a.Place histological slide in microscope.
-
b.Under low magnification (i.e., 10× or 20× Objective lens) identify the CA1 region of the slice. The specific microscope settings depend on the quality of the labeling and the choice of fluorescent markers used.
-
c.Apply a small amount (1–2 drops) of immersion medium and exchange objective lens.Note: Use the appropriate immersion medium for your objective lens, according to manufacturer’s guidelines.
-
d.Under high magnification identify the str. pyramidale layer of CA1.
-
e.Using the volume imaging function of your confocal microscope set the top (slice surface) and bottom (end of intense labeling) extents of your 3-dimensional volume.
-
f.Set the z-step size to 0.13 μm.
-
g.Establish optimal laser power and detector gain settings for start and end of volume.
-
h.If microscope is capable, set these values to automatically modulate over the slice depth.
-
i.Acquire image stack at 2,048 × 2,048 resolution under 2× zoom, which gives a pixel width of 0.04 μm (Figure 7A).
-
j.During acquisition modulate the detector gain and laser power to generate consistent image quality through volume.Note: The images presented here were collected using a LSM800 confocal microscope (Zeiss, Germany). The specific microscope settings were 1.2% of maximum power from a continuous spectrum laser and 599 V gain on the PMT detectors.CRITICAL: For appropriate volume rendering of AIS structure under diffraction limited light microscopy a voxel with vertices of 0.04 × 0.04 × 0.13 μm is optimal.
-
a.
-
34.
Full neuron morphology can be examined and reconstructed using a similar method, albeit with larger step sizes (0.5–1 μm) and no zoom, as described previously (Booker et al., 2014).
Note: Due to the axial point-spread function produced by imaging with longer wavelength fluorophores and in the depth of tissue smaller step sizes are not required.
Figure 7.

Imaging of AnkyrinG immunolabeling for deconvolution and 3-dimensional reconstruction
(A) A flattened z stack of high magnification and resolution images of AnkyrinG (AnkG, green pseudocolor) in the CA1 pyramidal layer before deconvolution. DAPI is shown for comparison. Scale bar: 10 μm. (A′) A blow up image of an example AIS from A. Scale bar: 10 μm.
(B) Flattened z stack of the same image series following deconvolution. (B′) The same example AIS, but following deconvolution. Note the sharper edges of the morphology and clearer association of AnkyrinG to the plasma membrane.
(C) Three-dimensional reconstruction and rendering of the same AIS following deconvolution. Note the spine located on the AIS (ar). Scale bar: 10 μm.
Expected outcomes
The protocol described here allows for multiple recordings from identified CA1 pyramidal cells both before and after a prolonged treatment with a depolarizing stimulus and the appropriate osmotic control (i.e., 15 mM KCl and 15 mM NaCl, respectively) as we have recently employed (Booker et al., 2020a). This approach allows for a highly correlated physiological characterization of the effects of long-term treatment, overcoming cell-to-cell or slice-to-slice variability that is inherent to recording from different cells for each treatment. Following recording, the brief fixation and immunohistochemistry protocol used then allows for visualization of recorded neurons and the AIS in the same slices, thus allowing for correlated structural analysis of the same tissue that has undergone this depolarizing treatment. Approximately 1–2 neurons per slice are able to be successfully re-patched, as described here.
From whole-cell patch-clamp recordings it is possible to obtain detailed electrophysiological measurements of cell excitability, such as active firing properties, action potential kinetics, and passive membrane properties (Figure 8). By incorporating a fluorescent dye into the intracellular recording solution, this allows for identification of the recorded neurons following treatment of the slice outside of the recording chamber, and facilitates repeated whole-cell recording of the same cells (Figure 4). Based on this protocol and our recent study (Booker et al., 2020a), these repeated patch-clamp recordings minimally affect cellular physiology as indicated by minimal change in excitability when treated with the physiologically inert 15 mM NaCl osmotic control (Table 1, Figure 8A) and is useful in identifying physiological changes following 15 mM KCl treatment (Figure 8B).
Figure 8.

