

Abstract
In this study, we focus on the molecular mechanisms associated with the A57G (Ala57-to-Gly57) mutation in myosin essential light chains (ELCs), found to cause hypertrophic cardiomyopathy (HCM) in humans and in mice. Specifically, we studied the effects of A57G on the super-relaxed (SRX) state of myosin that may contribute to the hypercontractile cross-bridge behavior and ultimately lead to pathological cardiac remodeling in transgenic Tg-A57G mice. The disease model was compared to Tg-WT mice, expressing the wild-type human ventricular ELC, and analyzed against Tg-Δ43 mice, expressing the N-terminally truncated ELC, whose hearts hypertrophy with time but do not show any abnormalities in cardiac morphology or function. Our data suggest a new role for the N terminus of cardiac ELC (N-ELC) in modulation of myosin cross-bridge function in the healthy as well as in HCM myocardium. The lack of N-ELC in Tg-Δ43 mice was found to significantly stabilize the SRX state of myosin and increase the number of myosin heads occupying a low-energy state. In agreement, Δ43 hearts showed significantly decreased ATP utilization and low actin-activated myosin ATPase compared with A57G and WT hearts. The hypercontractile activity of A57G-ELC cross-bridges was manifested by the inhibition of the SRX state, increased number of myosin heads available for interaction with actin, and higher ATPase activity. Fiber mechanics studies, echocardiography examination, and assessment of fibrossis confirmed the development of two distinct forms of cardiac remodeling in these two ELC mouse models, with pathological cardiac hypertrophy in Tg-A57G, and near physiologic cardiac growth in Tg-Δ43 animals.
Keywords: hypertrophic cardiomyopathy, myosin essential light chain, super-relaxed state, Tg-A57G mice, Tg-Δ43 mice
Introduction
Myosin essential light chains (ELCs; also known as alkali light chains) play an important role in muscle contraction, yet their specific functions in various muscle types: cardiac, skeletal, or smooth muscles, are not well understood [1]. In fast-twitch skeletal muscles, two ELC variants are expressed by the MYL1 gene [2], but due to a differential splicing event, they differ by a ~ 43-amino acid (aa)-long NH2 terminus (N-ELC). In smooth muscle, only the shorter ELC is present, whereas the exclusive expression of the long ELC (produced by the MYL3 gene) containing the N-ELC is observed in the heart [1]. The question that we address in this study regards the cardiac-specific expression of the N-ELC and its role in regulating myosin motor function in the normal heart and in cardiomyopathy disease associated with A57G (Ala57→Gly57) mutation in MYL3, encoding the human ventricular ELC wild-type protein (ELC-WT). The mutation was discovered in two unrelated Korean families and one Japanese man with hypertrophic cardiomyopathy (HCM) and was associated with a classic asymmetric septal hypertrophy and sudden cardiac death (SCD) [3].
A modeling study of Aydt et al. [4] pictured the N-ELC as a rod-like 91 Å-long extension that can bridge the myosin head with the actin filament, highlighting the importance of this ELC region for actin–myosin interaction, force development, and cardiac muscle contraction. Investigation by Kazmierczak et al. [5] revealed that genetic ablation of the N-ELC in transgenic (Tg) Δ43 mice, expressing truncated human ventricular ELC mutant (Δ43), made Δ43 hearts prone to hypertrophy, but no signs of histopathology or fibrosis were observed, and the mice did not manifest any signs of functional abnormalities by magnetic resonance imaging. Furthermore, no differences in steady-state parameters of contraction in skinned papillary muscles (PM) were recorded (Fmax and pCa50) in Tg-Δ43 versus Tg-WT mice, reinforcing the notion that Tg-Δ43 mice do not show pathological-like hypertrophy [6]. In contrast, mice expressing the human ventricular A57G-ELC mutant showed pathological cardiac morphology including extensive disorganization of myocytes and interstitial fibrosis, and diminished function (lower Fmax, increased Ca2+ sensitivity) compared with Tg-WT mice [6–8]. Exposure to strenuous exercise via swimming led to hypertrophy and pathological cardiac remodeling only in Tg-A57G animals, with no changes in Tg-Δ43 hearts, which maintained a nonpathological cardiac phenotype [6]. These humanized models of ELC-mediated cardiac remodeling, expressing human ventricular ELC variants, were further analyzed in the current report with the focus on the super-relaxed (SRX) state, a novel concept in the field of myosin that has not been explored for any of myosin ELC mutations before.
