

Abstract
Despite being among the most characterized G protein-coupled receptors (GPCRs), adenosine receptors (ARs) have always been a difficult target in drug design. To date, no agonist other than the natural effector and the diagnostic regadenoson has been approved for human use. Recently, the structure of the adenosine A1 receptor (A1R) was determined in the active, Gi protein complexed state; this has important repercussions for structure-based drug design. Here, we employed supervised molecular dynamics simulations and mutagenesis experiments to extend the structural knowledge of the binding of selective agonists to A1R. Our results identify new residues involved in the association and dissociation pathway, they suggest the binding mode of N6-cyclopentyladenosine (CPA) related ligands, and they highlight the dramatic effect that chemical modifications can have on the overall binding mechanism, paving the way for the rational development of a structure-kinetics relationship of A1R agonists.
Keywords: G protein coupled receptors, GPCRs, adenosine A1 receptor, A1R, supervised molecular dynamics, SuMD, mutagenesis experiments, binding
1. Introduction
X-ray and cryo-electron microscopy (cryo-EM) structures are determined in equilibrium conditions. This is not representative of the physiological environment, in which the concentration of endogenous effectors and exogenous drugs in the proximity of their target continuously change due to pharmacokinetics. It follows that the affinity of a ligand, which is measured in steady state conditions, is not descriptive of its in vivo binding. The mechanism through which a molecule binds to its biological target, on the other hand, does not change with the local concentration. This consideration has produced increasing attention toward the binding kinetics as a key aspect to consider in structure-based drug design (SBDD). Understanding the binding and unbinding pathways of congeneric ligands would unlock the development of favorable structure-kinetics relationships (SKR) and foster rational guidance for medicinal chemistry modifications.
G protein-coupled receptors (GPCRs), which are a target for roughly 34% of the FDA approved drugs,1 represent an invaluable reservoir for therapeutic strategies; however, the majority of the GPCRome is currently not drugged.2 One of the prototypical GPCR family still orphan of therapeutics is the adenosine receptors (ARs). Adenosine acts ubiquitously on four different ARs (e.g., A1R, A2AR, A2BR, and A3R) contributing to the broad range of purinergic signaling.3,4 The main effect mediated by adenosine comprises of the inhibition or stimulation of adenylyl cyclase, the activation of phospholipase C, intracellular Ca2+ regulation, and the mitogen-activated protein kinases (MAPK) pathways.5 Agonist-activated A1R couples to inhibitory G proteins (Gi/o) to trigger numerous physiological effects. For example, it produces negative chronotropic and inotropic effects in the heart,6 reduces the neuronal firing rate by blocking neurotransmitter release in the central nervous system (CNS), and inhibits lipolysis and renin release.7
Therapeutic approaches based on stimulating the A1R could pave the way for hitherto unavailable tools to treat the central nervous system (CNS) and cardiovascular diseases.5,8 From this standpoint, the SBDD of novel agonists can exploit the recent cryo-EM structure of A1R in complex with both Gi2 and adenosine.9 Like all the other GPCRs, A1R presents a transmembrane domain (TMD) formed by seven α-helixes spanning the cytosolic membrane and shaping the orthosteric and the intracellular Gi protein binding sites (Figure S1a). Three intracellular loops (ICL1–3) and three extracellular loops (ECL1–3) interconnect the TM helices. ECL2, the longest A1R loop, orients almost perpendicularly to the plane of the membrane both in the active9 and inactive10,11 A1R states, in contrast to ECL2 of A2AR, which is almost parallel to the membrane.10 ECL2 is important for A1R ligands;12 it has been implicated in the intermediate states that anticipate the orthosteric complex, therefore acting as a selectivity filter. Moreover, positive allosteric modulators can to bind to it.13,14 The key orthosteric interactions between adenosine and A1R (Figure S1b) are hydrogen bonds with residues N2546.55, S2777.42, H2787.43, and a π–π interaction that involves F171ECL2. AR agonists bearing small N6-cycloalkyl groups, like the N6-cyclopentyladenosine (CPA, Figure 1), display A1R selectivity.15 It is proposed that the ligand selectivity for A1R is driven by a small hydrophobic pocket underneath ECL3, due to the presence of T2707.35 in place of M2707.35 (A2A).16,9,10
Figure 1.

A1R agonists considered. Adenosine is the endogenous effector; NECA is a nonselective exogenous ARs agonist; CPA represents a prototypical A1R selective agonist; and HOCPA and BnOCPA are A1R selective analogues of CPA.
Here, we extensively studied the A1R recognition of adenosine, 5′-N-carboxamidoadenosine (NECA), CPA, and the recently characterized agonists HOCPA and BnOCPA17 (Figure 1). Binding and unbinding pathways were simulated by means of supervised molecular dynamics (SuMD)18,19 and the outcomes tested in mutagenesis experiments to identify A1R residues involved along the route toward and from the orthosteric site. We propose the binding conformation of N6-cyclopenthyl agonists and how chemical modifications could impact the binding mechanisms of these selective A1R agonists. Our results can be framed within the dynamic nature of ligand–receptor complex formations and highlight the dramatic effect on drug binding kinetics triggered by chemical substitutions.
2. Methods
2.1. Experimental Methods
2.1.1. Compounds
Adenosine, NECA ((2S,3S,4R,5R)-5-(6-aminopurin-9-yl)-N-ethyl-3,4-dihydroxyoxolane-2-carboxamide), CPA, were purchased from Sigma-Aldrich and dissolved in dimethyl-sulfoxide (DMSO). HOCPA and BnOCPA was synthesized as described in Knight et al.17 (compounds 6 and 7, respectfully). CA200645, a high affinity AR xanthine amine congener (XAC) derivative containing a polyamide linker connected to the BY630 fluorophore, was purchased from HelloBio (Bristol, U.K.) and dissolved in DMSO. The concentration of DMSO was maintained to 1.1% for NanoBRET ligand-binding experiments using CA200645.
2.1.2. Generation of Mutant A1R Constructs
The NanoLluc(Nluc)-tagged human A1R pcDNA3.1+ construct used to generate stable HEK 293 cell lines was kindly gifted to us by Stephen Hill and Stephen Briddon (University of Nottingham). Mutations within the A1R were made using the QuikChange Lightening Site-Directed Mutagenesis Kit (Agilent Technologies) in accordance with the manufacturer’s instructions. All oligonucleotides used for mutagenesis were designed using the online Agilent Genomics “QuikChange Primer Design” tool and purchased from Merck. All constructs were confirmed by in-house Sanger sequencing.
2.1.3. Cell Culture and Transfections
HEK 293 cells in a single well of a 6-well plate (confluency ≥80%) were transfected with 2 μg of DNA using polyethylenimine (PEI, 1 mg/mL, MW = 25 000 g/mol) (Polysciences Inc.) at a DNA–PEI ratio of 1:6 (w/v). Briefly, DNA and PEI were added to separate sterile tubes containing 150 mM sodium chloride (NaCl) (total volume 50 μL), allowed to incubate at room temperature for 5 min, mixing together, and incubating for a further 10 min prior to adding the combined mix dropwise to the cells. 48 h post-transfection, stable Nluc-A1R expressing HEK 293 cells were selected using 600 μg/mL Geneticin (Thermo Fisher Scientific) whereby the media was changed every 2 days. HEK 293 cell lines were routinely cultured in DMEM/F-12 GlutaMAX (Thermo Fisher Scientific) supplemented with 10% FBS (F9665, Sigma-Aldrich).
2.1.4. Analysis of A1R Cell-Surface Expression Using Flow Cytometry
HEK 293 cells and WT or mutant A1R expressing HEK 293 cells were harvested using a nonenzymatic cell dissociation solution (Sigma-Aldrich) and washed with PBS prior to counting. A total of 0.5 × 106 cells were washed three times in flow buffer (PBS supplemented with 1% BSA and 0.03% sodium azide) before resuspending in 50 μL of flow buffer containing anti-Nluc polyclonal primary antibody raised in rabbit (kindly gifted by Promega) at 1:100 dilution and incubated at room temperature for 1 h. All samples were washed three times with flow buffer and resuspended in 50 μL of flow buffer containing Allophycocyanin (APC)-conjugated anti-Rabbit IgG (Thermo Fisher Scientific, 31984) at 1:150 dilution and incubated for 1 h at room temperature in the dark. The cells received a final three washes and were resuspended in 300 μL of flow buffer.
