

Abstract
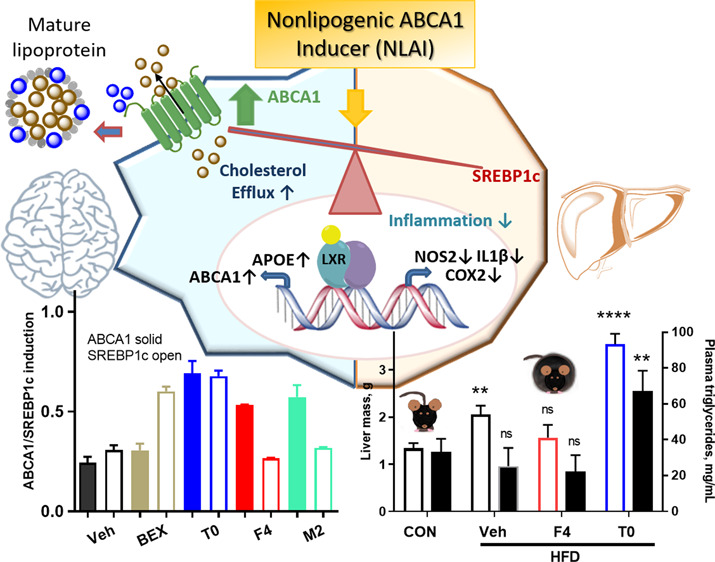
Selective liver X receptor (LXR) agonists have been extensively pursued as therapeutics for Alzheimer’s disease and related dementia (ADRD) and, for comorbidities such as type 2 diabetes (T2D) and cerebrovascular disease (CVD), disorders with underlying impaired insulin signaling, glucose metabolism, and cholesterol mobilization. The failure of the LXR-focused approach led us to pursue a novel strategy to discover nonlipogenic ATP-binding cassette transporter A1 (ABCA1) inducers (NLAIs): screening for ABCA1-luciferase activation in astrocytoma cells and counterscreening against lipogenic gene upregulation in hepatocarcinoma cells. Beneficial effects of LXRβ agonists mediated by ABCA1 include the following: control of cholesterol and phospholipid efflux to lipid-poor apolipoproteins forming beneficial peripheral HDL and HDL-like particles in the brain and attenuation of inflammation. While rare, ABCA1 variants reduce plasma HDL and correlate with an increased risk of ADRD and CVD. In secondary assays, NLAI hits enhanced cholesterol mobilization and positively impacted in vitro biomarkers associated with insulin signaling, inflammatory response, and biogenic properties. In vivo target engagement was demonstrated after oral administration of NLAIs in (i) mice fed a high-fat diet, a model for obesity-linked T2D, (ii) mice administered LPS, and (iii) mice with accelerated oxidative stress. The lack of adverse effects on lipogenesis and positive effects on multiple biomarkers associated with T2D and ADRD supports this novel phenotypic approach to NLAIs as a platform for T2D and ADRD drug discovery.
Keywords: ABCA1, diabetes, Alzheimer’s, cholesterol, phenotypic drug discovery
Introduction
Sporadic or late-onset Alzheimer’s disease and related dementia (ADRD) constitute a present and growing health crisis in the aging population. Equally, the increasing prevalence of obesity is a major risk factor for the development of chronic metabolic diseases including type 2 diabetes (T2D), which is also a risk factor for COVID19 mortality. T2D is a comorbidity with ADRD and a major risk factor for cardiovascular and cerebrovascular disease (CVD) causing cerebral infarcts that impact cognition.1 Specifically, impaired insulin signaling and glucose metabolism, mitochondrial dysfunction, inflammation, dyslipidemia, and impaired cholesterol mobilization may be common underlying pathogenic promoters of dementia in T2D and ADRD.2 Impaired insulin signaling contributes to AD pathogenesis even in patients without overt diabetes.3 Thus, therapeutic approaches targeting one or more underlying promoters of T2D are likely to be beneficial in treatment or prevention of CVD and ADRD.
APOE4 is the strongest genetic risk factor for ADRD and is an independent risk factor for T2D and CVD.4,5 ApoE is a component of high-density lipoprotein (HDL) and HDL-like particles that transport cholesterol and other lipids in the blood and brain, respectively.6 The main cholesterol transporter from cell to lipoprotein is ATP-binding cassette transporter A1 (ABCA1).7 ABCA1 is central to reverse cholesterol transport (RCT), a process in which cholesterol is exported from tissues to HDL particles to return to the liver, where it is metabolized or excreted. In the brain, ABCA1 adds cholesterol to HDL-like particles for distribution to various cell types or for efflux across the blood-brain barrier. Thus, ABCA1 is critical for proper maintenance of cholesterol homeostasis in the brain.8 Unsurprisingly, reduced ABCA1 activity or expression correlates with CVD, T2D, and AD risk.
Cell and rodent models of total or tissue-specific ABCA1 knockdown show the following: increased foam cell formation and inflammation associated with atherogenesis in CVD, impaired insulin signaling, and AD-related cognitive deficits and brain pathology.9,10 Carriers of ABCA1 loss-of-function mutations (Tangier disease) likewise are predisposed to atherosclerosis, heart disease, and impaired insulin secretion.11,12ABCA1 variants affect plasma HDL and risk of CVD and T2D.13 GWAS has led to evidence of associations of common ABCA1 variants with AD.14 In a recent report, a novel loss-of-function mutation in ABCA1 (N1800H) was associated with high risk of AD and CVD in a large Danish cohort. This variant affects plasma HDL and cholesterol efflux and is associated with low plasma levels of apoE.15
Multiple studies have demonstrated the association between decreased ABCA1, insulin signaling, and T2D in both human patients and animal models.16,17 ABCA1 upregulation is expected positively to influence insulin signaling and inflammation in the brain and periphery and, therefore, to be of potential therapeutic utility in T2D and ADRD. To discover and develop Non-Lipogenic ABCA1 Inducers (NLAIs), we developed a phenotypic approach to identify compounds selectively inducing ABCA1 over unwanted upregulation of hepatic lipogenic genes.
ABCA1 and apoE are under transcriptional control of nuclear receptors (NR), specifically liver X receptors (LXRs). LXR forms transcriptionally repressed heterodimeric complexes, with either a retinoid X receptor (RXR) or a peroxisome-proliferator-activated receptor (PPAR), which are derepressed (activated) by agonist binding.18,19 The RXR agonist bexarotene (Bex) was reported to clear oligomeric Aβ, the likely proximal neurotoxic form of Aβ; however, Bex may cause hypertriglyceridemia and hypercholesterolemia, increasing the risk of cardiovascular and liver disease.20 PPARγ agonists are used clinically in treatment of T2D, and LXR agonists have shown promise in models of T2D and ADRD.21,22 However, lipogenesis is an inherent risk.
Sterol regulatory element-binding protein 1c (SREBP1c) plays a key role in the induction of lipogenesis by the liver. Studies with isoform-specific LXR knockout mice show that SREBP1c-mediated effects are largely controlled via LXRα, which is highly expressed in the liver, while other effects can be mediated by either LXRα or β.23 The nonselective LXR agonist T0901317 (T0) demonstrated reversal of insulin resistance in mice; however, this was accompanied by hyperlipidemia, as would be expected for a potent pan-LXR agonist.21 In rhesus monkeys, T0 positively affected ABCA1, apoE, and CSF Aβ but with significant adverse effects on liver fat and triglycerides. In clinical trials, a selective LXRβ agonist upregulated LXR target genes ABCA1 and ABCG1.24
The potential of LXRβ agonists in T2D and ADRD has led to a substantial effort to develop potent, selective LXRβ agonists; however, this has not translated to successful clinical trials.25 Phenotypic drug discovery of NLAIs provides an alternative strategy that we describe for the first time herein. Screening of a 10,000 compound library led to identification of NLAI hits that upregulated ABCA1, without unwanted elevation of lipogenic genes. The translational validation of this approach was demonstrated in primary cell cultures and animal models, observing enhanced cholesterol mobilization, attenuated inflammation, and improved biomarkers of glucose metabolism, without lipogenesis. This novel strategy provides a platform for NLAI hit identification as a basis for optimization and further study in preclinical models of T2D and ADRD.