The contraction of the heart is a tightly regulated process that depends on the Ca2+ and ATP-regulated interaction between the actin-containing thin filaments and the myosin-containing thick filaments [9]. During cardiac muscle contraction/relaxation, myosin can be characterized by three states: an active/force-producing state, a disordered relaxed state (DRX), and a super-relaxed state (SRX) [10,11]. In DRX, myosin cross-bridges protrude into the interfilament space but are restricted from binding to actin and producing force, while in SRX, they display an ordered head arrangement along the thick filament axis and a highly inhibited ATP turnover rate [10,12]. The presence of the folded state of myosin, where the heads interact with each other and with the part of myosin heavy chain (MHC) and form so-called interacting head motif (IHM) structures [13], is hypothesized to be the origin of SRX [14]. In the heart, SRX serves as a modulator of cardiac energy utilization, involved in decreasing metabolic rate in both normally functioning myocardium and during times of stress, for example, cardiomyopathy [12].
The scientific premise of this study builds on the reports from our and other laboratories demonstrating that the cardiac-specific N-ELC may play an important role in regulating myosin motor function and force production in muscle [5,8,15–21]. We hypothesize that cardiac N-ELC works as a molecular linker/switch of the actin–myosin interaction by regulating the SRX-to-DRX transition and hence the proportion of myosin heads that are available for the interaction with actin. We further hypothesize that the clinical phenotype of hypercontractility associated with HCM may depend on the excessive number of myosin heads that transition from SRX to DRX and to force-producing state, resulting in premature left ventricular (LV) contractions compared to healthy myocardium. To address these hypotheses, we used our developed ELC-humanized mouse models of HCM (Tg-A57G) [7] along with Tg-Δ43 mice, lacking the N-ELC [5], and the results were compared to age- and sex-matched WT-ELC mice. Specifically, the effect of each ELC modification in the hearts of mice was analyzed in relation to the SRX-DRX equilibrium and the mutation-specific alterations of heart function in vivo that contribute to the development of pathological-versus physiologic-like hypertrophy observed in Tg-A57G and Tg-Δ43 mice, respectively.
Results
Cross-bridge behavior is antagonistically regulated in Tg-A57G versus Tg-Δ43 mice
