

Abstract

The calpain–cathepsin hypothesis posits a key role for elevated calpain-1 and cathepsin-B activity in the neurodegeneration underlying neurotrauma and multiple disorders including Alzheimer’s disease (AD). AD clinical trials were recently halted on alicapistat, a selective calpain-1 inhibitor, because of insufficient exposure of neurons to the drug. In contrast to neuroprotection, the ability of calpain-1 and cathepsin-B inhibitors to protect the blood–brain barrier (BBB), is understudied. Since cerebrovascular dysfunction underlies vascular dementia, is caused by ischemic stroke, and is emerging as an early feature in the progression of AD, we studied protection of brain endothelial cells (BECs) by selective and nonselective calpain-1 and cathepsin-B inhibitors. We show these inhibitors protect both neurons and murine BECs from ischemia–reperfusion injury. Cultures of primary BECs from ALDH2–/– mice that manifest enhanced oxidative stress were sensitive to ischemia, leading to reduced cell viability and loss of tight junction proteins; this damage was rescued by calpain-1 and cathepsin-B inhibitors. In ALDH2–/– mice 24 h after mild traumatic brain injury (mTBI), BBB damage was reflected by significantly increased fluorescein extravasation and perturbation of tight junction proteins, eNOS, MMP-9, and GFAP. Both calpain and cathepsin-B inhibitors alleviated BBB dysfunction caused by mTBI. No clear advantage was shown by selective versus nonselective calpain inhibitors in these studies. The lack of recognition of the ability of calpain inhibitors to protect the BBB may have led to the premature abandonment of this therapeutic approach in AD clinical trials and requires further mechanistic studies of cerebrovascular protection by calpain-1 inhibitors.
Keywords: calpain-1, cathepsin-B, brain endothelial cells, TBI, oxidative stress
The “calpain–cathepsin” hypothesis describes the coordinated and dysregulated proteolytic actions of the cysteine proteases, calpain-1 and cathepsin-B, causing neurodegeneration in multiple disorders including Alzheimer’s disease and related dementia (ADRD).1−3 Calpain-1, a calcium-regulated cysteine protease, is abnormally activated in the early pathogenesis of neurodegenerative diseases and in neurotrauma, such as ischemic stroke and traumatic brain injury (TBI).4 The efficacy of small molecules that inhibit calpain-1 has been reported widely in vivo, including in models of TBI,5,6 ischemic stroke,7,8 and ADRD.9,10 Calpain-1 is regarded as a validated therapeutic target; indeed, alicapistat, a selective ketoamide calpain-1 inhibitor, entered clinical trials for ADRD and mild cognitive impairment. These trials, reporting out in late 2019, demonstrated no treatment-associated adverse events across five different and diverse study arms even with very high plasma drug exposure.11 Unfortunately, the trial was abandoned, as it was concluded that drug exposure of neurons in the CNS was too low to obtain a pharmacodynamic effect.
Damage to the cerebrovasculature is inherent in TBI and ischemic stroke, and cerebrovascular dysfunction is increasingly seen as a critical feature in the onset and progression of ADRD.12−14 Therefore, we set out to determine if the calpain–cathepsin hypothesis is applicable to brain endothelial cells (BECs) and consequently, if calpain and cathepsin inhibitors protect the blood–brain barrier (BBB) in neurotrauma. A positive result would require reassessment of both (1) the need for neuronal exposure of calpain-1 inhibitors for clinical efficacy and (2) the role of cerebrovascular protection in the many preclinical studies showing benefits of calpain-1 and cathepsin-B inhibitors.
A normal physiological function of calpain-1 in the brain is to maintain neuroplasticity through regulated proteolytic processing of proteins that are important for neuronal function and are fundamentally intertwined with neurodegenerative pathology: substrates include proteins involved in long-term potentiation,15 tau kinases, neuronal cytoskeleton regulators (spectrin),3 and glial fibrillary acidic protein (GFAP).16,17 Moreover, calpain overactivation indirectly permeabilizes lysosomes causing the release of cathepsin-B; this second cysteine protease exacerbates lysosomal membrane disruption and causes mitochondrial damage, releasing pro-apoptotic factors including cytochrome c, caspase-9, and caspase-3. Cathepsin-B is upregulated and localized extra-lysosomally in animal models of TBI, in neurotrauma patients,18 and in post-mortem AD brains.19 Thus, calpain-1 and cathepsin-B are hypothesized to be underlying contributors in neurodegenerative diseases.
Cerebrovascular dysfunction has recently emerged as a key contributor to neurodegenerative diseases and ADRD.13,14 A “two-hit vascular hypothesis” suggests that a primary feature in ADRD pathogenesis is BBB dysfunction leading to neuronal injury and pathogenic accumulation of toxic proteins (Aβ, tau) as a “second hit”.20 This hypothesis is supported by studies implicating BBB dysfunction as an early feature in the progression of neurodegenerative disease;13,21 however, it is still debated whether BBB dysfunction is the cause or consequence of disease. In contrast to studies on neurons and neuronal function, considerably less attention has been paid to the roles of calpain-1 and cathepsin-B in component cells of the BBB, notably BECs. Herein, we provide the first study in BECs comparing selective and nonselective inhibitors of calpain-1 and cathepsin-B, which have long been proposed as therapeutics for treatment of neurodegenerative diseases, including ADRD.
Oxidative damage mediated by lipid peroxidation products (LPP) has been shown to play a significant role in mild cognitive impairment (MCI), ADRD, and TBI.22−24 Furthermore, ischemic stroke is well-known to induce BBB-dysfunction, which has been linked with lipid peroxidation, hyperactivation of calpain-1 and matrix metalloproteinase-9 (MMP-9), and downregulation of the tight junction protein occludin in cerebral microvessels.25ALDH2–/– mice, lacking the activity of mitochondrial aldehyde dehydrogenase-2 (Aldh2) to detoxify aldehyde LPP, show amplified responses to neurotrauma and a significant response to mild TBI (mTBI).26ALDH2–/– mice and BECs derived from these mice therefore provided excellent model systems to study the putative protective effects of calpain-1 and cathepsin-B inhibitors on the cerebrovasculature. We observed that ischemia–reperfusion injury in BECs from ALDH2–/– mice results in exacerbated responses including loss of the tight junction proteins occludin and ZO-1. We also observed that brain tissues of ALDH2–/– mice subject to mTBI show enhanced calpain-1 activity, upregulation of MMP-9, loss of tight junction proteins, and exacerbated endothelial dysfunction reflected by loss of endothelial NO synthase (eNOS). Notably, in this study, we observed for the first time that brain levels of eNOS are lower with oxidative stress, further reduced with neurotrauma, and restored by calpain-1 inhibitors.
The calpain–cathepsin hypothesis suggests that a nonselective inhibitor that inhibits both calpain-1 and cathepsin-B may be superior to a selective calpain-1 inhibitor. Nonselective inhibitors often inhibit off-target cysteine proteases.2 Although off-target binding may lead to adverse effects, off-target proteases include caspases that are also bona fide targets for neuroprotection.27 Similarly, in antiviral therapy of COVID-19, off-target inhibition by calpain-1 inhibitors of cathepsin-L and viral cysteine proteases may be opportunistically exploited.28 Both selective and nonselective inhibitors have shown efficacy in preclinical models of neurotrauma and AD.8−10,29 Herein we compare responses to a novel, highly selective calpain-1 inhibitor (an analogue of alicapistat), a nonselective inhibitor, and an inhibitor commonly used as specific for cathepsin-B (Figure 1A). We demonstrate that selective and nonselective calpain inhibitors protect BECs and the BBB from ischemia and neurotrauma, extending the calpain–cathepsin hypothesis from neurons to the cerbrovasculature.
Figure 1.

Enzyme inhibition by selective and non-selective calpain-1 and cathepsin-B inhibitors. (A) Structures of inhibitors used in this study. (B) Selectivity of compounds at 1 μM against calpain-1 (CAPN1), cathepsin-B (CTSB), cathepsin K (CTSK), and papain. (C) Dose–response curves for calpain-1 inhibitors 1–5 showing pIC50. (D) Quantification of western blots of HT22 cells cotreated with 10 μM inhibitors (n.t. = nontreated) and 20 μM ionomycin for 40 min. The calpain-1 proteolysis product, SBDP 145 kDa, was normalized to the housekeeping protein, actin, with equal protein loaded in all lanes of immunoblots. *, p < 0.05 compared to nontreated control; #, p < 0.05, and ##, p < 0.01, compared to cells treated with 20 μM ionomycin. All data are represented as mean ± SD from at least n = 6 replicates.
Results
Characterization of Calpain/Cathepsin Inhibitors
To provide a toolkit of selective and nonselective inhibitors, compounds were assayed for selective inhibition of papain and recombinant human calpain-1, cathepsin-B, and cathepsin-K (Figure 1). Papain was used as a generic cysteine protease, representative of lysosomal and secreted cysteine proteases, while cathepsin-K served as an off-target cathepsin-class cysteine protease. The ketoamide referred to as calpain inhibitor XI (CaXI) is an example of a reversible covalent inhibitor of calpain-1. As seen in Figure 1B, at 1 μM, CaXI is both an effective inhibitor of calpain-1 and cathepsin-K and also modestly inhibits (∼20%) both papain and cathepsin-B. This exemplifies the ambiguity that can result from the use of calpain-1 inhibitors as specific probes of calpain-1.