Results
Bioassay Development and Hit Validation for NLAI Discovery
Effective phenotypic screening requires a rigorous, validated screen and counterscreen. By screening in astrocytoma CCF-STTG1 cells stably transduced with an ABCA1 promoter linked to a luciferase response element (LRE), we identified hits that induce ABCA1 expression in a CNS cell model of astrocyte function. To remove hits with unwanted lipogenic effects in hepatocytes, we constructed a counterscreen in HepG2 hepatocellular carcinoma cells stably transduced with an LRE downstream of a SREBP1c promoter. SREBP1c is a transcription factor that acts as a master regulator of lipogenesis in the liver.26 The concentration–response of T0 in these assays emphasizes the lipogenic properties of T0 resulting from potent agonism of both LXRα and LXRβ (Figure 1A): T0 is widely used as a positive control, because it induces a maximal activation of LXRs. The primary assays were validated by measuring the Z′-factor (>0.6) using 1 μM T0 as a positive control. We screened 10,000 compounds from the 55,000 compound ChemDiv SMART Library, excluding PAINS and chemically intractable compounds and balancing the diversity of chemical scaffolds with inclusion of several representative analogs for each chemical scaffold.27,28 Screening was conducted at 10 μM in the CCF/ABCA1-luc primary screen. We also screened in the presence of submaximal T0 (50 nM) to explore the utility of this adaptation to the primary assay in identifying hits that potentiated T0 effects at LXR (Figure S1A–C). We identified 760 hit compounds, defined by a ≥1.5× increase of ABCA1-luc activity over the vehicle, of which 680 were deselected, owing to a >1.0× increase in SREBP1c-luc induction over the vehicle control in HepG2 cells (Figure S1D,E). Repurchase/resynthesis, dose–response assays in CCF cells, and cytotoxicity in CCF and HepG2 cells were used for hit validation and selection, with final validation requiring lack of toxicity in HepG2 3D spheroid cultures (Figure S2A–C). Using this workflow, 20 hit chemotypes were validated, including hits F4, M2, and L7 (Scheme 1). The combination of primary screen and counterscreen was designed to yield NLAIs with the potential to induce ABCA1 in the CNS while minimizing lipogenesis in the liver.
Figure 1.

NLAIs increased ABCA1 without affecting lipogenic gene induction. Response of primary screen (A: CCF-ABCA1) and counterscreen (B: HepG2-SREBP1c) to the NLAIs F4 and M2 was compared to benchmark LXR agonist T0 and C) concentration–response for T0 alone. D, E) Response, normalized to the vehicle, of primary screen (D) and counterscreen (E) to HTS hits (F4, M2; 10 μM), RXR agonist (BEX; 10 μM), endogenous LXR agonist (24S-OHC; 10 μM), and exogenous LXR agonists (T0, GW3965; 10 μM), alone or in combination, with significant increases observed in treatment groups versus the vehicle. F) Comparison of induction of ABCA1 (solid bars) and SREBP1c (open bars) by exogenous hits and benchmark compounds, normalized to the combination of T0+BEX. G) Modulation of mRNA for ABCA1 in CCF cells and for FAS and SREBP1c in HepG2 cells after treatment by T0, Bex, and hits F4 and M2. Data analyzed by one-way ANOVA with Dunnett’s multicomparison analysis: **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05.
Scheme 1. Chemical Structures and Abbreviations of Chemical Probes and NLAI Hits.

The response in the primary assays to hits F4 and M2 was compared with T0 (Figure 1A–C), LXRβ selective agonist GW3965 (GW), endogenous LXR ligand 24-hydroxycholesterol (24S-OHC), and RXR agonists Bex and LG100268 (LG) (Figure 1D,E; Scheme 1). It is important to note that charcoal-stripped media was used throughout to exclude interference from endogenous nuclear receptor ligands. The maximal response in the primary assay was observed for the combination of LXR (T0) and RXR (Bex) agonists. Normalizing to this maximal response, the NLAI hits F4 and M2 showed clear selectivity for induction of ABCA1 versus SREBP1c, in marked contrast to the lack of selectivity shown by LXR agonists and the reverse selectivity of RXR agonists (Figure 1F). Reporter activation data in these cell lines was confirmed by mRNA measurement of ABCA1 in CCF cells and lipogenic genes (SREBF1 and FASN encoding SREBP1c and FAS, respectively) in HepG2 cells (Figure 1G).
The ABCA1 transporter mediates cholesterol efflux from astrocytes to form HDL-like particles; therefore, we measured cholesterol efflux from CCF cells that were loaded with BODIPY-cholesterol. Cells were treated for 24 h with NLAI hits F4, M2, or L7, Bex, or T0 as a positive control (all 5 μM). Cholesterol efflux was measured at 8 h after the addition of unlipidated recombinant apoA-I (25 μg/mL) as the exogenous lipid acceptor and 24 h after addition of test compounds. We observed a significant increase in cholesterol efflux when astrocytes were treated overnight with NLAI hits F4 and M2, and interestingly, both F4 and M2 were equipotent to T0 in stimulating cholesterol efflux (Figure 2A,B; Figure S2F). Similar induction of cholesterol efflux was seen in J774 macrophages treated with T0 or F4 (Figure S2D).
Figure 2.

NLAIs enhanced cholesterol efflux and expression of cholesterol transport genes. A) Representative images of BODIPY-cholesterol in CCF cells treated with T0, Bex, and F4, before and after addition of acceptor ApoAI (also see Figure S2F). B) Quantitation of BODIPY-cholesterol efflux to ApoAI for benchmark LXR and RXR agonists and hits F4, M2, and L7, showing significance relative to the DMSO vehicle control. C) ABCA1, ABCG1, and APOE mRNA levels measured by RT-PCR in primary human astrocytes after treatment with validated hits or benchmark agonists (all at 5 μM), relative to the DMSO vehicle control. All fold changes are significantly different from the vehicle (p < 0.05) except those marked by ns. D) Comparison of hits F4 and M2 with T0 (all at 5 μM) in glial cell cultures from mice expressing human APOE4 showing significance of induction of ABCA1, ABCG1, APOE, and PGC1A mRNA relative to the DMSO vehicle control. Data analyzed by one-way ANOVA with Dunnett’s multicomparison analysis relative to the DMSO vehicle control: *** p < 0.001; ** p < 0.01; * p < 0.05.
Hit Profiling in Vitro Reveals Positive Biomarker Modulation and Target Engagement
Phenotypic screening may reveal hits with different targets and diverse mechanisms; therefore, profiling of hits in CCF and in HepG2 cells was used to explore transcriptional footprints associated with a positive phenotype and to aid in hit prioritization. Pathway-focused PCR arrays related to lipid and cholesterol metabolism, neuroinflammation, and glucose homeostasis are relevant to pathogenesis and progression of ADRD and T2D. Representative hit profiling (Figure S3A,B) in a lipid-focused array confirmed upregulation of ABCA1, ABCG1, and APOE by F4. In an LDL-related panel of genes encoding apolipoproteins and LDL receptor, as well as genes involved in immune and other LDL-related functions, treatment of HepG2 cells with F4 showed upregulation of PPAR target genes such as CXCL16, an atheroprotective scavenger receptor that specifically binds to oxidized LDL (OxLDL).29 In addition, F4 treatment showed upregulation of genes, such as LRP1, LRP1/11, and -/12, important in cholesterol mobilization and inflammatory response. Hits were further profiled using arrays of diabetes associated genes, with selected observations followed up with RT-PCR. F4 treatment modulated mitochondrial genes such as PGC1α and PGC1β in CCF cells and activated expression of glucose transporters (GLUT1 and GLUT4) in CCF cells and GLUT4 in HepG2 cells. GLUT4, the main insulin-responsive glucose transporter, is often downregulated in T2D.30 F4 treatment also increased the expression of insulin sensitizing genes such as IRS and IGF-1. The array analysis supported our phenotypic approach, with potentially beneficial gene modulations observed, in particular by hit F4 (Figure S3B).