The recently discovered SRX state of cardiac myosin serves as an innovative research tool to study the molecular basis of the energetic state of the heart at the level of sarcomere in health and in cardiac disease [10,22]. HCM is commonly associated with an increase in energy cost of tension generation through inefficient or excessive ATP usage [23]. This may come about by shifting myosin heads out of the energy-conserving SRX state into the disordered DRX or active states, where ATP usage is greatly increased [12]. To detemine the effect of ELC mutations on the SRX state of myosin, quantitative epifluorescence measurements were performed on skinned PM as described by Yadav et al. [24]. This method takes advantage of known kinetics of the cross-bridge cycle and a fluorescent nucleotide mantATP, an analogue of ATP that binds strongly to myosin but with a 2.5-fold increase in fluorescence emission compared with dark ATP. This increase in fluorescence is lost upon dissociation from myosin as mant-nucleotide is exchanged for ATP [12]. Skinned mouse cardiac PM fibers from Tg-WT, Tg-A57G, and Tg-Δ43 mice (diameter of ~ 100 µm) were incubated in a relaxing solution containing 250 µM mantATP. Then, the fibers were mounted in a flow cell of the IonOptix apparatus and subjected to nucleotide exchange assay using 4 mm dark ATP-relaxing solution. Decay in fiber fluorescence was recorded as the mant-nucleotides were released from myosin and replaced by dark ATP [25]. Figure 1A–D shows the time-dependent average fluorescence decay curves, including simulated single-exponential curves of the fast and slow phases of decay for Tg-WT (blue trace), Tg-A57G (green trace), and Tg-Δ43 (red trace) genotypes. The curves were fitted to a double-exponential equation:
where P1 and P2 (in %) are the relative fractions of fiber fluorescence (amplitudes) of DRX and SRX states and are characterized by the fast T1 and slow T2 fluorescence lifetimes (in seconds), respectively (Fig. 1E; Table 1). Specifically, P1 and P2 illustrate the population of myosin heads remaining in fast relaxed or slow super-relaxed states. As shown in Table 1, PM obtained from Tg-A57G showed a significant increase in the proportion of myosin heads in the DRX state (P1 = 70%) compared with WT (P1 = 60%). This difference is coupled to a significant decrease in the proportion of myosin heads existing in SRX in A57G (P2 = 28%) compared with WT (38%). Interestingly, deletion of the N-ELC in Tg-Δ43 animals resulted in a significant increase in fraction of myosin heads existing in SRX (P2 = 48%) and a corresponding decrease in DRX (P1 = 50%) compared with WT fibers (Fig. 1E). No significant differences were noted in lifetimes of both phases, T1 and T2, among all tested genotypes (Table 1).
