

Abstract
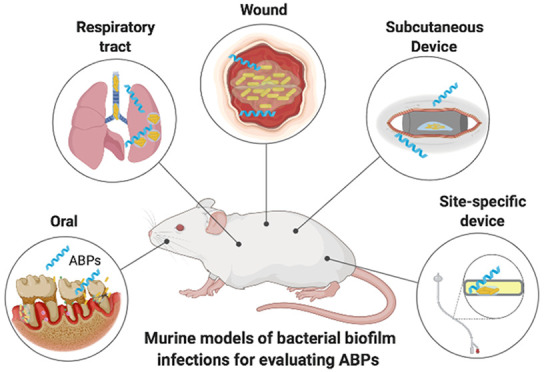
Biofilm-forming bacteria may be 10–1000 times more resistant to antibiotics than planktonic bacteria and represent about 75% of bacterial infections in humans. Antibiofilm treatments are scarce, and no effective therapies have been reported so far. In this context, antibiofilm peptides (ABPs) represent an exciting class of agents with potent activity against biofilms both in vitro and in vivo. Moreover, murine models of bacterial biofilm infections have been used to evaluate the in vivo effectiveness of ABPs. Therefore, here we highlight the translational potential of ABPs and provide an overview of the different clinically relevant murine models to assess ABP efficacy, including wound, foreign body, chronic lung, and oral models of infection. We discuss key challenges to translate ABPs to the clinic and the pros and cons of the existing murine biofilm models for reliable assessment of the efficacy of ABPs.
Keywords: biofilm infections, antibiofilm peptides, murine models
Introduction
Biofilms are aggregates of microorganisms in which cells are frequently embedded in a self-produced matrix of extracellular polymeric substances (EPS) that adhere to each other and/or a surface.1 Biofilm formation leads to increased resistance of the aggregate inhabitants to abrupt environmental changes, allowing them to live under hostile conditions.2 Consequently, biofilm-forming bacteria may be 10–1000 times more resistant to antibiotics than planktonic bacteria and represent about 75% of bacterial infections in humans.3 This resistance may be boosted by several genetic and phenotypic factors, including administration of antibiotics at sublethal doses, horizontal resistance gene transfer, and EPS production.4,5
Although many studies have focused on biofilms, there are still no antibiotics exclusively developed for this type of infection.6 Antibiotics routinely used for the treatment of planktonic bacterial infections are also applied against bacterial biofilms, often leading to antibiofilm therapy failure, as biofilms may be more tolerant to traditional antibiotic therapy.7 Moreover, biofilm cells can become recalcitrant, which hinders their eradication and treatment effectiveness.7 Therefore, new approaches for biofilm treatment, including organic acids, bacteriophages, and photoinactivation, have been extensively studied.8 Moreover, antimicrobial peptides (AMPs) are promising alternatives for the treatment of biofilm infections.9−11
Here, we will use the term antibiofilm peptides (ABPs) for all AMPs that are active against bacterial biofilms.12 ABPs of either natural or synthetic origin present amphipathic and cationic characteristics and sequences of less than 50 amino acid residues. Moreover, apart from preventing biofilm formation, ABPs have been shown to eradicate preformed biofilms.12,13 It is also worth noting that some ABPs’ activity is orthogonal versus biofilms, meaning that they are not active against bacteria in their planktonic mode of growth. The mechanisms of action of ABPs have been divided into different groups.14 The most well-known mechanisms of action include triggering changes in the bacterial membrane potential and membrane rupture.15 ABPs can also block cell signaling and communication by interfering with biofilm-related gene expression.16 The degradation of the biofilm polysaccharide matrix has also been reported as one of the mechanisms by which ABPs act on biofilms.17 Moreover, ABPs have been shown to interfere with the stress response.18
Diverse systems have been adopted to evaluate the antibiofilm activities of different compounds in vitro, as extensively reviewed by Coenye and Nelis (2010).19 Although numerous studies have evaluated ABPs in vitro (e.g., microtiter plate-based and flow displacement biofilm), only a few have reported in vivo strategies for studying biofilms. Given the relevance of bacterial biofilm infections worldwide, here we highlight the translational potential of ABPs and provide an overview of the different clinically relevant mouse models available to assess efficacy. This review is divided by mouse models for accessing ABPs antibiofilm activity, thus favoring the comparison of lead peptide candidates for each model and pinpointing methodological differences for a given animal model and how it can interfere with ABP activities. Therefore, for all subtopics, we provide a brief description of each model for evaluating ABPs followed by a detailed description of the positive and negative outcomes obtained for this class of antibiofilm agents.
Wound Models
Skin wound models are among the most commonly used strategies to evaluate antibiofilm compounds (Table 1). There are two approaches used for the formation of biofilms in murine skin wounds.20 The first strategy involves causing skin damage (e.g., scarification/abrasions, pressure-induced ischemic, surgical excision, or burns) and, subsequently, infecting the injured region with biofilm-forming bacteria.21 The second strategy inoculates high-density biofilm-forming bacteria subcutaneously, leading to the formation of abscesses and wounds.22 The main clinically relevant bacterial strains used individually or in consortium in both approaches are Staphylococcus aureus, Staphylococcus epidermidis, and Pseudomonas aeruginosa.23,24
Table 1. Studies Using In Vivo Bacterial Biofilm Infection Models to Evaluate ABPs.
| murine host | bacteria | bacterial load | bacterial route | peptide | treatment route | dose effect | analysis | time exp. | results | ref | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wound | skin scarification an abrasion | female CD-1 IGS mice | P. aeruginosa (PA14) | ∼107 CFU in 50 μL | inoculated with a pipet tip | [Lys]7-Pol-CP-NH2 | topic injection | 64 μmol L–1 (single dose 1 day postinfection) | bacterial load and body weight | 2/4 days | skin infections were sterilized 4 days after infection | (28) |
| female CD-1 mice | P. aeruginosa (PAO1) | 5.5 × 107 CFU in 20 μL | inoculated with a pipet tip | (P)PAP-A3 | topic injection | 50 μM in 20 μL (single dose 1 day postinfection) | bacterial load | 4 days | 4-log CFU reduction | (30) | ||
| female CD-1 mice | P. aeruginosa (PA14) | 5 × 107 CFU in 20 μL | inoculated with a pipet tip | EcDBS1R5 | topic injection | 64 μM (single dose 1 day postinfection) | bacterial load | 2 days | 2-log CFU reduction | (31) | ||
| female CD-1 mice | P. aeruginosa (PA14) | 5 × 106 CFU in 20 μL | inoculated with a pipet tip | PaDBS1R6F10 | topic injection | 64 μM (single dose 1 day postinfection) | bacterial load | 2/4 days | 3-log CFU reduction 4 days postinfection | (32) | ||
| female BALB/c | MRSA (bioluminescent Xen31) | 108 CFU mL–1 in 40 μL | inoculated with a pipet tip | RP557 | topic | 0.2% in 40 μL (single dose 4 h postinfection) | bioluminescence of viable cells and body weight | 7 days | 2-log CFU reduction 7 days postinfection; decreased body weight loss | (27) | ||
| female CD-1 IGS mice | P. aeruginosa (PA14) | 5 × 107 CFU in 50 μL | inoculated with a pipet tip | mastoparan-R1 and R4 | topic injection | 64 μM in 20 μL (single dose 1 day postinfection) | bacterial load | 2/4 days | 2-log CFU reduction at day 2 postinfection | (29) | ||
| pressure ulcer | male SPF Wistar rats | P. aeruginosa (PAO1) | 1.6–2.4 × 105 CFU mL–1 in 0.1 mL | intracutaneous and intramuscular injection and topical | QS autoinducer quantification (3OC12-HSL) | 3/7 days | correlation between autoinducer concentration and bacterial counts was observed | (21) | ||||
| surgical | male C57Bl6/J mice | S. aureus (CFS101) S. epidermidis (CFS201) | preformed biofilm | applied wound | RIP | topic injection | 1 mg mL–1 in 0.1 mL (single dose 3 days postinfection) | bacterial load, histology, and Gram’s stain | 9 days | sterilized skin infections post 7 days and delayed wound healing effects post 9 days | (33) | |
| female BALB/c mice | S. aureus (ATCC 25923) P. aeruginosa (ATCC 9027) | 1000:1 and grown on a polycarbonate filter | preformed biofilm applied to the wound | DRGN1 | topic | 20 μg in 20 μL (every 2 days until the 6th day) | bacterial load, wound healing and histological | 6 days | promoted the migration of keratinocytes, reduced bacterial load, and improved healing | (23) | ||
| iron burn | male ICR mice | MRSA (ATCC 43300) | 4 × 108 CFU mL–1 in 50 μL | inoculated into burn | WRL3 | topic | 4 μg mL–1 in 50 μL (twice a day for 14 days) | bacterial load, histological, cytokines dosage, chemotaxis | 14 days | MRSA proliferation control, bacterial load reduction and healing induction | (26) | |
| cutaneous abscess | female CD-1 mice | S. aureus (HG001 bioluminescent) | 5 × 107 CFU in 50 μL | injected right flank of the back | DJK-5 | i.p intra-abscess | 6 mg kg–1 in 50 μL (single dose before infection)3 | bacterial load, imaging system and abscess area measure | 5 days | 10.2-fold CFU reduction after 5 days and reduced dermonecrosis | (34) | |
| 3 mg kg–1 in 50 μL (single 1 h postinfection) | bacterial load, abscess area measure, histological and weight loss/gain | 3 days | lesion size 4.6-fold smaller, 8.4-fold fewer bacteria, less tissue damage and weight gain | |||||||||
| female CD-1 mice | P. aeruginosa (LESB58) | 5 × 107 CFU in 50 μL | injected right flank of the back | DJK-5 | i.p intra-abscess | 4 mg kg–1 in 50 μL (single dose before infection) | bacterial load and abscess area measure | 3 days | 2.2-fold smaller abscess lesions | (34) | ||
| 4 mg kg–1 in 50 μL (single dose 2 h postinfection) | 3 days | 2.2-fold smaller abscess lesions and 2.7-fold smaller lesions | ||||||||||
| female outbred CD-1 | P. aeruginosa (LESB58) | 5 × 107 CFU mL–1 in 50 μL | injected right flank of the back | DJK-5 with hyaluronic acid–based nanogels | intra-abscess | 3–6 mg mL–1 in 50 μL (single dose 1 h postinfection) | bacterial load and abscess lesion measure | 3 days | bacterial load reduction | (35) | ||
| female outbred CD-1 | S. aureus (LAC) | 5 × 107 CFU mL–1 in 50 μL | injected right flank of the back | peptide 73 | intra-abscess | 5 mg kg–1 in 50 μL (single dose 1 h postinfection) | bacterial load and abscess lesion measure | 3 days | 8.9-fold bacterial load reduction and 80% abscess reduction | (36) | ||
| peptide 73-C micelles | ||||||||||||
| female outbred CD-1 | P. aeruginosa (LESB58) | 5 × 107–2 × 1010 CFU mL–1 in 100 μL | injected right flank of the back | DJK5 | intra-abscess | (3, 0,25 and 10 mg kg–1 in 100 μL (single dose 1 h postinfection) | bacterial load and abscess lesion measure | 3 days | bacterial load reduction and abscess sizes; improved activities in synergism with antibiotics | (37) | ||
| A. baumannii (Ab5075) | HHC-10 | |||||||||||
| E. faecium | IDR-1002 | |||||||||||
| K. pneumoniae (KPLN649) | IDR-1018 | |||||||||||
| E. coli (E38) | ||||||||||||