Target engagement was rigorously validated for NLAI hits in human primary astrocytes (E3/E4 genotype) (Figure 2C and Figure S2E). We further investigated the effect of F4 and M2 in mouse primary astrocytes, derived from EFAD mice expressing human APOE4.31 The mRNA response to F4 and M2 treatment was significant and comparable to the response to T0 for ABCA1 and ABCG1 expression levels and superior to T0 for APOE expression. F4 also induced a significant upregulation of PGC1α (Figure 2D). Similarly, protein levels of ABCA1 increased following treatment with F4 or M2 in both CCF-STTG1 cells and mouse primary astrocytes derived from EFAD mice (Figure S4), corroborating the observed gene induction. Finally, we challenged human primary astrocytes with 1 μg/mL LPS and treated with test compounds at 10 μM. Interestingly, both F4 and M2 attenuated expression of pro-inflammatory biomarkers including TNFα, IL-1β, IL-6, CCL5, and CXCL10 with efficacy comparable to T0 (Figure S3C).
Identification of Nuclear Receptors as NLAI Targets
Phenotypic screening for NLAI hits is target agnostic; however, agonists for nuclear receptors, notably LXRs and PPARs, are anticipated to have NLAI activity. With the aim of identifying potential protein binding partners, we first screened NLAI hits using luciferase-reporter assays for agonist activity at human LXRβ, PPARα, and PPARγ in CHO cells from Indigo Biosciences. This cell line stably expresses a hybrid receptor comprising the N-terminal Gal4 DNA-binding domain fused to the ligand-binding domain of the specific nuclear receptor. Increases in LXRβ transactivation activity were detected in cells exposed to F4 and M2 (10 μM) (Figure 3A), while other NLAI hits elicited significant increases in PPARγ transactivation activity (Figure S5).
Figure 3.

Target identification for NLAIs at various nuclear receptors. A) LXRβ, PPARγ, and PPARα transactivation by NLAI hits: F4, L7, M2, and L5 displayed LXRβ agonistic activity in the luciferase reporter assay, whereas other NLAIs (see Figure S5) displayed PPARγ agonist activity. B) Recruitment of coactivator TRAP220/DRIP2 to human LXRα-LBD by NLAI hits, T0, or GW, as measured by coregulator recruitment TR-FRET (CRT). C) Recruitment of coactivator D22 to human LXRβ-LBD by NLAI hits, T0, or GW, as measured by CRT. CRT data expressed as the mean ratio of the emission signal at 520 nm and the signal at 495 nm, averaged across n = 3 independent experiments, and normalized to maximal coactivator recruitment. D) Tabulation of calculated potency and efficacy from CRT data: (*) to calculate LXRα potency for NLAI hits L5 and L7, maximal efficacy was constrained at 100%.
To further investigate LXRs as a target of NLAI hits, we measured binding to LXRα-LBD and LXRβ-LBD using Coregulator Recruitment TR-FRET (CRT) compared to the benchmark LXR agonists T0, a pan-agonist, and the LXRβ selective agonist GW. Recruitment of coactivator peptides leading to an increase in TR-FRET signal is induced by LXR ligand binding. For LXRα, T0 is a potent full agonist, whereas GW is a potent partial agonist, and all NLAI hits are weak agonists. The data for F4 and M2 are compatible with partial agonist activity with respect to recruitment of the coactivator TRAP220 peptide (Figure 3B,D): at 10 μM, F4 and M2 induced coactivator recruitment at approximately 50% of that induced by T0, and both NLAIs were approximately 20-fold less potent than T0. GW was more potent than T0 with similar efficacy in recruitment of the D22 coactivator to LXRβ: EC50 = 47 nM and Emax = 105% (Figure 3C,D). The potency of F4 and M2 at LXRβ was 2–5 μM with F4 showing similar efficacy to GW. At 10 μM, F4 induced coactivator recruitment to LXRβ at 80% of that induced by T0 and GW. Regarding potency, none of GW, F4, or M2 showed significant LXR selectivity; however, GW and F4 showed selective efficacy toward LXRβ, a strategy previously reported in development of LXRβ selective agents.25
Hit Profiling in Vivo Shows Target Engagement without Triglyceride Elevation
Treatment of C57BL/6 (WT) mice for 7 days is sufficient for preliminary assessment of target engagement (elevated ABCA1) and unwanted lipogenesis (liver triglyceride elevation). Dosing with T0 was guided by multiple literature studies and informed dosing of F4 and M2. In accord with the literature, T0 significantly elevated ABCA1 and SREBP1c in liver tissues and also significantly increased mRNA for ABCA1 and ABCG1 in brain tissues (Figure 4A–C). Administration of F4 at the higher dose (30 mg/kg) caused significant elevation of brain and liver ABCA1. Plasma and brain concentrations of F4 30 min after oral delivery (200–250 ng/mL) were compatible with these observations; however, M2 was not assayed in this study. Small but significant increases in liver SREBP1c were observed with NLAI treatment (Figure 4C); however, this did not translate to a significant increase in liver or plasma triglycerides on treatment with F4, in contrast to T0 (Figure 4D); moreover neither NLAI caused a significant increase in FAS and SCD expression in the liver. The significant increase in SREBP1c, the master regulator of lipogenic gene expression in the liver, and downstream genes such as FAS and SCD was mirrored by an increase in triglyceride levels for mice treated with T0 and illustrates the lipogenic effects that the NLAI discovery strategy was designed to avoid (Figure 4D).
Figure 4.

In vivo pharmacodynamic readouts revealed target engagement without increased triglyceride levels. Mice were dosed daily (p.o.) for 7 days with the vehicle or the indicated doses of F4, M2, or T0. A,B) Fold induction of ABCA1 and ABCG1 mRNA relative to the vehicle control was analyzed in brain tissues (A) and liver (B). C) Fold induction of lipogenic gene mRNA relative to the vehicle control was measured in liver. D) Plasma and liver triglycerides were quantified after oral dosing in response to T0 and F4 treatment. Data analyzed by one-way ANOVA with Dunnett’s multicomparison analysis: **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05.
In Mice on HFD, NLAI Hit F4 Is Strongly Differentiated from pan-LXR Agonist T0
Having shown that F4 did not induce lipogenesis in short-term treatment, this NLAI was further tested in mice administered HFD. Mice placed on HFD are an obesogenic model of T2D, with observed phenotypes including weight gain and insulin resistance and inflammation.32 The HFD mouse model also provides robust readouts of multifactorial mechanisms causative in T2D and ADRD.33 Mice were fed HFD for 4 weeks before treatment with F4 or T0 for a further 4 weeks at a dose (30 mg/kg/day p.o.) selected based on the previous 7-day study. After 8 weeks on HFD, the vehicle treated mice showed an increase in body weight, liver weight, and plasma triglycerides, with a significant decrease in HDL (Figure 5A–C). HFD mice that received T0 showed an increase in body weight compared to both the vehicle and F4 treated mice with a significant difference at 8 weeks (Figure 5A). T0 treatment induced liver steatosis with significantly increased liver weight, plasma triglycerides, and LDL and total cholesterol; whereas F4 treatment elicited no significant increases relative to vehicle treated mice, with the exception of HDL, which was restored by F4 to levels not significantly different from healthy control mice (Figure 5B,C). Similar observations were noted in liver sections of HFD-fed mice, where a marked accumulation of fat droplets was observed as compared with control mice fed standard chow. This phenotype was exacerbated by T0 treatment but rescued by F4 (Figure S7).
Figure 5.

Response to F4 treatment in a choline-deficient high-fat diet mouse model. Male C57BL/6 mice were fed choline-deficient HFD for 8 weeks, with F4 or T0 (p.o. 30 mg/kg/day), or the vehicle administered from weeks 5–8 of diet. A) Weight gain was significantly attenuated at 8 weeks in the F4 group versus T0 group. B) Liver weight and plasma triglycerides were significantly increased in the T0 group but not in the F4 treatment group, with overt steatohepatitis observed in the T0 treatment group. C) Decreased HDL-cholesterol due to HFD was significantly restored by F4 and T0 treatment, while LDL and total cholesterol increased only in the T0 treatment group. D–F) Well-known paradoxical decreases in lipogenic genes in the liver caused by HFD were observed, with significant increases in FASN and SCD in the T0 treatment group. G) Brain levels of ABCA1 and APOE were significantly upregulated by F4 and T0 treatment relative to the vehicle. H) Liver levels of ABCA1 and APOE were significantly downregulated by HFD, with F4 treatment significantly restoring levels relative to T0. Data analyzed by one-way ANOVA with Dunnett’s or Tukey’s multicomparison analysis: **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05.
