

Abstract

Hepatic steatosis (fatty liver) is a severe liver disease induced by the excessive accumulation of fatty acids in hepatocytes. In this study, we developed reliable in silico models for predicting hepatic steatosis on the basis of an in vivo data set of 1041 compounds measured in rodent studies with repeated oral exposure. The imbalanced nature of the data set (1:8, with the “steatotic” compounds belonging to the minority class) required the use of meta-classifiers—bagging with stratified under-sampling and Mondrian conformal prediction—on top of the base classifier random forest. One major goal was the investigation of the influence of different descriptor combinations on model performance (tested by predicting an external validation set): physicochemical descriptors (RDKit), ToxPrint features, as well as predictions from in silico nuclear receptor and transporter models. All models based upon descriptor combinations including physicochemical features led to reasonable balanced accuracies (BAs between 0.65 and 0.69 for the respective models). Combining physicochemical features with transporter predictions and further with ToxPrint features gave the best performing model (BAs up to 0.7 and efficiencies of 0.82). Whereas both meta-classifiers proved useful for this highly imbalanced toxicity data set, the conformal prediction framework also guarantees the error level and thus might be favored for future studies in the field of predictive toxicology.
Introduction
Hepatic steatosis (HS; also termed “fatty liver”) is a well-known and often observed condition in the human population and characterized by the accumulation of lipids/fat in the liver. HS can progress to steatohepatitis and irreversible stages of liver disease including fibrosis, cirrhosis, hepatocellular carcinoma, and death.
Alcoholic liver disease (ALD), directly caused by alcohol misuse, is the primary cause of fatty liver disease, and the second leading cause is nonalcoholic fatty liver disease (NAFLD). NAFLD can result from different exposure conditions such as high-fat diets, exposure to industrial chemicals and environmental pollutants,1 or pharmaceuticals.2,3 The progression of NAFLD might also be a result of insulin resistance, changes in microbiota, or predisposing genetic variants resulting in a disturbed lipid homeostasis.4
Typically, the pathological manifestation of HS is characterized by an excessive accumulation of triglycerides in vacuoles in the cytosol of hepatocytes, leading to macrovesicular or microvesicular steatosis. Macrovesicular steatosis usually shows single large vacuoles in the cytoplasm of hepatocytes with nuclear displacement, whereas microvesicular steatosis is characterized by small diffuse lipid droplets.5
Several mechanisms are in discussion to be involved in fatty degeneration of the liver. Recently, Angrish et al. summarized these mechanisms as an imbalance between four key events (KEs), namely hepatic fatty acid (FA) uptake, increased de novo FA and lipid synthesis, decreased hepatic FA oxidation, and/or decreased hepatic secretion of very low-density lipoproteins.6
Drugs like valproic acid, for example, impair mitochondrial β-oxidation by binding to coenzyme-A, which leads to a disruption of lipid metabolism and finally results in microvesicular steatosis.7,8 In extreme cases, drug withdrawals due to hepatic toxicity can be the consequence.9
Several nuclear receptors like peroxisome proliferator-activated receptor (PPAR) α are in discussion to be involved in the development of steatosis as well as mitochondria-derived oxidative stress.10 The information on which molecular initiating events (MIEs) lead to an adverse outcome (AO) via one or several key events (KE) is stored in so-called adverse outcome pathways (AOPs).11 Such modular assemblies of KEs and key event relationships (KERs) in the framework of an AOP also allow to fuse several independent AOPs into a more comprehensive, integrated, and biologically realistic AOP network. It is, however, not in the scope of this article to review all potential mechanisms and KEs that trigger HS, and reference is given to the AOP-Wiki,12 WikiPathways13 (WP4011), and recent reviews.14−16
Recently, the information on involved MIEs from a comprehensive AOP network (a synthesis of six different AOPs for steatosis) was utilized to build quantitative structure–activity relationship (QSAR) models for the MIEs17 based on in vitro data extracted from ToxCast.18 In total, sufficient data for six nuclear receptors, peroxisome proliferator-activated receptors (PPAR α and γ), pregnane X receptor (PXR), aryl hydrocarbon receptor (AhR), liver X receptor (LXR), and nuclear factor (erythroid-derived 2)-like 2 (Nrf2), are available in the open domain and could be leveraged for this purpose.17
Other predictive models estimating the potential of a compound to cause liver toxicity have been reported previously, for example, addressing hyperbilirubinemia19 or drug-induced liver injury (DILI).20
One possibility is to focus on liver transporters, which have been reported to be involved in drug–drug interactions and liver toxicity, such as P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), MRP2-4, bile salt export pump (BSEP), and hepatic organic anion transporting polypeptides (OATP1B1, OATP1B3, and OATP2B1). Models predicting the potential of a compound to interact with one of these transporters are available from, for example, the Vienna LiverTox Workspace.21
Very recently, other groups have published in silico models to predict hepatic steatosis, but based on relatively small data sets or with a focus on drugs/drug metabolites only. For example, the models developed by Cotterill et al.22 are based on a relatively small data set of 207 compounds measured in vivo (half of which have been annotated as in vivo steatosis positive). Shin et al.23 used a list of 165 drugs and 223 drug metabolites with toxicity annotations retrieved from PharmaPendium,24 Elsevier’s collection of drug approval documents, and extracted data for informing drug development decisions.
The main objective of the presented study was the development of QSAR/machine learning (ML) models which predict the potential of a given compound to cause liver steatosis in the human organism based on in vivo data and covering a large chemical space. The development of QSAR models requires large training data sets (TR) and test data sets (TS), including steatotic and nonsteatotic compounds. As human in vivo data were not available, we extracted this information from high-quality databases comprising in vivo animal studies with repeated oral exposure. Information on chemicals/pesticides were obtained from the RepDose,25 ToxRefDB,26 and Hazard Evaluation Support System (HESS) database.27 The apical adverse effects described in these preclinical studies did not allow to distinguish between marcrovesicular or microvesicular liver steatosis nor indicate the underlying mechanism.
The envisaged models should therefore be able to generalize and allow reliable predictions independent of the concrete MIE(s) and KE(s) being triggered by a particular compound. Due to the high structural diversity of the data set, the extraction of relevant features that can well separate the active from the inactive class is aggravated. Another challenging aspect in this study is caused by the imbalanced nature of the in vivo data set, comprising an imbalance ratio of approximately 1:8 with the active (steatotic) class being the minority class. We tackled this challenge by applying both bagging with stratified under-sampling (hence called “stratified bagging”; SB) as well as Mondrian conformal prediction (CP) as meta-classifiers (an additional processing step that is performed before and after the actual base-classifier sees the data, respectively; SB and CP are both explained in more detail in the Data and Methods section).
Most importantly, in this study the influence of the inclusion of mechanistic information (from the steatosis AOP and literature knowledge) on model performance was inspected. By training models with/without predictions from nuclear receptor (NR) models and/or transporter models as part of the features for model building, it should be possible to prioritize certain feature blocks and consequently determine the influence of specific protein targets in mediating HS. The models were trained in an automated fashion via iterative combination of descriptor blocks. KNIME workflows and python scripts are publicly available from https://github.com/BZdrazil/Steatosis_prediction.
Results
Composition of the Data Sets and Chemical Space Analysis
The initial data set of in vivo measurements for HS in rodents (collected from RepDose,25 ToxRefDB,26 and HESS DB27) is composed of 120 active and 921 inactive compounds after data curation, representing an imbalance ratio of 1:8. By splitting off a validation set/test set (TS) composed of 30% of the data set entries and maintaining the ratio of the active vs inactive class (stratified sampling), the final training set (TR) was made up of 727 compounds (86 in vivo steatosis positives/641 in vivo steatosis negatives; Figure 1). Visualizing TR vs TS in a two-dimensional t-distributed stochastic neighbor embedding (t-SNE)28 plot helped to verify that the TS compounds have been selected in an unbiased way, representing the full range of chemical space like the TR compounds (Supplementary Figure S1).
Figure 1.