| S. aureus (LAC) | ||||||||||||
| foreign body site specific devices | urinary tract stents | female Wistar rat | S. aureus Smith diffuse (SD) | 2 × 107 CFU mL–1 | injected into the bladder | RIP | stents impregnated with RIP | 1 μg mL –1 30 min immediately before implantation | bacterial load of stent and urine | 5 days | 2-log CFU reduction in stent and urine | (43) |
| central venous catheters | male Wistar rat | S. aureus (SD) | 1 × 106 CFU in 0.1 mL | injected into CVC after 30 min treatment | citropin 1.1 | via CVC | before infection, single dose 10 μg mL–1 in 0.1 mL | bacterial load of peripheral blood and catheters/venous tissues | 24 and 9 days | Reduced CFU counts in peripheral blood cultures and CVC and synergy with antibiotics bacteremia was eliminated | (42) | |
| BMAP-28 | 4-log CFU mL–1 reduction and bacteremia in monotherapy and synergy with antibiotics decreased to 6-log and bacteremia was not detected | (46) | ||||||||||
| E. faecalis S.aureus (SD) | IB-367 | bacterial load in biofilms was decreased; bacteremia was not detected during synergistic treatment with antibiotics | (47) | |||||||||
| periprosthetic joint | female B57BL/6 J mice Jackson | S. aureus (SH1000) | 10 μL with 1 × 106 CFU | injected joint space | WLBU2 | i.p. | 0.01 mg kg–1 and 10 mg kg–1 24 h after and twice a day for 3 days | biofilm mass of implant and femur homogenate | 3 days | decreased biofilm mass of implant and tissue | (49) | |
| foreign body subcutaneous devices | urethral stent | male Wistar rat | P. aeruginosa (ATCC 27853) | (1 mL) containing 2 × 107 CFU mL–1 | injected graft surface | tachyplesin III | ureteral stent | 10 mg L–1 Coated implanted | bacterial load of stent | 5 days | 3-log CFU reduction in monotherapy and 5-log reduction in synergy with antibiotics | (50) |
| catheter needle | BALB/c nude mouse | P. aeruginosa (3241) | 5 × 108 CFU mL–1 | preformed biofilm on a catheter needle 24 h | HPA3NT3-A2 | catheter | 50 μg mL–1 added to the biofilm formed into catheter | histological analyses skin, spleen, and kidney | 7 days | less epidermis, dermis, or hypodermis damage at the implantation site | (54) | |
| BALB/c mice | P. aeruginosa (MDR) | 1 × 108 CFU mL–1 | preformed biofilm on a catheter 24 h | EC1-17KV | at the implant site | 10 mg kg–1 once daily for three continuous days | bacterial load and pathological examination | 3 days | 3.5- and 2.7-log unit CFU reductions in the subcutaneous tissue and catheter surface, respectively, and protective effect against the inflammatory injury | (53) | ||
| female C57BL/6 mice | S. aureus (USA300 LAC) | 20 μL with 103 CFU/catheter | injected into the lumen of the catheter | 17tF-W | catheter lumen | 250 μg 2, 24, and 48 h postinfection, 50 μL per site | bacterial load of tissue and catheter/chemokine levels | 3 days | eliminated the burden in both mouse-embedded catheters and their surrounding tissues, suppressed the level of chemokine TNFα, and boosted chemokines MCP-1, IL-17A and IL-10 | (55) | ||
| titanium coating | female SPF BALB/c mice and Sprague–Dawley rat | S. aureus | 100 μL of 107 and 105 CFU per site | injected into site biomaterial bed | cys-melimine | titanium coating | built into the titanium disc inserted in the back of the animals | bacterial load | 5 and 7 days | bacterial load reduction in both models | (51) | |
| Dacron grafts | male Wistar rat | MRSA and MRSE | 2 × 107 CFU mL–1 | injected into the graft surface | DD13-RIP | peptide binding to Dacron | 50 μg mL–1 for 0.5 and 5 h | bacterial load | 7 days | bacterial load reduction of staphylococcal associated with graft at the lowest dose | (56) | |
| silicone implants | male SPF BALB/c | P. aeruginosa (PAO1) | initial concentration of OD600 = 0.1 in LB medium | preformed biofilm into 4 mm silicone tubes for 12h | melittin | injected at the incision site | 50 mg kg–1 after 1 day of incision | bacterial load, tissue damage and biofilm colonization | 3 days | bacterial load reduction and prevented tissue damage and inflammation in synergy with antibiotics | (52) | |
| silicone sheets | female BALB/c | P. aeruginosa (ATCC 15442) | 1 mL of 105 CFU mL–1 | preformed biofilm into PDMS sheets (0.2 × 0.6 cm2) for 48 h | CS-PEG-LK13 | injected in situ | 6.7 mg kg–1 once a day after 24 h surgery | bacterial load and pathology of the tissue. | 4 days | improved healing, eradication of biofilm, tissues (muscle and skin) reduced inflammation and production of IL-6 and TNF-α | (57) | |
| respiratory | alginate | female Lewis rats | P. aeruginosa (NH57388A) | 1010 CFU mL–1 | intratracheal instillation | Novispirin G10 | instillation into the lower left lung | 0.1 mg mL–1 in 0.1 mL | bacterial load in the lung pathology and histology and cytokines | 3, 5, 7, and 10 days | bacterial load reduction of the lung, inflammatory cytokines, and a decrease in inflammation | (63) |
| agar bead coated | rats | P. aeruginosa mucoid (PAO1) | 104 CFU mL–1 | preformed biofilm agar beads intratracheally | HBCM2, HBCM3, HBCPα-2 and HB71 | intratracheally instillation or nebulization | 100 g in 100 μL 3 days after inoculation once daily or 5 mg mL–1 once daily for 3 days | bacterial load in the lung | 3 days | bacterial load reduction in the lung | (60) | |
| bacterial solution | female BALB/c | P. aeruginosa (PAO1) | 8 × 106 UFC in 20 μL | intranasally | P5 and P6.2 | intranasally | 10 mg kg–1 30 minutes after infection. | bacterial load and cytokines in the lung | 20 h | bacterial load reduction in the lung and decrease in pro-inflammatory cytokines | (65) | |
| female C57BL/6 | P. aeruginosa (C1) | 1 × 106 CFU in 40 μL | intranasally | ZY4 | intravenously | 2, 4, and 8 mg kg–1, 1 h postbacteria inoculation twice a day for 3 days | bacterial load in the lung, blood cytokines and lung histopathology | 72 h | bacterial load reduction of 90% of the lung, alleviated lung inflammation, reduced infiltration of inflammatory cells and up-regulation of cytokine levels (IL-6, TNF-α, IL-1β, and IL-10) | (66) | ||
| female wild-type C57BL/6J | P. aeruginosa (PAO1) | ∼3 × 106 CFU per mouse in 50 μL | intratracheally | Esc (1–21), Esc (1–21)-1c, LL-37 | intratracheally | 0.1 mg kg–1 2 and 12 h after | bacterial load, cytokines and airway-epithelia associated genes | 24 h | bacterial load reduction of lung, reduced leukocyte recruitment, and attenuated inflammatory response | (67) | ||
| female wild-type C57BL/6J | P. aeruginosa (PAO1) | ∼3 × 106 CFU in 50 μL | intratracheally | WLBU2 WLBU2- D8 | ontratracheally | 0.05 mg kg1– in 50 μL 1 h after infection | bacterial load and histopathology of the lungs | bacterial load reduction in the lung and reduced inflammatory response; bacterial load reduction in the lung and reduced inflammatory response; therapeutic index greater than 140 | (68) | |||
| C57BL/6J mice | K. pneumoniae | 1 × 105 CFU | intranasal instillation | IK8L | vein injection | 20 mg kg1– 4 h before infection | bacterial load, survival, biophotonic imaging, cytokines and lung histopathology | 50 h | bacterial load reduction in the lung, reduced lung injury, longer survival, TNF-α, IL-6, and IL-1β decrease | (72) | ||
| oral | dental caries | weaned Sprague–Dawley rats | S. mutans (UA159) | mid logarithmic culture | oral (every day for 5 days)a,b | LN-7 | topic on the molars | 32 μM, twice a day for 6 weeks | Caries scores by Keyes system and macroscopic tissue lesions | 72 days | slight dentinal lesions in the sulcal surface reducing the occurrence of severe dental lesion | (78) |
| weaned Sprague–Dawley rats | S. mutans (UA159) | mid logarithmic culture | orally (once a day)a,b | GH12 | topic on the molars | 8 mg L–1 three times a day for 3 weeks | Caries scores by Keyes system, mucosal tissues histopathology and bacterial load by qPCR of saliva and scraped plaque. | 21 days | lower score for both smooth-surface and sulcal-surface lesions | (82) | ||
| periodontitis | BALB/cByJ mice | S. gordonii (DL-1) and P. gingivalis (ATCC 33277) | 109–107 CFU in 1 mL of 2% carboxymethylcellulose | orally (five times in total, every 2 days)a | BAR | orally | 3.4 μM every 2 days for 5 days together with P. gingivalisc | bacterial load by PCR of oral cavity and alveolar bone loss | 80 days | inhibition of P. gingivalis S. gordonii biofilms interaction, reduced alveolar bone loss | (85) | |
| BALB/cByJ mice | S. gordonii (DL-1) and P. gingivalis (ATCC 33277) | 109–107 CFU in 1 mL of 2% carboxymethylcellulose | orally (five times in total, every 2 days)a | BAR | orally | 0.7 μM or 3.4 μM every 2 days for 5 days together with P. gingivalisc | bacterial load, alveolar bone loss, and maxillary molar histology | 80 days | biofilm formation and periodontitis inhibition, reduced inflammatory process, and lower alveolar bone loss | (89) | ||
| male Sprague–Dawley | P. gingivalis (W83) | 1 × 109 CFU mL–1 in 1.5 mL | orally (twice times every day for 4 weeks)d | Nal-P-113 | topic into periodontal pockets | 6.25, 25, 100, or 400 μg mL 2 h later infection twice times every day for 4 weeks | alveolar bone loss, bacterial colonization, and distribution by scanning electron microscopy and qPCR; histopathologic of periodontal tissue and Western blot detection of gingival tissue | 45 days | alveolar bone loss inhibition; decreased bacterial load in biofilms; IL-1β and TNF-α decreased in the periodontal tissue | (92) | ||
Previous treatment with antibiotics to deplete the oral microbiome.
Offered cariogenic diet ad libitum throughout the experiment.
Observation for 47 days and then euthanasia.
Placement of a ligature around the first molars from the rats before infection.
The bacterial inoculum can vary according to the expected infection severity (Tables 1 and 2), ranging from acute to chronic. The latter mimics biofilm infections in humans most accurately.25 Antibiofilm activity is evaluated by bacterial recovery from the infected wound. The infection site is excised, macerated, and serially diluted to determine the number of biofilm-forming bacteria, expressed as colony-forming units (CFU) per gram (CFU g–1) of tissue.26 Nevertheless, other approaches have also been used to evaluate ABP effectiveness (Table 1 and 2), including (i) analyses of the infectious process and healing through real-time imaging with an in vivo imaging system (IVIS), along with wound size measurement with the aid of calipers and photographs;23,27 (ii) histological analysis (e.g., hematoxylin eosin) and histochemistry to evaluate the tissue regeneration process; (iii) analysis of genetic signatures associated with biofilm formation (e.g., pslD, mucC, and quorum sensing (QS) related genes); (iv) evaluation of underlying organs (e.g., liver, lung, kidneys); and (v) evaluation of inflammatory patterns (e.g., IL-6, IL-10, and TNF-α) (Figure 1).21,23,26,27
Table 2. ABP Sequences, Physicochemical Properties, and In Vivo Potentiala.
