

Abstract
Previously, we identified a mechanism of inflammation control directed by ribosomal protein L13a and “GAIT” (Gamma Activated Inhibitor of Translation) elements in target mRNAs and showed that its elimination in myeloid cell-specific L13a knockout mice (L13a KO) increased atherosclerosis susceptibility and severity. Here, we investigated the mechanistic basis of this endogenous defense against atherosclerosis. We compared molecular and cellular aspects of atherosclerosis in high-fat diet (HFD)-fed L13a KO and intact (control) mice. HFD treatment of control mice induced release of L13a from 60S ribosome, formation of RNA-binding complex, and subsequent GAIT element-mediated translational silencing. Atherosclerotic plaques from HFD-treated KO mice showed increased infiltration of M1 type inflammatory macrophages. Macrophages from KO mice showed increased phagocytic activity and elevated expression of LDL receptor and pro-inflammatory mediators. NanoString analysis of the plaques from KO mice showed upregulation of a number of mRNAs encoding inflammatory proteins. Bioinformatics analysis suggests the presence of the potential GAIT elements in the 3′UTRs of several of these mRNAs. Macrophage induces L13a/GAIT-dependent translational silencing of inflammatory genes in response to HFD as an endogenous defense against atherosclerosis in ApoE−/− model.
Keywords: atherosclerosis, GAIT, inflammation, L13a, translational control
1 |. INTRODUCTION
Previous studies from our laboratory using cell1–9 and animal models10–12 identified and characterized a novel mechanism of inflammation control in macrophages and monocytes that is dependent on ribosomal protein L13a. This involves phosphorylation-dependent release of L13a from the 60S ribosomal subunit,3 after which it assembles into a multi-protein RNA-binding complex (the IFN-gamma-activated-inhibitor of translation or “GAIT” complex). The Gamma Activated Inhibitor of Translation (GAIT) complex contains four proteins: glutamyl-prolyl-tRNA synthetase (GluProRS), NS1-associated protein-1 (NSAP-1), glyc-eraldehyde 3-phosphate dehydrogenase (GAPDH), and ribosomal protein L13a.5 L13a as a part of GAIT complex interacts with eIF4G of the eIF4F complex there by inhibits translation initiation by blocking the recruitment of the 43S ribosomal subunit.6 Binding of the GAIT complex to GAIT elements4 present in the 3′ untranslated region (3’UTR) of target mRNAs (eg, CCL22, CXCL13, CCR4, CCL8, CCL21, CCR3, and CCR6)8 blocks translation initiation.6 Mice with myeloid cell-specific genetic deficiency (knockout, KO) of L13a showed more severe disease than controls in several models of inflammation-associated pathogenesis, including lipopolysaccharide (LPS)-induced endotoxemia,10 dextran sodium sulfate (DSS)-induced colitis,12 and high-fat diet (HFD)-induced atherosclerosis.11 Thus, ribosomal protein L13a plays a critical role in restraining macrophage inflammation, thereby providing an endogenous defense against many inflammatory diseases.
Atherosclerosis is caused by the build-up of plaque within arterial vessel walls. This process is driven by chronic inflammation as well as uncontrolled inflammation initiated from vascular leukocytes.13–15 Multiple studies suggest that resolving the sustained inflammation mediated by vascular macrophages16–18 may be a potential therapeutic avenue for cardiovascular diseases. The first clinical trial of canakinumab (a therapeutic monoclonal antibody targeting interleukin-1β) clearly demonstrated the potential of anti-inflammatory therapy against cardiovascular disease independent of lipid-lowering.19 Despite this proof-of-principle, additional work is needed to fully understand endogenous mechanisms of inflammation control to the extent that they can be exploited for therapeutic gain.
The focus of our laboratory in this area has been to characterize the molecular and cellular mediators of inflammation-suppressing L13a/GAIT element-directed translation silencing in macrophages. In this study, we examined this system in the context of atherosclerosis using myeloid cell-specific L13a KO mice (L13a Flox+/+ Cre+/+) bred onto the apolipoprotein E KO (ApoE−/−) background (referred to throughout as KO mice). HFD-induced atherosclerosis in these animals was compared to that in corresponding L13a-intact mice (L13a Flox+/+ ApoE−/−, referred to as control). Our previous work in this model showed that the KO mice were significantly more susceptible to development of atherosclerosis than control mice, perhaps due to loss of translational control of several chemokine and chemokine receptors.11 However, this study did not reveal the consequences of myeloid cell-specific L13a depletion on cells within atherosclerotic plaques or the molecular mechanisms underlying L13a-dependent translational control of target genes in vascular macrophages.
In the current work, we sought to address these questions through further detailed examination of KO vs control animals under atherosclerosis-promoting or normal conditions (HFD or chow feeding, respectively). We confirmed that HFD treatment induces both atherosclerosis and dissociation of L13a from the 60S ribosome, and that HFD-induced atherosclerosis is more severe in the absence of L13a expression in macrophages. Cells from KO mice were found to have a number of phenotypic changes that likely contribute to disease promotion. For example, plaque-resident macrophages showed greater M1 type pro-inflammatory polarization (vs M2 wound healing/anti-inflammatory polarization) in KO mice. High-throughput analysis of gene expression in laser microdissected plaques of HFD-fed mice identified a number of genes with elevated mRNA levels specifically in KO mice and potential GAIT elements in their 3′ UTRs. Finally, we showed that lysates of macrophages from HFD-treated control, but not KO, mice are capable of directing GAIT element-dependent translational silencing in a cell-free system. These results provide significant new insights into the molecular basis of L13a-dependent translation control as a physiological defense against atherosclerosis and highlight the potential for therapeutic targeting of this mechanism.
2 |. MATERIALS AND METHODS
2.1 |. Animals and animal treatments
The mice used for this study were L13a Flox+/+ apoE−/− (homozygous for the Floxed L13a gene on the apoE null background (L13a intact), referred to throughout this report as “control” mice) and L13a Flox+/+ Cre+/+ apoE−/− (homozygous for myeloid cell-specific L13a deletion on the apoE null background, referred to as knockout or “KO” mice). These mouse strains were generated and characterized in our laboratory as described previously.11 Animals were maintained in accordance with guidelines of the Institutional Animal Care and Use Committee (IACUC). Age- and sex-matched mice were used for all experiments. For “high-fat diet” (HFD) treatment, mice were fed a Western-type lipid-rich diet containing 0.2% of cholesterol and providing 42% of calories as fat (Catalog # TD88137, Harlan Teklad) for 14 weeks unless a different time period is indicated. As a control treatment and for general maintenance and propagation of mice, the animals were fed regular chow diet (considered low-fat).
2.2 |. Aortic atherosclerosis quantification
Age- and sex-matched KO and control mice were fed a HFD for 14 weeks and then euthanized according to the IACUC-approved protocols. After euthanization, hearts and aortic arches were removed from the body and bisected perpendicular to the heart axis just below the atrial tips and then embedded in Tissue-Tek OCT Compound (VWR Scientific). Sections 10 μm thick were collected starting from where the atrioventricular valves were visible. Lesion/plaque areas were visualized by staining with Oil red O for lipid content. The entire intimal lesion area in each section was manually traced and quantitated using ImageJ software (NIH) in a blinded fashion. The mean lesion area per section was determined.
2.3 |. Immunohistochemistry
After mice were euthanized, hearts were gently perfused through the left ventricle with cold phosphate-buffered saline (PBS). Perfused hearts were removed with about 1 mm of proximal aorta attached, and the portion distal to the tips of the auricles was excised and discarded. The remaining portion of the heart with the aortic root was immediately fixed in acid methanol (60% methanol and 10% acetic acid) for 24 hours before paraffin embedding. Five μm thick sections were processed for high-temperature antigen retrieval and paraffin removal using Trilogy reagent (Cell Marque) in a pressure cooker. Endogenous peroxidase activity was blocked by incubation with 0.3% of H2O2 in 80% of methanol, and nonspecific protein interactions were blocked by incubation with a serum-free protein block (Catalog # X090930–2, Agilent). The following antibodies were used for immunostaining: Rabbit polyclonal anti-alpha smooth muscle Actin antibody (ab5694, Abcam, 1:250 dilution), Rat monoclonal anti-Ly-6G (Catalog # ab25377, BioXCell, 1:50 dilution), Rat monoclonal anti-Mac2 (Catalog # CL8942AP, Cedarlane Laboratories, 1:1500 dilution); Rabbit polyclonal anti-iNOS antibody (Catalog # ab3523, Abcam, 1:250 dilution); Goat polyclonal anti-CD206 (Catalog # AF2535, R&D Systems, 1:250 dilution). For detection, Rat-on-Mouse HRP-Polymer Kit (Catalog # RT517H, Biocare Medical) or Goat-on-Rodent HRP-Polymer Kit (Catalog # GHP516H, Biocare Medical) was used. For quantification, at least three sections per animal were analyzed for each staining. Digital images were acquired using a Nikon Eclipse 55i microscope and a CCD camera (Nikon DS-U2) and analyzed with Image J software (National Institute of Health).
2.4 |. Preparation of aortic cell suspensions
For generation of single cell suspensions of aortic cells, aortas were dissected from euthanized mice and minced, then incubated with collagenase A (1 μg/mL) (Roche) for 2 hours at 37°C. The cell suspension was passed through a 70 μm cell strainer to remove undigested fragments and then centrifuged at 1000 g for 10 minutes at 4°C. After centrifugation, the cell pellet was recovered and washed twice with ice-cold PBS. Cells were then used for total RNA isolation by Trizol.
2.5 |. Preparation and phenotypic differentiation of bone marrow-derived macrophages
Femurs were excised from euthanized mice and the bone marrow was flushed into culture medium and dispersed. Bone marrow cells were cultured in complete RPMI 1640 medium containing 20% L929 cell conditioned medium for 10 days at 37°C in a humidified incubator with 5% of CO2. For experiments, bone marrow-derived macrophages (BMDM) were removed from differentiation dishes using cold sterile phosphate-buffered saline (PBS) and subsequently cultured in complete RPMI 1640 medium. After 10 days mature BMM was differentiated into M1 type inflammatory macrophages by incubation with 50 ng/mL of LPS from E coli O11:B4 (Catalog # L4391, Sigma-Aldrich) and 50 ng/mL recombinant mouse IFN-γ (Catalog # PMC4033, Invitrogen) for 24 hours.
2.6 |. Total RNA isolation
Total RNA was extracted from aortic cells or in vitro polarized macrophage cells using TRIzol (Invitrogen) according to the manufacturer’s protocol. After acid-phenol-chloroform extraction and isopropyl alcohol precipitation, the resulting RNA pellet was washed with ice-cold 80% of ethanol and redissolved in nuclease-free water. The concentration of the prepared RNA was determined by absorbance at 260 nm before storage at −70°C.
2.7 |. Analysis of gene expression by real-time quantitative RT-PCR
Total RNA was reverse transcribed into cDNA using a High-Capacity cDNA Reverse Transcription Kit (Catalog # 4368814, Applied Biosystems) according to the manufacturer’s instructions. Real-time quantitative PCR analysis was performed using SYBR Green PCR Master Mix (Catalog # 4309155, Applied Biosystems). Transcript levels were determined through the comparative CT method (ΔΔCT method) and normalized to GAPDH (mouse) transcript levels. All PCR reactions were performed in triplicate. All individual PCR reactions were performed four times (n = 4) in triplicate.