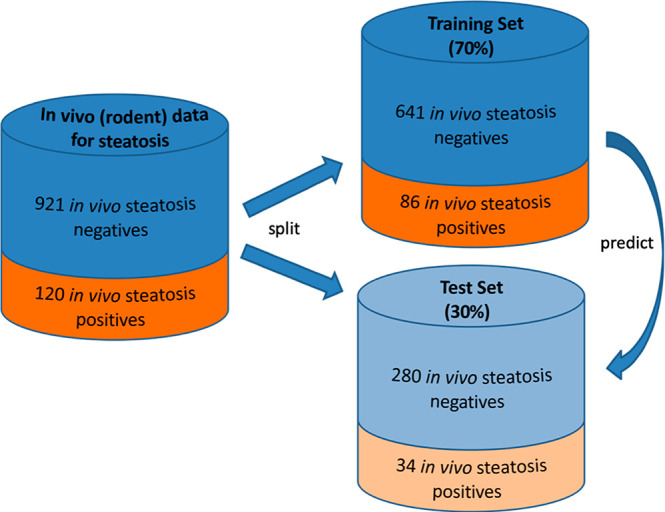
Composition of the data sets used in this study. The in vivo data set for repeated dose toxicity was split into training and test sets by maintaining the relative distribution of active vs inactive compounds.
Manual inspection of the training and test set compounds revealed a great structural diversity of the compounds within the in vivo steatosis data set. With the aim to quantify as well as qualitatively describe this structural diversity, we performed a similarity analysis of test vs training set compounds and an analysis of feature distribution among the active vs inactive class of compounds as well as an analysis of enriched scaffolds in the data set.
Checking for similarity of each test set compound to the closest similar compound in the training set revealed that >85% of the test set compounds do possess a very low similarity to training set compounds (Tanimoto coefficient <0.6; Supplementary Figure S2). This is an important finding since it suggests robustness of the established models when applied to new chemical matter (that will likely be also dissimilar to the training set compounds).
Notably, the inactive class of compounds seems to be the main contributor to high chemical diversity in our data set, since the range of values for seven out of eight calculated common physicochemical properties (partition coefficient/lipophilicity, topological polar surface area, molecular weight, number of rotatable bonds, number of hydrogen-bond donors and acceptors, and number of aromatic rings) appears to be much larger for this class (Figure 2). However, visually inspecting the difference of the mean and median values of the active vs inactive class of the inspected properties (Figure 2, Supplementary Table S1) as well as testing for statistical significance of the distributions by the Kolmogorov–Smirnov test revealed that only for SlogP and atomic molecular weight (AMW), the two classes are actually statistically different (p-value <0.05).
Figure 2.

Violin plots showing the physicochemical property distribution in the in vivo steatosis data set (including TR and TS). The distributions for steatosis negative and positive compounds are plotted separately (0, steatosis negative; 1, steatosis positive). SlogP, partition coefficient; TPSA, topological polar surface area; AMW, atomic molecular weight; RotB, number of rotatable bonds; HBD, number of hydrogen-bond donors; HBA, number of hydrogen bond acceptors; Fsp3, fraction of sp3-hybridized carbons; AromRings, number of aromatic rings; QED, quantitative estimate of drug-likeness.
Regarding the general features related to oral bioavailability, steatosis positive compounds appear to be on average more lipophilic (median SlogP: 2.7 vs 2.2 for positives vs negatives), to possess a higher molecular weight (median AMW: 293 vs 206), to show a higher number of rotatable bonds/greater flexibility (median RotB: 3 vs 2), a lower number of hydrogen-bond donor (HBD) (median HBD: 0 vs 1), and a higher number of hydrogen-bond acceptor (HBA) (median HBA: 3 vs 2) than the larger class of steatosis negative compounds (Supplementary Table S1).
The mean and median values for the number of aromatic rings (AromRings) and the fraction of hybridized carbon atoms (Fsp3) are in the same range for both classes (Supplementary Table S1), but AromRings in some cases show much more extreme values (up to 10 aromatic rings) for the inactive class. In general, the data set (both classes) shows a relatively high degree of aromaticity and thus higher planarity of the compounds (also demonstrated by the relatively low mean and median Fsp3 values (around 0.4) for both classes (Supplementary Table S1).
Looking a bit closer into the class of steatosis positive compounds (120 compounds) also reveals that most of the positive compounds appear as drug-like according to the rule-of-five, since the interquartile ranges appear below the R5 thresholds (MW < 500; logP ≤ 5; HBD ≤ 5; HBA ≤ 10; Supplementary Figure S3). Calculating the quantitative estimate of drug-likeness (QED) as defined by Bickerton et al.29 also reflects this trend of a relatively high drug-likeness in the data set with both the mean and median QED values being around 0.54 for both the steatotic and nonsteatotic class (Supplementary Table S1).
An orthogonal measure for the structural diversity of a chemical data set is delivered by analyzing the frequency of core scaffolds. By applying the Murcko scaffold algorithm,30 316 different Murcko scaffolds were identified within the in vivo data set made up of 1041 compounds in total. This translates into an average scaffold-to-compound ratio of 0.30, indicating a very large structural diversity of the data set. A more detailed analysis of scaffold frequency is presented in Figure 3, showing the most frequent scaffolds in our data set (occurring in at least 20 different compounds). Strikingly, only the benzene scaffolds can be found with very high frequency (approximately 58% of the data set compounds), but any other scaffold is not more prevalent than in 5% of compounds at maximum, indicating again a high structural diversity of the training and test set (Figure 3). With respect to the nature of those relatively enriched scaffolds, most of them contain aromatic ring systems (pyridine, biphenyl, pyrimidine, diphenyl ether, naphthalene, and 1,3,5-triazine), which confirms the finding of a high degree of planarity/aromaticity within the data set.
Figure 3.

Bar plot showing the most frequent Bemis–Murcko scaffolds (occurring in at least 20 compounds) in the in vivo steatosis data set (TR and TS). Absolute numbers of compounds possessing the respective scaffold (upper numbers) and relative percentages of frequencies within the data set (lower numbers) are indicated. The proportion of acyclic compounds in the data set is indicated by the outer right bar.
Development of In Silico Profilers for Hepatic Steatosis
In order to provide an unbiased summary of enriched chemical (sub)structures in the steatosis positive class which probably could serve as in silico profilers (structural alerts) for HS, we carried out enrichment analyses of scaffolds and ToxPrint chemotypes31 in the positive vs negative class of the curated in vivo data set (including both TR and TS).
The enrichment analyses accounted for the imbalance of the data set by calculating the relative frequency of a certain scaffold/substructure. Fisher’s exact test was used for extracting the statistical significance of the different frequencies in the active vs inactive class.
Applying such frequency analysis at the level of Murcko scaffolds (as described by Türkova et al.)32 revealed that only three scaffolds appear to be significantly enriched (p-value ≤0.05). However, for these cases, the number of compounds possessing the respective scaffold in the active class is rather small, with 1-(2-phenylethyl)-1H-1,2,4-triazole occurring most often (five unique compounds; Supplementary Table S2).
At the level of substructural fragments by using the ChemoTyper program,31 21 structural fragments are statistically significantly (p-values ≤0.05) enriched in the positive class vs the negative class (Supplementary Table S3) when considering only fragments with an occurrence in at least 5 active compounds (92 ToxPrint chemotypes). Eight of these structural alerts are at the same time occurring in at least 10% of compounds in the active class. Interestingly, and six of these eight highlighted substructural patterns include halogens (see Supplementary Table S3).
Halo-alkanes and halo-alkenes have the potential to induce toxicity and cancer depending on how well they can form a stable carbon-centered radical. The more halide groups they possess, the more likely a radical formation will occur since the electron-withdrawing capacity from the carbon center will increase. Reactive intermediates can further bind to macromolecules and disrupt lipid metabolism leading to HS.33
Iterative Descriptor Combination via a Semiautomatic Machine Learning Pipeline
The main aim of this study was the generation of a highly predictive binary classification model for predicting HS of chemicals with unknown toxicity. In order to find the most favorable amalgamation of features which would give the “best model”, we built a KNIME workflow which allows iterative combination of different descriptor sets in an automated fashion. In addition, model generation and validation (based on internal and external validation) were fully automatized as part of this workflow, delivering a summary table of the respective model parameters for assessing the different model accuracies (such as sensitivity, specificity, balanced accuracy, and Matthews correlation coefficient) (Figure 4). The workflow includes an in house established meta-node for performing SB in an iterated and automated fashion as a technique to overcome problems of an imbalanced data set distribution (of actives and inactives). The workflow is openly available from https://github.com/BZdrazil/Steatosis_prediction and can be adjusted for other use cases.
Figure 4.

Illustration of the semiautomatic KNIME workflow used for model generation with the SB modeling framework.