| peptide name | sequence | a.a.b | MWc | pId | net charge | H (%)e | μHf | in vivo potential | ref |
|---|---|---|---|---|---|---|---|---|---|
| [Lys]7-Pol-CP-NH2 | ILGTILKLLKSL-NH2 | 12 | 1311.71 | 10.00 | 2 | 80.2 | 0.764 | skin scarification an abrasion | (28) |
| (P)PAP-A3 | PIMYKVPLIRKKSLRRTLSERGLLKDFLKKHNLNPARKYFPQWKAPTL | 48 | 5792.03 | 11.17 | 11 | 34.1 | 0.122 | (30) | |
| EcDBS1R5 | PMKKLKLALRLAAKIAPVW | 19 | 2147.78 | 11.33 | 5 | 57.9 | 0.413 | (31) | |
| PaDBS1R6F10 | KKLRLKIAFK | 10 | 1244.63 | 11.33 | 5 | 23.3 | 0.021 | (32) | |
| RP557 | RFCWKVCYKGICFKKCK | 17 | 2140.71 | 9.62 | 6 | 58.9 | 0.060 | (27) | |
| mastoparan-R1 | KILKRLAAKIKKIL | 14 | 1636.19 | 11.39 | 6 | 36.9 | 0.775 | (29) | |
| mastoparan-R4 | INLKKLAARIKKKI | 14 | 1637.13 | 11.39 | 6 | 20.4 | 0.472 | ||
| RIP | YSPWTNF-NH2 | 7 | 913.98 | 5.52 | n.d. | n.d. | n.d. | surgical urinary tract stents | (33, 43) |
| DRGN1 | PSKKTKPVKPKKVA | 14 | 1535.94 | 10.70 | 6 | –5.8 | 0.013 | surgical | (23) |
| WRL3 | WLRAFRRLVRRLARGLRR-NH2 | 18 | 2351.88 | 12.85 | 8 | 25.6 | 0.838 | iron burn | (26) |
| DJK-5 | vqwrairvrvir-NH2 | 12 | 1551.90 | 12.48 | 4 | 46.3 | 0.267 | cutaneous abscess | (34, 35, 37) |
| peptide 73 | RLWDIVRRWVGWL | 13 | 1755.10 | 11.70 | 2 | 81.5 | 0.753 | (36) | |
| peptide 73-C | RLWDIVRRWVGWLC | 14 | 1857.24 | 10.26 | 2 | 86.6 | 0.670 | ||
| HHC-10 | KRWWKWIRW-NH2 | 9 | 1444.75 | 12.02 | 4 | 75.6 | 0.672 | (37) | |
| IDR-1002 | VQRWLIVWRIRK-NH2 | 12 | 1653.05 | 12.30 | 4 | 66.7 | 0.201 | ||
| IDR-1018 | VRLIVAVRIWRR-NH2 | 12 | 1536.93 | 12.48 | 4 | 62.3 | 0.271 | ||
| citoprin 1.1 | GLFDVIKKVASVIGGL-NH2 | 16 | 1615.98 | 8.59 | 1 | 62.3 | 0.614 | CVC | (42) |
| BMAP-28 | GGLRSLGRKILRAWKKYGPIIVPIIRI-NH2 | 27 | 3074.84 | 12.02 | 7 | 55.7 | 0.522 | (46) | |
| IB-367 | RGGLCYCRGRFCVCVGR-CONH2 | 17 | 1905.31 | 9.37 | 4 | 53.0 | 0.204 | (47) | |
| WLBU2 | RRWVRRVRRWVRRVVRVVRRWVRR | 24 | 3398.0 | 13.08 | 13 | 14.1 | 0.789 | periprosthetic joint respiratory (bacterial solution) | (49, 68) |
| tachyplesin III | KWCFRVCYRGICYRKCR-NH2 | 17 | 2240.75 | 9.79 | 5 | 53.6 | 0.050 | urethral stent | (50) |
| HPA3NT3-A2 | AKRLKKLAKKIWKWK-NH2 | 15 | 1925.48 | 11.47 | 8 | 15.9 | 0.607 | catheter needle | (54) |
| EC1-17KV | GWWRRTVKKVRNAVRKV | 17 | 2139.6 | 12.48 | 7 | 13.8 | 0.657 | (53) | |
| 17tF-W | GX1KRlVQRlKDWlRKLV-NH2 | 17 | 2229.78 | n.d | 5 | n.d. | n.d. | (55) | |
| cys-melimine | CTLISWIKNKRKQRPRVSRRRRRRGGRRR | 29 | 3733.47 | 12.70 | 15 | –16.2 | 0.148 | titanium coating | (51) |
| DD13-RIP | ALWKTLLKKVLKAYSPWTNF-CONH2 | 20 | 2407.93 | 10.18 | 4 | 62.7 | 0.562 | Dacron grafts | (56) |
| melittin | GIGAVLKVLTTGLPALISWIKRKRQQ | 26 | 2847.49 | 12.02 | 5 | 51.1 | 0.394 | silicone implants | (52) |
| CS-PEG-LK13 | LKLLKKLLKKLKK | 13 | 1594.19 | 10.78 | 7 | 25.2 | 0.809 | silicone sheets | (57) |
| novispirin G10 | KNLRRIIRKGIHIIKKYG | 18 | 2206.76 | 11.75 | 7 | 23.3 | 0.645 | respiratory (alginate) | (63) |
| HBCM2 | KWKSFIKKLTKAAKKVVTTAKKPLIV-NH2 | 26 | 2955.75 | 10.90 | 9 | 31.5 | 0.491 | respiratory (agar bead coated) | (60) |
| HBCM3 | KWKKFIKSLTKSAAKTVVKTAKKPLIV-NH2 | 27 | 3042.83 | 10.90 | 9 | 30.1 | 0.394 | ||
| HBCPα-2 | KWKKFIKKIGIGAVLKVLTTGLPALKLTKK-NH2 | 30 | 3322.26 | 10.90 | 9 | 45.3 | 0.183 | ||
| HB71 | FAKKLAKKLKKLAKKLAK-COOH | 18 | 2055.71 | 10.90 | 9 | 5.1 | 0.714 | ||
| P5 | RIVQRIKKWLLKWKKLGY | 18 | 2356.98 | 11.26 | 7 | 45.5 | 0.58 | respiratory (bacterial solution) | (65) |
| P6.2 | GLLRKWGKKWKEFLRRVWK | 19 | 2515.09 | 11.75 | 7 | 32.8 | 0.793 | ||
| ZY4 | VCKRWKKWKRKWKKWCV-NH2 | 17 | 2377.00 | 10.74 | 9 | 32.8 | 0.745 | (66) | |
| Esc (1–21) | GIFSKLAGKKIKNLLISGLKG-NH2 | 21 | 2185.72 | 10.60 | 5 | 41.3 | 0.188 | (67) | |
| Esc (1–21)-1c | GIFSKLAGKKIKNlLIsGLKG-NH2 | 21 | 2185.72 | 10.60 | 5 | 41.3 | 0.188 | ||
| WLBU2-D8 | RRWVRRvRRWvRRvvRvvRRWvRR | 24 | 3398.0 | 13.08 | 13 | 14.1 | 0.789 | (68) | |
| LL-37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | 37 | 4493.32 | 10.61 | 6 | 20.1 | 0.521 | (67) | |
| IK8L | IRIKIRI | 7 | 911.20 | 12.01 | n.d. | n.d. | n.d. | (72) | |
| LN-7 | LRRWLRWLLRWMR-NH2 | 13 | 1941.42 | 12.60 | 5 | 74.8 | 0.875 | dental caries | (78) |
| GH12 | GLLWHLLHHLLH-NH2 | 12 | 1488.80 | 7.10 | 0 | 108.1 | 0.399 | (80, 82) | |
| BAR | NH2-LEAAPKKVQDLLKKANITVKGAFQLFS-COOH | 27 | 2958.54 | 9.83 | 3 | 34.8 | 0.434 | periodontitis | (85, 89) |
| Nal-P-113 | Ac-AKR-Nal-Nal-GYKRKF-Nal-NH2 | 12 | n.d | n.d | n.d | n.d | n.d | (92) |
The physicochemical properties when not disclosed by the original article were calculated on the Heliquest server (http://heliquest.ipmc.cnrs.fr/). Molecular weight and isoelectric points were obtained through the ProtParam Expasy server (https://web.expasy.org/protparam/). Lower case letters represent D-amino acids. X1: 4-t-butylphenylalanine. NAL: β-naphthylalanines. n.d.: not determined due to the presence of unnatural amino acid residues or chemical modifications or too short input sequences.
Amino acid residues.
Theoretical molecular weight.
Theoretical isoelectric point.
Hydrophobicity (%).
Hydrophobic moment.
Figure 1.

Murine biofilm wound models and proposed ABPs’ modes of action. Biofilm wound models can be obtained by performing lesions on the animal’s skin using different techniques (e.g., surgical excision, skin scarification, iron-burn, and pressure), followed by the bacterium inoculum. Moreover, this model can be established by the bacterium’s direct inoculum subcutaneously (e.g., abscess). ABP treatment routes include topical and systemic administration. ABPs can assist in tissue recovery through mobilizing keratinocytes, stimulating angiogenesis, and reducing dermonecrosis. They can also display direct bactericidal effects alone or in synergism with antibiotics through membrane-associated mechanisms or acting intracellularly. ABPs also block cell signaling and communication, thus regulating biofilm-related gene transcription, impairing biofilm formation. In preformed biofilms, ABPs cause EPS degradation, detaching bacterial cells from surfaces and interfering with biofilm morphology. Finally, many ABPs act as immunomodulators, avoiding an exacerbated response mediated by cells (e.g., neutrophil and macrophage), and assisting in recruiting cells (e.g., keratinocytes) that assist wound healing. All figures were made by the authors with a subscription version of BioRender.com.
Torres et al. (2018)28 reported a skin scarification mouse model to establish a P. aeruginosa abscess on mice dorsum to evaluate the anti-infective potential of short AMPs. The authors did not immunosuppress the mice and focused on evaluating bacterial load exclusively by applying a single superficial treatment. After 2 or 4 days, the wounded skin was excised, and CFU counts were used to evaluate the peptides’ anti-infective effect. Similar studies were conducted for computationally designed peptides (Table 2).29−32 More recently, a peptide named RP557 (Table 2) was shown to effectively eliminate biofilms in a murine abrasion model infected with methicillin-resistant S. aureus (MRSA). Only a 0.2% topical dose of RP557 was required to eliminate MRSA biofilms. Moreover, MRSA demonstrated low resistance to RP557 after several passages with sublethal concentrations.27
Considering the importance of QS for bacterial biofilms, Nakagami et al. (2008)21 reported a pressure-induced ischemic wound rat model using P. aeruginosa. The authors demonstrated the presence of QS molecules in the infected wounds and showed QS’s role in P. aeruginosa biofilm wound infections. Later, Schierle et al. (2009)33 developed a new cutaneous excision model infected with S. aureus and S. epidermidis biofilms, in which QS inducers were observed and related to Staphylococcal biofilm formation, leading to skin re-epithelialization and healing delay during the infection process. By contrast, S. aureus or S. epidermidis-infected wounds treated with RNAIII-inhibiting peptide (RIP) (Table 2) showed accelerated wound healing.33
In terms of polymicrobial biofilm infections, Chung et al. (2017)23 demonstrated the in vivo antibiofilm activity of DRGN-1 (Table 2), a peptide designed from the peptide VK25. The polymicrobial biofilm was grown in a polycarbonate filter and implanted over an incision made on the mouse dorsum. DRGN-1 was administrated topically and reduced the microbial load (2-log) at the infection site. It also induced keratinocyte migration to the injured region, substantially improving the healing process.23
Burn wounds can be colonized by a biofilm, aggravating healing. Ma et al. (2017)26 evaluated WRL3 (Table 2), a peptide that synergized with ceftriaxone against MRSA biofilms in vivo using a scald-burn wound model. This peptide demonstrated potent activity, inhibiting biofilm growth, reducing biofilm biomass, and promoting wound healing.26
Biofilm infections in wound sites are challenging to treat and usually lead to cutaneous abscess formation. Mansour et al. (2016)34 used a mouse model to evaluate ABPs’ activity against MRSA cutaneous abscesses. The peptide DJK-5 (Table 2) was investigated in systemic (6 or 4 mg kg–1) and cutaneous (3 or 4 mg kg–1) models for MRSA and P. aeruginosa. When administrated topically, DJK-5 reduced skin damage and significantly decreased the local bacterial load.34 Although DJK-5 revealed potent antibiofilm activity, studies have shown that this peptide can cause tissue damage and inflammation at 1.5 mg mL–1.35 To overcome this obstacle, Kłodzińska et al. (2019)35 evaluated the potential of DJK-5 encapsulated within hyaluronic acid–based nanogels. Similar antibiofilm activities were reported, and the encapsulated DJK-5 system led to 4-fold decreased toxicity compared with free DJK-5.35
Kumar et al. (2019)36 also used a cutaneous abscess model to evaluate the activity of aurein-derived peptides. Among the molecules evaluated, peptide 73 (Table 2) was 2.2-fold more efficient at reducing MRSA cutaneous abscesses than its parent peptide. The in vivo antibiofilm potential was further enhanced by incorporating cysteine residues to the C-terminus of peptide 73 and performing chirality changes or encapsulating peptide 73 in polymers (e.g., polyethylene glycol (PEG) or hyperbranched polyglycerol (HPG)).36 This cutaneous abscess model was used by Pletzer et al. (2018)37 in a high-throughput in vivo study with the peptides HHC-10, IDR-1002, IDR-1018, and DJK-5 (Table 2). The synergistic activity of these peptides in combination with conventional antibiotics was investigated against ESKAPE pathogens (Enterococcus faecium, S. aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P. aeruginosa, and Enterobacter species), leading to reduced abscess size independent of the mechanism of action of either antibiotics or ABPs.37
Most of the studies mentioned demonstrate that ABPs operate by decreasing bacterial load and triggering tissue healing processes.21,28,31,33 Although we focused on describing studies that exclusively reported antibiofilm activity in vivo, such studies can be challenging. This may occur due to many factors, including bacterial load, bacteria inoculation versus treatment with APBs, and administration route. For instance, treatment with peptides is often initiated at the same time or within minutes after bacterial inoculation.34,36 An alternative that can overcome this issue is the model proposed by Chung et al. (2017).23 They used a preformed matrix in bacterial biofilms that were attached to the wound site and demonstrated biofilm proliferation. Moreover, in skin abscess models, the authors demonstrated genetic patterns as the presence of genes involved in EPS biosynthesis (pslD) and upregulation of EPS alginate synthesis (mucC) that are present in biofilm formation.22 These models have assisted the identification of wound biofilm infection patterns (at the gene level) with greater representativeness and translational potential for preclinical tests.
The topical route is the most used and presents the most effective results when evaluating ABPs in vivo. The systemic route has also been used;21,23,26,29,34 however, the results obtained were not as promising as those from topical administration studies, as ABPs usually present low bloodstream stability, rendering them inactive.10 As described above and reported by Kłodzińska et al. (2019)35 and Kumar et al. (2019),36 nanoencapsulation strategies can improve the bioavailability of ABPs, favoring the translation of these antibiofilm agents into the clinic.