The following primers based on mouse sequences were used:
| Forward (sequence shown 5’ to 3’) | Reverse (sequence shown 5’ to 3’) | |
|---|---|---|
| Col1a1 | TGGAAACCCGAGGTATGCTT | CATTGCATTGCACGTCATCG |
| IL6 | GAGGATACCACTCCCAACAGACC | AAGTGCATCATCGTTGTTCATACA |
| iNOS | TTTGCTTCCATGCTAATGCGAAAG | GCTCTGTTGAGGTCTAAAGGCTCCG |
| TNFα | CCTGTAGCCCACGTCGTAGC | AGCAATGACTCCAAAGTAGACC |
| CD163 | TCCACACGTCCAGAACAGTC | CCTTGGAAACAGAGACAGGC |
| CD206 | GGTGTGGGCTCAGGTAGT | GTGGTGAGCTGAAAGGTGA |
| Arg1 | CAGAAGAATGGAAGAGTCAG | CAGATATGCAGGGAGTCACC |
| CCL2 | AACTGCATCTGCCCTAAGGT | GCTTGAGGTGGTTGTGGAAA |
| Cdkn1a | GGAACATCTCAGGGCCGAAA | CAATCTGCGCTTGGAGTGAT |
| Clec4n | CCAGCAGCTGAATGAGTCAC | TCGATCCATTGCCATTTGCC |
| Cxcl12 | GGAGGATAGATGTGCTCTGGAAC | AGTGAGGATGGAGACCGTGGTG |
| Eng | TAGCACCTTGTCCCAGGAAG | CAGTACAGAGGGCAGGACAA |
| Lcn2 | ATGTCACCTCCATCCTGGTCAG | GCCACTTGCACATTGTAGCTCTG |
| Pecam1 | GCAAGAAGCAGGAAGGACAG | ACTCTGACTGCAAGAGTGCT |
| S100a8 | TGGACATCAATAGTGACAATGC | GGCTGTCTTTGTGAGATGCC |
| Tap1 | TGGAGCCCACGATTTCATCT | GGTTCCCAGTCTCACCTACC |
| vwf | GCCAGTATGTTCTGGTGCAG | CCCTCATTTCCCACCAGGAT |
| GAPDH | TATGTCGTGGAGTCTACTGGT | GAGTTGTCATATTTCTCGT |
2.8 |. Flow cytometry
For flow cytometry, BMDM (106/sample) were first incubated with rat anti-mouse CD16/CD32 (BD Pharmingen) to block nonspecific binding of antibodies to FcγRs. The cells were then washed and stained first with PerCP Cy5.5-conjugated CD11b (BD Pharmingen). After this surface staining, cells were fixed, permeabilized, and stained for the intracellular antigen iNOS using PE-conjugated iNOS Monoclonal Antibody (eBioscience). PE-conjugated Rat IgG2a kappa (eBioscience) was used as an isotype control to exclude background staining. After antibody staining and washing, the cells were analyzed using a FACSCanto II flow cytometer (BD Biosciences). Data were collected for cells within gates set for leukocytes and were analyzed using FACSDiva (BD Biosciences) software. For sorting, cells were stained by FITC conjugated rat anti-mouse CD11b IgG2b (BD Pharmingen) and allophycocyanin-conjugated rat anti-mouse F4–80 IgG2b (Abd Serotec) and sorted by BD FACSAria II (BD Bioscience).
2.9 |. Native LDL uptake assay
BMDM were seeded at 30 000 cells/well in 96-well plates and incubated overnight at 37°C/5% of CO2. Cells were then treated with IFN-γ (500 U/mL) for 24 hours followed by incubation with DiI-labeled native LDL (15 μg/mL; Invitrogen, Catalog # L3482) for 4 hours at 37°C. To check specificity, cells were pretreated with 250 μg/mL unlabeled competitor (native LDL) for 1 hour. The cells were then washed with D-PBS and fluorescence was measured at 554/571 (Ex/Em) with a Victor2 multilabel counter (Perkin Elmer).
2.10 |. RNA EMSA
For EMSA experiments, 15 fmol of biotinylated CP GAIT RNA oilgo (5′-AAUGUUACUUUG GAAUGACUAUAAACAUU-3′) were incubated with macrophage cell lysates for 30 min on ice in a 20 μL reaction mixture containing 10 mM of HEPES (pH 7.3), 20 mM of KCl, 1 mM of MgCl2, 1 mM of DTT, 5% of glycerol, 10 μg yeast tRNA, and 40 U of RNasin (Promega). For competition experiments, 10- or 50-fold molar excess of unlabeled self or scrambled CP GAIT RNA oligos were added to the extract. RNA-protein complexes were separated on a 5% of native polyacrylamide gel. Complexes were transferred to a nylon membrane and detected by chemiluminescence using the Pierce Lightshift chemiluminescent RNA EMSA kit (Thermo Scientific, Catalog # 20158) following vendor’s protocol.
2.11 |. Phagocytosis assay
Phagocytosis was evaluated using the CytoSelect 96-Well Phagocytosis Assay (Cell Biolabs, San Diego, CA, USA, Catalog # CBA-224). BMDM were seeded at 50 000 cells/well in 96-well plates and incubated overnight at 37°C/5% of CO2. On the next day, cells were treated with IFN-γ (500 U/mL) for 4 hours. About 10 μL of Zymosan suspension was then added to each well and the plates were incubated for an additional 2 hours. Unbound particles were removed and phagocytosis was stopped by washing with cold serum-free medium. Internalized Zymosan particles were detected by a colorimetric method following the manufacturer’s protocol by measuring absorbance at 405 nm. To block phagocytosis, BMM were pretreated with Cytochalasin D for 1 hour at 37°C before addition of Zymosan particles.
2.12 |. Laser microdissection of plaques and nanostring nCounter gene expression analysis
Hearts were collected from euthanized mice and embedded in OCT compound. 10 μm serial sections from the aortic root area of the hearts were made as described above for “Aortic Atherosclerosis Quantification” under RNAse-free conditions. The sections were mounted onto PET metal-framed membrane slides (Leica, Buffalo Grove, IL) and prepared for laser microdissection (LMD) after staining with Arcturus Histogen solution (Life Technologies, Grand Island, NY). Using a Leica AS-LMD microscope, aortic plaque areas were located and marked for dissection. Cutout targets from aortic root sections were collected in a single cap containing RLT buffer (RNeasy Plus Micro kit, Qiagen). Total RNA was extracted from the plaque cutouts using the RNeasy Plus Micro Kit (Qiagen) per the manufacturer’s instructions. The concentration of total RNA was quantified by UV absorption at 260 nm using a NanoDrop 1000 spectrophotometer.
The NanoString nCounter gene expression system (NanoString Technologies, Seattle, WA) was used for expression profiling of mRNA species isolated from aortic plaques using the mouse specific nCounter® PanCancer Immune Profiling Panel which is composed of probes for 770 genes including 20 internal reference genes. Assays were performed according to the manufacturer’s instructions. Prior to analysis, the data were normalized using nSolver Analysis Software version 4.0 (NanoString Technologies) followed by background subtraction.
Gene expression profiling (NanoString) data were validated by quantitative real-time RT-PCR. RNA from laser microdissected plaques was first reverse transcribed to cDNA using the iScript Select cDNA Synthesis Kit (catalog # 170–8896, Biorad). The small quantities of cDNA were then preamplified by using PrimePCR Preamp Assays and SsoAdvanced Preamp Supermix (catalog # 172–5160, Biorad). The relative amounts of different genes in the pre-amplification reactions were then measured by quantitative real-time RT-PCR as described above.
2.13 |. Statistical analysis
All statistical analyses were performed using GraphPad Prism 5.0 software. The statistical significance of differences between groups was determined using two-tailed Student’s t tests with statistical significance attributed to P values ≤ .05. Mean values from at least three independent experiments are reported with error bars representing the standard deviation (SD). One-way ANOVA was used to determine statistical significance for more than two groups.
3 |. RESULTS
3.1 |. Promotion of atherosclerosis by myeloid-specific L13a depletion is accompanied by breakdown of the medial smooth muscle cell layer and accumulation of inflammatory leukocytes and cholesterol crystals in the plaques
Our previous work suggested that L13a-dependent translational silencing in macrophages serves as a physiological defense against atherosclerosis. This was demonstrated using the model of HFD induced atherosclerosis in ApoE−/− mice through comparison of KO (myeloid cell-specific L13a deletion, N = 20) and control (L13 intact, N = 30) mice on the ApoE−/− background.11 Here, in order to confirm our previous findings and gain additional insight into this defense mechanism, control, and KO mice (N = 5/group) were fed a HFD for 14 weeks and then evaluated with respect to phenotypic characteristics of atherosclerotic plaques indicative of disease severity. Since this HFD treatment is a repeat procedure of our previous study,11 we did not use a large group of animal for atherosclerosis measurement. First, we compared the size of lesions (plaques; revealed by Oil red O staining) in aortic root cross sections prepared from the two groups of mice (KO and control). Prior studies using the mouse model of HFD-induced atherosclerosis on the C57BL/6 ApoE−/− background showed differences in the disease susceptibilities between males and females.20,21 However, in our work, differences between the control and L13a KO mice on the C57BL/6 ApoE−/− background with HFD treatment were equivalent for both genders.11 Therefore, in the current experiment, we used only male mice when comparing between control and L13a KO because in this study the size of plaque in male is less variable than in females. As shown in Figure 1A, we found significantly larger aortic root lesions in the KO group of mice compared to the control group (both HFD-treated; mean lesion area = 11 × 104 μm2 vs 7 × 104 μm2, respectively, P < .05). This finding recapitulates our previous results,11 showing increased severity of HFD-induced atherosclerosis specifically in mice lacking L13a expression in myeloid cells.
FIGURE 1.