Analysis was conducted using a BD AccuriTM C6 Plus Flow Cytometer which is equipped with a blue (488 nm) and red (640 nm) laser, two light scatter detectors (FSC and SCC), and four fluorescence detectors (FL1 emission λ 530/30 nm, FL2 emission λ 585/40 nm, FL3 emission λ 570, and FL4 emission λ 675/25 nm). FL4 optical filters were chosen APC (excitation λ 633 nm and emission λ 660 nm). Unstained cells and HEK 293 cells without A1R expression were used as controls for autofluorescence and unspecific antibody binding, respectively. All data is collected by the flow cytometer, and analysis can be conducted at any time using the BD AccuriTM C6 software. The images for the figures were made in FlowJo (V7.6.5), an analysis platform for single-cell flow cytometry analysis.
2.1.5. BRET Assays for Binding
Saturation binding at WT and mutant A1R for the determination of CA200645 KD was conducted using NanoBRET. The Nluc acts as the BRET donor (luciferase oxidizing its substrate, furimazine), and CA200645 acted as the fluorescent acceptor. Here, HEK 293 cells stably expressing WT or mutant Nluc-A1R 24 h postplating (10 000 cells/well of a 96-well plate) where pretreated with 0.1 μM furimazine for 5 min prior to stimulated with CA200645 at a range of concentrations (0–300 nM). Filtered light emission at 450 nm and >610 nm (640–685 nm band-pass filter) was measured over a 30 min period using a Mithras LB 940 following stimulation with a range of CA200645 concentrations and the raw BRET ratio calculated (610 nm/450 nm). Nonspecific binding was determined by saturating concentrations of DPCPX (1 μM) and subtracted from the BRET ratio. CA200645 binding reached equilibrium after 5 min (Figure S2), and the BRET ratio was taken at 10 min and then fit to the “One site–Specific binding” model built into Prism.
As described previously,20 NanoBRET competition binding assays were conducted to determine the affinity (pKi) of various A1R compounds. Briefly, following a 5 min preincubation with 0.1 μM furimazine, cells were costimulated with CA200645 (used at 25 nM, as previously reported20,21) and increasing concentration of unlabeled ligand and emission at 450 nm and >610 nm immediately measured. The BRET ratio at 10 min poststimulation was fitted with the “one-site–Ki model” derived from the Cheng and Prusoff correction,22 built into Prism to determine affinity (pKi) values for all unlabeled agonists at the A1Rs. The determined KD of CA200645 at the mutant A1R was taken into account during analysis. Nonspecific binding was determined using a high concentration of unlabeled antagonist at 1 μM DPCPX.
2.1.6. Data Analysis
All experiments were conducted in duplicate (technical replicates) to ensure the reliability of single values. Statistical analysis, performed using Prism 8.0, was undertaken for experiments where the group size was at least n = 3, and these independent values are used to calculate statistical significance (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001) using a one-way ANOVA with a Dunnett’s post-test for multiple comparisons.
2.2. Computational Methods
2.2.1. Biological Targets and Ligands Force Field Parameters
All 10 systems (Table 1) were prepared for molecular dynamics (MD) simulations using the CHARMM3623,24/CGenFF 3.0.125−27 force field combination. Initial ligand force filed topology and parameter files were obtained from the ParamChem Web server.25 Adenosine and NECA are already well-parametrized in the CGenFF force field. Optimized parameters for HOCPA and BnOCPA from our previous work28 were used and transferred to CPA.
Table 1. Ten Systems Simulated Employing SuMD and SuMD Path Sampling.
| ligand | transition | no. of SuMD replicas | total SuMD path sampling |
|---|---|---|---|
| adenosine | binding | 9 | 5.18 μs |
| unbinding | 5 | 3.09 μs | |
| NECA | binding | 6 | 1.86 μs |
| unbinding | 6 | 7.10 μs | |
| CPA | binding | 9 | 4.34 μs |
| unbinding | 10 | 0.97 μs | |
| HOCPA | binding | 8 | 4.06 μs |
| unbinding | 6 | 2.58 μs | |
| BnOCPA | binding | 7 | 5.82 μs |
| unbinding | 6 | 4.92 μs |
2.2.2. Protein preparation
The active A1R structure (Table 1) was retrieved from the Protein Data Bank29 (PDB code 6D9H(9)). For SuMD binding simulations, the agonists were placed at least 30 Å from the binding site in five different systems. For SuMD unbinding, the experimental coordinates (PDB 6D9H) were used to simulate the adenosine, while representative frames (ligand orthosteric conformations close to the experimental bound adenosine on A1R) from the SuMD binding were used to start the SuMD unbinding of NECA, CPA, HOCPA, and BnOCPA. The A1R intracellular loop 3 (ICL3) was modeled using Modeler 9.19.30 For all the 10 systems (Table 1), hydrogen atoms were added by means of the pdb2pqr31 and propka32 software (considering a simulated pH of 7.0); the protonation of titratable side chains was checked by visual inspection. The resulting receptor was inserted in a square 90 Å × 90 Å 1-palmitoyl-2-oleyl-sn-glycerol-3-phosphocholine (POPC) bilayer (previously built by using the VMD Membrane Builder plugin 1.1, Membrane Plugin, version 1.1 at http://www.ks.uiuc.edu/Research/vmd/plugins/membrane/), through an insertion method.33 The receptor orientation was obtained by superposing the coordinates on the corresponding structure retrieved from the OPM database.34 Lipids overlapping the receptor transmembrane helical bundle were removed, and TIP3P water molecules35 were added to the simulation box by means of the VMD Solvate plugin 1.5 (Solvate Plugin, version 1.5. at http://www.ks.uiuc.edu/Research/vmd/plugins/solvate/). Finally, overall charge neutrality was reached by adding Na+/Cl– counterions up to the final concentration of 0.150 M), using the VMD Autoionize plugin 1.3 (Autoionize Plugin, version 1.3. at http://www.ks.uiuc.edu/Research/vmd/plugins/autoionize/).
2.2.3. Systems Equilibration and General MD Settings
The MD engine ACEMD36 was employed for both the equilibration and productive simulations. The equilibration of the membrane systems was achieved in isothermal–isobaric conditions (NPT) using the Berendsen barostat37 (target pressure 1 atm) and the Langevin thermostat38 (target temperature 300 K) with low damping of 1 ps–1. A four-stage procedure was performed (integration time step of 2 fs): first, clashes between protein and lipid atoms were reduced through 2000 conjugate-gradient minimization steps, then a 2 ns long MD simulation was run with a positional constraint of 1 kcal mol–1 Å–2 on protein and lipid phosphorus atoms. During the second stage, 20 ns of MD simulation was performed constraining only the protein atoms, while in the last equilibration stage, positional constraints were applied only to the protein backbone alpha carbons, for a further 20 ns. Globular protein equilibration was achieved in two steps: after 500 cycles of conjugate-gradient minimization, the system was simulated for 5 ns, employing an integration time step of 2 fs, in the isothermal–isobaric conditions (NPT).
Productive trajectories (Table S4) were computed with an integration time step of 4 fs in the canonical ensemble (NVT). The target temperature was set at 300 K, using a thermostat damping of 0.1 ps–1; the M-SHAKE algorithm39,40 was employed to constrain the bond lengths involving hydrogen atoms. The cutoff distance for electrostatic interactions was set at 9 Å, with a switching function applied beyond 7.5 Å. Long-range Coulomb interactions were handled using the particle mesh Ewald summation method (PME)41 by setting the mesh spacing to 1.0 Å.
