

Abstract
This piece addresses the urge to assess the evolutionary fate of SARS-CoV-2 in a post-vaccination phase. The possibilities of COVID-19 becoming endemic or extinct are weighed against verifiable properties of extant vaccines and observed genetic trends already apparent under the mild selection pressure exerted almost exclusively by social rules.
Keywords: SARS-CoV-2, COVID-19 vaccine, evolutionary change, structural biology, vaccine resistance, protein−protein interface
A Priori Plausible Post-Vaccination Scenarios for COVID-19
In the best post-vaccination scenario that we all hope for,1,2 SARS-CoV-2 will behave like smallpox or measles: it will get wiped out by the immunity promoted by the COVID-19 vaccines. Indeed, no vaccine-resistant strain of smallpox or measles has ever arisen, and the evolution of these diseases has been essentially stagnant.2 However, a very different scenario holds for pneumonia, for example, where the pathogen Streptococcus pneumoniae evolved a resistant strain to the conjugate vaccine (PCV7), prompting the time-consuming development of a new vaccine, PCV13, at a significant expenditure.3 Other microbes such as the ones causative of whooping cough (pertussis) are also known to have evolved vaccine-evading mutants.4 In the worst-case scenario, COVID-19 will become endemic, joining the class of diseases that includes malaria, trypanosomiasis (sleeping sickness), influenza, and AIDS, where relatively high evolutionary rates undermine efforts to induce lasting immune responses by means of vaccination.
Thus, there is obviously a staggering diversity of a priori scenarios in a post-vaccination stage. The pressing questions that arise are whether COVID-19 will be neutralized or become endemic and what would be the evolutionary outcome when the virus is under the selection pressure caused by vaccine-induced immune surveillance. To narrow down possibilities, we need a clear assessment of the efficacy of extant vaccines vis-à-vis the evolutionary change and an a priori assessment of the possible extent of evolutionary change to be expected from the virus under selection pressure.
A Pharmacological Imperative: Assessing the Evolution of SARS-CoV-2 under the Selection Pressure Arising in the Post-Vaccination Phase
Once we enter the post-vaccination stage, SARS-CoV-2 will be facing significant selection pressure, and somatic evolution will set in as the virus develops escape routes through the workings of natural selection. In principle, we have virtually no way to tell if and how the virus will respond to the selection pressure imposed by vaccine-induced immune surveillance.
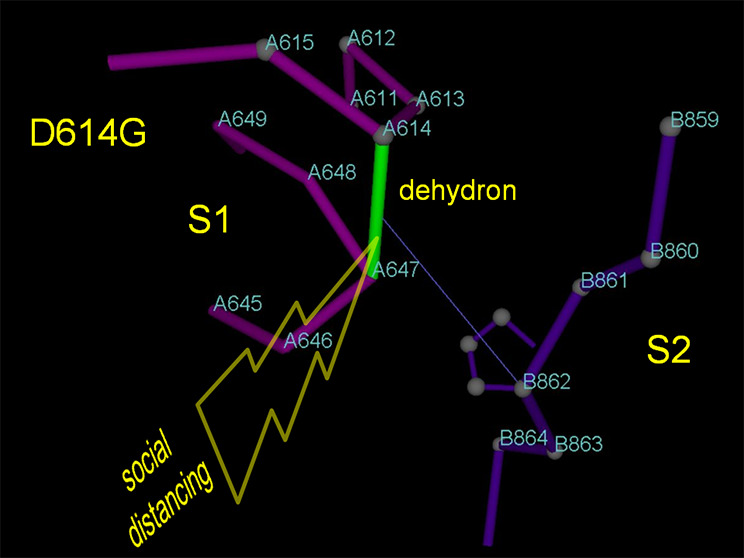
In reality, this may not be completely true. We have some hints of the evolutionary reaction of SARS-CoV-2 to selection pressure, although the pressure has not been of the magnitude that is expected in the post-vaccination stage. For example, social distance determined a fitness advantage for phenotypes with higher transmissibility that translates into higher survival rates. Thus, it is plausible that the dominant D614G mutation in the spike (S) protein5 would have arisen as a result of the selection pressure caused by social distancing. The structural impact of D614G is well-delineated, resulting in higher stabilization of the postcleavage S1/S2 interface.6 The net effect of this mutation is the enabling of synchronized anchoring (S1) and harpooning (S2) of the host cell,7 which results in higher virus transmissibility, thus countering the adverse impact of social distancing.