We pursued a series of ketoamides based upon published patents and computational docking studies.30 Of this series of compounds, 1–5 were observed to be potent and highly selective toward calpain-1 at 1 μM with minimal inhibitory activity against cathepsin-B, papain, and cathepsin-K (Figure 1B,C; Table S1). This series contains alicapistat (ABT-957; 1), the identity of which was not unambiguously revealed until 2019.31 The epoxides, E-64c and NYC-438, are irreversible inhibitors of calpain-1 and cathepsin-B. E-64c is a synthetic analog of E-64, a well-studied epoxide isolated from Aspergillus japonicas. NYC-438 was the product of optimization to increase selectivity toward calpain-1 versus generic cysteine proteases modeled by papain, and retains cathepsin-B activity (Figure 1D).10,32 At 1 μM, both E-64c and NYC-438 inhibited all cysteine proteases studied. Finally, CA-074, often referred to as a specific inhibitor of cathepsin-B was shown to inhibit cathepsin-K at 1 μM, but not to inhibit calpain-1 at this concentration.
In-cell inhibition of substrate proteolysis associated with the actions of specific cysteine proteases was studied in neuronal cells. Ionomycin, a calcium ionophore, was used to stimulate calpain-1 activation, thereby initiating spectrin proteolysis. After cotreatment with inhibitors, spectrin and spectrin breakdown products (SBDP) were quantified. SBDP 150 kDa are the result of proteolysis by caspase-3 and calpain-1, whereas the 145 kDa SBDP are specifically generated by calpain-1.33 Selective and nonselective calpain-1 inhibitors were able to inhibit formation of spectrin breakdown products after ionomycin stimulus (Figures 1D and S1). The effects of CA-074 were not significant, in contrast to the significant inhibitory effects of the calpain-1 inhibitors on formation of SBDP 145 kDa. Comparisons of the selective calpain-1 inhibitor, 5, with the nonselective calpain-1 inhibitor, NYC-438, were made throughout this study, with CA-074 and E64c included for comparison in selected experiments.
Neuroprotection in Ischemia/Reperfusion Injury
Neuroprotection was assessed in response to oxygen-glucose deprivation and reperfusion (OGD-R) in neuronal cells, a well-characterized cell culture model of ischemic stroke that closely mimics in vivo models of brain ischemia and reperfusion, in which both ischemic damage and damage due to reperfusion/reoxygenation contribute to neuronal loss.34 Cell viability of SH-SY5Y cultures exposed to 2 h “ischemia” and 24 h of “reperfusion” was assessed by MTS and LDH, which showed 82.9 ± 1.3% cell viability by MTS (Figure 2A) and 19.2 ± 1.2% LDH release (Figure 2B) compared to nontreated controls at 100% (MTS) and 0% (LDH), respectively. We intentionally selected an insult giving significant but relatively low cytotoxicity to avoid large populations of apoptotic or necrotic cells yet to provide sufficient dynamic range to test neuroprotectants. The ability of agents to protect against OGD-R was assessed with three treatment paradigms: pretreatment/protection (2 h prior to OGD), ischemia (coincident with OGD), and reperfusion (drug administered with glucose-reperfusion media). Results were normalized to vehicle-treated cells subjected to OGD-R (0% cell viability) and/or sulforaphane (1 μM) treatment (100% cell viability), since it is impossible to use an in-plate vehicle control not subject to OGD-R. Sulforaphane, a Nrf2 activator, elicits highly reproducible neuroprotection at a low concentration (1 μM) and neurotoxicity at a high concentration (10 μM) in all OGD-R paradigms, representing an excellent in-plate positive and negative control (Figure S2). We observed that in SH-SY5Y cells subject to OGD-R, calpain and cathepsin inhibitors (10 μM) were neuroprotective (Figure 2C–E).
Figure 2.

Neuroprotection in SH-SY5Y cells and primary human neurons in response to calpain/cathepsin inhibitors. Cell viability quantified by (A) MTS and (B) LDH release in SH-SY5Y cells exposed to OGD-R compared to control cells. (C–E) Cell viability quantified by MTS in SH-SY5Y cells following OGD-R after treatment with sulforaphane controls or calpain/cathepsin inhibitors (10 μM): pretreatment (t = −2 h, C), ischemia (t = 0, D), or reperfusion (t = 2 h, E). Cell viability quantified by (F) MTS and (G) LDH release in primary human hippocampal neurons exposed to OGD-R compared to control cells. (H) Cell viability quantified by MTS primary human hippocampal neurons following exposure to OGD-R with treatment of vehicle, sulforaphane or inhibitors (hashed 1 μM; solid 10 μM) in the ischemia paradigm (t = 0). Panels C–E were normalized to 1 μM sulforaphane treatment at 100% and vehicle control at 0%; panel H was normalized to 1 μM sulforaphane treatment at 100%. Data represent mean ± SD of at least n = 6 replicates analyzed by one-way ANOVA with Dunnett’s or Tukey’s multicomparison analysis. *, p < 0.05; **, p < 0.01; and ***, p < 0.001, ****, p < 0.0001 versus nontreated control.
Neuroprotection in response to OGD-R was confirmed in cultures of primary human hippocampal neurons, a more physiologically relevant model. The primary cells exhibited a similar response to cell lines when subjected to OGD-R: 82.9 ± 3.5% cell viability by MTS (Figure 2F) and 17.8 ± 1.5% LDH release (Figure 2G). Calpain and cathepsin inhibitors were added at the start of the induction of ischemia; all inhibitors showed significant concentration-dependent neuroprotection at 10 μM in this paradigm (Figure 2H), confirming observations in cell lines.
Loss of BEC Viability in Response to OGD-R and Lipid Peroxidation Products
With the anticipated neuroprotective profiles established in neuronal cell lines and primary cultures for our novel calpain-1 inhibitors and controls, our focus moved to brain endothelial cells (BECs). The BBB is composed primarily of pericytes, astrocytes and BECs, with BECs contributing strongly to BBB functional integrity.35 Cortical BECs isolated from wild-type (WT) mice showed an inversely proportional relationship between cell viability and increasing length of OGD-R (0, 1, 2, 4, and 6 h of OGD-R, Figure 3A). Significant loss in cell viability in BECs was first observed at 2 h of OGD-R (85.7 ± 2.6% cell viability).
Figure 3.

BECs are sensitive to oxidative stress and are protected by calpain inhibitors. (A) Cell viability quantified by MTS of BECs and SH-SY5Y cell cultures subjected to 0, 1, 2, 4, or 6 h of OGD-R. (B and C) Cell viability quantified by MTS of BECs and SH-SY5Y cells after 24 h of treatment with the lipid peroxidation products HNE (B) or ONE (C). (D) Cell death quantified by LDH release of BECs following 0, 0.5, 1, or 2 h of OGD-R. (E) LDH release of BECs following 2 h of OGD-R with cotreatment of vehicle, sulforaphane (1 μM), or inhibitors (1 μM hashed; 10 μM solid) normalized to vehicle control (0%). (F) Cell count of BECs following 2 h of OGD-R and cotreatment of vehicle, sulforaphane (1 μM), or inhibitors (1 μM hashed;10 μM solid), normalized to in-plate sulforaphane (100%) with significance tested relative to vehicle control. Data represent mean ± SD of at least n = 3 in 3 separate isolations analyzed by one-way ANOVA with Dunnett’s or Tukey’s multicomparison: *, p < 0.05; **, p < 0.01; and ***, p < 0.001, versus no OGD-R control; #, p < 0.05; ##, p < 0.01; and ###, p < 0.001, versus vehicle-treated.
Lipid peroxidation products, 4-hydroxynonenal (HNE) and 4-oxo-2-nonenal (ONE), are elevated in oxidative stress. Protein adduction by HNE correlates with many neurodegenerative states. HNE and ONE cause concentration-dependent cytotoxicity toward neuronal cells, with ONE more reactive and cytotoxic.26 Treatment of BECs with HNE or ONE compared to treatment of SH-SY5Y cells showed similar concentration dependence, with BECs possibly being more sensitive to higher HNE concentrations (Figures 3B,C). No sex differences were observed between BECs isolated from female versus male mice subjected to OGD-R (Figure S3).
To test the ability of calpain and cathepsin inhibitors to protect BECs, in simile with observations on neurons, we selected 2 h of ischemia followed by 24 h of reperfusion, under which conditions LDH release was significantly increased (Figure 3D). Under these conditions, in the “ischemia” treatment paradigm, calpain and cathepsin inhibitors (1 μM or 10 μM) protected BECs from ischemic damage as reflected by significant reduction in LDH release compared to vehicle control (Figure 3D): At 10 μM, all inhibitors significantly reduced LDH release (nontreated: 28.1 ± 3.3%; 5: 14.5 ± 4.1%; NYC-438: 14.5 ± 2.5%; CA-074:19.4 ± 2.3%). At the lower 1 μM concentration, sulforaphane, 5, and NYC-438 treatment caused significant reduction in LDH release. Proliferation of primary BECs was also measured by cell count (Figure 3E). Using the cytoprotective response to sulforaphane as a control (100% confluence), the selective (5) and nonselective (NYC-438) calpain-1 inhibitors were observed to elicit concentration-dependent cytoprotection (Figure 3F): At 10 μM, 5 and NYC-438 restored BECs to 95.1 ± 2.3 and 93.4 ± 3.1% of control, respectively. The selective cathepsin-B inhibitor, CA-074, did not significantly protect BEC cultures.
A decrease in the expression of tight junction proteins has been reported in the post-mortem brain tissue of ADRD patients and is a response to brain trauma and ischemia.36 We focused our measurements in BECs on two tight junction proteins: zona occludens 1 (ZO-1) and occludin. ZO-1 is a scaffold protein that anchors tight junction proteins to the actin cytoskeleton, while occludin is a plasma-membrane protein. Immunoblots revealed that levels of ZO-1 and occludin were significantly reduced after 2 h of OGD-R (88.3 ± 0.02 and 83.2 ± 0.03% respectively, Figure 4A–D). Co-treatment with 5 or NYC-438 (both 10 μM) significantly attenuated the loss in ZO-1 and occludin (Figure 4E,F). The response to CA-074 treatment did not reach significance. Selective calpain inhibitor 5, nonselective inhibitor NYC-438, and selective cathepsin-B inhibitor CA-074 all significantly inhibited formation of SBDP 150 kDa (Figure 4G); however, only calpain inhibitors significantly inhibited proteolysis of spectrin to SBDP 145 kDa (Figure 4H).