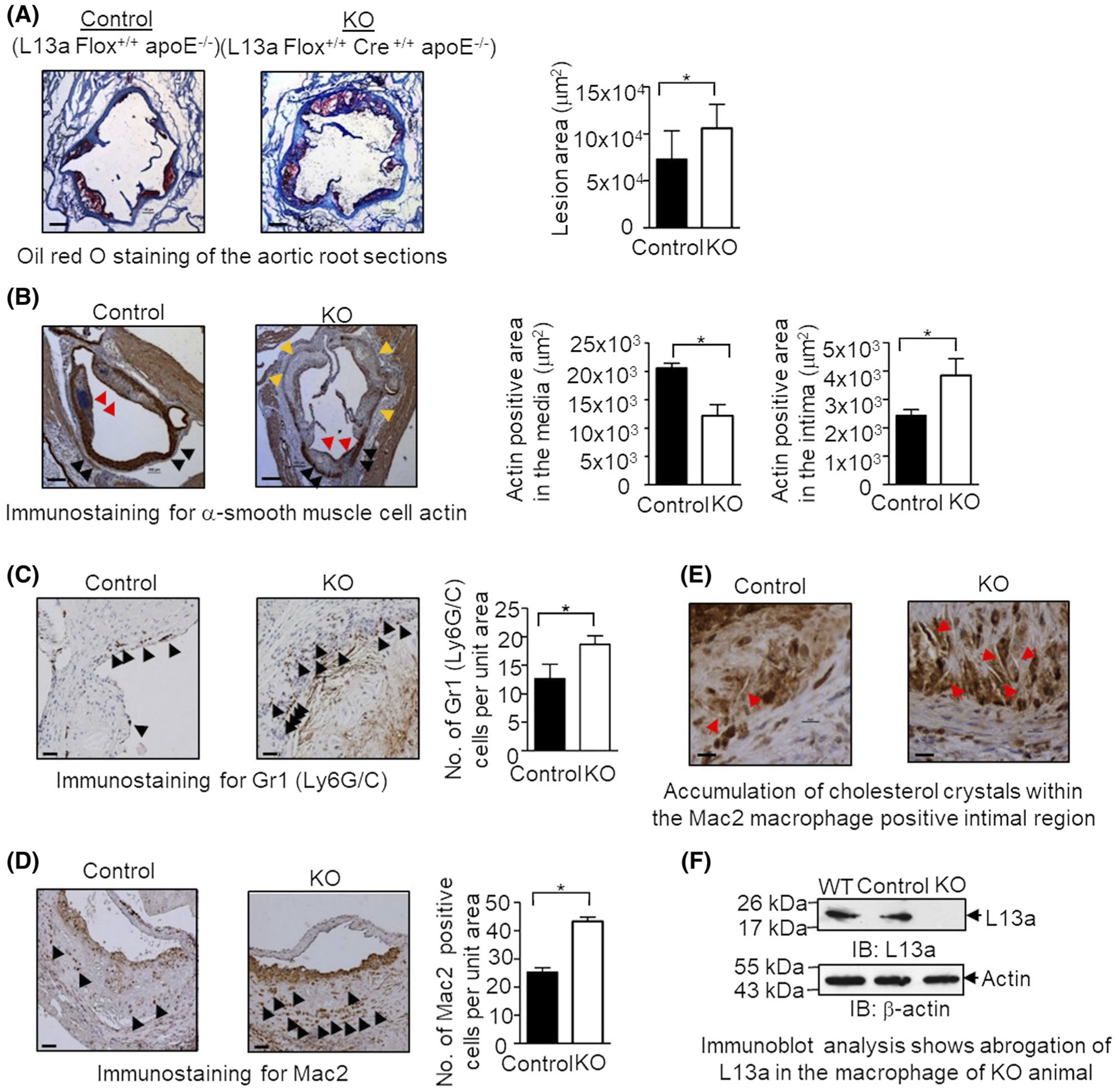
High-fat diet (HFD)-induced atherosclerosis in myeloid cell-specific L13a deficient mice is associated with larger plaques and greater number of immune cells. A, Quantification of atherosclerotic lesion size of aortic roots in mice with or without macrophage-specific L13a deletion on ApoE−/− background and representative images of oil red O staining; Left panel: control group (L13a Flox+/+ApoE−/−), Right panel: KO group (L13a Flox+/+LysMcre+/+ApoE−/−). Mice were fed HFD for 14 weeks before euthanization. B-D, Immunohistochemistry of aortic plaques with α-smooth muscle cell actin antibody for SMC (B), RB6–8C5 antibody for neutrophils (C), and anti-Mac2 antibody for macrophages (D). Scale bars: 100 μm. In (B), black arrows point medial smooth muscle cell layer, red arrows point smooth muscle cells accumulated in the plaque and yellow arrows point areas of medial smooth muscle cell layer breakdown. Quantifications of each cell types are shown. E, Abundance of cholesterol crystals in control and KO mice groups observed in Mac2 staining. Scale bars: 100 μm. F, Confirmation of macrophage-specific depletion of L13a in KO mice. Peritoneal macrophage lysates were analyzed by immunoblot with anti-L13a antibody followed by the reprobing with anti-beta actin antibody. Results are expressed as mean ± SD, n = 4. *P ≤ .05
Next, we assessed additional pathogenic hallmarks of atherosclerosis: thinning and breakdown of the medial smooth muscle cell (SMC) layer, migration of SMCs to the fibrous plaque of the intima,14 and infiltration of Mac2-positive macrophages22,23 and Gr1 (Ly6G/C)-positive neutrophils.24 Using immunostaining for α-actin to detect SMCs in aortic root sections, we found that KO animals showed both a significant reduction in the α-actin positive area in the medial region and a significant increase in α-actin staining in the intimal plaque as compared to control animals (Figure 1B). This is consistent with the disruption of the medial SMC layer in KO mice due to migration of SMCs from the media to the intima. As a feature of plaque vulnerability, we also measured collagen gene expression by quantitative RT-PCR of the plaque samples retrieved by LMD. We found significantly higher collagen expression in the plaque from the HFD- fed KO mice compared to the control mice (Figure S1A), thus, showing more vulnerable plaque. Next, we used immunostaining to evaluate the extent of infiltration of neutrophils (detected by anti-Gr1 mAb) and macrophages (detected by anti-Mac2 mAb) in aortic root sections from the HFD-fed KO and control mice. This showed significantly increased numbers of both macrophages and neutrophils in the KO mice compared to control mice (Figure 1C,D). Further analysis of the Mac2-positive intimal region revealed that lesions obtained from KO mice also had greater accumulation of cholesterol crystals than those from control mice (Figure 1E). Finally, total depletion of L13a in macrophages of the KO mice was confirmed by immunoblot analysis with anti-L13a antibody (Figure 1F). In a parallel experiment, monocytes and neutrophils were isolated from the blood of control and KO mice by Easy Sep mouse monocyte and neutrophil isolation kit (Stem cell Technologies) and tested for the depletion of L13a by immunoblot. In contrast to macrophages, this study showed partial depletion of L13a (Figure S1B). This result is consistent with the previous report showing the variation of LySM promoter activity within different myeloid cells.25 Taken together, these results confirm that myeloid cell-specific depletion of L13a leads to increased severity of atherosclerosis developing upon HFD treatment in the ApoE−/− mouse model.
3.2 |. Myeloid cell-specific deficiency of L13a promotes differentiation of macrophages in atherosclerotic plaques toward the M1 type inflammatory phenotype
Both classically activated pro-inflammatory M1-like and alternatively activated anti-inflammatory M2-like macrophages are present in human and mouse atheromas.15,26–28 Since L13a-dependent GAIT complex formation regulates production of a number of inflammatory cytokines by macrophages directly at the level of translation, we hypothesized that loss of L13a might influence macrophage reprograming and disrupt the equilibrium between M1 and M2 cell types. To test this possibility, we evaluated expression of previously established markers for M1 type inflammatory macrophages (iNOS26,29 and IL6 and TNF-α mRNAs28,30) and M2 type wound healing macrophages (CD20631,32 and CD16331) among bone marrow-derived macrophage (BMDM) from KO and control mice after IFN-γ treatment. This model is based on the known activity of IFN-γ as an extracellular signal that promotes differentiation of plastic BMDM populations toward the M1 phenotype.28,33 Thus, we isolated BMDM from the femurs of KO and control mice (fed regular chow), treated them with IFN-γ ex vivo, and then, used quantitative RT-PCR to measure the steady state mRNA levels of M1- and M2 type markers. We found small but significant upregulation (~1.5-fold; *P < .05) of all three M1 type inflammatory markers (IL6, iNOS, and TNFα) in the KO mice as compared to controls; however, changes in expression of M2 type markers (CD163 and CD206) were not significant (P > .05) (Figure 2A). In a parallel approach, using the same model of IFN-γ-treated BMDM from KO and control mice paired with fluorescence activated cell-sorting (FACS) analysis, we quantified CD11b and iNOS double-positive macrophages (ie, M1 type macrophages). As shown in Figure 2B, this revealed significantly greater abundance of CD11b and iNOS double-positive cells among IFN-γ-treated BMDM isolated from KO animals vs controls (26% vs 46%; *P < .05). These results demonstrate that absence of L13a results in enhanced IFN-γ-induced differentiation of BMDM toward the M1 inflammatory macrophage phenotype.
FIGURE 2.
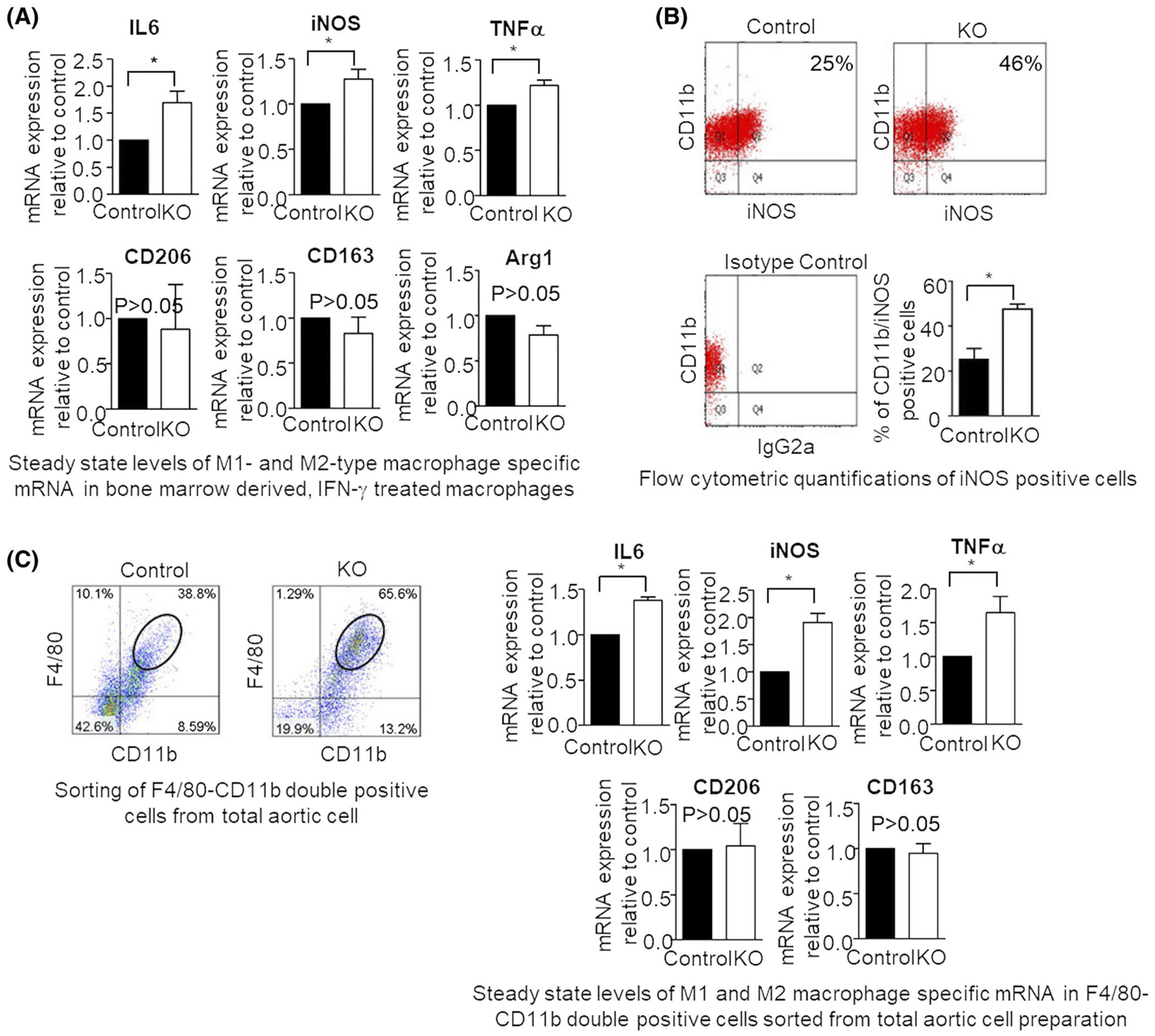
IFN-γ and HFD mediated stimulation results in similar changes in M1-type macrophage-specific gene induction in bone marrow-derived macrophages or total aortic cells, respectively. A, Real-time PCR analyses of the steady state levels of M1-type macrophage-specific IL6, iNOS, and TNFα mRNAs or M2-type macrophage-specific CD206, CD163, and Arg1 mRNAs were measured in the IFN-γ treated bone marrow-derived macrophages from control or L13a KO mice. mRNA levels were determined using comparative real-time RT-PCR and normalized to mouse GAPDH. B, Flowcytometric quantification of iNOS expression in IFN-γ treated macrophages derived from bone marrows of control and KO mice. C, steady state levels of M1- and M2-type macrophage-specific mRNAs in F4–80/CD11b double-positive cells sorted by FACS (Top panels, encircled) from total aortic cells prepared after 14-week HFD treatment. mRNA levels were determined through the comparative CT method and normalized to GAPDH. Results are expressed as mean ± SD, n = 3. *P ≤ .05 compared with the control group
It is well established that increased reprograming of the aortic macrophage population toward the M1 inflammatory phenotype plays a critical role in the pathogenesis of atherosclerosis and that this process is driven by environmental cues encountered by the cells.28 Based on this and our finding that L13a influences M1/M2 phenotypic changes in the model of IFN-γ-treated BMDM (see above), we sought to determine whether myeloid cell-specific L13a deficiency promotes reprograming of aortic macrophages toward the M1 inflammatory phenotype in the model of HFD-induced atherosclerosis in ApoE−/− animals. KO and control mice were fed a HFD for 14 weeks, after which total aortic cells were isolated. Considering the cellular heterogeneity of the total aortic cells the F480/CD11b double-positive leukocytes were sorted by FACS (left panel of Figure 2C) and evaluated for steady state levels of M1 and M2 type-associated marker mRNAs using quantitative RT-PCR. Similar to what was observed in the IFN-γ-treated BMDM model, we found significantly higher levels of M1 marker mRNAs (ie, IL6, iNOS, and TNF-α, ~1.5- to 2-fold, *P < .05) in HFD-treated KO mice compared to HFD-treated controls, but there was no significant difference in expression of M2 markers (CD206 and CD163) between the two groups (Figure 2C, P > .05).