Foreign Body Infection Model
Among the most commonly colonized medical devices are urinary catheters, cardiac pacemakers, dental implants, vascular prostheses, peritoneal dialysis catheters, stents, intrauterine devices, contact lenses, and breast implants.38 The presence of a foreign body significantly increases biofilm formation, as this exogenous structure offers an ideal surface for bacterial growth. Additionally, the foreign body can impair defense cells’ functions, including leukocytes, thus facilitating bacterial adhesion and growth.39
Animal models using foreign bodies can be divided into two broad groups, the (i) site-specific device models that are inserted into a given organ or are arranged in similar regions in humans (e.g., urinary stents, venous catheters, prosthetic implants) and (ii) subcutaneous device models (e.g., cage tissue, catheters subcutaneously) (Tables 1 and 2).38 Moreover, it is common to find studies evaluating ABPs’ stability in body fluids, including blood, urine, among others, prior to in vivo experiments. These in vitro assays allow a better understanding of ABPs’ behavior in different biological conditions, contributing to a more robust assessment of the possible mechanisms involved in their antibiofilm activities in vivo.
Site-Specific Device Models
The abiotic nature of medical devices favors biofilm proliferation, as a considerably lower bacterial load is sufficient to colonize these devices, triggering chronic infections and even systemic infections.40,41 In this model, the bacterial load is injected at the infection site after the device implantation surgery (e.g., central venous catheters (CVC), urinary catheter, and stent).42,43 ABPs have been used alone or in synergy with antibiotics before or after the infection is established (Tables 1 and 2). The antibiofilm potential of mono- and combination therapies is measured by bacterial recovery from the infection site or the portion of the device where bacteria were attached. Notably, in the case of the urinary stent case, bacteria can be detected in the urine, whereas in the CVC model, the peripheral blood can be collected and analyzed for quantifying bacterial load (Figure 2 A).42,43
Figure 2.

Foreign body murine models for biofilm infections and proposed ABPs modes of action. (A) Site-specific device model. The most common devices used to assess ABPs activity include catheter and urinary stents, central venous catheters (CVC), and periprosthetic implants, all surgically inserted. ABPs can be immobilized on the device’s surface, thus inhibiting biofilm formation for extended periods. This strategy can be used in association with conventional antibiotics. Some ABPs also inhibit bacterial communication (e.g., QS). Direct bacterial activity is also a common mechanism (e.g., ABPs alone or in synergism with antibiotics) for inhibiting biofilm formation. In preformed biofilms, ABPs can reduce biofilm biomass. Furthermore, direct bacterial activity is also a common mechanism. (B) Subcutaneous device models. In these models, biofilm is usually previously formed in the device (e.g., titanium disc, silicone beads, Dracon graft, and catheter). The infected device is inserted subcutaneously in the animal (mouse or rat), followed by ABP treatment. ABPs have demonstrated direct antibacterial activity in most cases with bacterial membrane disruption. The antibiofilm activity has also been achieved via EPS degradation, leading to biofilm dispersion and enabling synergism with antibiotics. Additionally, ABPs have been shown to penetrate the biofilms through water channels and disperse biofilm cells, followed by direct antibacterial effects. Immunomodulatory responses have also been observed, including reduced neutrophil migration and cytokine regulation (e.g., TNF-α, MCP1/CCL2 IL-17A, and IL-10). All figures were made by the authors with a subscription version of BioRender.com.
Many in vivo models have been proposed to help approximate experimental conditions from those found in urinary tract infections. ABPs have been widely evaluated as alternatives to combat urinary tract infections. For instance, the activity of teicoplanin combined with RIP has been assessed against S. aureus biofilms by coating urinary stents with peptide and surgically implanting them in the bladder of rats.43
The use of CVC is also associated with biofilms, aggravating difficult-to-treat infections.44 Antibiotics become ineffective against infected CVC because of biofilm attachment to the device wall.40 Therefore, several studies have used in vivo CVC models to investigate prospective antibiofilm compounds.45 For instance, to evaluate the activity of the peptide citropin1.1 (Table 2), Cirioni et al. (2016)42 used a CVC that was inserted into the jugular vein of rats and advanced to the superior vena cava. A pretreatment was performed by filling the catheters with citropin1.1 alone or in combination with antibiotics, followed by infection with S. aureus. The authors demonstrate that citropin1.1 alone and combined with minocycline and rifampicin reduced the bacterial load. The authors suggest that citropin1.1 acts by inhibiting S. aureus adherent cell growth, allowing antibiotics to act on planktonic bacterial cells, thus inhibiting biofilm formation.42
Similarly, a CVC model in rats was used to evaluate the activity of the cathelicidin BMAP-28 (Table 2). Catheters pretreated with BMAP-28 in conjunction with antibiotics (quinupristin/dalfopristin (Q/D), linezolid, and vancomycin) displayed a 4-log reduction in bacterial load with no observation of bacteremia.46 Moreover, Ghiselli et al. (2007)47 demonstrated that, by combining the peptide protegrin IB-367 with linezolid, a significant decrease in S. aureus and E. faecium bacterial load (and no bacteremia) was observed.47
Treatment of periprosthetic joint infections is scarce and usually requires surgical interventions and long-term antibiotic therapy, presenting high mortality rates.48 A periprosthetic joint infection mouse model was used to evaluate the activity of peptide WLBU2 (Table 2) in eliminating implant-associated biofilms.49 The bacterial load reduction was dose-dependent at 0.01 and 10 mg kg–1. Additionally, this peptide demonstrated low toxicity and high stability, making it a valuable candidate for antibiofilm treatment in prosthetic implants.49
Subcutaneous Device Models
Subcutaneous implant models are widely used in biofilm-related infections and to assess the efficacy of ABPs, since the surgical incision on the animal’s back is more accessible than the implantation of local devices.38 Various materials with different textures can be implanted (e.g., titanium, silicone, Dacron graft, and catheter), directly influencing bacterial load recovery and local inflammatory responses.40 In the foreign body subcutaneous device models, a surgical incision is made on the back of the animal, where the device is inserted subcutaneously (Figure 2 B). Most studies using ABPs in the context of subcutaneous device models evaluate their effects on preformed biofilms.50−53 Consequently, in these studies, it is imperative to guarantee that the bacteria are at the mature biofilm stage prior to treatment.
ABPs can also be immobilized on the device’s surface prior to the infection and surgery, with the overarching goal of preventing bacterial attachment and further biofilm formation. Recovered bacterial counts are typically utilized as a proxy for treatment effectiveness. However, histological analysis, imaging by IVIS, scanning microscopy, and inflammatory response detection are also used as critical parameters in these models (Table 1).50−53
Minardi et al. (2007)50 reported that tachyplesin III-coated urethral stents were used to eradicate P. aeruginosa infections in rat subcutaneous pouch models, inhibiting biofilm growth up to 1000 times compared to untreated controls.50 Similarly, Lee et al. (2019)54 showed that peptide HPA3NT3 (Table 2) reduced biofilm formation, tissue damage, and toxicity (Table 1). Using the same model, Ma et al. (2020)53 used a catheter infected with preformed P. aeruginosa biofilm, which was further inserted subcutaneously in mice to evaluate the antibiofilm activity of peptide EC1-17KV (Tables 1 and 2). This same model was used to demonstrate the antibiofilm potential of ABP 17tF-W (Tables 1 and 2), a peptide derived from LL-37, on MRSA biofilms. In addition to combating biofilms, this peptide exhibited immunomodulatory activity and resistance to proteases.55 Chen et al. (2016)51 described the activity of the ABP melamine (Table 2) as a titanium coating agent in a subcutaneous infection mouse model. The titanium surface was functionalized via a series of reactions that yielded a thioether linkage between the functionalized surface and the sulfhydryl group of melimine. The AMP-coated material significantly reduced biofilm formation by P. aeruginosa in both mouse and rat subcutaneous infection models and reduced the bacterial load by up to 2-log compared to the uncoated titanium surface (Table 1).
The model with Dacron grafts has also been used to evaluate ABPs. Soon after the Dacron grafts were pretreated with ABPs, including RIP, 13-residue dermaseptin derivative (DD13), and hybrid DD13-RIP, they were inserted in the subcutaneous pockets. All three peptides reduced the graft’s bacterial load (from 3 to 4-log), with DD13-RIP demonstrating the best results (5-log reduction) at a lower dose (10 mg L–1) (Tables 1 and 2). Moreover, these peptides demonstrated synergism with the antibiotic rifampicin, boosting its activity against resistant strains.56
Yu et al. (2020)52 demonstrated that supramolecular coassembly with mesoporous silica nanoparticles improved delivery of antibiotics and the peptide melittin. This strategy led to enhanced antibiofilm activity in vitro and in a preformed biofilm model of P. aeruginosa in subcutaneous silicone implants. Additionally, it prevented tissue damage and inflammation associated with the implant.52 Moreover, Ju et al. (2020)57 proposed a chitosan-polyethylene (CS-PEG) glycol-peptide (LK13 peptide) conjugate (CS-PEG-LK13) targeting the biofilm water channels and the negative charge of EPS (Figure 2B). The authors demonstrate that these characteristics allowed the penetration of this conjugate into biofilms and, subsequently, increased antibacterial activity compared to the LK13 (Table 2) alone. These conjugates’ effectiveness was also demonstrated in a murine model of subcutaneous implantation of silicone sheets infected with preformed P. aeruginosa biofilms.57
Respiratory Tract Chronic Infection Models
P. aeruginosa is considered the primary agent responsible for biofilm lung infection in cystic fibrosis.58,59 The biofilm mode of growth hinders complete eradication of the infection, leading to chronic inflammation of the subject’s airways.59 Some ABPs have been described for their activity in respiratory tract infection murine models (Tables 1 and 2).
In the CF murine model, bacteria may be inoculated through instillation, intranasally, or intratracheally.60−62 In these models, the infection severity is determined by the inoculum and inoculation frequency of bacteria. The establishment of chronic pulmonary infection models can be obtained by using bacteria carriers (e.g., alginate) produced by the bacterial strain itself or by bacterial incorporation onto agar beads (Table 1). In these cases, intratracheal instillation is the most appropriate route for bacterial inoculation.63,64
Some studies also use P. aeruginosa clinical isolates from CF patients (e.g., bacterial solution) to evaluate the efficacy of ABPs (Tables 1 and 2). In these models, the time between infection establishment and the end of the treatment is much shorter than when bacteria are incorporated into agar, alginate, or silicone. This shorter treatment time is because the animals get worse since the bacteria is directly inoculated, which can progress to severe acute respiratory syndrome (SARS), leading to animal death even before the treatment has been effective (Figure 3).
Figure 3.

Respiratory tract biofilm infection models and proposed ABPs modes’ of action. Respiratory tract biofilm infection models can be obtained by the incorporation of the bacteria in agar or alginate, which is instilled intratracheally, or a solution containing the biofilm-forming bacteria, which are inoculated directly into the animals’ nostrils or by inhalation. These models simulate a process similar to cystic fibrosis. In this process, both the innate and adaptive immune systems promote an exacerbated response mediated by immune cells (e.g., neutrophil, macrophage, dendritic cell, and lymphocytes), cytokines (e.g., IFN-γ, TNF-α, IL-4, IL-6, IL-17, and IL-10), and chemokines (e.g., CXCL-1 and CXCL-2). Therefore, ABPs have been used as immunomodulators. Some ABPs regulate the migration of inflammatory cells, including neutrophils, also acting by modulating the cytokine-mediated inflammatory response and reducing pro-inflammatory cytokines (e.g., IL-6 and TNF-α, IL-1β). These activities may be associated with both increased survival after treatment with ABPs and reduced lung tissue damage in treated animals. Additionally, these peptides demonstrate potent antibacterial activity (alone or in synergism with antibiotics) and antibiofilm, along with LPS neutralization. All figures were made by the authors with a subscription version of BioRender.com.
Song et al. (2005)63 reported a rat model in which the ABP novispirin G10 (Table 2) was administered intratracheally to treat P. aeruginosa mucoid biofilm lung infection. Compared to the control groups, the remaining bacteria in the lung between 3 and 5-days postinfection were reduced by 170 to 330 times in novispirin G10-treated mice (Table 1). Consistent with these results, in pulmonary pathological analysis, treated animals’ lungs showed milder lesions and lower cytokine-mediated responses.63
Zhang et al. (2005)60 performed a screening with 150 AMPs against clinical CF isolates, among which four peptides (HBCM2, HBCM3, HBCPα-2, and HB71) showed higher antibacterial and antibiofilm activity (Tables 1 and 2). These peptides were evaluated in a murine model of pulmonary biofilm infection with P. aeruginosa. The peptides HBCM2, HBCM3, HBCPα-2, and HB71 significantly reduced P. aeruginosa counts in the lung. Additionally, anti-inflammatory responses were also observed for HBCM2.60 More recently, Martínez et al. (2020)65 described two peptides, P5 and P6 (Table 2), with antibiofilm activity and effectiveness in treating pulmonary infection by P. aeruginosa. Moreover, these peptides also demonstrated anti-inflammatory activity and led to reduced pro-inflammatory cytokines in the lung.65
The cyclic peptide ZY4 (Table 2) described by Mwangi et al. (2019)66 demonstrated in vitro and in vivo effectiveness, high stability in plasma, and prolonged half-life. Additionally, this peptide inhibited MDR P. aeruginosa biofilm formation.66 The pulmonary infection model with MDR P. aeruginosa was used to evaluate ZY4 in vivo. Bacteria were inoculated intranasally, and the treatment was carried out intravenously. The authors observed that ZY4 reduced 90% of the lungs’ bacterial load at a concentration of 8 mg kg–1 (Table 1). Similarly, Chen et al. (2017)67 reported the in vivo therapeutic efficacy of the Esc(1–21)-1c peptides (Table 2) against P. aeruginosa-induced pulmonary infection in a mouse model after a single, low-dose intratracheal instillation. The authors also showed that the peptides reduced the lung bacterial burden by 2-log, with a concomitant reduction in leukocyte recruitment and attenuated inflammatory response.
