

Abstract
Stress-inducible phosphoprotein 1 (STI1) acts as a neuroprotective factor in the ischemic brain and its levels are increased following ischemia. Previous work has suggested that some of these STI1 actions in a stroke model depend on the recruitment of bone marrow-derived stem cells to improve outcomes after ischemic insult. However, STI1 can directly increase neuroprotective signaling in neurons by engaging with the cellular prion protein (PrPC) and activating α7 nicotinic acetylcholine receptors (α7nAChR). Given that α7nAChR activation has also been involved in neuroprotection in stroke, it is possible that STI1 can have direct actions on neurons to prevent deleterious consequences of ischemic insults. Here, we tested this hypothesis by exposing primary neuronal cultures to 1-h oxygen-glucose deprivation (OGD) and reperfusion and assessing signaling pathways activated by STI1/PrPC. Our results demonstrated that STI1 treatment significantly decreased apoptosis and cell death in mouse neurons submitted to OGD in a manner that was dependent on PrPC and α7nAChR, but also on the activin A receptor 1 (ALK2), which has emerged as a signaling partner of STI1. Interestingly, pharmacological inhibition of the ALK2 receptor prevented neuroprotection by STI1, while activation of ALK2 receptors by bone morphogenetic protein 4 (BMP4) either before or after OGD was effective in decreasing neuronal death induced by ischemia. We conclude that PrPC/STI1 engagement and its subsequent downstream signaling cascades involving α7nAChR as well as the ALK2 receptor may be activated in neurons by increased levels of STI1. This signaling pathway protects neurons from ischemic insults.
Keywords: ALK2, prion protein, STIP1, Stress-inducible phosphoprotein 1, stroke, α7nAChR
Stress-inducible phosphoprotein I (STIP1, STI1) is a cochaperone for heat-shock protein 70 (Hsp70) and Hsp90 that has also extracellular functions, acting as a ‘cytokine-like’ molecule (Lackie et al. 2017). As a co-chaperone, STI1 helps with the transfer of client proteins from Hsp70 to Hsp90 by binding onto both simultaneously via its tetratricopeptide repeat domains to maintain protein quality control (Lassle et al. 1997; Schmid et al. 2012). Similarly to Hsp70, Hsp90 (Clayton et al. 2005; Lancaster and Febbraio 2005) and some of their co-chaperones, such as cyclophilin A (Suzuki et al. 2006), STI1 is secreted by cells, including glia in the nervous system (Fonseca et al. 2012; Beraldo et al. 2013; Hajj et al. 2013). STI1 secretion is carried out, at least in part, by exosomes (Hajj et al. 2013), and extracellular STI1 can signal to increase neuronal protection and differentiation (Zanata et al. 2002; Lopes et al. 2005; Caetano et al. 2008; Beraldo et al. 2010, 2013).
Extracellular STI1 interacts with the prion protein (PrPC) with high-affinity (KD ~ 100 nM; Zanata et al. 2002; Ostapchenko et al. 2013; Maciejewski et al. 2016), triggering calcium signaling in neurons that depends on α7 nicotinic acetylcholine receptors (α7nAChR; Beraldo et al. 2010; Ostapchenko et al. 2013). STI1-induced calcium influx can activate protein kinase A protecting retinal and hippocampal neurons against staurosporine-induced programmed cell death (Zanata et al. 2002; Lopes et al. 2005; Beraldo et al. 2010). In addition, STI1/PrPC engagement in primary neurons can induce differentiation through activation of the extracellular signal-regulated kinase 1 and 2, and protein synthesis by activation of phosphoinositide 3-kinase-Akt-mediated mechanistic target of rapamycin (Lopes et al. 2005; Roffe et al. 2010). STI1 is also secreted by a number of tumor cells (Wang et al. 2010, 2017b). In an ovarian tumor, secreted STI1 has been shown to signal via PrPC or via ALK2 receptors (Activin A receptor, type 1 or ACVR1; Tsai et al. 2012). Secreted STI1, acting in an autocrine and paracrine fashion, can bind and activate ALK2 to induce cell proliferation via SMAD-regulated transcription of target genes (Tsai et al. 2012). Both autocrine and paracrine actions of STI1 have been observed in tumors by activation of ALK2 and PrPC (Chen et al. 2017; Wang et al. 2017b).
PrPC, a glycosylphosphatidylinositol-anchored protein, seems to function as a main organizer of signal platforms at the cellular surface (Linden et al. 2008; Martins et al. 2010). In ischemic conditions, PrPC expression is increased in both human and mouse tissues (McLennan et al. 2004). Interestingly, PrPC-null mice show increased infarct volume following ischemia (Weise et al. 2006). On the other hand, transgenic mice with increased PrPC expression present decreased injury and dysfunction when compared to control animals, an observation that was mirrored by adenovirus-mediated over-expression of PrPC (Shyu et al. 2005; Weise et al. 2008). STI1 expression is also increased under ischemic conditions as a result of the direct binding of the hypoxia-inducible factor-1α onto the STI1 gene promoter (Lee et al. 2013). STI1 has been shown to recruit bone marrow-derived stem cells into ischemic brains and promote their proliferation in a PrPC-dependent way, ultimately facilitating neurological recovery following stroke (Lee et al. 2013). STI1 also directly prevents neuronal death in a PrPC-dependent manner in response to oxygen/glucose deprivation, which models ischemic conditions in cell culture (OGD; Beraldo et al. 2013). Accordingly, decreased expression of STI in heterozygous knockout mice increases ischemic damage in the middle cerebral artery occlusion model of stroke (Beraldo et al. 2013).
Although increased STI1 levels improve the outcomes in models of stroke (Beraldo et al. 2013; Lee et al. 2013), the mechanism by which STI1 protects neurons against ischemic insults is poorly understood. Furthermore, whether α7nAChRs participate in this process remains unknown. Interestingly, α7nAChRs have been shown to protect brain tissue in different models of ischemic and hemorrhagic stroke (Krafft et al. 2013; Sun et al. 2013), by modulating neuroinflammation (Han et al. 2014), neurogenesis (Wang et al. 2017a), and by activating neuroprotective signaling pathways (Dajas-Bailador et al. 2000; Kihara et al. 2001; Beraldo et al. 2010). Hence, understanding whether STI1 can endogenously activate these and other signaling pathways in ischemic neurons is relevant for the future development of STI1-based compounds. Here, we show that STI1 can prevent ischemia-induced apoptosis and that the neuroprotection by STI1 is also dependent on α7nAChRs and ALK2 receptors. Interestingly, bone morphogenetic protein 4 (BMP4), an agonist of ALK receptors, could also protect neurons against OGD and this effect was abolished in α7nAChR knockout mice, suggesting cross-talk between these two pathways. Our experiments reveal that STI1 activates multiple signaling pathways that cooperate to increase neuronal resilience in neurons subjected to ischemic insult.