At the transcriptional level, HFD-induced lipid accumulation in the liver causes a downregulation of lipogenic genes,34 which was not influenced by F4 treatment, whereas T0 induced significant upregulation of FAS and SCD (Figure 5D,E). Additionally, F4 treatment induced upregulation of RCT-associated SR-BI significantly relative to the T0 group (Figure 5F). Recapitulating observations after short-term treatment of WT mice, both T0 and F4 upregulated ABCA1 and APOE in brain tissues (Figure 5G). The disruption of liver homeostasis by HFD is reflected by the significant downregulation of ABCA1 and APOE in the liver, which was not significantly influenced by T0 treatment but was partially rescued by F4 (Figure 5H). In general, T0 exacerbated the effects of HFD on liver biomarkers, causing hepatosteatosis; whereas, the response to F4 treatment was neutral, showing no lipogenic effects or body/liver weight gain. Interestingly, treatment of HFD mice with F4 did rescue HDL and levels of ABCA1 and APOE in the liver, in addition to upregulating SR-BI. For further development of NLAIs as therapeutics for T2D and metabolic syndrome, study in additional animal models is needed. However, the observations in HFD mice confirm the ability of the primary screen and counterscreen to identify NLAI hits with the desired nonlipogenic phenotype.
In Vivo Anti-Inflammatory and Procognitive Phenotype
ABCA1 and APOE are under transcriptional control by LXRs and other NRs with which LXRs form heterodimeric complexes. LXR agonists not only directly inhibit proinflammatory gene expression but also indirectly regulate macrophage function via cholesterol mobilization.35 LPS injection provides a well-characterized mouse model of acute systemic and central inflammation.36 We have observed that ALDH2 KO mice lacking expression of mitochondrial aldehyde dehydrogenase-2 (Aldh2) show an amplified response to LPS and other insults, providing a useful preclinical model for drug discovery.37 Aldh2 is a major enzymic contributor to detoxification of lipid peroxidation products (e.g., 4-hydroxynonenal), which are known to contribute to enhanced oxidative stress in aging and are elevated in T2D38 and in ADRD brains.39 Pro-inflammatory genes induced by acute LPS (4 mg/kg i.p. 4 h prior to sacrifice) were elevated relative to control mice not treated with LPS, and ALDH2 KO mice were more sensitive to LPS treatment. Pretreatment with F4 (Figure 6A–C) or T0 (data not shown) (10 mg/kg) for 3 days significantly attenuated the effects of LPS on TNFα, COX2, CCL5, CXCL10, and CCL2 brain inflammatory markers, relative to the vehicle treated group. Attenuation of LPS-induced cytokines has been reported: via activation of NRs, including LXR, upregulation of ABCA1, or via LRP-1 mediated mechanisms.36,40
Figure 6.

F4 suppressed inflammation in two mouse models of acute (LPS) and chronic (HFD) inflammation. A–C) WT and ALDH2–/– (KO) mice were administered LPS to induce expression of pro-inflammatory markers and chemokines in the brain, with responses significantly attenuated by pretreatment with F4. D–F) HFD increased brain expression of pro-inflammatory genes, which was reversed by F4 treatment. G) KO mice administered HFD showed attenuation of pro-inflammatory gene expression when cotreated with F4. H) Discrimination Index of KO mice and WT littermates in NOR task before initiating HFD (Pre) and after 1 and 2 weeks on HFD showing a decline in performance and rescue by F4. Data analyzed by one-way ANOVA with Dunnett’s or Tukey’s multicomparison analysis: **** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05.
The HFD model of obesity-induced T2D induces a chronic pro-inflammatory response in tissues.33 WT and ALDH2 KO mice fed HFD showed significant increases in inflammatory markers in the brain, which were attenuated by administration of F4 in WT (Figure 6D–F) and KO mice (Figure 6G). In WT mice, significant increases in NOS2, COX2, and CCL5 were observed in HFD-fed compared to control mice, which in the case of NOS2 and CCL5 was significantly attenuated by treatment with either F4 or T0 (8 weeks on HFD with drug treatment weeks 4–8). In ALDH2 KO mice fed HFD for 4 weeks, cotreatment with F4 led to reduction in levels of TNFα, COX2, and IL6 in the brain, reaching significance for TNFα and COX2 (Figure 6G). Since ALDH2 KO mice show significant cognitive deficits in the Novel Object Recognition task (NOR) from 4M–14M of age,37 we tested 3M-old KO mice alongside WT littermates in NOR at three time points: before initiating HFD, after 1 week, and 2 weeks on HFD (Figure 6H). In some mouse strains, HFD causes cognitive impairment,41 and we observed in NOR in ALDH2 KO mice fed HFD for 2 weeks, a significant deficit measured by the discrimination index (DI = time with novel object – time with familiar object ÷ total time). DI = 0 reflects equal time spent with novel and familiar objects, which occurs if the mouse has no memory of the familiar object. Treatment with F4 reversed the cognitive deficit in NOR at 2 weeks in ALDH2 KO mice. Further studies are needed to reinforce these data.
Discussion
ABCA1 is the main cholesterol efflux transporter, mediating RCT, the clearance of cholesterol from tissues by HDL particles via the liver. RCT protects against cholesterol deposition, risk of coronary events, and CVD.7 The efflux of cholesterol and phospholipids to extracellular apolipoprotein acceptors, notably apoA1 and apoE, creates HDL and HDL-like particles and is driven by the sequential actions of ABCA1 and ABCG1.42 Direct links have been drawn from attenuated ABCA1 function and poorly lipidated apoE4 particles to Aβ clearance in AD and may underlie the increased AD risk in APOE4 carriers.43,44 In mice, loss of ABCA1 results in decreased levels of apoE in the brain, CSF, and plasma and in increased brain amyloid burden.45,46 In humans, the loss-of-function ABCA1 N1800H variant is associated with low plasma levels of apoE and with a high risk of AD and CVD.13,15 The role of ABCA1 in AD extends beyond Aβ clearance and includes roles in inflammation, insulin signaling, and glucose metabolism.9,10,36,47 ABCA1 is critical for maintenance of cholesterol homeostasis, and reduced ABCA1 activity contributes to pathogenesis of not only AD but also CVD and T2D. The identification of small molecule NLAIs (Non-Lipogenic ABCA1 Inducers) is therefore a therapeutic strategy for treatment of multiple diseases.
The expression of ABCA1 and ABCG1 is transcriptionally driven by members of the NR families: LXR, RXR, and PPAR. The key limitation of RXR agonists lies with the upregulation of multiple gene products, notably lipogenic genes leading to triglyceride elevation. In FAD-Tg mice, several LXR agonists have been reported to improve cognitive function and/or reduce brain levels of Aβ;22,48−50 however, this approach is again limited by upregulation of lipogenic genes, including SREBP1c and FAS. For example, T0 showed beneficial effects in an FAD-Tg mouse model but with significant negative effects on liver fat and triglycerides.25 LXR agonists have been reported that do not induce lipogenic genes (Figure S6).25,51 An LXRβ-selective agonist significantly increased brain ABCA1 and ApoE and lowered brain Aβ in FAD-Tg mice without an increase in liver triglycerides.25
However, given the failure, to date, of LXRβ-selective agonists in clinical trials, we chose to develop a novel, alternative phenotypic strategy to discover small molecule NLAIs that upregulate ABCA1 in astrocytes without upregulating SREBP1c in hepatocytes. The high-throughput primary screens in astrocytoma and hepatocarcinoma cell lines yielded hits that delivered the desired phenotype in primary cells and mouse models, that is upregulation of ABCA1 without the concomitant lipogenic effects in the liver leading to triglyceride elevation and steatosis. Demonstration of the desired NLAI phenotype in vivo indicates that this drug discovery strategy may yield therapeutic agents with a safe therapeutic window for treatment of T2D and AD. Our approach is novel and differentiated from previous work using CCF-STTG1 cells to screen for compounds increasing apoE levels and counterscreening against hits with LXR agonist activity.52
Screening of a 10,000 compound library for hits that activated ABCA1-luc and counterscreening against SREBP1c-luc activation gave a hit rate of ∼0.5%, representing 20 hit chemotypes as candidate NLAIs. We chose to profile the validated hits in PCR microarrays relevant to cholesterol metabolism, inflammation, and glucose homeostasis. On this basis, hits F4 and M2 were prioritized for further study. Comparison of hit compounds with LXR and RXR agonists demonstrated that the fold activation by NLAI hits was similar to the endogenous LXR agonist 24S-OHC. Most importantly, F4 and M2 showed superior selectivity for ABCA1 elevation versus SREBP1c compared to benchmark LXR and RXR agonists.