Here, we described the use of WLBU2 to treat biofilm-associated periprosthetic implant infections. WLBU2 has also been used to treat P. aeruginosa infections in a murine model of pulmonary infection. This peptide reduces pulmonary bacterial load and inflammation, with a single dose of 0.05 mg kg–1 instilled directly into the animals’ lungs (Table 1). Additionally, this peptide is effective against bacteremia induced by P. aeruginosa in vivo. Recently, it was demonstrated the structural optimization of WLBU2 by D-amino acids insertion, thus improving this peptide’s stability and reducing its toxicity in vivo.68
K. pneumoniae can also form a biofilm in tissues such as the lungs.69 Additionally, this bacterium is widely found in respiratory devices (e.g., mechanical ventilation system),70 resulting in pulmonary infections in the lower respiratory tract. In a study by Guilhen et al. (2019),71 a robust murine pneumonia model was established, revealing that free-floating bacteria dispersed from K. pneumoniae biofilms are associated with this bacterium colonization capacity, also compromising the host’s immune system.71
In this context, a murine model of pneumonia was used to assess the ABP’s activity, IK8L (Table 2).72 In that study, the authors monitored the pulmonary infection evolution with bioluminescent bacteria instillation. Moreover, the antibiofilm effect of IK8L was accessed over time using a biphotonic imaging (Caliper’s Xenogen IVIS XRII optical imaging technology) system. IK8L inhibited biofilm formation and modulated the inflammatory response mediated by inflammatory cytokines (e.g., TNF-α, IL-6, and IL-1β) in the bronchoalveolar fluid (Table 1). Furthermore, IK8L interfered with signaling proteins (e.g., STAT3, JAK2, and ERK1/2) that regulate IL-6, significantly reducing the animal’s mortality.72
Oral Infections Models
Dental caries is formed by diverse biofilms from pathogenic and commensal microorganisms. This disease is driven by diet and microbiota–matrix interactions that occur on the oral surface.73 Moreover, ABPs have been explored as potent oral therapeutic agents.74,75
Dental caries is commonly reproduced in animal models using recently weaned rats (Figure 4A). To establish a dental infection, previous treatment with antibiotics is necessary to eliminate the existing microbiome. Subsequently, the animals are fed with cariogenic diets and, in parallel, they receive the bacteria orally (e.g., Streptococcus mutans) in a period of 5–7 days, daily.76,77 The infection can be confirmed by sowing oral samples. The topical treatment with the peptides is carried out on the teeth, daily, for 30–45 days. At the end of the experiment, the animals’ mandibles and molars are excised to determine the carious lesions. Scanning electron microscopy analyses are also used in some studies (Table 1).76−78
Figure 4.

Oral biofilm infection model and proposed ABPs modes of action. (A) Dental caries infection model. In this model, some important factors are considered, including the use of newly weaned animals, pretreatment with antibiotics, confirmation of oral microbial depletion, and cariogenic diets in association with oral S. mutans infection. After the infection is confirmed, the topical treatment with ABPs is initiated. ABPs used in this model have been capable of preventing and eradicating biofilm-associated caries. ABPs can prevent tooth caries by inhibiting bacteria adherence to the tooth surface. In addition, ABPs also present direct antibacterial activity, significantly killing cariogenic pathogens before biofilm formation. When it comes to preformed biofilms, ABPs can interfere with the biofilm’s structure by avoiding EPS synthesis and reducing biofilm biomass. (B) Periodontitis murine model. Periodontitis mainly consists of inflammation of the periodontium due to biofilm formation. Particularly, S. gordonni and P. gingivalis have been shown to play a crucial role in this infection. Usually, the primary colonization of S. gordonni is followed by the infection with P. gingivalis, using carboxymethylcellulose (CMC) as a vehicle. P. gingivalis can also be used alone to induce oral infection. However, in this case, there is a subgingival thread in the first molars from the mice to allow biofilm formation. ABPs have demonstrated the ability to prevent biofilm formation by acting directly on free-floating bacteria or eradicating preformed biofilms. ABPs can also modulate cytokines regulation (e.g., IL-17, IL-1β, TNF-α), which significantly contributes to reducing alveolar bone loss, one of the main aggravating factors associated with periodontitis. All figures were made by the authors with a subscription version of BioRender.com.
Recently, the dental caries model described above was used to evaluate the LN-7 peptide (Table 2).78 This peptide is derived from two bacteriocins, called reutericin 6 and/or gassericin A, which had known antibacterial and antibiofilm activities in vitro. In animal models, LN-7 was capable of significantly reducing dental injuries, at 32 μM, by efficiently suppressing the development of dental caries promoted by S. mutans biofilms.78
Similarly, the ABP GH12 (Table 2) has been reported for its potent antibacterial activity and antibiofilm activities, also reducing EPS and lactic acid in vitro.79−81 This peptide (at 8 mg L–1), when evaluated in a caries animal model infected with S. mutans not only reduced the incidence but also the severity of caries in rats (Table 1).82 More recently, this same ABP was evaluated at a higher dose (64 mg L–1), leading to the regulation of the dental plaque microbiota. It was observed that the abundance of commensal species was positively regulated, whereas cariogenic bacteria were negatively regulated. In addition, more accurate analyses, including the assessment of sulcal caries and dental surface, revealed that such damages were controlled by GH12 under cariogenic conditions.83
Other types of oral pathologies can be associated with biofilm formation. For example, periodontitis is triggered by an oral inflammatory dysfunction caused, mainly, by microbial biofilms formation in the subgingival region.84,85 Biofilm formation in periodontitis is driven by interactions between different bacteria, including Porphyromonas gingivalis and oral streptococci species (e.g., Streptococcus oralis and Streptococcus gordonii).84,86,87 Therefore, in vivo models have been established to mimic periodontitis in mice (Figure 4 B). Infections with periodontitis-related bacteria (e.g., S. gordonii, P. gingivalis) are performed orally and can be confirmed by oral sowing or PCR analysis. The treatment can be carried out topically either for prevention or to eliminate an established biofilm infection. At the end of the experiment, the animals are euthanized and the skull is excised for alveolar bone loss analysis of the maxilla.85,88,89
Some ABPs have been tested in the above-mentioned periodontitis model (Tables 1 and 2). Numerous studies have shown that P. gingivalis biofilm formation requires an interaction with oral streptococci. Based on this, Daep et al. (2006)84 developed a synthetic peptide, denominated BAR (Table 2), capable of inhibiting P. gingivalis and S. gordonii interaction, thus preventing biofilm formation in vitro.84 Years later (2011),85 those authors used a periodontitis mouse model in which both S. gordonii and P. gingivalis were used to simulate oral human periodontitis.85 As a result, the BAR peptide showed promising in vivo activity by significantly preventing alveolar bone loss (Table 1), which is known to be the main consequence in periodontitis.85
More recently, that same peptide (BAR) was immobilized on poly(lactic-co-glycolic acid) nanoparticles surface, configuring nanoparticle–peptide complex BAR-modified NPs (BNPs).89 This nanoformulation led to in vivo antibiofilm potential in a S. gordonii and P. gingivalis infection mouse model at lower doses (0.7 μM) than nonformulated BAR (3.4 μM).89 In addition, BNPs have been shown to significantly reduce bone loss and IL-17 expression (Table 1),89 an important gingival inflammatory mediator in P. gingivalis associated infections. Therefore, BNPs have proved to be strong candidates for the prevention and prophylaxis of P. gingivalis biofilms in periodontitis. This nanoformulation strategy could be applied, for example, for mouthwashes or gels development.89
Another ABP, named Nal-P-113 (Table 2), has also demonstrated preventive periodontitis activity in rats’ lower molars. Previous studies have already demonstrated that Nal-P-113 is stable in saliva and presents antibacterial and antibiofilm activity.90,91 When evaluated in a periodontitis rat model, this peptide showed preserved biological potential toward pathogenic bacteria, also demonstrating anti-inflammatory activity by modulating cytokines production (IL-1β and TNF-α) and reducing alveolar bone loss (Table 1).92 Interestingly, in that study,92 biofilm formation was demonstrated using a 0.2 mm wire inserted in the dentogingival region of the lower first molars from rats. At the end of the experiment, this wire was removed and evaluated for biofilm formation through scanning electron microscopy. This strategy allowed the evaluation of the direct in vivo antibiofilm properties of Nal-P-113 against P. gingivalis associated with cocci bacteria and Bacillus brevis. Additionally, this technique allowed adherent bacteria recovery in their biofilm state and, through absolute quantitative real-time PCR analyses, it was possible to quantify the bacteria.92 The peptide Nal-P-113 has already been submitted to clinical studies for the treatment of patients with chronic periodontitis, showing promising results in the inhibition of periodontal pathogens (e.g., P. gingivalis, Treponema denticola, Fusobacterium nucleatum, and S. gordonii), also effectively countering oral biofilms.93
Challenges in Translating ABPs to the Clinic
Preclinical studies are enabled by the development of robust animal models that are clinically significant.94 When it comes to biofilm infections, it is often difficult to demonstrate biofilm formation and maintenance accurately and convincingly. However, as biofilms are notoriously present in chronic infections, different animal models have been used to evaluate antibiofilm compounds in preclinical trials, as described above.95
Most studies with ABPs include basic research and/or proof-of-concept studies. Consequently, there is a discrepancy between the volume of published studies and preclinical and clinical trials using ABPs. Currently (October 2020), according to the Biofilm-active AMPs Database (BaAMPs), a total of 221 ABPs have shown antibiofilm potential toward 116 different target microorganisms. However, none of these ABPs have yet reached advanced clinical trials. The delay in translating antibiofilm drug candidates into the clinic was recently reviewed by Rumbaugh et al. (2020),95 revealing that most clinical studies for treating biofilms involve the repurposing of drugs or combination of FDA-approved drugs.
By searching for antibiofilm preclinical and clinical trials in ClinicalTrails.gov (filters: bacterial infections; biofilm), only 14 studies were found, none of which involved ABPs. One of the main limitations when translating ABPs to the clinic is the use of appropriate in vivo models for preclinical screening. Studies using ABPs present significant differences among them, most of which are due to the lack of experimental standardization.96 Such differences include the bacterial load used, which can widely change the outcome of the study, since the use of a low bacterial load may lead to false-positive results in the treated animal group.
Some alternatives have been explored to overcome these obstacles. Many studies have adopted colorimetric reagents to monitor and demonstrate biofilm development, including using bioluminescent bacterial strains.97 Moreover, a new biofilm infection model of shoulder implants has been reported, in which a noninvasive tracking of the biofilm was performed through optical images.98 Additionally, specific biofilm features can be investigated to confirm their proper establishment, including Gac regulatory pathways, EPS production, and QS signaling.62
Although previous studies have already demonstrated the role of extracellular DNA (eDNA) in biofilm formation and increased resistance to antibiotics,99,100 its role and location in vivo have not yet been fully clarified.100,101 Recently, transmission electron microscopy in conjunction with laser confocal scanning was used to evaluate the interactions between P. aeruginosa biofilms and polymorphonucleated (PMNs) cells, also shedding light on the role and location of eDNA in a murine implant model.102 Furthermore, a new model of murine keratitis biofilm was recently established and features such as the extracellular matrix were identified through fluorescence electron microscopy and transmission from the animals’ cornea.103
In biofilms associated with chronic infections, a recent work has reported more robust models to achieve chronicity in animals.38 Bayes et al. (2016)64 developed a murine model of pulmonary biofilm infection adapted from agar beads using a clinical mucoid strain of P. aeruginosa. The authors demonstrated the transition from transient infection of airways to chronic infection. The animals were kept for 2 weeks to reproduce some of the characteristics observed in humans, including variable bacterial clearance, endobronchial infection, development of antipseudomonal antibodies, and low mortality in the acute infection phase.
