

Abstract
The adaptive immune system recognizes tumor antigens at an early stage to eradicate cancer cells. This process is accompanied by systemic proliferation of the tumor antigen–specific T lymphocytes. While detection of asymptomatic early-stage cancers is challenging due to small tumor size and limited somatic alterations, tracking peripheral T cell repertoire changes may provide an attractive solution to cancer diagnosis. Here, we developed a deep learning method called DeepCAT to enable de novo prediction of cancer-associated T cell receptors (TCRs). We validated DeepCAT using cancer-specific or non-cancer TCRs obtained from multiple major histocompatibility complex I (MHC-I) multimer-sorting experiments and demonstrated its prediction power for TCRs specific to cancer antigens. We blindly applied DeepCAT to distinguish over 250 patients with cancer from over 600 healthy individuals using blood TCR sequences and observed high prediction accuracy, with area under the curve (AUC) ≥ 0.95 for multiple early-stage cancers. This work sets the stage for using the peripheral blood TCR repertoire for noninvasive cancer detection.
INTRODUCTION
Most malignancies can be cured when diagnosed early (1). Organ-specific imaging assessment, including colonoscopy, breast mammogram, and low-dose lung computerized tomography (CT) scan, are widely used to detect limited types of early-stage cancers (2). Blood tests monitoring selected cancer biomarkers, including prostate-specific antigen, CA-125, and CA-153, have also been investigated in clinical studies (3, 4), yet none reached the high specificity required for population-level screening (5). The rapid development of single-cell and high-throughput sequencing technologies has advanced detection methods relying on cell-free DNA (cfDNA) or circulating tumor cells in liquid biopsies (6–8). Although they demonstrated exciting potential for cancer diagnosis, cfDNA-based methods rely on preselected panels of cancer somatic mutations, and the identification of circulating tumor cells usually relies on a few epithelial biomarkers (9) or cell morphological changes (10), which might be subjective and nonspecific. A recent study further revealed that most mutations found in plasma cfDNA are derived from white blood cells instead of cancer (11), thus putting the specificity of cfDNA-based approaches into question. Imaging scans, cancer biomarkers, cfDNA, and circulating tumor cells all depend on tumor-associated molecules, and the detection of earlier stage tumors is difficult for all methods.
In this work, we explore the feasibility of using the immune repertoire as an independent cancer diagnosis modality. T cells reactive to tumor antigens are central mediators of cancer immunity and key targets of immunotherapies (12–14). With immunoediting (15), the T cell repertoire is expected to undergo cancer-specific changes during tumor progression. However, because most cancer antigens are unknown (16), identification of cancer-associated T cells remains difficult, and currently, there is no diagnostic method to monitor signals in the T cell repertoire. To bridge this gap, we hypothesized that cancer-associated T cell receptors (caTCRs) may share common biochemical signatures allowing for their de novo identification. This hypothesis was supported by the previous observation of higher usage of hydrophobic residuals both in the immunogenic epitopes (17) and in the tumor-infiltrating T lymphocyte (TIL) receptors (18), and a recent study on consistent biophysicochemical motifs in breast or colorectal TILs that are absent from normal tissues (19).
RESULTS
DeepCAT method for de novo prediction of caTCRs
In this work, we developed a deep learning method to de novo predict caTCRs in patient blood. To generate the training set, we applied the TCR Repertoire Utilities for Solid Tissue or the TRUST algorithm (18, 20) to extract the complementarity-determining region 3 (CDR3) of the TCRs from approximately 4200 tumor RNA-sequencing (RNA-seq) samples in The Cancer Genome Atlas (TCGA) covering 32 cancer types (table S1) (21). TRUST is a computational method developed to perform highly sensitive de novo assembly of TCR hypervariable CDR3 regions from bulk tissue short-read RNA-seq data. We excluded public sequences (22) that are also found in healthy donors (23) and obtained over 43,000 complete productive β chain CDR3 sequences as the training data. TCRs encoded by the remaining sequences were hypothesized to be specific to the tumor microenvironment (18) and more likely to be cancer-associated (24–26). Non-cancer control TCRs were collected from blood β chain CDR3 sequences in a cohort of young healthy donors (table S2) (23). About half of the healthy donors carried human cytomegalovirus (HCMV) infection, which is commonly found in adults (27).
Biochemical features of the 20 amino acids (AAs) have been summarized from protein structure studies, referred to as the AA indices (28). We discovered that a subset of indices displayed different distributions in the caTCRs (fig. S1), which served as their predictive markers. Deep convolutional neural networks (CNNs) can be powerful tools to study functional genomics and protein structures (29–31) and can identify hidden patterns to solve difficult classification problems and achieve better performance than traditional methods (32). Therefore, we developed a Deep CNN Model for Cancer-Associated TCRs (DeepCAT) to learn the representative features (Fig. 1). In this model, each CDR3 sequence was first converted into a two-dimensional image with principal components analysis (PCA) encoding to integrate the AA indices and passed onto two consecutive one-dimensional convolutional layers each with 8 and 16 filters. Random dropouts at a rate of 40% were applied to the dense layer to prevent overfitting. The output layer generated a probability of cancer association. Because CDR3s with different lengths usually form distinct loop structures to interact with the antigens (33, 34), we built five models each for lengths 12 through 16. These lengths covered 83% of the total peripheral TCR repertoire (fig. S2, A and B). DeepCAT was trained with threefold cross-validation and achieved an average of 80% accuracy for de novo prediction of caTCR using single βCDR3 sequences, as measured by the area under the receiver operating characteristic (ROC) curves (AUC) (fig. S2, C and D).
Fig. 1. Schematic illustration of the DeepCAT workflow.
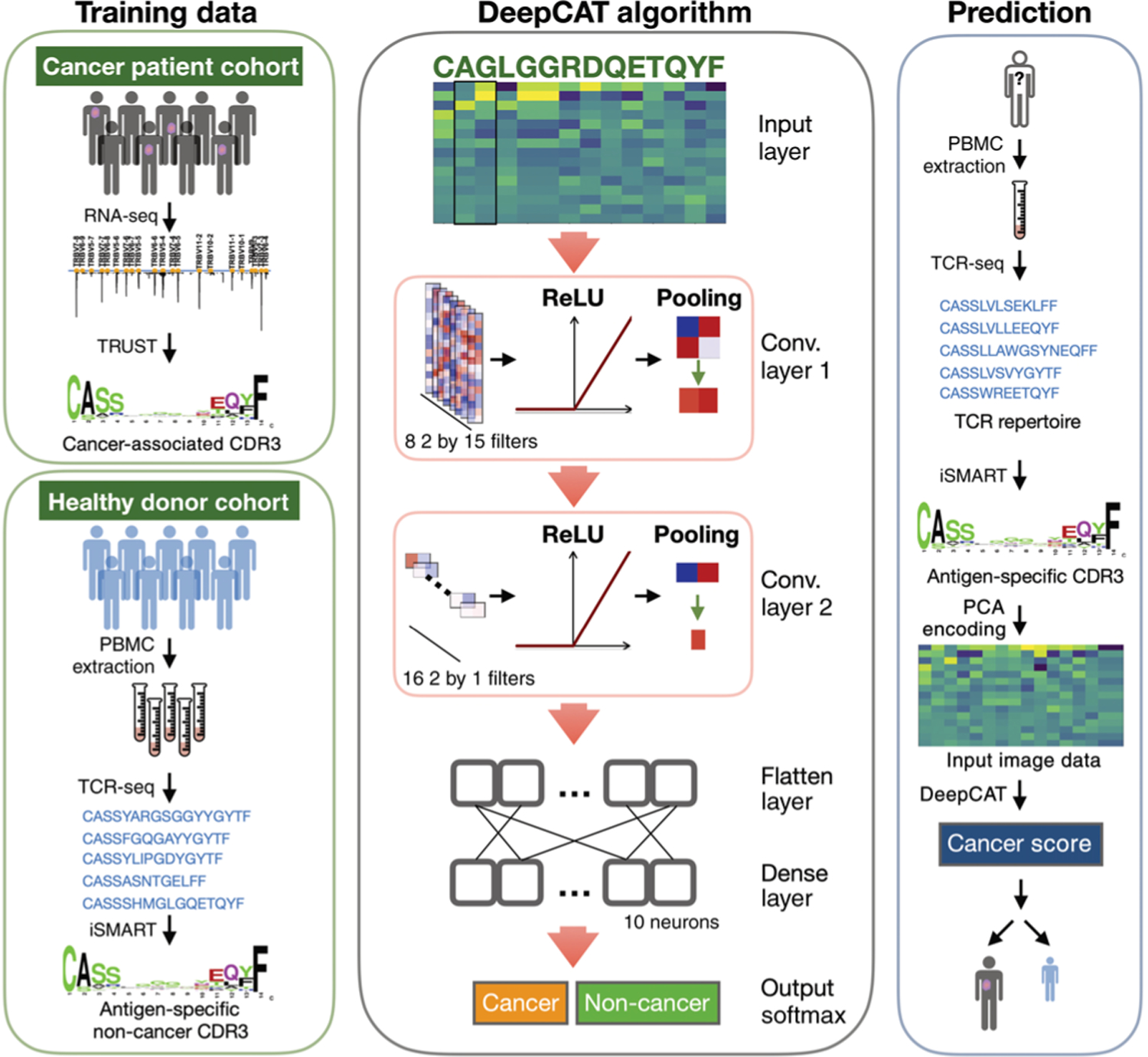
The method is divided into three parts: training data generation, CNN algorithm development, and cancer score estimation for independent TCR-seq samples. caTCRs were collected from TCGA tumor RNA-seq samples with TRUST, excluding the public CDR3s also found in healthy donors. Non-cancer TCRs were collected from peripheral blood TCR-seq samples from a cohort of young healthy donors, by selecting the most frequent clonotypes (top 10,000 TCRs ranked by their clonal abundance in a repertoire) and performing antigen-specific clustering. Both datasets were converted to two-dimensional images with PCA encoding to train the deep learning model, which consists of two convolutional layers followed by a dense layer and emits a probability of cancer relatedness as output. The trained classifiers are readily applicable to new blood TCR-seq samples, which have been clustered and PCA-encoded, to obtain a final cancer score. TRUST performs de novo assembly of the TCR hypervariable CDR3 regions using unselected bulk tissue RNA-seq data. iSMART performs pairwise alignment on the CDR3 AA sequences and identifies highly similar sequence clusters to represent their antigen specificity.
Performance evaluation of DeepCAT
It is conceivable that specific cancer types may be associated with certain human leukocyte antigen (HLA) alleles and that DeepCAT was simply learning the HLA-related sequence signatures. To rule this out, we tested on the three most prevalent alleles: HLA-A*02:01 (A2; allele frequency = 42%), HLA-B*07:02 (B7; 20%), and HLA-C* 07:01 (C7; 25%). HLA allele frequency estimations for TCGA samples were obtained from a previous study (35). For each allele, we split the training data into TCRs from patients carrying at least one copy of that allele or TCRs from the noncarriers. We trained CNN models within the DeepCAT framework using only TCRs from the noncarriers and used the same models to predict the probability of cancer association for TCRs derived from the allele carriers. If the performance of DeepCAT was attributable to HLA sharing, then caTCRs from allele carriers would be expected to have reduced prediction accuracy, because their features were not covered in the training data. In contrast, we observed the same accuracies for all tested alleles (fig. S3), indicating that the DeepCAT prediction of caTCRs relied on features other than shared HLA alleles.