Importantly, testing different descriptor types was also used for evaluating the potential influence of a particular descriptor block for predicting steatosis. In the case of the use of predictions from transporter and NR models as feature blocks, a potential mechanistic role of these proteins in triggering the onset or development of steatosis could be deduced if the differences in predictive power were significant (see next subchapter for concrete results).
Predictive Models for Hepatic Steatosis: Stratified Bagging and Conformal Prediction as Valuable Strategies to Deal with Highly Imbalanced Data Sets
A random forest (RF) classifier34 was trained on a set of 727 compounds (= TR) with associated class labels (in vivo steatosis positive or negative) by using different combinations of descriptor blocks: 26 RDKit physicochemical properties,35 ToxPrint substructure fingerprints,31 predictions from in vitro NR, and hepatic transporter models. The incentive to include information from in vitro models by using their binary predictions as a feature vector (in the form of a bit string) is to investigate if a potential mechanistic link of these protein targets to the development of HS exists. A set of transcription factors has been previously identified as MIEs linked to the discussed AO, and efforts to model steatosis by using a consensus of in vitro NR models were reported in literature.17 Since NR proteins have been shown to be involved in the transcription of drug transporters,36 alteration in nuclear transcription factor activation may result in altered expression of transporters.37 In addition, direct impairment of a liver transporters’ function (e.g., through inhibition of the transporter by small molecules) might induce liver toxicity (such as hepatosteatosis, nonalcoholic fatty liver disease, cholestasis, etc.). A similar approach was already tested for predicting hyperbilirubinemia by including the information from in vitro inhibition models for OATP1B1 and OATP1B3.19 Since in this previous work no improvement of the predictive models could be observed in terms of model accuracy, we tested here the inclusion of additional transporter models (especially ABC transporter models) as well as NR models.
All descriptor combinations were tested iteratively and with two different methods for handling the imbalanced distribution of steatotic vs nonsteatotic compounds in our data set: bagging with stratified under-sampling (SB) and conformal prediction (CP). In addition, our validation procedure was performed in a two-step fashion: including internal validation and the prediction of a TS that was split off from the TR data set before model building. Since the combination of the base classifier (RF) with SB or CP allows it, we sampled this procedure 64 and 100 times, respectively, and calculated the average and median predictions, respectively, from these independent runs.
The necessity of including algorithms to counterbalance problems occurring when performing ML with (highly) unbalanced data sets is demonstrated by the performance achieved for the baseline model (RF classifier, RDKit descriptors; see Table 1). Whereas the specificity (true negative rate) of the model is extremely high (0.98 and 0.99 for TR and TS predictions, respectively), it completely fails to predict the toxic class (sensitivity: 0.02 and 0.09 for TR and TS). It is therefore important to not rely on merely decent balanced accuracies (BA) of the generated models, but in addition to always study the true positive and true negative rate separately.
Table 1. Results from SB Models Compared to the Baseline Model (RDKit_baseline)a.
| descriptors | validation set | sensitivity | specificity | BA | MCC |
|---|---|---|---|---|---|
| RDKit_baseline | TR | 0.02 | 0.98 | 0.50 | 0.02 |
| TS | 0.09 | 0.99 | 0.54 | 0.18 | |
| RDKit | TR | 0.51 | 0.74 | 0.63 | 0.18 |
| TS | 0.62 | 0.77 | 0.69 | 0.27 | |
| 5 selected RDKit | TR | 0.50 | 0.73 | 0.61 | 0.16 |
| TS | 0.52 | 0.78 | 0.65 | 0.21 | |
| ToxPrint | TR | 0.43 | 0.80 | 0.61 | 0.17 |
| TS | 0.47 | 0.78 | 0.63 | 0.19 | |
| transporter prediction | TR | 0.39 | 0.73 | 0.56 | 0.09 |
| TS | 0.52 | 0.76 | 0.64 | 0.20 | |
| NR prediction | TR | 0.41 | 0.63 | 0.52 | 0.03 |
| TS | 0.46 | 0.61 | 0.53 | 0.04 | |
| RDKit + ToxPrint | TR | 0.51 | 0.77 | 0.64 | 0.20 |
| TS | 0.57 | 0.79 | 0.68 | 0.26 | |
| RDKit + transporter prediction | TR | 0.54 | 0.74 | 0.64 | 0.20 |
| TS | 0.64 | 0.76 | 0.70 | 0.28 | |
| RDKit + NR prediction | TR | 0.53 | 0.75 | 0.64 | 0.20 |
| TS | 0.58 | 0.78 | 0.68 | 0.25 | |
| ToxPrint + transporter prediction | TR | 0.45 | 0.80 | 0.62 | 0.19 |
| TS | 0.54 | 0.79 | 0.67 | 0.24 | |
| ToxPrint + NR prediction | TR | 0.41 | 0.80 | 0.61 | 0.16 |
| TS | 0.48 | 0.78 | 0.63 | 0.19 | |
| transporter prediction + NR prediction | TR | 0.48 | 0.64 | 0.56 | 0.08 |
| TS | 0.52 | 0.63 | 0.58 | 0.10 | |
| RDKit + ToxPrint + transporter prediction | TR | 0.51 | 0.78 | 0.64 | 0.21 |
| TS | 0.59 | 0.79 | 0.69 | 0.27 | |
| RDKit + transporter prediction + NR prediction | TR | 0.53 | 0.74 | 0.64 | 0.20 |
| TS | 0.60 | 0.78 | 0.69 | 0.27 | |
| ToxPrint + transporter prediction + NR prediction | TR | 0.43 | 0.80 | 0.62 | 0.18 |
| TS | 0.51 | 0.80 | 0.65 | 0.23 | |
| RDKit + ToxPrint + NR prediction | TR | 0.50 | 0.78 | 0.64 | 0.21 |
| TS | 0.53 | 0.81 | 0.67 | 0.25 | |
| RDKit + ToxPrint + transporter prediction + NR prediction | TR | 0.49 | 0.79 | 0.64 | 0.21 |
| TS | 0.57 | 0.80 | 0.69 | 0.27 |
Performances on training set (TR) and test set (TS) are shown. BA is balanced accuracy, and MCC is Matthews correlation coefficient.
Stratified Bagging Models
With respect to overall model performances, balanced accuracies for TS predictions with the SB models are ranging from 0.53 (MCC = 0.04; models built with NR predictions as descriptors) to 0.70 (MCC = 0.28) for the different models built upon different descriptor combinations (for TR predictions BAs range from 0.52 to 0.64). Whereas the sole use of NR predictions, ToxPrint features, or transporter predictions as descriptors does not give good model performances, the combination of these features with physicochemical descriptors (RDKit) leads to good model performances, with the best model being achieved by combining RDKit descriptors with transporter predictions (BA = 0.70). However, a model built on the basis of RDKit descriptors alone is performing reasonably well overall (TS predictions: BA = 0.69/MCC = 0.27), and the observed performance differences are not statistically significant, though it can be observed that there is a tendency that best performing models include transporter predictions. Looking closer at the predictive capacities of the SB models for the separate classes, it becomes obvious that the SB models generally are still better at predicting the nonsteatotic class, but that some descriptors/descriptor combinations seem to better be suited for achieving good sensitivities. Interestingly, the best overall performing SB model—RDKit, features in combination with transporter predictions—is also the one with the highest sensitivities for both TR predictions (sensitivity = 0.54) and TS predictions (sensitivity = 0.64).
Further, the contribution of the predictions of the individual transporter inhibition models was investigated. Because the influence of an individual transporter feature block to the overall model performance would be hard to detect if a large number of other descriptors are being deployed, only the five most important physicochemical descriptors were filtered out and combined with individual transporter predictions. An analysis of descriptor importance and intercorrelation of features, combined with knowledge gained previously by analyzing the mean and median values of the respective features comparing the inactive (nonsteatotic) and active (steatotic) class, revealed that a combination of SlogP (lipophilicity), molecular weight (AMW), number of rotatable binds (NumRotatableBonds), HBDs, and topological polar surface area (TPSA) delivers a reasonable reduced descriptor set (TS prediction for the SB model: BA = 0.65/MCC = 0.21). Combining these five features with the predictions of any of the transporter models increases model performance up to a BA of 0.66–0.68 (MCC = 0.23–0.25; see Supplementary Table S4) with none of the individual transporters outperforming the others.
