

Abstract
Per- and polyfluoroalkyl substances (PFAS) have been released into the environment for decades, yet contributions of air emissions to total human exposure, from inhalation and drinking water contamination via deposition, are poorly constrained. The atmospheric transport and fate of a PFAS mixture from a fluoropolymer manufacturing facility in North Carolina was investigated with the Community Multiscale Air Quality (CMAQ) model applied at high resolution (1 km) and extending ~150 km from the facility. Twenty-six explicit PFAS compounds, including GenX, were added to CMAQ using current best estimates of air emissions and relevant physicochemical properties. The new model, CMAQ-PFAS, predicts 5% by mass of total emitted PFAS and 2.5% of total GenX are deposited within ~150 km of the facility, with the remainder transported out. Modeled air concentrations of total GenX and total PFAS around the facility can reach 24.6 ng m−3 and 8,500 ng m−3 but decrease to ~0.1 ng m−3 and ~10 ng m−3 at 35 km downwind, respectively. We find that compounds with acid functionality have higher deposition due to enhanced water solubility and pH-driven partitioning to aqueous media. To our knowledge, this is the first modeling study of the fate of a comprehensive, chemically resolved suite of PFAS air emissions from a major manufacturing source.
Graphical Abstract
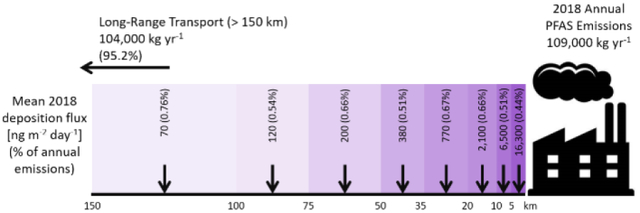
1. Introduction
Per- and polyfluoroalkyl substances (PFAS) are an evolving class of industrially-produced compounds, with unique chemical structures estimated to number in the thousands1. They are used extensively in consumer products and for industrial applications2 and as a result, have been found throughout the environment3–6. Much research has focused on the occurrence of PFAS in ground, surface, and drinking water7, with PFOA (perfluorooctanoic acid, a well-known PFAS)8–11 contaminated drinking water correlating positively with human blood PFAS levels12–14. More recently, soil and water contamination for two specific PFAS, PFOA and HFPO-DA (hexafluoropropylene oxide dimer acid, CAS 13252–13-6, often referred to as GenX and discussed below), has been attributed, at least in part, to atmospheric deposition15–17, leading to the study of PFAS air emissions. The magnitude of direct human exposure to airborne PFAS due to inhalation or other uptake mechanismsis largely unknown, although previous studies have shown positive correlations between indoor dust levels of PFAS and human blood concentrations18.
PFAS released to the environment, or their terminal perfluorinated degradation products, persist in soil and water 7 or more years after release17 due to the strong C-F bond19 that resists degradation. The atmosphere, meanwhile, does not facilitate local, long-term PFAS accumulation, but rather provides a medium for long-range transport20–23, resulting in low surface concentrations (e.g. < 1 pg m−3 24) that are difficult to measure. Due to low concentrations, many ambient air studies focus on the area immediately surrounding production facilities8–11, 25.
One of these production facilities, The Chemours Company (formerly DuPont) Washington Works in West Virginia, USA, has been the focus of multiple air concentration- and deposition-based studies of PFOA. More recently, The Chemours Company Fayetteville Works, located ~25 km south of Fayetteville, North Carolina, USA, has been identified as a source of PFAS to the Cape Fear River26–28, although high PFAS concentrations upstream suggest direct water discharge from the facility is not the only PFAS source to the Cape Fear27, 29. The Fayetteville Works produces and uses PFAS compounds of varying carbon chain length and functionality including HFPO-DA. HFPO-DA was introduced to replace legacy compounds like PFOA and has been found in the Cape Fear River26 and in treated drinking water taken from the river27. HFPO-DA is used at other Chemours sites, and concentrations in soil and surface water surrounding those facilities suggest a role for air transport and deposition17, 30. Investigations at the Fayetteville Works found water wells surrounding the facility, including many upstream and on the opposite side of the Cape Fear river as the facility, were contaminated with GenX and other PFAS31. Near-field air dispersion modeling results indicate air emissions could be transported to locations near the facility, resulting in well contamination31, 32.
In this work, we leverage the production process-level, chemically explicit PFAS emission rates reported by Chemours Fayetteville Works to inform a regional-scale chemical transport model, the Community Multiscale Air Quality (CMAQ) model, and predict ambient air concentrations and deposition fluxes for one simulation year. We use existing methods to estimate chemical properties like vapor pressure and water solubility for 26 explicit PFAS. We then evaluate the resulting model, CMAQ-PFAS, with deposition measurements from NC Department of Environmental Quality (NC DEQ) measurement sites surrounding the facility and explore relationships between predicted deposition pathways and compound specific physicochemical properties. Finally, we highlight important gaps in our understanding that require further research.
2. Methods:
2.1. Emissions:
The PFAS-specific emission report submitted by The Chemours Company to NC DEQ31 provides, process-level emission rates of 53 PFAS compounds for the year 2017 (Table S1), along with hydrofluoric acid (HF) which is a hazardous air pollutant (HAP) listed in the National Emissions Inventory (NEI). Total PFAS emissions at the Fayetteville Works facility were 109,393 kg yr−1 (Fig. 1A). The top three compounds by emitted mass have 3 or fewer carbons and make up 82% (89,722 kg yr−1) of the total emissions (Fig. 1A). The emissions report listed species by name, an acronym, and CAS. Some CAS were not identifiable, presumably because they are novel and proprietary, but only account for 5.1 kg in 2017 or <0.005% of emissions by mass. Figure 1B classifies the 50 species with the lowest emission rate (“other” in Fig. 1A) by number of functional groups (Table S2), which indicates the structural complexity. We denote simple fluorocarbons and chlorofluorocarbons as compounds without functional groups (e.g. CF3H and C2F3Cl3). These are expected to be the least susceptible to oxidation and phase change. ~8% of emissions by mass have 2 or more functional groups. The structural complexity and diversity of functionality underscores the challenge of understanding the role of atmospheric chemistry in the fate of PFAS air emissions.
Figure 1.