To test whether DeepCAT correctly predicted caTCRs specific to unseen tumor antigens, we mixed cancer-specific TCRs with non-cancer TCRs for validation (Fig. 2A). Two recent datasets of major histocompatibility complex class I (MHC-I) multimer-sorted T cells with known specificity were used for this analysis. The first one consisted of tetramer-sorted TCRs (36) specific to 268 epitopes derived from neoantigens, cancer-associated antigens, and oncogenic viruses (Epstein-Barr, hepatitis B and C, and herpes simplex viruses) (37–39). The second dataset (10x Genomics) consisted of flow-sorted single T cells from peripheral blood mononuclear cell (PBMC) samples, where barcoded dextramers of 44 cancer or non-cancer epitopes were used to sort the T cells. Because most of the epitopes in the first dataset (36) were cancer-associated, we used TCRs collected from independent healthy donors as controls. For the 10x Genomics dataset, TCRs specific to a nononcogenic virus (influenza A) were used as controls. DeepCAT was blindly applied to predict the probability of cancer association for each TCR. DeepCAT was able to distinguish caCDR3s of different antigen categories when compared to control sequences (Fig. 2B) or influenza-specific TCRs (Fig. 2C). Most AUC values were between 0.5 and 0.8. This result was expected, because the training accuracy was around AUC = 0.8. For multiple comparisons, we observed higher prediction accuracy for longer CDR3s, which may be related to the observation that longer CDR3s are enriched in patients with cancer and likely to be cancer-associated (18). To examine potential data leakage, we checked both datasets for overlapping sequences in the training data (103,553 TCRs, including both tumor and control). In total, we found 0 and 112 CDR3s from the Zhang et al. (36) and 10x datasets, respectively. Because we used cross-validation to train the model, it was unlikely that such a small proportion of sequences (0.11%) would bias the prediction.
Fig. 2. Independent validation of DeepCAT predictions using cancer-specific TCRs.
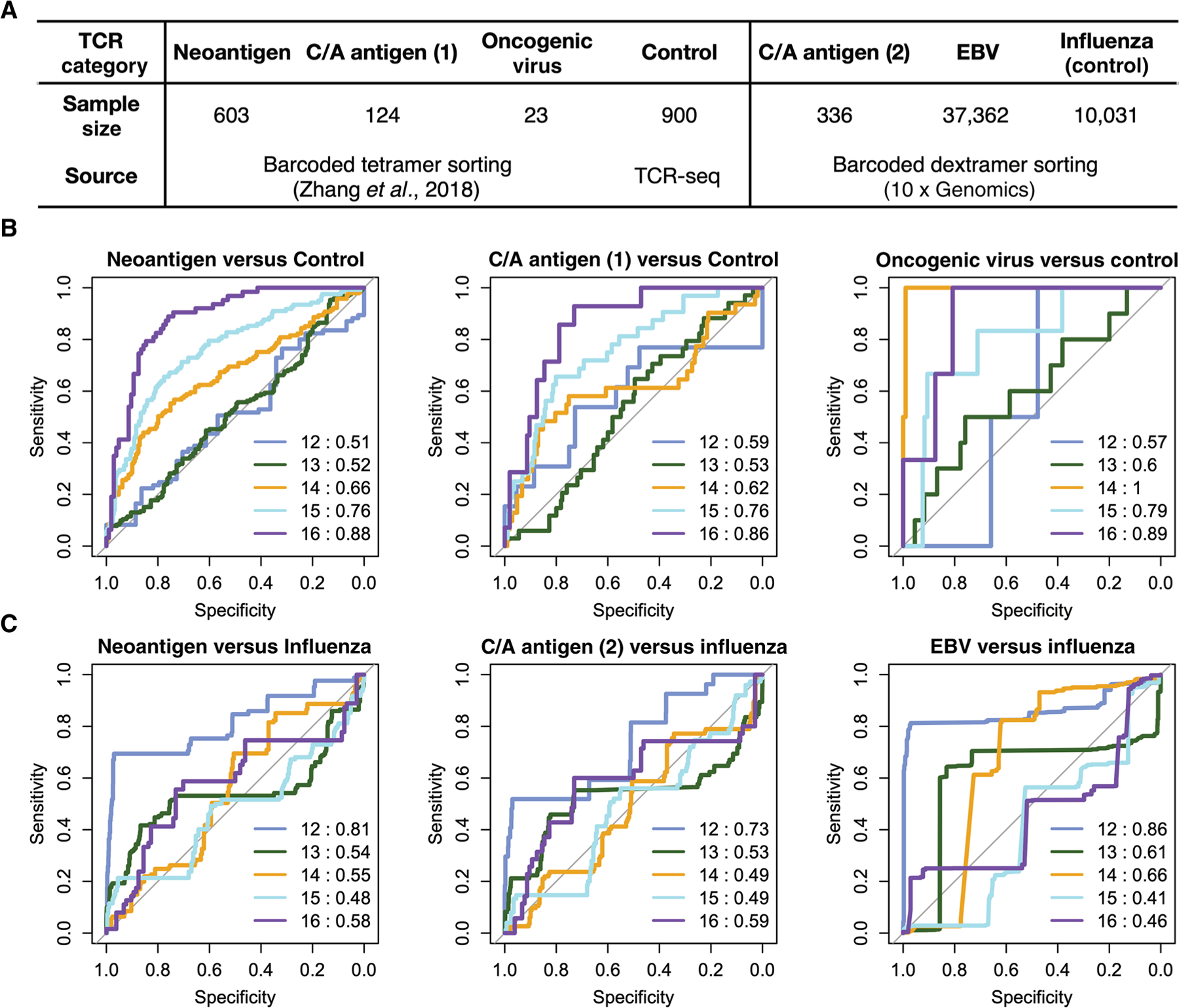
(A) Summary of the sample size and data generation methods (36) for each TCR category of the two validation experiments (36). (B) ROC curves showing the performance of DeepCAT to distinguish each category of cancer TCRs from the control ones, with control TCRs obtained from healthy donors’ TCR-seq data (23). (C) ROC curves of DeepCAT performance to distinguish each category of TCRs from the control, where the control sequences were influenza-specific TCRs obtained from the second experiment. C/A stands for cancer-associated antigens, which come from immunogenic epitopes from common autoantigens, including MART-1, NY-ESO-1, MAGE-A1, and PMEL (gp100). The title at the top of each panel indicates the antigen specificities of the two groups of TCRs being compared, with the antigen categories matched to the first row in (A). Line colors indicate CDR3 length, with the corresponding AUC values labeled in the legends. Different CNN models were applied to CDR3 sequences with different lengths ranging from 12 to 16. For all the PBMC cohorts, no prior treatment was reported in the original literature.
Definition of cancer score as a TCR repertoire index
From the above results, we made three observations: (i) DeepCAT prediction was independent of HLA alleles; (ii) DeepCAT was able to predict TCRs specific to cancer (neo)antigens not present in the training data (Fig. 2, B and C); and (iii) although it was trained on TILs and PBMC TCRs, DeepCAT could differentiate cancer-specific TCRs sorted from blood (Fig. 2C). Because the peripheral blood repertoire of patients with cancer contains a sizeable fraction of tumor-reactive T cells (40), we postulated that DeepCAT could be used for noninvasive cancer detection. We averaged the DeepCAT predictions for TCRs in a repertoire and introduced an additional index, cancer score, to measure the content of caTCRs by averaging the output cancer-association probabilities for each input TCR. The use of mean instead of median was based on the distribution of caTCR probabilities (fig. S4), where the outliers were expected to be real caTCRs instead of noise in the repertoire. Cancer score is a summary statistic of an immune repertoire, which is comparable to species diversity measurements that have also been used to analyze this type of data in clinical studies (41–44). We investigated nine quantities, including cancer score and eight diversity indices, and observed that cancer score was the only measurement not affected by sequencing depth, a known confounding variable in TCR-sequencing (TCR-seq) data analysis (fig. S5, A and B) (45). This was a desired quality, because it allowed direct comparisons of cancer scores across different TCR-seq sample cohorts, an analysis not appropriate for the other indices. Therefore, we decided to explore the feasibility of using our cancer score as a diagnostic criterion.
Evaluation of cancer score as a cancer predictor
Although the prediction power for individual caTCRs was not uniformly high, aggregating multiple TCRs in a repertoire by cancer score might achieve better performance. We therefore estimated scores of blood TCR-seq samples from 13 clinical studies, including 8 cohorts of patients with early- or late-stage cancer and 5 cohorts of healthy or virus-infected individuals (table S2). To be conservative, we avoided cancer types associated with oncogenic viruses because these samples might be confounded by the higher oncogenic virus signals in DeepCAT. All the TCR-seq data were generated using the immunoSEQ platform from Adaptive Biotechnologies. We used the Emerson et al. cohort (n = 176) with age-matched (35 to 70 years) blood donors as controls. Within this range, we observed no association between age and cancer score (fig. S5C), indicating that despite the age differences between cohorts, it was legitimate to use the donor samples as control. We observed higher scores in all the cancer cohorts with PBMC samples collected before treatments (Fig. 3A). We also included five independent TIL cohorts as validation and observed higher scores than in the PBMC samples, consistent with the enrichment of cancer-associated T cells in the tumor microenvironment. In contrast, except for the HCMV-infected individuals, who showed a slight increase, all non-cancer cohorts had equal or lower values, as measured by ratio of means. As a predictive biomarker, cancer score reached near-perfect accuracy (AUC ≥ 0.99) for breast, pancreatic, ovarian, colorectal cancers, and melanoma (Fig. 3B and fig. S6). On the other hand, the AUCs for glioblastoma, bladder, and lung cancer cohorts were low, with AUCs between 0.71 and 0.83 (fig. S7A). This observation was unexpected, at least for the patients with lung cancer given the high AUC (>0.99) for the lung TIL samples (Fig. 3B). We noticed that all three cohorts came from treatment-refractory patients selected for immunotherapies (46), who have undergone multiple pretreatments (47, 48), including neoadjuvant or postsurgical chemo-/radiotherapies (fig. S7B). It is possible that these cytotoxic therapies may deplete proliferating lymphocytes in the immune system (49), lower the content of effector T cells in the blood repertoire, and alter the estimation. In contrast, the blood samples collected without prior treatment, including the breast, pancreatic, and ovarian cancer cohorts, consistently yielded higher cancer scores.
Fig. 3. Cancer score is a robust predictor for multiple cancer types.
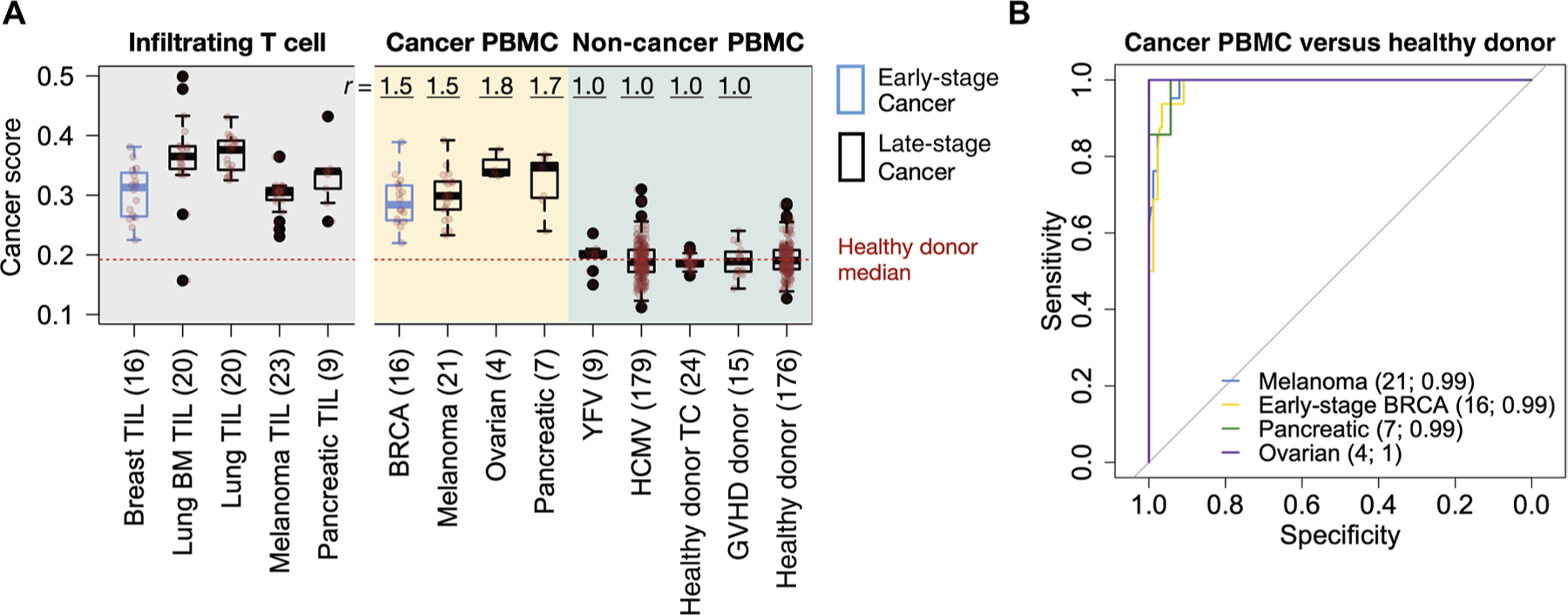
(A) Cancer score distributions across diverse disease cohorts displayed as boxplots, with original data overlaid as translucent red points. Numbers in parentheses in the x-axis label are sample sizes for each cohort. Healthy donor TC refers to healthy subjects from a time-course analysis (50); graft versus host disease (GVHD) donor denotes the healthy donors in a bone marrow transplantation study (88); YFV is the YFV vaccination cohort (87). Tumor-infiltrating T cell (TIL) cohorts (14, 52, 91, 93) were also included on the left-hand side of the panel as validation. Lung BM stands for lung cancer brain metastasis (91). To visualize the magnitude of cancer score signals, we displayed the ratio of the mean (r), defined as the mean score of the given cancer cohort, to the mean of the donor cohort (23). BRCA is the abbreviation for breast cancer. (B) ROC curves measuring the performance of cancer scores as a single predictor for cancer status, with PBMC repertoires of 176 healthy donors being used as control. Sample size and AUC for each cohort were labeled in parentheses in the figure. Detailed information for each sample cohort in this figure is available in table S2.