Another mutation in the receptor binding domain (RBD), N501Y, has become a defining feature (together with deletions Δ69 and Δ70) in a new highly virulent strain that is quickly spreading in England.8 It is hard at this stage to assess what specific factors select for this mutation or what has it evolved in response to. It is entirely possible that it emerged in response to the selection pressure determined by social distancing rules, which in all likelihood select for higher transmissibility.
From a structural perspective focusing on PDB-reported complexes, the residue at position 501 is paired through a hydrogen bond with Y41 in the hACE2 (human angiotesin convertase enzyme 2) host-cell receptor. Both asparagine (N) and its substituent tyrosine (Y) can behave dually as proton donors or acceptors, but the substitution N501Y brings about additional possibilities of interaction based on the additional quadrupole moment of the benzene ring in tyrosine, such as pi-cation interaction engaging the polarized bridging hydrogen,9 benzene ring stacking, etc. Thus, the effect of N501Y substitution on the net stability of the virus–receptor interface cannot be easily discerned in the absence of isothermal titration calorimetry measurements. Additional factors would need to the weighed such as the larger excluded volume effect from the bulkier tyrosine, the precise side chain positioning across the interface, and the thermodynamic cost of the burial of the large dipole moment in asparagine. In the unlikely case that N501Y destabilizes the virus–receptor interface, it may well be that evolution is selecting for another host in response to social distancing, but that would be completely at odds with the fast pace at which the strain is spreading among humans in England. On the other hand, under the same selection pressure, the N501Y substitution more likely brings about extra stabilization of the virus–receptor interface that would result in higher infectivity arising from more efficacious transmission, a far more plausible scenario. In any case, this mutation entails some danger because it occurs in the highly antigenic RBD and hence may forestall the vaccine-induced immune response when the latter focuses solely on RBD, as it is the case with some vaccines.
Equally decisive for the assessment of the plausibility of vaccine escape routes in principle available to the virus are the antigenic patterns promoting the induced immune surveillance. An antigen presenting a single epitope may be overcome by a virus mutation that destabilizes the epitope–antibody interface (as in the case of the N501Y mutation of the “English strain”), whereas an antigen with multiple epitopes cannot be so readily defused, requiring the highly improbable conjunction of evolutionary events that must result in multiple mutations.
The biotechnology platforms for the COVID-19 vaccines are broadly diverse:1 from an inactivated virus (Wuhan Institute of Biological Products, Sinopharm), to a proprietary recombinant adenovirus vector (Gamaleya’s Sputnik V or ChAdOx1 nCoV-19 AZD1222, University of Oxford and AstraZeneca), or mRNA within nanoparticle formulation (BNT162b1, Pfizer and BioNTech or mRNA-1273, Moderna and NIAID). They typically present an antigen consisting of the S protein with the exception of BNT162b1, which encodes for the expression of the RBD portion of the S protein. With the information at hand and based on the previous argument, it is unlikely that the virus may eventually develop resistance even to the Pfizer vaccine because multiple epitopes were molecularly identified that are recognized by the BNT162b1-induced CD8+ T cells. However, mutations such as N501Y in the antigenic RBD region are likely to have an impact, especially on BNT162b1.
There are reasons to be cautiously optimistic regarding the evolution of SARS-CoV-2 in the post-vaccination phase, even though the virus has proven to be evolutionarily responsive (mutations D614G and N501Y) even to relatively mild selection pressure imposed by social rules. The reasons for cautious optimism in regards to evolution-proof vaccines are based on judicious assumptions. The assumptions are expected to have been corroborated in the clinical trials and may be listed as follows:
The COVID-19 vaccines induce antibodies that recognize multiple viral epitopes in a first line of attack, so the possibility of evolving immediate evasion routes is remote.