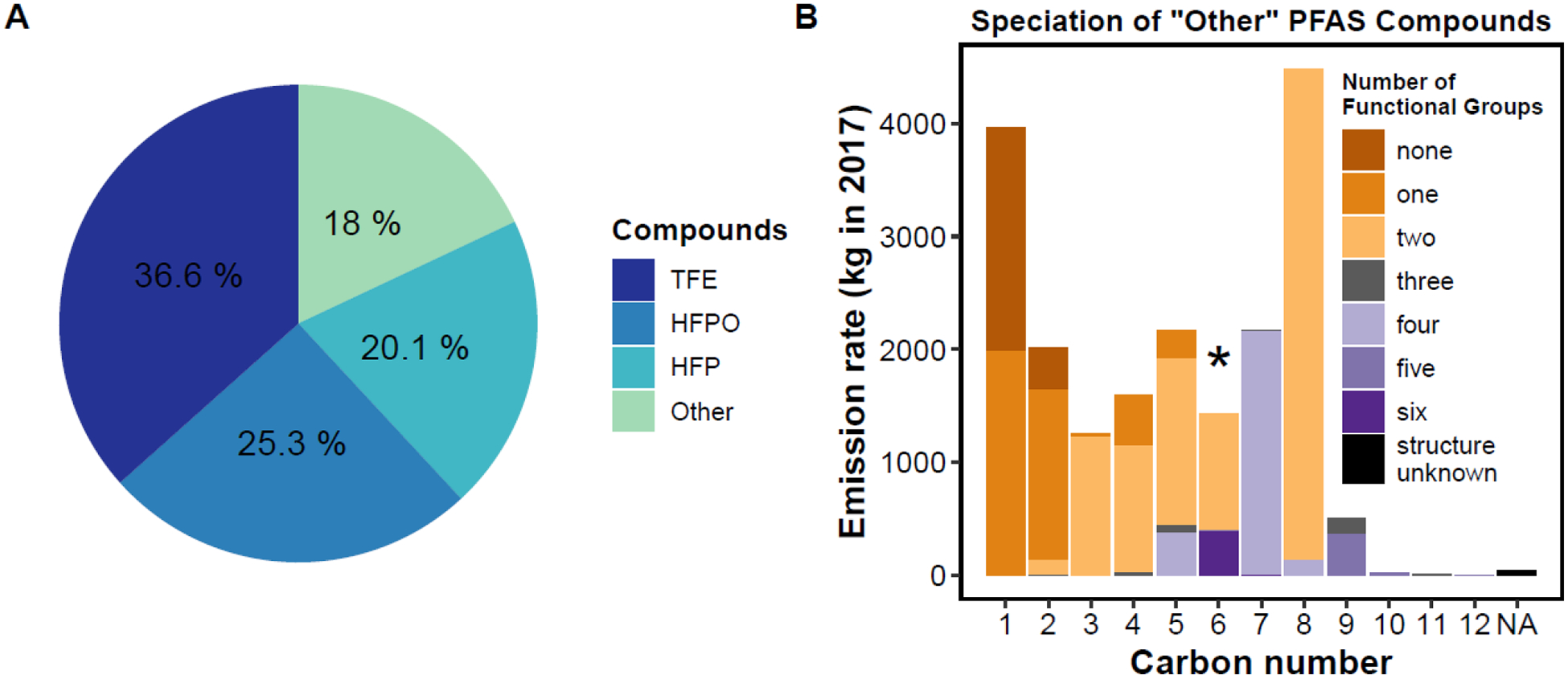
(A) Pie chart of facility emissions, with the lowest 50 by mass grouped into “other”. (B) Facility emissions for the “other” category by carbon number, categorized by number of functional groups. Compounds with unknown structure are grouped in the “NA” bin. The * indicates the location of total GenX (HFPO-DA + HFPO-DAF, gas + particle).
GenX is a trademark name often applied to HFPO-DA (hexafluoropropylene oxide dimer acid) or the anion of HFPO-DA(aq) which is measured in water (Fig. S1)33. The anion HFPO-DA(aq) can also be generated from the hydrolysis of the acyl fluoride, HFPO-DAF (hexafluoropropylene oxide dimer acid fluoride), in water34 (Fig. S1). Throughout this work “total GenX” refers to the sum of HFPO-DA (304.6 kg yr−1) and HFPO-DAF (725.1 kg yr−1) in both the gas and condensed phases.
2.2. Physical properties:
Physicochemical properties are used by CMAQ to predict gas-particle partitioning and influence rates of dry (removal of gases and particles to soil, vegetation, and other surfaces in the absence of precipitation) and wet (removal via rain, snow, or fog, etc.) deposition. The Open structure-activity/Property Relationship App (OPERA) model35 was used to estimate physicochemical properties (Table S2) and can be accessed via https://comptox.epa.gov/dashboard or https://qed.epacdx.net/cts/pchemprop/input. Few measurements of these properties exist for PFAS, therefore the model results were used for each species property for consistency. PFAS functionalities are highly diverse, therefore both solubility- and vapor pressure-driven partitioning to condensed water and organic particles were explored. The saturation vapor concentration, C*36, 37, was used to predict condensation to the particulate organic phase, . The predicted C* values range from 2.0×104 to 3.6×1012 μg m−3 (Table S2) which can vary up to 7 orders of magnitude where measurements exist for comparison (Table S3), although OPERA was shown to most accurately predict C* for a series of PFAS compared to other common property estimation models38. The enthalpy of vaporization (ACD/labs, http://www.chemspider.com/), used to adjust the volatility to atmospheric temperature, range from 17.2 to 56.8 kJ mol−1. Henry’s Law constants were estimated with OPERA to model solubility driven condensation to the aqueous phase of atmospheric particles, cloud liquid water, and aqueous ground surfaces (e.g. bodies of water, leaf stomata, etc.), ranging from insoluble to slightly soluble (8.6 × 10−3 to 4.2 × 106 M atm−1). This result is broadly consistent with the hydrophobic nature of the fluorinated tails. We assume all PFAS compounds are emitted in the gas phase (See PFAS Emissions in the SI). The role of surface interactions of PFAS compounds with other atmospheric constituents, possibly resulting in micelles or surface coated particles for example, is unknown. PFAS are designed for their surfactant properties and it is unclear to what extent those properties impact their own phase partitioning or the partitioning of other compounds.
2.3. CMAQ-PFAS configuration
CMAQ v5.3.239 was applied to a 260 km × 244 km domain across Eastern North Carolina and Northeastern South Carolina, USA, with 1 km × 1 km horizontal resolution and ~1 minute time resolution (Fig. S2). The model was run for January through December 2018, coinciding with NC DEQ’s deposition measurements of total GenX surrounding the facility (section 2.5). Meteorological inputs were simulated using the Weather Research and Forecasting model (WRF), Advanced Research WRF core (ARW) version 4.1.140 (see WRF Modeling in the SI). Boundary conditions were processed from a series of nested CMAQ simulations from a hemispheric domain at 108 km × 108 km, through a continental U.S. domain at 12 km × 12 km, and finally through an Eastern U.S. domain at 4 km × 4 km. Gas-phase chemical kinetics were simulated via a modified version of Carbon Bond 6 (CB6r3)41 and inorganic aerosol formation (AERO7) was consistent with Nolte et al.42. Non-PFAS anthropogenic emissions (e.g. NOx, SOx, VOCs, etc) were based on the 2014 National Emissions Inventory (NEI) version 143, projected to 2018. Biogenic vapor emissions were calculated online in CMAQ using the Biogenic Emission Inventory System (BEIS) v3.6.144. The Surface Tiles Aerosol and Gaseous Exchange (STAGE) module45 was used to account for the parallel deposition to heterogeneous land-surface types within each model grid cell. CMAQ showed reasonable performance for prediction of conventional pollutants like NOx, O3, and PM2.5 (Table S4).
The Base CMAQ-PFAS simulation (“Base”) treats explicitly the 26 compounds with annual emissions greater than 45 kg (109,190 kg, 99.8% of emitted mass) and lumps the remaining 24 compounds as one surrogate compound (PFASOTHER) with mole-weighted parameters (Table S5). PFAS emissions are allocated in space and time using CMAQ’s buoyant plume rise algorithm, a record of the relevant facility processes running each day of the year, and an estimate of incremental reductions achieved during 2018 due to installed controls (see PFAS emissions in the SI). The physicochemical properties for each model compound (Tables S2–S3, S6) are used to calculate the partitioning between the gas- and aerosol-phase components as well as the dissolution in cloud droplets, and uptake to dry and aqueous land-surface types. Gas-phase and heterogeneous chemical reactions are not considered in this implementation of CMAQ-PFAS, nor are aerosol surface interactions between PFAS and non-PFAS constituents.
2.4. Sensitivity to acyl fluoride hydrolysis
The conversion of acyl fluorides to carboxylic acids in the bulk aqueous phase can occur nearly spontaneously34. The hydrolysis of one acyl fluoride, CF3COF (PAF, Table S1) has been measured in air46 and modeled in chemical transport models as in-cloud processes47–49. More recently, the hydrolysis of a larger molecular weight acyl fluoride was incorporated into a box model50. The feasibility and impact of hydrolysis of a wide structural array of acyl fluorides in the gas phase or on aerosol particles has yet to be studied, yet the transformation of acyl fluorides to carboxylic acids may drastically alter their solubility. For example, the standard Henry’s Law constant for HFPO-DAF is predicted to be 0.05 M atm−1 compared to the effective Henry’s Law constant for HFPO-DA of 2.3 × 1010 M atm−1 at pH of 3. To bound the impact of potential gas-phase hydrolysis of acyl fluorides on total PFAS deposition, one sensitivity case (“CarbAcid”) assumes eight of the acyl fluorides reported by the facility are emitted as their corresponding carboxylic acids (Table S6). Thus, total GenX emissions are assumed to be in the form of HFPO-DA for the CarbAcid case, representing instantaneous hydrolysis.