Because the adaptive immune repertoire is a dynamic system, we evaluated how the random fluctuations affected cancer scores using PBMC samples from healthy donors over 1 year (50). In the three individuals examined, we observed small longitudinal changes of scores, with SDs <0.015 for all individuals (fig. S8, A and B). The mean score for healthy donors was 0.193 (ranging from 0.127 to 0.286), and that for patients with treatment-naïve cancer was 0.302 (ranging from 0.220 to 0.392) (Fig. 3A), which is more than 3 SDs higher than healthy donors. These results suggested that a healthy individual might not have a cancer score as high as that of patients with cancer owing to the random noise in the immune repertoire.
We also noticed that the distributions of caTCR probabilities from the five length models in DeepCAT are not the same (fig. S9A). Further, patients with cancer and healthy donors have different CDR3 length distributions (fig. S9B). To evaluate whether this difference affected the performance of cancer score, we implemented an in silico experiment to simulate 100 “patients with cancer” and 100 “healthy donors.” Assuming that the TCRs of the patients and donors were sampled from the same pool of TCR sequences, we tested whether it was possible to observe higher scores in the patients simply due to CDR3 length differences. On the contrary, the simulated cancer scores were not higher for the patients with cancer (fig. S9C), suggesting that our observation of higher scores in the patient cohorts (Fig. 3A) was not an artifact of the difference in the CDR3 length distributions.
Technical dissection of DeepCAT to evaluate the prediction power of cancer score
Having established cancer score as a potential cancer predictor, we next conducted an investigation on the technical factors that may contribute to its prediction power. DeepCAT consisted of two critical components: deep CNN model and feature construction with PCA encoding. To understand how CNN contributes to the performance of cancer score, we replaced it with a non–deep learning approach, Adaptive Boosting (AdaBoost), which is one of the best-performing traditional methods that combine weak classifiers into a strong one (51). To study the importance of feature construction, we explored two alternative input types: (i) raw data of 544-AA biochemical features (raw) and (ii) PCA-encoded features (PCA). We examined the prediction accuracy of all four combinations of machine learning and input methods: (i) CNN + PCA (DeepCAT), (ii) AdaBoost + PCA, (iii) CNN + raw, and (iv) AdaBoost + raw.
The treatment-naïve cancer PBMC cohorts and healthy donors (n = 176) (Fig. 3A) were analyzed to estimate the AUCs for each combination. Prediction accuracies of CNN + PCA/raw were higher than those of AdaBoost + PCA/raw (fig. S10, A to D), indicating that the CNN model’s ability to build nonlinear mapping with higher complexity is superior to the traditional approach. PCA encoding only slightly (1%) increased the performance for AdaBoost, but increased the AUC by 6% for the CNN (fig. S10E). This result suggested that proper feature construction of input data may further improve the performance of CNN models. The outcomes of this in silico experiment are potentially informative for future development of deep neural networks to investigate genomics data, because encoding protein and DNA sequences is nontrivial and usually necessary for this type of analysis.
Evaluation of cancer score as an early detection criterion
The breast cancer cohort in the above analyses consisted of 16 stage I or II patients (52), indicating that even at early stages, the cancer-associated alterations of the peripheral TCR repertoire were sufficient for disease diagnosis. To validate this observation, we collected blood samples from four independent cohorts of patients with treatment-naïve early-stage cancer, including kidney (renal cell carcinoma), ovarian, pancreatic, and lung cancers (table S3). Currently, there is no effective early detection method for kidney, ovarian, or pancreatic cancer, and most of the early-stage cases were incidentally diagnosed during examinations over other chief complaints. TCR repertoire sequencing was performed on these samples using the same commercial pipeline as above. To exclude the potential bias of using only one control cohort, we combined another four non-cancer patient cohorts as healthy controls (n = 56). Higher scores were observed for all the cancer cohorts compared to the control samples, with a trend toward higher scores for patients with more advanced ovarian tumors (P = 0.002, Mann-Kendall trend test) (Fig. 4A). This observation is potentially due to the increased antigen release during tumor progression. In contrast to the treatment-refractory lung cancer samples (fig. S7A), the AUC for the patients with treatment-naïve lung cancer reached 0.95, thus supporting our hypothesis that cytotoxic pretreatments can lower the cancer score. Notably, we also observed a significant trend (P = 0.001, Mann-Kendall test) of cancer scores to rise with increasing severity of pancreatic tumors, from benign cysts to early- and late-stage pancreatic cancers. This result implied that the cancer score might be used to differentiate benign from malignant lesions, although a larger cohort is needed to confirm this finding. Using the score as a predictor, we observed high AUCs for all cancer types tested (Fig. 4B). At 98% specificity (SP), it reached 73% sensitivity (SN) for all early-stage samples combined (Fig. 4C), superior to the current blood-based biomarkers, including CA-153 (SN = 19%, SP = 95%) (53), CA-125 (SN = 46%, SP = 98%) (54), and PSA (SN = 21%, SP = 94%) (55). It also reached the same, if not higher, accuracy as recent methods using cell-free DNA (cfDNA) methylation (7) or circulating tumor DNA (ctDNA) mutations (fig. S11, A and B) (6). To evaluate the utility of the cancer score in the general population, we also estimated the positive predictive value (PPV) and negative predictive value (NPV) at various cutoffs with a complete cancer prevalence of 3.66% (56), using in silico simulations. We observed PPV 0.447 ± 0.076 (mean ± SD) and NPV 0.986 ± 0.005 at cutoff = 0.27 (fig. S12). These numbers are considered satisfactory for early-stage cancer detection according to recent reviews (57, 58).
Fig. 4. Cancer score predicts early-stage cancers with high accuracy.
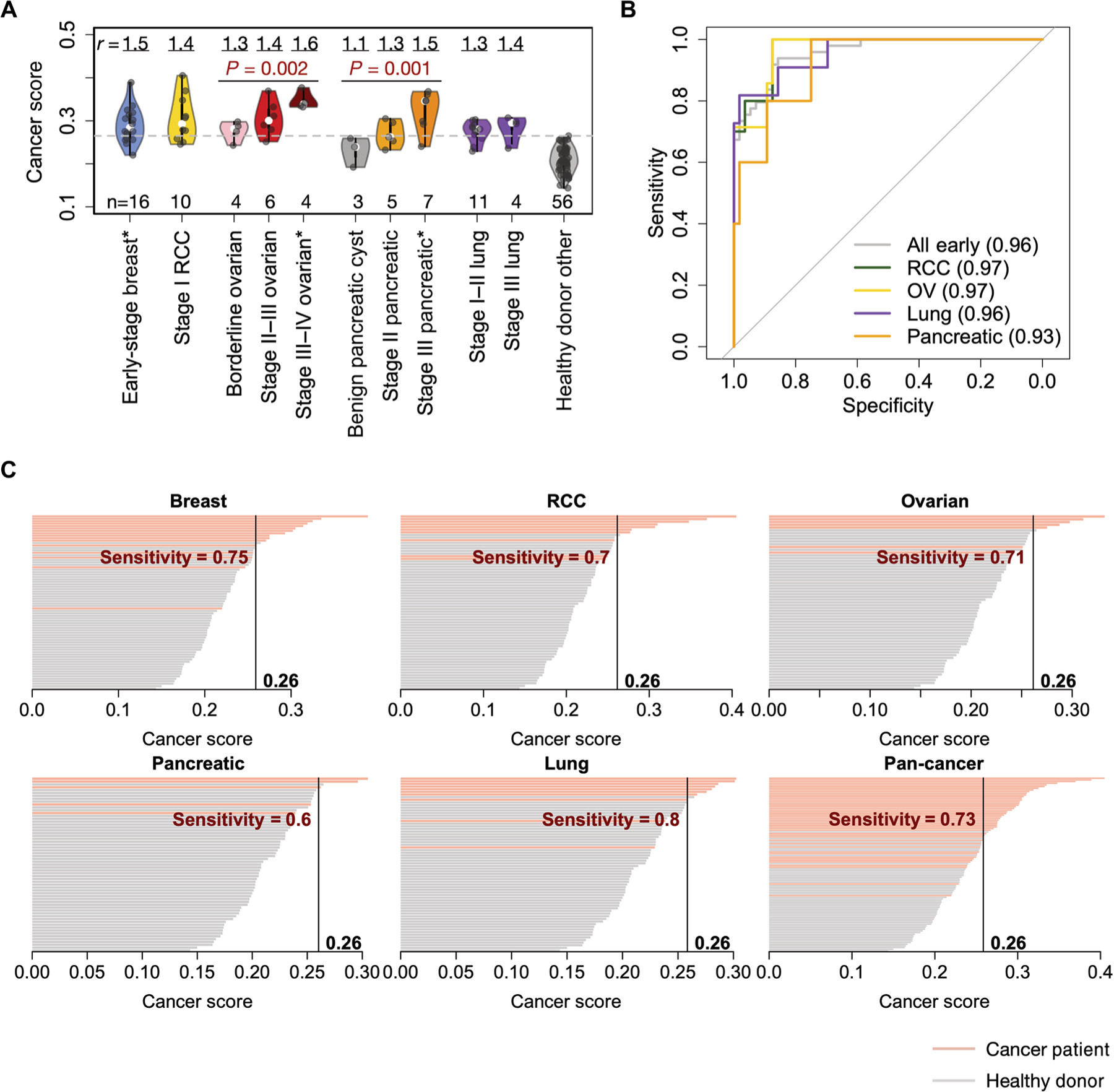
(A) Violin plots of cancer scores from five disease types with one or more stages available. TCR-seq data (generated by the Adaptive Biotechnologies platform) for early-stage RCC, early- to mid-stage ovarian, benign or early-stage pancreatic, and early-stage lung cancer (or cyst) samples were obtained in this study. All blood samples were collected before treatment and were sequenced with gDNA. Statistical significance of the increasing trend of cancer scores for ovarian and pancreatic cancer samples was evaluated with Mann-Kendall trend test. Mean ratio r value displayed on the top of each cohort is defined the same way as in Fig. 3A, except that the denominator is the mean score of the combined additional non-cancer cohort (healthy donor other). Asterisks (*) after the x-axis labels indicate reused cohorts from the analysis in Fig. 3A. (B) ROC curves with cancer score as a single predictor for each of the early-stage sample cohorts (except for breast cancer) and for the combined set of five early-stage cancer types (breast, ovarian, kidney, pancreatic, and lung). (C) Waterfall plots showing the cancer scores of patients with cancer and healthy individuals for five early-stage diseases and their combination, with cutoff points at 98% specificity. Sample sizes used to calculate each curve are as follows: breast (n = 16), kidney (n = 10), ovarian (n = 7, borderline + stage II), lung (n = 10), pancreatic (n = 5), and all early (n = 48). A total of 56 non-cancer samples from four additional cohorts (50, 87, 88, 95) were used as controls. Detailed information for each sample cohort in this figure is available in table S2.