The trans-endothelial migration of monocytes to the intimal plaque is driven by the encountered milieu of chemokines and their cognate receptors.34,35 Our earlier works suggest direct activation of protein synthesis of a cohort of chemokine receptors in animal model of myeloid cell-specific L13a depletion10 and severe atherosclerosis.11 M1 type inflammatory macrophages and their inflammatory outputs play a critical role in the pathogenesis of unstable plaques during atherogenesis.28,36 Therefore, we investigated whether over representation of M1 type inflammatory macrophages contribute to the severity of atherosclerosis. Immunohistochemical staining of aortic root cross sections with antibodies against iNOS (M1 marker) shows that in both control and KO mouse, iNOS positive cells are distributed throughout the atheroma. We detected iNOS positive cells in the intimal plaque region as well as around media/adventitia and quantification of these showed significantly higher abundance in the KO compared to the control (~2-fold; *P < .05) (Figure 3A). To ensure the identity of iNOS expressing macrophages, we also added Mac2 staining in adjacent serial sections to show same immuno-localization of both Mac2 and iNOS positive macrophages in the atheroma (lower panel of Figure 3A). More detail quantification of the Mac2-positive cells are shown in Figure 1D. Immunostaining for CD206 (M2 marker) showed no significant difference (P > .05) between the HFD-fed KO mice and control (Figure 3B).
FIGURE 3.
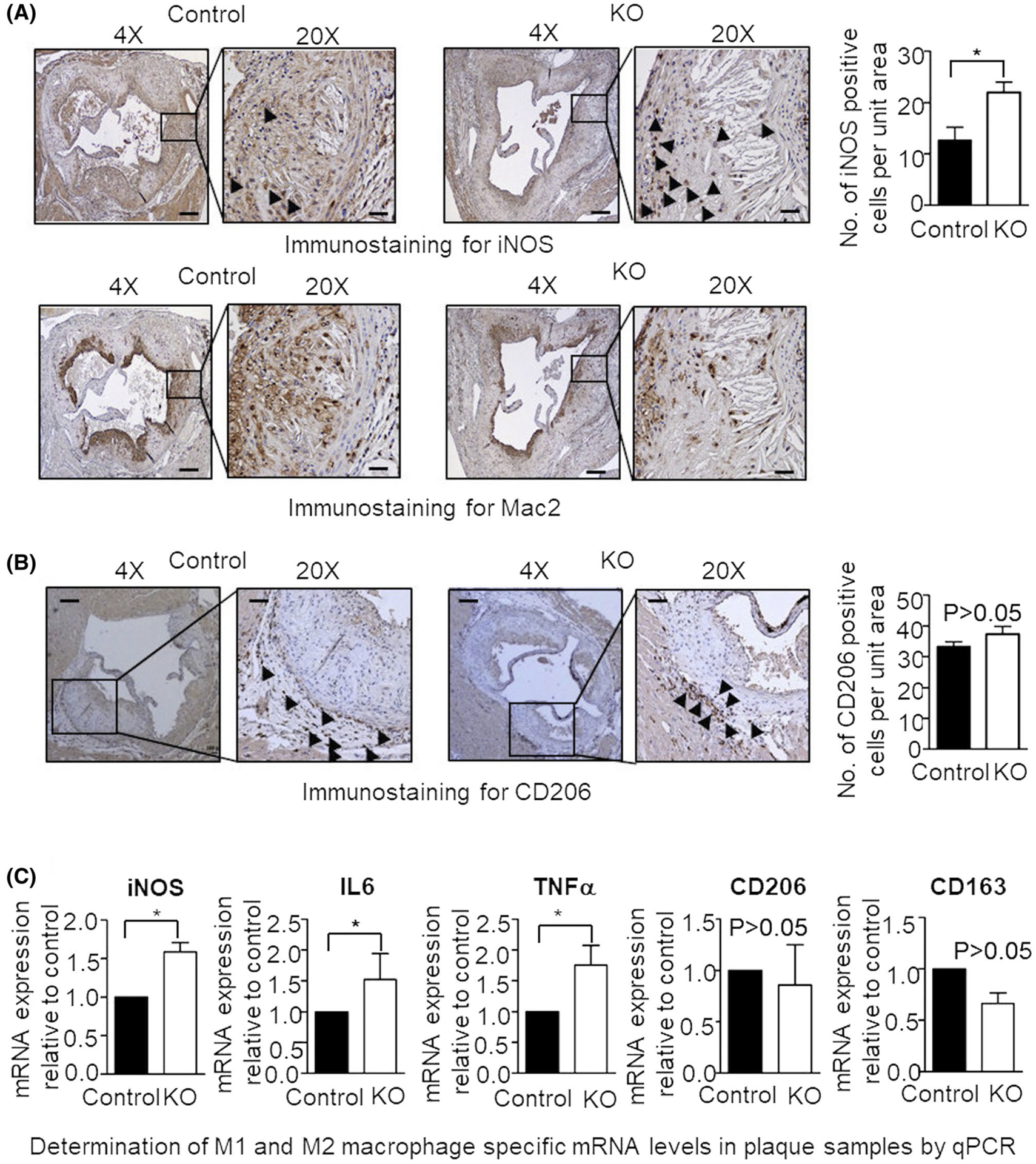
HFD results in increased expression of M1 macrophage-specific markers in the atherosclerotic plaque of myeloid cell-specific L13a deficient mice. A and B, Immunohistochemical detection of iNOS (A), Mac2 (A) and CD206 (B) expressing cells in the aortic root sections prepared from control and KO mice. Quantifications for iNOS and CD206 are shown; Mac2 staining of the adjacent serial section shows the presence of macrophages in the same area. Scale bars: 100 μm. C, steady state levels of iNOS and other M1- and M2-type macrophage-specific mRNAs in aortic plaque cells were measured by real-time quantitative PCR. To generate cDNA template, mRNA obtained from laser microdissected cuts of aortic plaques were reverse transcribed. The levels of target mRNAs were normalized with mouse GAPDH mRNA and were presented as mean ± SD, n = 3. *P ≤ .05, compared with the control group
To confirm the M1/M2 phenotypical differentiation of macrophages specifically in atherosclerotic plaques, quantitative RT-PCR was used to measure marker transcript levels in plaques harvested by LMD from HFD-treated KO and control mice. Consistent with the immunostaining results described above (Figure 3A,B), this RT-PCR analysis showed significantly higher levels of M1 type inflammatory specific markers (~1.7- to 2-fold; *P < .05), but not M2 type wound healing markers (P > .05) in atherosclerotic lesions of the HFD-treated KO mice compared to those from HFD-treated control mice (Figure 3C). Taken together, these findings clearly illustrate that myeloid cell-specific depletion of L13a promotes reprograming of macrophages in atherosclerotic plaques toward the M1 type inflammatory phenotype that is associated with pathogenesis of the disease.
3.3 |. Genetic depletion of L13a in myeloid cells results in increased abundance of native LDL receptor and upregulation of phagocytic activity
Internalization of modified lipoproteins by scavenger receptors displayed on the surface of macrophages (eg, SR-A, CD36, CD68) plays a critical role in the process of lipid uptake that initiates formation of foam cells and leads to atherosclerosis.37 However, an alternative, scavenger receptor-independent,38 mechanism of lipid uptake, and foam cell formation mediated by native low-density lipoprotein (LDL) has also been reported.39 Supporting the potential importance of this pathway, several recent studies demonstrated a significant role of inflammatory stress in the native LDL uptake and LDL receptor (LDLR) mediated pathways of foam cell formation40 and in disruption of a LDLR negative feedback loop.41
Here, we investigated the relevance of pathways of modified lipoprotein uptake (mediated by scavenger receptors) and native LDL uptake (mediated by LDLR) in atherogenesis promoted by the physiological inflammation caused by myeloid cell-specific depletion of L13a. This was accomplished by evaluating LDLR and scavenger receptor expression levels and phagocytic activity in aortic plaque and total aortic cells from HFD-treated control and KO animals as well as IFN-γ treated BMDM from chow (low-fat) diet-fed control and KO animals. Significant upregulation of LDLR in KO mice was observed at both the mRNA level (by real-time quantitative RT-PCR, Figure 4A) and at the protein level (by immunoblot analysis, Figure 4B). However, there was no significant difference in expression of mRNAs for scavenger receptors CD36, CD68, and SR-A between control and KO mice, regardless of whether aortic plaque cells, total aortic cells, or BMDM were evaluated (Figure S1C). The upregulation of LDLR is also supported by our follow-up studies involving direct quantification of unmodified DIL (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine per-chlorate)-labeled LDL uptake. Using BMDM model of control and KO mice we found significantly higher LDL uptake of the macrophages from KO mice (Figure 4C).
FIGURE 4.