2.5. Measurements:
The NC DEQ established 5 rain water collection sites within 3.5 km of the Chemours facility (Fig. S3), results from which are posted publicly31 (Table S7). The sites N, NW, and SE of Chemours were equipped with 5-gallon HDPE buckets for collection of weekly composite wet/dry deposition samples. The sites NE and SW of Chemours were equipped with samplers (NCON Model 110) to collect weekly discrete wet and dry samples. These samplers had rain sensors that moved a cover from one position to the other to collect either wet or dry samples depending on whether precipitation was occurring. We combine both NCON-observed and CMAQ-predicted estimates of dry and wet deposition.
Analysis began with measuring and recording the total collected sample volume. Dry samplers were rinsed with 600 mL of PFAS-free water. From this rinse, 100 mL was reserved for refrigerated archiving, and two 250 mL aliquots were added to PFAS-free HDPE plastic sample bottles for shipment to the analysis lab. The samples were analyzed by HPLC-MS/MS following EPA Method 537.151 with isotope dilution for calibration for a subset of PFAS compounds and specifically HFPO-DA. Because acyl fluoride hydrolysis is rapid in bulk solution34 and results in HFPO-DAF being converted to HFPO-DA, model predictions of HFPO-DA and HFPO-DAF were summed to estimate total GenX model deposition (section 2.1).
3. Results & Discussion:
3.1. CMAQ-PFAS evaluation
The deposition measurements of GenX made by NC DEQ are compared to the CMAQ-PFAS model outputs for 2018 in Figure 2. Modeled results are excluded before measurements began or when sampling errors occurred. Both modeled and measured results below the measurement Limit of Quantification (LOQ), which varied depending on sampler type, were set to zero. Measurements were weekly composites, so modeled data was summed across weeks. Measured and modeled weekly values were divided by 7 to calculate the average daily deposition.
Figure 2.
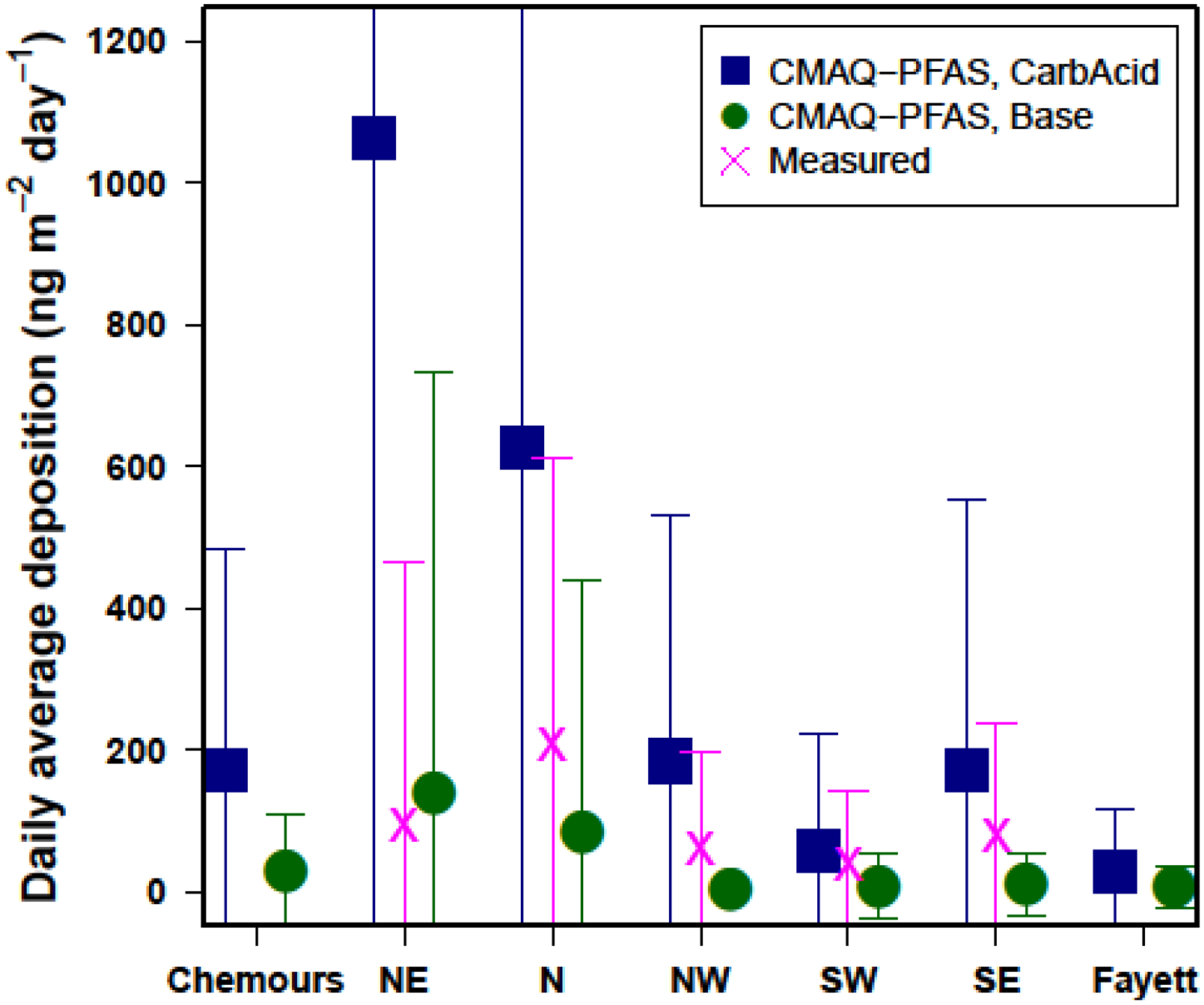
Daily averaged observed GenX deposition at 5 NC DEQ sampling sites paired with daily averaged deposition predictions for the base CMAQ-PFAS model configuration. The CarbAcid CMAQ model configuration is also shown and represents a bounding scenario for the hydrolysis rate of the acyl fluoride. Model predictions include the deposition of total GenX (HFPO-DA + HFPO-DAF, gas + particle). Also shown for comparison are CMAQ-PFAS predictions at the Chemours Fayetteville Works and in Fayetteville, NC, the nearest urban area. Error bars represent the standard deviation of data at each site.
The Base model predicts an average GenX deposition rate across all sites of 48.6 ng m−2 day−1 (maximum = 3,333 ng m−2 day−1), which is about half the measured value of 97.5 ng m−2 day−1 (maximum = 2,119 ng m−2 day−1). The episodic nature of deposition is reflected in the low frequency of deposition measurements above the LOQ (78 out of 239 measurements, Table S7). This results in large standard deviation across all sites (error bars in Fig. 2) and makes it difficult to confirm decreases in deposition from the implementation of controls throughout 2018 (Table S7). The model captures the spatial variability among the sites (r=0.54). Overall, the model performs well, especially considering that detailed knowledge of novel PFAS is still emerging in the environmental science community. Discrepancies between the predicted and observed deposition values could be due to uncertainties in meteorology, emissions, property estimation, or sample collection method (see Model-Measurement Discrepancies in the SI). See section 3.3 for discussion of the CarbAcid model results.