Independent validation of cancer score using mRNA TCR-seq cohorts
All of the above analyses were based on TCR-seq datasets produced by Adaptive Biotechnologies, which uses genomic DNA (gDNA) to profile the CDR3 regions. To rule out the possible yet unknown biases induced by a single platform, we conducted an independent validation with TCR-seq samples generated on a different platform (iRepertoire) using mRNA (59) instead of gDNA. We collected three PBMC sample cohorts, including 17 patients with metastatic renal cell carcinoma, 11 patients with glioma (60), and 225 healthy donors (control). Similar to the DNA-based method, no association of RNA-based cancer score with library size or patient age was observed in the control cohort (fig. S13). We reproduced the observation that cancer scores are generally higher for patients with cancer compared to controls (Fig. 5A), although the baseline of healthy donors was different from that seen with the DNA-based method. For renal cell carcinoma and glioma cohorts, we observed AUCs of 0.84 and 0.87, respectively (Fig. 5, B and C). The reduced prediction power was expected because a subset of these patients had received multiple regimes of cytotoxic therapies before blood collection. Nonetheless, we cannot rule out the possibility that the different T cell clonotype quantification by the RNA-based method might alter the results. Together, these results independently validated the ability of the cancer score to differentiate patients with cancer from healthy individuals. The cancer score estimations of all the samples, including both the gDNA and mRNA cohorts, are available as a supplementary dataset (data file S1).
Fig. 5. Independent validation of cancer score performance using RNA-based TCR-seq cohorts.
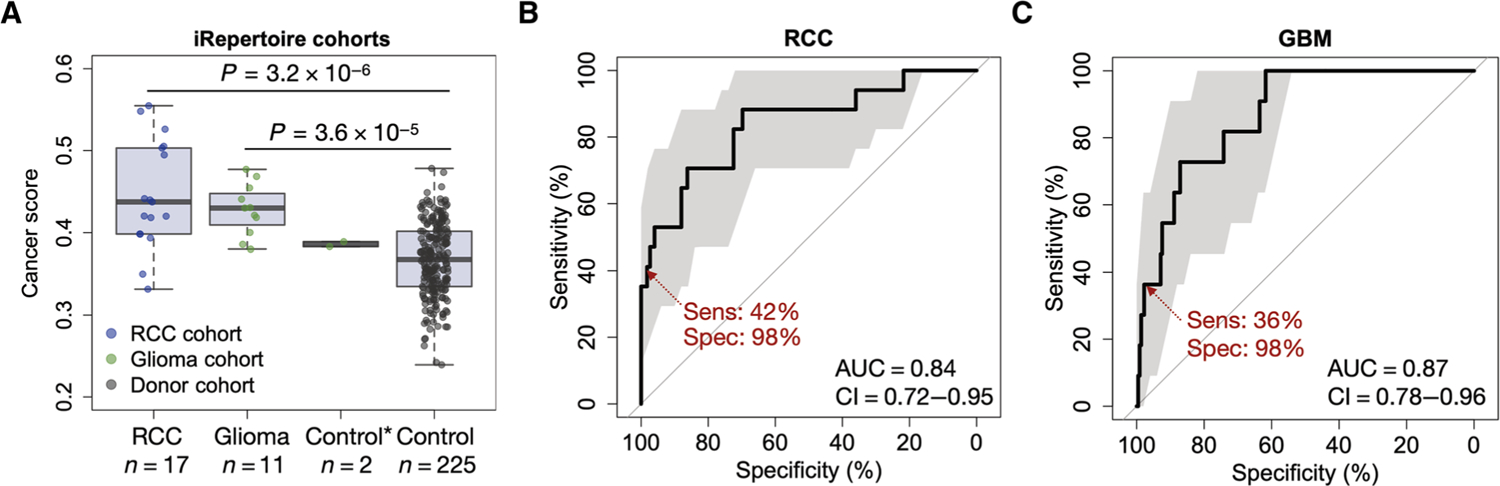
(A) Cancer score distributions for cohorts of patients with renal cell carcinoma (RCC) and glioma compared to healthy individuals from the iRepertoire TCR-seq cohort. Two-sided Wilcoxon rank-sum test was performed to evaluate the statistical significance of the difference between cancer and normal cohorts. *The control sample in the glioma cohort consisted of two replicates of one healthy donor. (B) ROC curve of the cancer score distinguishing patients with RCC from the healthy donors. (C) ROC curve of the cancer score distinguishing patients with glioma from the healthy donors; 95% confidence intervals (CIs) were estimated from stratified bootstraps and marked in translucent gray color.
Influence of non-cancer chronic inflammatory conditions to cancer score
Chronic inflammation is common in the general population, which includes chronic viral infection, autoimmune disorders, cancer, etc. To investigate how non-cancer chronic inflammatory conditions affect the cancer score, we further analyzed three cohorts with such conditions, including HCMV infection (23), rheumatoid arthritis (RA) (61), and multiple sclerosis (MS) (62). All three cohorts contained both patient samples and healthy controls. However, unlike the HCMV cohort, the other two could not be compared to the other samples in our previous analyses, because the RA cohort (61) used flow-sorted CD8+ T cells and the MS cohort (62) was profiled using 5′ RACE [rapid amplification of complementary DNA (cDNA) ends] with mRNA. Different TCR data generation procedures may produce systematic differences in cancer score estimation. For all three cohorts, cancer scores were increased in patients with inflammatory conditions, but this increase (ratio of means) did not reach the magnitude seen in the patients with cancer (fig. S14 and Fig. 3B). In conclusion, preexisting chronic inflammatory conditions slightly increase cancer scores, which may result in a reduction in diagnostic specificity when the cancer score is applied to the individuals with such conditions.
DISCUSSION
In this work, we designed a computational framework to gauge the content of caTCRs in a blood repertoire. We derived the training data from TIL sequences of multiple cancers and from healthy donors with or without HCMV infection to develop a deep learning method to predict the caTCRs, and we validated the predictions using MHC-I multimer-sorted T cells with known specificity. Because of the hypervariability of the CDR3 regions, it was unexpected to observe this predictability. One possible explanation is that the qualities of the tumor microenvironment impose selective pressure on TCRs with certain biochemical signatures. For example, it is known that increased acidosis of cancer cells (63) may alter the conformation of histidine (His) in the protein structure (64). Consistent with this idea, we observed 1.7-fold enrichment (P < 2.2 × 10−16, Fisher’s exact test) for His usage in the TIL sequences compared to TCRs obtained from healthy donors. It is also possible that tumor tissue–resident T cells are specific for distinct, shared antigens, with biased usage of certain AAs in the TCRs (65). These T cells can be detected when they reenter the circulation (66). Furthermore, recent studies have suggested that embedding microbial homology alters the efficacy of T cell priming to selected neoantigen targets, which might influence the usage of TCRs in the tumor microenvironment as well (67–70). Thus, we speculate that the reproducible cancer signals in the blood TCR repertoire may result from multiple conserved mechanisms of tumor-immune interactions that lead to biased selection of the biochemical features of the infiltrating T cells.
Given the ability of the immune system to clear transformed cells under healthy conditions, there is a concern as to whether DeepCAT picks up these signals and reports cancer in a nonspecific way. We observed that cancer scores of all healthy donor cohorts are generally lower than those of the patients with cancer. At the time of the blood draw, premalignant cells might exist in the donors (71), but the numbers of these cells are expected to be low; thus, no “cancer” would be diagnosed. The TCR repertoire would recognize and eliminate the premalignant cells, but this response would be transient and would not persistently increase the cancer scores. A related question follows: What is the earliest time point for DeepCAT to diagnose cancer? The T cell repertoire is under constant turnover driven by antigen presentation and clearance. Effective elimination of premalignant cells would lead to effector to memory T cell differentiation and tissue homing, a process where most effector T cells will be removed from the circulation (72). However, when the immune system and cancer reach the “equilibrium” or “escape” states (15), cancer-associated T cells would be constantly generated and would accumulate after they become exhausted instead of apoptotic (73). We speculate that this is the stage when cancer is potentially diagnosable via the TCR repertoire. Consistent with this, a recent study using single-cell RNA-seq to investigate patients with early-stage colorectal cancer confirmed the presence of cancer-associated T cells in the blood (74), which expressed putative exhaustion markers programmed death 1 and lymphocyte activation gene 3 (75). This observation may provide the basis for cancer score elevation in patients with early-stage malignancies.
As an attempt to make pan-cancer diagnosis through the peripheral TCR repertoire, our study has several limitations. First, it cannot determine the tissue of origin, which is a common limitation of detection methods based on liquid biopsies (6). Because different cancers present tissue-specific antigens (16), it is anticipated that future generations of caTCR datasets and the development of machine learning methods will likely overcome this problem. Alternatively, it might be possible to use TCR clustering (76) to predict cancer localization for new patients using the TCR-seq samples of existing patients with known cancer types, provided that sufficient reference samples covering diverse diseases are available. Second, because of the clinical challenges in diagnosing asymptomatic early-stage cancers, all the validation cohorts were relatively small. Future collection of more samples will be needed to benchmark the prediction accuracy of the cancer score. Third, we only used the TCR β chain to perform model training, caTCR prediction, and cancer score estimation, because most datasets do not have paired αβ chain information. Combined use of both chains for model training and prediction is expected to further increase the accuracy. Last, our analysis showed that chronic inflammatory conditions might affect cancer score estimations. This caveat, however, might be alleviated by exhaustive examination of each patient’s medical history to exclude chronic viral infections and common autoimmune disorders. Last, although our method yielded high prediction power for multiple cancer types, its performance in the general population remains untested, and the full clinical applications can only be explored using large prospective clinical cohorts.
Our method offers several advances over traditional approaches. We demonstrated the feasibility of antigen-independent de novo predictions for cancer-associated T cells and repeatedly validated the methodology using flow-sorted cancer-specific TCR data. This finding may help to prioritize therapeutically relevant TCR receptors in future cancer immunotherapies. The cancer score introduced in this work can gauge the caTCRs in an immune repertoire and distinguish patients with cancer from individuals without cancer. Notably, although using averaged caTCR probabilities to calculate the cancer score reached satisfactory performance, this approach remains a first attempt that could be refined to increase prediction power. Further, we demonstrated that cancer score achieves high prediction accuracy even for early-stage malignancies, where the performances of other methods are typically worse (6, 7). Besides higher accuracy, the cancer score does not rely on preselected panels of genetic or epigenetic features, making it more generalizable than cfDNA-based approaches. In addition, this method only requires T cells from a small amount of peripheral blood, with a data generation cost of less than $200, which is lower than most screening tests, making it a competitive diagnostic choice. Notably, the sensitivities at cutoff 0.26 (98% specificity) for different early-stage cancer types were highly consistent, suggesting the feasibility of using a unified cancer score threshold for disease detection. The cancer score is not intended to replace the current diagnostic methods at this time. Rather, future efforts should be made to explore whether the combined use of the cancer score with existing screening modalities, such as breast mammogram, lung CT scan, gastrointestinal endoscopy, or pelvic ultrasonography, can improve diagnostic accuracy in patients.