2.2.4. Supervised MD (SuMD) Protocol
The supervised molecular dynamics (SuMD) is an adaptive sampling method42 for speeding up the simulation of binding18,43 and unbinding processes.19,44 In the simplest SuMD implementation, sampling is gained without the introduction of any energetic bias, by applying a tabu-like algorithm to monitor the distance between the centers of mass (or the geometrical centers) of the ligand and the predicted binding site or the receptor. However, the supervision of a second metric of the system can be considered.44 A series of short unbiased MD simulations are performed, and after each simulation, the distances (collected at regular time intervals) are fitted to a linear function. If the resulting slope is negative (for binding) or positive (for unbinding), the next simulation step starts from the last set of coordinates and velocities produced; otherwise, the simulation is restarted by randomly assigning the atomic velocities.
2.2.5. Settings for SuMD Binding to the A1R
To simulate the agonists’ binding to the A1R (Table 1, Videos S1–S4), the distance between the centroid of the ligand and the centroid of the orthosteric residues N2546.55, F171ECL2, T2777.42, and H2787.43 was supervised during 500 ns long time windows until it reached a value less than 4 Å. Then?
2.2.6. Settings for SuMD Unbinding from the A1R
For the SuMD unbinding (Table 1, Videos S1–S4), different from the SuMD binding algorithm, the length (Δt) of the short simulations increases along the dissociation pathway, according to the formula:
| 1 |
Δt0 is the duration of the very first MD time window, and Nti is a factor that is picked from three user-defined values (Nt1, Nt2, and Nt3), according to the last ligand–protein distance detected. At the end of each MD run, the ligand–protein distance (rL) is compared to three distance threshold values (D1, D2, and D3, also defined by the user), allowing a decision on the value of Nti factor according to the following conditions:
| 2 |
| 3 |
| 4 |
| 5 |
Values of 3 Å, 5 Å, and 8 Å were used for D1, D2, and D3, respectively, while Nt1, Nt2, and Nt3 were set to 2, 4, and 8 (SuMD time windows of 100 ps, 200 ps, 400 ps, and 800 ps).
2.2.7. SuMD Path Sampling Protocol
Further MD sampling (SuMD path sampling, Table 1) was performed using the outputs from each SuMD replica (Videos S1–S4) for both binding and unbinding. Each trajectory was aligned on the protein α carbon atoms, and the frames were clustered according to the ligand root-mean-square deviation (RMSD) to the starting positions (bin of 1 Å). A frame from each group was randomly extracted and used as a starting point for 20 ns long classic MD simulations. The number of classic MD simulations run varies according to the number of bins and in turn with the linearity of the SUMD pathways. The BnOCPA binding28 and the adenosine SuMD binding and unbinding19 trajectories used to seed classic MD simulations were the same as our previous work.
2.2.8. Analysis of the MD Trajectories
Only the MD trajectories from the SuMD path sampling were analyzed, as they should favor a relaxation of the systems toward local energy minima poorly sampled through the SuMD simulations. Interatomic contacts and root-mean-square deviations (RMSD) were computed using VMD.45 Contacts were considered productive if the distance between two atoms was less than 3.5 Å. Ligand–protein hydrogen bonds were detected using the GetContacts scripts tool (https://getcontacts.github.io), setting a hydrogen bond donor–acceptor distance of 3.3 Å and an angle value of 150° as geometrical cutoffs. Contacts and hydrogen bond persistency are quantified as the percentage of frames (considering all the frames obtained by merging the different replicas) in which protein residues formed contacts or hydrogen bonds with the ligand. The computation takes into account direct and water-mediated interactions.
Distances between atoms were computed using PLUMED 2.3.46 The molecular mechanics generalized Born surface area (MM/GBSA) energy was computed with the MMPBSA.py47 script (AmberTools17 suite at http://ambermd.org/), after transforming the CHARMM psf topology files to an Amber prmtop format using ParmEd (documentation at http://parmed.github.io/ParmEd/html/index.html). We preferred the MM/GBSA approach over the molecular Poisson–Boltzmann surface area (MM/PBSA) because binding and unbinding paths are not compatible with a grid method.48
2.3. Numbering System
Throughout the manuscript, the Ballesteros–Weinstein residues numbering system for the GPCRs49 is adopted as a superscript.
3. Results
3.1. N6-Cyclopentyl Agonists Bind to A1R with Similar Fashion
ARs ligands bearing an alkyl ring in position N6 (on the adenine scaffold) or C8 (xanthine scaffold) display increased affinity for A1R over A2AR10. The reason for this is attributed to T2707.35 (A1R) in place of M2707.35 (A2AR), which shapes a hydrophobic subpocket underneath ECL3 that partially accommodates the lipophilic substituent. While the inactive structures of A1R and A2AR in complex with the xanthine antagonist PSB3610 have univocally shown this structural aspect of the selectivity, no confirmation from crystallography or cryo-EM studies is yet available for ARs agonists.
Besides reproducing the adenosine cryo-EM binding conformation into A1R (Figure S3, Video S1),9 our simulations sampled the orthosteric binding mode of NECA observed on A2AR (Figure S3, Video S2).50 In light of this reliability, we propose the likely binding mode of CPA, HOCPA, and BnOCPA (Figure 2, Video S3 and Video S4). As expected, the adenine ring forms a bidentate hydrogen bond with N2546.55 and a π–π stacking with F171ECL2, while the N6-cyclopentyl ring inserts in the hydrophobic pocket under ECL3, interacting with T2707.35 and L2536.54 (Figure 2). Mutagenesis experiments confirmed the importance of L2536.54 (Table 2) for the affinity of the agonists. BnOCPA is proposed to bind to A1R with same features of the smaller ligands CPA and HOCPA (Figure 2b, Video S4). However, during the simulations the oxybenzyl group showed28 high flexibility and explored three different orientations (Figure 2b). In two of these conformations, BnOCPA interacted with A1R residue L2586.59 (mode B, Figure 2b) and Y2717.36 (mode C, Figure 2b). Binding assays with mutants L2586.59A, F2586.59A, F2586.59T, and Y2717.36A (Table 2) confirmed that these two residues are likely involved in the orthosteric complex with BnOCPA. However, a role during agonist association and dissociation events cannot be ruled out, as also the affinity for CPA and HOCPA (which are not involved in contacts with L2586.59 and Y2717.36 in the bound state, Figure 2a) was affected (Table 2).
Figure 2.

Binding modes of the agonists according to SuMD path sampling simulations. (a) CPA (magenta) and HOCPA (pink) engage A1R with the same orientation as adenosine (tan stick); the N6-cyclopentyl group interacts with L2536.54 and T2576.58. (b) BnOCPA orients the oxybenzyl group in three different orientations. Hydrogen bonds with N254ECL2 are shown as dashed lines, while hydrophobic contacts are depicted as cyan transparent surfaces.
Table 2. NanoBRET Saturation- And Competition-Binding Assays in WT and Mutant Nluc-A1Ra.