An effective NLAI must induce cholesterol efflux from astrocytes, which was modelled in CCF and J774 cell lines. The NLAI activity of hits in cell lines was confirmed by PCR in primary human astrocytes and astrocytes from FAD-Tg mice carrying human APOE4. Upregulation of APOE and ABCG1 was also observed with T0, F4, and M2.
The in vitro data showing selectivity for induction of ABCA1, APOE, and ABCG1 versus induction of lipogenic genes is supportive of an improved in vivo safety profile compared to the LXR agonist T0. Since T0 is known to induce lipogenic genes and induce triglycerides in mice administered drug orally over 7 days, comparison was made with hits M2 and F4 in WT mice on normal mice chow. Liver and plasma triglycerides were significantly increased by T0, whereas F4 had no effect on triglyceride levels. Significant upregulation of peripheral ABCA1 and ABCG1 was induced by oral administration of F4, with less dramatic effects in brain tissues.
We observed that four NLAI hits acted as weak LXR ligands in LXR-CRT assays, and F4, in simile with the LXRβ-selective agonist GW, elicited a full response at LXRβ and a partial agonist response at LXRα. However, several NLAI hits showed no significant activation of LXRβ, demonstrating that NLAI activity and ABCA1-induced cholesterol efflux can occur without direct agonist activity at LXR. Potential mechanisms of ABC activation beyond direct NR agonism have recently been reviewed.53
LXR agonists directly inhibit proinflammatory gene expression and indirectly regulate macrophage function via ABC-mediated cholesterol mobilization, thus supporting the anti-inflammatory M2 phenotype.35 Phagocytosis of lipid-rich apoptotic cells by macrophages and microglia requires cholesterol to be mobilized and cleared by HDL particles. The anti-inflammatory effects of NLAIs were validated in two mouse models and may result from LXRβ agonism and ABCA1 upregulation. LXRβ silencing in microglia was reported to suppress phagocytic uptake of fibrillar Aβ42.54 ABCA1 upregulation and LXR agonism have been reported to facilitate phagocytosis by astrocytes and to promote Aβ phagocytosis and clearance.55,56
Mice fed HFD provide a useful preclinical model for obesity-linked T2D, which has translational value in drug discovery.33 Since C57BL/6 mice on HFD are highly susceptible to adiposity and liver inflammation, we used a 4-week drug treatment to compare the effects of F4 to those of T0. Due to cross talk between the CNS and periphery, HFD is expected to cause a pro-inflammatory response in mouse brain. We hypothesized and subsequently observed from measurement of cytokines, chemokines, and inflammatory mediators that this response was significantly attenuated by both F4 and T0 treatment. These effects may be due both to direct action in the brain and peripheral anti-inflammatory activity. T0 caused significant increases in lipogenic genes, triglycerides, and both body and liver weight in HFD-fed mice, whereas response to F4 was not significantly different from the vehicle and was beneficial with regards to HDL, SIRT1, and SR-B1, compatible with the ability of F4 to induce cholesterol mobilization and clearance via RCT.
ALDH2 KO mice lack mitochondrial Aldh2, an enzyme that detoxifies lipid peroxidation products leading to oxidative stress and a mild cognitive deficit from 4 months of age.37 In HFD-fed mice, lipid peroxidation products and protein adducts are elevated; furthermore, Aldh2 has been reported to protect mice from HFD-induced adverse effects.57 We observed that pro-inflammatory markers in the brain of ALDH2 KO mice fed HFD were attenuated by administration of F4.
In summary, a novel, phenotypic screening workflow, supported by secondary assays, was established to identify NLAI hits that would upregulate ABCA1 without inducing lipogenic genes in the liver. This strategy was validated by observations in four mouse models on one NLAI hit that functions, in part, as a weak LXR agonist. This strategy provides the basis for identification of NLAI hits for further optimization as therapeutics for T2D and/or AD.
Materials and Methods
Animals
In vivo studies were compliant with NIH guidelines for the use and care of laboratory animals. All mice were housed in a temperature-controlled room (22–24 °C) with a 12-h light, 12-h dark cycle and were allowed free access to food and water according to the University of Illinois at Chicago Animal Care Committee (ACC protocol #17-029). Male C57BL/6J mice were purchased from Jackson Laboratories and were aged 12–14 weeks at study onset. ALDH2 KO mice originally generated by gene targeting knockout by Kitagawa et al. were backcrossed with C57BL/6 mice for more than 10 generations and bred in-house. Male ALDH2 KO and WT littermates were studied at 10–12 weeks of age at study onset. In all studies, test compounds were administered by oral gavage (p.o.), formulated in 9% PEG-400 and 0.5% Tween-80. At the end of each treatment paradigm, mice were anesthetized, with blood collected from posterior vena cava. Following transcardial perfusion with ice-cold PBS, tissue samples were removed, flash-frozen in liquid nitrogen, and then stored at −80 °C until use (except for liver samples used for histology as detailed below). Plasma was separated from cellular blood components by centrifugation and kept at −80 °C until lipid analysis.
Animal Experiments
HFD Studies
Male C57BL/6 mice were fed with choline-deficient high-fat diet (HFD) (60% kcal from fat) formulated by Research Diets (Diet; A06071302). Food and water were provided ad libitum, with food intake and body weights determined twice weekly. After 4 weeks of HFD, animals were divided into three groups (n = 5) to receive 30 mg/kg T0, 30 mg/kg F4 test compound, or the vehicle control. After an additional 4 weeks of diet and drug coadministration, animals were sacrificed for histological and biochemical analysis. A second study was conducted with male ALDH2 KO mice provided HFD ad libitum, with 10 mg/kg F4 or vehicle treatment (n = 5) coadministered from day 1 of HFD and continuing for 4 weeks. A third control group of male C57BL/6 mice provided HFD over the same period was also used. Acute Inflammation study: Male ALDH2 KO and WT mice were administered 10 mg/kg F4, 10 mg/kg T0, or the vehicle for 3 days. On day 3, mice were also injected i.p. with 4 mg/kg LPS (Sigma-Aldrich) and then sacrificed 4 h later. Target Engagement study: Male C57BL/6 were treated daily for 1 week with test compounds (F4, M2, and T0) at 10 mg/kg and 30 mg/kg or the vehicle (n = 4). On the final day, mice were sacrificed 4 h after the last dose.
Cell Culture
Astrocytoma cell line CFF-STTG1 (CCF)
and hepatoma cell line HepG2 were purchased from ATCC. All cell lines
were maintained as detailed by the manufacturer. Briefly, HepG2 were
maintained in Dulbecco’s modified eagle’s medium (DMEM,
Gibco) low glucose supplemented with 10% fetal bovine serum (FBS,
Gibco) and 1% penicillin/streptomycin (P/S) (Gibco) at 37 °C
and 5% CO2. CCF-STTG1 were maintained in DMEM/F12 nutrient
mix (Gibco) supplemented with 10% FBS (Gibco) and 1% P/S and cultured
at 37 °C and 5% CO2. To generate CCF cells with stable expression
of inducible ABCA1-LXRE constructs (CCF-ABCA1), cells were infected
with lentiviral particles encoding pGreenFire1-LXRE-in-ABCA1 Lentivector
(SBI SystemBioscience). This vector encodes a luciferase gene linked
to the minimal promoter region of ABCA1 that contains four consensus
LXRE DR4 binding sites. Similarly, HepG2 were transduced with pGreenFire1-LXRE-in-SREBP1c
(HepG2-SREBP1c). Transduced cells were selected with 2 μg/mL
puromycin. Puromycin-resistant stable cells were further expanded
into several clones. The Z′ factor of each
clone was calculated as described by Zhang and colleagues with a value
> 0.5 indicating an excellent assay with high statistical reliability.58 The Z′ factor was determined,
in three independent experiments, from 48 replicate wells containing
either 1 μM T0 (positive control) or no ligand (DMSO solvent
negative control). The Z′ factor equation
is 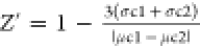 , where σc1 and σc2 are the
standard deviations of the positive and negative control wells in
the assay plate, and μc1 and μc2 are mean values for the
positive and negative control wells in the assay plate. Highly responding
cell clones with Z′ > 0.7 after T0 treatment
were retained for high-throughput screen (HTS), further expanded,
and frozen in liquid N2 to enable HTS with cells at an
identical passage number.