The vaccines typically elicit a robust expansion of CD4+ T cells and especially CD8+ T cells capable of recognizing multiple epitopes and capable of orchestrating a lasting adaptive immune response.
The vaccines block virus replication and transmission because induced immune elements interfere directly with virus recognition of the hACE2 receptor.
The vaccines protect against all SARS-CoV-2 serotypes.
The vaccine-induced immune response is expected to be directed solely at RBD epitopes, since other antigenic regions are likely blocked through camouflaging glycosylation. On the other hand, mutations at RBD that may compromise antibody affinity are unlikely unless the virus chooses to give up its anchoring receptor.
Discussion
While not explicitly stated,1 it is expected that the conditions listed above have all been verifiably fulfilled by all extant COVID-19 vaccines or corroborated before inoculation in an extension of the respective phase 3 clinical trials. To an extent, the Pfizer vaccine with its RBD antigen may prove to be an exception, as hinted by the mutation N501Y that arises under far milder selection pressure.8 This mutation in the RBD region of the spike may partially forestall the induced immunosurveillance that focuses precisely on RBD recognition.
In general, the conditions listed above should be sufficient but probably not strictly necessary to preclude an evolutionary route of escape, in spite of the fact that the virus has proven responsive to even mild selection forces involving changes in social behavior. Thus, while an endemic scenario is still on the cards, we can be reasonably confident of its low likelihood.
The author declares no competing financial interest.
References
- World Health Organization . Draft landscape of COVID-19 candidate vaccines. https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed December 20, 2020).
- Kennedy D. A.; Read A. F. (2020) Monitor for COVID-19 vaccine resistance evolution during clinical trials. PLoS Biol. 18, e3001000 10.1371/journal.pbio.3001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger D. M.; Malley R.; Lipsitch M. (2011) Serotype replacement in disease after pneumococcal vaccination. Lancet 378, 1962–1973. 10.1016/S0140-6736(10)62225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octavia S.; Maharjan R. P.; Sintchenko V.; Stevenson G.; Reeves P. R.; Gilbert G. L.; et al. (2011) Insight into evolution of Bordetella pertussis from comparative genomic analysis: evidence of vaccine-driven selection. Mol. Biol. Evol. 28, 707–715. 10.1093/molbev/msq245. [DOI] [PubMed] [Google Scholar]
- Korber B.; Fischer W. M.; Gnanakaran S.; Yoon H.; Theiler J.; Abfalterer W.; Hengartner N.; Giorgi E. E.; Bhattacharya T.; Foley B.; Hastie K. M.; Parker M. D.; Partridge D. G.; Evans C. M.; Freeman T. M.; de Silva T. I.; McDanal C.; Perez L. G.; Tang H.; Moon-Walker A.; Whelan S. P.; LaBranche C. C.; Saphire E. O.; Montefiori D. C. (2020) Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182, 1–16. 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández A. (2020) Structural impact of mutation D614G in SARS-CoV-2 spike protein: enhanced infectivity and therapeutic opportunity. ACS Med. Chem. Lett. 11, 1667–1670. 10.1021/acsmedchemlett.0c00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández A. (2020) Defusing SARS-CoV-2: Emergency Brakes in a Vaccine Failure Scenario. ACS Pharm. Transl. Sci. 3, 1425–1426. 10.1021/acsptsci.0c00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler S. (2020) What We Know About the New Covid-19 Strain in England. Wall Street J. https://www.wsj.com/articles/what-we-know-about-the-new-covid-19-strain-in-england-11608423416 (accessed December 19, 2020). [Google Scholar]
- Fernández A. (2016) Physics at the Biomolecular Interface: Fundamentals for Molecular Targeted Therapy; Springer International Publishing, Switzerland. [Google Scholar]