MATERIALS AND METHODS
Study design
The goal of this study was to develop a cancer diagnostic score, based on the blood TCR repertoire data. This goal was achieved through the development and validation of a machine learning method, which predicted the caTCRs in a TCR repertoire. Its output was used to predict the cancer status of any given patient. Four categories of datasets were analyzed and/or generated in this study: (i) training and testing data for the machine learning model, (ii) additional cancer TCR or non-caTCR for independent model validation, (iii) TCR-seq data from cancer or non-cancer cohorts for performance evaluation, and (iv) additional TCR-seq data from cancer or non-cancer cohorts for independent validation. Most of the study cohorts in this work were public datasets, except for the fourth category, where we produced TCR-seq data using early-stage cancer samples collected in this study. The details of the machine learning method and different data categories are described below.
Public data accession
A total of 9702 tumor RNA-seq samples were obtained from TCGA. Level 2 BAM files aligned to hg19 human reference genome by MapSplice for tumor gene expression were downloaded from the Genomic Data Commons legacy archive (https://portal.gdc.cancer.gov/legacy-archive/search/f), and the TCR CDR3 sequences were extracted as previously described (18). TCR repertoire sequencing data of both cancer and non-cancer cohorts (table S2) were downloaded from Adaptive Biotechnologies immuneACCESS online database (https://clients.adaptivebiotech.com/immuneaccess) or from Gene Expression Omnibus. Dextramer-sorted antigen-specific single-cell sequencing data of 44 epitopes were downloaded from the 10x Genomics website (https://support.10xgenomics.com/single-cell-vdj/datasets).
DeepCAT workflow
Training data generation
We applied TRUST to call CDR3s from the TCGA RNA-seq samples. TRUST is a de novo assembly algorithm to extract fragmented or complete immune receptor hypervariable CDR3 regions from bulk tissue RNA-seq data (18). We have previously demonstrated that TRUST has high sensitivity to call TCRs from shallow coverage RNA-seq samples (20), with high specificity when compared to true-positive TCR-seq data (77). Because fragmented CDR3s are not appropriate clonal markers, we only used complete sequences beginning with the last cysteine (C) in the variable gene and ending with the phenylalanine (F) in the FGXG motif in the joining gene according to the IMGT (ImMunoGeneTics) nomenclature (78). The nonproductive sequences containing a stop codon between C and F were excluded. More than 80% of TCRs in the TIL repertoire are not tumor reactive (79). To remove public TCRs that are also found in individuals without cancer, we generated a reference dataset by pooling the top 20,000 most abundant CDR3s from each of the TCR-seq samples in a large cohort of healthy individuals (denoted as G) (23). If any CDR3 in the TRUST calls was found in G, it was removed. The resulting 43,702 unique β chain CDR3 sequences were expected to be nonpublic and cancer-associated (caTCRs) and were used to train and evaluate the CNN model. The healthy individual cohort here is the first batch (n = 666) in the Emerson et al. 2017 study (23).
We used the second batch of TCR repertoire data from 120 young healthy donors in the same study (23) to construct the non-cancer CDR3 control sequences. These two batches contained independent individuals, and thus, we do not expect sequences in the second batch to affect the downstream analysis using the first batch as controls. To balance the sample sizes of the two training classes, instead of using all the data, we randomly sampled 50 individuals from the second batch and selected the TCRs with clonal frequencies ≥4 times the minimum in the repertoire. This strategy was to ensure the selection of effector/memory T cell population and exclusion of most naïve T cells (80). Selected CDR3s were merged together (n = 170,286), and we clustered these sequences to select antigen-specific TCR groups using iSMART as previously described (81). This step identified a total of 59,851 antigen-specific non-cancer TCRs. Consistently throughout this work, we applied iSMART to the TCR repertoire sequencing data before pattern recognition or cancer score estimation (see below).
PCA encoding
The current AA index database documented 544 biochemical indices from previous literature (28), which could be used as surrogates of the functional and structural impact for AAs. We excluded 13 features for which some AAs do not have values. Z-transformation was applied to each of the remaining 531 indices to normalize them into the same scale. PCA was performed on the resulting dataset to obtain a 20-by-20 scoring matrix. The top 15 scores explained over 95% of total data variation. For each AA, we used the vectors PC1 to PC15 to represent its biochemical signatures. Notably, the PCA encoding layer was frozen and not updated during model training. Because the original 544 vectors were very large, PCA served as dimension reduction to limit the number of parameters in convolutional filters and to prevent overfitting.
Specification of the CNN
For CDR3s with length L, L = 12, 13, …, 16, the input layer consisted of images (as two-dimensional tensors) with dimensions 15 by L. The input tensor was then sent for a convolutional layer with eight 15-by-2 filters, where a rectified linear unit (ReLU) activation and a maximum pooling layer with width 2 and stride 1 were subsequently applied. With the length of filter equal to the width of the image, DeepCAT performed one-dimensional convolution along the CDR3 sequence. A second convolutional layer was applied on top, with 16 1-by-2 filters, followed by the same additional ReLU and pooling layers. No padding was allowed in either convolutional layer. A dense layer with 10 units was added on top of the convolutional layers, and the final output layer applied softmax functions to export probabilities related to class labels, which were cancer or non-cancer.
There are approaches to combine sequences with different lengths into one model. One way is zero padding, which is adding zeros to the end of the shorter sequences to enforce the same length. This approach is commonly used for one-hot encoding, where non-letter positions are naturally filled with zeros (82), but may not be suitable to PCA encoding, because the added zeros will alter the distribution of input data. Another solution is recurrent neural network (RNN), which is commonly used in language-processing tasks. However, RNN models, such as long short-term memory networks (83), are typically optimized to analyze long sequences of text or words, whereas CDR3 regions are very short. In addition, RNN models introduce dependencies to neurons within the same layer (84), which increases the complexity of the network and imposes unknown risk for overfitting. On the basis of these concerns, we chose to use a very simple CNN model with a small number of filters, which gave satisfactory results in our downstream analysis.
Model training and evaluation
Cancer (n = 30,000) and non-cancer (n ~ 60,000) CDR3s were label-encoded (1 for cancer and 0 for non-cancer). We performed 20 runs of cross-validation for model training and evaluation. For each run, we randomly selected two-thirds of both cancer and non-cancer CDR3s, split by different lengths, and trained each of the five models for 20,000 steps, at a learning rate of 0.001. Forty percent random dropout was applied to the dense layer to avoid overfitting. The output model was then evaluated using the remaining one-third of the data to test for accuracy and to estimate the AUCs for each model. The resulting AUC values for each run were very similar, and we performed an additional training run using randomly selected 20,000 cancer and 40,000 non-cancer samples as the final model for downstream analysis.
Estimation of cancer score with TCR repertoire data
In this work, all the TCR repertoire sequencing data were generated by the Adaptive Biotechnologies immunoSEQ platform. After downloading the raw data from immuneACCESS, we first removed the following types of low-quality calls from the CDR3 AA sequences: (i) sequence length was <10 or >24; (ii) sequence contained nonstandard characters (*, +, X); (iii) sequence was not starting from cysteine (C) or not ending with phenylalanine (F); and (iv) variable gene locus was not solved. After removal of low-quality calls, the remaining CDR3s were decreasingly ordered by clonotype frequencies, and the following columns were selected for clustering analysis: CDR3 AA, variable gene, and clonotype frequency. For each sample, we selected the top 10,000 sequences. If the data contained fewer than 10,000 CDR3s, then all sequences were selected. Here, we enforced the same number of top CDR3s, instead of selection based on frequencies to increase the comparability between different datasets. The cutoff of 10,000 sequences was set to include most of the high abundant clonotypes that are likely to be effector/memory cells, while excluding low-frequency naïve cells. Inclusion of naïve T cells would result in increased noise, because they might be inactivated tumor-specific T cells in healthy individuals.
We previously developed iSMART to detect antigen-specific T cell groups by clustering CDR3s based on their sequence similarity. Antigen specificity is based on research, indicating that T cells with similar CDR3 motifs are likely to recognize the same antigen (85). In brief, iSMART performs pairwise alignment on highly similar CDR3 sequences using the BLOSUM62 matrix and reports TCR clusters based on a preselected cutoff value (default, 7.5). Both CDR3 sequence and variable gene information were used in the method to ensure high specificity. iSMART achieved higher specificity than previous methods, benchmarked using TCR sequences specific to different antigens (81). In this work, we consistently applied the default parameters in iSMART to process all the TCR repertoire sequencing data (including individuals with and without cancer). In practice, we found that application of iSMART clustering before cancer probability estimation could improve the signal/noise ratio, which is because antigen-experienced TCRs in a blood repertoire may carry similar sequence motifs, whereas naïve TCRs are more random and cannot be clustered.
Trained DeepCAT classifiers were then applied to the processed TCR data to obtain probability estimations for each CDR3 sequence, without changing any parameter. The final cancer score was the mean value of all the probabilities for cancer association. It is possible that a TCR cluster contained several CDR3s with identical AA sequences. This is due to the codon degeneracy of DNA to protein, where different TCRs were selected to antagonize the same antigen. Therefore, we treated them as different observations. In theory, there are multiple ways to combine models with nonrandom predictions to improve the performance, a concept known as ensemble classifier. In our analysis, we aimed to make this combination as simple as possible, without introducing additional parameters to prevent overfitting. Therefore, we used the averaged probability as output. Other summary statistics, such as median, could also be used. In practice, we chose sample mean ad hoc for the analysis of independent TCR-seq samples and refrained from using other statistics post hoc to avoid overfitting. No cutoff on the DeepCAT output cancer probabilities was applied to assign TCRs to cancer or non-cancer groups, because an arbitrarily selected cutoff might also impose the unnecessary risk of overfitting.
Combinations of different feature encoding and machine learning methods
AdaBoost with raw features
The current AA index database (28) documented 544 biochemical indices from previous protein structure studies, which can be used as surrogates of the functional and structural impact for AAs. From the training data, we selected CDR3 sequences with length L between 12 and 16 AAs. The total feature set was the union of features from all AAs. We used nL to denote the number of CDR3s with length L for cancer CDR3s (derived from TCGA data) and kL for the number for non-cancer CDR3s from VDJdb.
We first subsampled 50% of all the sequences from both cancer and non-cancer TCRs and used the remaining half of the data for cross-validation. For each feature, we compared the 0.5nL cancer observations with the 0.5kL non-cancer ones. If the fold change (cancer over non-cancer) was smaller than 1.1, then this feature was removed. Let S denote the number of features left. In the above setting, we have a total of 0.5 × (nL + kL) CDR3 sequences (samples) and S features, with known sample labels (0.5nL with label 1 and 0.5kL with label −1). Let Y denote the sample label vector with length 0.5 × (nL + kL) and X denote the feature matrix with dimension 0.5 × (nL + kL) by S. On the basis of our analysis (fig. S1), we determined that the prediction power for individual features is weak. Therefore, we applied the Adaptive Boosting algorithm, an ensemble learning approach that is able to aggregate weak classifiers into a stronger one.
Model training was completed using the adaboost() function in R package JOUSBoost (86), with 50 rounds of boosting and a tree depth of 10. We selected parameters based on the criteria of minimizing the number of training cycles (rounds) and the complexity of classification tree (depth) while minimizing cross-validation errors. Cross-validation errors were calculated by applying the trained classifier for CDR3 length L (denoted as TL) to the independent validation data with known class labels. We ran the subsampling 10 times and selected the one with the best cross-validation value. The above procedure was repeated for L = 12, 13, 15, and 16, but not for L = 14, where fourfold cross-validation was applied because we found that this setting achieved smaller cross-validation error. Therefore, in total, five classifiers were trained and were denoted as T12–16. We applied TCRboost to define cancer score in the same way as DeepCAT.
AdaBoost with PCA encoding
Using the 544-by-20 AA index matrix, we performed PCA after removal of missing values (531 features remaining) and selected the top 15 PCs (PC1 to PC15, ranked by variance explained). We modified the AdaBoost method by using the 15 PCs to construct ensemble tree classifiers. The final cancer score was defined the same way as DeepCAT.