, where σc1 and σc2 are the
standard deviations of the positive and negative control wells in
the assay plate, and μc1 and μc2 are mean values for the
positive and negative control wells in the assay plate. Highly responding
cell clones with Z′ > 0.7 after T0 treatment
were retained for high-throughput screen (HTS), further expanded,
and frozen in liquid N2 to enable HTS with cells at an
identical passage number.
High-Throughput Screen
CCF-ABCA1 and HepG2-SREBP1c cells were plated in a 96-well format in culture media containing 10% charcoal-stripped FBS (Gibco) for 24 h to avoid interference from endogenous agonists. Subsequently, cells were treated at a 10 μM test compound for 24 h and then lysed with an addition of 20 μL of passive lysis buffer (Promega) followed by freezing at −80 °C for 10 min. Luciferase activities were determined as fold induction compared with activities of the vehicle control using the Luciferase Reporter Assay system (Promega) according to the manufacturer’s protocol as measured on the Synergy Neo2 Hybrid Multi-Mode plate reader (BioTek Synergy). Hit validation was performed by a concentration–response assay to determine EC50s, using newly purchased compounds (purity > 98%) from different vendors (ChemDiv, Enamine, etc.) or from in-house scale-up synthesis.
CRT Assays
LanthaScreen time-resolved fluorescence energy transfer (TR-FRET) LXRα-Coactivator Assay Kit (PV4655; ThermoFisher Scientific) and LXRβ-Coactivator Assay Kit (PV4658; ThermoFisher Scientific) were used to determine binding and agonist activity at LXRα and LXRβ, respectively.
Cholesterol Efflux
CCF-STTG1 cells were plated in 96-well plates at the desired cell density and grown for 24 h. Cells were loaded with 25 μg/mL fluorescent BODIPY-cholesterol (542/563) (Invitrogen), treated with test compounds, and incubated overnight. Cells were washed three times with PBS containing 2% fatty acid-free (FFA) BSA, equilibrated in 0.2% FFA-BSA for 30 min, and then imaged to measure cholesterol uptake. Media was switched serum free media (lipoprotein deficient) containing 0.2% FFA-BSA and test compounds. Cellular cholesterol efflux was induced by adding 25 μg/mL apoA-I (Calbiochem). Cell efflux was monitored for 6 h using a Celigo Imaging Cytometer (Nexcelom Bioscience). The percentage of cholesterol efflux was calculated as percent of fluorescence intensity at 6 h relative to fluorescence intensity measured at time zero.
Gene Expression Analysis
Total RNA was extracted from animal tissues using TRIZOL reagent (Invitrogen) and from cell pellets using the RNeasy Plus kit (Qiagen) per the manufacturer’s instructions. Total RNA samples (2 μg) were reverse-transcribed to cDNA with Superscript III (Invitrogen) per the manufacturer’s instructions. Expression profiles were then obtained using a real-time quantitative polymerase chain reaction (qPCR) performed on an ABI Prism 7300 Real Time PCR System (Applied Biosystems) with Quantity One software. Gene expression levels were determined using the ΔΔCt method normalized to reference genes Actb and Hprt.
Statistical Analysis
The GraphPad Prism 8 software package (GraphPad Software, USA) was used to perform statistical analysis. All data were presented as the mean ± SD unless otherwise noted. One-way analysis of variance (ANOVA) with appropriate posthoc tests (3+ groups) and Student’s t test (2 groups) were used to calculate statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001.
Glossary
Abbreviations
- ABC
ATP-binding cassette transporter
- AD
Alzheimer’s disease
- ADRD
Alzheimer’s disease and related dementia
- ALDH2
aldehyde dehydrogenase 2
- Aβ
amyloid-β
- BBB
blood-brain barrier
- CNS
central nervous system
- CRT
Coregulator Recruitment TR-FRET
- CSF
cerebrospinal fluid
- CVD
cardiovascular and cerebrovascular disease
- GWAS
genome-wide association study
- HDL
high density lipoprotein
- HFD
high-fat diet
- LAM
lipid droplet-accumulating microglia
- LDL
low density lipoprotein
- LRE
luciferase response element
- LXR
liver X receptors
- MCI
mild cognitive impairment
- NR
nuclear receptors
- PAINS
pan-assay interference compounds
- PGC1
PPARG coactivator
- PPAR
peroxisome-proliferator-activated receptors
- RCT
reverse cholesterol transport
- RXR
retinoid X receptor
- T2D
Type 2 diabetes
- WT
wild type
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00149.
Details of experimental methodology; Table 1, genes and primers; figures referenced within this manuscript; and Figure S8, timelines for all animal experiments (PDF)
Author Contributions
M.B.A. designed, directed, conducted, analyzed, and interpreted data from in vitro and in vivo studies and contributed to manuscript preparation. C.T.L. performed chemistry scale up for F4 compound for in vivo studies and assisted with in vitro studies. K.R. assisted with HTS screening and data analysis. S.H.L. assisted with in vivo studies. B.T.L. and M.J.L. provided project oversight. G.R.J.T. conceived and coordinated the overall project and manuscript preparation.
The funding sources were as follows: NIH R21AG044682 (G.R.J.T., M.J.L.), Chicago Biomedical Consortium HTS Award HTS-025 (M.B., M.J.L., G.R.J.T.), NIH UL1TR002003 (UICentre), NIH T32AG57468 (C.T.L./S.H.L.), and American Heart Association 20PRE35150022 (C.T.L.). B.T.L. is supported by NIH R01DK104927 and Department of Veterans Affairs, VHA, Office of Research and Development, VA merit (Grant no. 1I01BX003382).
The authors declare the following competing financial interest(s): G.R.J.T. is an inventor on patents owned by the University of Illinois.