Figure 4.

Calpain and cathepsin-B inhibitors mitigate loss of tight junction proteins in BECs subjected to OGD-R. Representative immunoblots of BECs subjected to 2 h of OGD-R versus control cells probed with antibodies against ZO-1 (A) and occludin (C). Quantitative analysis of these immunoblots (B and D, respectively). (E–H) Quantitative analysis of BECs treated with inhibitors (10 μM) followed by 2 h of OGD-R relative to control cells without OGD-R probed for ZO-1 (E), occludin (F), or SBDP (150 kDa, G, and 145 kDa, H). All protein levels were normalized relative to the housekeeping protein, β-actin. Data represent mean ± SD of at least n = 3 separate BEC isolations analyzed by one sample t-test and Wilcoxon test or one-way ANOVA with Dunnett’s or Tukey’s multicomparison analysis. Equal protein amounts were loaded in all lanes of immunoblots. ***, p <.001 versus no OGD-R control. #, p < 0.05; ##, p < 0.01; ###, p < 0.001, versus OGD-R vehicle-treated.
Amplified Response to OGD-R of BECs from ALDH2–/– Mice Is Rescued by Inhibitors
Mitochondrial aldehyde dehydrogenase 2 (Aldh2) is a major detoxifier of the lipid peroxidation products, HNE and ONE. Genetic knockout in the ALDH2–/– mouse model causes exacerbated oxidative stress, accelerated cognitive impairment, and increased sensitivity to neurotrauma.26 We anticipated that BECs from ALDH2–/– mice, with diminished ability to detoxify HNE and ONE would be more susceptible to OGD-R, since we observed BEC cultures to be sensitive to HNE and ONE (Figure 3). We anticipated further that the BBB of these mice would show amplified susceptibility to neurotrauma. BECs were isolated from WT and ALDH2–/– mice and subjected to OGD for increasing times, using cell count and LDH release to measure cytotoxicity.
ALDH2–/– BECs exhibited significantly lower cell count (confluence) than WT BECs post-OGD-R (WT versus ALDH2–/– @ 1 h: 89.1 ± 4.7% versus 72.1 ± 4.0%, Figure 5A,B). The exacerbated response of ALDH2–/– BECs to OGD-R was also seen in LDH release (WT versus ALDH2–/– @ 0.5 h: 16.7 ± 1.0 versus 3.2 ± 1.6%; @ 1 h: 27.9 ± 12.5 versus 11.3 ± 1.7%; @ 2 h: 35.5 ± 3.2 versus 28.1 ± 3.3%; Figure 5C). No sex differences were observed with the ALDH2–/– BECs (Figure S3). LDH release was significantly reduced on treatment with calpain inhibitors, 5 and NYC-438, after 1 h of OGD-R in the ALDH2–/– BECs; however, CA-074 was without effect (Figure 5D; 5: 8.6 ± 0.8%, NYC-438: 9.6 ± 0.5%).
Figure 5.

Calpain and cathepsin inhibitors mitigate loss of cell viability and tight junction proteins in ALDH2–/– BECs. Representative images (A) and quantitative analysis of (B) cell count (confluence normalized as %) or (C) LDH release of WT and ALDH2–/– BECs subjected to 0, 30, 60, and 120 min of OGD-R. (D) LDH release of ALDH2–/– BECs after 1 h of OGD-R and cotreatment of 1 μM sulforaphane or 10 μM inhibitors. (E and F) Quantitative analysis of immunoblots of ALDH2–/– BECs subjected to 1 h of OGD-R and nontreated control probed with antibodies against ZO-1 (E) or occludin (F). (G–J) Quantitative analysis of immunoblots of BECs treated with inhibitors (10 μM) followed by 1 h of OGD-R versus nontreated controls probed for ZO-1 (G), occludin (H), or SBDP (145 kDa and 150 kDa analyzed in I and J, respectively). Data represent mean ± SD of at least n = 3 separate isolated cell preps analyzed by one-way ANOVA with Dunnett’s or Tukey’s multicomparison analysis: *, p < 0.05; **, p < 0.01; and ***, p < 0.001, versus no OGD-R control; #, p < 0.05; ##, p < 0.01; and ###, p < 0.001, versus OGD-R vehicle-treated.
BECs from ALDH2–/– mice were also more susceptible to loss of tight junction proteins caused by OGD-R compared to those from WT littermates. Normalized to control BECs, OGD-R produced a 21 and 12% loss of ZO-1 in ALDH2–/– and WT BECs, respectively (Figure 5E; 0.79 ± 0.4 versus 0.88 ± 0.2). Normalized to control BECs, OGD-R produced a 36 and 17% loss of occludin in ALDH2–/– and WT BECs, respectively (Figure 5F; 0.64 ± 0.4 versus 0.83 ± 0.3). Treatment of ALDH2–/– BECs with 5 significantly attenuated OGD-induced loss of both tight junction proteins (Figure 5G,H). Treatment with NYC-438 and CA-074 also significantly restored levels of ZO-1, although the observed increase in occludin levels relative to vehicle treated BECs did not reach significance. Proteolysis to give SBDPs was inhibited by the calpain inhibitors but not by the cathepsin-B inhibitor CA-074 (Figure 5I,J).
BBB Dysfunction from mTBI is Amplified in ALDH2–/– Mice
Since BECs from ALDH2–/– mice demonstrated enhanced sensitivity to OGD-R, we sought to explore the in vivo response of the BBB in ALDH2–/– mice to neurotrauma. We have previously reported that ALDH2–/– mice are sensitive to a single closed-head weight drop with a 30 g weight on the right hemisphere, an mTBI model.26 WT and ALDH2–/– mice underwent mTBI followed by assessment of BBB permeability with sodium fluorescein (NaFl), a widely used method.37 NaFl was injected i.p., 30 min before sacrifice, which occured 24 h post-mTBI, and the left and right (contralateral and ipsilateral) hemispheres were collected. Quantification revealed a significant increase in NaFl extravasation induced by mTBI in the WT mice in the ipsilateral hemisphere (ipsilateral: WT = 1.0 ± 0.13 μg/uL, WT + mTBI = 1.46 ± 0.20 μg/uL, contralateral: WT = 1.0 ± 0.11 μg/uL, WT + mTBI = 0.91 ± 0.11 μg/uL, Figure 6A). In the absence of mTBI, ALDH2–/– mice exhibited an increase in absolute NaFl extravasation compared to WT mice (ipsilateral: 1.67 ± 0.33 μg/uL, contralateral: 1.56 ± 0.22 μg/uL), compatible with BBB dysfunction caused by innate oxidative stress and lack of lipid peroxidation product detoxification. The combination of underlying oxidative stress with mTBI exacerbated the NaFl extravasation in the ipsilateral hemisphere without effect on the control hemisphere (ALDH2–/– + mTBI; ipsilateral: 2.1 ± 0.21 μg/uL, contralateral: 1.47 ± 0.09 μg/uL).
Figure 6.

BBB-dysfunction post-mTBI is exacerbated by oxidative stress. (A) Sodium fluorescein (NaFl) extravasation measured 24 h post-mTBI in the ipsilateral (right) and contralateral (left) hemispheres. (B–F) Quantitative analysis of immunoblots of WT and ALDH2–/– right brain hemispheres 24 h post-mTBI probed for ZO-1 (B), occludin (C), claudin-5 (D), MMP-9 (E), or eNOS (F). (G) Representative immunoblots of WT and ALDH2–/– brain hemispheres 24 h post-mTBI and null controls. Data represent mean ± SD of n = 7–10 animals analyzed by one-way ANOVA with Dunnett’s or Tukey’s multicomparison analysis. All protein was normalized to the housekeeping protein, β-actin; 50 μg of protein was loaded in each well. *, p <.05, and **, p <.001, versus WT null control.
As significant changes in NaFl extravasation were only observed in the ipsilateral hemisphere after mTBI, we measured changes in protein levels associated with BBB breakdown in these tissues (ZO-1, occludin, claudin-5, MMP-9, and endothelial nitric oxide synthase (eNOS)). While mTBI and ALDH2–/– alone had no effect on ZO-1 or occludin, the combination of enhanced oxidative stress with neurotrauma significantly reduced protein levels at 24 h post-mTBI (ZO-1: WT = 1.0 ± 0.06, WT + mTBI = 0.93 ± 0.08, ALDH2–/– = 0.96 ± 0.1, ALDH2–/– + mTBI = 0.65 ± 0.06; occludin: WT = 1.0 ± 0.05, WT + mTBI = 0.87 ± 0.8, ALDH2–/– = 1.06 ± 0.7, ALDH2–/– + mTBI = 0.74 ± 0.04, Figure 6B,C,G). Claudin-5, an integral membrane protein, reported to be decreased significantly in ADRD and other neurodegenerative disorders,36 remained unchanged (Figure 6D,G).