Example recordings from before and after 15 mM KCl depolarization or 15 mM NaCl osmotic controls and analysis criteria
(A) Top: Whole-cell patch-clamp recording from a CA1 PC, and the resulting voltage measured in response to depolarizing stimuli (25 pA steps, 500 ms), the 400 pA maximum firing step and the rheobase current trace is shown (gray). Traces are shown for control (i.e., first recording) and after 3 h treatment with 15 mM NaCl (i.e., second recording). Bottom: The response of this cell to a small −10 pA, 500 ms current step. The control trace is underlain in the 15 mM NaCl trace (gray).
(B) A recording from a CA1 PC but before and after 15 mM KCl, according to the same scheme as (A).
(C) Example trace of −10 pA step with fitting of monoexponential curve (gray overlain) to determine membrane time constant and setting of baseline (green dashed lines) and peak cursors to measure peak voltage response (ΔVm) to determine input resistance.
(D) Rheobase current step with peak cursors (red dashed lines) flanking the first AP fired used to analyze action potential kinetics.
(E) Representation of action potential kinetic measurements: red horizontal line, half-height duration; vertical green arrow, action potential amplitude from threshold; ascending and descending blue arrows, action potential maximum rise and decay rates, respectively.
Table 1.
Summary of intrinsic physiological properties of wild-type CA1 pyramidal cells recorded before and after 15 mM NaCl osmotic control
| Recording 1 | Recording 2 | p value | |
|---|---|---|---|
| Resting membrane potential (mV) | −61.8 ± 3.4 | −60.5 ± 2.1 | 0.7169 |
| Input resistance (MΩ) | 168 ± 20 | 167 ± 20 | 0.9463 |
| Membrane time constant (ms) | 27.2 ± 3.4 | 25.9 ± 2.0 | 0.6906 |
| Rheobase (pA) | 146 ± 17 | 149 ± 20 | 0.8696 |
| Voltage threshold (mV) | −40.9 ± 1.7 | −40.2 ± 1.4 | 0.7379 |
| Action potential amplitude (mV) | 119 ± 2 | 113 ± 3 | 0.1538 |
| Half-height duration (ms) | 1.00 ± 0.05 | 1.07 ± 0.07 | 0.2810 |
| Maximum action potential frequency (Hz) | 36.5 ± 3.4 | 38.3 ± 4.2 | 0.6801 |
Data are shown as the mean ± SEM from 10 CA1 pyramidal cells from 5 mice. Statistics are reported from Student’s 2-tailed paired t tests. These data have been re-analyzed from recordings reported previously (Booker et al., 2020a).
Provided that good slice quality is maintained (Figure 4B), then it is possible to record multiple cells per slices before and after treatment. However, the technical ability of the experimenter will determine the number of cells that can be recorded in a given slice within the shortest possible time frame. An important consideration is to compare the values obtained in starting recordings from those performed without the time constraints of repeated whole-cell patch-clamp experiments (i.e., under true control conditions). It is critical that either vehicle or osmotic controls are included in any such experiment, to discount the effect of repeated whole-cell recording on the specific physiological parameter being measured.
Following recordings, our protocol provides a method to perform high-resolution imaging of the AIS from slices that have undergone depolarizing treatment and recording. If labeling has been successful it is expected that highly intense labeling for AnkyrinG is observed in the proximal portion of stratum oriens in CA1 (Figures 5, 6, and 7). When viewed under high-power magnification this labeling is due to the presence of a high number of immunolabeled AIS that often emerge from the base of the CA1 pyramidal cell somata and extend ∼20–40 μm into stratum oriens. It is expected that the AIS begins as a wide structure (2–3 μm) and tapers to a fine ending (<0.5 μm), forming a conical structure, spines are sometimes observed emanating from the AIS. Occasionally, the AIS emerges from a proximal dendrite (Thome et al., 2014), as observed by weak NeuN labeling that fills the proximal dendritic cytoplasm. Based on this quality of labeling, it is possible to reconstruct the AIS for analysis of its length, surface area, width, and starting location relative to the soma in 3-dimensions (Figure 7).
Quantification and statistical analysis
Electrophysiology analysis
Analysis of electrophysiological data was performed using Stimfit (Guzman et al 2014).