Fig. 1.
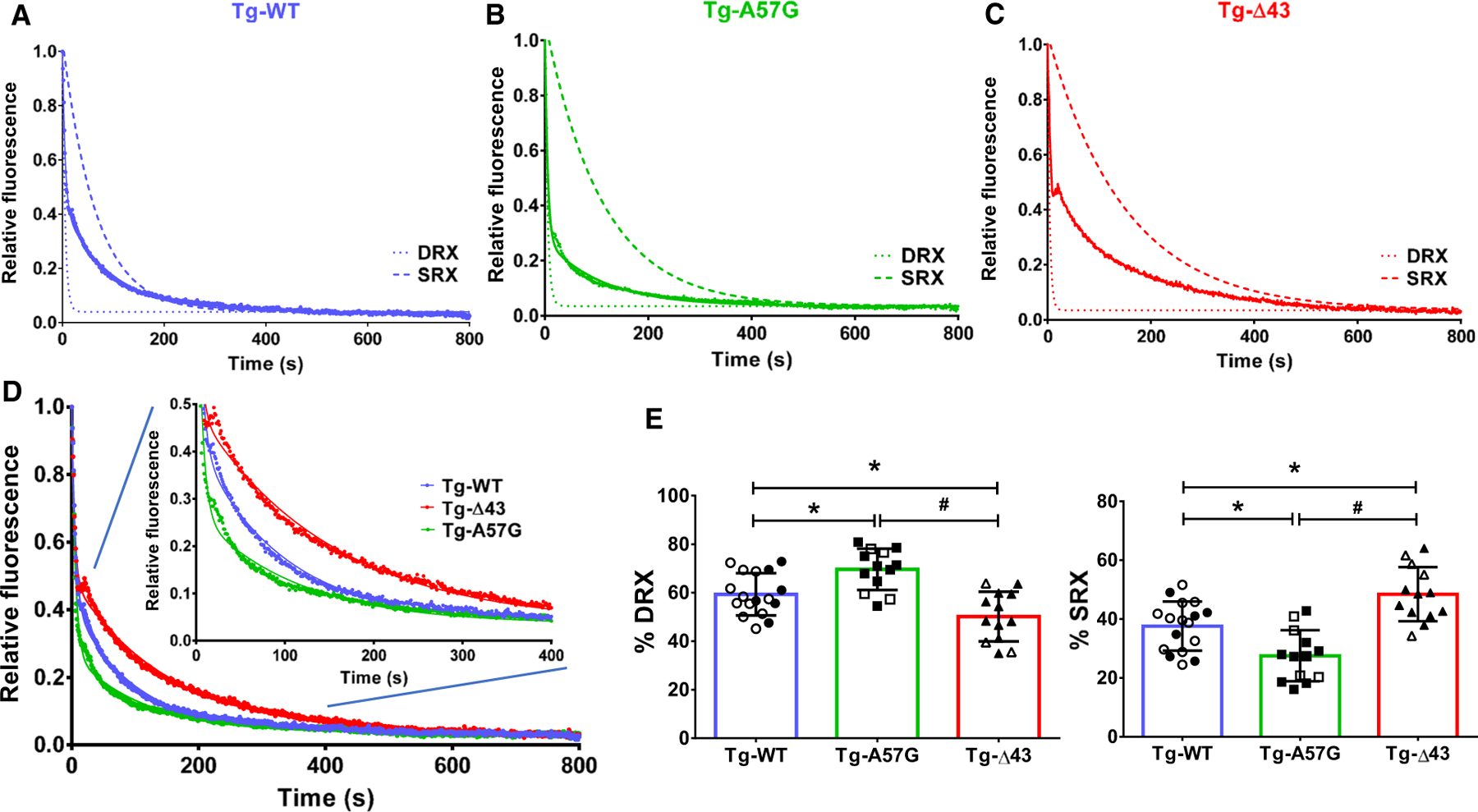
Single nucleotide turnover studies. Fluorescence decay curves of mantATP release in skinned papillary muscle (PM) fibers from: (A) Tg-WT (blue trace), n = 17 fibers, isolated from 5 female (F) and 3 male (M) mice; (B) Tg-A57G (green trace), n = 13 fibers (3F and 3M); and (C) Tg-Δ43 (red trace), n = 13 fibers (4F and 5M). PM were first incubated in 250 µm mantATP and subsequently chased with a solution containing 4 mm ATP. Decay traces were fitted to a double-exponential equation, yielding the fraction of myosin heads in DRX (P1) and SRX (P2) states, and respective lifetimes of fast (T1) and slow (T2) phases. Simulated single-exponential decays for DRX and SRX are depicted with dashed curves. (D) Overlapped fluorescence decay traces for Tg-WT, Tg-A57G, and Tg-Δ43 PM fibers. Inset depicts fluorescence decay traces collected from 0 to 400 s. (E) Proportion of myosin heads (in %) in DRX and SRX states for each group of mice. Open symbols depict PM fibers from F, while closed symbols depict PM fibers from M mice. Note that the % DRX is increased in Tg-A57G and decreased in Tg-Δ43. This is coupled to a decrease in % SRX in Tg-A57G and increase in Tg-Δ43. Data are mean T SD, in which n represents the number of fibers and *P < 0.05 depicts significance between Tg-WT and Tg-A57G/Tg-Δ43 and #P < 0.05 between Tg-A57G and Tg-Δ43, one-way ANOVA with Tukey’s multiple comparison test.
Table 1.
The super-relaxed (SRX) state of myosin measured by mantATP/ATP chase assays in skinned papillary muscles from Tg-WT, Tg-A57G, and Tg-Δ43 mice. P1 and P2 represent fractions (in %) of myosin cross-bridges in the disordered relaxed (DRX) and SRX states, respectively, with fast T1 and slow T2 lifetimes. 1/T1 and 1/T2 represent the rates of ATP turnover in DRX and SRX states, respectively. Values are means T SD for n = # fibers.
| Parameter | Tg-WT | Tg-A57G | Tg-Δ43 |
|---|---|---|---|
| P1 (%) | 59.33 ± 8.66 | 69.61 ± 8.51*,# | 50.13 ± 10.25* |
| P2 (%) | 37.60 ± 8.34 | 27.52 ± 8.67*,# | 48.45 ± 9.21* |
| T1 (s) | 6.25 ± 2.97 | 5.92 ± 3.94 | 4.23 ± 2.00 |
| T2 (s) | 117.70 ± 68.22 | 116.00 ± 80.89 | 193.40 ± 143.70 |
| n = # fibers (#F and #M mice) | 17 (5F and 3M) | 13 (3F and 3M) | 13 (4F and 5M) |
P < 0.05 Tg-A57G/Tg-Δ43 versus Tg-WT.;
P < 0.05 Tg-A57G versus Tg-Δ43, one-way ANOVA with Tukey’s multiple comparison test.