CNN with raw features
We implemented the same CNN network as DeepCAT, with the same number of convolutional layers and the same number of filters for each layer. We encoded each AA with 531 features that do not contain any missing values. Therefore, the input image had a dimension of 531 by L, where L is the length of the CDR3 sequence. The dimensions of the filters of the first CNN layer were 531 by 2. Five models were trained for CDR3s with L = 12, 13, …, 16, using the same training data as DeepCAT. Cancer score was calculated as the mean of model predictions for a given TCR repertoire.
CNN with PCA encoding
This is the original DeepCAT method described in the DeepCAT method for de novo prediction of caTCRs.
Training additional CNN models using TCRs from noncarriers of specific HLA alleles
To demonstrate that HLA alleles do not bias the prediction of DeepCAT, we trained additional models using the same framework of DeepCAT to test for three common HLA alleles: A2, B7, and C7. For each allele, we selected TCRs from individuals who do not carry the allele (noncarrier TCR) as tumor training data and those from individuals carrying the allele (carrier TCR) as tumor testing data. The numbers of sequences for the three alleles are as follows: A2 (training = 16,824; testing = 11,534), B7 (22,854; 5504), and C7 (21,150; 7208). To train the model, we input tumor training data and the 40,000 training control TCRs derived from healthy donors (described above) and conducted fivefold cross-validation and trained for 20,000 steps to optimize the parameters. ROC curves were generated using the predicted probabilities for each training TCR against the control ones. The CNN model was then applied to the evaluation dataset containing the tumor testing data and the additional 20,000 control TCR sequences. Prediction accuracy for the testing TCRs was measured using AUC values and visualized using ROC curves.
Independent validation of DeepCAT using tetramer- or dextramer-sorted TCR data
Both the Zhang et al. cohort (36) and the 10x Genomics single-cell cohort contained flow-sorted antigen-specific T cells derived from donor PBMC samples. The first cohort used barcoded tetramers carrying 268 epitopes from cancer, viral, or self-proteins. The second cohort applied dextramer sorting and obtained TCRs specific to 44 common epitopes from cancer or viral targets. We directly applied DeepCAT to predict the probability of cancer association for each CDR3 with length 12 to 16. The outcomes were compared to the control sequences. For the first cohort, to obtain true-negative (non-cancer control) sequences, we randomly selected 10 additional samples from the Emerson et al. 2017 (23) batch 2 cohort with 120 young healthy donors, excluding the 50 samples used to train DeepCAT, and performed the same clustering analysis to acquire the antigens pecific TCR groups. In total, iSMART grouped 1761 TCRs as extra non-cancer sequences. To avoid data leakage, we removed sequences that overlap with the training data and obtained 900 CDR3s as control. For the second cohort, we directly used dextramer-sorted TCR sequences specific to influenza virus in the 10x Genomics dataset as non-cancer control. DeepCAT was blindly applied to the TCR sequences in both validation experiments, with prediction accuracy measured by ROC curves and AUC values. Neoantigen-specific T cells from the Zhang et al. dataset (36) were compared with influenza-specific TCRs from the 10x Genomics dataset in Fig. 2C.
In silico simulation of cancer scores for evaluation of different CDR3 length distributions
CDR3 length distributions were estimated using the iSMART clustered sequences from four control cohorts (23, 50, 87, 88) and from cohorts of patients with cancer (14, 42, 46, 52, 89–94). We implemented in silico simulations to test how these differences affect cancer score estimation under the null hypothesis that the TCRs from individuals with and without cancer come from the same pool of TCR sequences; thus, there are no “baseline” differences of caTCR probabilities. Specifically, we simulated 100 “patients with cancer” and 100 “normal individuals.” Each individual had 500 TCRs, with lengths following the distributions of caTCR probabilities and with different CDR3 lengths for cancer or normal (fig. S9B). The numbers for each CDR3 length were sampled using the Multinomial sampler in R. For each individual, we sampled the number of length L CDR3s following the caTCR length distribution of the healthy donor (fig. S9A). For example, if the individual with cancer subject #1 has 72 sequences with length 16, we sampled 72 numbers from the caTCR probabilities estimated from length 16 CDR3s. We used the same caTCR probabilities for both individuals with cancer and healthy controls under the null hypothesis.
Postprocessing of DeepCAT predictions from TCR repertoire data and ROC analysis
Because each cohort of TCR-seq samples was designed differently, we applied a consensus approach to select the PBMC and TIL samples to maximize comparability. The DeWitt et al. cohort (87) for yellow fever virus (YFV) has day 1 and day 14 samples after vaccination of healthy volunteers, and we used day 14 samples because they were expected to carry the signatures of YFV infection. For cancer cohorts with longitudinal samplings, including Tumeh et al., Robert et al., and Snyder et al. (14, 42, 92), we used TIL or PBMC samples at the earliest time point possible, which was either before immunotherapy or after the first cycle of treatment if pretreatment samples were not available. The Emerson et al. 2017 cohort (23) consisted of 666 individuals, which were further split into HCMV+ and HCMV− donors. The latter were used as normal controls. We further excluded patients younger than 35 years, to match the ages of most of the cohorts of patients with cancer. Libraries with low (total template count ≤ 40,000) total templates were also excluded, because they were potentially low-quality samples with insufficient gDNA.
We calculated the median differences of cancer score values between each diseased cohort and control, evaluated statistical significance using Wilcoxon rank-sum test, and corrected P values using the Benjamini-Hochberg procedure, with cutoff false discovery rate = 0.05 for significance. To evaluate the prediction power of cancer scores, we selected each cohort with sample size n ≥ 3, compared to the age-matched healthy donors, and calculated area under the ROC curves.
Clinical sample collection and processing
In this work, early-stage cancer was consistently defined as stage I or II according to the TNM staging system. Informed consent was obtained from patients with renal clear cell carcinoma (RCC) or ovarian cancer receiving treatments at University of Texas (UT) Southwestern Medical Center, under protocol number STU 012011–190. PBMC samples of patients with pancreatic ductal adenocarcinoma or benign pancreatic cysts were collected by the Pancreatic Cancer Prevention Program at UT Southwestern Medical Center. All peripheral blood samples were collected before surgeries or scheduled neoadjuvant treatments and were stored in EDTA tubes. For RCC samples, plasma was removed after separation, with mononuclear cells isolated and cryopreserved in 90% phosphate-buffered saline and 10% dimethyl sulfoxide. For patients with ovarian cancer, whole-blood samples were stored at −80°C until gDNA isolation. For patients with pancreatic cancer, buffy coat samples were stored at −80°C until DNA isolation and library construction.
gDNA isolation and immune repertoire sequencing
gDNA was isolated from 50 µl of RCC mononuclear cells and 200 µl of ovarian cancer whole blood using the DNeasy Blood and Tissue Kit (catalog no. 69504, Qiagen) following the manufacturer’s guidelines. gDNA concentration was measured using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific). The purity of gDNA was determined by measuring the 260- to 280-nm absorbance ratio. Optimal purity was expected to be in the range of 1.7 to 2.0. The integrity of the gDNA samples was assessed for evidence of degradation using agarose gel electrophoresis. Appropriate quality gDNA was expected to migrate predominantly above 10 kb on agarose gels. All samples passed DNA purity and integrity quality controls. Twenty samples of gDNA were sent to Adaptive Biotechnologies for targeted TCR β chain repertoire sequencing using immunoSEQ at survey sequencing depth. Raw TCR reads were processed with immunoSEQ Analyzer for CDR3 assembly, variable/joining gene calling, and clonal frequency estimations.
Description of the additional healthy donor control cohorts
For the analyses in Fig. 4, to test whether the cancer score can reproducibly distinguish patients with cancer from healthy individuals, we collected TCR-seq data from PBMC samples in the public domain. Four cohorts were included in this analysis: DeWitt et al. 2015 (YFV vaccination, n = 9) (87), Chu et al. 2019 (time course first time point, n = 3, also used in fig. S8) (50), Kanakry et al. 2016 (donors for bone marrow transplantation, n = 15) (88), and DeWitt et al. 2018 (active tuberculosis-infected individuals, n = 29) (95). For the last cohort, there were 33 individuals sequenced, and 4 were removed because of low coverage (total template counts ≤ 40,000). PubMed IDs for all donor cohorts are available in table S2.
In silico simulations to estimate PPV and NPV
The Surveillance, Epidemiology, and End Results Program database (https://seer.cancer.gov/csr/1975_2017/) reported a 3.66% complete prevalence of all cancers combined. We sampled 300 cancer scores from a total of 522 scores combining five healthy donor cohorts (23, 24, 50, 87, 88, 95), including HCMV-infected individuals. We then replaced 3.66% of the population with scores sampled from the early-stage cancer samples. The mixed vector contained cancer scores representing the expected distribution in the general population. PPV was estimated as the number of true patients with cancer divided by the number of total positive calls, at a given cancer score cutoff. NPV was the fraction of true-negative individuals with cancer scores lower than the cutoff. We scanned a range of cutoffs, from 0.24 to 0.30, with 0.01 increments. The above analysis was repeated 1000 times to estimate statistical uncertainty.
Description of the Johns Hopkins University lung cancer cohort
Patients with early-stage lung cancer were recruited from Johns Hopkins University Hospital (96). The cohort included 11 patients with stages I and II cancer and 4 patients with stage III cancer. Peripheral blood samples were collected before any treatment, and TCR-seq data were generated by Adaptive Biotechnologies with the platform described above.
Description and analysis of the RNA-based iRepertoire cohorts
We obtained mRNA-based TCR-seq data from iRepertoire, including 17 patients with metastatic RCC and a healthy donor cohort containing 225 individuals without a history of cancer (97). Peripheral blood samples of all individuals were profiled using the iRepertoire platform. No HCMV serotyping was performed, and this cohort was expected to contain individuals with or without HCMV infection. Notably, this platform uses cDNA reverse-transcribed from mRNA of the TCR β chain transcripts with a different quantification method from Adaptive Biotechnologies. The iRepertoire glioma samples were accessed from GSE79338. The cohort contained 15 individuals, 14 of whom were patients with glioma and 1 healthy control. We used the blood TCR-seq data from the patients with glioma for cancer score inference. For each sample, we ordered TCR clones by decreasing read counts (each read covered a complete CDR3 region) and selected the top 10,000 sequences. iSMART was performed on each of the selected TCR-seq samples to obtain TCR clusters. Cancer scores were estimated using DeepCAT on iSMART outputs.
Statistical analysis
DeepCAT was written in python3, with model construction and training performed using the tensorflow package (version 1.4). All statistical analyses and visualization were performed using R, the statistical programming language (version 3.2.2) (98). Two sample tests were performed using two-sided Wilcoxon rank-sum test. If multiple tests were performed for a single analysis, then we used the Benjamini-Hochberg procedure to correct for false discovery rate. For all the boxplots displayed in the figures, the middle line defines the median value, with borders of the boxes indicating the first (25%) and third (75%) quartiles of the data. Lower and upper whiskers corresponded to Q1 − 1.5 interquartile range (IQR) and Q3 + 1.5 IQR. ROC curves and AUC values were obtained using the R package pROC. Ecological diversity indices were calculated using the vegan package. His enrichment analysis was performed using all the TIL and donor training TCRs, where the first and last three AAs were removed. Pooling all the AAs for tumor and control, we counted the numbers of His and performed Fisher’s exact test to evaluate the statistical significance for the enrichment.
Supplementary Material
Data file S1. DeepCAT estimation of cancer scores across all the sample cohorts.
Fig. S1. Biochemical features of caTCRs showing significant differences from non-cancer TCRs.
Fig. S2. CDR3 length distribution and prediction accuracy of CNN models for each CDR3 length.
Fig. S3. DeepCAT prediction is independent of HLA allele types.
Fig. S4. Distribution of raw DeepCAT outputs for patients with cancer and healthy donors.
Fig. S5. Cancer score estimation is independent of library size or patient age.
Fig. S6. Ninety-five percent confidence interval plots for ROC curves and waterfall plots for the cohorts of patients with cancer in Fig. 3.
Fig. S7. ROC analysis for all public PBMC cohorts with both treated and treatment-naïve patients.
Fig. S8. Variation of cancer scores in the time course cohort.