Deposition measurements do not exist on the facility grounds or in Fayetteville (“Fayett”), although the modeled results are shown in Figure 2 for comparison. Predicted deposition at the Chemours facility, where emissions are elevated due to ejection from the stack, is equal to or significantly lower than deposition at the sampling sites immediately downwind after some down-mixing has occurred. The deposition predicted in Fayetteville, the nearest urban area, is in the range of the measurements at the sampling sites.
3.2. Total GenX deposition and concentration
The modeled deposition of total GenX (HFPO-DA + HFPO-DAF, gas + particle) is regionally distributed and highest near the facility in the Base model (Fig. 3). Note the non-linear color scale illustrates the maximum deposition value (1,549 ng m−2 day−1) which is two orders of magnitude larger than the 99th percentile (9.8 ng m−2 day−1). Both dry and wet deposition are important loss pathways for total GenX due to its effective Henry’s Law constant (Fig. S4). Dry deposition dominates particularly at locations farther from the source (Fig. S4, bottom). The mean domain-wide deposition estimated by CMAQ-PFAS, 1.1 ng m−2 day−1, integrated over a decade of production52 suggests a mean deposition of ~4,200 ng m−2 of total GenX to the environment and a maximum deposition of 5,650 μg m−2 near the facility, consistent with accumulation observed elsewhere for total GenX and other PFAS17. Similar studies to those performed at the Washington Works facility in WV17 could be performed to investigate whether GenX has accumulated in the soil to the extent our simulations suggest is possible.
Figure 3.
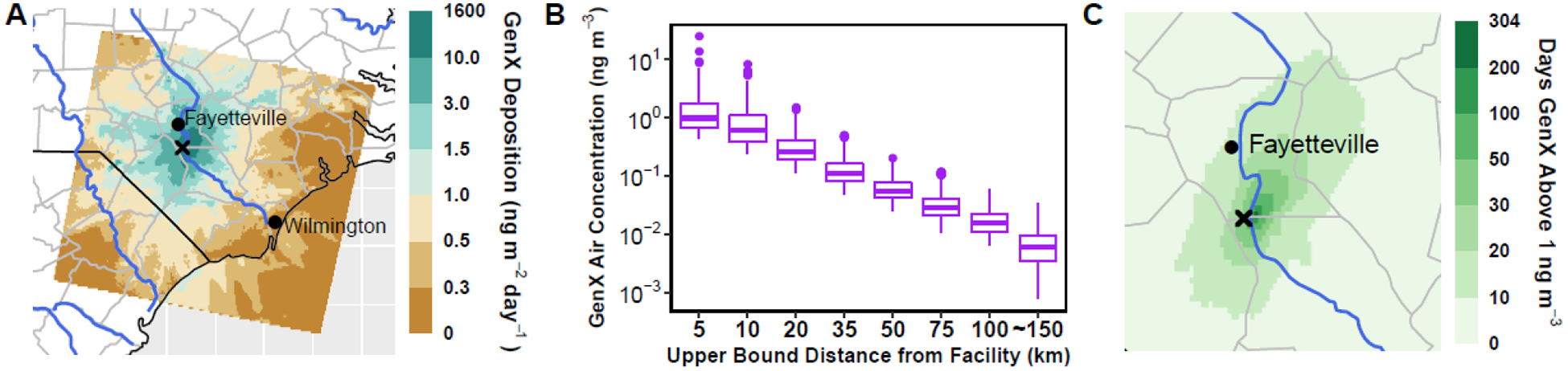
(A) The annual accumulated deposition of total GenX (HFPO-DA + HFPO-DAF). (B) The annually averaged air concentration of total GenX binned as a function of distance from the facility. (C) The cumulative time in 2018 that the simulated total GenX concentration was predicted above 1 ng m−3. Note the non-linear color scales on panels A and C.
Annual average air concentrations of total GenX near the facility can reach 24.6 ng m−3, although decrease to an average of 1.9 ng m−3 at 5 km, and then further decrease with an e-folding distance (i.e. the distance at which the concentration drops by a factor of e (~63.2%)) of 25.5 km (Fig. 3B). These values are broadly consistent with previous measurements and modeling predictions of single compounds around fluoropolymer production facilities with air concentrations ranging from ~1–1,000 ng m−3 and deposition rates of 55 – 38,000 ng m−2 day−1 9–11, 25.
While 50% of the annual-averaged concentrations are on the order of 0.001–0.01 ng m−3, the hourly averaged concentration for any location within the domain can be much higher (Fig. S5). To quantify the influence of plumes which cause episodic elevated concentrations, the cumulative time (in days) each model grid cell is above an arbitrary threshold of 1 ng m−3 is shown in Figure 3C. Most of the domain has 10 or fewer days at or above 1 ng m−3, while the area immediately surrounding the facility consistently exceeds this threshold (up to 304 days per year). Further research on health impacts of inhalation exposure is needed to determine whether these total GenX concentrations pose a health risk.
3.3. The role of acyl fluoride hydrolysis
Carboxylic acid products of acyl fluoride hydrolysis more favorably partition to the particle phase under conditions of increased pH and particle liquid water content. The eastern US contains relatively high levels of aerosol liquid water compared to other parts of the United States, consistent with a relatively humid continental atmosphere53. Eastern North Carolina contains a local maximum in ammonia emissions which contribute to elevated aerosol water and pH as shown in Figure S6A and S6B, respectively. HFPO-DA partitioning is dynamic throughout the domain, ranging from almost entirely in the gas phase to almost entirely in the particle phase (Fig. 4A), correlating with pH and particle liquid water content. In contrast, HFPO-DAF is entirely in the gas phase across the domain (Fig. 4B and S7).
Figure 4.
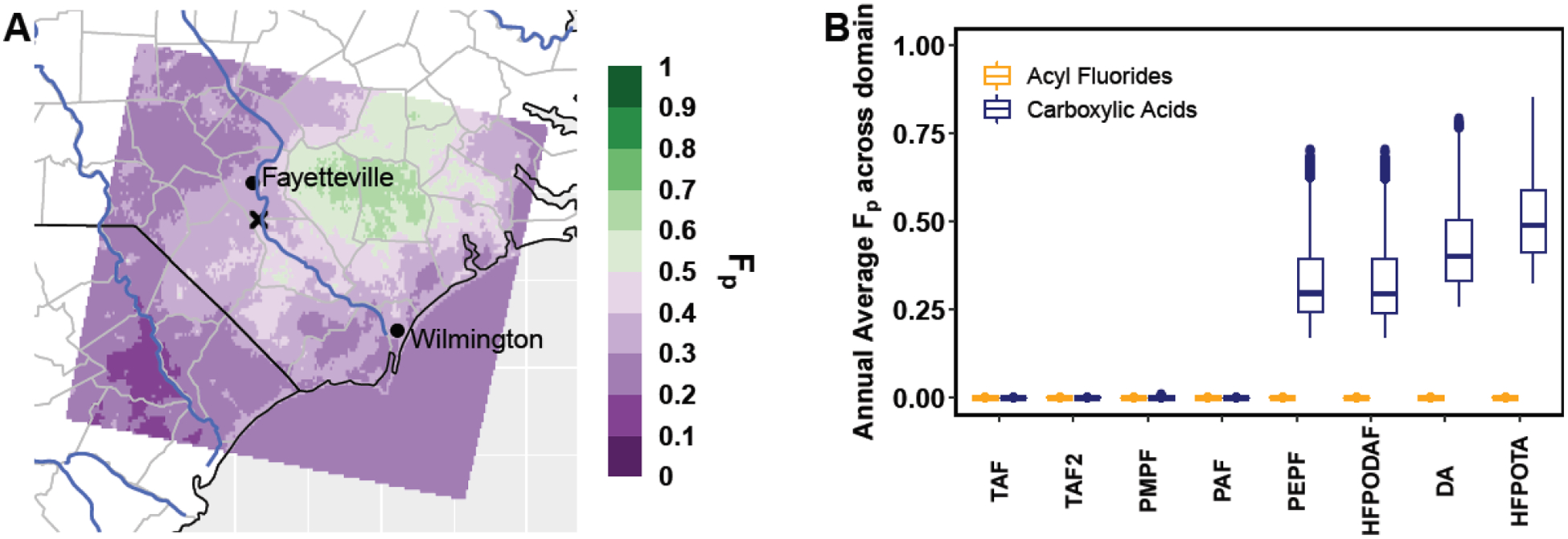
(A) The annually averaged fraction of HFPO-DA in the particle phase (Fp), and (B) the particle-phase fraction (Fp) for 8 acyl fluoride containing compounds explicitly implemented into the model for the Base simulation (yellow) and that of their analogous carboxylic acid forms (blue). Compounds with the lowest effective Henry’s Law constants at a pH of 3 are on the left, with the highest on the right.