Materials and methods
Materials
Recombinant mouse STI1 (His6-STI1) was generated and purified as described previously (Zanata et al. 2002; Soares et al. 2013; Maciejewski et al. 2016). Staurosporine (cat#PHZ1271; Invitrogen, Burlington, ON L7L 5Z1 Canada) was used as a positive control to induce apoptotic cell death (Chae et al. 2000). Methyllycaconitine citrate (MLA; Abcam, Cambridge, UK) was used to inhibit (Bertrand et al. 1997) and PNU 282987 (cat#ab120558; Abcam) to activate α7nAChR signaling (Hajos et al. 2005). LDN 193189 (cat#6053; Tocris, Burlington, Canada), a small molecule inhibitor of activin A receptor 1 (bone morphogenetic protein type 1 receptors), was used to inhibit ALK2 receptors (Cuny et al. 2008). BMP4 (cat#SRP3298; Sigma-Aldrich, Oakville, Ontario, Canada) dissolved in 10 mM citric acid was used as an agonist for ALK2 receptors (Chaikuad et al. 2012).
Animals
Hippocampal and cortical neurons were harvested from both male and female mouse embryos at embryonic day 17 and maintained in dishes coated with poly-l-lysine (Sigma-Aldrich) for 7 days (DIV) as described previously (Beraldo et al. 2013; Ostapchenko et al. 2013). The cells were maintained at 37°C and 5% CO2 in Neurobasal medium (Invitrogen) supplied with 1× penicillin/streptomycin mix (Invitrogen), 2 mM l-glutamine (Invitrogen), and 1× B27 supplement (Invitrogen). Half of the medium was replaced every 2–3 days. Prnp0/0 mice in a C57BL/6 background were kindly donated by Dr. Frank Jirik, University of Calgary (Tsutsui et al. 2008), and were maintained in our facility. Briefly, Prnp0/0 mice (RRID:MGI:2174709, Zurich I strain; Bueler et al. 1992) were backcrossed for several generations (N = 7–8) into C57BL/6 mice (Khosravani et al. 2008; Tsutsui et al. 2008). The mice were received in our laboratory and we continued to cross them with C57BL/6J mice obtained from the Jackson Laboratory (RRID:IMSR_JAX:000664). α7nAChR knockout in a C57BL/6J background (RRID:MGI:3050953; Orr-Urtreger et al. 1997) were obtained from the Jackson Laboratory and bred in our facility. Procedures were conducted in accordance with approved animal use protocols at the University of Western Ontario (2016/104) following Canadian Council of Animal Care and National Institutes of Health guidelines. Mice were maintained on a standard 12-h light cycle (7 am lights on, 7 pm lights off). The colony rooms were typically maintained at a temperature of 22°C with food and water provided ad libitum.
A new caspase 3 activity reporter mouse line named Apo mice (Nicholls et al. 2017) was used to identify live neurons undergoing apoptosis. This transgenic reporter line takes advantage of the bimolecular fluorescence complementation technique (Hu et al. 2002) instead of more laborious FRET imaging to generate fluorescence in response to caspase 3 activity. Namely, the fluorophore monomeric-Cerulean (Rizzo et al. 2004) is split into two pieces; neither piece is fluorescent on its own, but together the fragments reconstitute the fully functional fluorescent protein. The N- and C-terminal portions of Cerulean are targeted to separate subcellular compartments. In addition, both fragments contain a PEST protein degradation sequence, which shortens protein half-life to approximately 1 h. Nuclear export and nuclear localization signals and PEST sequences are linked to the N- and C-Cerulean portions by DEVDG, caspase 3/7 cleavage sequences, and once caspase 3/7 is activated, the N and C-Cerulean portions are reconstituted with the help of complementary inteins. Apoptosis, therefore, can be quantified by Cerulean fluorescence in cells obtained from these reporter mice. Cultures from a minimum of four individual embryos of both sexes were obtained and analyzed for each OGD experiment. Fluorescence was quantified by using an FV1000 confocal microscope (Olympus, Richmond Hill, Ontario, Canada) equipped with 10×/0.3 or 20×/0.4 dry objectives. Cerulean fluorescence was detected using excitation at 405 nm and emission at 470–485 nm. Integrated fluorescence was divided by total cell number and normalized to the control cells that were not subjected to OGD using ImageJ software (NIH, Bethesda, MD, USA).
Oxygen–glucose deprivation
Original media were replaced with a glucose-free Neurobasal medium (Invitrogen) with 1× penicillin/streptomycin mix (Invitrogen), 2 mM l-glutamine (Invitrogen), and 1× B27 supplement (Invitrogen). The cells were then placed in an incubator with 0% oxygen for 1 h. Following OGD, the original medium with glucose was restored and the cells were allowed to recover under normal conditions for 3, 12, or 24 h.
Immunofluorescence microscopy
Primary hippocampal neuronal cultures grown on glass coverslips for 7 days were fixed using 2% paraformaldehyde and permeabilized using phosphate-buffered saline (PBS) containing 0.5% Triton X-100 (Sigma-Aldrich). The cells were then blocked using 2% bovine serum albumin in PBS containing 0.1% Triton X-100 (PBS-T) before applying the primary antibodies against NeuN (1 : 200, cat#ab104224, RRID:AB_10711040) and ALK2 (1 : 200, cat#ab60157, RRID:AB_940117, both Abcam). After overnight incubation, the cultures were washed with PBS-T and incubated for 1 h with secondary antibodies, followed by nuclear staining with Hoechst 33342 dye (Invitrogen). Coverslips were mounted using Immu-mount medium (ThermoFisher, Mississauga, Ontario, Canada) and the images were collected using an LSM 510 confocal microscope (Zeiss, Toronto, Ontario, Canada), equipped with a 10×/0.3 objective.
Live/dead assay
A live/dead mammalian cell viability assay (Invitrogen) was used to determine cell death as described previously (Soares et al. 2013; Beraldo et al. 2016). Briefly, the cultures were incubated with the calcein-AM/ethidium homodimer mix according to the manufacturer’s instructions in the original medium for 45 min and then washed three times with Krebs-Ringer HEPES buffer. Images were collected from at least six random fields per dish using an LSM 510 confocal microscope (Zeiss), equipped with a 10×/0.3 objective, using 488 nm excitation and emission filters 505–525 nm (for calcein) and 650 nm LP (for ethidium homodimer). Cell death levels were quantified as the percentage of dead cells relative to the total number of cells. For all the Live/dead assays the experimenter was blinded during imaging acquisition and quantification. No randomization was performed for assignment of cell culture treatments.
Statistical analysis
The study was not pre-registered. A pre-study power analysis was not conducted, but for all experiments data from at least four independent embryos were obtained, which is the standard in the field. Integrated fluorescence and cell number in cultures were obtained using Measure and Cell Counter instruments in ImageJ software. All data were expressed as the mean ± standard error, and complete datasets were analyzed by one-way anova or two-way anova following by Tukey’s post hoc test using GraphPad 5.0 (Prism, La Jolla, CA, USA).