-
1.Passive membrane properties were measured from the voltage response to a small hyperpolarizing current step (−10 pA, Figures 8A and 8C):
-
a.Membrane time constant was determined by fitting of a monoexponential curve to the initial voltage response using the “fit function to data” tool.
-
b.Peak cursors were set to the final 100 ms of the current step allowing to determine the steady state voltage response to the current step and subsequently calculate input resistance according to Ohm’s Law.
-
a.
-
2.AP kinetics and AP discharge were quantified from the voltage response to a family of depolarizing current steps from −100 to +400 pA in 25 pA increments.
-
a.AP kinetics were analyzed for the first AP elicited and measured from threshold set as 20 mV/ms:
- b.
-
c.AP discharge was quantified by selecting all traces and using the “batch analysis” tool to count the number of threshold crossings for each current injection step.
-
a.
AIS length analysis
-
3.
Import microscope image files to FIJI (Image J).
-
4.
AISs can be reliably measured by scrolling through the 3D z stack and manually tracing each AIS from the proximal to distal end. Tracing can be achieved using the segmented line tool: double click at the proximal end of the AIS to initiate tracing, use single clicks to mark points throughout the length of the AIS and allow for changes in direction, double click again at the distal end of the AIS to terminate the region of interest (ROI).
Note: Avoid tracing AISs that are cut at the surface of the slice or at the edges of the field of the image (Figures 6B and 6C).
-
5.
Once an AIS has been traced it can be added to the ROI manager by pressing “T” on the keyboard.
-
6.
Repeat steps 4 and 5 until a suitable number of AISs have been traced (in our study we analyzed 50 per image and 2 images per treatment condition for each mouse). This creates a list of ROIs correspondent to the traced AISs.
-
7.
Select all ROIs in ROI manager and measure AIS lengths using the “Measure” button.
Note: ROIs and respective measurements can be saved as .zip and .csv files respectively for future reference.
Note: Alternative semi-automated methods for AIS measurement have been employed by others (Grubb and Burrone, 2010, Gutzmann et al., 2014, Pan-Vazquez et al., 2020). While these provide a time efficient alternative to manual tracing of individual AISs their accuracy should be validated against manual measurements for your own labeling and imaging conditions.
Three-dimensional reconstruction of AIS
The AIS was reconstructed in 3 dimensions to allow for surface rendering, similar to previous reports (Favuzzi et al., 2019).
-
8.Deconvolve images using Huygens Essential software package.
-
a.Import microscope images (including header file information) into Huygens Essential software (Figure 7A).
-
b.Set optical settings according to preparation of slides, i.e., refractory index of embedding medium (1.46 for Vectashield Hardset mounting medium).
-
c.Set deconvolution settings:
-
i.Automatic estimation.
-
ii.1 μm area radius.
-
iii.100 maximum iterations.
-
iv.20 signal to noise ratio.
-
v.0.1 quality threshold.
-
vi.All other values are automatically loaded from image metadata.
-
i.
-
a.
Note: These settings are optimized for our imaging conditions and optimization should be performed for each sample prior to reconstruction (Figure 7B).
-
9.Reconstruct AIS volumes.
-
a.Import deconvolved images into Imaris analysis software.
-
b.Render surfaces using the Surfaces tool.
-
i.0.3 surfaces detail.
-
ii.1 μm background subtraction.
-
iii.Quality threshold was automatically set.
-
i.
-
c.The Surfaces Split function was applied to split the reconstruction into individual AIS surface objects.
-
d.The volume and surface area values of each AIS were measured automatically and reported.
-
a.
Statistical analysis of electrophysiology data
Given the paired nature of experiments from control and treatment conditions, paired statistical analysis should be performed to take into account the power obtained by performing repeated patch-clamp recordings.
Power analysis to calculate the required sample sizes to detect a difference in rheobase between control and treated recordings revealed a paired t test would require a sample size of 16 cells to detect a difference between groups, while an unpaired T test would require a sample size of 44 cells per group. Power calculation was performed assuming 95% power, 95% confidence interval and based on the standard deviation and mean rheobase values from control and KCl treated slices in WT mice published in our previous study (control: 159 ± 54 pA, KCl: 221 ± 99 pA, mean ± SD, Booker et al 2020a).