The difference in the fraction in the particle phase (Fp) between the acyl fluoride and carboxylic acid compounds has profound implications for their removal. The only significant removal pathway for HFPO-DAF is gas-phase dry deposition, while HFPO-DA is removed by all deposition pathways (i.e. wet and dry deposition of both particles and gases, Fig. S8A). This in turn affects the spatial pattern of deposition (Fig. S8, B & C). In contrast, Moreno et al.32 found that varying the Henry’s law constant for HFPO-DA did not affect the overall spatial pattern of deposition. This inconsistency highlights the importance of process-level deposition models such as CMAQ for understanding complex PFAS behaviors.
The effect of the enhanced solubility on total deposition and model performance is shown in Figure 2 where the deposition at sampling sites can vary greatly between the two model runs. The CarbAcid model consistently overestimates the measurements as a result of the increased solubility when HFPO-DAF is assumed to be emitted as HFPO-DA. That the Base simulation generally underestimates the measurements suggests the reality is somewhere between these two cases. Either the emitted ratio of HFPO-DAF to HFPO-DA is lower than assumed in the Base model, and/or HFPO-DAF converts to HFPO-DA, rapidly (occurring within the first ~5 km of transport), but not instantaneously (as assumed in the CarbAcid case). More information on the rate of acyl fluoride hydrolysis under atmospherically relevant conditions is necessary.
As in the case of HFPO-DA and HFPO-DAF, the relative change in Henry’s Law constant between the carboxylic acid and acyl fluoride forms of 7 other acyl fluorides affects their model-predicted deposition rates. The annually averaged domain wide Fp for all 8 acyl fluorides are shown in Figure 4D for the Base and CarbAcid cases, with the lowest effective Henry’s Law constants at a pH of 3 (Table S6) on the left to the highest on the right. All the acyl fluorides partition strongly to the gas phase in the Base simulation. When they are instead emitted in the model as carboxylic acids, those with the highest effective Henry’s Law constants (> 106 M atm−1) partition substantially to the particle phase, which accelerates their deposition. Understanding the feasibility and timescale of hydrolysis, which will likely differ for each acyl fluoride, is vital for understanding the fate of these compounds.
3.4. Total PFAS
Despite the interest in GenX from the Fayetteville Works facility, total GenX emissions account for 1% of the total PFAS emissions by mass and only 2 (HFPO-DA and HFPO-DAF) of the total 53 individual compounds emitted from the facility (Fig. 5, A and B). The ambient air concentrations and total deposition of the entire suite of PFAS emitted from this facility are enhanced relative to total GenX (Fig. S9) and generally scale with emissions. The predicted total PFAS concentrations reach an annual average maximum of 8.5 μg m−3 with a significantly lower domain-wide median of 0.004 μg m−3 (mean = 0.01 μg m−3). At 5 km from the facility the annual average air concentration of total PFAS is 0.66 μg m−3, and decreases further with an e-folding distance of 24.5 km. At 35 km from the facility, the air concentration drops to approximately 0.01 μg m−3. Total PFAS deposition reaches a maximum value of 245,000 ng m−2 day−1 although, as with total GenX, the 99th percentile of the data is two orders of magnitude lower (2,170 ng m−2 day−1). These deposition rates are similar to previously observed rates of 2–3,400 ng m−2 day−1 for groups of PFAS54, 55. In addition, the exponential decay of surface concentrations and deposition downwind of a fluoropolymer manufacturing facility is also consistent with previous findings17, 25, 30, 56. CMAQ-PFAS predicts that only 4.8% of total PFAS and 2.6% of total GenX emissions will deposit within approximately 150 km of the facility (Fig. S10), with the majority of mass depositing via dry deposition (Table S8). The rest of the mass emitted from the facility is transported further in the U.S. and beyond, consistent with previous work suggesting long-range transport of HFPO-DA57.
To understand the variability among individual compounds, the total annual deposition within the domain as a percent of emissions were binned by carbon number in Figure 5. In the Base simulation, total PFAS deposition is minimal within the domain, 4.8% of the emission rate, increasing slightly to 5.8% in the CarbAcid simulation. The carbon number-specific percent deposited varies between 1.6 – 19.5% for the Base case, increasing to 19.8% for the CarbAcid case, with significant relative increases for carbon numbers 1, 4, and 6. While the carbon number-specific percent deposited can approach 20%, ~85% of PFAS emissions by mass have 2–3 carbons, which have a carbon number-specific percent deposited of 3.4–6.5% (Base case), resulting in a total PFAS percent deposited of 4.8%. CMAQ-PFAS does not predict a clear trend of the relative strength of deposition on carbon number for this domain and mix of PFAS. The relative importance of deposition pathways changes slightly between the Base and CarbAcid models; dry deposition accounts for 99.3% of deposition in the Base simulation versus 94.4% in the CarbAcid results. This sensitivity further highlights the importance of the composition of emissions and likely transformation products.
Figure 5.