Results
Extracellular STI1 decreases apoptosis and neuronal death induced by OGD
Previous experiments indicated that STI1-activated signaling can protect neurons against apoptotic insults, such as treatment with staurosporine (Lopes et al. 2005; Beraldo et al. 2010) or OGD (Beraldo et al. 2013). To investigate the mechanisms by which STI1 protects neurons against OGD-induced cell death, hippocampal neurons obtained from a caspase 3 reporter mouse were submitted to 1-h OGD treatment and reperfusion in the presence or absence of 1 μM STI1. Caspase 3 activity and cell death were measured in the same fields using the fluorescence from the mCerulean protein and ethidium homodimer 1 dye, respectively. Cells were imaged at 3, 12, and 24 h of reperfusion. Increased caspase 3 fluorescence was observed in neurons after 1-h OGD exposure as early as 3 h after treatment (Fig. 1a and b). Cerulean fluorescence decreased at 12 and 24 h after OGD. Interestingly, treatment of neurons with STI1 significantly decreased caspase-induced fluorescence (Fig. 1b). Furthermore, cell death in post-OGD cultures pre-treated with STI1 was also significantly lower than in cultures with no STI1 added at all three time points (Fig. 1c). To test whether STI1 prevents neuronal death in a PrPC-dependent fashion, wild-type and Prnp0/0 (homozygous for PrPC gene deletion) hippocampal neurons were treated with 1 μM STI1 and submitted to 1-h OGD and 24-h reperfusion. Treatment with STI1 significantly decreased cell death induced by OGD in wild-type neurons (Fig. 1d and e). However, STI1 was unable to rescue cell death in Prnp0/0 cultures (Fig. 1d and e).
Fig. 1.

Caspase 3 activity and cell death in neurons treated with stress-inducible phosphoprotein 1 (STI1) and oxygen-glucose deprivation (OGD). Hippocampal neurons were treated with 1 μM STI1 for 30 min and then subjected to 1 h OGD. (a–c) Apo mouse hippocampal neurons from four different embryos were imaged after 3, 12, or 24 h reperfusion. (a) Representative images of mCerulean (green) and ethidium homodimer 1 (DEAD, red) fluorescence merged with transmitted light channel. Caspase 3 activity in Apo mouse hippocampal neurons was quantified in arbitrary units as fluorescence per cell normalized to the control. Cell death was quantified as percentage of total cells that were dead. (d and e) Wild-type (n = 4) and Prnp0/0 (n = 4) hippocampal neurons were treated with 1 μM STI1 for 30 min and then subjected to 1 h OGD followed by 24 h reperfusion. (d) Representative images of calcein (LIVE, green) and ethidium homodimer 1 (DEAD, red) fluorescence. (e) Quantification was performed as described in Methods. Results are presented as mean ± SEM; data were analyzed and compared by one-way anova (b and c) or two-way anova (e) and Tukey’s post hoc test. ****p < 0.0001, ***p < 0.001 compared to control. Scale bar = 100 μm.
STI1 engages α7nAChR for neuroprotection
Our previous work suggested that STI1 can engage a complex containing PrPC and α7nAChR in hippocampal neurons for neuronal signaling and neuroprotection against staurosporine-mediated cell death (Beraldo et al. 2010). However, in other neuronal types and cancer cells STI1 can activate signaling independent of PrPC, α7nAChR, or both (Arruda-Carvalho et al. 2007; Tsai et al. 2012; Santos et al. 2013). To determine the potential role for α7nAChR in STI1-mediated neuroprotection, wild-type hippocampal neurons were treated with various concentrations of the α7nAChR-selective antagonist MLA for 30 min prior to the treatment with 1 μM STI1 and 1-h OGD/24-h reperfusion. MLA treatment inhibited STI1-induced survival of neurons during OGD (Fig. 2a). To extend these pharmacological experiments, we then used cultured hippocampal neurons obtained from α7nAChR-knockout mice. Hippocampal neurons were treated with 1 μM STI1 followed by 1-h OGD/24-h reperfusion. STI1 neuroprotection against OGD, present in wild-type cultures, was lost in α7nAChR-knockout hippocampal neurons (Fig. 2b). These results together suggest that STI1 engages PrPC/α7nAChRs to rescue neurons against OGD/reperfusion-induced cell death.
Fig. 2.
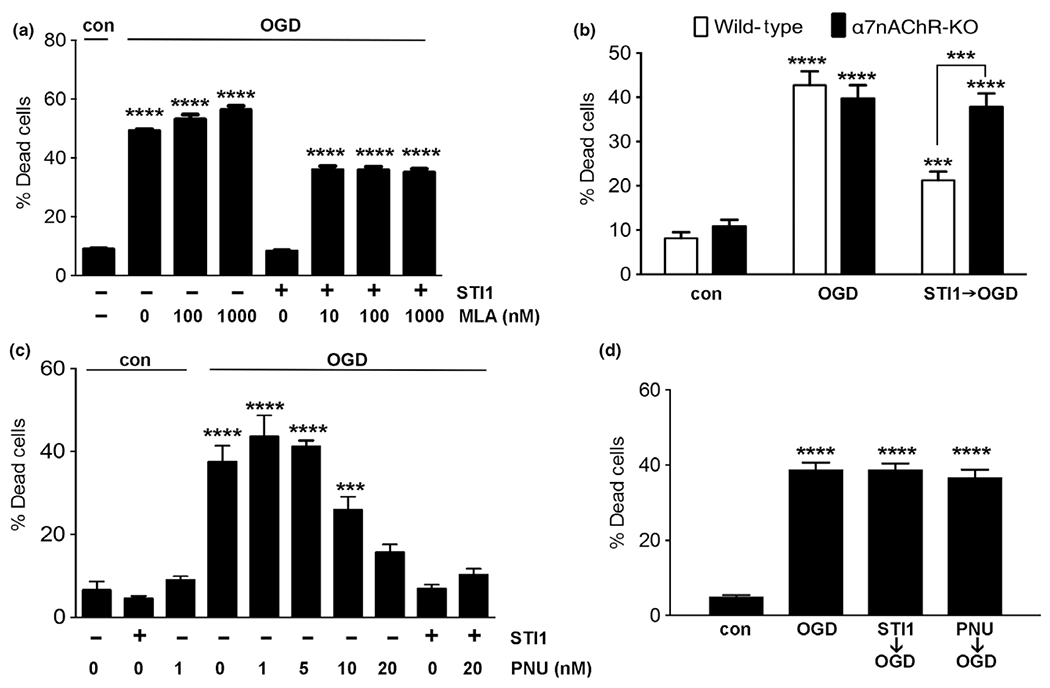
Effect of activation or inhibition of α7nAChR receptors on stress-inducible phosphoprotein 1 (STI1) neuroprotection against oxygen-glucose deprivation (OGD). (a) Wild-type hippocampal neurons were treated with MLA (2–1000 nm) and/or STI1 (1 μM) for 30 min and then subjected to 1 h OGD. (b) Wild-type hippocampal neurons and α7nAChR-KO neurons were treated with 1 μM STI1 for 30 min and then subjected to 1 h OGD. Wild-type (c) and α7nAChR-KO (d) hippocampal neurons were treated with STI1 (1 μM) and/or PNU 282987 (PNU, 0–20 μM) for 30 min and then subjected to 1 h OGD. Cultures were imaged by confocal microscopy after 24 h of reperfusion and analyzed as described in Methods. Results are presented as mean ± SEM; data were analyzed and compared by one-way anova (a, c and d) or two-way anova (b) and Tukey’s post hoc test. ****p < 0.0001, ***p < 0. 001.