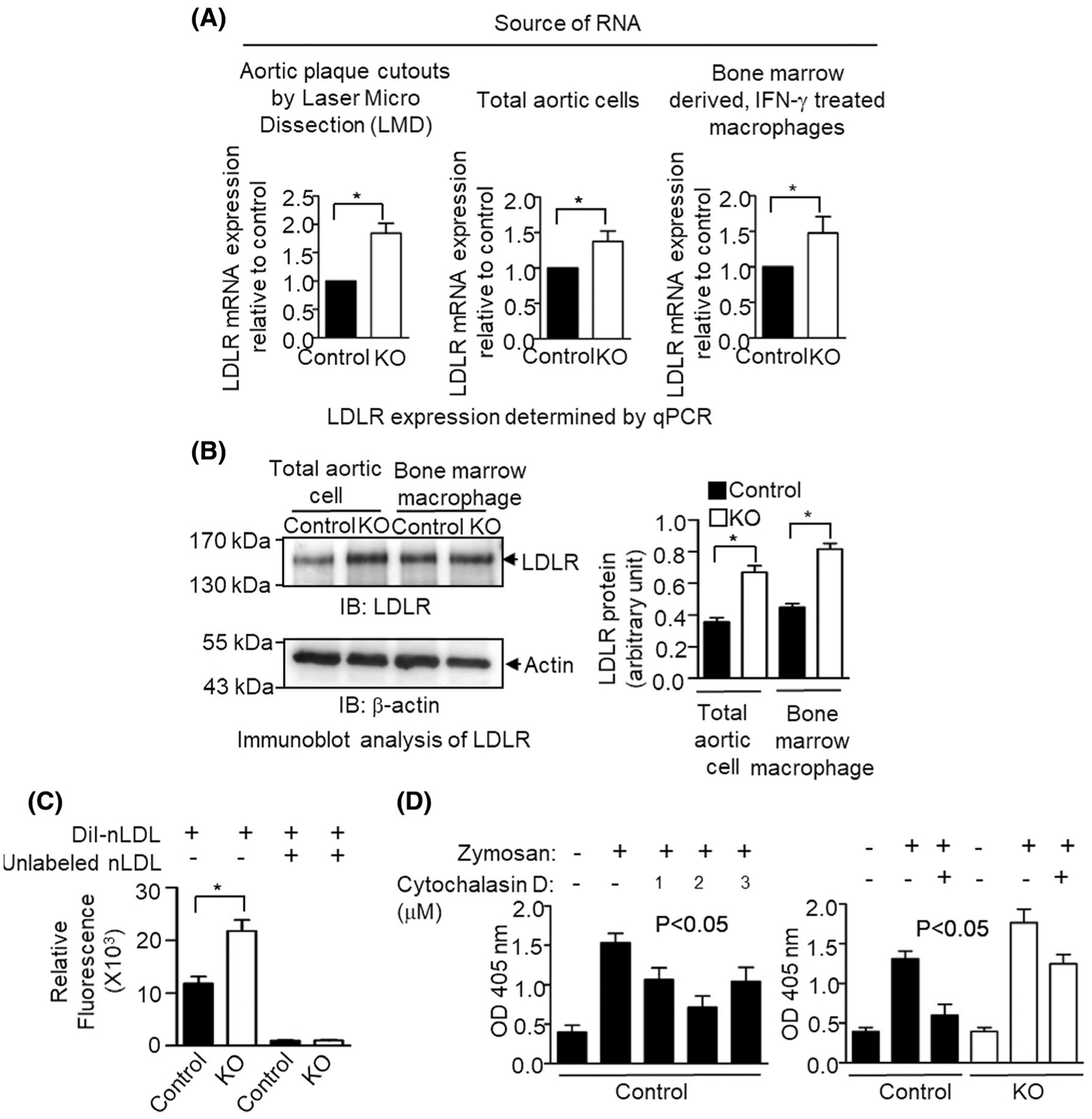
Effect of inflammatory stimulation on low-density lipoprotein receptor (LDLR) expression and phagocytic activity in L13a deficient mouse macrophage. A, Mice were kept on a HFD for 14 weeks, after which LDLR mRNA levels were determined by real-time quantitative RT-PCR in RNA preparations from laser microdissected aortic plaque cutouts and total aortic cells. Simultaneously, LDLR mRNA levels were also checked in IFN-γ stimulated bone marrow-derived macrophage cells. B, Left, Western blot analysis of LDL receptor protein expression. Lysates were prepared from either IFN-γ stimulated bone marrow-derived macrophages or total aortic cells from HFD-fed control and KO mice. Protein lysates were subjected to Western blot by anti LDLR antibody. Right, Densities of the LDLR bands in the blot after normalization with the internal invariable control beta-actin. C, Internalization of native low-density lipoprotein (nLDL) by bone marrow-derived macrophages. IFN-γ treated control and L13a KO macrophages were incubated with DiI-labeled nLDL for 4 hours at 37°C before the determination of relative fluorescence. Specificity of internalization was checked by preincubating cells with unlabeled nLDL before adding Dil-nLDL. Data represent mean ± SD, n = 3, * P ≤ .05. D, Increased phagocytic activity of L13a deficient bone marrow-derived macrophages (BMM). Left panel, determination of phagocytosis inhibitor (Cytochalasin D) concentration for BMM. 50 000 control BMM cells/well were seeded overnight in a 96-well plate. Cytochalasin D was used to pretreat for 1 hour at 37°C before addition of Zymosan particles at 50:1 ratio. Phagocytosis was stopped after 30 minutes and the amount of engulfed Zymosan particles was determined. Right panel, control, and L13a deficient BMM were pretreated with IFN-γ for 24 hours before adding zymosan particles in the presence or absence of Cytochalasin D. In A and B, results are expressed as mean ± SD, n = 3. *P ≤ .05 compared with the control group. In C, 1-way ANOVA was used for statistical analysis
Widespread phagocytosis of modified lipoproteins and apoptotic cells by macrophages plays a complex role in the pathogenesis of atherosclerosis. This process can have either pro- and anti-atherogenic effects through context-dependent activation or resolution of inflammation, respectively.42,43 To assess the effect of L13a deficiency on the phagocytic activity of macrophages, we performed phagocytosis assays with BMDM using Zymosan as a substrate. First, we confirmed the specificity of phagocytosis by testing its sensitivity to Cytochalasin D (Figure 4D left panel). Next, we compared Cytochalasin D-sensitive phagocytosis in BMDM from KO and control mice that were treated ex vivo with IFN-γ prior to addition of Zymosan. This showed a significant increase in the phagocytic activity of BMDM isolated from KO animals compared to those from control mice (Figure 4D right panel). Together, these results indicate that genetic depletion of L13a in macrophages results in increased expression of LDLR (but not scavenger receptors important for modified lipoprotein internalization) and increased phagocytic activity of macrophages. Both of these increases are likely to be associated with promotion of atherosclerosis.
3.4 |. Identification of proatherogenic genes that are upregulated in atherosclerotic plaques from HFD-treated L13a KO mice compared to L13a-intact controls using high-throughput gene expression profiling
To gain additional insight into the mechanisms underlying the enhancement of atherosclerosis observed in HFD-fed apoE−/− mice with myeloid-specific L13a deficiency (Figure 1 and Ref. [11]) and potentially identify novel mediators, we assessed gene expression on a broad scale using RNA NanoString analysis. Atherosclerotic plaques were harvested by LMD from the aortic root of HFD-fed control and KO mice (see eg, Figure 5A). Gene expression profiling was performed on RNA isolated from the plaques using a commercially available panel of 770 immune-related genes & 20 internal reference genes (nCounter® PanCancer Immune Profiling Panel, NanoString Technologies). As illustrated by plotting the NanoString data in volcano plots (Figure 5B), this analysis identified 10 out of the 770 genes that showed greater than 2-fold increased expression in atherosclerotic plaques of HFD-fed KO mice compared to plaques from HFD-fed controls. These genes were CCL2/MCP-1 (chemokine C-C motif ligand 2/monocyte chemotactic protein-1), Cdkn1a (cyclin-dependent kinase inhibitor 1a), Clec4n/Dectin-2 (c-type lectin receptors), CXCL12 (chemokine C-X-C motif ligand 12), Eng (endoglin), LCN2 (lipocalin 2), Pecam1/CD31 (platelet endothelial cell adhesion molecule 1/cluster of differentiation 31), S100a8 (S100 calcium-binding protein A8), Tap 1 (transporter associated with antigen processing 1), and Vwf (Von-Willebrand Factor). KO-specific upregulation of these genes was independently validated by real-time PCR, with all 10 genes showing significantly increased expression (P < .05) in atherosclerotic plaques isolated from HFD-fed KO mice compared to those from HFD-fed controls (Figure 5C). Notably, as described below (see Discussion), proatherogenic roles were defined for all of these genes in previous studies.
FIGURE 5.
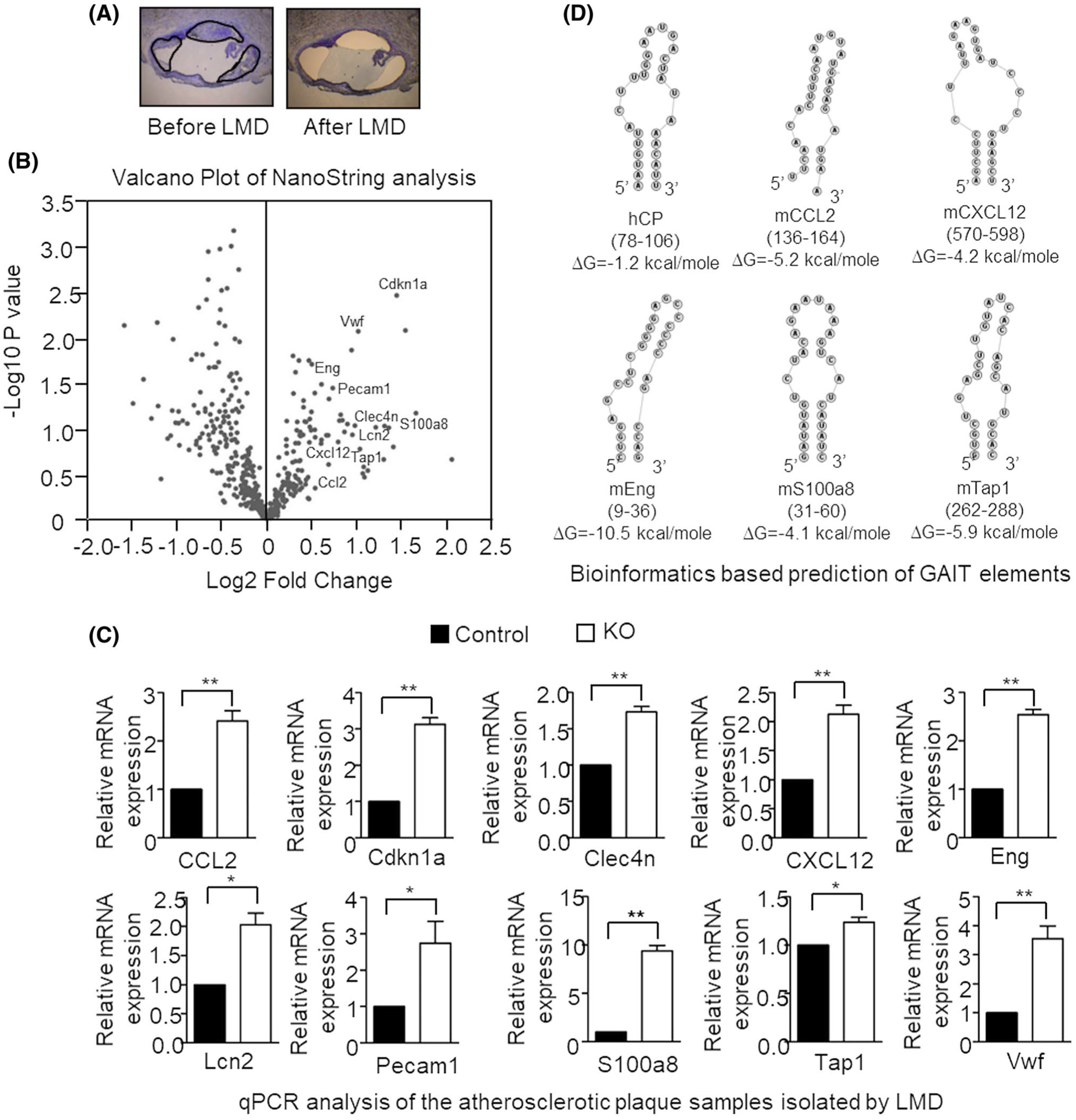
Analysis of gene expression in the atherosclerotic lesions of L13a-deficient mice by laser capture microdissection (LMD) coupled with NanoString nCounter Analysis. A, representative pictures of aortic root sections before and after LMD. Sections of the plaque to be cut out were marked. B, volcano plot of NanoString analysis of mRNAs in laser microdissected atherosclerotic plaques from L13a KO animal compared with control animal after 14 weeks of Western-type diet. n = 10. C, upregulation of 10 genes identified in NanoString were validated by real-time quantitative RT-PCR analysis. cDNA templates were made from total RNA isolated from LMD cutouts. Results are expressed as mean ± SD, n = 3. *P ≤ .05; **P ≤ .01. D, Bioinformatics-based prediction of the GAIT elements in the 3′ UTRs of mouse CCL2, CXCL12, Eng, S100a8, and Tap1. RNA structure and free energy predictions were made using “FOLDALIGN” and “RNAstructure” programs. From among the potential folding patterns identified for each RNA element, those with the lowest free energy and highest similarity to the canonical ceruloplasmin (Cp) GAIT element are shown
Specific genes are subject to L13a-dependent translational silencing due to the presence of GAIT sequence elements in the 3′UTRs of their mRNAs. We previously identified functional GAIT elements in a number of chemokine and chemokine receptor mRNAs using ex vivo (cellular),8 in vivo (murine),10–12 bioinformatics and structure-(NMR) based analyses.9 Interestingly, however, the GAIT elements in these newly identified targets9 did not share any sequence homology with the canonical GAIT element of the ceruloplasmin (Cp) mRNA,4 indicating that secondary structure is the defining feature of GAIT elements that directs assembly of the L13a-associated translation-blocking complex. Since the GAIT pathway is controlled by an autoregulatory negative feedback mechanism,44 we hypothesized that some of the mRNAs found to be upregulated in atherogenic plaques from HFD-treated L13a KO mice might harbor GAIT elements. To search for potential GAIT elements, the 10 mRNAs identified by NanoString analysis (see above) were tested for folding homology against the human Cp GAIT element using “Foldalign”45–47 (http://foldalign.ku.dk) and “RNA structure” (http://rna.urmc.rochester.edu/RNAstructure.html) software platforms. Predicted stem-loop structures with similarity to the Cp GAIT element were identified in 5 of the 10 tested mRNAs (CCl2, CXCl12, Eng, S100a8, and Tap1; Figure 5D). Out of these five in silico identified potential GAIT elements, we have experimentally validated the GAIT elements of CCL2, S100a8, and Tap1 as functionally active (see Figure 6D). Future NMR-based studies will test the actual conformations of these GAIT elements. Together, these studies demonstrate that myeloid cell-specific depletion of L13a and subsequent abrogation of the GAIT element-mediated pathway of translational suppression results in wide-spread upregulation of inflammatory genes directly in the cellular mass of atherosclerotic plaques.