Supplementary Material
References
- Vagelatos N. T.; Eslick G. D. (2013) Type 2 diabetes as a risk factor for Alzheimer’s disease: the confounders, interactions, and neuropathology associated with this relationship. Epidemiol Rev. 35, 152–160. 10.1093/epirev/mxs012. [DOI] [PubMed] [Google Scholar]
- Chornenkyy Y.; Wang W.-X.; Wei A.; Nelson P. T. (2019) Alzheimer’s disease and type 2 diabetes mellitus are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 29, 3–17. 10.1111/bpa.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman A.; Pike C. J. (2014) Alzheimer’s disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr. Diabetes Rep. 14, 476. 10.1007/s11892-014-0476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie F.; Fitzpatrick A. L.; Lopez O. L.; Kuller L. H.; Peila R.; Newman A. B.; Launer L. J. (2008) Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch. Neurol. 65, 89–93. 10.1001/archneurol.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary R.; Likidlilid A.; Peerapatdit T.; Tresukosol D.; Srisuma S.; Ratanamaneechat S.; Sriratanasathavorn C. (2012) Apolipoprotein E gene polymorphism: effects on plasma lipids and risk of type 2 diabetes and coronary artery disease. Cardiovasc. Diabetol. 11, 36. 10.1186/1475-2840-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filou S.; Lhomme M.; Karavia E. A.; Kalogeropoulou C.; Theodoropoulos V.; Zvintzou E.; Sakellaropoulos G. C.; Petropoulou P. I.; Constantinou C.; Kontush A.; Kypreos K. E. (2016) Distinct Roles of Apolipoproteins A1 and E in the Modulation of High-Density Lipoprotein Composition and Function. Biochemistry 55, 3752–3762. 10.1021/acs.biochem.6b00389. [DOI] [PubMed] [Google Scholar]
- Wang S.; Smith J. D. (2014) ABCA1 and nascent HDL biogenesis. Biofactors 40, 547–554. 10.1002/biof.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasinska J. M.; Rinninger F.; Lutjohann D.; Ruddle P.; Franciosi S.; Kruit J. K.; Singaraja R. R.; Hirsch-Reinshagen V.; Fan J.; Brunham L. R.; Bissada N.; Ramakrishnan R.; Wellington C. L.; Parks J. S.; Hayden M. R. (2009) Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J. Neurosci. 29, 3579–3589. 10.1523/JNEUROSCI.4741-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz N. F.; Cronican A. A.; Saleem M.; Fauq A. H.; Chapman R.; Lefterov I.; Koldamova R. (2012) Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J. Neurosci. 32, 13125–13136. 10.1523/JNEUROSCI.1937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eck M.; Bos I. S.; Kaminski W. E.; Orso E.; Rothe G.; Twisk J.; Bottcher A.; Van Amersfoort E. S.; Christiansen-Weber T. A.; Fung-Leung W. P.; Van Berkel T. J.; Schmitz G. (2002) Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc. Natl. Acad. Sci. U. S. A. 99, 6298–6303. 10.1073/pnas.092327399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseki M.; Matsuyama A.; Nakatani K.; Inagaki M.; Nakaoka H.; Kawase R.; Yuasa-Kawase M.; Tsubakio-Yamamoto K.; Masuda D.; Sandoval J. C.; Ohama T.; Nakagawa-Toyama Y.; Matsuura F.; Nishida M.; Ishigami M.; Hirano K.; Sakane N.; Kumon Y.; Suehiro T.; Nakamura T.; Shimomura I.; Yamashita S. (2009) Impaired insulin secretion in four Tangier disease patients with ABCA1 mutations. J. Atheroscler. Thromb. 16, 292–296. 10.5551/jat.E599. [DOI] [PubMed] [Google Scholar]
- Oram J. F. (2002) Molecular basis of cholesterol homeostasis: lessons from Tangier disease and ABCA1. Trends Mol. Med. 8, 168–173. 10.1016/S1471-4914(02)02289-X. [DOI] [PubMed] [Google Scholar]
- Frikke-Schmidt R.; Nordestgaard B. G.; Stene M. C.; Sethi A. A.; Remaley A. T.; Schnohr P.; Grande P.; Tybjaerg-Hansen A. (2008) Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. Jama 299, 2524–2532. 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- Lupton M. K.; Proitsi P.; Lin K.; Hamilton G.; Daniilidou M.; Tsolaki M.; Powell J. F. (2013) The role of ABCA1 gene sequence variants on risk of Alzheimer’s disease. J. Alzheimer's Dis. 38, 897–906. 10.3233/JAD-131121. [DOI] [PubMed] [Google Scholar]
- Nordestgaard L. T.; Tybjaerg-Hansen A.; Nordestgaard B. G.; Frikke-Schmidt R. (2015) Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimer's Dementia 11, 1430–1438. 10.1016/j.jalz.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Tang C.; Kanter J. E.; Bornfeldt K. E.; Leboeuf R. C.; Oram J. F. (2010) Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J. Lipid Res. 51, 1719–1728. 10.1194/jlr.M003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D. C.; Albrecht C.; Pavitt D.; Paul V.; Pourreyron C.; Newman S. P.; Godsland I. F.; Valabhji J.; Johnston D. G. (2011) Type 2 diabetes is associated with reduced ATP-binding cassette transporter A1 gene expression, protein and function. PLoS One 6, e22142 10.1371/journal.pone.0022142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata M.; Tsujita M.; Hossain M. A.; Akita N.; Gonzalez F. J.; Staels B.; Suzuki S.; Fukutomi T.; Kimura G.; Yokoyama S. (2009) On the mechanism for PPAR agonists to enhance ABCA1 gene expression. Atherosclerosis 205, 413–419. 10.1016/j.atherosclerosis.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanarasimha M.; Davis A. M.; Soman F. L.; Rider S. D. Jr.; Hostetler H. A. (2014) Ligand-regulated heterodimerization of peroxisome proliferator-activated receptor alpha with liver X receptor alpha. Biochemistry 53, 2632–2643. 10.1021/bi401679y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai L. M.; Koster K. P.; Luo J.; Lee S. H.; Wang Y. T.; Collins N. C.; Ben Aissa M.; Thatcher G. R.; LaDu M. J. (2014) Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 289, 30538–30555. 10.1074/jbc.M114.600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G.; Liang Y.; Broderick C. L.; Oldham B. A.; Beyer T. P.; Schmidt R. J.; Zhang Y.; Stayrook K. R.; Suen C.; Otto K. A.; Miller A. R.; Dai J.; Foxworthy P.; Gao H.; Ryan T. P.; Jiang X. C.; Burris T. P.; Eacho P. I.; Etgen G. J. (2003) Antidiabetic action of a liver x receptor agonist mediated by inhibition of hepatic gluconeogenesis. J. Biol. Chem. 278, 1131–1136. 10.1074/jbc.M210208200. [DOI] [PubMed] [Google Scholar]
- Riddell D. R.; Zhou H.; Comery T. A.; Kouranova E.; Lo C. F.; Warwick H. K.; Ring R. H.; Kirksey Y.; Aschmies S.; Xu J.; Kubek K.; Hirst W. D.; Gonzales C.; Chen Y.; Murphy E.; Leonard S.; Vasylyev D.; Oganesian A.; Martone R. L.; Pangalos M. N.; Reinhart P. H.; Jacobsen J. S. (2007) The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell. Neurosci. 34, 621–628. 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Quinet E. M.; Savio D. A.; Halpern A. R.; Chen L.; Schuster G. U.; Gustafsson J. A.; Basso M. D.; Nambi P. (2006) Liver X receptor (LXR)-beta regulation in LXRalpha-deficient mice: implications for therapeutic targeting. Mol. Pharmacol. 70, 1340–1349. 10.1124/mol.106.022608. [DOI] [PubMed] [Google Scholar]
- Katz A.; Udata C.; Ott E.; Hickey L.; Burczynski M. E.; Burghart P.; Vesterqvist O.; Meng X. (2009) Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J. Clin. Pharmacol. 49, 643–649. 10.1177/0091270009335768. [DOI] [PubMed] [Google Scholar]
- Stachel S. J.; Zerbinatti C.; Rudd M. T.; Cosden M.; Suon S.; Nanda K. K.; Wessner K.; DiMuzio J.; Maxwell J.; Wu Z.; Uslaner J. M.; Michener M. S.; Szczerba P.; Brnardic E.; Rada V.; Kim Y.; Meissner R.; Wuelfing P.; Yuan Y.; Ballard J.; Holahan M.; Klein D. J.; Lu J.; Fradera X.; Parthasarathy G.; Uebele V. N.; Chen Z.; Li Y.; Li J.; Cooke A. J.; Bennett D. J.; Bilodeau M. T.; Renger J. (2016) Identification and in Vivo Evaluation of Liver X Receptor beta-Selective Agonists for the Potential Treatment of Alzheimer’s Disease. J. Med. Chem. 59, 3489–3498. 10.1021/acs.jmedchem.6b00176. [DOI] [PubMed] [Google Scholar]
- Eberle D.; Hegarty B.; Bossard P.; Ferre P.; Foufelle F. (2004) SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848. 10.1016/j.biochi.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Dahlin J. L.; Nissink J. W.; Strasser J. M.; Francis S.; Higgins L.; Zhou H.; Zhang Z.; Walters M. A. (2015) PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. 58, 2091–2113. 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erve J. C. (2006) Chemical toxicology: reactive intermediates and their role in pharmacology and toxicology. Expert Opin. Drug Metab. Toxicol. 2, 923–946. 10.1517/17425255.2.6.923. [DOI] [PubMed] [Google Scholar]