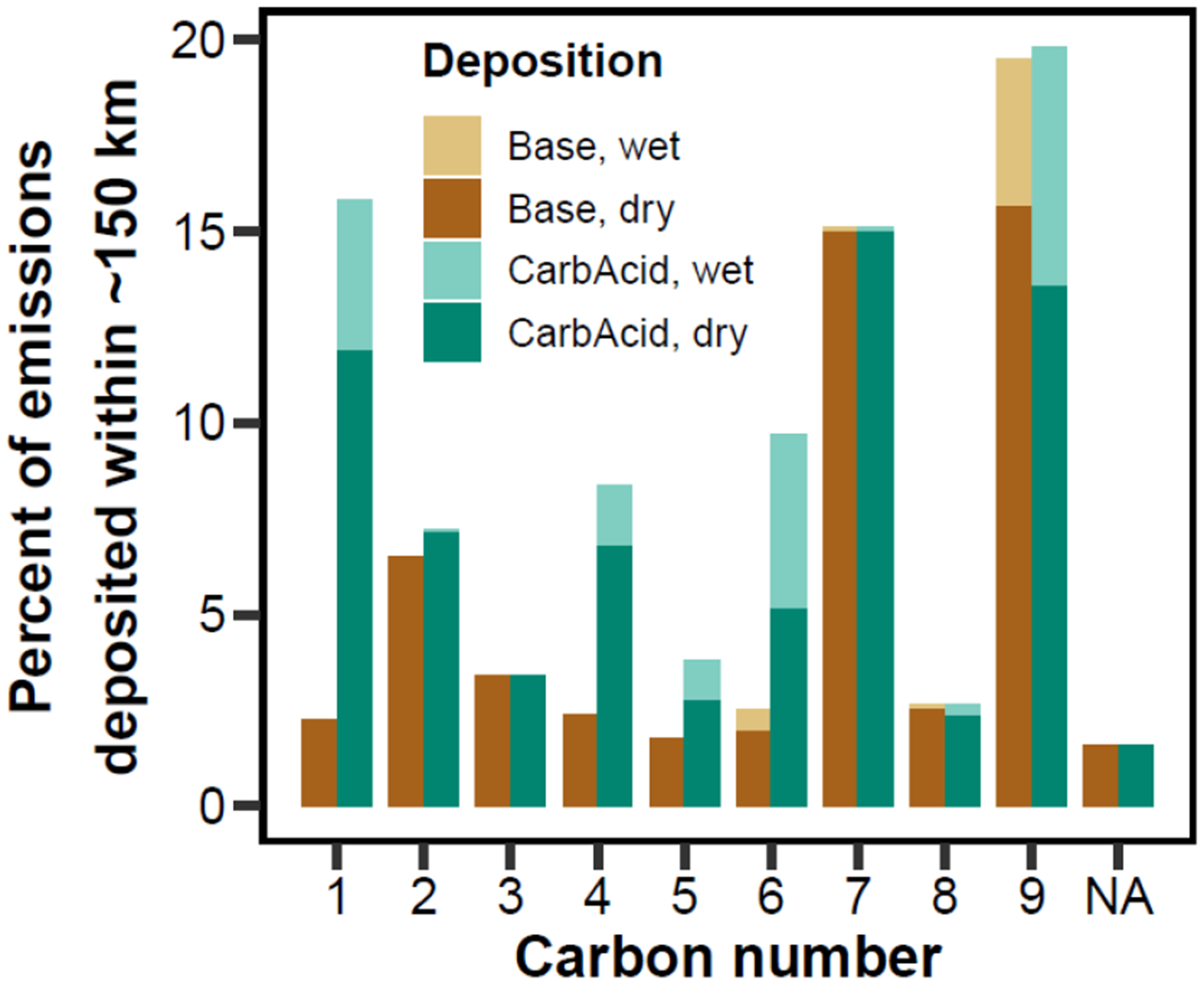
Annual-averaged deposition as a percent of emissions of all explicitly modeled compounds and the “PFASOTHER” compound (classified as NA) within the model domain, as a function of carbon number and grouped via wet and dry deposition.
4. Impacts:
This study focuses on the air emissions of one PFAS manufacturing facility with relatively well-characterized speciated emissions in order to understand how chemical structure and abundance modulates regional-scale atmospheric concentrations and deposition of PFAS. The approaches developed could be extended to study additional compounds and PFAS facilities as data become available. In addition, the model can be expanded to treat the atmospheric chemical degradation products of emitted PFAS, as well as PFAS surfactant effects at the aerosol-air, cloud-air, and aerosol organic-aqueous interfaces which may have significant implications for gas-particle partitioning of conventional pollutants, cloud formation, and secondary aerosol processing, especially in concentrated PFAS plumes.
In order to extend our analysis to other PFAS air emission sites or to a broader, regional or continental accounting of the impacts of numerous PFAS air emission sources on distant receptors, high-quality emission rates and speciation inputs will be required. The exact number and location of significant PFAS air emission sources in the U.S. is uncertain, and as PFAS manufacturers operate proprietary processes for which emission speciation is often kept confidential, the emissions speciation and rate are usually unknown. This work utilized detailed emissions data shared by the facility with the state regulatory agency, and data with this level of detail is rarely available. Furthermore, the composition of PFAS emissions can have dramatic impacts on the predicted atmospheric lifetime and the spatial distribution of deposition to ecosystems, and present unknown risk of exposure via inhalation or other uptake mechanisms. The CarbAcid sensitivity simulation demonstrates the importance of acknowledging the variability among compounds as well as the uncertainty in the rate of relevant chemical transformations. The difference in predicted fate of two structurally similar compounds, HFPO-DA and HFPO-DAF, illustrates the potential consequences of ignoring chemical identity. Finally, considering the outsized role of dry deposition to total deposition predicted herein (Fig. 5 and Table S8), there is a pressing need for both higher spatiotemporal resolution and discrete dry deposition measurements that are reflective of deposition to various environmental surfaces (e.g. surface waters, vegetation, soils). Measurements of physicochemical properties are also rare and necessary for modeling deposition processes.
We have shown that the new model, CMAQ-PFAS, when informed by estimated emissions from a fluoropolymer manufacturer, captures observed deposition fluxes of total GenX within a factor of 10 on average for the 5 measurement sites. Ambient air concentrations of 24.6 ng m−3 are predicted at the facility, which decreases with distance to ~0.1 ng m−3 35 km from the facility. We have further characterized the ambient air concentrations and domain-wide deposition of all of the individual PFAS compounds emitted by the facility and find that the annually averaged total PFAS air concentrations reach a maximum of 8.5 μg m−3 near the facility and the majority of PFAS emissions (~95 % by mass) are transported more than ~150 km away. Lastly, we find that the uncertainty related to acyl fluoride hydrolysis to carboxylic acid products in the atmosphere is relevant for predicting and constraining PFAS deposition within ~150 km from the facility, especially in the vicinity of the plant. This work makes important steps towards both explicitly understanding the fate of PFAS air emissions from one manufacturing facility and building a methodology that quantifies regional-scale impacts of PFAS air emissions in general.
Supplementary Material
Acknowledgements
We thank Nancy Jones, Tom Anderson, Michael Pjetraj, Gary Saunders, Steve Hall, and Brent Hall at North Carolina Department of Environmental Quality for helpful discussions as well as emissions and deposition data. We thank The Chemours Company for discussions and data relevant for emissions timing and controlled reductions throughout 2018. We thank Andrew Lindstrom, Ray Merrill, Chris Nolte, John Offenberg, Theran Riedel, Jeff Ryan, Donna Schwede, Madeleine Strum, Mark Strynar, and Alan Vette at the U.S. EPA for helpful discussions and review. The U.S. EPA through its Office of Research and Development supported the research described here. It has been subjected to Agency administrative review and approved for publication, but may not necessarily reflect official Agency policy.
Footnotes
Supporting Information
Additional information on WRF modeling and emissions allocation. Several tables including the compounds emitted from the facility with their identifiers, physical properties, and emissions information. Tables on stack parameters, standard pollutant evaluation, WRF meteorological evaluation, and weekly deposition measurements. Figures denoting GenX-relevant compounds (HFPO-DA, HFPO-DAF, etc), CMAQ-PFAS domain, measurement locations. Several figures investigating deposition, air concentration, and phase state of GenX and total PFAS.
References
- 1.OECD (The Organisation for Economic Co-operation and Development) Toward a new comprehensive global database of per- and polyfluoroalkyl substances (PFASs): summary report on updating the OECD 2007 list of per and polyfluoroalkyl substances (PFASs); https://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV-JM-MONO(2018)7&doclanguage=en 2018.