Conformal Prediction Models
The CP models at the significance level 0.3 (30% errors accepted) are valid (meaning that the number of errors it commits does not exceed the chosen confidence level) for all models accounting for small statistical fluctuations except for TS predictions of four models. For the model based on ToxPrint features combined with NR predictions (steatotic class), the respective validity reached only a value of 0.68. Models based on ToxPrint features alone, in combination with Transporter predictions, as well as the model based on transporter and NR predictions in combination show a validity for the nonsteatotic class of 0.69, respectively (Table 2 and Supplementary Figure S4).
Table 2. Results from CP Models at the Significance Level 0.3a.
| descriptors | validation set | validity steatotic class | validity nonsteatotic class | efficiency | sensitivity | specificity | BA | MCC |
|---|---|---|---|---|---|---|---|---|
| RDKit | TR | 0.73 | 0.72 | 0.69 | 0.62 | 0.59 | 0.61 | 0.14 |
| TS | 0.74 | 0.71 | 0.75 | 0.67 | 0.61 | 0.64 | 0.18 | |
| 5 selected RDKit | TR | 0.77 | 0.73 | 0.71 | 0.67 | 0.61 | 0.64 | 0.18 |
| TS | 0.76 | 0.79 | 0.71 | 0.67 | 0.7 | 0.69 | 0.24 | |
| ToxPrint | TR | 0.77 | 0.71 | 0.71 | 0.67 | 0.59 | 0.63 | 0.17 |
| TS | 0.71 | 0.69 | 0.74 | 0.63 | 0.57 | 0.6 | 0.13 | |
| transporter prediction | TR | 0.86 | 0.72 | 0.41 | 0.76 | 0.28 | 0.52 | 0.03 |
| TS | 0.88 | 0.76 | 0.44 | 0.82 | 0.43 | 0.62 | 0.18 | |
| NR prediction | TR | 0.76 | 0.72 | 0.52 | 0.57 | 0.46 | 0.52 | 0.02 |
| TS | 0.79 | 0.74 | 0.5 | 0.61 | 0.48 | 0.54 | 0.06 | |
| RDKit + ToxPrint | TR | 0.78 | 0.73 | 0.69 | 0.68 | 0.61 | 0.64 | 0.19 |
| TS | 0.74 | 0.71 | 0.82 | 0.71 | 0.65 | 0.68 | 0.24 | |
| RDKit + transporter prediction | TR | 0.73 | 0.73 | 0.69 | 0.63 | 0.6 | 0.62 | 0.16 |
| TS | 0.74 | 0.71 | 0.77 | 0.69 | 0.62 | 0.65 | 0.2 | |
| RDKit + NR prediction | TR | 0.74 | 0.72 | 0.69 | 0.65 | 0.6 | 0.62 | 0.16 |
| TS | 0.76 | 0.74 | 0.73 | 0.71 | 0.63 | 0.67 | 0.23 | |
| ToxPrint + transporter prediction | TR | 0.78 | 0.71 | 0.72 | 0.7 | 0.6 | 0.65 | 0.2 |
| TS | 0.74 | 0.69 | 0.77 | 0.67 | 0.6 | 0.63 | 0.17 | |
| ToxPrint + NR prediction | TR | 0.8 | 0.73 | 0.69 | 0.71 | 0.61 | 0.66 | 0.2 |
| TS | 0.68 | 0.71 | 0.76 | 0.61 | 0.62 | 0.61 | 0.15 | |
| transporter prediction + NR prediction | TR | 0.8 | 0.72 | 0.54 | 0.61 | 0.48 | 0.55 | 0.06 |
| TS | 0.79 | 0.69 | 0.6 | 0.73 | 0.47 | 0.6 | 0.14 | |
| RDKit + ToxPrint + transporter prediction | TR | 0.81 | 0.71 | 0.69 | 0.72 | 0.58 | 0.65 | 0.2 |
| TS | 0.74 | 0.72 | 0.82 | 0.71 | 0.65 | 0.68 | 0.24 | |
| RDKit + transporter prediction + NR prediction | TR | 0.77 | 0.74 | 0.67 | 0.67 | 0.61 | 0.64 | 0.18 |
| TS | 0.71 | 0.73 | 0.76 | 0.67 | 0.64 | 0.65 | 0.21 | |
| ToxPrint + transporter prediction + NR prediction | TR | 0.79 | 0.71 | 0.71 | 0.71 | 0.59 | 0.65 | 0.20 |
| TS | 0.71 | 0.70 | 0.80 | 0.66 | 0.62 | 0.64 | 0.18 | |
| RDKit + ToxPrint + NR prediction | TR | 0.76 | 0.71 | 0.73 | 0.66 | 0.61 | 0.63 | 0.17 |
| TS | 0.74 | 0.74 | 0.79 | 0.69 | 0.67 | 0.68 | 0.24 | |
| RDKit + ToxPrint + transporter prediction + NR prediction | TR | 0.78 | 0.72 | 0.7 | 0.65 | 0.61 | 0.63 | 0.17 |
| TS | 0.74 | 0.73 | 0.8 | 0.7 | 0.65 | 0.68 | 0.23 |
Performances on training set (TR) and test set (TS) are shown. BA is balanced accuracy, and MCC is Matthews correlation coefficient.
Evaluating the performances of the models on the basis of BA and efficiency (only single label predictions), just three models show poor performances, namely models built with NR predictions as features or transporter predictions as features, or the combination of both (see Table 2 and Supplementary Figure S5). For models built on the basis of ToxPrint features, BAs for the TR and TS are not overwhelming (0.63 and 0.6), but the efficiency is generally reaching higher values than for the other just mentioned descriptor sets (0.71 and 0.74). All other models are performing comparably well with not much difference regarding their predictive performances. Like in the case of SB models, the best model performances overall are achieved by combining RDKit features with other features, such as the “RDKit + ToxPrint model” and the “RDKit + ToxPrint + transporter model”, which are both showing the best combination of values for efficiency and BA (0.82 and 0.68, respectively). This example also shows the usefulness of CP per se, since the method allows to detect the influence of ambiguous compounds which are assigned to the “both” or “empty” class, respectively. Whereas no real difference in performances can be detected on the basis of the BAs comparing, for example, models built on the basis of the five selected RDKit features and the just mentioned best performing models (“RDKit + ToxPrint model” and the “RDKit + ToxPrint + transporter model”), we can see much better performances for the best models when inspecting the respective efficiencies (0.71 for the “5 selected RDKit model” vs 0.82 for the TS predictions of these best models). Refining the models by these additional features obviously helped to increase the percentage of unambiguous class assignments (single class predictions).
Analyzing the distribution of compounds with different class assignments (steatotic, nonsteatotic, and both) in chemical space by performing a t-SNE projection in two-dimensions on the basis of the RDKit descriptors shows a fair separation of the steatotic (“high”) and nonsteatotic (“low”) classes in chemical space (Figure 5 and Supplementary Figure S6). Compounds predicted as belonging to the “both” category tend to be closer positioned to instances from the steatotic class. This shows that the classifier is missing information for those compounds (assigned to the “both” class) needed to assign a single label to these instances, but would rather assign them to the steatotic class if a better discriminative power was given.
Figure 5.

Two-dimensional t-SNE projection of the in vivo steatosis data set (TR and TS compounds): The chemical space projection is based on 26 physicochemical RDKit descriptors. The color code indicates compounds which have been predicted by the CP framework based on these RDKit features (at significance level 0.3) to belong to either the “high” (= steatotic), “low”(= nonsteatotic), or “both” classes (colored in light blue, green, and brown, respectively).
Avoiding an unreasonably high number of compounds belonging to the “both” class can also be achieved by the set significance level that the model is supposed to achieve. Accepting a lower error rate (as in the case of significance levels lower than 0.3) leads to a steady increase of compounds being assigned to the “both” class for the TR as well as the TS data set (Supplementary Figure S7).
Interestingly, although leading to poorly performing models overall, models built on the basis of only transporter predictions are showing the highest sensitivities (0.76 and 0.82 for TR and TS) and steatotic class validities (0.86 and 0.88 for TR and TS) among all CP models. This behavior might also explain the benefits of including transporter predictions, however this effect is not clearly visible from a statistical standpoint when comparing model performances.
Compared to the SB models, CP models are in general showing higher sensitivities: 0.39–0.64 for the various SB models (mean sensitivity = 0.51) and 0.57–0.82 for CP models (mean sensitivity = 0.68). Since in Mondrian CP the conformity function is applied to the calibration instances separately for the active and inactive class, validity can be guaranteed for each separate class label. Thus, the CP framework works well for highly imbalanced data sets and gives good predictive power also for the minority class.