- Barlic J.; Zhu W.; Murphy P. M. (2009) Atherogenic lipids induce high-density lipoprotein uptake and cholesterol efflux in human macrophages by up-regulating transmembrane chemokine CXCL16 without engaging CXCL16-dependent cell adhesion. J. Immunol. 182, 7928–7936. 10.4049/jimmunol.0804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan C.; Liu M.; Xu H.; Tang W.; Liu J.; Hou L.; Li L. (2016) Decreased expression of GLUT4 in male CG-IUGR rats may play a vital role in their increased susceptibility to diabetes mellitus in adulthood. Acta Biochim. Biophys. Sin. 48, 872–882. 10.1093/abbs/gmw088. [DOI] [PubMed] [Google Scholar]
- Youmans K. L.; Tai L. M.; Nwabuisi-Heath E.; Jungbauer L.; Kanekiyo T.; Gan M.; Kim J.; Eimer W. A.; Estus S.; Rebeck G. W.; Weeber E. J.; Bu G.; Yu C.; Ladu M. J. (2012) APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem. 287, 41774–41786. 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner R.; Scholmerich J.; Bollheimer L. C. (2007) High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity 15, 798–808. 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- Heydemann A. (2016) An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2902351. 10.1155/2016/2902351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L.; Wang Q.; Yu Y.; Zhao F.; Huang P.; Zeng R.; Qi R. Z.; Li W.; Liu Y. (2009) Leptin contributes to the adaptive responses of mice to high-fat diet intake through suppressing the lipogenic pathway. PLoS One 4, e6884 10.1371/journal.pone.0006884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A.; Hong C.; Rong X.; Zhu X.; Tarling E. J.; Hedde P. N.; Gratton E.; Parks J.; Tontonoz P. (2015) LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 4, e08009 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasinska J. M.; de Haan W.; Franciosi S.; Ruddle P.; Fan J.; Kruit J. K.; Stukas S.; Lutjohann D.; Gutmann D. H.; Wellington C. L.; Hayden M. R. (2013) ABCA1 influences neuroinflammation and neuronal death. Neurobiol. Dis. 54, 445–455. 10.1016/j.nbd.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Knopp R. C.; Lee S. H.; Hollas M.; Nepomuceno E.; Gonzalez D.; Tam K.; Aamir D.; Wang Y.; Pierce E.; BenAissa M.; Thatcher G. R. J. (2020) Interaction of oxidative stress and neurotrauma in ALDH2(−/−) mice causes significant and persistent behavioral and pro-inflammatory effects in a tractable model of mild traumatic brain injury. Redox Biol. 32, 101486. 10.1016/j.redox.2020.101486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokuni S.; Yamada S.; Kashima M.; Ihara Y.; Yamada Y.; Tanaka T.; Hiai H.; Seino Y.; Uchida K. (2000) Serum 4-hydroxy-2-nonenal-modified albumin is elevated in patients with type 2 diabetes mellitus. Antioxid. Redox Signaling 2, 681–685. 10.1089/ars.2000.2.4-681. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A.; Reed T.; Perluigi M.; De Marco C.; Coccia R.; Cini C.; Sultana R. (2006) Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 397, 170–173. 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Joseph S. B.; Castrillo A.; Laffitte B. A.; Mangelsdorf D. J.; Tontonoz P. (2003) Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 9, 213–219. 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- Knight E. M.; Martins I. V.; Gumusgoz S.; Allan S. M.; Lawrence C. B. (2014) High-fat diet-induced memory impairment in triple-transgenic Alzheimer's disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol. Aging 35, 1821–1832. 10.1016/j.neurobiolaging.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W. S.; Rahmanto A. S.; Kamili A.; Rye K. A.; Guillemin G. J.; Gelissen I. C.; Jessup W.; Hill A. F.; Garner B. (2007) Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 282, 2851–2861. 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- Wahrle S. E.; Jiang H.; Parsadanian M.; Legleiter J.; Han X.; Fryer J. D.; Kowalewski T.; Holtzman D. M. (2004) ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993. 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Rawat V.; Wang S.; Sima J.; Bar R.; Liraz O.; Gundimeda U.; Parekh T.; Chan J.; Johansson J. O.; Tang C.; Chui H. C.; Harrington M. G.; Michaelson D. M.; Yassine H. N. (2019) ApoE4 Alters ABCA1Membrane Trafficking in Astrocytes. J. Neurosci. 39, 9611–9622. 10.1523/JNEUROSCI.1400-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle S. E.; Jiang H.; Parsadanian M.; Hartman R. E.; Bales K. R.; Paul S. M.; Holtzman D. M. (2005) Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242. 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- Koldamova R.; Staufenbiel M.; Lefterov I. (2005) Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J. Biol. Chem. 280, 43224–43235. 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- Key C. C.; Liu M.; Kurtz C. L.; Chung S.; Boudyguina E.; Dinh T. A.; Bashore A.; Phelan P. E.; Freedman B. I.; Osborne T. F.; Zhu X.; Ma L.; Sethupathy P.; Biddinger S. B.; Parks J. S. (2017) Hepatocyte ABCA1 Deletion Impairs Liver Insulin Signaling and Lipogenesis. Cell Rep. 19, 2116–2129. 10.1016/j.celrep.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkin J. J.; Stukas S.; Hirsch-Reinshagen V.; Namjoshi D.; Wilkinson A.; May S.; Chan J.; Fan J.; Collins J.; Wellington C. L. (2010) ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J. Biol. Chem. 285, 34144–34154. 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz N. F.; Cronican A.; Pham T.; Fogg A.; Fauq A. H.; Chapman R.; Lefterov I.; Koldamova R. (2010) Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J. Neurosci. 30, 6862–6872. 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmierlo T.; Rutten K.; Dederen J.; Bloks V. W.; van Vark-van der Zee L. C.; Kuipers F.; Kiliaan A.; Blokland A.; Sijbrands E. J.; Steinbusch H.; Prickaerts J.; Lutjohann D.; Mulder M. (2011) Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol. Aging 32, 1262–1272. 10.1016/j.neurobiolaging.2009.07.005. [DOI] [PubMed] [Google Scholar]
- El-Gendy B. E. M.; Goher S. S.; Hegazy L. S.; Arief M. M. H.; Burris T. P. (2018) Recent Advances in the Medicinal Chemistry of Liver X Receptors. J. Med. Chem. 61, 10935–10956. 10.1021/acs.jmedchem.8b00045. [DOI] [PubMed] [Google Scholar]
- Seneviratne U.; Huang Z.; Am Ende C. W.; Butler T. W.; Cleary L.; Dresselhaus E.; Evrard E.; Fisher E. L.; Green M. E.; Helal C. J.; Humphrey J. M.; Lanyon L. F.; Marconi M.; Mukherjee P.; Sciabola S.; Steppan C. M.; Sylvain E. K.; Tuttle J. B.; Verhoest P. R.; Wager T. T.; Xie L.; Ramaswamy G.; Johnson D. S.; Pettersson M. (2020) Photoaffinity Labeling and Quantitative Chemical Proteomics Identify LXRbeta as the Functional Target of Enhancers of Astrocytic apoE. Cell Chem Biol 10.1016/j.chembiol.2020.09.002. [DOI] [PubMed] [Google Scholar]
- Frambach S.; de Haas R.; Smeitink J. A. M.; Rongen G. A.; Russel F. G. M.; Schirris T. J. J. (2020) Brothers in Arms: ABCA1- and ABCG1-Mediated Cholesterol Efflux as Promising Targets in Cardiovascular Disease Treatment. Pharmacol. Rev. 72, 152–190. 10.1124/pr.119.017897. [DOI] [PubMed] [Google Scholar]
- Gao T.; Jernigan J.; Raza S. A.; Dammer E. B.; Xiao H.; Seyfried N. T.; Levey A. I.; Rangaraju S. (2019) Transcriptional regulation of homeostatic and disease-associated-microglial genes by IRF1, LXRbeta, and CEBPalpha. Glia 67, 1958–1975. 10.1002/glia.23678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H.; Spinrad A.; Weiner A.; Matcovitch-Natan O.; Dvir-Szternfeld R.; Ulland T. K.; David E.; Baruch K.; Lara-Astaiso D.; Toth B.; Itzkovitz S.; Colonna M.; Schwartz M.; Amit I. (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- Morizawa Y. M.; Hirayama Y.; Ohno N.; Shibata S.; Shigetomi E.; Sui Y.; Nabekura J.; Sato K.; Okajima F.; Takebayashi H.; Okano H.; Koizumi S. (2017) Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 8, 28. 10.1038/s41467-017-00037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S.; Li L.; Zhang Y. L.; Zhang L.; Lu J.; Guo S.; Liang N.; Ge J.; Zhu M.; Tao Y.; Wu Y. C.; Yin H. (2019) Acetaldehyde dehydrogenase 2 interactions with LDLR and AMPK regulate foam cell formation. J. Clin. Invest. 129, 252–267. 10.1172/JCI122064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. (1999) A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screening 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.