| pKid |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cell-surface expressionb | n | KDc | n | CPA | n | BnOCPA | n | HOCPA | n | adenosine | n | |
| WT | 100 ± <0.1 | 8 | 68.6 ± 7.5 | 5 | 6.95 ± 0.02 | 3 | 6.21 ± 0.04 | 4 | 6.37 ± 0.03 | 4 | 5.90 ± 0.08 | 4 |
| F81.31A | 53 ± 12** | 3 | 90.7 ± 21.5 | 3 | 7.34 ± 0.08* | 4 | 6.37 ± 0.02 | 3 | 6.95 ± 0.07** | 3 | 6.55 ± 0.17*** | 3 |
| Q91.32A | 21 ± 2**** | 3 | 60.6 ± 5.4 | 3 | 6.41 ± 0.09** | 3 | 5.44 ± 0.06**** | 3 | 5.71 ± 0.05*** | 3 | 5.07 ± 0.11**** | 3 |
| Y121.35A | 120 ± 19 | 3 | 75.8 ± 6.0 | 3 | 6.50 ± 0.06** | 4 | 5.44 ± 0.06**** | 4 | 5.78 ± 0.10*** | 4 | 5.35 ± 0.05*** | 4 |
| I692.64A | 158 ± 7*** | 3 | 147.2 ± 13.4* | 3 | 4.94 ± 0.06**** | 3 | 4.72 ± 0.04**** | 3 | 4.34 ± 0.11**** | 3 | 4.64 ± 0.13**** | 3 |
| N702.65A | 86 ± 9 | 3 | 51.6 ± 3.7 | 3 | 6.97 ± 0.12 | 3 | 5.92 ± 0.06* | 3 | 6.05 ± 0.09 | 3 | 5.83 ± 0.05 | 3 |
| N148ECL2A | 13 ± 1**** | 3 | 70.7 ± 13.2 | 3 | 7.20 ± 0.13 | 3 | 6.15 ± 0.03 | 3 | 6.15 ± 0.05 | 4 | 5.71 ± 0.07 | 3 |
| I175ECL2A | 23 ± 3**** | 3 | 153.2 ± 27.6** | 3 | 6.30 ± 0.06**** | 3 | 5.60 ± 0.05**** | 3 | 5.66 ± 0.02**** | 4 | 5.00 ± 0.11**** | 3 |
| T2576.58A | 114 ± 19 | 4 | 128.8 ± 16.0 | 3 | 7.30 ± 0.03** | 3 | 6.77 ± 0.05**** | 4 | 6.94 ± 0.05**** | 3 | 5.85 ± 0.09 | 3 |
| L2536.54A | 160 ± 24*** | 3 | 104.1 ± 19.3 | 3 | 6.17 ± 0.06**** | 4 | 5.39 ± 0.10**** | 4 | 5.49 ± 0.13**** | 3 | 5.40 ± 0.06** | 3 |
| L2586.59A | 82 ± 2 | 3 | 118.3 ± 20.6 | 3 | 6.34 ± 0.07**** | 6 | 6.03 ± 0.05*** | 5 | 5.85 ± 0.08**** | 5 | 5.66 ± 0.08 | 5 |
| H264ECL3A | 21 ± 6**** | 3 | 84.1 ± 4.6 | 3 | 6.33 ± 0.09*** | 3 | 5.60 ± 0.01**** | 3 | 6.02 ± 0.18 | 3 | 6.21 ± 0.05 | 3 |
| K265ECL3A | 68 ± 13 | 3 | 54.7 ± 8.6 | 3 | 6.59 ± 0.13 | 3 | 6.21 ± 0.05 | 4 | 6.01 ± 0.12 | 4 | 5.58 ± 0.09 | 3 |
| S2677.32A | 90 ± 6 | 3 | 89.9 ± 11.7 | 3 | 6.66 ± 0.06* | 3 | 5.85 ± 0.01*** | 3 | 5.99 ± 0.05*** | 3 | 5.66 ± 0.08 | 3 |
| Y2717.36A | 171 ± 10**** | 3 | 94.4 ± 11.1 | 3 | 5.82 ± 0.03**** | 3 | 5.14 ± 0.07**** | 4 | 5.15 ± 0.07**** | 3 | 5.15 ± 0.05*** | 3 |
| L2586.59T | 77 ± 18 | 3 | 200.4 ± 24.6**** | 3 | 6.39 ± 0.07 | 3 | 5.69 ± 0.04*** | 4 | 5.69 ± 0.04**** | 4 | 5.45 ± 0.05 | 4 |
| L2586.59F | 131 ± 2 | 4 | 123.0 ± 17.3 | 3 | 6.40 ± 0.10 | 3 | 6.12 ± 0.05**** | 3 | 5.79 ± 0.08*** | 3 | 5.47 ± 0.04 | 3 |
| L2586.59G | 15 ± 4**** | 3 | 143.3 ± 37.5* | 3 | ||||||||
CA200645 equilibrium dissociation constant (KD) and compound affinity (pKi) at WT and mutant Nluc-A1R, as determined by NanoBRET saturation and competition ligand-binding assays, respectively. Data are expressed as mean ± SEM obtained in n separate experiments. All individual experiments were conducted in duplicate. Statistical significance (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001) compared to WT was determined by one-way ANOVA with the Dunnett’s post-test.
Mean fluorescence intensity of APC (% wild-type (WT)). Cell-surface expression of WT or mutant Nluc-A1R in HEK 293 cells was determined by flow cytometry. Cells were incubated with anti-Nluc antibody followed by APC-conjugated antirabbit IgG secondary antibody and the fluorescence detector FL4 (emission λ 675/25 nm) used to detect APC fluorescence.
CA200645 equilibrium dissociation constant (KD) as determined by NanoBRET saturation binding assays.
Compound affinity (pKi) (mean ± SEM) determined through NanoBRET competition-binding assays in WT/mutant Nluc-A1R stably expressing HEK 293 cells. The resulting concentration-dependent decrease in BRET ratio at 10 min was used to calculate pKi. Compound affinity could not be determined (−) for L253G Nluc-A1R given the low cell-surface expression and reduced binding affinity of CA200645 (KD).
3.2. Simulations Suggest the Binding and Unbinding Paths of A1R Agonists
SuMD and SuMD path sampling delivered detailed insights on the possible transitory metastable states that the agonists experienced along the route to, and from, the orthosteric site (Figure 3, Tables S1–S4, Figures S5 and S6; Video S1–Video S4). While the endogenous agonist adenosine reached the orthosteric site with very limited intermediate interactions with ECL2 (Figure 3b, Table S1, Figure S5, Video S1), the other ligands formed contacts with this extracellular vestibule, with CPA and BnOCPA most involved in the metastable states (Figure 3d,h, Table S1, Figure S5, Video S3, Video S4). As a general view, the increase in lipophilicity at the N6 position (CPA, HOCPA, and BnOCPA, Figure 3d,f,h, Table S1, Figure S5) and the 5′ position (NECA, Figure 3, Figures S4a and S5, Table S1) favored intermediate interactions with ECL2. The importance of ECL2 for the binding of NECA, CPA, and the antagonist DPCPX to A1R has been recently demonstrated.12,51 Our simulations suggest I175ECL2 and E172ECL2 as involved in interactions during both the binding and unbinding (Figure 3d, Figures S4 and S6), while E170ECL2, I167ECL2, N159ECL2, and W156ECL2 engaged NECA during the binding (Figures S4a and S6). L149ECL2, on the other hand, was not involved in direct interactions with the ligands, suggesting an important role in stabilizing the overall secondary structure of ECL2 due to its position at the base of the loop helix. Besides the aforementioned residues, further A1R side chains were involved along the simulated binding paths for all the agonists considered (Tables S1 and S2 and Figure S5).
Figure 3.

Simulated binding and unbinding paths of the agonists and the relative position of the A1R mutants tested. (a) Two side views of the A1R (white transparent ribbon); residues considered for the mutations (Table 2) are shown as gray sticks. The cryo-EM bound adenosine (tan stick) is reported as reference. (b–i) Left-hand panels, position of the agonist centroid during simulations, colored according to the interaction energy with A1R (white ribbon); residues mutated (Table 2) are shown as sticks and colored according to effect on the affinity (red, decreased affinity; green, increased affinity; black, unaltered affinity). Right-hand panels, A1R-agonist contacts plotted onto the protein surface and colored according to the contacts occupancy. (b) Adenosine binding simulations, (c) adenosine unbinding simulations, (d) CPA binding simulations, (e) CPA unbinding simulations, (f) HOCPA binding simulations, (g) HOCPA unbinding simulations, (h) BnOCPA binding simulations, (i) BnOCPA unbinding simulations.
SuMD unbinding routes (Figure 3c,e,g,i, Video S1–Video S4) had only limited overlap with the binding routes (Figure 3b,d,f,h, Video S1–Video S4). The agonists, indeed, established frequent interactions with the top of TM1, TM2, and TM7 (Figure 3c,e,g,i, Figures S4b and S6, Tables S2 and S4). However, in analogy with the binding simulations, CPA, HOCPA, and BnOCPA (all bearing an N6 hydrophobic moiety) showed a major involvement of ECL2 (Tables S3 and S4, Figure S6; Video S3 and Video S4). Many of the A1R residues involved in both agonist association (Tables S1 and S2, Figure S5) and dissociation (Tables S3 and S4, Figure S6) are part of the orthosteric site. It is therefore not surprising that mutagenesis experiments already pointed out their importance.12,52−56
Interestingly, our unbiased nonequilibrium simulations pointed out several residues, located outside the orthosteric site, involved along the binding and unbinding routes. These residues comprise F81.31, Q91.32, Y121.35, I692.64, N702.64, N148ECL2, I175ECL2, L2536.54, T2576.58, L2586.59, H264ECL3, K265ECL2, S2677.32, Y2717.36 (Figure 3a). Mutagenesis experiments were designed and performed to confirm computational predictions (Figure 3, Table 2, Table S5).