- 2.Lindstrom AB; Strynar MJ; Libelo EL, Polyfluorinated Compounds: Past, Present, and Future. Environmental Science & Technology 2011, 45 (19), 7954–7961. [DOI] [PubMed] [Google Scholar]
- 3.Ghisi R; Vamerali T; Manzetti S, Accumulation of perfluorinated alkyl substances (PFAS) in agricultural plants: A review. Environmental Research 2019, 169, 326–341. [DOI] [PubMed] [Google Scholar]
- 4.Harrad S; de Wit CA; Abdallah MA-E; Bergh C; Björklund JA; Covaci A; Darnerud PO; de Boer J; Diamond M; Huber S; Leonards P; Mandalakis M; Östman C; Haug LS; Thomsen C; Webster TF, Indoor Contamination with Hexabromocyclododecanes, Polybrominated Diphenyl Ethers, and Perfluoroalkyl Compounds: An Important Exposure Pathway for People? Environmental Science & Technology 2010, 44 (9), 3221–3231. [DOI] [PubMed] [Google Scholar]
- 5.Houde M; De Silva AO; Muir DCG; Letcher RJ, Monitoring of Perfluorinated Compounds in Aquatic Biota: An Updated Review. Environmental Science & Technology 2011, 45 (19), 7962–7973. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita N; Kannan K; Taniyasu S; Horii Y; Petrick G; Gamo T, A global survey of perfluorinated acids in oceans. Mar. Pollut. Bull 2005, 51 (8–12), 658–668. [DOI] [PubMed] [Google Scholar]
- 7.Rahman MF; Peldszus S; Anderson WB, Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: A review. Water Research 2014, 50, 318–340. [DOI] [PubMed] [Google Scholar]
- 8.Shin HM; Vieira VM; Ryan PB; Detwiler R; Sanders B; Steenland K; Bartell SM, Environmental Fate and Transport Modeling for Perfluorooctanoic Acid Emitted from the Washington Works Facility in West Virginia. Environmental Science & Technology 2011, 45 (4), 1435–1442. [DOI] [PubMed] [Google Scholar]
- 9.Paustenbach DJ; Panko JM; Scott PK; Unice KM, A Methodology for Estimating Human Exposure to Perfluorooctanoic Acid (PFOA): A Retrospective Exposure Assessment of a Community (1951–2003). Journal of Toxicology and Environmental Health, Part A 2006, 70 (1), 28–57. [DOI] [PubMed] [Google Scholar]
- 10.Barton CA; Butler LE; Zarzecki CJ; Flaherty J; Kaiser M, Characterizing perfluorooctanoate in ambient air near the fence line of a manufacturing facility: Comparing modeled and monitored values. Journal of the Air & Waste Management Association 2006, 56 (1), 48–55. [DOI] [PubMed] [Google Scholar]
- 11.Barton CA; Zarzecki CJ; Russell MH, A Site-Specific Screening Comparison of Modeled and Monitored Air Dispersion and Deposition for Perfluorooctanoate. Journal of the Air & Waste Management Association 2010, 60 (4), 402–411. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman K; Webster TF; Bartell SM; Weisskopf MG; Fletcher T; Vieira VM, Private Drinking Water Wells as a Source of Exposure to Perfluorooctanoic Acid (PFOA) in Communities Surrounding a Fluoropolymer Production Facility. Environmental Health Perspectives 2011, 119 (1), 92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emmett EA; Shofer FS; Zhang H; Freeman D; Desai C; Shaw LM, Community exposure to perfluorooctanoate: Relationships between serum concentrations and exposure sources. Journal of Occupational and Environmental Medicine 2006, 48 (8), 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotlarz N; McCord J; Collier D; Lea CS; Strynar M; Lindstrom AB; Wilkie AA; Islam JY; Matney K; Tarte P; Polera ME; Burdette K; DeWitt J; May K; Smart RC; Knappe DRU; Hoppin JA, Measurement of Novel, Drinking Water-Associated PFAS in Blood from Adults and Children in Wilmington, North Carolina (vol 128, 077005, 2020). Environmental Health Perspectives 2020, 128 (8), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis KL; Aucoin MD; Larsen BS; Kaiser MA; Hartten AS, Transport of ammonium perfluorooctanoate in environmental media near a fluoropolymer manufacturing facility. Chemosphere 2007, 67 (10), 2011–2019. [DOI] [PubMed] [Google Scholar]
- 16.ITRC (Interstate Technology & Regulatory Council), PFAS Technical and Regulatory Guidance Document and Fact Sheets PFAS-1. Interstate Technology & Regulatory Council, P. T, Ed. Washington, D.C., 2020. [Google Scholar]
- 17.Galloway JE; Moreno AVP; Lindstrom AB; Strynar MJ; Newton S; May AA; Weavers LK, Evidence of Air Dispersion: HFPO–DA and PFOA in Ohio and West Virginia Surface Water and Soil near a Fluoropolymer Production Facility. Environmental Science & Technology 2020, 54 (12), 7175–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makey CM; Webster TF; Martin JW; Shoeib M; Harner T; Dix-Cooper L; Webster GM, Airborne Precursors Predict Maternal Serum Perfluoroalkyl Acid Concentrations. Environmental Science & Technology 2017, 51 (13), 7667–7675. [DOI] [PubMed] [Google Scholar]
- 19.O’Hagan D, Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev 2008, 37 (2), 308–319. [DOI] [PubMed] [Google Scholar]
- 20.Armitage JM; MacLeod M; Cousins IT, Modeling the Global Fate and Transport Of Perfluorooctanoic acid (PFOA) and Perfluorooctanoate (PFO) Emitted from Direct Sources Using a Multispecies Mass Balance Model (vol 43, pg 1134, 2009). Environmental Science & Technology 2009, 43 (16), 6438–6439. [DOI] [PubMed] [Google Scholar]
- 21.Jahnke A; Berger U; Ebinghaus R; Temme C, Latitudinal Gradient of Airborne Polyfluorinated Alkyl Substances in the Marine Atmosphere between Germany and South Africa (53° N−33° S). Environmental Science & Technology 2007, 41 (9), 3055–3061. [DOI] [PubMed] [Google Scholar]
- 22.Dreyer A; Weinberg I; Temme C; Ebinghaus R, Polyfluorinated Compounds in the Atmosphere of the Atlantic and Southern Oceans: Evidence for a Global Distribution. Environmental Science & Technology 2009, 43 (17), 6507–6514. [DOI] [PubMed] [Google Scholar]
- 23.Thackray CP; Selin NE; Young CJ, A global atmospheric chemistry model for the fate and transport of PFCAs and their precursors. Environmental Science: Processes & Impacts 2020, 22 (2), 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yarwood G; Kemball-Cook S; Keinath M; Waterland RL; Korzeniowski SH; Buck RC; Russell MH; Washburn ST, High-resolution atmospheric modeling of fluorotelomer alcohols and perfluorocarboxylic acids in the north American troposphere. Environmental Science & Technology 2007, 41 (16), 5756–5762. [DOI] [PubMed] [Google Scholar]
- 25.Chen H; Yao Y; Zhao Z; Wang Y; Wang Q; Ren C; Wang B; Sun H; Alder AC; Kannan K, Multimedia Distribution and Transfer of Per- and Polyfluoroalkyl Substances (PFASs) Surrounding Two Fluorochemical Manufacturing Facilities in Fuxin, China. Environmental Science & Technology 2018, 52 (15), 8263–8271. [DOI] [PubMed] [Google Scholar]
- 26.Strynar M; Dagnino S; McMahen R; Liang S; Lindstrom A; Andersen E; McMillan L; Thurman M; Ferrer I; Ball C, Identification of Novel Perfluoroalkyl Ether Carboxylic Acids (PFECAs) and Sulfonic Acids (PFESAs) in Natural Waters Using Accurate Mass Time-of-Flight Mass Spectrometry (TOFMS). Environmental Science & Technology 2015, 49 (19), 11622–11630. [DOI] [PubMed] [Google Scholar]
- 27.Sun M; Arevalo E; Strynar M; Lindstrom A; Richardson M; Kearns B; Pickett A; Smith C; Knappe DRU, Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina. Environmental Science & Technology Letters 2016, 3 (12), 415–419. [Google Scholar]
- 28.McCord J; Strynar M, Identification of Per- and Polyfluoroalkyl Substances in the Cape Fear River by High Resolution Mass Spectrometry and Nontargeted Screening. Environmental Science & Technology 2019, 53 (9), 4717–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayama S; Strynar MJ; Helfant L; Egeghy P; Ye X; Lindstrom AB, Perfluorinated Compounds in the Cape Fear Drainage Basin in North Carolina. Environmental Science & Technology 2007, 41 (15), 5271–5276. [DOI] [PubMed] [Google Scholar]
- 30.Brandsma SH; Koekkoek JC; van Velzen MJM; de Boer J, The PFOA substitute GenX detected in the environment near a fluoropolymer manufacturing plant in the Netherlands. Chemosphere 2019, 220, 493–500. [DOI] [PubMed] [Google Scholar]
- 31.NC DEQ GenX Investigation. 2018. https://deq.nc.gov/news/key-issues/genx-investigation/.