MMP-9 and eNOS are key proteins responsible for endothelial cell integrity and function. MMP-9 is a versatile zinc-containing endopeptidase that has been reported to be increased in TBI and ADRD, wherein it degrades both tight junction proteins and basement membrane proteins.38,39 In brain tissues of mice subjected to mTBI and control hemispheres, active MMP-9/total MMP-9 levels were significantly increased with mTBI and oxidative stress alone; however, the interaction of ALDH2–/– and mTBI did not significantly increase this ratio (WT: 1.0 ± 0.1; WT + mTBI: 1.5 ± 0.1; ALDH2–/–: 1.6 ± 0.2; ALDH2–/– + mTBI: 1.6 ± 0.2, Figure 6E,G). A key function of the endothelium is the production and release of NO through the activity of eNOS, which controls vascular tone and co-regulates neuronal function in the brain with the neuronal NOS isoform. NO is vital for the regulation of vascular homeostasis and healthy cognitive function in the central nervous system (CNS). In neurodegenerative diseases and neurotrauma, eNOS activity is significantly decreased, a key indicator of endothelial dysfunction.40 After mTBI, levels of eNOS protein were significantly reduced in WT mice (WT: 1.0 ± 0.2, WT + mTBI: 0.82 ± 0.12, Figure 6F,G). The background oxidative stress in ALDH2–/– mice also resulted in lower eNOS levels; however, in this case, mTBI significantly exacerbated loss of eNOS (ALDH2–/–: 0.60 ± 0.23, ALDH2–/– + mTBI: 0.30 ± 0.17).
Calpain Inhibitors Rescue Mice from mTBI-Induced Damage
Having demonstrated that ALDH2–/– mice are susceptible to neurotrauma leading to significant disruption of proteins associated with BBB dysfunction, we tested the protective effects of selective (5) and nonselective (NYC-438) calpain inhibitors in this animal model. ALDH2–/– mice were administered inhibitors (10 mg/kg i.p.) at 1 h post-mTBI. Ipsilateral hemispheres were collected 24 h post-mTBI and analyzed by immunoassay. Both inhibitors significantly attenuated the loss of ZO-1 (5: 1.3 ± 0.1, NYC-438: 1.7 ± 0.3) and occludin (5:1.3 ± 0.2, NYC-438: 1.4 ± 0.1) (Figure 7A,B). Moreover, while claudin-5 levels were not sensitive to mTBI, treatment with calpain inhibitors led to significant increases in expression of claudin-5 (5: 2.5 ± 0.2, NYC-438: 2.0 ± 0.2, Figure 7C). Furthermore, selective calpain-1 inhibition significantly reduced active MMP-9, while the nonselective inhibitor had no effect (5: 0.7 ± 0.1, NYC-438: 1.4 ± 0.2, Figure 7D). Finally, both inhibitors attenuated the loss of eNOS induced by mTBI (5: 1.25 ± 0.04, NYC-438: 1.23 ± 0.23, Figure 7E).
Figure 7.

Calpain inhibitors reverse or attenuate loss of proteins linked to BBB dysfunction and block proteolysis of spectrin and GFAP after neurotrauma. Quantitative analysis of immunoblots of ipsilateral hemispheres of WT and ALDH2–/– mice 24 h post-mTBI or null treatment, showing effect of calpain inhibitors (10 mg/kg i.p.), probed for ZO-1 (A), Occludin (B), Claudin-5 (C), MMP-9 (active/total, D), SBDP 145 kDaA (E), eNOS (F), GFAP (G), and GFAP-BDP (H). Data represents mean ± SD of n = 7–10 animals analyzed by one-way ANOVA with Dunnett’s or Tukey’s multicomparison analysis. All protein was normalized to the housekeeping protein, β-actin. 50 μg of protein was loaded in each well. *, p < 0.05; **, p < 0.01; and ***, p < 0.001 versus WT null control; #, p < 0.05; ##, p < 0.01; and ###, p < 0.001 versus ALDH2–/– + mTBI.
The calpain-1 breakdown product, SBDP 145 kDa, was significantly elevated in the ALDH2–/– mice in response to mTBI (WT: 1.0 ± 0.1, ALDH2–/– + mTBI: 2.1 ± 0.5, Figure 7F). SBDPs are biomarkers of neurodegenerative severity.3 Treatment with both calpain inhibitors significantly reduced SBDP 145 kDa levels to those observed in WT mice in the absence of neurotrauma and confirmed that both selective and nonselective compounds are effective calpain inhibitors in vivo (5: 0.83 ± 0.63, NYC-438: 0.93 ± 0.20, Figure 7F).
GFAP and its associated breakdown products (GFAP-BDP) are biomarkers of inflammation induced by oxidative stress and neurotrauma.41 GFAP is also a substrate of calpain-1 proteolysis.16 Brain levels of both GFAP and GFAP-BDP were significantly increased by mTBI in ALDH2–/– mice, reflecting the activation of calpain-1 and induction of neuroinflammation (GFAP: WT = 1.0 ± 0.2, ALDH2–/– + mTBI = 1.4 ± 0.3; GFAP-BDP: WT = 1.0 ± 0.2, ALDH2–/– + mTBI = 1.6 ± 0.9, Figure 7G,H). As anticipated, both calpain inhibitors significantly attenuated cleavage of GFP to block GFAP-BPD formation, whereas only NYC-438 lowered levels of GFAP significantly (GFAP: 5 = 1.5 ± 0.3, NYC-438 = 0.99 ± 0.49; GFAP-BDP: 5 = 0.74 ± 0.45, NYC-438 = 0.49 ± 0.24).
Levels of tight junction proteins after neurotrauma were not only restored by calpain inhibitor treatment but also appeared to be elevated compared to the null treatment WT group (Figure 7). Therefore, to further interrogate the effects of calpain inhibitors in the absence of neurotrauma, we conducted a second study incorporating treatment of WT mice with 5 and NYC-438. At the transcriptional level, only small changes in mRNA were observed (Figure S4). NYC-438 significantly increased brain levels of ZO-1 and claudin-5 under all treatments, including in WT mice not subject to mTBI (Figure 8A,B). Selective calpain inhibitor 5 also increased levels of these tight junction proteins, but the data did not reach significance for all groups. Occludin levels did not conform to this pattern, with no significant changes induced by calpain inhibitor treatment (with the exception of the ALDH2–/–group subject to neurotrauma; Figure 8C). Brain levels of eNOS were elevated by calpain inhibitors, with most treatment groups responding significantly (Figure 8D). With the exception of occludin, the levels of BBB-associated proteins were elevated by calpain inhibitor treatment in the presence and absence of neurotrauma; however, in the ALDH2–/– group subject to neurotrauma, protein levels were consistently, significantly restored by drug treatment.
Figure 8.

Tight junction proteins and eNOS levels are regulated by calpain inhibitors. Quantitative analysis of immunoblots of ipsilateral hemispheres of WT and ALDH2–/– mice 24 h post-mTBI with or without 5 or NYC-438 (10 mg/kg i.p.) and probed with ZO-1 (A), occludin (B), claudin-5 (C), and eNOS (D). Data represent mean ± SD of n = 7–10 animals analyzed by one-way ANOVA with Tukey’s multicomparison analysis. All protein was normalized to the housekeeping protein, β-actin; 50 μg of protein was loaded in each well. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Discussion
The “calpain–cathepsin hypothesis” has been stated in various nuanced versions; however, the common theme is that calpain-1 hyperactivation leading to lysosomal rupture releases cathepsins, notably cathepsin-B, with calpain/cathepsin activity causing the neuronal cell death that underlies neurodegeneration in TBI, ischemic stroke, and AD. In many versions, oxidative stress is an initiator of calpain hyperactivation and HNE protein modification contributes to lysosomal rupture. There is overwhelming evidence supporting the calpain–cathepsin hypothesis in preclinical studies of calpain-1 and cathepsin-B inhibitors in models of neurodegeneration including TBI, stroke and AD.5−10,29,42 Furthermore, studies have shown that calpain and cathepsin inhibitors may have an extended therapeutic window, as delay of treatment to 2–22 h after TBI or stroke-induced injury can provide neuroprotection.18,43,44
In contrast to genetic manipulation, pharmacological approaches to calpain-1 and cathepsin-B inhibition are less able to differentiate the effects of calpain versus cathepsin inhibition, because of the modest selectivity of most small molecule inhibitors. Furthermore, many inhibitors also block other relevant cysteine proteases, including cathepsins and caspases; for example, the selective cathepsin-B inhibitor, CA-074 inhibits cathepsin K (Figure 1). Proteolysis of spectrin to SBDP 145 kDa by calpain-1 was able to differentiate CA-074 from selective (5) and nonselective (NYC-438) calpain-1 inhibitors in vitro. Measurement of SBDP 145 kDa, a biomarker for TBI,45 was also used in our subsequent in vivo studies. With a novel, highly selective calpain-1 inhibitor in hand (5), we first replicated the neuroprotective effects of calpain-1 and cathepsin-B inhibitors in a neuronal cell line and primary human neuronal cultures in response to ischemia-reperfusion injury (Figure 2). As expected, all inhibitors were neuroprotective, in support of the calpain–cathepsin hypothesis.
Beyond neuroprotection, the calpain–cathepsin hypothesis has been relatively poorly explored. Protection by calpain inhibitors of the BBB and BECs would preserve tight junction proteins and support endothelial integrity, providing a mechanism of high relevance to neurodegenerative diseases and neurotrauma, including ischemic stroke, TBI, and AD. Reports indicate that BBB dysfunction, in particular BEC dysfunction and disruption of tight junction proteins, can precede neurodegeneration;12,46 therefore preserving BBB functionality is a viable therapeutic strategy for neurodegenerative diseases. Loss of primary BEC viability was observed in response to ischemia–reperfusion injury or addition of the lipid peroxidation products HNE and ONE. Under the mild OGD-R conditions optimized for modest, but significant loss of cell viability, ischemia–reperfusion injury resulted in significant loss of the tight junction proteins ZO-1 and occludin and significantly increased spectrin proteolysis. We observed that selective and nonselective calpain-1 inhibitors preserved BEC cell viability, attenuated loss of tight junction proteins, and inhibited spectrin breakdown, supporting extension of the calpain–cathepsin hypothesis to protection of the neurovasculature. In BECs, in contrast to neurons, a selective cathepsin-B inhibitor was inferior to the calpain inhibitors.