3.3. Mutagenesis Experiments Reveal novel A1R Residues Involved in the Binding of Agonists
3.3.1. Surface Expression of the A1R Alanine Mutants and CA200645 KD
The cell-surface expression of WT and mutant A1R, as determined by mean fluorescence intensity of APC (% WT), was comparable to WT for Y121.35A, N702.64A, T2576.58A, L2586.59A, K265ECL2A, S2677.32A, L2586.59T, and L2586.59F A1R. When compared to WT, the cell-surface expression was determined to be significantly reduced for F81.31A, Q91.32A, N148ECL2A, I175ECL2A, H264ECL2A, and L2586.59G, whereas it was significantly increased for I692.65A, L2536.54A, and Y2717.36A. This enhanced or reduced mutant A1R cell-surface expression did not correlate with changes in CA200645 equilibrium dissociation constant (KD). For example, the KD for CA200645 determined in L2586.59T was significantly increased when compared to WT but showed comparable cell-surface expression. I175 ECL2A and L2586.59G also showed an increased KD but had a reduced cell-surface expression when compared to WT.
Importantly, the determined changes in CPA, BnOCPA, HOCPA, or adenosine affinity (pKi) were determined to be independent of changes in the cell-surface expression. For example, despite the significantly elevated cell-surface expression of Y2717.36A, the determined compound affinity of all four tested compounds was significantly reduced when compared to that determined at WT. This is likely due to the high receptor expression in our system and/or reserve. The only exception was L2586.59G, whereby compound pKi could not be determined due to the likely combined effect of low cell-surface expression and reduced CA200645 affinity.
3.3.2. General Effects of Alanine Substitutions on Agonists Affinity
A number of mutations enhanced affinity, while other either decreased affinity or had no overall effect. The mutated residues can be divided into three groups according to their positions relative to the (un)binding paths sampled during the simulations.
3.3.3. TM1, TM2, and TM7 Residues
The A1R residues located at the top of TM1, TM2, and TM7 (Figure 3a) were generally involved during both the simulated binding (Figure 3b,d,f,h, Figures S5 and S6) and unbinding (Figure 3c,e,g,i, Figure S5). Adenosine, CPA, and HOCPA displayed decreased affinity to A1R mutants Q91.32A and Y121.35A (Table 2) and enhanced the binding to F81.31A compared to the WT, in agreement with the transitory interactions formed with TM1 during simulations (Figure 3b–g, Tables S1–S4, Figure S5 and S6). The affinity for the agonists diminished on Q91.32A and Y121.35A, but BnOCPA was not significantly affected by F81.31A. This is apparently in disagreement with simulations, which proposed BnOCPA as the agonist more prone to form metastable states in the proximity of F1.318 during the dissociation from the receptor (Figure 3i). However, the fact that BnOCPA was the ligand most prone to interact with F8 also during the association (Figure 3h) may suggest a degree of compensation between binding and unbinding. That is, the variation of the binding rate can be compensated by an opposite change in the unbinding rate (and vice versa) resulting in unchanged affinity. This could also be the case of N702.65A, which did not alter the affinity of the agonists, except for BnOCPA (Table 2). Binding and unbinding simulations suggested that N702.65 forms numerous interactions with the ligands (Tables S1–S4, Figures S5 and S6). The adjacent residue I692.64, instead, when mutated to alanine (I692.64A) significantly decreased the affinity of all the agonists (Table 2). Besides being involved in frequent interactions with the agonists (Tables S1–S4), I692.64 is packed in hydrophobic contacts with TM3, possibly stabilizing the neighboring part of ECL2.
Moving to TM7, the agonists NECA, CPA, HOCPA, and BnOCPA, but not adenosine, displayed diminished binding to the S2677.32A (Table 2, Table S5), while all of them lost affinity to Y2717.36A (Table2, Table S5). The bulky Y2717.36 side chain, which occupies an important position at the interface between the orthosteric site and the extracellular vestibule, could participate in numerous intermediate interactions along the (un)binding routes.
3.3.4. TM6 Residues
The three A1R residues mutated on TM6 (L2536.54, T2576.58, and L2586.59, Figure 3a) are part of different protein environments. L2536.54 shapes part of the hydrophobic pocket underneath ECL3 partially responsible for A1/A2A ligands selectivity (along with L2697.34 and T2707.35). All the agonists displayed diminished affinity to L2536.54A (Table 2), with the N6-substituted agonists CPA, HOCPA, and BnOCPA most affected, in line with the hydrophobic interactions occurring in the bound state (Figure 2). Interestingly, T2576.58A increased the affinity of CPA, HOCPA, and BnOCPA. This could be due to an increase in lipophilicity of the protein environment surrounding the cyclopentyl group either in the bound complex or along the unbinding pathways, (Figure 3e,g,i). NECA showed reduced affinity to T2576.58A (Table S5, Figure S4), confirming the importance of hydrophobic N6-substituents for interactions with the top of TM6. L2586.59, which is located at the interface with the membrane, is one of the TM5 and TM6 residues shaping a saddle between ECL2 and ECL3, where agonists tended to form metastable interactions along the simulated (un)binding paths (Figure 3b–i, Figures S5 and S6). All the agonists (excepted adenosine) displayed a reduced affinity for L2586.59A and L2586.59T, suggesting that the bulkier ligands may be more prone to interact with this part of A1R (BnOCPA was the only ligand proposed to interact at some extent with L2586.59 in mode B, Figure 2b). The affinity decrease (Table 2) displayed by HOCPA and BnOCPA for L2586.59F likely excludes a destabilization of the neighbor protein structure, as residue F2586.59 WT A2AR does not change the conformation of the top of TM6.
3.3.5. ECL2 and ECL3 Residues
As I175ECL2 formed numerous interactions with the agonists during the simulations (Figure 3b–i, Tables S1 and S2, Figures S5 and S6), not surprisingly the I175ECL2A mutation reduced the affinity of all the ligands (Table 2, Table S5). I175ECL2 could contribute to keeping the aromatic side chain of F171ECL2 in an appropriate conformation for interacting with the ligands’ adenine ring in the orthosteric complex (Figure S1). On the other hand, N148ECL2A did not affect the affinity of the agonists, despite the frequent interactions during the binding simulations (Table 2, Figure 3b,d,f,h, Tables S1 and S2, Figures S5 and S6).
While H264ECL2A showed diminished affinity (Table 2) for CPA and BnOCPA (which have the most lipophilic N6-group, Figure 1), none of the tested ligands were significantly affected by K265ECL2A. As K265ECL2 is part of a stable salt bridge with E172ECL2, K265ECL2A is expected to affect the affinity of the ligands due to the reduced hindrance of the orthosteric site. The subtype A2AR bears A265ECL2 in place of K265ECL2 and the residue involved in the salt bridge with E169ECL2 (corresponding to E172ECL2 in A1R) is H264ECL2. The A2AR H264ECL2A mutant does not display a modified affinity for the antagonists ZM241385, despite the increase in off rate.57 This could indicate a kinetic compensation due to a faster binding to H264ECL2A (A2AR) and K265ECL2A (A1R). A less bulky alanine side chain, indeed, would favor ligand binding and the unbinding to a similar extent.