Activation of α7nAChR protects neurons against OGD
Previous work suggested that α7nAChRs may provide a new pathway to interfere with outcomes in hemorrhagic stroke (Krafft et al. 2013) and activation of nicotinic receptors can protect neurons against OGD (Hejmadi et al. 2003). To further investigate the potential for activation of α7nAChR to protect neurons against OGD-induced cell death, we used PNU 282987, a selective agonist for α7nAChR. Treatment with variable concentrations of PNU 282987 for 30 min prior to exposure to OGD rescued neuronal death in wild-type mouse hippocampal cultures in a dose-dependent way (Fig. 2c). Furthermore, we tested whether this effect of PNU 282987 was selective for α7nAChRs. Indeed, similar to STI1, PNU 282987 was unable to prevent OGD-induced neuronal death in α7nAChR-knockout neuronal cultures (Fig. 2d).
ALK2 receptor inhibition blocks STI1 neuroprotection
Previous experiments have shown that STI1 can also modulate signaling pathways in dorsal root ganglia neurons (Santos et al. 2013), neurons (Arruda-Carvalho et al. 2007), and cancer cells (Tsai et al. 2012) independently of α7nAChRs. In ovarian cancer, ALK2 activation in response to STI1 induces cell proliferation (Tsai et al. 2012). To further test whether other signaling pathways can be activated by STI1 we focused on ALK2, a receptor that is still poorly studied in the brain. Expression profile obtained from Allen Mouse Brain Atlas (Lein et al. 2007) indicates selective expression of ALK2 receptors in the hippocampus (http://mouse.brain-map.org/experiment/show/69514714) (Fig. 3a). Staining of cultured mouse hippocampal neurons for ALK2 showed membrane expression in neurons (Fig. 3b). To determine the potential involvement of ALK2 in STI1-mediated neuronal protection against OGD, neurons were treated with an ALK2 small molecule inhibitor, LDN 193198, and 1 μM STI1 prior to exposure to OGD. ALK2 inhibition did not increase neuronal death in the absence of STI1, however, the compound prevented STI1 neuroprotection in a dose-dependent manner (Fig. 3c). To further test the possibility that ALK2 activation can modulate cell death in response to ischemia reperfusion, we used the ALK2 ligand BMP4. Treatment of neurons with BMP4 for 30 min significantly decreased cell death induced by ischemia reperfusion in a dose-dependent way (Fig. 3d). To test the possibility of cross-talk between ALK2, α7nAChR, and the prion protein, we bypassed STI1 by using BMP4 and PNU 282987 to activate their respective receptors and compared their effects in wild-type, α7nAChR-knockout, and Prnp0/0 neurons. Surprisingly, when we investigated the effects of BMP4 in the absence of α7nAChRs, we found that the neuroprotection afforded by BMP4 (100 ng/mL) was completely abolished in α7nAChR-knockout neurons (Fig. 3e), suggesting a role for α7nAChRs in the rescuing effect of BMP4 during OGD. In contrast, in the absence of PrPC both BMP4 and PNU 282987 were still able to prevent OGD-induced neuronal death (Fig. 3f), suggesting that PrPC is not required to directly modulate these two receptors.
Fig. 3.
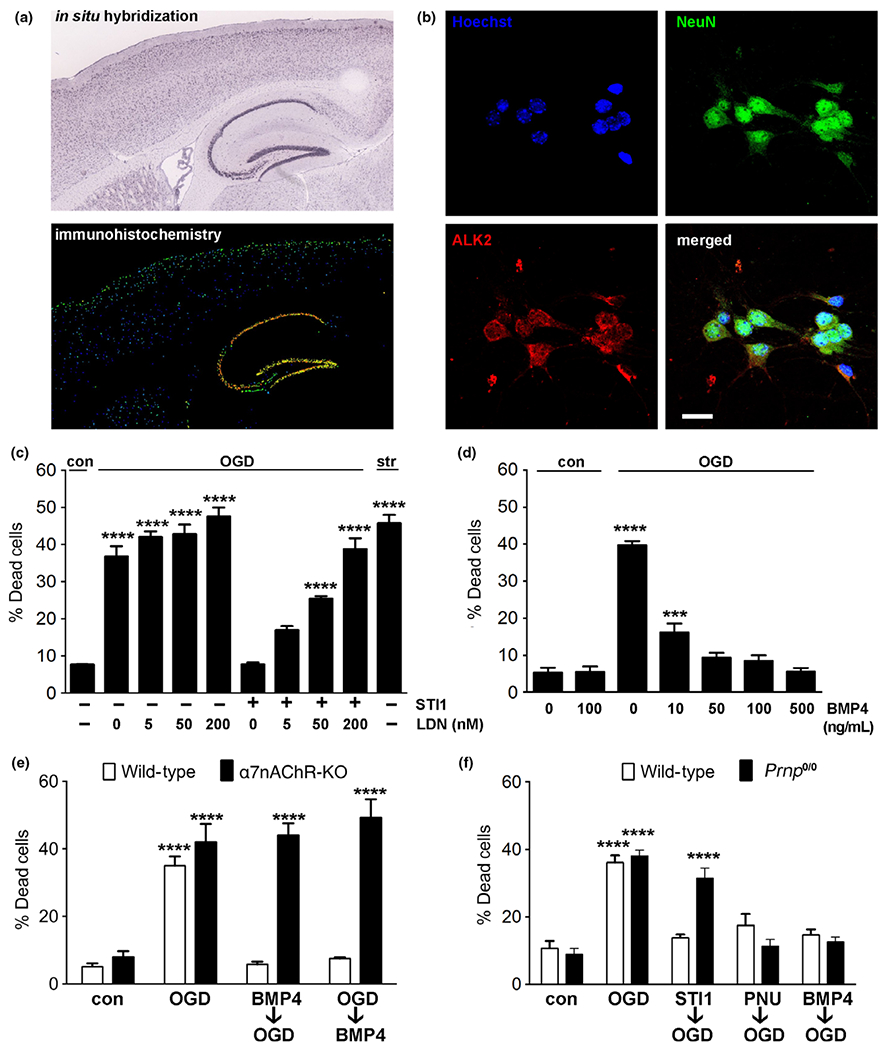
ALK2 receptors are involved in the neuroprotective effects of stress-inducible phosphoprotein 1 (STI1) against oxygen-glucose deprivation (OGD). (a) An Allen Mouse Brain Atlas image showing in situ hybridization for the ALK2 gene acvr1 (top) and ALK2 immunostaining (bottom) on a mouse brain sagittal section. Warmer colors indicate higher gene expression. (b) Wild-type murine E17 primary hippocampal neurons after 7 days in culture were stained with nuclear marker Hoechst (blue), antibody against mature neuronal marker NeuN (green), and ALK2 antibody (red). ALK2 is enriched in the cell membrane in NeuN-positive cells. Scale bar = 20 μm. (c) Wild-type hippocampal neurons were treated with LDN 193189 (LDN, 0–200 nM) and/or 1 μM STI1 for 30 min and then subjected to 1 hOGD. Staurosporine(200 nM) was used as a positive control. (d) Wild-type hippocampal neurons were treated with 1 μM STI1 and/or the ALK2 agonist BMP4 (0–500 ng/mL) for 30 min and then subjected to 1 h OGD. (e) Wild-type and α7nAChR-KO hippocampal neurons were treated with 0 or 100 ng/mL BMP4 30 min before (BMP4→OGD) or immediately after being subjected to 1 h OGD (OGD→BMP4). (f) Wild-type and Prnp0/0 hippocampal neurons were treated with 1 μM STI1, 20 μM PNU 282987 (PNU), or 100 ng/mL BMP4 and then subjected to 1 hOGD. After 24 hreperfusion the cultures (n = 4) were imaged and analyzed as described in Methods. Results are presented as mean ± SEM; data were analyzed by one-way anova (c and d) or two-way anova (e) and Tukey’s post hoc test. ****p < 0.0001, ***p < 0.001.