We have previously shown that ALDH2–/– mice have elevated oxidative stress through the inability to detoxify HNE, ONE, and other lipid peroxidation products, resulting in significant responses to mTBI, a mild neurotrauma.26 This is a clinically relevant model, since HNE and its protein adducts are elevated in AD brains and in other neurodegenerative diseases.47 Before testing calpain and cathepsin inhibitors in ALDH2–/– mice, we first chose to study the response of BECs from ALDH2–/– mice to ischemia–reperfusion injury. As anticipated, the elevated oxidative stress associated with loss of Aldh2 in these BECs led to ischemia–reperfusion-mediated loss of cell viability, loss of tight junction proteins, and spectrin proteolysis, exacerbated when compared to WT BECs. Calpain-1 and cathepsin-B inhibitors were protective against ischemia-reperfusion injury in ALDH2–/– BECs; however, in this system the performance of the selective calpain-1 inhibitor was superior, possibly owing to this being a highly potent reversible inhibitor and the amplified injury in ALDH2–/– BECs.
Having demonstrated that the in vitro neurotrauma modeled by OGD-R in ALDH2–/– BECs causes loss of tight junction proteins and spectrin proteolysis, we sought to extend these observations to an in vivo neurotrauma model. In studying the effects of mTBI in ALDH2–/– mice, additional biomarkers of BBB integrity and endothelial function were measured, including fluorescein extravasation and expression of activated MMP-9 and eNOS. Supporting the use of this mTBI model and consistent with the role of oxidative stress in vascular dysfunction, we observed significant increases in fluorescein extravasation and activated MMP-9 and significant loss of ZO-1, occludin, and eNOS only in the ipsilateral hemispheres of ALDH2–/– mice subject to mTBI. NO production by eNOS is essential for maintaining vascular homeostasis and cognition during aging and is reported to be decreased in neurodegenerative diseases.40 Moreover, recent reports propose that the decrease in eNOS actually drives the progression of ADRD and TBI.48,49 We found that ALDH2–/– mice have significantly elevated fluorescein extravasation and activated MMP-9 and reduced levels of eNOS, even in the absence of mTBI. Levels of eNOS in the ipsilateral brain hemisphere of ALDH2–/– mice subject to mTBI were measured at only 30% of WT mice without mTBI. The observations on tight junction proteins, fluorescein extravasation, eNOS and activated MMP-9 confirm the significant loss of BBB function and integrity in ALDH2–/– mice 24 h post mTBI.
Treatment of mice with calpain inhibitors at 1 h post-mTBI led to changes in biomarkers consistent with protection of the BBB. Inhibitors blocked the calpain-mediated proteolysis of spectrin to SBDP 145 kDa and the calpain-mediated proteolysis of GFAP to GFAP-BDP. Both SBDP 145kDA and GFAP-BDP are elevated in TBI.17,45,50 Elevation of MMP-9 activity is a downstream consequence of calpain-1 hyperactivation8 and was blocked by calpain inhibitor treatment. Calpain-1 activity is well-reported to be increased in activated glial cells after neurotrauma;16,17 however, while both 5 and NYC-438 blocked GFAP proteolysis, only the nonselective inhibitor restored total GFAP, suggesting that calpain-independent anti-inflammatory effects may contribute to attenuation of astrocyte activation in response to mTBI. Finally, the direct biomarkers of BBB damage and endothelial dysfunction, eNOS, occludin, and ZO-1, were significantly restored on treatment with either calpain inhibitor. Intriguingly, claudin-5 levels were not perturbed by either mTBI nor loss of Aldh2; however, treatment with calpain inhibitors significantly increased the levels of claudin-5 in brain ipsilateral hemispheres. Previous studies using nonselective calpain inhibitors have implicated ZO-1 as a substrate of calpain-1,51 and eNOS degradation has been reported to be mediated by calpain-1.52 The ability of calpain inhibitors to increase levels of tight junction protein and eNOS in the absence of neurotrauma requires further study.
The stimulus for this work was our own discovery and development of NYC-438, a potent and irreversible inhibitor of calpain-1 and cathepsin-B. NYC-438 was optimized to increase calpain-1 potency over papain, thus reducing nonspecific cysteine protease inhibition and retaining potent inhibition of calpain-1 and cathepsin-B. NYC-438 reversed cognitive deficits and restored long-term potentiation in the APP/PS1 familial Alzheimer’s disease transgenic mouse (FAD-Tg) model; however, in this model efficacy was independent of effects on Aβ neuropathology.30,32 Since the brain bioavailability of NYC-438 is low, we hypothesized that the beneficial effects of NYC-438 may be mediated by protection of the BBB and BEC functionality. For comparison, we synthesized a selective calpain-1 inhibitor, 5, an analogue of the AbbVie drug candidate alicapistat (ABT-957). During the course of the studies described herein, AbbVie published the results of the Phase 1 clinical trials of alicapisat for AD.11 Somewhat surprisingly, alicapistat was also reported to have relatively low brain bioavailability.
The Phase 1 clinical trial results for alicapistat showed no dose-limiting toxicities (DLTs) in the broad populations studied, including multiple groups of healthy young and old men and women and a group of men and women meeting NINCDS/ADRDA criteria for probable AD and prescribed an acetylcholinesterase (AChE) inhibitor. Peak plasma concentrations reached approximately 4 μM without DLTs. However, the trial was terminated, because of the failure to reach the pharmacodynamic end point in 17 healthy men aged 25–45 years with regular sleep habits. This failure was attributed to the low brain bioavailability: The concentration of ABT-957 achieved in the CSF at the highest dose (<20 nM) was less than half of the in vitro IC50 for calpain inhibition. It is important to review the trial end point, interruption of REM sleep, which was selected based upon preclinical animal studies in which sleep perturbations were observed for ABT-957 and were proposed to be a functional consequence of increased extracellular acetylcholine (ACh).53 AbbVie have used amnesia induced by the muscarininc Ach-receptor blocker scopolamine in preclinical development of calpain inhibitors; we have shown 5 and a nonselective calpain inhibitor to be effective in this assay.30 However, neither REM sleep nor scopolamine-induced amnesia provide a test of the calpain–cathepsin hypothesis.
In this work, we have successfully extended the calpain–cathepsin hypothesis as it relates to neuroprotection to demonstrate protection of BECs and the BBB from oxidative stress and neurotrauma delivered both in vitro and in vivo. Calpain inhibition was demonstrated by measurement of proteolytic products and was concomitant with attenuation of biomarkers associated with oxidative stress and neurotrauma, with these biomarkers often restored to normal levels. The results demonstrate that both selective and nonselective calpain inhibitors are able to protect the neurovasculature as reflected by tight junction proteins and eNOS and MMP-9 activity.
The overwhelmingly positive data on the actions of calpain inhibitors in preclinical models of neurotrauma, combined with our novel observations on protection of BECs and the BBB, should stimulate more detailed mechanistic studies. Importantly, NaFl is a substrate of the transporters multidrug-resistance-associated protein 2 (MRP2) and organic anion transporter 3 (OAT3),54 raising the possibility that the paracellular leak associated with NaFl underestimates the true magnitude of BBB permeability. Regulation of transporter proteins by calpain and cathepsin in neurotrauma may contribute to the observed efficacy of their inhibitors. The impact of calpain inhibition on assembly and localization of tight junction proteins also requires further interrogation, beyond measurement of protein levels, since such organization is critical to controlling paracellular permeability at the BBB.55,56
Given the very strong safety signal seen in clinical trials for the calpain inhibitor alicapistat, further study of this drug class is warranted, with a particular focus on the neurovasculature and protection of the BBB in neurotrauma and neurodegeneration.
Materials and Methods
Chemicals
Calpain inhibitor XI and CA-074 were purchased from Sigma. NYC-438, 5, and E64d were synthesized and characterized according to the literature.30,32
Animals
All animal care and procedures were conducted with approved institutional animal care protocols and in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All animal protocols were approved by the University of Illinois at Chicago Institutional Animal Care and Use Committee (ACC no. 18–034 and ACC no. 18–017). ALDH2–/– and WT littermates were generated and maintained according to previous literature.26
Enzymatic Assays
Full-length human calpain-1 (210 nM, Sigma) or papain (Carica papaya, 236 nM, Sigma) was added to a buffer solution of 100 mM NaCl, 50 mM HEPES, pH 7.6, 1 mM TCEP, and inhibitor, then incubated at 30 °C for 10 min prior to the addition of Suc-LLVY-AMC substrate (30 μM, Enzo Life Sciences). Calpain-1 reactions also contained 1 mM CaCl2. Recombinant human cathepsin-B (10 μM, R & D Systems) was added to a buffer solution of 25 mM MES, 5 mM DTT, and pH 5.0 and incubated at room temperature for 15 min. The activated cathepsin-B (11 nM) was diluted with 25 mM MES, pH 5.0, and inhibitor, then incubated at 30 °C for 10 min prior to the addition of Z-LR-AMC substrate (10 μM, R & D Systems). Recombinant human cathepsin K (269 pM, Enzo Life Scienes) was added to a solution of 50 mM Na acetate, 2.5 mM EDTA, 1 mM DTT, 0.01% Triton X-100, pH 5.5, and inhibitor, then incubated at 30 °C for 10 min prior to the addition of Z-FR-AMC (10 μM). Papain reactions were carried out in microtiter 96-well plates, while the cathepsin-B, cathepsin K, and calpain-1 reactions were conducted in Corning 384-well low-volume plates. All reactions were performed at 30 °C, and relative fluorescence was monitored over time (excitation and emission of 346 and 444 nm, respectively). The initial rate of all reactions were normalized to the initial rate of each enzyme with no inhibitor present, and the data were represented as percent enzyme activity with standard deviation (SD). Compounds were dissolved in DMSO and kept below 2% in all experiments. IC50 curves were determined by varying the concentration of the inhibitor and plotting the percent enzyme activity on Prism 7.0 and compiling nonlinear fit regression curves. Reversibility was determined in a dilution assay with a 1:20 fold dilution. Directly following the 10 min incubation period, and prior to fluorescent reading, 1:20 of the compound-protease complex was diluted in substrate. The nondiluted and diluted reactions were monitored simultaneously. Reactions with similar activity were classified as irreversible, while reactions with increased protease activity, as compared to the undiluted control, were classified as reversible.