From a statistical standpoint, although there are slight variations among the CP models, no descriptor set is performing significantly better than another at a 95% confidence interval (with Bonferroni correction; see Supplementary Figure S8).
Discussion
In this paper, the generation of reliable ML models for predicting HS based on small molecules tested in vivo in rodents is described. The best performing models developed in this investigation enable the classification of chemicals with unknown toxicity into “steatotic” or “nonsteatotic” compounds with a balanced accuracy of up to 0.7 (MCC = 0.28). Models built within the CP framework in addition allow compounds to be classified as ambiguous (by falling into the “both” category). To the best of our knowledge, these are the first published models built on basis of in vivo data for predicting HS.
The integration of in vitro data into these models has been tested by utilizing predictions from in silico models (that were built on basis of in vitro data) for certain protein targets which have been recognized as being linked to HS. Combining different feature blocks, including predictions from several NR models and transporter models, did not allow to select one model that statistically outperforms others, but identified a few poorly performing models (models with features made up of solely NR or transporter predictions). Including physicochemical properties as part of the feature set seems beneficial as well as adding predictions from the transporter in vitro models as features, since all of the (numerically) “best” models are including this combination.
Since the in vivo data set possesses a very broad chemical diversity of compounds, it is very difficult to identify a definitive list of important descriptors able to well separate steatotic from nonsteatotic compounds. Since this is a target-agnostic data set, potentially including steatotic compounds triggering diverse branches of the steatosis AOP, it is likely that multiple modes of action are being leveraged contributing to the chemical diversity of the data set. Several MIEs were identified to be involved in the published AOP, including mainly the activation of NR by small molecules.
Although observed performance differences are statistically insignificant, our investigations suggest to further inspect the role of transporters in causing HS and eventually include them into newer AOPs for HS.
Investigations on class membership of model predictions and chemical space analysis indicate that substantial information is still missing for the classifier to reliably assign single labels to a substantial number of the compounds, which are predicted to fall into the “both” category. Such information can be easily added, once more knowledge about the more precise AOP becomes available. The modular structure of the developed KNIME workflows allows for an easy addition of features (and also the python scripts for CP models are easily adaptable).
This finding is, however, also an indication for a preferential use of CP as the method of choice for such challenging classification tasks where (a) an in vivo toxicity end point is to be predicted, (b) classes are not represented equally, and (c) the known mechanistic information (e.g., AOP) indicate multiple (branches of a) regulatory pathway(s) to be involved.
In conclusion, we have observed that both meta-classifiers (SB and CP) are proving useful for handling the highly imbalanced nature of the toxicity data set, but the CP framework allows for “predictions with confidence”, which are of particular relevance in the field of predictive toxicology where the guaranteed error level allows firm decisions to be made about the toxicity of a chemical compound.
Data and Methods
TR and TS Preparation
In vivo data from rodent studies with repeated oral exposure were extracted from RepDose,25 ToxRefDB,26 and HESS database27 (as being available from the OECD toolbox). Compounds were classified as steatotic positive in cases where liver steatosis was reported in at least one high-quality preclinical rodent study with repeated exposure and oral application. Study types include subacute to chronic duration. Subsequently, the data set underwent some chemical curation using the following protocol:
Removal of all inorganic compounds according to chemical formula in MOE 2014.0938
Removal of salts and compounds containing metals (organometallic compounds)
Removal of compounds having atoms for which some descriptors cannot be calculated (such as tellurium or selenium) were identified and discarded using an in-house MOE SVL script
Standardization of chemical structures using the Atkinson standardization protocol (available at https://github.com/flatkinson/standardiser)
Removal of duplicates and permanently charged compounds using MOE 2014.0938
The final data set consists of 1041 unique compounds (120 steatosis positives and 921 steatosis negatives). The data set was further randomly split into a 70% training set (TR; 727) and 30% test set (TS) while retaining the initial ratio of in vivo positive/negative compounds in the two sets (stratified sampling). The TR consists of 86 steatosis positive and 641 steatosis negative compounds, whereas the TS is composed of 34 positive and 280 negative compounds.
T-Distributed Stochastic Neighbor Embedding
Two-dimensional t-SNE plots were generated in KNIME by using the “t-SNE (L. Jonsson)” node and choosing the following parameters: iterations: 3000; θ: 0.5; and perplexity: 100. Visuals were created with the “Scatter Plot” node. Prior to dimensionality reduction, the 26 RDKit physicochemical features were subjected to a Z-score normalization (“Normalizer” node).
Analysis of Feature Distribution
Physicochemical descriptors were calculated in KNIME39 (“RDKit Descriptor Calculation” node), and violin and box plots were generated in R40 (v3.5.2).
Analysis of Drug-likeness
The quantitative estimate of drug-likeness (QED)29 was caluculated in KNIME by using the “Qed caculator” node of the “Silicos-it” nodes package.41
Kolmogorov–Smirnov Test
The statistical significance of the differences of the distribution of physicochemical properties of the active vs inactive class compounds was analyzed by performing a two-sample Kolmogorov-Smirnow Test in R40 (v3.5.2) with the “ks.test” function.
Frequency Analysis of Substructural Patterns
The prevalence of scaffolds and chemical substructures in the active class of the data set was analyzed by extracting Bemis–Murcko scaffolds30 as well as ToxPrint31 fingerprints from the compounds in the in vivo steatosis data set. For the extraction and frequency analysis of scaffolds, a workflow published in Türková et al.32 was utilized. ToxPrint is a public set of chemotypes encoded in the XML-based substructure definition language CSRML.42 Statistical significance of the enrichment of a particular scaffold or ToxPrint chemotype in the active class was calculated by applying Fisher’s exact test for count data, calculated in R40 (v3.5.2) with the “fisher.test” function.
In Vitro data for NR Activation/Deactivation
In vitro data sets for nuclear receptor (NR) activation or deactivation were kindly provided by Gadaleta et al. and described in ref (17). Nine different NRs were identified as potential MIEs upstream of the adverse effect (i.e., hepatic steatosis), including the peroxisome proliferator-activated receptors (PPARα, PPARβ, PPARγ), the constitutive androstane receptor (CAR), the pregnane X receptor (PXR), the aryl hydrocarbon receptor (AhR), the liver X receptor (LXR), the nuclear factor (erythroid-derived 2)-like 2 (Nrf2), and the farnesoid X receptor (FXR) by collecting data from AOP-Wiki. Out of these MIEs, only those providing a sufficient amount of high-quality data and weight of evidence supporting them were modeled, and up- and down-regulation assays (for evaluating agonistic and antagonistic activity) were considered separately. Thus, nine different in vitro data sets were finally extracted from ToxCast18 and used in this study for modeling NR activation or deactivation: PXR_up, PXR_down, AhR_up, AhR_down, LXR_up, LXR_down, PPARgamma_up, PPARalpha_up, and Nrf2_up.
In the respective NR data sets, approximately 500–600 compounds were overlapping with the in vivo steatosis data set (TR and TS) and were therefore removed from the NR data sets. The resulting number of compounds used for modeling the NR end points can be found in Supplementary Table S5.
In Vitro NR Models and Transporter Models
The nine NR data sets served as building predictive models by using different basic descriptor sets: RDKit35 physicochemical properties, ECFP6 circular fingerprints (both available from KNIME), and ToxPrint chemotypes.42 As most of these data sets are highly imbalanced (Supplementary Table S5), we used the SB approach as the meta-classifier in combination with RF as a base classifier (as described in the Machine Learning Algorithms and Unbalanced Learning Techniques section) to build predictive models for the nine NR data sets. Model statistics for the different descriptor sets and the nine NR models are provided in Supplementary Table S6. The best model (highlighted in Supplementary Table S6) was chosen respectively for predicting compounds from the in vivo steatosis data set as NR positive or negative, and the binary string (9 bits) further served as one of the descriptors for building the in vivo steatosis models in the subsequent step.
Transporter models including a P-gp, BCRP, BSEP, MRP3, and MRP4 inhibition model were provided by the Vienna LiverTox Workspace (https://livertox.univie.ac.at/). A general hepatic OATP inhibition model (including inhibition data for OATP1B1, OATP1B3, and OATP2B1) was taken from Türková et al.32 These models also served for predicting compounds from the in vivo steatosis data set, and the predictions were taken as a combined binary string to be used as input features for model generation of the same.