4. Discussion
The association and dissociation of a ligand from its receptor is a multistep process, characterized by intermediate metastable states that anticipate (e.g., during the association) or follow (e.g., during the dissociation) the bound state. In this complex scenario, further complicated by more than one possible pathway, it is difficult to rationalize the role played by GPCR metastable binding sites. Preliminary interactions with the EC vestibules, for example, act as selectivity filters by favoring the recognition of a chemotype over other molecules,58,59 in some cases driving the affinity and the selectivity.60 However, if the stability of an intermediate state is similar to the orthosteric one, segregation may occur, that is, the displayed ligand affinity for the receptor diminishes.61
For the A1R, as a general view, the agonist’s binding and unbinding occurred through two major pathways, namely, Path A and Path B (Figure 4a). Path A comprises residues located at the top of TM5, TM6 (L2536.54, T2576.58, and L2586.59) and on the distal region of ECL2 (I175ECL2 and N148ECL2), while Path B is well-defined by residues on the top of TM1, TM2, TM7 (F81.31, Q91.32, Y121.35, I692.64, N702.65, S2677.32, and Y2717.36). The analysis of the position of the residues in terms of path and distance from the orthosteric site, the occupancy of the contacts formed during the binding and unbinding, and the effect of the mutation to alanine on the pKi, highlights a complicated pattern (Figure 4b). The endogenous agonist adenosine, which is the smallest ligand considered here, was the least affected by mutations; this is consistent with the relatively straight (un)binding pathways sampled during MD simulations (Figure 3a,b). The mutation of the hydrophobic residues L2536.54, I175ECL2, and I692.64, that are close to the orthosteric site, decreased the pKi due to reduced apolar contacts with the ligands (Figures S5 and S6). T2576.58A, which augmented the hydrophobicity of the pocket underneath ECL3, increased the binding affinity of all the agonists bar adenosine, in line with the role of the N6 hydrophobic substituent. The other mutation, beside T2576.58 A, that increased the pKi (BnOCPA excepted) was F81.31A (Figure 4b). F81.31 is located far away from the orthosteric site and is involved in the contacts between TM1 and TM7; therefore, its mutation to alanine could have local effects on the neighboring side chains or the flexibility of the TM1 N-terminal.
Figure 4.
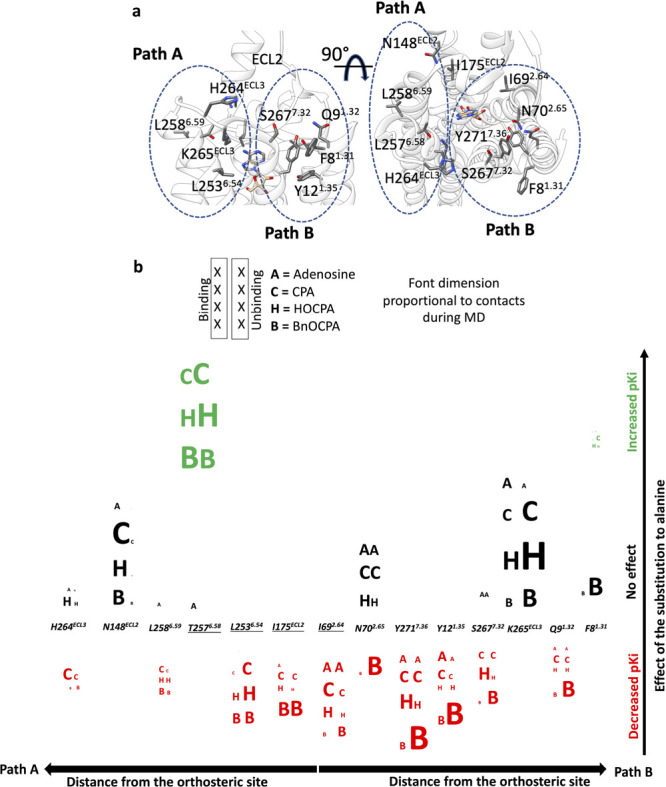
Scrambled plot of the mutated A1R residues according to the distance from the orthosteric site along Path A or Path B (a) and the effect of the mutation on the pKi. The contacts formed with the agonists are represented as letters of dimension proportional to the occupancy; contacts during the binding are reported in the first column, while contacts from unbinding are reported in the second column; residues in close proximity of the orthosteric site are underlined (b).
Strikingly, the mutation of N148ECL2, located distant from the orthosteric site along Path A and highly involved during the binding (Figure 4b, Figure 3) did not change the pKi of any of the agonists, in line with previous findings.12 We speculate that its effect on the overall kinetics of binding is neglectable because it is not part of the secondary hydrophobic binding site on ECL2 that has been associated with A1R allosterism13,62 and highlighted also in our simulations (Figure 3). Moving to residues located along Path B, N702.65A negatively affected only BnOCPA pKi. Notably, BnOCPA was highly prone to hydrogen bond with N702.65 during the unbinding, but N702.65 was less involved during the binding (Figures S5 and S6). This unbalance was not mirrored by the other agonists, which instead interacted with N702.65 during binding and unbinding and therefore experienced a sort of compensation, with the final effect of not affecting the pKi. This binding/unbinding compensation is evident in the case of K265ECL3A. K265ECL2 forms a salt bridge with E172ECL2 that is likely to contribute to the (un)binding kinetics of A1R ligands, in analogy with the A2R salt bridge formed between H264ECL3 and E169ECL2 57. It follows that the disruption of the K265ECL2–E172ECL2 salt bridge by mutating K265ECL2 to alanine should speed up both the association, as an agonist find a more accessible orthosteric site, and the dissociation, because of less hindrance for the transition to the EC vestibules.
4.1. Toward the Definition of the Structure-Binding/Unbinding Path Relationships of A1R Agonists
The (un)binding kinetics have a major impact on both pharmacodynamics63−65 and pharmacokinetics66 of a drug. Even small structural modifications within a congeneric series of ligands modify the kinetics through modified on and off rates.67 The reason for this lies in the enormously higher number of different intermediate states that a ligand can experience along (un)binding paths compared to the orthosteric complexes, where it is restrained by intermolecular interactions and steric hindrances. Protein–ligand recognition events have very complex energy landscapes,68−71 and small changes in either the structure of the ligand or the protein can alter the nature and the position of the transition states along the (un)binding routes.72 To investigate this aspect on the A1R agonists, we compared the A1R interaction patterns between adenosine, CPA, or BnOCPA (Figure 5) to understand how the introduction of the N6 cyclopentyl group on the adenosine scaffold and the introduction of the oxybenzyl group on CPA (BnOCPA) could alter the overall (un)binding mechanisms.
Figure 5.

Structural modification of A1R agonists led to different simulated binding and unbinding mechanisms. (a and b) Heatmap showing the contact difference, during SuMD path sampling, between adenosine and CPA, which differ for the N6-cyclipentyl group (highlighted as a dashed circle line); A1R blue surfaces indicates more contacts formed with adenosine, while red surfaces are indicative of more contacts with CPA. (a) Binding simulations and (b) unbinding simulations. (c and d) Heatmap showing the contacts difference, during SuMD path sampling, between CPA and BnOCPA, which differ for the oxybenzyl group (highlighted as a dashed circle line); A1R blue surfaces indicate more contacts formed with CPA, while red surfaces are indicative of more contacts with BnOCPA. (c) Binding simulations and (d) unbinding simulations.
4.1.1. N6-Cycloalkyl Ring on the Adenosine Scaffold
In general, CPA formed more intermediate interactions than adenosine with the extracellular vestibule of A1R (Figure 5a,b). The presence of the N6-cycloalkyl substituent produced mode contacts with ECL2 and residues located at the top of TM1, TM5, TM6, TM7, and ECL3 (Figure 5a,b). This scenario is in good agreement with mutagenesis experiments (Table 2) showing CPA affinity (but not adenosine’s affinity) was significantly affected by T2576.58A, L2586.59A, H264ECL3A, and S2677.32A, all located in the proximity of the extracellular vestibule.
4.1.2. 2-Oxybenzyl Group on CPA N6-Cycloalkyl Ring
The presence of this lipophilic moiety (BnOCPA was the bulkiest ligand considered) favored more interactions with the top of TM5 and TM6 during association (Figure 5c) and with the top of TM1 and TM2 during dissociation (Figure 5d). CPA, on the other hand, was more prone to interact with the ECL2 (Figure 5c,d). The changes in the barycenter of the contacts during unbinding due to the 2-oxybenzyl group could explain the unique profile that BnOCPA showed on A1R mutants F81.31A and N702.65A (Table 2). BnOCPA, indeed, was the only agonist significantly affected by N702.65A and the only one not affected by F81.31A. We speculate that the F81.31A mutation affects both BnOCPA binding and unbinding (Figure 5c,d), leading to compensation and therefore an unmodified pKi compared to A1R wt (Table 2).