FIGURE 6.
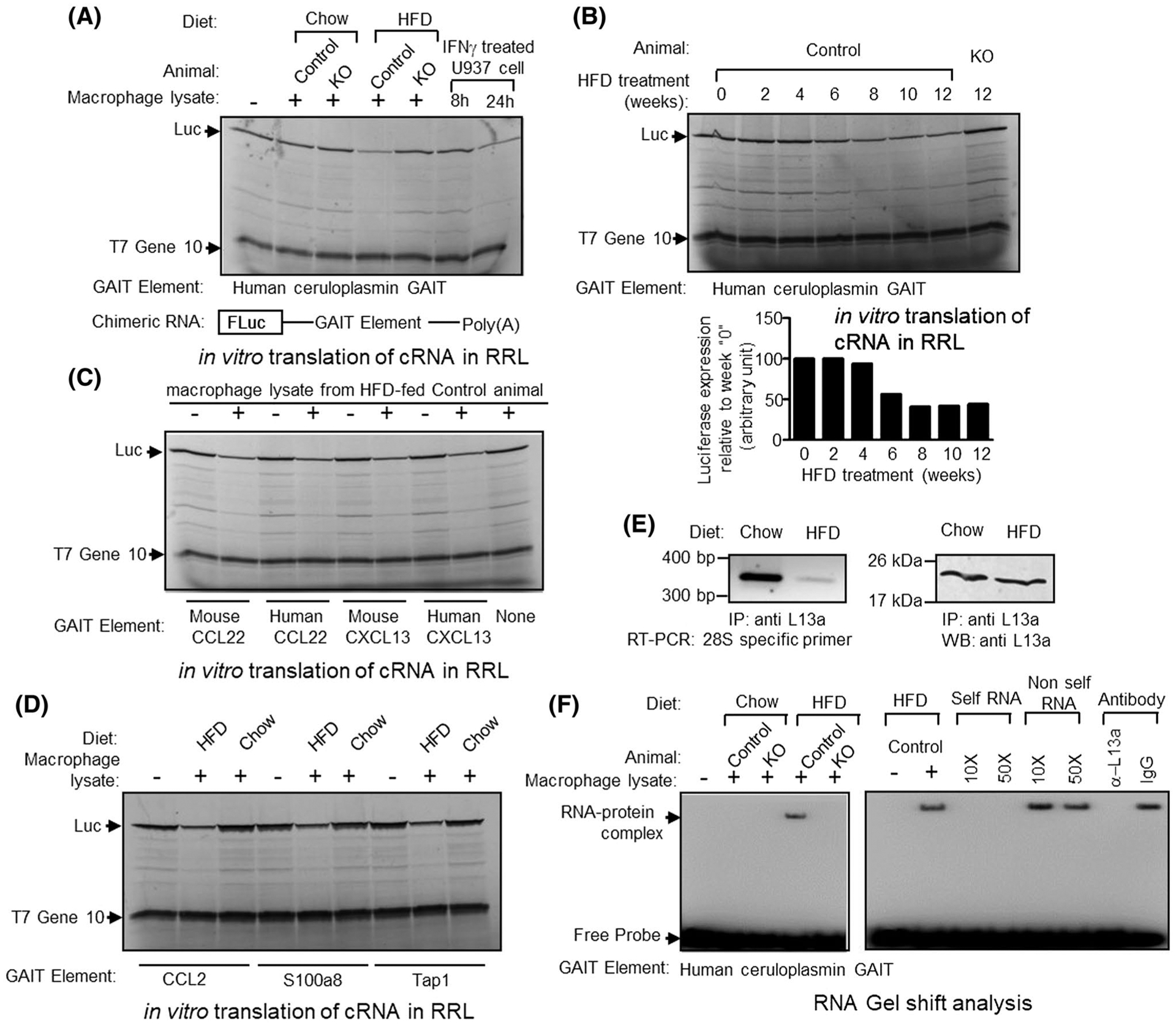
Macrophage lysate from HFD-fed mouse can direct translational silencing of Firefly Luciferase (Fluc) reporter mRNA, driven by human or mouse GAIT elements. A, Top panel, Human ceruloplasmin (Cp) GAIT element containing Fluc mRNA were translated in vitro in rabbit reticulocyte lysate (in the presence of [35S]-Methionine). Macrophage lysate prepared from chow or Western-type diet-fed control or L13a KO animal were added to the translation mixture as indicated above each lane. As control, extracts prepared from U937 cells treated for 8 or 24 hours with IFN-γ were added to the translation reactions as indicated. Luciferase and T7 gene 10 protein (included as a specificity control) were indicated by arrows. Bottom panel, schematic diagram of GAIT element containing Fluc reporter gene construct. Different human or mouse GAIT elements were cloned in the 3′ UTR of Fluc and in vitro transcribed to generate mRNA template. B, peritoneal macrophage lysates prepared from control mice treated with HFD for 0–12 weeks, were added to the translation reaction as described in A. C, species-independent activation of human and mouse CCL22 and CXCL13 GAIT mediated translational silencing by HFD-treated control mice’s peritoneal macrophage lysate. Human and murine CCL22 or CXCL13 GAIT containing Fluc reporter mRNAs were subjected to in vitro translation as described in A in presence of 12-week HFD-treated control mice peritoneal macrophage lysate. D, In vitro translation silencing assay with the predicted GAIT elements from CCL2, S100a8, and Tap1 3′UTR (Figure 5D). E, Reduced association of L13a with the 60S ribosomal subunit in the macrophages from HFD-treated mice. F, Macrophage lysate from HFD but not chow diet-fed control mice forms complex with human Cp GAIT element (left panel). Competition with unlabeled self-RNA (10- and 50-fold molar excess) and preincubation with L13a antibody abolishes complex formation (right panel)
3.5 |. Proatherogenic HFD treatment of ApoE−/− mice activates L13a-dependent GAIT element-mediated translational silencing activity
As described above, our previous work with the murine model of HFD-induced atherosclerosis demonstrated that myeloid cell-specific deficiency of L13a promotes atherosclerosis by abrogating translation control-dependent suppression of inflammation.11 However, this study did not address whether HFD treatment could, in itself, turn on GAIT element-mediated translational control as an endogenous defense against atherosclerosis. To directly test this possibility, we prepared lysates from peritoneal macrophages harvested from KO and control mice fed either regular chow or a HFD and tested their effect on translational silencing in a reconstituted cell-free system in which a reporter RNA harboring a functional GAIT element4 in its 3′UTR (bottom part of Figure 6A) is translated by rabbit reticulocyte lysate. Addition of macrophage lysates from HFD-fed control (L13a intact) mice to this cell-free system resulted in translational silencing of the GAIT element containing reporter mRNA (Figure 6A), however, in the same system translation of a T7gene10 reporter RNA without the GAIT element remain insensitive. In contrast, macrophage lysates from HFD-fed KO mice or from chow-fed control or KO mice had no effect in this experiment system. This demonstrates activation of L13a-dependent translational silencing activity specifically in response to pro-atherogenic conditions (HFD treatment on ApoE−/− background). For a diet-induced activity, it is also important to know the duration of the diet treatment required to turn on the activity. Therefore, using the same cell-free system, we tested macrophage lysates from control mice fed a HFD for 2, 4, 6, 8, 10, or 12 weeks for their effect on reconstituted translational silencing. This showed that at least 6 weeks of HFD treatment is required to turn on the L13a-dependent GAIT element-mediated translational silencing activity in macrophages (Figure 6B).
We previously found that HFD-treated myeloid cell-specific L13a KO mice showed increased steady state levels of CCL22, CXCL13, and many other chemokine ligands in their serum compared to similarly treated L13a-intact control mice.11 This, together with the presence of functional GAIT elements in the mRNAs encoding CCL22 and CXCL13,9 suggests that HFD treatment might activates L13a-dependent GAIT element-mediated translational silencing of these chemokine ligands to resolve inflammation as an endogenous defense mechanism. To directly test this, we evaluated the effect of macrophage lysates prepared from HFD-treated control (L13a intact) mice on reconstituted translation of reporter mRNAs harboring the GAIT elements from human and mouse CCL22 and CXCL13 mRNAs. As shown in Figure 6C, HFD treatment induces an activity in macrophages that mediates GAIT element-directed translational silencing in a species-independent manner (reporters containing human and mouse GAIT elements were similarly affected by the mouse macrophage lysates). However, translation of T7gene10 RNA without the GAIT element remains insensitive. On the contrary, RNA NanoString analysis of the atherosclerotic plaques from the HFD-fed KO mice showed increased steady state level of 5 mRNAs harboring in silico identified potential GAIT elements in the 3′UTRs (Figure 5D). To directly test the functional activity of these elements, we used parallel experiment involving reporter luciferase RNAs harboring these five potential GAIT elements under test in reconstituted cell-free system. Lysates from peritoneal macrophages harvested from HFD-fed mice (not from chow diet-fed mice) to this cell-free system resulted in translational silencing of the reporter mRNA harboring in silico identified GAIT elements from CCL2, S100a8, and Tap 1 (Figure 6D).