Fig. S9. CDR3 length distribution of healthy individuals and patients with cancer.
Fig. S10. ROC curves for cancer scores predicted using four different combinations.
Fig. S11. Evaluation of prediction accuracy for early-stage cancers in two recent studies.
Fig. S12. Boxplots showing the positive and negative prediction values at different cancer score cutoffs.
Fig. S13. Cancer score association with library size or age using the RNA-based samples.
Fig. S14. Cancer score distributions of patients with non-cancer chronic conditions compared against healthy controls.
Table S1. Summary information of caTCRs obtained from TCGA data.
Table S2. Summary of datasets used in this study.
Table S3. Clinical information for TCR-seq samples generated in this study.
Acknowledgments:
We thank J. Han (HudsonAlpha Institute and iRepertoire) for sharing the PBMC TCR-seq samples for healthy donors and the RCC samples profiled using the iRepertoire platform; D. Pardoll, K. Smith, and H. Ji (Johns Hopkins) for sharing the early-stage lung cancer samples profiled with Adaptive Biotechnologies; and T. Aguilera and the Pancreatic Cancer Prevention Program at UT Southwestern Medical Center for providing the PBMC samples from patients with early-stage pancreatic cancer or benign cyst. We also thank C. Arteaga and J. Minna for useful discussions during manuscript preparation and S. Rajaram for useful suggestions to help the methodology development.
Funding:
This work is supported by CPRIT RR170079 (B.L.) and NCI SPORE 1P50CA196516 (J.B.).
Footnotes
Competing interests: J.L. has an advisory affiliation with Clovis Oncology. B.L. is the inventor on patent application (133892–020601/PCT) submitted by UT Southwestern Medical Center that covers “Computerized system and method for antigen-independent de novo prediction of cancer-associated TCR repertoire.” All other authors declare that they have no competing interests.
Data and materials availability:
Source code of DeepCAT and AdaBoost and the training and testing datasets are available at https://github.com/s175573/DeepCAT. All the raw TCR-seq data generated in this study and the post-processed datasets associated with this study are available at Zenodo DOI: 10.5281/zenodo.3894880.
REFERENCES AND NOTES
- 1.Siegel RL, Miller KD, Jemal A, Cancer Statistics, 2017. CA Cancer J. Clin 67, 7–30 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Fass L, Imaging and cancer: A review. Mol. Oncol 2, 115–152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez J, Thompson IM, Prostate-specific antigen: A review of the validation of the most commonly used cancer biomarker. Cancer 101, 894–904 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Jacobs I, Bast RC Jr., The CA 125 tumour-associated antigen: A review of the literature. Hum. Reprod 4, 1–12 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Badgwell D, Bast RC Jr., Early detection of ovarian cancer. Dis. Markers 23, 397–410 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A, Hruban RH, Wolfgang CL, Goggins MG, Molin MD, Wang TL, Roden R, Klein AP, Ptak J, Dobbyn L, Schaefer J, Silliman N, Popoli M, Vogelstein JT, Browne JD, Schoen RE, Brand RE, Tie J, Gibbs P, Wong HL, Mansfield AS, Jen J, Hanash SM, Falconi M, Allen PJ, Zhou S, Bettegowda C, Diaz LA Jr., Tomasetti C, Kinzler KW, Vogelstein B, Lennon AM, Papadopoulos N, Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, Zuzarte PC, Borgida A, Wang TT, Li T, Kis O, Zhao Z, Spreafico A, Medina TDS, Wang Y, Roulois D, Ettayebi I, Chen Z, Chow S, Murphy T, Arruda A, O’Kane GM, Liu J, Mansour M, McPherson JD, O’Brien C, Leighl N, Bedard PL, Fleshner N, Liu G, Minden MD, Gallinger S, Goldenberg A, Pugh TJ, Hoffman MM, Bratman SV, Hung RJ, De Carvalho DD, Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 563, 579–583 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Alix-Panabieres C, Pantel K, Circulating tumor cells: Liquid biopsy of cancer. Clin. Chem 59, 110–118 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, Roederer M, Davis MM, Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med 5, 677–685 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Park S, Ang RR, Duffy SP, Bazov J, Chi KN, Black PC, Ma H, Morphological differences between circulating tumor cells from prostate cancer patients and cultured prostate cancer cells. PLOS ONE 9, e85264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, Abida W, Juluru K, De Bruijn I, Hou C, Venn O, Lim R, Anand A, Maddala T, Gnerre S, Satya RV, Liu Q, Shen L, Eattock N, Yue J, Blocker AW, Lee M, Sehnert A, Xu H, Hall MP, Santiago-Zayas A, Novotny WF, Isbell JM, Rusch VW, Plitas G, Heerdt AS, Ladanyi M, Hyman DM, Jones DR, Morrow M, Riely GJ, Scher HI, Rudin CM, Robson ME, Diaz LA Jr., Solit DB, Aravanis AM, Reis-Filho JS, High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med 25, 1928–1937 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD, Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA, Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schreiber RD, Old LJ, Smyth MJ, Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T, Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Chowell D, Krishna S, Becker PD, Cocita C, Shu J, Tan X, Greenberg PD, Klavinskis LS, Blattman JN, Anderson KS, TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc. Natl. Acad. Sci. U.S.A 112, E1754–E1762 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Li T, Pignon JC, Wang B, Wang J, Shukla SA, Dou R, Chen Q, Hodi FS, Choueiri TK, Wu C, Hacohen N, Signoretti S, Liu JS, Liu XS, Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet 48, 725–732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostmeyer J, Christley S, Toby IT, Cowell LG, Biophysicochemical motifs in T-cell receptor sequences distinguish repertoires from tumor-infiltrating lymphocyte and adjacent healthy tissue. Cancer Res 79, 1671–1680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Li T, Wang B, Dou R, Zhang J, Liu JS, Liu XS, Ultrasensitive detection of TCR hypervariable-region sequences in solid-tissue RNA-seq data. Nat. Genet 49, 482–483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomczak K, Czerwinska P, Wiznerowicz M, The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol 19, A68–A77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madi A, Poran A, Shifrut E, Reich-Zeliger S, Greenstein E, Zaretsky I, Arnon T, Laethem FV, Singer A, Lu J, Sun PD, Cohen IR, Friedman N, T cell receptor repertoires of mice and humans are clustered in similarity networks around conserved public CDR3 sequences. eLife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emerson RO, DeWitt WS, Vignali M, Gravley J, Hu JK, Osborne EJ, Desmarais C, Klinger M, Carlson CS, Hansen JA, Rieder M, Robins HS, Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet 49, 659–665 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA, Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, Robbins PF, Sette A, Appella E, Rosenberg SA, Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J. Immunol 154, 3961–3968 (1995). [PubMed] [Google Scholar]
- 26.Stevanovic S, Draper LM, Langhan MM, Campbell TE, Kwong ML, Wunderlich JR, Dudley ME, Yang JC, Sherry RM, Kammula US, Restifo NP, Rosenberg SA, Hinrichs CS, Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J. Clin. Oncol 33, 1543–1550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cannon MJ, Schmid DS, Hyde TB, Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol 20, 202–213 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Kawashima S, Kanehisa M, AAindex: Amino acid index database. Nucleic Acids Res 28, 374 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alipanahi B, Delong A, Weirauch MT, Frey BJ, Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol 33, 831–838 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Hou J, Adhikari B, Cheng J, DeepSF: Deep convolutional neural network for mapping protein sequences to folds. Bioinformatics 34, 1295–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou J, Troyanskaya OG, Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krizhevsky A, Sutskever I, Hinton G, Imagenet classification with deep convolutional neural networks. Advances in neural information processing system 25 (NIPS) (2012).
- 33.Garcia KC, Adams EJ, How the T cell receptor sees antigen—A structural view. Cell 122, 333–336 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Wucherpfennig KW, Gagnon E, Call MJ, Huseby ES, Call ME, Structural biology of the T-cell receptor: Insights into receptor assembly, ligand recognition, and initiation of signaling. Cold Spring Harb. Perspect. Biol 2, a005140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, Sougnez C, Cibulskis K, Kiezun A, Hacohen N, Brusic V, Wu CJ, Getz G, Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol 33, 1152–1158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang SQ, Ma KY, Schonnesen AA, Zhang M, He C, Sun E, Williams CM, Jia W, Jiang N, High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol 36, 1156–1159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raza SA, Clifford GM, Franceschi S, Worldwide variation in the relative importance of hepatitis B and hepatitis C viruses in hepatocellular carcinoma: A systematic review. Br. J. Cancer 96, 1127–1134 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shillitoe EJ, Silverman S Jr., Oral cancer and herpes simplex virus—A review. Oral Surg. Oral Med. Oral Pathol 48, 216–224 (1979). [DOI] [PubMed] [Google Scholar]
- 39.Young LS, Rickinson AB, Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 4, 757–768 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, Trebska-McGowan K, Wunderlich JR, Yang JC, Rosenberg SA, Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med 22, 433–438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greiff V, Bhat P, Cook SC, Menzel U, Kang W, Reddy ST, A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status. Genome Med 7, 49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, Mok S, Huang RR, Cochran AJ, Comin-Anduix B, Koya RC, Graeber TG, Robins H, Ribas A, CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res 20, 2424–2432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robins H, Immunosequencing: Applications of immune repertoire deep sequencing. Curr. Opin. Immunol 25, 646–652 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Venturi V, Kedzierska K, Turner SJ, Doherty PC, Davenport MP, Methods for comparing the diversity of samples of the T cell receptor repertoire. J. Immunol. Methods 321, 182–195 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Laydon DJ, Bangham CR, Asquith B, Estimating T-cell repertoire diversity: Limitations of classical estimators and a new approach. Philos. Trans. R. Soc. Lond. B Biol. Sci 370, 20140291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, Heguy A, Imai N, Gnjatic S, Emerson RO, Zhou XK, Zhang T, Chachoua A, Demaria S, Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med 24, 1845–1851 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blonski M, Taillandier L, Herbet G, Maldonado IL, Beauchesne P, Fabbro M, Campello C, Goze C, Rigau V, Moritz-Gasser S, Kerr C, Ruda R, Soffietti R, Bauchet L, Duffau H, Combination of neoadjuvant chemotherapy followed by surgical resection as a new strategy for WHO grade II gliomas: A study of cognitive status and quality of life. J. Neurooncol 106, 353–366 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Grossman HB, Natale RB, Tangen CM, Speights VO, Vogelzang NJ, Trump DL, deVere White RW, Sarosdy MF, Wood DP Jr., Raghavan D, Crawford ED, Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N. Engl. J. Med 349, 859–866 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Rebe C, Ghiringhelli F, Cytotoxic effects of chemotherapy on cancer and immune cells: How can it be modulated to generate novel therapeutic strategies? Future Oncol 11, 2645–2654 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Chu ND, Bi HS, Emerson RO, Sherwood AM, Birnbaum ME, Robins HS, Alm EJ, Longitudinal immunosequencing in healthy people reveals persistent T cell receptors rich in highly public receptors. BMC Immunol 20, 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Freund Y, Schapire R, A decision-theoretic generalization of online learning and an application to boosting. J. Comput. Syst. Sci 55, 119–139 (1997). [Google Scholar]
- 52.Beausang JF, Wheeler AJ, Chan NH, Hanft VR, Dirbas FM, Jeffrey SS, Quake SR, T cell receptor sequencing of early-stage breast cancer tumors identifies altered clonal structure of the T cell repertoire. Proc. Natl. Acad. Sci. U.S.A 114, E10409–E10417 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wojtacki J, Dziewulska-Bokiniec A, Skokowski J, Ciesielski D, Evaluation of CA 15–3 tumor marker in the diagnosis of breast cancer. A pilot study. Neoplasma 41, 213–216 (1994). [PubMed] [Google Scholar]
- 54.Havrilesky LJ, Whitehead CM, Rubatt JM, Cheek RL, Groelke J, He Q, Malinowski DP, Fischer TJ, Berchuck A, Evaluation of biomarker panels for early stage ovarian cancer detection and monitoring for disease recurrence. Gynecol. Oncol 110, 374–382 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Ankerst DP, Thompson IM, Sensitivity and specificity of prostate-specific antigen for prostate cancer detection with high rates of biopsy verification. Arch. Ital. Urol. Nefrol. Androl 78, 125–129 (2006). [PubMed] [Google Scholar]
- 56.National Cancer Institute, SEER statistics of cancer prevalence (2016).