Molecular Descriptors for In Vivo Steatosis Models
We employed four sets of molecular descriptors: physicochemical descriptors (RDKit),35 ToxPrint chemotypes,42 predictions from the nine nuclear receptor models, and predictions from hepatic transporter models (as described in the previous section). The RDKit feature block includes the following 26 physicochemical properties: SlogP, SMR, LabuteASA, TPSA, AMW, ExactMW, NumLipinskiHBA, NumLipinskiHBD, NumRotatableBonds, NumHBD, NumHBA, NumAmideBonds, NumHeteroAtoms, NumHeavyAtoms, NumAtoms, NumRings, NumAromaticRings, NumSaturatedRings, NumAliphaticRings, NumAromaticHeterocycles, NumSaturatedHeterocycles, NumAliphaticHeterocycles, NumAromaticCarbocycles, NumSaturatedCarbocycles, NumAliphaticCarbocycles, and FractionCSP3.
These different descriptor blocks were further iteratively combined by employing all possible combinations (each descriptor block individually, combinations of two or three descriptor blocks, and all descriptor blocks together).
Machine Learning Algorithms and Unbalanced Learning Techniques
Random Forest (RF)34 was used as a base-classifier for SB and CP models developed in this study. The number of trees was arbitrarily set to 100 (default), since it has been shown that the optimal number of trees is usually 64–128, while further increasing the number of trees does not necessarily improve the model’s performance.43
In order to overcome the problem of data imbalance, we used bagging with stratified under-sampling (SB)44,45 and Mondrian CP.46 These methods have proven to be among the best performing methods for dealing with imbalanced data sets.47,48 SB is a ML technique that is based on an ensemble of models developed using multiple training data sets sampled from the original training set. It uses minority class samples to create the training set of positive samples using a traditional bagging approach (resampling with replacement), and after that, randomly selects the same number of samples from the majority class. Thus, the total bagging training set size was double the number of the minority class molecules. Several models are then built and predictions averaged in order to produce a final ensemble model output. Because of random sampling, about 37% of the molecules are not selected and left out in each run. These samples create the “out-of-the-bag” sets, which are used for testing the performance of the final model.49 Although a small set of samples are selected each time, a majority of molecules contributed to the overall bagging procedure, since the data sets were generated randomly. Further, an earlier study by Tetko et al.49 showed that larger numbers of models per ensemble (e.g., 128, 256, 512 and 1024) did not significantly increase the balanced accuracy of models. Thus, in this study, we built a total of 64 models per ensemble. All models using RF in combination with SB were developed and deployed by using the data analytics platform KNIME.39
Mondrian CP on the other hand, belongs to a group of predictors called confidence predictors.50 CP is a framework allowing the incorporation of any ML algorithm that as output provides a ranking of the investigated compounds.50 An attractive feature of CP is that, provided the data set fulfills the exchangeable criterion, the output is always valid for which there exists a mathematical proof by Vovk et al.50 This means that the user can set a level (percentage) of errors that is acceptable and CP will return this level of errors at most. CP will also, at the same time, provide the user with information for each prediction, that is, for each compound, whether the prediction is reliable or not.
For a classification problem, a set of class labels is assigned to new compounds through comparison to a calibration set with known labels (experimental classes). In this study with two classes (steatotic and nonsteatotic), this means that two separate calibrations sets are used. One set with experimentally known steatotic calibration compounds and another set with experimentally known nonsteatotic calibration compounds. If the outcome for the new compound is higher than the set significance level and, thus, similar to the prediction outcomes on either of the two calibration sets, the new compound is assigned that class label and given a CP p-value for that class. Consequently, for a binary classification problem, there are four possible outcomes. A new compound can be labeled with either of the two classes, or it could be assigned both labels (both classification) or neither one (empty classification). For an illustrative example of how conformal prediction is carried out, we refer the reader to Norinder et al.51
We used RF34 as the underlying model for our predictors. CP models were developed using Python, Scikit-learn52 version 0.17, and the nonconformist package version 1.2.5 (https://github.com/donlnz/nonconformist). Binary classification models were built using the RandomForestClassifier in Scikit-learn with 100 trees, and all other options set at default. Conformal predictions were performed using the ProbEstClassifierNC and IcpClassifier functions in the nonconformist package with options for class conditional (Mondrian) conformal predictions enabled: icp = IcpClassifier(nc, condition = lambda x: x[1]).
Model Performance Assessment
The performance of each classification model was assessed on the basis of the sensitivity (true positive rate; eq 1), specificity (true negative rate; eq 2), accuracy (eq 3), balanced accuracy (correct classification rate; eq 4), and MCC (eq 5) calculated for the TS (30% of initial in vivo data set). For a highly imbalanced data set, accuracy may be misleading, thus we considered balanced accuracy (which considers both sensitivity and specificity) and MCC as a more appropriate performance measure to compare different classifiers for their ability to handle imbalanced data sets.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
where TP is true positive, TN is true negative, FP is false positive, and FN is false negative.
The performance of the conformal predictor was additionally measured by its validity (percentage of correct classifications for each class) and efficiency (percentage of single label classifications) when employed on the TS. A conformal predictor is said to be valid if the percentage of errors does not exceed the set significance level. Thus, a prediction is considered correct if it includes the correct class label, which means that both predictions are always correct and empty predictions never are (i.e., always erroneous). Validity was determined for the active (aka steatotic) and inactive (aka nonsteatotic) class separately. In order to compare results from SB and CP, we have to consider that validity includes the “both” classifications (since they are per definition always correct), whereas sensitivity and specificity are only calculated on single label predictions. In CP, there exists a trade-off between the validity of the model and the efficiency. For the final predictions from CP, we applied the aggregated conformal prediction method described by Carlsson et al.53
The training set was randomly divided into a proper training set and calibration set using 70% and 30% of the training data, respectively. This whole process was repeated 64 times in case of SB and 100 times in case of CP, each time storing the predictions on the test compounds. For the SB, the consensus of all models was chosen as a way to evaluate the test set (mean), and for CP models, the median predicted CP p-value for each compound was calculated and used for class assignment in accordance with the set significance level.
Data, Code, and Model Availability
Precalculated descriptors for the whole data set (1141 entries) and labels for steatotic positive and negative compounds are provided as Supplementary File S1. For the public fraction of the compound data set (512 compounds), CAS registry numbers as well as chemical structures (in smiles format) are provided in addition to the class label (Supplementary File S2). In addition, we made all KNIME workflows and python scripts used to build the herein discussed models publicly available in an open GitHub repository (https://github.com/BZdrazil/Steatosis_prediction) and are providing example models by using the SB and CP meta-classifiers (based on 26 RDKit descriptors).
Glossary of ML Specific Terms
In supplements (Supplementary Table S7) we provide a glossary describing specific terms used in this manuscript in order to make the methodology easily understandable for a broader readership outside the cheminformatics community.
Acknowledgments
The EU-ToxRisk project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 681002. This work received funding from the Austrian Science Fund (FWF) (grants P 29712 and W1232 - MolTag). We thank Gerhard Ecker for his valuable input to the design of the study.
Glossary
Abbreviations
- ALD
alcoholic liver disease
- AhR
aryl hydrocarbon receptor
- AO
adverse outcome
- AOP
adverse outcome pathway
- CAR
constitutive androstane receptor
- CP
conformal prediction
- FA
fatty acid
- FXR
farnesoid X receptor
- HESS
Hazard Evaluation Support System
- KE
key event
- KER
key event relationship
- LXR
liver X receptor
- MIE
molecular initiating event
- ML
machine learning
- NAFLD
nonalcoholic fatty liver disease
- NR
nuclear receptor
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- PCA
principal component analysis
- PPAR
peroxisome proliferator-activated receptor
- PXR
pregnane X receptor
- QSAR
quantitative structure–activity relationship
- RF
random forest
- SB
bagging with stratified under-sampling
- TR
training data set
- TS
test data set
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.0c00511.