Cell Culture
Human neuroblastoma SH-SY5Y cells (ATTC CRL-2266) and mouse hippocampal HT22 cells (Kind gift from Dr. Dargusch, Salk Institute) were cultured in DMEM/F12 and DME, respectively, and supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin at 37 °C in a humidified atmosphere of 5% CO2. Primary human hippocampal cells (catalog no. 1540) were purchased from ScienCell Research Laboratories (California), seeded and grown according to manufacturer’s instructions. For cytotoxicity studies, cells were seeded at 2 × 104 cells per well in a 96-well plate in low serum media (1% fetal bovine serum). After overnight incubation, cells were administered with varying concentrations of compounds for defined periods of time. Cell viability was determined by MTS using CellTilter 96 Aqueous One Solution Cell Proliferation Assay or MTT (methylthiazolyldiphenyl-tetrazolium bromide, Millipore Sigma) and LDH release using CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega) according to the manufacturer’s instructions. Untreated cells served as a negative control as DMSO concentrations were below 0.5%. The experiments were performed in triplicate. For immunoblots, cells were cultured up to 95% confluence in 6-well plates and treated with varying concentrations of compounds for defined periods of time. At termination of treatment, cells were washed twice with ice-cold PBS and protein was extracted by adding ice-cold RIPA buffer (Sigma) supplemented with protease and phosphatase inhibitors (Calbiochem). Cells were scraped, and the cell lysate was centrifuged at 10 000 × rpm for 10 min at 4 °C.
Primary Cortical Endothelial Cell Isolation
Cortices were dissected from 3–8 week old mice, chopped, and centrifuged in minimal essential media (ThermoFisher) at 1000g for 10 min at 4 °C. The supernatant was removed, and the pellet was triturated in papain (17U per brain) and DNase (84U per brain, LK003178 and LK003172, respectively, Worthington Biochemical) using a 19G needle before being placed in a 37 °C water bath for 15 min. Following incubation, the sample was triturated with a 21G needle and mixed vigorously with 2 parts 25% BSA (A2153–50G, Sigma, solubilized in HBSS–/–), before centrifugation at 3880g for 15 min at 4 °C. After removing the supernatant, the pellet was resuspended in 1 mL of endothelial cell medium (M1166, Cell Biologics) with heparin (5.5U/mL, H3149, Sigma) and centrifuged for 5 min at 1000g at 4 °C. The pellet was then resuspended in endothelial cell medium + heparin and plated onto plates coated with 0.005% collagen (C8919, Sigma), 1–2 μg/mL laminin (L2020, Sigma), and 50 μg/mL fibronectin (F0895, Sigma). The following day, the plates were washed twice with HBSS+/+, and media was replaced with endothelial cell medium and puromycin (4 μg/mL) as a selection agent for 72 h. Cells were used at the second passage for all studies.
Oxygen Glucose Deprivation
SH-SY5Y cells were plated in a 96-well plate at 4 × 104 cells per well and cultured overnight. To trigger “ischemia”, cell media was replaced with glucose-free media (DMEM, no glucose, no glutamine, no phenol red, Gibco catalog no. A1443001) and incubated in a hypoxic chamber (95% N2, 5% CO2, <0.5% O2). After 2 h, cells were reperfused with 5% CO2 and replenished with glucose-containing media. Cell viability was monitored after 24 h of “reperfusion”, quantified by MTS and confirmed via LDH release. BEC cells were plated in clear-bottomed black plates coated with 0.005% collagen (C8919, Sigma), 1–2ug/mL laminin (L2020, Sigma), and 50 ug/mL fibronectin (F0895, Sigma). To measure confluence, at termination of the experiment, cells were treated with 5 μM Cell Tracker Red CMTPX (C34552, ThermoFisher) for 30 min, then washed and fixed with 4% paraformaldehyde and 0.1% Triton X-100 in PBS for 5 min. Cells were then washed and treated with 5 uM Hoeschst 33342 (Invitrogen) in Fluorobrite DMEM (A1896701, Fisher). Images were captured on the BZ-X700 microscope (Keyence), and confluence was quantified using Keyence software. All OGD-R studies were performed in the absence of fetal bovine serum and endothelial growth supplements.
Closed Head mTBI
The previously reported protocol was followed as described.26 Animals were dosed 1 h post-mTBI with 5 or NYC-438 (4% Tween 80/4% DMSO/92% diH2O) at 10 mg/kg via i.p. injection.
Protein Extraction from Tissue
At the termination of experiment, mice were sacrificed with CO2 and perfused with ice-cold PBS at a rate of 5 mg/mL for 3 min. Brains were collected with cerebellum removed, and then split into right and left hemispheres, stored in 1.5 mL tubes, and flash-frozen in dry ice before storing at −80 °C. The next day, brains were removed from the freezer, weighed, and homogenized using a BeadBug microtube homogenizer (Sigma). Briefly, 2× volume of ice-cold lysis buffer (25 mM HEPES pH 7.0, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 0.1% SDS supplemented with protease and phosphatase inhibitors [Roche Diagnostics]) was added to a 2 mL bead tube with beads (3× tissue mass). Brain samples were homogenized three times at 3800 rpm for 10 s with a rest of 1 min in between. The homogenized brain was then centrifuged at 1000 rpm at 4 °C for 15 min. The supernatant was then moved to another tube and homogenized again at the same speed and time. The samples were stored on ice for the entirety of the protein extraction.
Immunoblots
Protein concentration was determined using the Pierce BCA-200 Protein Assay Kit (ThermoFisher). Lysates were stored at −20 °C before use. Equal amounts of protein were prepared with 6× loading buffer (0.375 M Tris (pH 6.8), 12% SDS, 60% glycerol, 0.6 M DTT, 0.06% bromophenol blue), heated at 95 °C for 5 min, cooled to RT before loading onto NuPage 4–12% Bis-Tris Protein Gels. The gels were transferred onto PVDF membranes using the iBLOT2 (ThermoFisher), blocked in 5% nonfat milk for 1 h, probed overnight with the primary antibody at 4 °C, washed three times with TBS-T, probed with an HRP-linked secondary antibody (1:1000 in blocking buffer) for 1 h, washed three times with TBS-T, then visualized using SuperSignal west Fempto Chemiluminescence substrate (ThermoFisher) on the Azure c400 series. Immunoblots were quantified using Azure Biosystems Technology. All values were normalized to expression levels of beta-actin. Antibodies: anti-spectrin (MAB1622, Millipore), anti-ZO-1 (bs-1329R-TR, Bioss), anti-occludin (66378–1-Ig, Proteintech), anti-claudin-5 (4C3C2, ThermoFisher), anti-eNOS (bs-0163R, Bioss), anti-map2 (ab5392, Abcam), anti-MMP9 (ab38898, abcam), anti-GFAP (Z0334, Dako), and HRP-conjugated anti-β-actin (HRP-60008, Proteintech).
Sodium Fluorescein Extravasation
Mice were injected with 5 mg/kg of 2% NaFl in sterile dH2O via i.p. injection 30 min prior to sacrifice. Mice were sacrificed with CO2, and 600–800 μL of blood was collected from the right ventricle of the heart prior to perfusion with ice-cold PBS at a rate of 6 mL per min for 3 min. Plasma was centrifuged at 1500g for 15 min at 4 °C. Following perfusion, the mice were decapitated, and brain hemispheres were collected and stored at −80 °C prior to biochemical analysis. Plasma was analyzed immediately on a 96-well black plate using the Synergy Neo2 plate reader, measuring absorbance at an excitation wavelength of 440 nm and emission of 525 nm. Each hemisphere was weighed and homogenized in PBS. An equal amount of 60% trichloroacetic acid was added to precipitate proteins, left on ice for 30 min, then centrifuged at 18 000g for 10 min at 4 °C. Supernatants were measured in a 96-well black plate. The samples were protected from light throughout the entire experiment either by using light-sensitive 1.5 mL Eppendorf tubes, covering them in aluminum foil, or working in a dark room. Sodium fluorescein extravasation was quantified as follows:
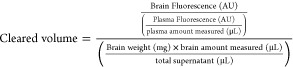 |
RNA Extraction and Real-Time qPCR
Following perfusion, whole brains were extracted and separated into two hemispheres (ipsilateral and contralateral) and immediately stored in 1.5 mL tubes at −80 °C for at least 1 day. Hemispheres were removed from −80 °C, thawed on ice for 10–15 min and homogenized with 1 mL of Trizol using a hand-held pestle homogenizer. The tubes were then spun at 10 000g for 10 min at 4 °C to remove debris. Supernatant was removed and placed in a fresh tube with 0.200 μL of chloroform. Samples were vigorously vortexed for 15 s then incubated at room temperature for 2 min before centrifugation of 12 000g for 15 min at 4 °C. The upper aqueous phase containing RNA was transferred and isolated using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s instructions. First-strand cDNA synthesis was synthesized with 2 μg of total RNA using the SuperScript III First-Strand Synthesis System for qRT-PCR (Invitrogen) according to manufacturer’s instructions. Inflammation primers were acquired from Applied Biosystems. Each PCR reaction was carried out using the Taqman Gene Expression Master Mix (Applied Biosystems) on the StepOnePlus Real-Time PCR system (Life Technologies). ΔΔCT values for each gene were normalized to the expression level of β-actin in each sample.