In addition, we evaluated the functional consequences of ELC genetic manipulations on the enzymatic activities of mutant myosins and their ability to interact with actin using myosin preparations purified from right and left ventricles of Tg-A57G, Tg-Δ43, and Tg-WT mice using the actin-activated myosin ATPase assays, as described earlier [26]. The specific ATPase readings were plotted as a function of increasing F-actin concentration, from 0 to 15 µM (Fig. 2A). Consistent with previously published results [15], Tg-A57G myosin demonstrated a significantly higher actin-activated ATPase activity: Vmax = 0.72 ± 0.10 s−1 compared with Tg-WT: Vmax = 0.62 ± 0.08 s−1. On the other hand, deletion of the N-ELC in Tg-Δ43 mice led to a significant decrease in Vmax = 0.45 ± 0.09 s−1 compared with Tg-WT myosin (Fig. 2B). Since Vmax represents the velocity of the cross-bridge cycling, results from this experiment suggest that the A57G mutation speeds up the cross-bridge turnover rate of the actomyosin cycle, while Δ43-ELC slows it down. The Vmax data may also indicate an increased number of A57G myosin heads and a reduced number of Δ43 heads capable of interacting with actin. The latter interpretation of ATPase results is in accord with the SRX data showing a significant increase in the proportion of A57G myosin heads in the DRX state compared with WT, while the Δ43-ELC heads demonstrated a significant increase in the SRX state (Table 1). These results also reveal significant differences in the energetic state of the heart between these two mouse models, with Tg-A57G hearts showing increased ATP utilization and Tg-Δ43 hearts characterized by low ATP demand/usage. Interestingly, Km values, depicting actin concentrations needed to reach half Vmax, were not statistically different among the genotypes (Km in µm: 0.71 ± 0.09 for Tg-A57G, 0.65 ± 0.12 for Tg-Δ43, and 0.84 T 0.22 for Tg-WT) (Fig. 2C).
Fig. 2.