In addition to these innovative in vivo models, well-established in situ screening can lead to more efficient products in vivo by reducing the unnecessary use of animals.104 Techniques with 3D skin, cell culture from different tissues, and ex vivo models can be relevant alternatives for determining the activity of ABPs against biofilms.9 Taken together, all these techniques contribute to the establishment of the 3Rs (Replacement, Reduction, and Refinement) aiming at the welfare of animals with less invasive techniques and the refinement of techniques to obtain clinically significant results for novel drug candidates specifically developed for biofilm infections.
Conclusions and Prospects
The multifactorial nature of biofilm infections and multidrug resistance poses a significant challenge when developing novel effective drugs. Therefore, the combination of multifactorial therapies with improved efficiency is needed. The selection of robust murine models that accurately mimic biofilm infections directly and significantly improves any study associated with ABPs. Indeed, many of these studies fail at the stage of selecting an appropriate animal model, thus compromising subsequent preclinical work. Moreover, even if the in vivo model adopted is adequate and the experiment is well designed, most studies to date have lacked pharmacokinetics and pharmacodynamics data. Another limiting factor for translating ABPs to the clinic is their high cost of synthesis. For preclinical and clinical trials, large amounts of raw peptide material are needed and, depending on the candidate and its therapeutic dose, these tests may not be viable. In summary, although some limitations still need to be overcome, here we describe the most appropriate and significant murine models for antibiofilm evaluation of peptide-based drug candidates and highlight recent advances that have contributed significantly to the evaluation of the lead ABPs described to date.
Acknowledgments
This work was supported by grants from Fundação de Apoio à Pesquisa do Distrito Federal (FAPDF), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento e Tecnológico (CNPq), and Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT), Brazil. C.d.l.F-N. holds a Presidential Professorship at the University of Pennsylvania, is a recipient of the Langer Prize by the AIChE Foundation, and acknowledges funding from the Institute for Diabetes, Obesity, and Metabolism, the Penn Mental Health AIDS Research Center of the University of Pennsylvania, and the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM138201.
The authors declare no competing financial interest.
References
- Vert M.; Doi Y.; Hellwich K.-H.; Hess M.; Hodge P.; Kubisa P.; Rinaudo M.; Schué F. (2012) Terminology for Biorelated Polymers and Applications (IUPAC Recommendations 2012). Pure Appl. Chem. 84, 377–410. 10.1351/PAC-REC-10-12-04. [DOI] [Google Scholar]
- de la Fuente-Núñez C.; Reffuveille F.; Fernández L.; Hancock R. E. W. (2013) Bacterial Biofilm Development as a Multicellular Adaptation: Antibiotic Resistance and New Therapeutic Strategies. Curr. Opin. Microbiol. 16, 580–589. 10.1016/j.mib.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Dieltjens L.; Appermans K.; Lissens M.; Lories B.; Kim W.; Van der Eycken E. V.; Foster K. R.; Steenackers H. P. (2020) Inhibiting Bacterial Cooperation Is an Evolutionarily Robust Anti-Biofilm Strategy. Nat. Commun. 11, 107. 10.1038/s41467-019-13660-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed M. N.; Abdelsamad A.; Wassermann T.; Porse A.; Becker J.; Sommer M. O. A.; Høiby N.; Ciofu O. (2020) The Evolutionary Trajectories of P. aeruginosa in Biofilm and Planktonic Growth Modes Exposed to Ciprofloxacin: Beyond Selection of Antibiotic Resistance. npj Biofilms Microbi. 6, 28. 10.1038/s41522-020-00138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader S. M.; Vaubourgeix J.; Nathan C. (2020) Biology of Antimicrobial Resistance and Approaches to Combat It. Sci. Transl. Med. 12, eaaz6992. 10.1126/scitranslmed.aaz6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo H.; Allan R. N.; Howlin R. P.; Hall-Stoodley L.; Stoodley P. (2017) Targeting Microbial Biofilms: Current and Prospective Therapeutic Strategies. Nat. Rev. Microbiol. 15, 740–755. 10.1038/nrmicro.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderosa A. D.; Totsika M.; Fairfull-Smith K. E. (2019) Bacterial Biofilm Eradication Agents: A Current Review. Front. Chem. 7, 824. 10.3389/fchem.2019.00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes G.; Webber M. A. (2017) Novel Approaches to the Treatment of Bacterial Biofilm Infections. Br. J. Pharmacol. 174, 2237–2246. 10.1111/bph.13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Breij A.; Riool M.; Cordfunke R. A.; Malanovic N.; de Boer L.; Koning R. I.; Ravensbergen E.; Franken M.; van der Heijde T.; Boekema B. K.; Kwakman P. H. S.; Kamp N.; El Ghalbzouri A.; Lohner K.; Zaat S. A. J.; Drijfhout J. W.; Nibbering P. H. (2018) The Antimicrobial Peptide SAAP-148 Combats Drug-Resistant Bacteria and Biofilms. Sci. Transl. Med. 10, eaan4044. 10.1126/scitranslmed.aan4044. [DOI] [PubMed] [Google Scholar]
- Dostert M.; Belanger C. R.; Hancock R. E. W. (2019) Design and Assessment of Anti-Biofilm Peptides: Steps Toward Clinical Application. J. Innate Immun. 11, 193–204. 10.1159/000491497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr C. G.; Ghimire J.; Guha S.; Hoffmann J. P.; Wang Y.; Sun L.; Landreneau B. N.; Kolansky Z. D.; Kilanowski-Doroh I. M.; Sammarco M. C.; Morici L. A.; Wimley W. C. (2020) Synthetic Molecular Evolution of Host Cell-Compatible, Antimicrobial Peptides Effective against Drug-Resistant, Biofilm-Forming Bacteria. Proc. Natl. Acad. Sci. U. S. A. 117, 8437–8448. 10.1073/pnas.1918427117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletzer D.; Hancock R. E. W. (2016) Antibiofilm Peptides: Potential as Broad-Spectrum Agents. J. Bacteriol. 198, 2572–2578. 10.1128/JB.00017-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Somma A.; Moretta A.; Canè C.; Cirillo A.; Duilio A. (2020) Antimicrobial and Antibiofilm Peptides. Biomolecules 10, 652. 10.3390/biom10040652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasir M.; Willcox M. D. P.; Dutta D. (2018) Action of Antimicrobial Peptides against Bacterial Biofilms. Materials 11, 2468. 10.3390/ma11122468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K.; Zendo T.; Sugimoto S.; Iwase T.; Tajima A.; Yamada S.; Sonomoto K.; Mizunoe Y. (2013) Effects of Bacteriocins on Methicillin-Resistant Staphylococcus aureus Biofilm. Antimicrob. Agents Chemother. 57, 5572–5579. 10.1128/AAC.00888-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Núñez C.; Korolik V.; Bains M.; Nguyen U.; Breidenstein E. B. M.; Horsman S.; Lewenza S.; Burrows L.; Hancock R. E. W. (2012) Inhibition of Bacterial Biofilm Formation and Swarming Motility by a Small Synthetic Cationic Peptide. Antimicrob. Agents Chemother. 56, 2696–2704. 10.1128/AAC.00064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libardo M. D. J.; Bahar A. A.; Ma B.; Fu R.; McCormick L. E.; Zhao J.; McCallum S. A.; Nussinov R.; Ren D.; Angeles-Boza A. M.; Cotten M. L. (2017) Nuclease Activity Gives an Edge to Host-Defense Peptide Piscidin 3 over Piscidin 1, Rendering It More Effective against Persisters and Biofilms. FEBS J. 284, 3662–3683. 10.1111/febs.14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Núñez C.; Reffuveille F.; Haney E. F.; Straus S. K.; Hancock R. E. W. (2014) Broad-Spectrum Anti-Biofilm Peptide That Targets a Cellular Stress Response. PLoS Pathog. 10, e1004152. 10.1371/journal.ppat.1004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenye T.; Nelis H. J. (2010) In vitro and In vivo Model Systems to Study Microbial Biofilm Formation. J. Microbiol. Methods 83, 89–105. 10.1016/j.mimet.2010.08.018. [DOI] [PubMed] [Google Scholar]
- Wu Y.-K.; Cheng N.-C.; Cheng C. M. (2019) Biofilms in Chronic Wounds: Pathogenesis and Diagnosis. Trends Biotechnol. 37, 505–517. 10.1016/j.tibtech.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Nakagami G.; Sanada H.; Sugama J.; Morohoshi T.; Ikeda T.; Ohta Y. (2008) Detection of Pseudomonas aeruginosa Quorum Sensing Signals in an Infected Ischemic Wound: An Experimental Study in Rats. Wound Repair Regen. 16, 30–36. 10.1111/j.1524-475X.2007.00329.x. [DOI] [PubMed] [Google Scholar]
- Pletzer D.; Mansour S. C.; Wuerth K.; Rahanjam N.; Hancock R. E. W. (2017) New Mouse Model for Chronic Infections by Gram-Negative Bacteria Enabling the Study of Anti-Infective Efficacy and Host-Microbe Interactions. mBio 8, e00140. 10.1128/mBio.00140-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E. M. C.; Dean S. N.; Propst C. N.; Bishop B. M.; van Hoek M. L. (2017) Komodo Dragon-Inspired Synthetic Peptide DRGN-1 Promotes Wound-Healing of a Mixed-Biofilm Infected Wound. npj Biofilms Microbiomes 3, 9. 10.1038/s41522-017-0017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen T. R.; Aasholm M. S.; Rudkjøbing V. B.; Saunders A. M.; Bjarnsholt T.; Givskov M.; Kirketerp-Møller K.; Nielsen P. H. (2010) The Bacteriology of Chronic Venous Leg Ulcer Examined by Culture-Independent Molecular Methods. Wound Repair Regen. 18, 38–49. 10.1111/j.1524-475X.2009.00561.x. [DOI] [PubMed] [Google Scholar]
- Roberts A. E. L.; Kragh K. N.; Bjarnsholt T.; Diggle S. P. (2015) The Limitations of in vitro Experimentation in Understanding Biofilms and Chronic Infection. J. Mol. Biol. 427, 3646–3661. 10.1016/j.jmb.2015.09.002. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Han J.; Chang B.; Gao L.; Lu Z.; Lu F.; Zhao H.; Zhang C.; Bie X. (2017) Membrane-Active Amphipathic Peptide WRL3 with in vitro Antibiofilm Capability and in vivo Efficacy in Treating Methicillin-Resistant Staphylococcus aureus Burn Wound Infections. ACS Infect. Dis. 3, 820–832. 10.1021/acsinfecdis.7b00100. [DOI] [PubMed] [Google Scholar]
- Woodburn K. W.; Jaynes J. M.; Clemens L. E. (2019) Evaluation of the Antimicrobial Peptide, RP557, for the Broad-Spectrum Treatment of Wound Pathogens and Biofilm. Front. Microbiol. 10, 1688. 10.3389/fmicb.2019.01688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M. D. T.; Pedron C. N.; Higashikuni Y.; Kramer R. M.; Cardoso M. H.; Oshiro K. G. N.; Franco O. L.; Junior P. I. S.; Silva F. D.; Junior V. X. O.; Lu T. K.; Fuente-Nunez C. de la. (2018) Structure-Function-Guided Exploration of the Antimicrobial Peptide Polybia-CP Identifies Activity Determinants and Generates Synthetic Therapeutic Candidates. Commun. Biol. 1, 221. 10.1038/s42003-018-0224-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro K. G. N.; Cândido E. S.; Chan L. Y.; Torres M. D. T.; Monges B. E. D.; Rodrigues S. G.; Porto W. F.; Ribeiro S. M.; Henriques S. T.; Lu T. K.; de la Fuente-Nunez C.; Craik D. J.; Franco O. L.; Cardoso M. H. (2019) Computer-Aided Design of Mastoparan-like Peptides Enables the Generation of Nontoxic Variants with Extended Antibacterial Properties. J. Med. Chem. 62, 8140–8151. 10.1021/acs.jmedchem.9b00915. [DOI] [PubMed] [Google Scholar]