5. Conclusion
GPCR–ligand complexes form and dissociate through multistep mechanisms. The importance of intermediate metastable states along the (un)binding paths in modulating the overall affinity of ARs agonist and GPCRs ligands in general is gradually emerging.12,60,61 Here, we combined unbiased nonequilibrium MD simulations and mutagenesis experiments to study the dynamic binding and unbinding of A1R agonists. The in silico analysis suggested several A1R residues involved on regions so far poorly investigated.73 Mutagenesis experiments generally confirmed the computational prediction and allow mapping novel receptor vestibules involved in the association or dissociation of the agonists. For binding, the A1R ECL2 was involved in numerous preliminary contacts as well as the top of TM6, TM1, TM2, and TM7. The dissociation from A1R followed similar but distinct routes, with ECL2 generally less engaged (especially by the more hydrophilic adenosine and NECA). The chemical modifications that increase the agonists’ selectivity toward A1R (e.g., the introduction of N6-cycloalkyl groups) also changed the (un)binding mechanism, favoring the interactions with the extracellular vestibules in general and the hydrophobic allosteric pocket located on ECL2 in particular.
MD simulations and mutagenesis experiments are frequently combined to deliver structural insights on end point protein–ligand complexes; however, to the best of our knowledge,74−76 this is the first time that the whole process of formation/dissociation of several GPCR ligands is mapped with a combined in silico and in vitro approach. Our results pave the way for the rationalization of SKR for A1R, agonists, with potential repercussion on the design of long-awaited clinical agents.
Acknowledgments
C.A.R. is grateful for a Royal Society Industry Fellowship. Funding from Leverhulme Trust (Grant RPG-2017-255, C.A.R. and G.L. to fund K.B. and G.D.) is acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00195.
Additional characterization data (PDF)
Merged SuMD simulations of adenosine binding (left side) and unbinding (right side). (MP4)
Merged SuMD simulations of NECA binding (left side) and unbinding (right side). (MP4)
Merged SuMD simulations of CPA binding (left side) and unbinding (right side). (MP4)
Merged SuMD simulations of BnOCPA binding (left side) and unbinding (right side). (MP4)
Author Present Address
⊥ K.B.: Sosei Heptares, Granta Park Steinmetz Building, Cambridge, CB21 6DG, U.K.
Author Contributions
# G.D. and K.B. contributed equally. G.L. and C.A.R. also contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Hauser A. S.; Attwood M. M.; Rask-Andersen M.; Schiöth H. B.; Gloriam D. E. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discovery 16 (12), 829–842. 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K.; Insel P. A. (2018) G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs?. Mol. Pharmacol. 93 (4), 251–258. 10.1124/mol.117.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. (2006) Purinergic signalling. Br. J. Pharmacol. 147 (S1), S172–S181. 10.1038/sj.bjp.0706429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth S.; Brito R.; Mukherjea D.; Rybak L. P.; Ramkumar V. (2014) Adenosine receptors: expression, function and regulation. Int. J. Mol. Sci. 15 (2), 2024–2052. 10.3390/ijms15022024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K. A.; Gao Z.-G. (2006) Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discovery 5 (3), 247–264. 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen M.; Eckle T.; Eltzschig H. K. (2009) Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PLoS One 4 (8), e6784 10.1371/journal.pone.0006784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.-F.; Eltzschig H. K.; Fredholm B. B. (2013) Adenosine receptors as drug targets--what are the challenges?. Nat. Rev. Drug Discovery 12 (4), 265–286. 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. (2017) Purinergic signalling: therapeutic developments. Front. Pharmacol. 8, 661. 10.3389/fphar.2017.00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper-Joyce C. J.; Khoshouei M.; Thal D. M.; et al. (2018) Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 558 (7711), 559–563. 10.1038/s41586-018-0236-6. [DOI] [PubMed] [Google Scholar]
- Cheng R. K.Y.; Segala E.; Robertson N.; Deflorian F.; Dore A. S.; Errey J. C.; Fiez-Vandal C.; Marshall F. H.; Cooke R. M. (2017) Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 25 (8), 1275–1285.e4. 10.1016/j.str.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Glukhova A.; Thal D. M.; Nguyen A. T.; Vecchio E. A.; Jorg M.; Scammells P. J.; May L. T.; Sexton P. M.; Christopoulos A. (2017) Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 168 (5), 867–877.e13. 10.1016/j.cell.2017.01.042. [DOI] [PubMed] [Google Scholar]
- Nguyen A. T. N.; Baltos J.-A.; Thomas T.; et al. (2016) Extracellular loop 2 of the adenosine A1 receptor has a key role in orthosteric ligand affinity and agonist efficacy. Mol. Pharmacol. 90 (6), 703–714. 10.1124/mol.116.105007. [DOI] [PubMed] [Google Scholar]
- Nguyen A. T. N.; Vecchio E. A.; Thomas T.; et al. (2016) Role of the second extracellular loop of the adenosine A1 receptor on allosteric modulator binding, signaling, and cooperativity. Mol. Pharmacol. 90 (6), 715–725. 10.1124/mol.116.105015. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Bhattarai A.; Nguyen A. T. N.; Christopoulos A.; May L. T. (2018) Structural basis for binding of allosteric drug leads in the adenosine A1 receptor. Sci. Rep. 8 (1), 16836. 10.1038/s41598-018-35266-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller C. E.; Jacobson K. A. (2011) Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochim. Biophys. Acta, Biomembr. 1808 (5), 1290–1308. 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate C. G.; García-Nafría J.; Lee Y.; Bai X.; Carpenter B. (2018) Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. eLife 10.7554/eLife.35946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight A.; Hemmings J. L.; Winfield I.; et al. (2016) Discovery of novel adenosine receptor agonists that exhibit subtype selectivity. J. Med. Chem. 59 (3), 947–964. 10.1021/acs.jmedchem.5b01402. [DOI] [PubMed] [Google Scholar]
- Cuzzolin A.; Sturlese M.; Deganutti G.; et al. (2016) Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 56 (4), 687–705. 10.1021/acs.jcim.5b00702. [DOI] [PubMed] [Google Scholar]
- Deganutti G.; Moro S.; Reynolds C. A. (2020) A Supervised Molecular Dynamics Approach to Unbiased Ligand-Protein Unbinding. J. Chem. Inf. Model. 60, 1804. 10.1021/acs.jcim.9b01094. [DOI] [PubMed] [Google Scholar]
- Barkan K.; Lagarias P.; Stampelou M.; et al. (2020) Pharmacological characterisation of novel adenosine A3 receptor antagonists. Sci. Rep. 10 (1), 20781. 10.1038/s41598-020-74521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart L. A.; Johnstone E. K. M.; Wheal A. J.; et al. (2015) Application of BRET to monitor ligand binding to GPCRs. Nat. Methods 12 (7), 661–663. 10.1038/nmeth.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung-Chi C.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22 (23), 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34 (25), 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; et al. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14 (1), 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. (2012) Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 52 (12), 3144–3154. 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. (2012) Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 52 (12), 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; He X.; Vanommeslaeghe K.; MacKerell A. D. (2012) Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 33 (31), 2451–2468. 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall M.; Hill E.; Huckstepp R. (2020) Non-opioid analgesia based on Gα signalling bias. BioRxiv 10.1101/2020.04.04.023945. [DOI] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; et al. (2000) The protein data bank. Nucleic Acids Res. 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; et al. (2006) Comparative protein structure modeling using Modeller. Curr. Protoc Bioinformatics 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky T. J.; Nielsen J. E.; McCammon J. A.; Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665. 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M. H. M.; Søndergaard C. R.; Rostkowski M.; Jensen J. H. (2011) PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pK Predictions. J. Chem. Theory Comput. 7 (2), 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Sommer B. (2013) Membrane Packing Problems: A short Review on computational Membrane Modeling Methods and Tools. Comput. Struct. Biotechnol. J. 5, e201302014 10.5936/csbj.201302014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize M. A.; Lomize A. L.; Pogozheva I. D.; Mosberg H. I. (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22 (5), 623–625. 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79 (2), 926. 10.1063/1.445869. [DOI] [Google Scholar]