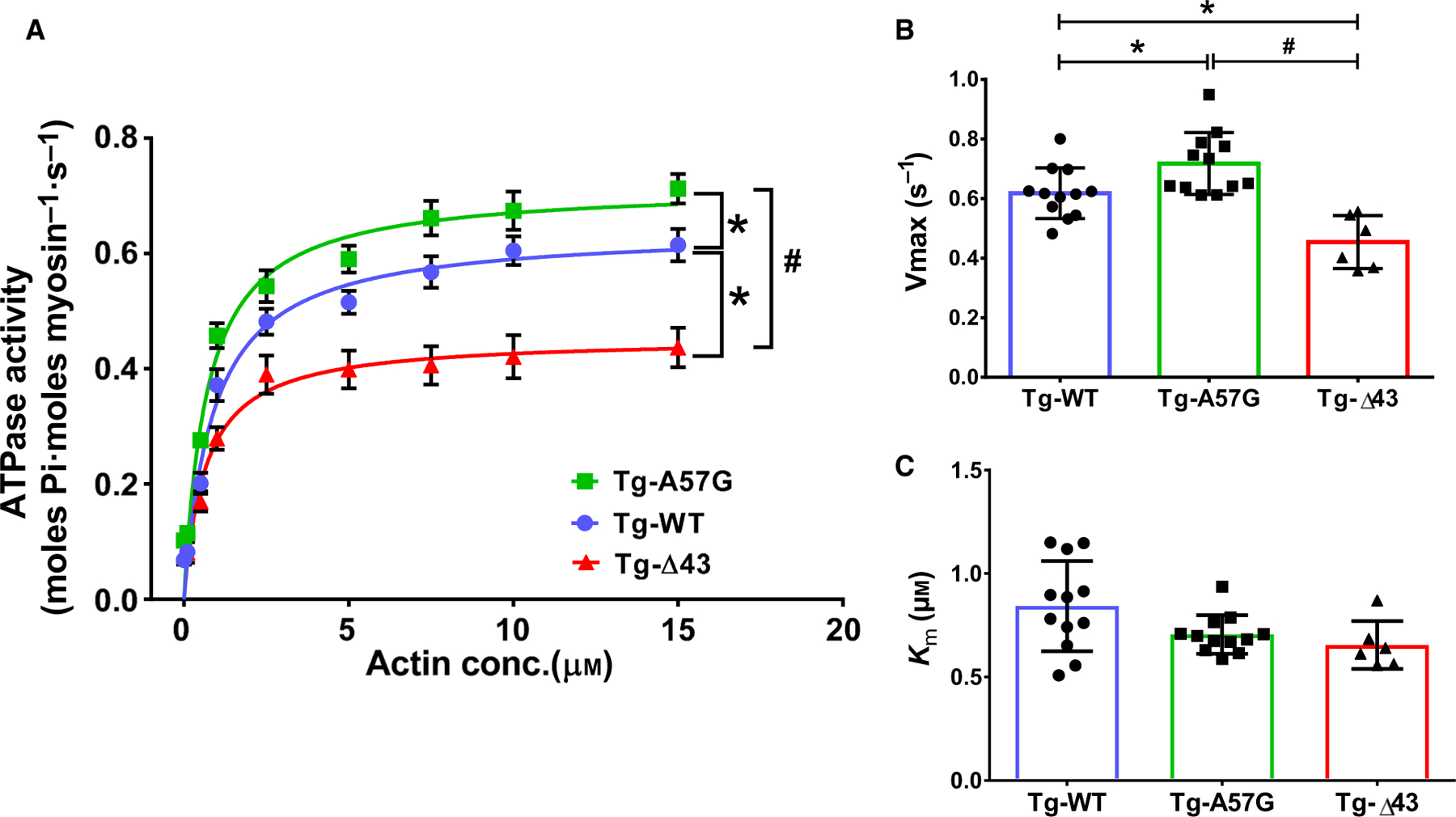
Actin-activated ATPase activity assays performed on myosin purified from left and right ventricles of Tg-A57G and Tg-Δ43 compared with Tg-WT mice (A), yielding Vmax (B) and Km (C), derived from the Michaelis–Menten equation. Four to five hearts from mice of each group of both sexes were used to generate one batch of myosin, and the assays (performed in triplicate) were repeated on two to four different batches of myosin. Data are mean ± SEM (A) or T SD (B; C) for n = 12 experiments (Tg-A57G), n = 6 experiments (Tg-Δ43), and n = 12 experiments (Tg-WT). *P < 0.05 depicting significance between Tg-WT and Tg-A57G/Tg-Δ43, and #P < 0.05 between Tg-A57G and Tg-Δ43, one-way ANOVA with Tukey’s multiple comparison test.
The HCM-linked A57G mutation, but not Δ43-ELC, alters force and Ca2+ transients in cardiac PM
We have also examined the cross-bridge contraction/relaxation kinetics in intact PM from Tg-A57G and Tg-Δ43 mice and compared with age- and sex-matched Tg-WT (Fig. 3). Using the IonOptix Calcium and Contractility Recording System, force transients were recorded in PM of SL = ~ 2.2 µm and subjected to 8 V at 1 Hz stimulus [26]. No differences were seen in the rate of contraction among the genotypes (Fig. 3A). Likewise, no differences were noted in the rates of force relaxation in Tg-A57G or Tg-Δ43 versus Tg-WT mice (Fig. 3B; Table 2). However, Tg-A57G muscles displayed significantly faster relaxation times compared with Tg-Δ43 mice, as demonstrated by shorter t25, t50, and t75 values depicting the time (in ms) from peak force to 25%, 50%, and 75% of the baseline and revealing differences in their cycling characteristics (Fig. 3B; Table 2). The maximal force developed by PM in these mice, calculated using the physiologic cross-sectional area of each muscle (PM mass/PM density/PM length) and expressed in mN·mm−2 [27], was not different among the genotypes (Fig. 3C).