- 32.Moreno AV P. Modeling Atmospheric Transport of Perfluorinated Alkyl Substances from Chemours Facilities Using CALPUFF View The Ohio State University, 2019. [Google Scholar]
- 33.Hopkins ZR; Sun M; DeWitt JC; Knappe DRU, Recently Detected Drinking Water Contaminants: GenX and Other Per- and Polyfluoroalkyl Ether Acids. Journal American Water Works Association 2018, 110 (7), 13–28. [Google Scholar]
- 34.Bunton CA; Fendler JH, The Hydrolysis of Acetyl Fluoride. The Journal of Organic Chemistry 1966, 31 (7), 2307–2312. [Google Scholar]
- 35.Mansouri K; Grulke CM; Judson RS; Williams AJ, OPERA models for predicting physicochemical properties and environmental fate endpoints. Journal of Cheminformatics 2018, 10 (1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pankow JF, An Absorption Model of Gas/Paritcle Partitioning of Organic Compounds in the Atmosphere. Atmospheric Environment 1994, 28 (2), 185–188. [Google Scholar]
- 37.Donahue NM; Robinson AL; Stanier CO; Pandis SN, Coupled partitioning, dilution, and chemical aging of semivolatile organics. Environmental Science & Technology 2006, 40 (8), 2635–2643. [DOI] [PubMed] [Google Scholar]
- 38.Lampic A; Parnis JM, Property Estimation of Per- and Polyfluoroalkyl Substances: A Comparative Assessment of Estimation Methods. Environ. Toxicol. Chem 2020, 39 (4), 775–786. [DOI] [PubMed] [Google Scholar]
- 39.US EPA Office of Research and Development CMAQ, v5.3.1; 2019.
- 40.Skamarock WC, Klemp JB, Dudhia J, Gill DO, Liu Z, Berner J, Wang W, Powers JG, Duda MG, Barker DM, Huang X-Y A Description of the Advanced Research WRF Version 4. NCAR Tech. Note NCAR/TN-556+STR; 2019; p 145. [Google Scholar]
- 41.Luecken DJ; Yarwood G; Hutzell WT, Multipollutant modeling of ozone, reactive nitrogen and HAPs across the continental US with CMAQ-CB6. Atmospheric Environment 2019, 201, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nolte CG; Appel KW; Kelly JT; Bhave PV; Fahey KM; Collett JL Jr; Zhang L; Young JO, Evaluation of the Community Multiscale Air Quality (CMAQ) model v5.0 against size-resolved measurements of inorganic particle composition across sites in North America. Geosci. Model Dev 2015, 8 (9), 2877–2892. [Google Scholar]
- 43.U.S. EPA 2014 National Emissions Inventory, Version 1 Technical Support Document; https://www.epa.gov/air-emissions-inventories/2014-national-emission-inventory-nei-report US Environmental Protection Agency: 2016. [Google Scholar]
- 44.Bash JO; Baker KR; Beaver MR, Evaluation of improved land use and canopy representation in BEIS v3.61 with biogenic VOC measurements in California. Geoscientific Model Development 2016, 9 (6), 2191–2207. [Google Scholar]
- 45.Appel KW; Bash JO; Fahey KM; Foley KM; Gilliam RC; Hogrefe C; Hutzell WT; Kang D; Mathur R; Murphy BN; Napelenok SL; Nolte CG; Pleim JE; Pouliot GA; Pye HOT; Ran L; Roselle SJ; Sarwar G; Schwede DB; Sidi FI; Spero TL; Wong DC, The Community Multiscale Air Quality (CMAQ) Model Versions 5.3 and 5.3.1: System Updates and Evaluation. Geosci. Model Dev. Discuss 2020, 2020, 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.George C; Saison JY; Ponche JL; Mirabel P, Kinetics of mass transfer of carbonyl fluoride, trifluoroacetyl fluoride, and trifluoroacetyl chloride at the air/water interface. The Journal of Physical Chemistry 1994, 98 (42), 10857–10862. [Google Scholar]
- 47.Kotamarthi VR; Rodriguez JM; Ko MKW; Tromp TK; Sze ND; Prather MJ, Trifluoroacetic acid from degradation of HCFCs and HFCs: A three-dimensional modeling study. J. Geophys. Res.-Atmos 1998, 103 (D5), 5747–5758. [Google Scholar]
- 48.Wang ZY; Wang YH; Li JF; Henne S; Zhang BY; Hu JX; Zhang JB, Impacts of the Degradation of 2,3,3,3-Tetrafluoropropene into Trifluoroacetic Acid from Its Application in Automobile Air Conditioners in China, the United States, and Europe. Environmental Science & Technology 2018, 52 (5), 2819–2826. [DOI] [PubMed] [Google Scholar]
- 49.Kazil J; McKeen S; Kim SW; Ahmadov R; Grell GA; Talukdar RK; Ravishankara AR, Deposition and rainwater concentrations of trifluoroacetic acid in the United States from the use of HFO-1234yf. J. Geophys. Res.-Atmos 2014, 119 (24), 14059–14079. [Google Scholar]
- 50.Thackray CP; Selin NE, Uncertainty and variability in atmospheric formation of PFCAs from fluorotelomer precursors. Atmospheric Chemistry and Physics 2017, 17 (7), 4585–4597. [Google Scholar]
- 51.Shoemaker J; Tettenhorst D, Method 537.1: Determination of Selected Per- and Polyfluorinated Alkyl Substances in Drinking Water by Solid Phase Extraction and Liquid Chromatography/Tandem Mass Spectrometry (LC/MS/MS) U.S. Environmental Protection Agency, O. o. R. a. D., National Center for Environmental Assessment, Ed. Washington, DC, 2018. [Google Scholar]
- 52.DuPont, DuPont™ GenX Processing Aid for Making Fluoropolymer Resins: Setting a new industry standard for sustainable replacement technology. In https://bladenonline.com/wp-content/uploads/2017/06/Chemours_GenX_Brochure_Final_07July2010.pdf, 2010.
- 53.Carlton AG; Turpin BJ, Particle partitioning potential of organic compounds is highest in the Eastern US and driven by anthropogenic water. Atmos. Chem. Phys 2013, 13 (20), 10203–10214. [Google Scholar]
- 54.Chen H; Zhang L; Li MQ; Yao YM; Zhao Z; Munoz G; Sun HW, Per- and polyfluoroalkyl substances (PFASs) in precipitation from mainland China: Contributions of unknown precursors and short-chain (C2-C3) perfluoroalkyl carboxylic acids. Water Research 2019, 153, 169–177. [DOI] [PubMed] [Google Scholar]
- 55.Dreyer A; Matthias V; Weinberg I; Ebinghaus R, Wet deposition of poly- and perfluorinated compounds in Northern Germany. Environ. Pollut 2010, 158 (5), 1221–1227. [DOI] [PubMed] [Google Scholar]
- 56.Washington JW; Rosal CG; McCord JP; Strynar MJ; Lindstrom AB; Bergman EL; Goodrow SM; Tadesse HK; Pilant AN; Washington BJ; Davis MJ; Stuart BG; Jenkins TM, Nontargeted mass-spectral detection of chloroperfluoropolyether carboxylates in New Jersey soils. Science 2020, 368 (6495), 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joerss H; Xie Z; Wagner CC; von Appen W-J; Sunderland EM; Ebinghaus R, Transport of Legacy Perfluoroalkyl Substances and the Replacement Compound HFPO-DA through the Atlantic Gateway to the Arctic Ocean—Is the Arctic a Sink or a Source? Environmental Science & Technology 2020, 54 (16), 9958–9967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.