Statistical analysis
GraphPad Prism 8 software package (GraphPad Software, USA) was used to perform statistical analysis. All data were presented as the mean ± SD unless otherwise noted. One-way analysis of variance (ANOVA) with appropriate post hoc tests (3+ groups) and Student’s t-test (2 groups) were used to calculate statistical significance: *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
Acknowledgments
Ragda Izar is thanked for technical support.
Glossary
Abbreviations
- ADRD
Alzheimer’s disease and related dementia
- ALDH2
aldehyde dehydrogenase 2
- BBB
blood–brain barrier
- BEC
brain endothelial cells
- CCH
calpain–cathepsin hypothesis
- CNS
central nervous system
- eNOS
endothelial nitric oxide synthase
- GFAP
glial fibrillary acidic protein
- GFAP-BDP
glial fibrillary acidic protein breakdown products
- HNE
4-hydroxynonenal
- IS
ischemic stroke
- MCI
mild cognitive impairment
- MMP-9
matrix metalloprotease 9
- mTBI
mild traumatic brain injury
- NO
nitric oxide
- ONE
oxo-2-nonenal
- OGD-R
oxygen glucose deprivation and reperfusion
- OS
oxidative stress
- SBDP
spectrin breakdown products
- TBI
traumatic brain injury
- WT
wildtype
- ZO-1
zona occludens-1
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.0c00217.
Further details of experimental methodology, table of genes and primers used in rt-qPCR experiments, and additional figures (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Specific contributions are listed as follows: R.C.K. conducted enzyme assays, in vitro and in vivo assays and drafted and revised the manuscript. A.J. and I.G. synthesized compounds. O.D. maintained all animals and conducted in vivo assays. G.R.J.T. coordinated the overall project, with significant input from L.T., and manuscript preparation and revision.
NIH UL1TR002003 (UICentre), NIH T32AG57468 (RCK).
The authors declare the following competing financial interest(s): G.R.J.T. is an inventor on patents assigned to UIC.
Supplementary Material
References
- Yamashima T. (2016) Can ‘calpain-cathepsin hypothesis’ explain Alzheimer neuronal death?. Ageing Res. Rev. 32, 169–179. 10.1016/j.arr.2016.05.008. [DOI] [PubMed] [Google Scholar]
- Siklos M.; BenAissa M.; Thatcher G. R. (2015) Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 5, 506–519. 10.1016/j.apsb.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czogalla A.; Sikorski A. F. (2005) Spectrin and calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal cells. Cell. Mol. Life Sci. 62, 1913–1924. 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson E.; Alafuzoff I.; Blennow K.; Blomgren K.; Hall C. M.; Janson I.; Karlsson I.; Wallin A.; Gottfries C. G.; Karlsson J. O. (1990) Calpain and calpastatin in normal and Alzheimer-degenerated human brain tissue. Neurobiol. Aging 11, 425–431. 10.1016/0197-4580(90)90009-O. [DOI] [PubMed] [Google Scholar]
- Posmantur R.; Kampfl A.; Siman R.; Liu J.; Zhao X.; Clifton G. L.; Hayes R. L. (1997) A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience 77, 875–888. 10.1016/S0306-4522(96)00483-6. [DOI] [PubMed] [Google Scholar]
- Thompson S. N.; Carrico K. M.; Mustafa A. G.; Bains M.; Hall E. D. (2010) A pharmacological analysis of the neuroprotective efficacy of the brain- and cell-permeable calpain inhibitor MDL-28170 in the mouse controlled cortical impact traumatic brain injury model. J. Neurotrauma 27, 2233–2243. 10.1089/neu.2010.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. C.; Goto Y.; Lanzino G.; Soleau S.; Kassell N. F.; Lee K. S. (1994) Neuroprotection with a calpain inhibitor in a model of focal cerebral ischemia. Stroke 25, 663–669. 10.1161/01.STR.25.3.663. [DOI] [PubMed] [Google Scholar]
- Tsubokawa T.; Solaroglu I.; Yatsushige H.; Cahill J.; Yata K.; Zhang J. H. (2006) Cathepsin and calpain inhibitor E64d attenuates matrix metalloproteinase-9 activity after focal cerebral ischemia in rats. Stroke 37, 1888–1894. 10.1161/01.STR.0000227259.15506.24. [DOI] [PubMed] [Google Scholar]
- Medeiros R.; Kitazawa M.; Chabrier M. A.; Cheng D.; Baglietto-Vargas D.; Kling A.; Moeller A.; Green K. N.; LaFerla F. M. (2012) Calpain inhibitor A-705253 mitigates Alzheimer’s disease-like pathology and cognitive decline in aged 3xTgAD mice. Am. J. Pathol. 181, 616–625. 10.1016/j.ajpath.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Fa M.; Zhang H.; Staniszewski A.; Saeed F.; Shen L. W.; Schiefer I. T.; Siklos M. I.; Tapadar S.; Litosh V. A.; Libien J.; Petukhov P. A.; Teich A. F.; Thatcher G. R.; Arancio O. (2015) Novel Selective Calpain 1 Inhibitors as Potential Therapeutics in Alzheimer’s Disease. J. Alzheimer's Dis. 49, 707–721. 10.3233/JAD-150618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lon H. K.; Mendonca N.; Goss S.; Othman A. A.; Locke C.; Jin Z.; Rendenbach-Mueller B. (2019) Pharmacokinetics, Safety, Tolerability, and Pharmacodynamics of Alicapistat, a Selective Inhibitor of Human Calpains 1 and 2 for the Treatment of Alzheimer Disease: An Overview of Phase 1 Studies. Clin. Pharmacol. Drug Dev. 8, 290–303. 10.1002/cpdd.598. [DOI] [PubMed] [Google Scholar]
- Sweeney M. D.; Sagare A. P.; Zlokovic B. V. (2018) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation D. A.; Sweeney M. D.; Montagne A.; Sagare A. P.; D’Orazio L. M.; Pachicano M.; Sepehrband F.; Nelson A. R.; Buennagel D. P.; Harrington M. G.; Benzinger T. L. S.; Fagan A. M.; Ringman J. M.; Schneider L. S.; Morris J. C.; Chui H. C.; Law M.; Toga A. W.; Zlokovic B. V. (2019) Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 25, 270–276. 10.1038/s41591-018-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturria-Medina Y.; Sotero R. C.; Toussaint P. J.; Mateos-Perez J. M.; Evans A. C. (2016) Alzheimer’s Disease Neuroimaging, I. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 7, 11934. 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer I.; Romano-Clarke G.; Grynspan F. (1991) Calpain-mediated proteolysis of microtubule associated proteins MAP1B and MAP2 in developing brain. Neurochem. Res. 16, 891–898. 10.1007/BF00965538. [DOI] [PubMed] [Google Scholar]
- Lee Y. B.; Du S.; Rhim H.; Lee E. B.; Markelonis G. J.; Oh T. H. (2000) Rapid increase in immunoreactivity to GFAP in astrocytes in vitro induced by acidic pH is mediated by calcium influx and calpain I. Brain Res. 864, 220–229. 10.1016/S0006-8993(00)02180-6. [DOI] [PubMed] [Google Scholar]
- McMahon P. J.; Panczykowski D. M.; Yue J. K.; Puccio A. M.; Inoue T.; Sorani M. D.; Lingsma H. F.; Maas A. I.; Valadka A. B.; Yuh E. L.; et al. (2015) Measurement of the glial fibrillary acidic protein and its breakdown products GFAP-BDP biomarker for the detection of traumatic brain injury compared to computed tomography and magnetic resonance imaging. J. Neurotrauma 32, 527–533. 10.1089/neu.2014.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook G. R.; Yu J.; Sipes N.; Pierschbacher M. D.; Hook V.; Kindy M. S. (2014) The cysteine protease cathepsin B is a key drug target and cysteine protease inhibitors are potential therapeutics for traumatic brain injury. J. Neurotrauma 31, 515–529. 10.1089/neu.2013.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein H. G.; Keilhoff G. (2018) Putative roles of cathepsin B in Alzheimer’s disease pathology: The good, the bad, and the ugly in one?. Neural Regener. Res. 13, 2100–2101. 10.4103/1673-5374.241457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic B. V. (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 12, 723–738. 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuolo J.; Gliozzi M.; Musolino V.; Scicchitano M.; Carresi C.; Scarano F.; Bosco F.; Nucera S.; Ruga S.; Zito M. C.; et al. (2018) The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 19, 2693. 10.3390/ijms19092693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield D. A.; Reed T.; Perluigi M.; De Marco C.; Coccia R.; Cini C.; Sultana R. (2006) Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 397, 170–173. 10.1016/j.neulet.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Pratico D.; Sung S. (2004) Lipid peroxidation and oxidative imbalance: early functional events in Alzheimer’s disease. J. Alzheimer's Dis. 6, 171–175. 10.3233/JAD-2004-6209. [DOI] [PubMed] [Google Scholar]