So far, we have shown that STI1 addition prior to OGD activated neuroprotective pathways via α7nAChR and ALK2. However, considering the high demand for pharmacological intervention in post-stroke scenarios, it is important to test whether STI1 can activate neuroprotective pathways and direct prevent neuronal death after the onset of ischemia. For this, we compared treatments of neuronal cultures with STI1, PNU 282987, or BMP4 prior to inducing ischemia and immediately after the OGD treatment. STI1 and PNU 282987 prevented neuronal death only if neurons are treated prior to the OGD exposure, but not when the neurons were exposed to them during reperfusion (Fig. 4a). Interestingly, BMP4 was equally effective to protect neurons against OGD-induced cell death when it was used either prior to OGD or during the reperfusion period (Figs 3e and 4b). Of note, prevention of neuronal death by BMP4 was attenuated not only by the ALK2 selective small molecule inhibitor, LDN 193189, but also by inhibition of α7nAChRs by MLA (Fig. 4b). The latter result corroborates the findings in α7nAChR-knockout neurons (Fig. 3e).
Fig. 4.
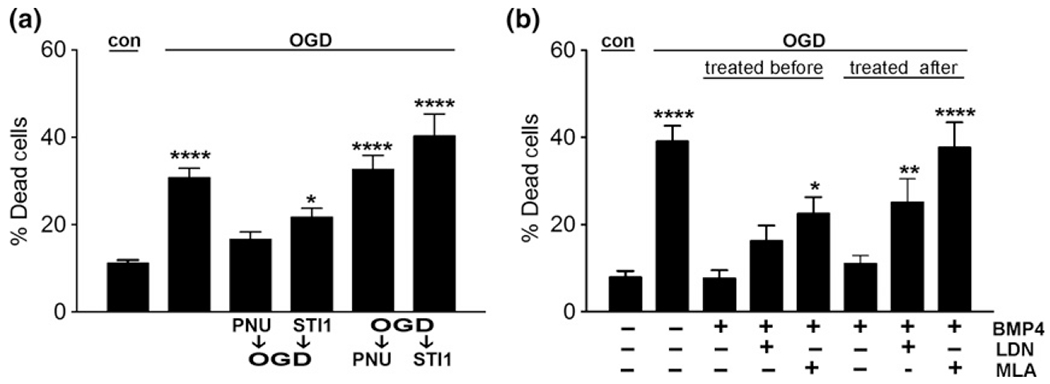
Modulation of α7nAChR and ALK2 pathways during oxygen-glucose deprivation (OGD) and reperfusion. (a) Wild-type hippocampal neurons were treated with 1 μM stress-inducible phosphoprotein 1 (STI1) or 20 μM PNU 282987 (PNU) before or immediately after 1 h OGD followed by reperfusion. (b) Wild-type hippocampal neurons were treated with 10 nM methyllycaconitine citrate (MLA), 200 nM LDN 193189 (LDN), and/or 100 ng/mL BMP4 before or immediately after 1 h OGD followed by reperfusion. Cultures (n = 4) underwent reperfusion for 24 h and were imaged and analyzed as described in Methods. Results are presented as mean ± SEM; data were analyzed and compared by one-way anova (a and b) and Tukey’s post hoc test. ****p < 0.0001, **p < 0.01, *p < 0.05 compared to control.
Discussion
Our experiments provide new insights on how extracellular STI1 directly protects neurons against ischemic insults via PrPC and introduces a new therapeutic target in ischemia, the ALK2 receptor. The prion protein functions as a scaffold to regulate multiple signaling pathways involved in neuronal protection and toxicity (Linden et al. 2008; Martins et al. 2010). In ischemic tissue, expression of the prion protein has been suggested to be neuroprotective (Shyu et al. 2005; Weise et al. 2006, 2008). PrPC expression is increased after ischemia in mice, which correlates with the severity of brain lesion (Weise et al. 2004; Mitsios et al. 2007). Remarkably, STI1 is also up-regulated in ischemic brains from mice and humans (Lee et al. 2013), and STI1 secretion from astrocytes is increased after OGD treatment (Beraldo et al. 2013; Lee et al. 2013). Reduced levels of STI1 increased stroke-mediated brain damage, whereas increased STI1 levels in vitro and in vivo protected against ischemia and improved functional recovery (Beraldo et al. 2013; Lee et al. 2013). While STI1-induced migration of peripheral blood stem cells to the site of ischemic injury is an important mechanism for STI1 anti-ischemic action (Lee et al. 2013), extracellular STI1 can also protect neurons directly against a number of insults that trigger apoptosis (Beraldo et al. 2010, 2013; Soares et al. 2013). STI1 induces the endocytosis of PrPC and these two molecules are quickly directed to distinct compartments after internalization (Caetano et al. 2008). Despite the change in PrPC and STI1 levels in ischemia, increased or decreased STI1 levels do not affect the expression of PrPC (Beraldo et al. 2015). These data together indicate the potential importance of PrPC/STI1 complex not only in physiological conditions, as previously demonstrated (Lopes et al. 2005; Coitinho et al. 2007; Caetano et al. 2008; Beraldo et al. 2010, 2013; Martins et al. 2010; Roffe et al. 2010), but also in response to ischemic insults and other types of cellular stresses.