- Pane K.; Cafaro V.; Avitabile A.; Torres M. D. T.; Vollaro A.; De Gregorio E.; Catania M. R.; Di Maro A.; Bosso A.; Gallo G.; Zanfardino A.; Varcamonti M.; Pizzo E.; Di Donato A.; Lu T. K.; de la Fuente-Nunez C.; Notomista E. (2018) Identification of Novel Cryptic Multifunctional Antimicrobial Peptides from the Human Stomach Enabled by a Computational–Experimental Platform. ACS Synth. Biol. 7, 2105–2115. 10.1021/acssynbio.8b00084. [DOI] [PubMed] [Google Scholar]
- Cardoso M. H.; Cândido E. S.; Chan L. Y.; Der Torossian Torres M.; Oshiro K. G. N.; Rezende S. B.; Porto W. F.; Lu T. K.; de la Fuente-Nunez C.; Craik D. J.; Franco O. L. A (2018) Computationally Designed Peptide Derived from Escherichia coli as a Potential Drug Template for Antibacterial and Antibiofilm Therapies. ACS Infect. Dis. 4, 1727–1736. 10.1021/acsinfecdis.8b00219. [DOI] [PubMed] [Google Scholar]
- Cândido E. S.; Cardoso M. H.; Chan L. Y.; Torres M. D. T.; Oshiro K. G. N.; Porto W. F.; Ribeiro S. M.; Haney E. F.; Hancock R. E. W.; Lu T. K.; de la Fuente-Nunez C.; Craik D. J.; Franco O. L. (2019) Short Cationic Peptide Derived from Archaea with Dual Antibacterial Properties and Anti-Infective Potential. ACS Infect. Dis. 5, 1081–1086. 10.1021/acsinfecdis.9b00073. [DOI] [PubMed] [Google Scholar]
- Schierle C. F.; De la Garza M.; Mustoe T. A.; Galiano R. D. (2009) Staphylococcal Biofilms Impair Wound Healing by Delaying Reepithelialization in a Murine Cutaneous Wound Model. Wound Repair Regen. 17, 354–359. 10.1111/j.1524-475X.2009.00489.x. [DOI] [PubMed] [Google Scholar]
- Mansour S. C.; Pletzer D.; de la Fuente-Núñez C.; Kim P.; Cheung G. Y. C.; Joo H.-S.; Otto M.; Hancock R. E. W. (2016) Bacterial Abscess Formation Is Controlled by the Stringent Stress Response and Can Be Targeted Therapeutically. EBioMedicine 12, 219–226. 10.1016/j.ebiom.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kłodzińska S. N.; Pletzer D.; Rahanjam N.; Rades T.; Hancock R. E. W.; Nielsen H. M. (2019) Hyaluronic Acid-Based Nanogels Improve in vivo Compatibility of the Anti-Biofilm Peptide DJK-5. Nanomedicine 20, 102022. 10.1016/j.nano.2019.102022. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Pletzer D.; Haney E. F.; Rahanjam N.; Cheng J. T. J.; Yue M.; Aljehani W.; Hancock R. E. W.; Kizhakkedathu J. N.; Straus S. K. (2019) Aurein-Derived Antimicrobial Peptides Formulated with Pegylated Phospholipid Micelles to Target Methicillin-Resistant Staphylococcus aureus Skin Infections. ACS Infect. Dis. 5, 443–453. 10.1021/acsinfecdis.8b00319. [DOI] [PubMed] [Google Scholar]
- Pletzer D.; Mansour S. C.; Hancock R. E. W. (2018) Synergy between Conventional Antibiotics and Anti-Biofilm Peptides in a Murine, Sub-Cutaneous Abscess Model Caused by Recalcitrant ESKAPE Pathogens. PLoS Pathog. 14, e1007084. 10.1371/journal.ppat.1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaux D.; Chauhan A.; Rendueles O.; Beloin C. (2013) From in vitro to in vivo Models of Bacterial Biofilm-Related Infections. Pathogens 2, 288–356. 10.3390/pathogens2020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Moser C.; Wang H. Z.; Høiby N.; Song Z. J. (2015) Strategies for Combating Bacterial Biofilm Infections. Int. J. Oral Sci. 7, 1–7. 10.1038/ijos.2014.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatoon Z.; McTiernan C. D.; Suuronen E. J.; Mah T. F.; Alarcon E. I. (2018) Bacterial Biofilm Formation on Implantable Devices and Approaches to Its Treatment and Prevention. Heliyon 4, e01067. 10.1016/j.heliyon.2018.e01067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart P. S.; Bjarnsholt T. (2020) Risk Factors for Chronic Biofilm-Related Infection Associated with Implanted Medical Devices. Clin. Microbiol. Infect. 26, 1034–1038. 10.1016/j.cmi.2020.02.027. [DOI] [PubMed] [Google Scholar]
- Cirioni O.; Giacometti A.; Ghiselli R.; Kamysz W.; Orlando F.; Mocchegiani F.; Silvestri C.; Licci A.; Chiodi L.; Lukasiak J.; Saba V.; Scalise G. (2006) Citropin 1.1-Treated Central Venous Catheters Improve the Efficacy of Hydrophobic Antibiotics in the Treatment of Experimental Staphylococcal Catheter-Related Infection. Peptides 27, 1210–1216. 10.1016/j.peptides.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Cirioni O.; Ghiselli R.; Minardi D.; Orlando F.; Mocchegiani F.; Silvestri C.; Muzzonigro G.; Saba V.; Scalise G.; Balaban N.; Giacometti A. (2007) RNAIII-Inhibiting Peptide Affects Biofilm Formation in a Rat Model of Staphylococcal Ureteral Stent Infection. Antimicrob. Agents Chemother. 51, 4518–4520. 10.1128/AAC.00808-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gominet M.; Compain F.; Beloin C.; Lebeaux D. (2017) Central Venous Catheters and Biofilms: Where Do We Stand in 2017?. APMIS 125, 365–375. 10.1111/apm.12665. [DOI] [PubMed] [Google Scholar]
- Danese P. N. (2002) Antibiofilm Approaches: Prevention of Catheter Colonization. Chem. Biol. 9, 873–880. 10.1016/S1074-5521(02)00192-8. [DOI] [PubMed] [Google Scholar]
- Cirioni O.; Giacometti A.; Ghiselli R.; Bergnach C.; Orlando F.; Mocchegiani F.; Silvestri C.; Licci A.; Skerlavaj B.; Zanetti M.; Saba V.; Scalise G. (2006) Pre-Treatment of Central Venous Catheters with the Cathelicidin BMAP-28 Enhances the Efficacy of Antistaphylococcal Agents in the Treatment of Experimental Catheter-Related Infection. Peptides 27, 2104–2110. 10.1016/j.peptides.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Ghiselli R.; Giacometti A.; Cirioni O.; Mocchegiani F.; Silvestri C.; Orlando F.; Kamysz W.; Licci A.; Nadolski P.; Della Vittoria A.; Łukasiak J.; Scalise G.; Saba V. (2007) Pretreatment with the Protegrin IB-367 Affects Gram-Positive Biofilm and Enhances the Therapeutic Efficacy of Linezolid in Animal Models of Central Venous Catheter Infection. JPEN, J. Parenter. Enteral Nutr. 31, 463–468. 10.1177/0148607107031006463. [DOI] [PubMed] [Google Scholar]
- Zmistowski B.; Karam J. A.; Durinka J. B.; Casper D. S.; Parvizi J. (2013) Periprosthetic Joint Infection Increases the Risk of One-Year Mortality. J. Bone Joint Surg. Am. 95, 2177–2184. 10.2106/JBJS.L.00789. [DOI] [PubMed] [Google Scholar]
- Mandell J. B.; Deslouches B.; Montelaro R. C.; Shanks R. M. Q.; Doi Y.; Urish K. L. (2017) Elimination of Antibiotic Resistant Surgical Implant Biofilms Using an Engineered Cationic Amphipathic Peptide WLBU2. Sci. Rep. 7, 18098. 10.1038/s41598-017-17780-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minardi D.; Ghiselli R.; Cirioni O.; Giacometti A.; Kamysz W.; Orlando F.; Silvestri C.; Parri G.; Kamysz E.; Scalise G.; Saba V.; Giovanni M. (2007) The Antimicrobial Peptide Tachyplesin III Coated Alone and in Combination with Intraperitoneal Piperacillin-Tazobactam Prevents Ureteral Stent Pseudomonas Infection in a Rat Subcutaneous Pouch Model. Peptides 28, 2293–2298. 10.1016/j.peptides.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chen R.; Willcox M. D. P.; Ho K. K. K.; Smyth D.; Kumar N. (2016) Antimicrobial Peptide Melimine Coating for Titanium and Its in vivo Antibacterial Activity in Rodent Subcutaneous Infection Models. Biomaterials 85, 142–151. 10.1016/j.biomaterials.2016.01.063. [DOI] [PubMed] [Google Scholar]
- Yu Q.; Deng T.; Lin F.-C.; Zhang B.; Zink J. I. (2020) Supramolecular Assemblies of Heterogeneous Mesoporous Silica Nanoparticles to Co-Deliver Antimicrobial Peptides and Antibiotics for Synergistic Eradication of Pathogenic Biofilms. ACS Nano 14, 5926–5937. 10.1021/acsnano.0c01336. [DOI] [PubMed] [Google Scholar]
- Ma L.; Ye X.; Sun P.; Xu P.; Wang L.; Liu Z.; Huang X.; Bai Z.; Zhou C. (2020) Antimicrobial and Antibiofilm Activity of the EeCentrocin 1 Derived Peptide EC1–17KV via Membrane Disruption. EBioMedicine 55, 102775. 10.1016/j.ebiom.2020.102775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-K.; Mereuta L.; Luchian T.; Park Y. (2019) Antimicrobial Peptide HPA3NT3-A2 Effectively Inhibits Biofilm Formation in Mice Infected with Drug-Resistant Bacteria. Biomater. Sci. 7, 5068–5083. 10.1039/C9BM01051C. [DOI] [PubMed] [Google Scholar]
- Narayana J. L.; Mishra B.; Lushnikova T.; Golla R. M.; Wang G. (2019) Modulation of Antimicrobial Potency of Human Cathelicidin Peptides against the ESKAPE Pathogens and in vivo Efficacy in a Murine Catheter-Associated Biofilm Model. Biochim. Biophys. Acta, Biomembr. 1861, 1592–1602. 10.1016/j.bbamem.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaban N.; Gov Y.; Giacometti A.; Cirioni O.; Ghiselli R.; Mocchegiani F.; Orlando F.; D’Amato G.; Saba V.; Scalise G.; Bernes S.; Mor A. (2004) A Chimeric Peptide Composed of a Dermaseptin Derivative and an RNA III-Inhibiting Peptide Prevents Graft-Associated Infections by Antibiotic-Resistant Staphylococci. Antimicrob. Agents Chemother. 48, 2544–2550. 10.1128/AAC.48.7.2544-2550.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X.; Chen J.; Zhou M.; Zhu M.; Li Z.; Gao S.; Ou J.; Xu D.; Wu M.; Jiang S.; Hu Y.; Tian Y.; Niu Z. (2020) Combating Pseudomonas Aeruginosa Biofilms by a Chitosan-PEG-Peptide Conjugate via Changes in Assembled Structure. ACS Appl. Mater. Interfaces 12, 13731–13738. 10.1021/acsami.0c02034. [DOI] [PubMed] [Google Scholar]
- Stefani S.; Campana S.; Cariani L.; Carnovale V.; Colombo C.; Lleo M. M.; Iula V. D.; Minicucci L.; Morelli P.; Pizzamiglio G.; Taccetti G. (2017) Relevance of Multidrug-Resistant Pseudomonas aeruginosa Infections in Cystic Fibrosis. Int. J. Med. Microbiol. 307, 353–362. 10.1016/j.ijmm.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Ciofu O.; Tolker-Nielsen T.; Jensen P. Ø.; Wang H.; Høiby N. (2015) Antimicrobial Resistance, Respiratory Tract Infections and Role of Biofilms in Lung Infections in Cystic Fibrosis Patients. Adv. Drug Delivery Rev. 85, 7–23. 10.1016/j.addr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Parente J.; Harris S. M.; Woods D. E.; Hancock R. E. W.; Falla T. J. (2005) Antimicrobial Peptide Therapeutics for Cystic Fibrosis. Antimicrob. Agents Chemother. 49, 2921–2927. 10.1128/AAC.49.7.2921-2927.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardirossian M.; Pompilio A.; Crocetta V.; De Nicola S.; Guida F.; Degasperi M.; Gennaro R.; Di Bonaventura G.; Scocchi M. (2016) In vitro and in vivo Evaluation of BMAP-Derived Peptides for the Treatment of Cystic Fibrosis-Related Pulmonary Infections. Amino Acids 48, 2253–2260. 10.1007/s00726-016-2266-4. [DOI] [PubMed] [Google Scholar]
- Harrington N. E.; Sweeney E.; Harrison F. (2020) Building a Better Biofilm - Formation of in vivo-like Biofilm Structures by Pseudomonas aeruginosa in a Porcine Model of Cystic Fibrosis Lung Infection. Biofilm 2, 100024. 10.1016/j.bioflm.2020.100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z.; Wu H.; Mygind P.; Raventos D.; Sonksen C.; Kristensen H.-H.; Høiby N. (2005) Effects of Intratracheal Administration of Novispirin G10 on a Rat Model of Mucoid Pseudomonas aeruginosa Lung Infection. Antimicrob. Agents Chemother. 49, 3868–3874. 10.1128/AAC.49.9.3868-3874.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes H. K.; Ritchie N.; Irvine S.; Evans T. J. A (2016) Murine Model of Early Pseudomonas aeruginosa Lung Disease with Transition to Chronic Infection. Sci. Rep. 6, 35838. 10.1038/srep35838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez M.; Polizzotto A.; Flores N.; Semorile L.; Maffía P. C. (2020) Antibacterial, Anti-Biofilm and in vivo Activities of the Antimicrobial Peptides P5 and P6.2. Microb. Pathog. 139, 103886. 10.1016/j.micpath.2019.103886. [DOI] [PubMed] [Google Scholar]