- Nunomura A.; Perry G.; Aliev G.; Hirai K.; Takeda A.; Balraj E. K.; Jones P. K.; Ghanbari H.; Wataya T.; Shimohama S.; Chiba S.; Atwood C. S.; Petersen R. B.; Smith M. A. (2001) Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Abdullahi W.; Tripathi D.; Ronaldson P. T. (2018) Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am. J. Physiol Cell Physiol 315, C343–C356. 10.1152/ajpcell.00095.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopp R. C.; Lee S. H.; Hollas M.; Nepomuceno E.; Gonzalez D.; Tam K.; Aamir D.; Wang Y.; Pierce E.; BenAissa M.; Thatcher G. R. J. (2020) Interaction of oxidative stress and neurotrauma in ALDH2(−/−) mice causes significant and persistent behavioral and pro-inflammatory effects in a tractable model of mild traumatic brain injury. Redox Biol. 32, 101486. 10.1016/j.redox.2020.101486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J.; Lauretti E.; Pratico D. (2017) Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3beta kinase: implications for Alzheimer’s disease. Mol. Psychiatry 22, 1002–1008. 10.1038/mp.2016.214. [DOI] [PubMed] [Google Scholar]
- Sacco M. D., Ma C., Lagarias P., Gao A., Townsend J. A., Meng X., Dube P., Zhang X., Hu Y., and Kitamura N.. et al. (July 27, 2020) Structure and inhibition of the SARS-CoV-2 main protease reveals strategy for developing dual inhibitors against M(pro) and cathepsin L. bioRxiv (Biophysics), DOI: 10.1101/2020.07.27.223727. [DOI] [PMC free article] [PubMed]
- Hook V. Y.; Kindy M.; Hook G. (2008) Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J. Biol. Chem. 283, 7745–7753. 10.1074/jbc.M708362200. [DOI] [PubMed] [Google Scholar]
- Jastaniah A.; Gaisina I. N.; Knopp R.; Thatcher G. R. (2020) Synthesis of alpha-Ketoamide-based Stereoselective Calpain-1 Inhibitors as Neuroprotective Agents. ChemMedChem 15, 2280. 10.1002/cmdc.202000385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantos K.; Kling A.; Mack H.; Hornberger W.; Moeller A.; Nimmrich V.; Lao Y.; Nijsen M. (2019) Discovery of ABT-957:1-Benzyl-5-oxopyrrolidine-2-carboxamides as selective calpain inhibitors with enhanced metabolic stability. Bioorg. Med. Chem. Lett. 29, 1968–1973. 10.1016/j.bmcl.2019.05.034. [DOI] [PubMed] [Google Scholar]
- Schiefer I. T.; Tapadar S.; Litosh V.; Siklos M.; Scism R.; Wijewickrama G. T.; Chandrasena E. P.; Sinha V.; Tavassoli E.; Brunsteiner M.; et al. (2013) Design, synthesis, and optimization of novel epoxide incorporating peptidomimetics as selective calpain inhibitors. J. Med. Chem. 56, 6054–6068. 10.1021/jm4006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X. X.; Jeromin A. (2012) Spectrin Breakdown Products (SBDPs) as Potential Biomarkers for Neurodegenerative Diseases. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 1, 85–93. 10.1007/s13670-012-0009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasca C. I.; Dal-Cim T.; Cimarosti H. (2015) In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol. Biol. 1254, 197–210. 10.1007/978-1-4939-2152-2_15. [DOI] [PubMed] [Google Scholar]
- Daneman R.; Prat A. (2015) The blood-brain barrier. Cold Spring Harbor Perspect. Biol. 7, a020412 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y.; Shinohara M.; Shinohara M.; Yamazaki A.; Murray M. E.; Liesinger A. M.; Heckman M. G.; Lesser E. R.; Parisi J. E.; Petersen R. C.; Dickson D. W.; Kanekiyo T.; Bu G. (2019) Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain 142, 1077–1092. 10.1093/brain/awz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya M.; Ahishali B. (2011) Assessment of permeability in barrier type of endothelium in brain using tracers: Evans blue, sodium fluorescein, and horseradish peroxidase. Methods Mol. Biol. 763, 369–382. 10.1007/978-1-61779-191-8_25. [DOI] [PubMed] [Google Scholar]
- Weekman E. M.; Wilcock D. M. (2015) Matrix Metalloproteinase in Blood-Brain Barrier Breakdown in Dementia. J. Alzheimer's Dis. 49, 893–903. 10.3233/JAD-150759. [DOI] [PubMed] [Google Scholar]
- Pijet B.; Stefaniuk M.; Kostrzewska-Ksiezyk A.; Tsilibary P. E.; Tzinia A.; Kaczmarek L. (2018) Elevation of MMP-9 Levels Promotes Epileptogenesis After Traumatic Brain Injury. Mol. Neurobiol. 55, 9294–9306. 10.1007/s12035-018-1061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Cao Y.; Ma L.; Pei H.; Rausch W. D.; Li H. (2018) Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 10, 376. 10.3389/fnagi.2018.00376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J.; Hol E. M. (2011) GFAP in health and disease. Prog. Neurobiol. 93, 421–443. 10.1016/j.pneurobio.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Hook G.; Jacobsen J. S.; Grabstein K.; Kindy M.; Hook V. (2015) Cathepsin B is a New Drug Target for Traumatic Brain Injury Therapeutics: Evidence for E64d as a Promising Lead Drug Candidate. Front Neurol 6, 178. 10.3389/fneur.2015.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markgraf C. G.; Velayo N. L.; Johnson M. P.; McCarty D. R.; Medhi S.; Koehl J. R.; Chmielewski P. A.; Linnik M. D. (1998) Six-hour window of opportunity for calpain inhibition in focal cerebral ischemia in rats. Stroke 29, 152–158. 10.1161/01.STR.29.1.152. [DOI] [PubMed] [Google Scholar]
- Frederick J. R.; Chen Z.; Bevers M. B.; Ingleton L. P.; Ma M.; Neumar R. W. (2008) Neuroprotection with delayed calpain inhibition after transient forebrain ischemia. Crit. Care Med. 36, S481–485. 10.1097/CCM.0b013e31818a8ec8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike B. R.; Zhao X.; Newcomb J. K.; Posmantur R. M.; Wang K. K.; Hayes R. L. (1998) Regional calpain and caspase-3 proteolysis of alpha-spectrin after traumatic brain injury. NeuroReport 9, 2437–2442. 10.1097/00001756-199808030-00002. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Zhang L.; Pu H.; Mao L.; Hu X.; Jiang X.; Xu N.; Stetler R. A.; Zhang F.; Liu X.; et al. (2016) Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 7, 10523. 10.1038/ncomms10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F.; Tramutola A.; Butterfield D. A. (2017) Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radical Biol. Med. 111, 253–261. 10.1016/j.freeradbiomed.2016.10.490. [DOI] [PubMed] [Google Scholar]
- Austin S. A.; Santhanam A. V.; Hinton D. J.; Choi D. S.; Katusic Z. S. (2013) Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J. Neurochem. 127, 691–700. 10.1111/jnc.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad C.; Grande P. O.; Bentzer P. (2009) Hemodynamic and histological effects of traumatic brain injury in eNOS-deficient mice. J. Neurotrauma 26, 1953–1962. 10.1089/neu.2009.0955. [DOI] [PubMed] [Google Scholar]
- Metzger R. R.; Sheng X.; Niedzwecki C. M.; Bennett K. S.; Morita D. C.; Zielinski B.; Schober M. E. (2018) Temporal response profiles of serum ubiquitin C-terminal hydrolase-L1 and the 145-kDa alpha II-spectrin breakdown product after severe traumatic brain injury in children. J. Neurosurg Pediatr 22, 369–374. 10.3171/2018.4.PEDS17593. [DOI] [PubMed] [Google Scholar]
- Alluri H.; Grimsley M.; Anasooya Shaji C.; Varghese K. P.; Zhang S. L.; Peddaboina C.; Robinson B.; Beeram M. R.; Huang J. H.; Tharakan B. (2016) Attenuation of Blood-Brain Barrier Breakdown and Hyperpermeability by Calpain Inhibition. J. Biol. Chem. 291, 26958–26969. 10.1074/jbc.M116.735365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averna M.; Stifanese R.; De Tullio R.; Passalacqua M.; Salamino F.; Pontremoli S.; Melloni E. (2008) Functional role of HSP90 complexes with endothelial nitric-oxide synthase (eNOS) and calpain on nitric oxide generation in endothelial cells. J. Biol. Chem. 283, 29069–29076. 10.1074/jbc.M803638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kling A.; Jantos K.; Mack H.; Hornberger W.; Drescher K.; Nimmrich V.; Relo A.; Wicke K.; Hutchins C. W.; Lao Y.; Marsh K.; Moeller A. (2017) Discovery of Novel and Highly Selective Inhibitors of Calpain for the Treatment of Alzheimer’s Disease: 2-(3-Phenyl-1H-pyrazol-1-yl)-nicotinamides. J. Med. Chem. 60, 7123–7138. 10.1021/acs.jmedchem.7b00731. [DOI] [PubMed] [Google Scholar]
- Hawkins B. T.; Ocheltree S. M.; Norwood K. M.; Egleton R. D. (2007) Decreased blood-brain barrier permeability to fluorescein in streptozotocin-treated rats. Neurosci. Lett. 411, 1–5. 10.1016/j.neulet.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead J. J.; McCaffrey G.; Sanchez-Covarrubias L.; Finch J. D.; Demarco K. M.; Quigley C. E.; Davis T. P.; Ronaldson P. T. (2012) Tempol modulates changes in xenobiotic permeability and occludin oligomeric assemblies at the blood-brain barrier during inflammatory pain. Am. J. Physiol Heart Circ Physiol 302, H582–593. 10.1152/ajpheart.00889.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey G.; Willis C. L.; Staatz W. D.; Nametz N.; Quigley C. A.; Hom S.; Lochhead J. J.; Davis T. P. (2009) Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J. Neurochem. 110, 58–71. 10.1111/j.1471-4159.2009.06113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.