Fig. 3.
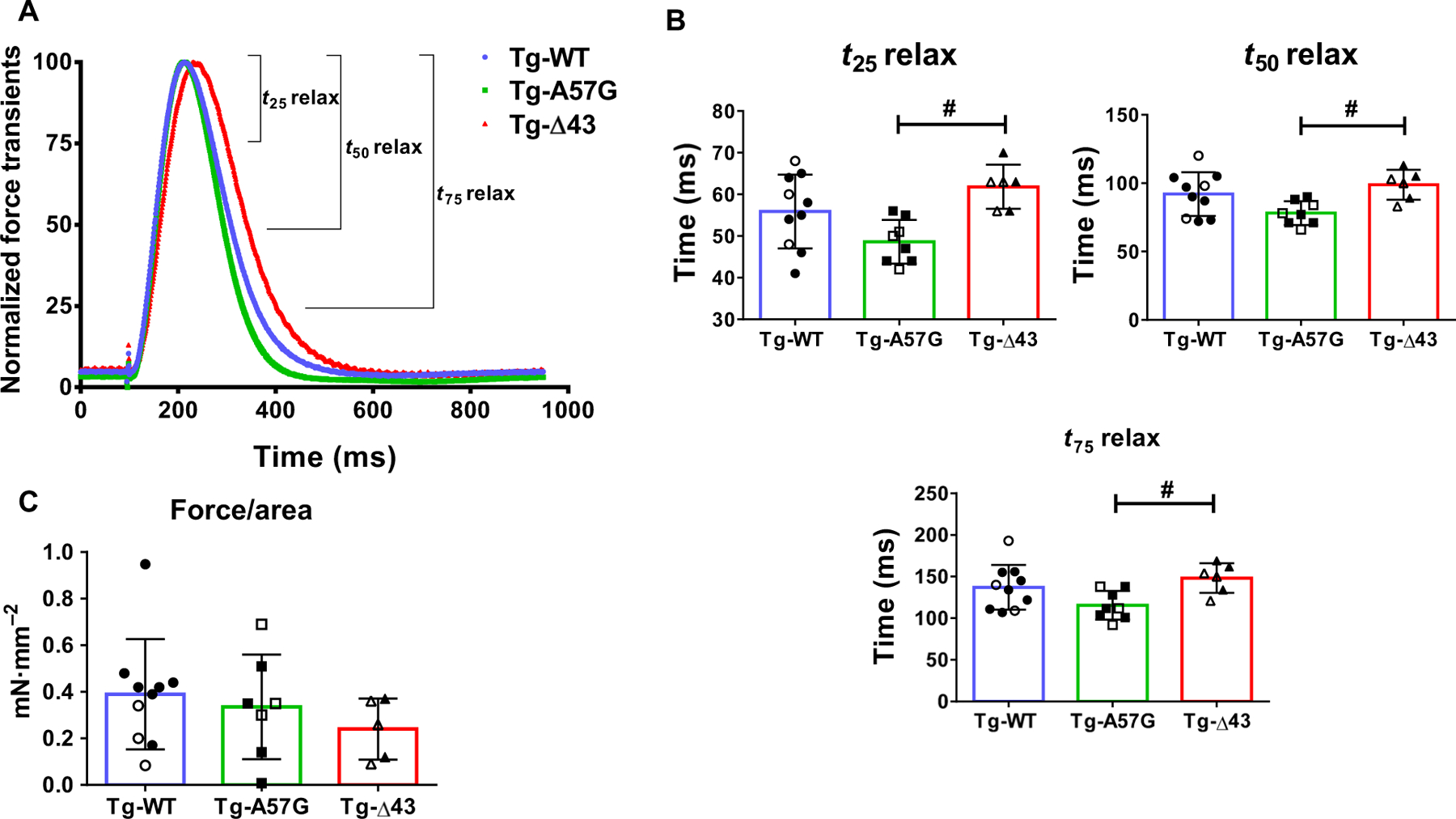
(A) Force transients in intact PM fibers from Tg-A57G and Tg-Δ43 compared with Tg-WT mice. Open symbols depict female (F), while closed symbols depict male (M) mice. (B) T25 relax, T50 relax, and T75 relax depict the time (in ms) from peak force to 25%, 50%, and 75% toward baseline. (C) Force (in mN·mm−2) was calculated as a ratio of peak force/physiologic cross-sectional area (see Materials and methods). Note faster relaxation times for Tg-A57G compared to Tg-Δ43 mice. Values are mean of n = 8 (3F and 5M) Tg-A57G, n = 6 (3F and 3M) Tg-Δ43, and n = 10 (3F and 7M) Tg-WT animals ± SD with #P < 0.05 depicting significance between Tg-A57G and Tg-Δ43, calculated by one-way ANOVA with Tukey’s multiple comparison test.
Table 2.
Intact papillary muscle fiber studies on Tg-WT, Tg-A57G, and Tg-Δ43 mice. Force and calcium transients times (T) from peak force/[Ca2+] to 25, 50, and 75 percent of relaxation/baseline. Values are mean T SD for n = # animals.
| Tg-WT | Tg-A57G | Tg-Δ43 | |
|---|---|---|---|
| n = # animals (#F and #M mice) | 10 (3F and 7M) | 8 (3F and 5M) | 6 (3F and 3M) |
| Force transients | |||
| T25 relax (ms) | 55.9 ± 8.8 | 48.6 ± 5.2# | 61.8 ± 5.3 |