Two-dimensional t-SNE projection showing the distribution of compounds belonging to the training set and test set in chemical space based on 26 RDKit physicochemical descriptors; bar chart showing the distribution of Tanimoto similarity of the test set compounds to the closest (most similar) training set compound; violin plots showing the physicochemical property distribution in the in vivo steatosis data set for the steatosis positive class; bar charts showing the validity of the steatotic class and the nonsteatotic class for the models built with the CP framework (at significance level 0.3); bar charts showing the mean efficiencies and mean balanced accuracies for the models built with the CP framework (at significance level 0.3); two-dimensional t-SNE projection of the in vivo steatosis data set excluding compounds predicted as falling into the “both” class; bar chart showing the mean number of compounds predicted to be falling into the “both” category when adjusting the significance levels: 0.15, 0.2, 0.25, and 0.3; bar charts showing counts of when the respective descriptor set is performing better than any other set; summary statistics for eight common physicochemical properties and the quantitative estimate of drug-likeness of the in vivo steatosis data set; frequency analysis of Murcko scaffolds in the positive class of the in vivo steatosis data set; frequency analysis of ToxPrint chemotypes in the positive class of the in vivo steatosis data set; results from SB models using five selected RDKit features plus predictions from individual transport inhibition models; composition of the final NR data sets; model performances for the nuclear receptor data sets when predicting the test set; glossary of specific terms used in ML applications (PDF)
Supplementary File S1: Data set including 1041 entries with all calculated descriptors and class labels (XLSX)
Supplementary File S2: Data set of 512 chemical compounds including CAS registry numbers, smiles code, and class label (XLSX)
Author Present Address
∥ National Center for Advancing Translational Sciences (NCATS), National Institute of Health (NIH), 9800 Medical Center Drive, Rockville, Maryland 20850, United States
Author Present Address
⊥ Department of Computer and Systems Sciences, Stockholm University, Box 7003, SE-164 07 Kista, Sweden; Department of Pharmaceutical Biosciences, Uppsala University, Box 591, SE-75124, Uppsala Sweden. MTM Research Centre, School of Science and Technology, Örebro University, SE-70182 Örebro, Sweden
Author Contributions
S.J. and U.N. performed the experiments and analyzed the results. S.E.E. extracted and curated the in vivo data set. B.Z. designed the study, drafted the manuscript, and performed parts of the analyses. The manuscript was written through contributions of all authors, and all authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Al-Eryani L.; Wahlang B.; Falkner K. C.; Guardiola J. J.; Clair H. B.; Prough R. A.; Cave M. (2015) Identification of Environmental Chemicals Associated with the Development of Toxicant-Associated Fatty Liver Disease in Rodents. Toxicol. Pathol. 43 (4), 482–497. 10.1177/0192623314549960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massart J.; Begriche K.; Moreau C.; Fromenty B. (2017) Role of Nonalcoholic Fatty Liver Disease as Risk Factor for Drug-Induced Hepatotoxicity. J. Clin Transl Res. 3, 212–232. 10.18053/jctres.03.2017S1.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessone F.; Dirchwolf M.; Rodil M. A.; Razori M. V.; Roma M. G. (2018) Review Article: Drug-Induced Liver Injury in the Context of Nonalcoholic Fatty Liver Disease - a Physiopathological and Clinical Integrated View. Aliment. Pharmacol. Ther. 48 (9), 892–913. 10.1111/apt.14952. [DOI] [PubMed] [Google Scholar]
- Arab J. P.; Arrese M.; Trauner M. (2018) Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol.: Mech. Dis. 13 (1), 321–350. 10.1146/annurev-pathol-020117-043617. [DOI] [PubMed] [Google Scholar]
- Patel V.; Sanyal A. J. (2013) Drug-Induced Steatohepatitis. Clin Liver Dis 17 (4), 533–546. 10.1016/j.cld.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrish M. M.; Kaiser J. P.; McQueen C. A.; Chorley B. N. (2016) Tipping the Balance: Hepatotoxicity and the 4 Apical Key Events of Hepatic Steatosis. Toxicol. Sci. 150 (2), 261–268. 10.1093/toxsci/kfw018. [DOI] [PubMed] [Google Scholar]
- Fromenty B.; Pessayre D. (1995) Inhibition of Mitochondrial Beta-Oxidation as a Mechanism of Hepatotoxicity. Pharmacol. Ther. 67 (1), 101–154. 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- Fromenty B.; Pessayre D. (1997) Impaired Mitochondrial Function in Microvesicular Steatosis. Effects of Drugs, Ethanol, Hormones and Cytokines. J. Hepatol. 26 (2), 43–53. 10.1016/S0168-8278(97)80496-5. [DOI] [PubMed] [Google Scholar]
- Siramshetty V. B.; Nickel J.; Omieczynski C.; Gohlke B.-O.; Drwal M. N.; Preissner R. (2016) WITHDRAWN—a Resource for Withdrawn and Discontinued Drugs. Nucleic Acids Res. 44 (D1), D1080–D1086. 10.1093/nar/gkv1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões I. C. M.; Fontes A.; Pinton P.; Zischka H.; Wieckowski M. R. (2018) Mitochondria in Non-Alcoholic Fatty Liver Disease. Int. J. Biochem. Cell Biol. 95, 93–99. 10.1016/j.biocel.2017.12.019. [DOI] [PubMed] [Google Scholar]
- Leist M.; Ghallab A.; Graepel R.; Marchan R.; Hassan R.; Bennekou S. H.; Limonciel A.; Vinken M.; Schildknecht S.; Waldmann T.; Danen E.; van Ravenzwaay B.; Kamp H.; Gardner I.; Godoy P.; Bois F. Y.; Braeuning A.; Reif R.; Oesch F.; Drasdo D.; Höhme S.; Schwarz M.; Hartung T.; Braunbeck T.; Beltman J.; Vrieling H.; Sanz F.; Forsby A.; Gadaleta D.; Fisher C.; Kelm J.; Fluri D.; Ecker G.; Zdrazil B.; Terron A.; Jennings P.; van der Burg B.; Dooley S.; Meijer A. H.; Willighagen E.; Martens M.; Evelo C.; Mombelli E.; Taboureau O.; Mantovani A.; Hardy B.; Koch B.; Escher S.; van Thriel C.; Cadenas C.; Kroese D.; van de Water B.; Hengstler J. G. (2017) Adverse Outcome Pathways: Opportunities, Limitations and Open Questions. Arch. Toxicol. 91 (11), 3477–3505. 10.1007/s00204-017-2045-3. [DOI] [PubMed] [Google Scholar]
- Collaborative Adverse Outcome Pathway Wiki (AOP-Wiki). https://aopwiki.org/ (accessed 2020-05-29).
- Slenter D. N.; Kutmon M.; Hanspers K.; Riutta A.; Windsor J.; Nunes N.; Mélius J.; Cirillo E.; Coort S. L.; Digles D.; Ehrhart F.; Giesbertz P.; Kalafati M.; Martens M.; Miller R.; Nishida K.; Rieswijk L.; Waagmeester A.; Eijssen L. M. T.; Evelo C. T.; Pico A. R.; Willighagen E. L. (2018) WikiPathways: A Multifaceted Pathway Database Bridging Metabolomics to Other Omics Research. Nucleic Acids Res. 46 (D1), D661–D667. 10.1093/nar/gkx1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N.; Borlak J. (2008) Molecular Mechanisms and Therapeutic Targets in Steatosis and Steatohepatitis. Pharmacol. Rev. 60 (3), 311–357. 10.1124/pr.108.00001. [DOI] [PubMed] [Google Scholar]
- Vinken M. (2015) Adverse Outcome Pathways and Drug-Induced Liver Injury Testing. Chem. Res. Toxicol. 28 (7), 1391–1397. 10.1021/acs.chemrestox.5b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breda S. G. J.; Claessen S. M. H.; van Herwijnen M.; Theunissen D. H. J.; Jennen D. G. J.; de Kok T. M. C. M.; Kleinjans J. C. S. (2018) Integrative Omics Data Analyses of Repeated Dose Toxicity of Valproic Acid in Vitro Reveal New Mechanisms of Steatosis Induction. Toxicology 393, 160–170. 10.1016/j.tox.2017.11.013. [DOI] [PubMed] [Google Scholar]
- Gadaleta D.; Manganelli S.; Roncaglioni A.; Toma C.; Benfenati E.; Mombelli E. (2018) QSAR Modeling of ToxCast Assays Relevant to the Molecular Initiating Events of AOPs Leading to Hepatic Steatosis. J. Chem. Inf. Model. 58 (8), 1501–1517. 10.1021/acs.jcim.8b00297. [DOI] [PubMed] [Google Scholar]
- Dix D. J.; Houck K. A.; Martin M. T.; Richard A. M.; Setzer R. W.; Kavlock R. J. (2007) The ToxCast Program for Prioritizing Toxicity Testing of Environmental Chemicals. Toxicol. Sci. 95 (1), 5–12. 10.1093/toxsci/kfl103. [DOI] [PubMed] [Google Scholar]
- Kotsampasakou E.; Escher S. E.; Ecker G. F. (2017) Linking Organic Anion Transporting Polypeptide 1B1 and 1B3 (OATP1B1 and OATP1B3) Interaction Profiles to Hepatotoxicity - The Hyperbilirubinemia Use Case. Eur. J. Pharm. Sci. 100, 9–16. 10.1016/j.ejps.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Kotsampasakou E.; Montanari F.; Ecker G. F. (2017) Predicting Drug-Induced Liver Injury: The Importance of Data Curation. Toxicology 389, 139–145. 10.1016/j.tox.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vienna LiverTox. https://livertox.univie.ac.at/ (accessed 2020-05-29).