PrPC can interact with several transmembrane receptors to modulate neuronal survival (Beraldo et al. 2010, 2011) or toxicity (You et al. 2012; Um et al. 2013). Notably, we and others have shown that PrPC interacts directly with mGluR5, a mechanism responsible for the toxicity of amyloid β peptides in Alzheimer’s disease (Beraldo et al. 2011, 2016; Um et al. 2013). The PrPC-mGluR5 complex also seems to contribute to alpha-synuclein toxicity (Ferreira et al. 2017). PrPC can also interact with α7nAChRs to mediate signaling by STI1 (Beraldo et al. 2010). Remarkably, activation of α7nAChRs decreases tissue damage by ischemic and hemorrhagic experimental stroke (Hejmadi et al. 2003; Krafft et al. 2013; Han et al. 2014).
Our results suggest that neurons are protected against ischemia reperfusion by STI1 at least partially via α7nAChRs. Treatment of neurons with MLA, a specific and competitive α7nAChR antagonist (Bertrand et al. 1997), abolished the neuroprotective effect of STI1 against OGD. The role of α7nAChR in STI1-mediated neuroprotection against OGD was confirmed in α7nAChR-knockout neurons, for which STI1 showed no protective effect. Moreover, wild-type neurons treated with α7nAChR agonist PNU 282987 had significantly lower OGD-induced cell death, an effect absent in α7nAChR-knockout neurons. In mice, PNU 282987 has been effective in attenuating damages from intracerebral hemorrhage (Hijioka et al. 2012) and ischemic stroke (Wang et al. 2017a). Importantly, previous studies have shown that administration of α7nAChR agonists to humans is well tolerated (Deutsch et al. 2008). Although we showed here that neuroprotection by STI1 against OGD in neurons is dependent on α7nAChR, activation of these receptors in non-neuronal cells in vivo is also known to have anti-inflammatory effects. Future studies should explore if the suppression of inflammatory cascades plays a role in the STI1-PrPC-α7nAChR mechanism of neuroprotection.
STI1 has also been shown to activate cell signaling pathways independent of PrPC. In ovarian cancer cells, secreted STI1 acts as a ligand for the ALK2 receptors leading to cancer cell proliferation (Tsai et al. 2012). Activins are members of the transforming growth factor β family and transduce their signals through type I and II receptor serine/threonine kinases (Lotinun et al. 2012; Link et al. 2016). Activin signaling pathways are involved in cellular proliferation, differentiation, and apoptosis. Interestingly the activin gene is transiently up-regulated in ischemic insults (Mukerji et al. 2007) and this up-regulation is associated with the neuroprotective effects of activin pathways in the nervous system (Tretter et al. 2000; Mukerji et al. 2007; Link et al. 2016). These receptors were previously detected in the hippocampus and we confirmed their cell surface expression in hippocampal neuronal cultures. Interestingly, in agreement with the recent evidence that STI1 can signal via ALK2 in tumor cells, we found that treatment of neurons with LDN 193189, a specific inhibitor of the intracellular kinase domain of the ALK2 receptor, important for its downstream signaling (Horbelt et al. 2015), significantly decreased STI1 protection against OGD. Following this observation, we tested whether direct activation of ALK2 receptors protects neurons against OGD. For this, we used BMP4, an established agonist for ALK2 (de Sousa Lopes et al. 2004; Medici et al. 2010). BMP4 is a family member of bone morphogenetic proteins that are involved in cellular functions such as growth and differentiation (Panchision and McKay 2002), as well as neuronal apoptosis during early development (Furuta et al. 1997). Treatment of neurons with BMP4 significantly decreased cell death following OGD, further suggesting that modulation of activin receptors protects against ischemia. Using PrP-null neurons, we established that whereas neuroprotection by STI1 depends on PrPC, both direct activation of ALK2 or α7nAChRs can bypass this PrPC-dependent step. Unexpectedly, neuroprotection against OGD by BMP4 was not observed in neurons from α7nAChR-knockout mice, and neuroprotection was attenuated by MLA, suggesting a functional interaction between ALK2 and α7nAChRs. Future studies should focus on potential mechanisms for this interaction.
Although our experiments provide initial insight on how pre-treatment with STI1 may protect neurons against ischemia, they also show that using STI1 or PNU 282987 during reperfusion is not as effective in providing neuroprotection. Because both STI1 and activation of α7nAChRs have demonstrated to be effective in vivo to prevent ischemic injury, it is possible that such treatments in vivo may elicit additional pathways in non-neuronal cells that are relevant for stroke. For example, STI1 appears to induce the migration of bone marrow-derived stem cells to the sites of injury, which are known to help in improving outcomes in stroke (Lee et al. 2013). Moreover, α7nAChR activation can also have effects on glia and immune responses, which may also contribute to the improvement observed in vivo (Han et al. 2014). In contrast, BMP4 could provide direct protection to neurons even after exposure to ischemic insult, suggesting a greater potential for translation beyond biochemical studies. Future experiments should focus on the potential use of BMP4 in models of stroke in vivo.
In summary, our experiments provide new light on the mechanisms by which STI1 can trigger signaling pathways to prevent apoptosis in response to ischemic insult (Fig. 5). We found that in addition to α7nAChRs, ALK2 receptors also seem to be involved in STI1 neuroprotection, and there is a functional cross-talk between these two pathways. Extracellular STI1 seems to afford neurons with the ability to become more resilient to apoptosis by activating a multiprotein signaling complex organized by PrPC.
Fig. 5.

Schematic of the proposed model of the α7nAChR, PrPC, ALK2, and their interaction with stress-inducible phosphoprotein 1 (STI1). Extracellular STI1 has been shown to act as a ligand for the glycosylphosphatidylinositol-anchored PrPC leading to calcium influx via the α7nAChR. Our data indicate that the ALK2 receptor plays a role in hippocampal neurons as a coreceptor for STI1 leading to neuroprotection in conjunction to PrPC with a possible cross-talk with α7nAChRs. Arrow heads indicate activation, while blunt heads indicate inhibition.
Acknowledgments and conflict of interest disclosure
This work was supported by the Canadian Institute of Health Research (MOP 136930, MOP 126000, and MOP 89919), the Canadian Foundation for Innovation, and the Ontario Research Fund (M.A.M.P., V.F.P.). The authors declare no competing financial interests. Marco Prado is a handling editor for the Journal of Neurochemistry.
All experiments were conducted in compliance with the ARRIVE guidelines.