- Harvey M. J.; Giupponi G.; Fabritiis G. D. (2009) ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 5 (6), 1632–1639. 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81 (8), 3684. 10.1063/1.448118. [DOI] [Google Scholar]
- Loncharich R. J.; Brooks B. R.; Pastor R. W. (1992) Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers 32 (5), 523–535. 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- Forester T. R.; Smith W. (1998) SHAKE, rattle, and roll: Efficient constraint algorithms for linked rigid bodies. J. Comput. Chem. 19, 102.. [DOI] [Google Scholar]
- Kr-utler V.; van Gunsteren W. F.; H-nenberger P. H. (2001) A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22 (5), 501–508. . [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103 (19), 8577. 10.1063/1.470117. [DOI] [Google Scholar]
- Deganutti G.; Moro S. (2017) Estimation of kinetic and thermodynamic ligand-binding parameters using computational strategies. Future Med. Chem. 9 (5), 507–523. 10.4155/fmc-2016-0224. [DOI] [PubMed] [Google Scholar]
- Sabbadin D.; Moro S. (2014) Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 54 (2), 372–376. 10.1021/ci400766b. [DOI] [PubMed] [Google Scholar]
- Atanasio S.; Deganutti G.; Reynolds C. A. (2020) Addressing free fatty acid receptor 1 (FFAR1) activation using supervised molecular dynamics. J. Comput.-Aided Mol. Des. 34, 1181. 10.1007/s10822-020-00338-6. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graphics 14 (1), 33. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. (2014) PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 185 (2), 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Miller B. R.; McGee T. D.; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. (2012) MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 8 (9), 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Wang E.; Sun H.; Wang J.; et al. (2019) End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 119 (16), 9478–9508. 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A., and Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors in Receptor Molecular Biology, Methods in Neurosciences, Vol. 25, pp 366–428, Elsevier, 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- Lebon G.; Warne T.; Edwards P. C.; et al. (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474 (7352), 521–525. 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters M. C.; Wisse L. E.; Dinaj A.; Vroling B.; Vriend G.; Ijzerman A. P. (2012) The role of the second and third extracellular loops of the adenosine A1 receptor in activation and allosteric modulation. Biochem. Pharmacol. 84 (1), 76–87. 10.1016/j.bcp.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Rivkees S. A.; Barbhaiya H.; IJzerman A. P. (1999) Identification of the adenine binding site of the human A1 adenosine receptor. J. Biol. Chem. 274 (6), 3617–3621. 10.1074/jbc.274.6.3617. [DOI] [PubMed] [Google Scholar]
- Palaniappan K. K.; Gao Z.-G.; Ivanov A. A.; et al. (2007) Probing the binding site of the A1 adenosine receptor reengineered for orthogonal recognition by tailored nucleosides. Biochemistry 46 (25), 7437–7448. 10.1021/bi7001828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpiaz A.; Townsend-Nicholson A.; Beukers M. W.; Schofield P. R.; IJzerman A. P. (1998) Thermodynamics of full agonist, partial agonist, and antagonist binding to wild-type and mutant adenosine A1 receptors. Biochem. Pharmacol. 56 (11), 1437–1445. 10.1016/S0006-2952(98)00202-0. [DOI] [PubMed] [Google Scholar]
- Kim J.; Wess J.; van Rhee A. M.; Schöneberg T.; Jacobson K. A. (1995) Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem. 270 (23), 13987–13997. 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q.; Lee B. X.; Glashofer M.; van Rhee A. M.; Jacobson K. A. (1997) Mutagenesis reveals structure-activity parallels between human A2A adenosine receptors and biogenic amine G protein-coupled receptors. J. Med. Chem. 40 (16), 2588–2595. 10.1021/jm970084v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segala E.; Guo D.; Cheng R. K. Y.; et al. (2016) Controlling the Dissociation of Ligands from the Adenosine A2A Receptor through Modulation of Salt Bridge Strength. J. Med. Chem. 59 (13), 6470–6479. 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- Dror R. O.; Pan A. C.; Arlow D. H.; et al. (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. U. S. A. 108 (32), 13118–13123. 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley M.; Wootten D.; Conner M. T.; et al. (2012) Lifting the lid on GPCRs: the role of extracellular loops. Br. J. Pharmacol. 165 (6), 1688–1703. 10.1111/j.1476-5381.2011.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Kaindl J.; Clark M. J.; et al. (2020) Binding pathway determines norepinephrine selectivity for the human β1AR over β2AR. Cell Res. 10.1038/s41422-020-00424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo E.; Hinz S.; Pellizzari V.; et al. (2020) A2A and A2B adenosine receptors: The extracellular loop 2 determines high (A2A) or low affinity (A2B) for adenosine. Biochem. Pharmacol. 172, 113718. 10.1016/j.bcp.2019.113718. [DOI] [PubMed] [Google Scholar]
- Kennedy D. P.; McRobb F. M.; Leonhardt S. A.; et al. (2014) The second extracellular loop of the adenosine A1 receptor mediates activity of allosteric enhancers. Mol. Pharmacol. 85 (2), 301–309. 10.1124/mol.113.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J. R.; May L. T.; Parton R. G.; Sexton P. M.; Christopoulos A. (2017) A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 13 (9), 929–937. 10.1038/nchembio.2431. [DOI] [PubMed] [Google Scholar]
- Sykes D. A.; Stoddart L. A.; Kilpatrick L. E.; Hill S. J. (2019) Binding kinetics of ligands acting at GPCRs. Mol. Cell. Endocrinol. 485, 9–19. 10.1016/j.mce.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Herenbrink C.; Sykes D. A.; Donthamsetti P.; et al. (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 7, 10842. 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl G.; Akerud T. (2013) Pharmacokinetics and the drug-target residence time concept. Drug Discovery Today 18 (15–16), 697–707. 10.1016/j.drudis.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Strasser A.; Wittmann H.-J.; Seifert R. (2017) Binding kinetics and pathways of ligands to gpcrs. Trends Pharmacol. Sci. 38 (8), 717–732. 10.1016/j.tips.2017.05.005. [DOI] [PubMed] [Google Scholar]
- Paul F.; Wehmeyer C.; Abualrous E. T.; et al. (2017) Protein-peptide association kinetics beyond the seconds timescale from atomistic simulations. Nat. Commun. 8 (1), 1095. 10.1038/s41467-017-01163-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner N.; Doerr S.; De Fabritiis G.; Noé F. (2017) Complete protein-protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling. Nat. Chem. 9 (10), 1005–1011. 10.1038/nchem.2785. [DOI] [PubMed] [Google Scholar]
- Huang D.; Caflisch A. (2011) The free energy landscape of small molecule unbinding. PLoS Comput. Biol. 7 (2), e1002002 10.1371/journal.pcbi.1002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson A. (2018) Mapping the ligand binding landscape. BioRxiv 10.1101/346817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deganutti G.; Zhukov A.; Deflorian F.; et al. (2017) Impact of protein-ligand solvation and desolvation on transition state thermodynamic properties of adenosine A2A ligand binding kinetics. Silico Pharmacol 5 (1), 16. 10.1007/s40203-017-0037-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jespers W.; Schiedel A. C.; Heitman L. H.; et al. (2018) Structural mapping of adenosine receptor mutations: ligand binding and signaling mechanisms. Trends Pharmacol. Sci. 39 (1), 75–89. 10.1016/j.tips.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Deganutti G.; Moro S.; Reynolds C. A. (2019) Peeking at G-protein-coupled receptors through the molecular dynamics keyhole. Future Med. Chem. 11 (6), 599–615. 10.4155/fmc-2018-0393. [DOI] [PubMed] [Google Scholar]
- Miao Y.; McCammon J. A. (2016) G-protein coupled receptors: advances in simulation and drug discovery. Curr. Opin. Struct. Biol. 41, 83–89. 10.1016/j.sbi.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vivo M.; Masetti M.; Bottegoni G.; Cavalli A. (2016) Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 59 (9), 4035–4061. 10.1021/acs.jmedchem.5b01684. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.