Nonetheless, to avoid potential pseudoreplication effects when multiple recordings per animal are performed, mixed effects modeling is the most appropriate method to test for statistical differences (LMER function, R Studio).
Statistical analysis of AIS length data
First, to assess AIS length distributions across treatment conditions we performed a Kolmogorov-Smirnov test, based on the average cumulative distribution. This was performed on cumulative distributions in which the AIS lengths from each animal were binned (e.g., into bins of 0.5–1 μm, dependent on variability). Once binned, each histogram was averaged between animals across bins, to allow direct comparison (Booker et al., 2020a).
For statistical comparison of AIS length under control and treatment conditions an average of AIS length should be obtained for each condition and each animal and a parametric or non-parametric test performed as appropriate. Ideally, a linear mixed effect model should be applied to compare AIS length across treatments, with “Treatment” as a fixed effect and “animal” and “slice/section” selected as random effects.
Limitations
There are a number of technical and conceptual limitations to the current protocol. First, there is an inherent risk of damage to the slice each time it is handled. This can be overcome by using a wide-mouth transfer pipette and ensuring that all pumps are turned off before introducing the slice to the chamber
Second, slice health deteriorates over time, thus limiting the length of treatment that can be tested. Adaptation of this protocol to organotypic slices might allow for longer treatment windows.
Moreover, it is not always possible to label the AIS of the recorded cell, depending how deep in the tissue the cell was located. Resectioning of the slices into thinner sections could circumvent this limitation but it might compromise recovery of cell morphology.
Another limitation is the low data throughput. Multiple cells per slice need to be recorded pre-treatment in order to maximize the chance of being able to patch from these again.
Finally, this approach may not be the most efficient for shorter pharmacological or plasticity treatments (<1 h), in which case continual recording may be more appropriate. This should be decided during the experimental design and take into account the above factors.
Troubleshooting
Problem 1
Recordings from CA1 pyramidal cells are not reliably of high quality and cells do not form reliable seals (steps 9 and 10).
Potential solution
The most likely cause is poor slice quality. To remedy this, decrease the time it takes to remove the brain and place in ice-cold sucrose-ACSF and take greater care when handling the brain during placement in slicing chamber. Healthy slices should display multiple ovoid neurons located near to the slice surface (Figure 4B).
Also, double check if the solution composition is correct. Check the pH and osmolarity of solutions used for recording, as equilibrated with carbogen and warmed at the time of recording. Ideal pH should be 7.3 to 7.4 when measured at 30°C and osmolarity should be 290–310 mOsm.
Problem 2
Following 3 h of depolarization or osmotic control, no fluorescently labeled cells are present in the slice (step 17).
Potential solution
Check the following possibilities:
Is the slice in the same orientation as it was during the first recording session? Try turning the slice over and check for labeled cells.
Is a reliable gigaohm seal formed following first recording? Confirm the presence of the gigaohm seal by monitoring electrode retraction in the seal-test mode of the amplifier. Under IR-DIC confirm that the cell is maintained in the slice and that the nucleus has not been accidentally withdrawn as well (step 12).
Are the cells sufficiently deep within the slice, to prevent damage at the surface, from being stored for an extended period? Try to record from neurons that are embedded deeper within the slice.
Slice quality is not sufficient to maintain slices at 35°C for 3 h. See Problem 1 for suggestions of improving slice quality.
Record from more cells. Not every neuron survives 3 h of treatment, despite best efforts. A minimum of 3 cells normally yields at least 1 cell following 3 h treatment.
Is the solution composition correct? Check the pH and osmolarity of solutions used for recording, as equilibrated with carbogen and warmed at the time of recording. Ideal pH should be 7.3 to 7.4 when measured at 30°C and osmolarity should be 290–310 mOsm
Problem 3
No labeling for AnkyrinG is present in the slice (step 32; Figure 5C).