Abbreviations used:
- Akt
protein kinase B
- ALK2
activin A receptor 1
- anova
analysis of variance
- BMDC
bone marrow-derived stem cells
- BMP4
bone morphogenetic protein 4
- BSA
bovine serum albumin
- ERK1/2
extracellular signal-regulated kinase 1 and 2
- Hsp70
heat-shock protein 70
- Hsp90
heat-shock protein 90
- MCAO
middle cerebral artery occlusion
- MLA
methyllycaconitine citrate
- mTOR
mechanistic target of rapamycin
- OGD
oxygen-glucose deprivation
- PI3K
phosphoinositide 3-kinase
- PrPC
cellular prion protein
- STI1
stress-inducible phosphoprotein 1
- α7nAChR
α7 nicotinic acetylcholine receptor
References
- Arruda-Carvalho M, Njaine B, Silveira MS, Linden R and Chiarini LB (2007) Hop/STI1 modulates retinal proliferation and cell death independent of PrPC. Biochem. Biophys. Res. Comm 361, 474–480. [DOI] [PubMed] [Google Scholar]
- Beraldo FH, Arantes CP, Santos TG, Queiroz NG, Young K, Rylett RJ, Markus RP, Prado MA and Martins VR (2010) Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem 285, 36542–36550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo FH, Arantes CP, Santos TG et al. (2011) Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 25, 265–279. [DOI] [PubMed] [Google Scholar]
- Beraldo FH, Soares IN, Goncalves DF et al. (2013) Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. 27, 3594–3607. [DOI] [PubMed] [Google Scholar]
- Beraldo FH, Thomas A, Kolisnyk B et al. (2015) Hyperactivity and attention deficits in mice with decreased levels of stress-inducible phosphoprotein 1 (STIP1). Dis. Model. Mech 8, 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beraldo FH, Ostapchenko VG, Caetano FA et al. (2016) Regulation of amyloid beta oligomer binding to neurons and neurotoxicity by the prion protein-mGluR5 complex. J. Biol. Chem 291, 21945–21955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand S, Devillers-Thiery A, Palma E, Buisson B, Edelstein SJ, Corringer PJ, Changeux JP and Bertrand D (1997) Paradoxical allosteric effects of competitive inhibitors on neuronal alpha7 nicotinic receptor mutants. NeuroReport 8, 3591–3596. [DOI] [PubMed] [Google Scholar]
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M and Weissmann C (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582. [DOI] [PubMed] [Google Scholar]
- Caetano FA, Lopes MH, Hajj GN et al. (2008) Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci 28, 6691–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae HJ, Kang JS, Byun JO, Han KS, Kim DU, Oh SM, Kim HM, Chae SW and Kim HR (2000) Molecular mechanism of staurosporine-induced apoptosis in osteoblasts. Pharmacol. Res 42, 373–381. [DOI] [PubMed] [Google Scholar]
- Chaikuad A, Alfano I, Kerr G et al. (2012) Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem 287, 36990–36998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Xu L, Su T et al. (2017) Autocrine STIP1 signaling promotes tumor growth and is associated with disease outcome in hepatocellular carcinoma. Biochem. Biophys. Res. Comm 493, 365–372. [DOI] [PubMed] [Google Scholar]
- Clayton A, Turkes A, Navabi H, Mason MD and Tabi Z (2005) Induction of heat shock proteins in B-cell exosomes. J. Cell Sci 118, 3631–3638. [DOI] [PubMed] [Google Scholar]
- Coitinho AS, Lopes MH, Hajj GN et al. (2007) Short-term memory formation and long-term memory consolidation are enhanced by cellular prion association to stress-inducible protein 1. Neurobiol. Dis 26, 282–290. [DOI] [PubMed] [Google Scholar]
- Cuny GD, Yu PB, Laha JK et al. (2008) Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett 18, 4388–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Lima PA and Wonnacott S (2000) The alpha7 nicotinic acetylcholine receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a Ca(2+) dependent mechanism. Neuropharmacology 39, 2799–2807. [DOI] [PubMed] [Google Scholar]
- Deutsch SI, Schwartz BL, Schooler NR, Rosse RB, Mastropaolo J and Gaskins B (2008) First administration of cytidine diphosphocholine and galantamine in schizophrenia: a sustained alpha7 nicotinic agonist strategy. Clin. Neuropharmacol 31, 34–39. [DOI] [PubMed] [Google Scholar]
- Ferreira DG, Temido-Ferreira M, Miranda HV et al. (2017) Alpha-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci 20, 1569–1579. [DOI] [PubMed] [Google Scholar]
- Fonseca AC, Romao L, Amaral RF, Assad Kahn S, Lobo D, Martins S, Marcondes de Souza J, Moura-Neto V and Lima FR (2012) Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience 200, 130–141. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW and Hogan BL (1997) Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124, 2203–2212. [DOI] [PubMed] [Google Scholar]
- Hajj GN, Arantes CP, Dias MV et al. (2013) The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell. Mol. Life Sci 70, 3211–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos M, Hurst RS, Hoffmann WE, Krause M, Wall TM, Higdon NR and Groppi VE (2005) The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetizedrats. J. Pharmacol. Exp. Ther 312, 1213–1222. [DOI] [PubMed] [Google Scholar]
- Han Z, Li L, Wang L, Degos V, Maze M and Su H (2014) Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J. Neurochem 131, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejmadi MV, Dajas-Bailador F, Barns SM, Jones B and Wonnacott S (2003) Neuroprotection by nicotine against hypoxia-induced apoptosis in cortical cultures involves activation of multiple nicotinic acetylcholine receptor subtypes. Mol. Cell Neurosci 24, 779–786. [DOI] [PubMed] [Google Scholar]
- Hijioka M, Matsushita H, Ishibashi H, Hisatsune A, Isohama Y and Katsuki H (2012) alpha7 Nicotinic acetylcholine receptor agonist attenuates neuropathological changes associated with intracerebral hemorrhage in mice. Neuroscience 222, 10–19. [DOI] [PubMed] [Google Scholar]
- Horbelt D, Boergermann JH, Chaikuad A, Alfano I, Williams E, Lukonin I, Timmel T, Bullock AN and Knaus P (2015) Small molecules dorsomorphin and LDN-193189 inhibit myostatin/GDF8 signaling and promote functional myoblast differentiation. J. Biol. Chem 290, 3390–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Toft D, Anselmo D and Wang X (2002) In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone proteins. J. Virol 76, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravani H, Zhang Y, Tsutsui S et al. (2008) Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Gen. Physiol 131, i5. [DOI] [PubMed] [Google Scholar]
- Kihara T, Shimohama S, Sawada H, Honda K, Nakamizo T, Shibasaki H, Kume T and Akaike A (2001) alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. Biol. Chem 276, 13541–13546. [DOI] [PubMed] [Google Scholar]
- Krafft PR, Caner B, Klebe D, Rolland WB, Tang J and Zhang JH (2013) PHA-543613 preserves blood-brain barrier integrity after intracerebral hemorrhage in mice. Stroke 44, 1743–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackie RE, Maciejewski A, Ostapchenko VG, Marques-Lopes J, Choy WY, Duennwald ML, Prado VF and Prado MAM (2017) The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Front. Neurosci 11, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster GI and Febbraio MA (2005) Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J. Biol. Chem 280, 23349–23355. [DOI] [PubMed] [Google Scholar]
- Lassle M, Blatch GL, Kundra V, Takatori T and Zetter BR (1997) Stress-inducible, murine protein mSTI1. Characterization of binding domains for heat shock proteins and in vitro phosphorylation by different kinases. J. Biol. Chem 272, 1876–1884. [DOI] [PubMed] [Google Scholar]