Using a cellular model of human monocytes, we previously showed that release of L13a from the 60S ribosome in response to inflammatory stimuli (eg, IFN-γ treatment) and formation of L13a-dependent RNA-binding complex on GAIT element triggers GAIT element-mediated translational silencing.3 To determine whether this same mechanism controls the process in the HFD-induced atherogenesis model, we compared association of L13a with the 60S ribosomal subunit in lysates of peritoneal macrophages from chow-fed vs HFD-treated control (L13 intact) mice. This was accomplished by subjecting anti-L13a immunoprecipitates to RT-PCR analysis using primers specific for 28S rRNA (a component of the 60S ribosomal subunit). Results of this experiment showed significantly reduced association of L13a with the 60S ribosome in macrophages from HFD-treated mice compared to those from chow-fed mice (Figure 6E). We then tested the ability of the macrophages harvested from HFD-treated mice to induce L13a-dependent RNA-binding activity on GAIT element. Result presented in the left panel of Figure 6F showed the ability of the lysates prepared from peritoneal macrophages harvested from HFD-treated control mice to form RNA-binding complex on GAIT element. In contrast, no RNA-binding complex was found when macrophages from HFD-treated L13a KO mice or macrophage from chow diet treated control mice were used. The specificity and L13a dependency of this RNA-binding complex were confirmed by abrogation of the binding by self-RNA competition and preincubation of the lysate with anti-L13a antibody (Figure 6F right panel). Thus, coincident with its promotion of atherosclerosis, HFD treatment of mice with an ApoE−/− background turns on L13a-dependent GAIT element-mediated translational silencing involving release of L13a from the 60S ribosome and the formation of RNA-binding complex.
4 |. DISCUSSION
Inflammation initiated from vascular macrophages plays a pivotal role in the initiation and progression of atherosclerosis.13–19 Altered epigenetic pathways in the vascular macrophages present in atherosclerotic plaques also trigger and sustain inflammation that fails to resolve.48 Using the well-established model of HFD-induced atherosclerosis in apoE−/− mice, we previously showed that genetic depletion of L13a in macrophages increases susceptibility to atherosclerosis, apparently by removing a natural mechanism of translational control of several chemokines and chemokine receptors.11 While this work suggested a myeloid cell-specific role for L13a as an endogenous mechanism for resolving inflammation, the cellular and molecular consequences of the absence of L13a in macrophages (eg, target gene expression) and how they influence characteristics of atherosclerotic plaques were not defined. The current study provides significant insights into these questions. First, we characterized in further detail the enhanced atherosclerosis observed upon HFD treatment of myeloid cell-specific L13a deficient (KO) mice compared to control (L13a intact) mice (both on ApoE−/− background). In addition to the increased size of vascular plaques at the aortic root in KO mice, we also observed changes in the cellular composition of the plaques. As quantitated on a smooth muscle α-actin staining in Figure 1B, the lesion causes thinning and rupture of the medial SMC layer and migration of SMCs to the intimal fibrous plaque, infiltration/proliferation of Gr1 (Ly6G/C)-positive neutrophils (Figure 1C), and Mac2-positive inflammatory macrophages (Figure 1D) and deposition of cholesterol crystals (Figure 1E). The inflammatory reaction expands into that adventitia which may facilitate enhanced infiltration of M1 positive inflammatory macrophages to the adventitia observed in Figure 3A. The adventitial component of atherosclerosis has recently been appreciated in mouse models and human coronaries with atherosclerosis.49–51
These changes all likely contribute to the promotion of atherosclerosis observed in the myeloid cell-specific L13a KO mice. Together, degeneration of the SMC layer in the media and accumulation of neutrophils and Mac2-positive macrophages may create an environment that favors unstable lesions.14,22–24 These cellular changes are also consistent with previous studies showing a strong association of the atherosclerosis severity with intra-plaque neutrophil numbers,24 lower smooth muscle content,52 and plaque fragility53,54 perhaps due to neutrophil-released matrix degradation enzymes (eg, MMP-2, MMP-9, proteinase-3, and elastase).54 The abundance of cholesterol crystals in the atherosclerotic plaques of the KO mice could perpetuate inflammation through their previously described ability to trigger the NLRP3-Caspase1-IL1β axis and stimulate M1 type differentiation of resident macrophages.55,56 In contrast to the widely accepted dogma that resident macrophages are terminally differentiated, and thus, incapable of proliferation, increasing evidence supports the possibility of macrophage proliferation independent of monocyte recruitment.57,58 Our results showing increased abundance of plaque-resident macrophages and neutrophils in the absence of L13a-dependent translational control are consistent with the notion that a currently unknown proliferation signal(s) in the plaque microenvironment could be a target of this anti-atherogenic regulatory mechanism.
Macrophages within atherosclerotic plaques are highly plastic and capable of switching between wide ranges of phenotypes, from pro-inflammatory M1 type to anti-inflammatory M2 type, in response to environmental cues (currently undefined) they encounter within the lesion.15,28 We examined the consequences of myeloid cell-specific L13a depletion on macrophage polarity by measuring expression of marker molecules defining the two ends of the phenotypic spectrum (iNOS, IL-6, TNF-α for M1 type vs CD206 and CD163 for M2 type) in KO and control mice fed a HFD for 14 weeks (thus, looking at “mature” plaques). This showed increased abundance of M1 type macrophages specifically in the KO mice (Figures 2 and 3). However, this experiment was not designed to reveal changes in the dynamics of the entire spectrum of macrophage polarity as atherosclerosis develops and progresses in the absence of L13a-dependent translational suppression. Future studies using genome-wide RNA sequencing of early- to late-stage atherosclerotic plaques from control and KO mice should yield valuable information on the signals that regulate macrophage plasticity during atherogenesis.
The current study also suggests that L13a-dependent mechanisms may directly or indirectly control LDL receptor (LDLR) expression and phagocytosis in macrophages to restrain atherogenesis. This is supported by our finding of increased expression of LDLR (but not SR-A or CD36 scavenger receptors) in atherosclerotic plaques, total aortic cells and bone marrow-derived macrophages (BMDM) from KO mice compared to control mice (Figure 4A,B). In consistence with this result we also found increased LDL uptake in the BMDM of the KO mice (Figure 4C). Based on the observation that, macrophage has limited LDLR expression, which is inhibited by excess cholesterol,59,60 scavenger receptor-mediated modified LDL uptake considered to be the major pathway for foam cell formation. However, several studies suggests that LDLR of murine peritoneal macrophage has the ability to actively metabolize unmodified atherogenic lipoproteins, such as β-very low-density lipoprotein (β-VLDL) and chylomicron remnants that induces transformation of macrophages into foam cells.61–63 Therefore, macrophage LDLR expression may have a relevant impact in modulation of foam cell formation. The significance of this observation is highlighted by prior studies demonstrating the role of LDLR in inflammation-induced unmodified LDL accumulation,40,41 foam cell formation39 and insensitivity of HFD-induced atherosclerosis in response to loss of SR-A and CD36 pathways.38 In addition, our finding of increased phagocytic activity of BMM from L13a-deficient mice vs controls (Figure 4D) is notable given the fact that phagocytosis is clearly involved in the pathogenesis of atherosclerosis, albeit with roles that are complex and currently not fully defined.42
The cellular mass of atherosclerotic plaques consists of highly plastic macrophages and numerous other cell types of myeloid origin as well as SMCs and endothelial cells. Alteration of the inflammatory genes in macrophages may transduce the signal to other cells and this cross talk between diverse cell types via the molecules they express ultimately governs the microenvironment and determines progression of the disease.15,28 It is becoming increasingly appreciated that atherosclerotic plaques are not simply masses of dead foam cells and other cellular debris, but contain live and proliferating cells that impact pathogenesis.57,58 In this context, we hypothesized that suppressing the expression of inflammatory genes directly within the cellular mass of plaques by an L13a-dependent mechanism could provide a natural defense against atherosclerosis. Consistent with this hypothesis, we identified substantial differences in the gene expression profiles of HFD-treated L13a KO and control (L13a intact) mice directly at the level of the cellular mass of atherosclerotic plaques (using high-throughput NanoString analysis with RNA from laser capture microdissection of aortic root sections; Figure 5B). Thus far, we have validated significant KO-specific increases in expression for 10 genes identified by NanoString analysis (Figure 5B,C), most of which have been previously shown to have proven roles in the pathogenesis of atherosclerosis. These include (i) von Willebrand factor (VWF), an endothelial-associated factor that facilitates platelet interactions and early atherogenesis64; (ii) Lipocalin (LCN) 2, which promotes polarization and migration of plaque macrophages65; (iii) Endoglin, an endothelial and SMC protein with a positive role in the pathogenesis of atherosclerosis66; (iv) S100a8, a protein that is expressed by inflammatory macrophages and promotes atherosclerosis67; (v) CCL2 (monocyte chemoattractant protein-1), a chemokine with a fundamental role in monocyte recruitment in the subendothelium (an early event in atherosclerosis)68,69; and (vi-vii) CXCL1270 and Pecam1,71 which are both endothelium cell-derived factors known to promote atherosclerosis and to be strongly associated with coronary artery diseases as shown by the presence of several SNPs in patients from multiple populations.72 It is important to note that the GAIT element-mediated atheroprotective mechanism that we have identified relies on L13a-dependent translational silencing of target genes.11 However, our data only shows increased abundance of the transcript levels for these genes in the absence of L13a (Figure 5B,C). As a part of our search for potential targets of L13a-dependent GAIT element-mediated translational control, our in silico analyses with FOLDALIGN and RNAstructure software tools identified potential GAIT elements in the 3′ UTRs of five of the 10 candidate targets identified by NanoString/RT-PCR (Figure 5D). Subsequent studies revealed the functional validation of the GAIT elements of CCL2, S100a8, and Tap1 (Figure 6D). However, based on studies deciphering the relationship between mRNA steady state levels and translation control, it is possible that absence of L13a-directed translational suppression could increase steady state mRNA levels by influencing mRNA stability.73,74 An additional or alternative possibility is that L13a-dependent GAIT element-mediated translational control may indirectly control transcription of these genes by targeting intermediate factor(s). Our future plans include studies aimed at deciphering the mechanism underlying how L13a-directed translational silencing impacts mRNA levels of particular genes and identifying the key molecular mediators that ultimately enhance the pathogenesis of atherosclerosis.
Another important facet of our work is the discovery that feeding ApoE−/− mice with a HFD not only induces inflammation that promotes atherosclerosis, but also triggers a program to resolve inflammation by activating L13a-dependent GAIT element-mediated translational silencing of chemokines and chemokine receptors. This is supported by our findings that activation of translational silencing was not observed upon chow diet feeding and that HFD treatment also triggers the release of L13a from 60S ribosome and the formation of L13a-dependent RNA-binding complex on GAIT element (Figure 6). Work by others using a cellular model of monocytes defined an essential role for the IFN-γ/DAP kinase Zip kinase axis in the process of phosphorylation-induced release of L13a and translational silencing.44 However, it is not currently known whether this signaling pathway also controls ribosomal release of L13a in macrophages in response to HFD treatment. Nevertheless, our results show that, similar to the resolution program triggered by other inflammatory stimuli of the innate immune system,75,76 HFD treatment in the ApoE−/− mouse model triggers a comparable resolution program through GAIT element-mediated translational silencing which provides a natural defense against atherosclerosis. In the future, pharmacological manipulation of this pathway through small molecule(s) capable of promoting L13a-dependent translational silencing could offer new therapeutic strategies to prevent or slow the progression of atherosclerosis.
Supplementary Material
Highlights.
High-fat diet (HFD)-induced L13a-dependent GAIT element-mediated translational silencing could be a natural defense against atherogenesis.
Treatment with HFD causes release of L13a from 60S ribosomal subunit, subsequent formation of L13a-dependent RNA-binding complex on GAIT element and translational silencing of mRNAs harboring GAIT elements at 3′UTRs.