- Mwangi J.; Yin Y.; Wang G.; Yang M.; Li Y.; Zhang Z.; Lai R. (2019) The Antimicrobial Peptide ZY4 Combats Multidrug-Resistant Pseudomonas aeruginosa and Acinetobacter baumannii Infection. Proc. Natl. Acad. Sci. U. S. A. 116, 26516–26522. 10.1073/pnas.1909585117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Mangoni M. L.; Di Y. P. (2017) In vivo Therapeutic Efficacy of Frog Skin-Derived Peptides Against Pseudomonas aeruginosa-Induced Pulmonary Infection. Sci. Rep. 7, 8548. 10.1038/s41598-017-08361-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Y. P.; Lin Q.; Chen C.; Montelaro R. C.; Doi Y.; Deslouches B. (2020) Enhanced Therapeutic Index of an Antimicrobial Peptide in Mice by Increasing Safety and Activity against Multidrug-Resistant Bacteria. Sci. Adv. 6, eaay6817. 10.1126/sciadv.aay6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C. N.; Clegg S. (2012) Klebsiella pneumoniae and Type 3 Fimbriae: Nosocomial Infection, Regulation and Biofilm Formation. Future Microbiol. 7, 991–1002. 10.2217/fmb.12.74. [DOI] [PubMed] [Google Scholar]
- Nair G. B.; Niederman M. S. (2015) Ventilator-Associated Pneumonia: Present Understanding and Ongoing Debates. Intensive Care Med. 41, 34–48. 10.1007/s00134-014-3564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilhen C.; Miquel S.; Charbonnel N.; Joseph L.; Carrier G.; Forestier C.; Balestrino D. (2019) Colonization and Immune Modulation Properties of Klebsiella pneumoniae Biofilm-Dispersed Cells. npj Biofilms Microbiomes 5, 25. 10.1038/s41522-019-0098-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S.; Gan C.; Li R.; Ye Y.; Zhang S.; Wu X.; Yang Y. Y.; Fan W.; Wu M. (2015) A Novel Chemosynthetic Peptide with β-Sheet Motif Efficiently Kills Klebsiella pneumoniae in a Mouse Model. Int. J. Nanomed. 10, 1045–1059. 10.2147/IJN.S73303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen W. H.; Burne R. A.; Wu H.; Koo H. (2018) Oral Biofilms: Pathogens, Matrix, and Polymicrobial Interactions in Microenvironments. Trends Microbiol. 26, 229–242. 10.1016/j.tim.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Wang Z.; Hancock R. E. W.; de la Fuente-Núñez C.; Haapasalo M. (2016) Treatment of Oral Biofilms by a D-Enantiomeric Peptide. PLoS One 11, e0166997. 10.1371/journal.pone.0166997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashper S. G.; Liu S. W.; Reynolds E. C. (2007) Antimicrobial Peptides and Their Potential as Oral Therapeutic Agents. Int. J. Pept. Res. Ther. 13, 505–516. 10.1007/s10989-007-9094-z. [DOI] [Google Scholar]
- Kim D.; Liu Y.; Benhamou R. I.; Sanchez H.; Simón-Soro Á.; Li Y.; Hwang G.; Fridman M.; Andes D. R.; Koo H. (2018) Bacterial-Derived Exopolysaccharides Enhance Antifungal Drug Tolerance in a Cross-Kingdom Oral Biofilm. ISME J. 12, 1427–1442. 10.1038/s41396-018-0113-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen W. H. (2013) Rodent Model in Caries Research. Odontology 101, 9–14. 10.1007/s10266-012-0091-0. [DOI] [PubMed] [Google Scholar]
- Liang J.; Liang D.; Liang Y.; He J.; Zuo S.; Zhao W. (2021) Effects of a Derivative of Reutericin 6 and Gassericin A on the Biofilm of Streptococcus mutans in vitro and Caries Prevention in vivo. Odontology 109, 53. 10.1007/s10266-020-00529-5. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang X.; Jiang W.; Wang K.; Luo J.; Li W.; Zhou X.; Zhang L. (2018) Antimicrobial Peptide GH12 Suppresses Cariogenic Virulence Factors of Streptococcus mutans. J. Oral Microbiol. 10, 1442089. 10.1080/20002297.2018.1442089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Fan Y.; Zhou Z.; Tu H.; Ren Q.; Wang X.; Ding L.; Zhou X.; Zhang L. (2017) De novo Synthetic Short Antimicrobial Peptides against Cariogenic Bacteria. Arch. Oral Biol. 80, 41–50. 10.1016/j.archoralbio.2017.03.017. [DOI] [PubMed] [Google Scholar]
- Tu H.; Fan Y.; Lv X.; Han S.; Zhou X.; Zhang L. (2016) Activity of Synthetic Antimicrobial Peptide GH12 against Oral Streptococci. Caries Res. 50, 48–61. 10.1159/000442898. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zeng Y.; Wang Y.; Li H.; Yu S.; Jiang W.; Li Y.; Zhang L. (2019) Antimicrobial Peptide GH12 Targets Streptococcus mutans to Arrest Caries Development in Rats. J. Oral Microbiol. 11, 1549921. 10.1080/20002297.2018.1549921. [DOI] [Google Scholar]
- Jiang W.; Wang Y.; Luo J.; Chen X.; Zeng Y.; Li X.; Feng Z.; Zhang L. (2020) Antimicrobial Peptide GH12 Prevents Dental Caries by Regulating Dental Plaque Microbiota. Appl. Environ. Microbiol. 86, e00527. 10.1128/AEM.00527-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daep C. A.; James D. M.; Lamont R. J.; Demuth D. R. (2006) Structural Characterization of Peptide-Mediated Inhibition of Porphyromonas gingivalis Biofilm Formation. Infect. Immun. 74, 5756–5762. 10.1128/IAI.00813-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daep C. A.; Novak E. A.; Lamont R. J.; Demuth D. R. (2011) Structural Dissection and in vivo Effectiveness of a Peptide Inhibitor of Porphyromonas gingivalis Adherence to Streptococcus gordonii. Infect. Immun. 79, 67–74. 10.1128/IAI.00361-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont R. J.; El-Sabaeny A.; Park Y.; Cook G. S.; Costerton J. W.; Demuth D. R. (2002) Role of the Streptococcus gordonii SspB Protein in the Development of Porphyromonas gingivalis Biofilms on Streptococcal Substrates. Microbiology 148, 1627–1636. 10.1099/00221287-148-6-1627. [DOI] [PubMed] [Google Scholar]
- Chopra A.; Bhat S. G.; Sivaraman K. (2020) Porphyromonas gingivalis Adopts Intricate and Unique Molecular Mechanisms to Survive and Persist within the Host: A Critical Update. J. Oral Microbiol. 12, 1801090. 10.1080/20002297.2020.1801090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker P. J.; Dixon M.; Roopenian D. C. (2000) Genetic Control of Susceptibility to Porphyromonas gingivalis-Induced Alveolar Bone Loss in Mice. Infect. Immun. 68, 5864–5868. 10.1128/IAI.68.10.5864-5868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud M. Y.; Steinbach-Rankins J. M.; Demuth D. R. (2019) Functional Assessment of Peptide-Modified PLGA Nanoparticles against Oral Biofilms in a Murine Model of Periodontitis. J. Controlled Release 297, 3–13. 10.1016/j.jconrel.2019.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.-Y.; Tu C. H.; Yip B.-S.; Chen H.-L.; Cheng H.-T.; Huang K.-C.; Lo H.-J.; Cheng J. W. (2011) Easy Strategy to Increase Salt Resistance of Antimicrobial Peptides. Antimicrob. Agents Chemother. 55, 4918–4921. 10.1128/AAC.00202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-Y.; Cheng J.-W.; Yu H.-Y.; Lin L.; Chih Y.-H.; Pan Y.-P. (2015) Efficacy of a Novel Antimicrobial Peptide against Periodontal Pathogens in Both Planktonic and Polymicrobial Biofilm States. Acta Biomater. 25, 150–161. 10.1016/j.actbio.2015.07.031. [DOI] [PubMed] [Google Scholar]
- Wang H.; Lin L.; Fu W.; Yu H.-Y.; Yu N.; Tan L.; Cheng J.; Pan Y. (2017) Preventive Effects of the Novel Antimicrobial Peptide Nal-P-113 in a Rat Periodontitis Model by Limiting the Growth of Porphyromonas gingivalis and Modulating IL-1β and TNF-α Production. BMC Complementary Altern. Med. 17, 426. 10.1186/s12906-017-1931-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Ai L.; Zhang Y.; Cheng J.; Yu H.; Li C.; Zhang D.; Pan Y.; Lin L. (2018) The Effects of Antimicrobial Peptide Nal-P-113 on Inhibiting Periodontal Pathogens and Improving Periodontal Status. BioMed Res. Int. 17, 1805793. 10.1155/2018/1805793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denayer T.; Stöhr T.; Van Roy M. (2017) Animal Models in Translational Medicine: Validation and Prediction. New Horiz. Transl. Med. 2, 5–11. 10.1016/j.nhtm.2014.08.001. [DOI] [Google Scholar]
- Rumbaugh K. P. (2020) How Well Are We Translating Biofilm Research from Bench-Side to Bedside?. Biofilm 2, 100028. 10.1016/j.bioflm.2020.100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer D. K.; Torres M. D. T.; Duay S. S.; Lovie E.; Simpson L.; von Köckritz-Blickwede M.; de la Fuente-Nunez C.; O’Neil D. A.; Angeles-Boza A. M. (2020) Antimicrobial Susceptibility Testing of Antimicrobial Peptides to Better Predict Efficacy. Front. Cell. Infect. Microbiol. 10, 326. 10.3389/fcimb.2020.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunniyi A. D.; Kopecki Z.; Hickey E. E.; Khazandi M.; Peel E.; Belov K.; Boileau A.; Garg S.; Venter H.; Chan W. Y.; Hill P. B.; Page S. W.; Cowin A. J.; Trott D. J. (2018) Bioluminescent Murine Models of Bacterial Sepsis and Scald Wound Infections for Antimicrobial Efficacy Testing. PLoS One 13, e0200195. 10.1371/journal.pone.0200195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard W. L.; Mosich G. M.; Smith R. A.; Hamad C. D.; Park H. Y.; Zoller S. D.; Trikha R.; McCoy T. K.; Borthwell R.; Hoang J.; Truong N.; Cevallos N.; Clarkson S.; Hori K. R.; van Dijl J. M.; Francis K. P.; Petrigliano F. A.; Bernthal N. M. (2020) Novel in vivo Mouse Model of Shoulder Implant Infection. J. Shoulder and Elbow Surg. 29, 1412–1424. 10.1016/j.jse.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allesen-Holm M.; Barken K. B.; Yang L.; Klausen M.; Webb J. S.; Kjelleberg S.; Molin S.; Givskov M.; Tolker-Nielsen T. A. (2006) Characterization of DNA Release in Pseudomonas aeruginosa Cultures and Biofilms. Mol. Microbiol. 59, 1114–1128. 10.1111/j.1365-2958.2005.05008.x. [DOI] [PubMed] [Google Scholar]
- Jung C.-J.; Hsu R.-B.; Shun C.-T.; Hsu C.-C.; Chia J.-S. (2017) AtlA Mediates Extracellular DNA Release, Which Contributes to Streptococcus mutans Biofilm Formation in an Experimental Rat Model of Infective Endocarditis. Infect. Immun. 85, e00252. 10.1128/IAI.00252-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover M. S.; Mishra M.; Deora R. (2011) Extracellular DNA Is Essential for Maintaining Bordetella Biofilm Integrity on Abiotic Surfaces and in the Upper Respiratory Tract of Mice. PLoS One 6, e16861. 10.1371/journal.pone.0016861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhede M.; Alhede M.; Qvortrup K.; Kragh K. N.; Jensen P. Ø.; Stewart P. S.; Bjarnsholt T. (2020) The origin of extracellular DNA in bacterial biofilm infections. Pathog. Dis. 78, ftaa018. 10.1093/femspd/ftaa018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce-Angulo D. G.; Bautista-Hernández L. A.; Calvillo-Medina R. P.; Castro-Tecorral F. I.; Aparicio-Ozores G.; López-Villegas E. O.; Ribas-Aparicio R. M.; Bautista-de Lucio V. M. (2020) Microscopic Characterization of Biofilm in Mixed Keratitis in a Novel Murine Model. Microb. Pathog. 140, 103953. 10.1016/j.micpath.2019.103953. [DOI] [PubMed] [Google Scholar]
- Doke S. K.; Dhawale S. C. (2015) Alternatives to Animal Testing: A Review. Saudi Pharm. J. 23, 223–229. 10.1016/j.jsps.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]