Potential solution
Are the confocal microscope imaging settings correct for the wavelengths required for excitation/emission of the used secondary antibodies? Does the confocal microscope detect the streptavidin labeling of the filled neurons? Confirm that the microscope is functioning correctly by imaging a known calibration standard (e.g., dye beads) or test samples in another microscope system (e.g., epifluorescent microscope)
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sam A. Booker, sbooker@ed.ac.uk.
Materials availability
All materials used in this protocol are commercially available.
Data and code availability
This study did not generate/analyze datasets/code.
Acknowledgments
The authors would like to thank our funders, the Simons Initiative for the Developing Brain (SFARI Grant: 529085) for facilitating our research. Second, we wish to thank Professors Peter C. Kind and David J.A. Wyllie and the members of their labs for help with developing these methods. Finally, our thanks to the IMPACT imaging facility at the University of Edinburgh for technical support with confocal imaging.
Author contributions
L.S.O. devised the protocol, performed experiments, and wrote the manuscript. S.A.B. devised the protocol, performed experiments, and wrote the manuscript. A,S, performed experiments and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Laura S. Oliveira, Email: l.oliveira@ed.ac.uk.
Sam A. Booker, Email: sbooker@ed.ac.uk.
References
- Booker S.A. Preparing acute brain slices from the dorsal pole of the hippocampus from adult rodents. J. Vis. Exp. 2020:e61669. doi: 10.3791/61699. [DOI] [PubMed] [Google Scholar]
- Booker S.A., Campbell G.R., Mysiak K.S., Brophy P.J., Kind P.C., Mahad D.J., Wyllie D.J. Loss of protohaem IX farnesyltransferase in mature dentate granule cells impairs short-term facilitation at mossy fibre to CA3 pyramidal cell synapses. J. Physiol. 2017;595:2147–2160. doi: 10.1113/JP273581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker S.A., De Oliveira L.S., Anstey N.J., Kozic Z., Dando O.R., Jackson A.D., Baxter P.S., Isom L.L., Sherman D.L., Hardingham G.E. Input-output relationship of ca1 pyramidal neurons reveals intact homeostatic mechanisms in a mouse model of fragile x syndrome. Cell Rep. 2020;32:107988. doi: 10.1016/j.celrep.2020.107988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker S.A., Song J., Vida I. Whole-cell patch-clamp recordings from morphologically-and neurochemically-identified hippocampal interneurons. J. Vis. Exp. 2014:e51706. doi: 10.3791/51706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favuzzi E., Deogracias R., Marques-Smith A., Maeso P., Jezequel J., Exposito-Alonso D., Balia M., Kroon T., Hinojosa A.J., Maraver E.F. Distinct molecular programs regulate synapse specificity in cortical inhibitory circuits. Science. 2019;363:413–417. doi: 10.1126/science.aau8977. [DOI] [PubMed] [Google Scholar]
- Grubb M.S., Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzmann A., Ergül N., Grossmann R., Schultz C., Wahle P., Engelhardt M. A period of structural plasticity at the axon initial segment in developing visual cortex. Front. Neuroanat. 2014;8:11. doi: 10.3389/fnana.2014.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman S.J., Schlögl A., Schmidt-Hieber C. Stimfit: quantifying electrophysiological data with Python. Front. Neuroinform. 2014;8:16. doi: 10.3389/fninf.2014.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E., Sakmann B. The patch clamp technique. Sci. Am. 1992;266:44–51. doi: 10.1038/scientificamerican0392-44. [DOI] [PubMed] [Google Scholar]
- Pan-Vazquez A., Wefelmeyer W., Sabater V.G., Neves G., Burrone J. Activity-dependent plasticity of axo-axonic synapses at the axon initial segment. Neuron. 2020;106:265–276.e6. doi: 10.1016/j.neuron.2020.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Method. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2013. R: A language and environment for statistical computing. [Google Scholar]
- Thome C., Kelly T., Yanez A., Schultz C., Engelhardt M., Cambridge S.B., Both M., Draguhn A., Beck H., Egorov A.V. Axon-carrying dendrites convey privileged synaptic input in hippocampal neurons. Neuron. 2014;83:1418–1430. doi: 10.1016/j.neuron.2014.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets/code.