- Lee SD, Lai TW, Lin SZ et al. (2013) Role of stress-inducible protein-1 in recruitment of bone marrow derived cells into the ischemic brains. EMBO Mol. Med 5, 1227–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I and Brentani RR (2008) Physiology of the prion protein. Physiol. Rev 88, 673–728. [DOI] [PubMed] [Google Scholar]
- Link AS, Zheng F and Alzheimer C (2016) Activin signaling in the pathogenesis and therapy of neuropsychiatric diseases. Front. Mol. Neurosci 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes MH, Hajj GN, Muras AG, Mancini GL, Castro RM, Ribeiro KC, Brentani RR, Linden R and Martins VR (2005) Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci 25, 11330–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotinun S, Pearsall RS, Horne WC and Baron R (2012) Activin receptor signaling: a potential therapeutic target for osteoporosis. Curr. Mol. Pharmacol 5, 195–204. [DOI] [PubMed] [Google Scholar]
- Maciejewski A, Ostapchenko VG, Beraldo FH, Prado VF, Prado MA and Choy WY (2016) Domains of STIP1 responsible for regulating PrPC-dependent amyloid-beta oligomer toxicity. Biochem. J 473, 2119–2130. [DOI] [PubMed] [Google Scholar]
- Martins VR, Beraldo FH, Hajj GN, Lopes MH, Lee KS, Prado MA and Linden R (2010) Prion protein: orchestrating neurotrophic activities. Curr. Issues Mol. Biol 12, 63–86. [PubMed] [Google Scholar]
- McLennan NF, Brennan PM, McNeill A et al. (2004) Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol 165, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R and Olsen BR (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med 16, 1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsios N, Saka M, Krupinski J et al. (2007) Cellular prion protein is increased in the plasma and peri-infarcted brain tissue after acute stroke. J. Neurosci. Res 85, 602–611. [DOI] [PubMed] [Google Scholar]
- Mukerji SS, Katsman EA, Wilber C, Haner NA, Selman WR and Hall AK (2007) Activin is a neuronal survival factor that is rapidly increased after transient cerebral ischemia and hypoxia in mice. J. Cereb. Blood Flow Metabol 27, 1161–1172. [DOI] [PubMed] [Google Scholar]
- Nicholls P, Pack T, Urs N et al. (2017) Mapping nonapoptotic caspase activity with a transgenic reporter in mice. bioRxiv 10.1101/196105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW and Beaudet AL (1997) Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci 17, 9165–9171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostapchenko VG, Beraldo FH, Mohammad AH, et al. (2013) The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-β oligomer toxicity. J Neurosci. 33, 16552–16564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchision DM and McKay RD (2002) The control of neural stem cells by morphogenic signals. Curr. Opin. Genet. Dev 12, 478–487. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B and Piston DW (2004) An improved cyan fluorescent protein variant useful for FRET. Nat. Biotechnol 22, 445–449. [DOI] [PubMed] [Google Scholar]
- Roffe M, Beraldo FH, Bester R et al. (2010) Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. NatlAcad. Sci. USA 107, 13147–13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos TG, Beraldo FH, Hajj GN et al. (2013) Laminin-gamma1 chain and stress inducible protein 1 synergistically mediate PrPC-dependent axonal growth via Ca2+ mobilization in dorsal root ganglia neurons. J. Neurochem 124, 210–223. [DOI] [PubMed] [Google Scholar]
- Schmid AB, Lagleder S, Grawert MA et al. (2012) The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 31, 1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu WC, Chen CP, Saeki K et al. (2005) Hypoglycemia enhances the expression of prion protein and heat-shock protein 70 in a mouse neuroblastoma cell line. J. Neurosci. Res 80, 887–894. [DOI] [PubMed] [Google Scholar]
- Soares IN, Caetano FA, Pinder J et al. (2013) Regulation of stress-inducible phosphoprotein 1 nuclear retention by protein inhibitor of activated STAT PIAS1. Mol. Cell Proteomics 12, 3253–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa Lopes SM, Roelen BA, Monteiro RM, Emmens R, Lin HY, Li E, Lawson KA and Mummery CL (2004) BMP signaling mediated by ALK2 in the visceral endoderm is necessary for the generation of primordial germ cells in the mouse embryo. Genes Dev. 18, 1838–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Jin K and Uteshev VV (2013) A type-II positive allosteric modulator of alpha7 nAChRs reduces brain injury and improves neurological function after focal cerebral ischemia in rats. PLoS ONE 8, e73581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Jin ZG, Meoli DF, Matoba T and Berk BC (2006) Cyclophilin A is secreted by a vesicular pathway in vascular smooth muscle cells. Circ. Res 98, 811–817. [DOI] [PubMed] [Google Scholar]
- Tretter YP, Hertel M, Munz B, ten Bruggencate G, Werner S and Alzheimer C (2000) Induction of activin A is essential for the neuroprotective action of basic fibroblast growth factor in vivo. Nat. Med 6, 812–815. [DOI] [PubMed] [Google Scholar]
- Tsai CL, Tsai CN, Lin CY, Chen HW, Lee YS, Chao A, Wang TH, Wang HS and Lai CH (2012) Secreted stress-induced phosphoprotein 1 activates the ALK2-SMAD signaling pathways and promotes cell proliferation of ovarian cancer cells. Cell Rep. 2, 283–293. [DOI] [PubMed] [Google Scholar]
- Tsutsui S, Hahn JN, Johnson TA, Ali Z and Jirik FR (2008) Absence of the cellular prion protein exacerbates and prolongs neuroinflammation in experimental autoimmune encephalomyelitis. Am. J. Pathol 173, 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Kaufman AC, Kostylev M et al. (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TH, Chao A, Tsai CL et al. (2010) Stress-induced phosphoprotein 1 as a secreted biomarker for human ovarian cancer promotes cancer cell proliferation. Mol. Cell Proteomics 9, 1873–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lu Z, Fu X et al. (2017a) Alpha-7 nicotinic receptor signaling pathway participates in the neurogenesis induced by ChAT-positive neurons in the subventricular zone. Transl. Stroke Res 8, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, You H, Qi J, Yang C, Ren Y and Cheng H (2017b) Autocrine and paracrine STIP1 signaling promote osteolytic bone metastasis in renal cell carcinoma. Oncotarget 8, 17012–17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise J, Crome O, Sandau R, Schulz-Schaeffer W, Bahr M and Zerr I (2004) Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett 372, 146–150. [DOI] [PubMed] [Google Scholar]
- Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I and Bahr M (2006) Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 37, 1296–1300. [DOI] [PubMed] [Google Scholar]
- Weise J, Doeppner TR, Muller T, Wrede A, Schulz-Schaeffer W, Zerr I, Witte OW and Bahr M (2008) Overexpression of cellular prion protein alters postischemic Erk1/2 phosphorylation but not Akt phosphorylation and protects against focal cerebral ischemia. Restor. Neurol. Neurosci 26, 57–64. [PubMed] [Google Scholar]
- You H, Tsutsui S, Hameed S et al. (2012) Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc. Natl Acad. Sci. USA 109, 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanata SM, Lopes MH, Mercadante AF et al. (2002) Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 21, 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]