- Cotterill J.; Price N.; Rorije E.; Peijnenburg A. (2020) Development of a QSAR Model to Predict Hepatic Steatosis Using Freely Available Machine Learning Tools. Food Chem. Toxicol. 142, 111494. 10.1016/j.fct.2020.111494. [DOI] [PubMed] [Google Scholar]
- Shin H. K.; Kang M.-G.; Park D.; Park T.; Yoon S. (2020) Development of Prediction Models for Drug-Induced Cholestasis, Cirrhosis, Hepatitis, and Steatosis Based on Drug and Drug Metabolite Structures. Front. Pharmacol. 11, 11. 10.3389/fphar.2020.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PharmaPendium. https://www.pharmapendium.com/login (accessed 2020-09-09).
- RepDose Database Fraunhofer ITEM QSAR. https://repdose.item.fraunhofer.de/ (accessed 2020-05-29).
- Watford S.; Ly Pham L.; Wignall J.; Shin R.; Martin M. T.; Friedman K. P. (2019) ToxRefDB Version 2.0: Improved Utility for Predictive and Retrospective Toxicology Analyses. Reprod. Toxicol. 89, 145–158. 10.1016/j.reprotox.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazard Evaluation Support System Integrated Platform (HESS), National Institute of Technology and Evaluation (NITE), Tokyo.https://www.nite.go.jp/en/chem/qsar/hess-e.html (accessed 2020-05-29).
- van der Maaten L.; Hinton G. (2008) Visualizing Data Using T-SNE. J. Mach. Learn. Res. 9, 2579–2605. [Google Scholar]
- Bickerton G. R.; Paolini G. V.; Besnard J.; Muresan S.; Hopkins A. L. (2012) Quantifying the Chemical Beauty of Drugs. Nat. Chem. 4 (2), 90–98. 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis G. W.; Murcko M. A. (1996) The Properties of Known Drugs. 1. Molecular Frameworks. J. Med. Chem. 39 (15), 2887–2893. 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- Yang C.; Tarkhov A.; Marusczyk J.; Bienfait B.; Gasteiger J.; Kleinoeder T.; Magdziarz T.; Sacher O.; Schwab C. H.; Schwoebel J.; Terfloth L.; Arvidson K.; Richard A.; Worth A.; Rathman J. (2015) New Publicly Available Chemical Query Language, CSRML, to Support Chemotype Representations for Application to Data Mining and Modeling. J. Chem. Inf. Model. 55 (3), 510–528. 10.1021/ci500667v. [DOI] [PubMed] [Google Scholar]
- Türková A.; Jain S.; Zdrazil B. (2019) Integrative Data Mining, Scaffold Analysis, and Sequential Binary Classification Models for Exploring Ligand Profiles of Hepatic Organic Anion Transporting Polypeptides. J. Chem. Inf. Model. 59, 1811–1825. 10.1021/acs.jcim.8b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders M. W. (2004) Glutathione-Dependent Bioactivation of Haloalkanes and Haloalkenes. Drug Metab. Rev. 36 (3–4), 583–594. 10.1081/DMR-200033451. [DOI] [PubMed] [Google Scholar]
- Breiman L. (2001) Random Forests. Machine Learning 45 (1), 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- RDKit. https://www.rdkit.org/ (accessed 2020-05-29).
- Scotto K. W. (2003) Transcriptional Regulation of ABC Drug Transporters. Oncogene 22 (47), 7496–7511. 10.1038/sj.onc.1206950. [DOI] [PubMed] [Google Scholar]
- Cobbina E.; Akhlaghi F. (2017) Non-Alcoholic Fatty Liver Disease (NAFLD) - Pathogenesis, Classification, and Effect on Drug Metabolizing Enzymes and Transporters. Drug Metab. Rev. 49 (2), 197–211. 10.1080/03602532.2017.1293683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc., Montreal, QC.
- Berthold M. R., Cebron N., Dill F., Gabriel T. R., Kötter T., Meinl T., Ohl P., Sieb C., Thiel K., and Wiswedel B.. KNIME: The Konstanz Information Miner. (2008) In Data Analysis, Machine Learning and Applications (Preisach C., Burkhardt H., Schmidt-Thieme L., and Decker R., Eds.) pp 319–326, Springer, Berlin. [Google Scholar]
- R: The R Project for Statistical Computing, https://www.r-project.org/ (accessed 2019-04-03).
- Kooistra A. J.; Vass M.; McGuire R.; Leurs R.; de Esch I. J. P.; Vriend G.; Verhoeven S.; de Graaf C. (2018) 3D-e-Chem: Structural Cheminformatics Workflows for Computer-Aided Drug Discovery. ChemMedChem 13 (6), 614–626. 10.1002/cmdc.201700754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ToxPrint - A Public Set of Chemotypes, MN-AM, Nurnburg, Germany. https://www.mn-am.com/products/toxprint (accessed 2020-05-29).
- Oshiro T. M., Perez P. S., and Baranauskas J. A.. How Many Trees in a Random Forest? (2012) In Machine Learning and Data Mining in Pattern Recognition (Perner P., Ed.) pp 154–168, Springer, Berlin. [Google Scholar]
- Kotsiantis S.; Kanellopoulos D.; Pintelas P. (2006) Handling Imbalanced Datasets: A Review. GESTS Int. Trans. Comput. Sci. Eng. 30, 25–36. [Google Scholar]
- Jain S.; Kotsampasakou E.; Ecker G. F. (2018) Comparing the Performance of Meta-Classifiers—a Case Study on Selected Imbalanced Data Sets Relevant for Prediction of Liver Toxicity. J. Comput.-Aided Mol. Des. 32 (5), 583–590. 10.1007/s10822-018-0116-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer G.; Vovk V. (2008) A Tutorial on Conformal Prediction. J. Mach. Learn. Res. 9, 371–421. [Google Scholar]
- Norinder U.; Boyer S. (2017) Binary Classification of Imbalanced Datasets Using Conformal Prediction. J. Mol. Graphics Modell. 72, 256–265. 10.1016/j.jmgm.2017.01.008. [DOI] [PubMed] [Google Scholar]
- Svensson F.; Norinder U.; Bender A. (2017) Modelling Compound Cytotoxicity Using Conformal Prediction and PubChem HTS Data. Toxicol. Res. 6 (1), 73–80. 10.1039/C6TX00252H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetko I. V.; Novotarskyi S.; Sushko I.; Ivanov V.; Petrenko A. E.; Dieden R.; Lebon F.; Mathieu B. (2013) Development of Dimethyl Sulfoxide Solubility Models Using 163 000 Molecules: Using a Domain Applicability Metric to Select More Reliable Predictions. J. Chem. Inf. Model. 53 (8), 1990–2000. 10.1021/ci400213d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conformal Prediction. (2005) In Algorithmic Learning in a Random World (Vovk V., Gammerman A., and Shafer G., Eds.) pp 17–51, Springer, Boston, MA. [Google Scholar]
- Norinder U.; Myatt G.; Ahlberg E. (2018) Predicting Aromatic Amine Mutagenicity with Confidence: A Case Study Using Conformal Prediction. Biomolecules 8 (3), 85. 10.3390/biom8030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F.; Varoquaux G.; Gramfort A.; Michel V.; Thirion B.; Grisel O.; Blondel M.; Müller A.; Nothman J.; Louppe G.; Prettenhofer P.; Weiss R.; Dubourg V.; Vanderplas J.; Passos A.; Cournapeau D.; Brucher M.; Perrot M.; Duchesnay É. (2011) Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830. [Google Scholar]
- Carlsson L., Eklund M., and Norinder U.. Aggregated Conformal Prediction. (2014) In Artificial Intelligence Applications and Innovations (Iliadis L., Maglogiannis I., Papadopoulos H., Sioutas S., and Makris C., Eds.) pp 231–240, Springer, Berlin. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.