- 57.Koo MM, Hamilton W, Walter FM, Rubin GP, Lyratzopoulos G, Symptom signatures and diagnostic timeliness in cancer patients: A review of current evidence. Neoplasia 20, 165–174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shapley M, Mansell G, Jordan JL, Jordan KP, Positive predictive values of >/=5% in primary care for cancer: Systematic review. Br. J. Gen. Pract 60, e366–e377 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang C, Sanders CM, Yang Q, Schroeder HW Jr., Wang E, Babrzadeh F, Gharizadeh B, Myers RM, Hudson JR Jr., Davis RW, Han J, High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proc. Natl. Acad. Sci. U.S.A 107, 1518–1523 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sims JS, Grinshpun B, Feng Y, Ung TH, Neira JA, Samanamud JL, Canoll P, Shen Y, Sims PA, Bruce JN, Diversity and divergence of the glioma-infiltrating T-cell receptor repertoire. Proc. Natl. Acad. Sci. U.S.A 113, E3529–E3537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Savola P, Kelkka T, Rajala HL, Kuuliala A, Kuuliala K, Eldfors S, Ellonen P, Lagstrom S, Lepisto M, Hannunen T, Andersson EI, Khajuria RK, Jaatinen T, Koivuniemi R, Repo H, Saarela J, Porkka K, Leirisalo-Repo M, Mustjoki S, Somatic mutations in clonally expanded cytotoxic T lymphocytes in patients with newly diagnosed rheumatoid arthritis. Nat. Commun 8, 15869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alves Sousa AP, Johnson KR, Ohayon J, Zhu J, Muraro PA, Jacobson S, Comprehensive analysis of TCR-β repertoire in patients with neurological immunemediated disorders. Sci. Rep 9, 344 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chiche J, Brahimi-Horn MC, Pouyssegur J, Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med 14, 771–794 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, Wakabayashi T, Hayasaka A, Kadono S, Miyazaki T, Haraya K, Sekimori Y, Kojima T, Nabuchi Y, Aso Y, Kawabe Y, Hattori K, Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol 28, 1203–1207 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Wirasinha RC, Singh M, Archer SK, Chan A, Harrison PF, Goodnow CC, Daley SR, αβT-cell receptors with a central CDR3 cysteine are enriched in CD8αα intraepithelial lymphocytes and their thymic precursors. Immunol. Cell Biol 96, 553–561 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Campbell DJ, Klicznik MM, Morawski PA, Hollbacher B, Varkhande SR, Motley SJ, Kuri-Cervantes L, Goodwin E, Rosenblum MD, Long SA, Brachtl G, Duhen T, Betts MR, Campbell DJ, Gratz IK, Human CD4+ CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci. Immunol 4, eaav8995 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, Wells DK, Cary CIO, Grbovic-Huezo O, Attiyeh M, Medina B, Zhang J, Loo J, Saglimbeni J, Abu-Akeel M, Zappasodi R, Riaz N, Smoragiewicz M, Kelley ZL, Basturk O; Australian Pancreatic Cancer Genome Initiative; Garvan Institute of Medical Research; Prince of Wales Hospital; Royal North Shore Hospital; University of Glasglow; St Vincent’s Hospital; QIMR Berghofer Medical Research Institute; University of Melbourne; Centre for Cancer Research; University of Queensland; Institute for Molecular Science; Bankstown Hospital; Liverpool Hospital; Royal Prince Alfred Hospital; Chris O’Brien Lifehouse; Westmead Hospital, Fremantle Hospital; St John of God Healthcare; Royal Adelaide Hospital; Finders Medical Centre Envoi Pathology; Princess Alexandria Hospital; Austin Hospital; Johns Hopkins Medical Institutes; ARC-Net Centre for Applied Research on Cancer, Gonen M, Levine AJ, Allen PJ, Fearon DT, Merad M, Gnjatic S, Iacobuzio-Donahue CA, Wolchok JD, DeMatteo RP, Chan TA, Greenbaum BD, Merghoub T, Leach SD, Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bever KM, Le DT, DNA repair defects and implications for immunotherapy. J. Clin. Invest 128, 4236–4242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buonaguro L, Mauriello A, Cavalluzzo B, Petrizzo A, Tagliamonte M, Immunotherapy in hepatocellular carcinoma. Ann. Hepatol 18, 291–297 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Fessler J, Matson V, Gajewski TF, Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 7, 108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cooper G, The development and causes of cancer, in The Cell: A Molecular Approach 2nd edition (2000).
- 72.Obar JJ, Lefrancois L, Memory CD8+ T cell differentiation. Ann. N. Y. Acad. Sci 1183, 251–266 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wherry EJ, Kurachi M, Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Y, Zheng L, Zhang L, Hu X, Ren X, Zhang Z, Deep single-cell RNA sequencing data of individual T cells from treatment-naïve colorectal cancer patients. Sci. Data 6, 131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA, Gupta RK, Curtis C, Bucktrout SL, Davis MM, Chang ALS, Chang HY, Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, Crawford JC, Clemens EB, Nguyen THO, Kedzierska K, La Gruta NL, Bradley P, Thomas PG, Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu X, Zhang J, Liu JS, Li B, Liu XS, Evaluation of immune repertoire inference methods from RNA-seq data. Nat. Biotechnol 36, 1034 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, Hadi-Saljoqi S, Sasorith S, Lefranc G, Kossida S, IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res 43, D413–D422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, Hirt C, Mezzadra R, Slagter M, Dijkstra K, Kluin RJC, Snaebjornsson P, Milne K, Nelson BH, Zijlmans H, Kenter G, Voest EE, Haanen J, Schumacher TN, Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med 25, 89–94 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Jenkins MK, Moon JJ, The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J. Immunol 188, 4135–4140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang H, Liu L, Zhang J, Chen J, Ye J, Shukla S, Qiao J, Zhan X, Chen H, Wu CJ, Fu YX, Li B, Investigation of antigen-specific T-cell receptor clusters in human cancers. Clin. Cancer Res 26, 1359–1371 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Dumoulin V, Visin F, A guide to convolution arithmetic for deep learning. arXiv:1603.07285 (2018).
- 83.Hochreiter S, Schimidhuber J, Long short-term memory. Neural. Comput 9, 1735–1780 (1997). [DOI] [PubMed] [Google Scholar]
- 84.Sherstinsky A, Fundamentals of recurrent neural network (RNN) and long short-term memory (LSTM) network. arXiv:1808.03314 (2018).
- 85.Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, Ji X, Han A, Krams SM, Pettus C, Haas N, Arlehamn CSL, Sette A, Boyd SD, Scriba TJ, Martinez OM, Davis MM, Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Olson M, JOUSBoost: Implements under/oversampling for probability estimation. R package version 2.1.0, (2017).
- 87.DeWitt WS, Emerson RO, Lindau P, Vignali M, Snyder TM, Desmarais C, Sanders C, Utsugi H, Warren EH, McElrath J, Makar KW, Wald A, Robins HS, Dynamics of the cytotoxic T cell response to a model of acute viral infection. J. Virol 89, 4517–4526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kanakry CG, Coffey DG, Towlerton AM, Vulic A, Storer BE, Chou J, Yeung CC, Gocke CD, Robins HS, O’Donnell PV, Luznik L, Warren EH, Origin and evolution of the T cell repertoire after posttransplantation cyclophosphamide. JCI Insight 1, e86252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Emerson RO, Sherwood AM, Rieder MJ, Guenthoer J, Williamson DW, Carlson CS, Drescher CW, Tewari M, Bielas JH, Robins HS, High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J. Pathol 231, 433–440 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hsu M, Sedighim S, Wang T, Antonios JP, Everson RG, Tucker AM, Du L, Emerson R, Yusko E, Sanders C, Robins HS, Yong WH, Davidson TB, Li G, Liau LM, Prins RM, TCR sequencing can identify and track glioma-infiltrating T cells after DC vaccination. Cancer Immunol. Res 4, 412–418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mansfield AS, Ren H, Sutor S, Sarangi V, Nair A, Davila J, Elsbernd LR, Udell JB, Dronca RS, Park S, Markovic SN, Sun Z, Halling KC, Nevala WK, Aubry MC, Dong H, Jen J, Contraction of T cell richness in lung cancer brain metastases. Sci. Rep 8, 2171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Snyder A, Nathanson T, Funt SA, Ahuja A, Buros Novik J, Hellmann MD, Chang E, Aksoy BA, Al-Ahmadie H, Yusko E, Vignali M, Benzeno S, Boyd M, Moran M, Iyer G, Robins HS, Mardis ER, Merghoub T, Hammerbacher J, Rosenberg JE, Bajorin DF, Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLOS Med 14, e1002309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR, T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol. Res 5, 978–991 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, Diaz LA Jr., Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DeWitt WS, Yu KKQ, Wilburn DB, Sherwood A, Vignali M, Day CL, Scriba TJ, Robins HS, Swanson WJ, Emerson RO, Bradley PH, Seshadri C, A diverse lipid antigen-specific TCR repertoire is clonally expanded during active tuberculosis. J. Immunol 201, 888–896 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang J, Ji Z, Caushi JX, El Asmar M, Anagnostou V, Cottrell TR, Chan HY, Suri P, Guo H, Merghoub T, Chaft JE, Reuss JE, Tam AJ, Blosser RL, Abu-Akeel M, Sidhom JW, Zhao N, Ha JS, Jones DR, Marrone KA, Naidoo J, Gabrielson E, Taube JM, Velculescu VE, Brahmer JR, Housseau F, Hellmann MD, Forde PM, Pardoll DM, Ji H, Smith KN, Compartmental analysis of T-cell clonal dynamics as a function of pathologic response to neoadjuvant PD-1 blockade in resectable non-small cell lung cancer. Clin. Cancer Res 26, 1327–1337 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Byrne-Steele M, Pan WJ, Public CDR3 sequence browser (www.pcdr3s.com). (2020).
- 98.R Core Team, R: A language and environment for statistical computing (2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. DeepCAT estimation of cancer scores across all the sample cohorts.
Fig. S1. Biochemical features of caTCRs showing significant differences from non-cancer TCRs.
Fig. S2. CDR3 length distribution and prediction accuracy of CNN models for each CDR3 length.
Fig. S3. DeepCAT prediction is independent of HLA allele types.
Fig. S4. Distribution of raw DeepCAT outputs for patients with cancer and healthy donors.
Fig. S5. Cancer score estimation is independent of library size or patient age.
Fig. S6. Ninety-five percent confidence interval plots for ROC curves and waterfall plots for the cohorts of patients with cancer in Fig. 3.
Fig. S7. ROC analysis for all public PBMC cohorts with both treated and treatment-naïve patients.
Fig. S8. Variation of cancer scores in the time course cohort.
Fig. S9. CDR3 length distribution of healthy individuals and patients with cancer.
Fig. S10. ROC curves for cancer scores predicted using four different combinations.
Fig. S11. Evaluation of prediction accuracy for early-stage cancers in two recent studies.
Fig. S12. Boxplots showing the positive and negative prediction values at different cancer score cutoffs.
Fig. S13. Cancer score association with library size or age using the RNA-based samples.
Fig. S14. Cancer score distributions of patients with non-cancer chronic conditions compared against healthy controls.
Table S1. Summary information of caTCRs obtained from TCGA data.
Table S2. Summary of datasets used in this study.
Table S3. Clinical information for TCR-seq samples generated in this study.
Data Availability Statement
Source code of DeepCAT and AdaBoost and the training and testing datasets are available at https://github.com/s175573/DeepCAT. All the raw TCR-seq data generated in this study and the post-processed datasets associated with this study are available at Zenodo DOI: 10.5281/zenodo.3894880.