Myeloid-specific loss of L13a causes pro-inflammatory differentiation of macrophages present in atherosclerotic plaque.
Loss of L13a in myeloid cells causes significant upregulation of several pro-atherosclerotic genes in the plaque.
ACKNOWLEDGMENTS
We thank Dr Jayeeta Dhar for help during the analysis of the NanoString data by volcano plots. We are grateful to Dr Patricia Stanhope Baker for help with article editing. This work was supported by National Institutes of Health (NIH) grants HL79164 (to B. Mazumder), PO1AI087586 (to WM Baldwin) and Shared instrumentation grant 1S10OD025252-01. B. Mazumder also acknowledges support from the Center for Gene Regulation in Health and Disease.
Funding information
National Heart, Lung, and Blood Institute (NHLBI), Grant/Award Number: HL79164; National Institute of Allergy and Infectious Diseases (NIAID), Grant/Award Number: AI087586; Cleveland State University, Center for Gene Regulation in Health and Disease
Abbreviations:
- BMDM
bone marrow-derived macrophages
- DSS
dextran sodium sulfate
- FACS
fluorescence activated cell sorting
- GAIT
gamma activated inhibitor of translation
- HFD
high-fat diet
- KO
knockout
- LDLR
low-density lipoprotein receptor
- LMD
laser microdissection
- LPS
lipopolysaccharide
- SMC
smooth muscle cell
Footnotes
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Mazumder B, Fox PL. Delayed translational silencing of ceruloplasmin transcript in gamma interferon-activated U937 monocytic cells: role of the 3′ untranslated region. Mol Cell Biol. 1999;19:6898–6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mazumder B, Seshadri V, Imataka H, Sonenberg N, Fox PL. Translational silencing of ceruloplasmin requires the essential elements of mRNA circularization: poly(A) tail, poly(A)-binding protein, and eukaryotic translation initiation factor 4G. Mol Cell Biol. 2001;21:6440–6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazumder B, Sampath P, Seshadri V, Maitra RK, DiCorleto PE, Fox PL. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell. 2003;115:187–198. [DOI] [PubMed] [Google Scholar]
- 4.Sampath P, Mazumder B, Seshadri V, Fox PL. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol Cell Biol. 2003;23:1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sampath P, Mazumder B, Seshadri V, et al. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004;119:195–208. [DOI] [PubMed] [Google Scholar]
- 6.Kapasi P, Chaudhuri S, Vyas K, et al. L13a blocks 48S assembly: role of a general initiation factor in mRNA-specific translational control. Mol Cell. 2007;25:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhuri S, Vyas K, Kapasi P, et al. Human ribosomal protein L13a is dispensable for canonical ribosome function but indispensable for efficient rRNA methylation. RNA. 2007;13:2224–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vyas K, Chaudhuri S, Leaman DW, et al. Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in gamma interferon-activated monocytes. Mol Cell Biol. 2009;29:458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basu A, Jain N, Tolbert BS, Komar AA, Mazumder B. Conserved structures formed by heterogeneous RNA sequences drive silencing of an inflammation responsive post-transcriptional operon. Nucleic Acids Res. 2017;45:12987–13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poddar D, Basu A, Baldwin WM 3rd, Kondratov RV, Barik S, Mazumder B. An extraribosomal function of ribosomal protein L13a in macrophages resolves inflammation. J Immunol. 2013;190:3600–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basu A, Poddar D, Robinet P, et al. Ribosomal protein L13a deficiency in macrophages promotes atherosclerosis by limiting translation control-dependent retardation of inflammation. ArteriosclerThromb Vasc Biol. 2014;34:533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poddar D, Kaur R, Baldwin WM 3rd, Mazumder B. L13a-dependent translational control in macrophages limits the pathogenesis of colitis. Cell Mol Immunol. 2016;13:816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Libby P Inflammation in atherosclerosis. Nature. 2002;420:868–874. [DOI] [PubMed] [Google Scholar]
- 14.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. [DOI] [PubMed] [Google Scholar]
- 15.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 20.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies-statement from ATVB council. Arterioscler Thromb Vasc Biol. 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Surra JC, Guillen N, Arbones-Mainar JM, et al. Sex as a profound modifier of atherosclerotic lesion development in apolipoprotein E-deficient mice with different genetic backgrounds. J Atheroscler Thromb. 2010;17:712–721. [DOI] [PubMed] [Google Scholar]
- 22.Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin J, Li H, Yang M, et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013;3:200–210. [DOI] [PubMed] [Google Scholar]
- 24.Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875–888. [DOI] [PubMed] [Google Scholar]
- 25.Shi J, Hua L, Harmer D, Li P, Ren G. Cre driver mice targeting macrophages. Methods Mol Biol. 2018;1784:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khallou-Laschet J, Varthaman A, Fornasa G, et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE. 2010;5:e8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leitinger N, Schulman IG. Phenotypic polarization of macrophages in atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:1120–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12:10–17. [DOI] [PubMed] [Google Scholar]
- 29.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nawaz A, Aminuddin A, Kado T, et al. CD206(+) M2-like macrophages regulate systemic glucose metabolism by inhibiting proliferation of adipocyte progenitors. Nat Commun. 2017;8:286–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM. Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci Rep. 2017;7:40144–40153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zernecke A, Weber C. Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc Res. 2010;86:192–201. [DOI] [PubMed] [Google Scholar]
- 35.Golledge J. Targeting chemokines in aortic aneurysm: could this be key to a novel therapy for a common problem? Arterioscler Thromb Vasc Biol. 2013;33:670–672. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani A, Garlanda C, Locati M. Macrophage diversity and polarization in atherosclerosis: a question of balance. Arterioscler Thromb Vasc Biol. 2009;29:1419–1423. [DOI] [PubMed] [Google Scholar]
- 37.Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26:1702–1711. [DOI] [PubMed] [Google Scholar]
- 38.Moore KJ, Kunjathoor VV, Koehn SL, et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kruth HS, Huang W, Ishii I, Zhang WY. Macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2002;277:34573–34580. [DOI] [PubMed] [Google Scholar]
- 40.Ye Q, Chen Y, Lei H, et al. Inflammatory stress increases unmodified LDL uptake via LDL receptor: an alternative pathway for macrophage foam-cell formation. Inflamm Res. 2009;58:809–818. [DOI] [PubMed] [Google Scholar]
- 41.Ye Q, Lei H, Fan Z, Zheng W, Zheng S. Difference in LDL receptor feedback regulation in macrophages and vascular smooth muscle cells: foam cell transformation under inflammatory stress. Inflammation. 2014;37:555–565. [DOI] [PubMed] [Google Scholar]
- 42.Schrijvers DM, De Meyer GR, Herman AG, Martinet W. Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res. 2007;73:470–480. [DOI] [PubMed] [Google Scholar]
- 43.Yurdagul A Jr, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front Cardiovasc Med. 2017;4:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukhopadhyay R, Ray PS, Arif A, Brady AK, Kinter M, Fox PL. DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol Cell. 2008;32:371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Havgaard JH, Lyngso RB, Stormo GD, Gorodkin J. Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics. 2005;21:1815–1824. [DOI] [PubMed] [Google Scholar]
- 46.Havgaard JH, Torarinsson E, Gorodkin J. Fast pairwise structural RNA alignments by pruning of the dynamical programming matrix. PLoS Comput Biol. 2007;3:1896–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torarinsson E, Havgaard JH, Gorodkin J. Multiple structural alignment and clustering of RNA sequences. Bioinformatics. 2007;23:926–932. [DOI] [PubMed] [Google Scholar]
- 48.Khyzha N, Alizada A, Wilson MD, Fish JE. Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med. 2017;23:332–347. [DOI] [PubMed] [Google Scholar]
- 49.Maiellaro K, Taylor WR. The role of the adventitia in vascular inflammation. Cardiovasc Res. 2007;75:640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramel D, Gayral S, Sarthou MK, Auge N, Negre-Salvayre A, Laffargue M. Immune and smooth muscle cells interactions in atherosclerosis: how to target a breaking bad dialogue? Front Pharmacol. 2019;10:1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moos MP, John N, Grabner R, et al. The lamina adventitia is the major site of immune cell accumulation in standard chow-fed apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2386–2391. [DOI] [PubMed] [Google Scholar]
- 52.Ionita MG, van den Borne P, Catanzariti LM, et al. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol. 2010;30:1842–1848. [DOI] [PubMed] [Google Scholar]
- 53.Naruko T, Ueda M, Haze K, et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–2900. [DOI] [PubMed] [Google Scholar]
- 54.Leclercq A, Houard X, Philippe M, et al. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J Leukoc Biol. 2007;82:1420–1429. [DOI] [PubMed] [Google Scholar]
- 55.Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rajamaki K, Lappalainen J, Oorni K, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE. 2010;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jenkins SJ, Ruckerl D, Cook PC, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52:223–261. [DOI] [PubMed] [Google Scholar]
- 60.Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc Natl Acad Sci U S A. 1979;76:333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koo C, Wernette-Hammond ME, Innerarity TL. Uptake of canine beta-very low density lipoproteins by mouse peritoneal macrophages is mediated by a low density lipoprotein receptor. J Biol Chem. 1986;261:11194–11201. [PubMed] [Google Scholar]
- 62.Ellsworth JL, Kraemer FB, Cooper AD. Transport of beta-very low density lipoproteins and chylomicron remnants by macrophages is mediated by the low density lipoprotein receptor pathway. J Biol Chem. 1987;262:2316–2325. [PubMed] [Google Scholar]
- 63.Koo C, Wernette-Hammond ME, Garcia Z, et al. Uptake of cholesterol-rich remnant lipoproteins by human monocyte-derived macrophages is mediated by low density lipoprotein receptors. J Clin Invest. 1988;81:1332–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu MD, Atkinson TM, Lindner JR. Platelets and von Willebrand factor in atherogenesis. Blood. 2017;129:1415–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oberoi R, Bogalle EP, Matthes LA, et al. Lipocalin (LCN) 2 mediates pro-atherosclerotic processes and is elevated in patients with coronary artery disease. PLoS ONE. 2015;10:e0137924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nachtigal P, Zemankova Vecerova L, Rathouska J, Strasky Z. The role of endoglin in atherosclerosis. Atherosclerosis. 2012;224:4–11. [DOI] [PubMed] [Google Scholar]
- 67.Averill MM, Kerkhoff C, Bornfeldt KE. S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler Thromb Vasc Biol. 2012;32:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gu L, Okada Y, Clinton SK, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281. [DOI] [PubMed] [Google Scholar]
- 69.Aiello RJ, Bourassa PA, Lindsey S, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1518–1525. [DOI] [PubMed] [Google Scholar]
- 70.Doring Y, van der Vorst EPC, Duchene J, et al. CXCL12 derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation. 2019;139:1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goel R, Schrank BR, Arora S, et al. Site-specific effects of PECAM-1 on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1996–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fava C, Montagnana M. Atherosclerosis is an inflammatory disease which lacks a common anti-inflammatory therapy: how human genetics can help to this issue. A narrative review. Front Pharmacol. 2018;9:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Radhakrishnan A, Green R. Connections underlying translation and mRNA stability. J Mol Biol. 2016;428:3558–3564. [DOI] [PubMed] [Google Scholar]
- 74.Wu Q, Medina SG, Kushawah G, et al. Translation affects mRNA stability in a codon-dependent manner in human cells, eLife. 8:45396–45417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. [DOI] [PubMed] [Google Scholar]
- 76.Ortega-Gomez A, Perretti M, Soehnlein O. Resolution of inflammation: an integrated view. EMBO Mol Med. 2013;5:661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.