| T50 relax (ms) | 92.0 ± 15.9 | 78.1 ± 8.6# | 98.8 ± 11.0 |
| T75 relax (ms) | 137.2 ± 26.8 | 115.6 ± 17.3# | 148.3 ± 17.9 |
| Force/area (mNmm−2) | 0.38 ± 0.23 | 0.33 ± 0.22 | 0.24 ± 0.13 |
| [Ca2+] transients | |||
| T25 relax (ms) | 88.1 ± 13.5 | 107.4 ± 23.7 | 90.8 ± 17.8 |
| T50 relax (ms) | 148.2 ± 22.1 | 194.3 ± 41.7* | 154.7 ± 31.7 |
| T75 relax (ms) | 241.2 ± 18.0 | 320.0 ± 80.0* | 247.3 ± 52.3 |
P < 0.05 Tg-A57G versus Tg-WT.;
P < 0.05 Tg-A57G versus Tg-Δ43, one-way ANOVA with Tukey’s multiple comparison test.
To investigate the effect of ELC mutations on Ca2+ homeostasis and sarcoplasmic reticulum Ca2+ release and uptake, intact PM from mice were loaded with Fura-2 Ca2+ indicator and [Ca2+] transients were recorded. In agreement with the hypercontractile behavior seen for A57G cross-bridges, [Ca2+] transients were significantly delayed in Tg-A57G mice compared with Tg-WT animals (Fig. 4A), suggesting a potential for HCM-A57G-induced diastolic dysfunction. The differences in t50 and t75 between Tg-A57G and Tg-WT muscles were statistically significant (Fig. 4B), while no differences were noted between Tg-Δ43 and Tg-WT mice, indicating no abnormalities in heart diastolic function in Tg-Δ43 animals (Fig. 4B; Table 2).
Fig. 4.
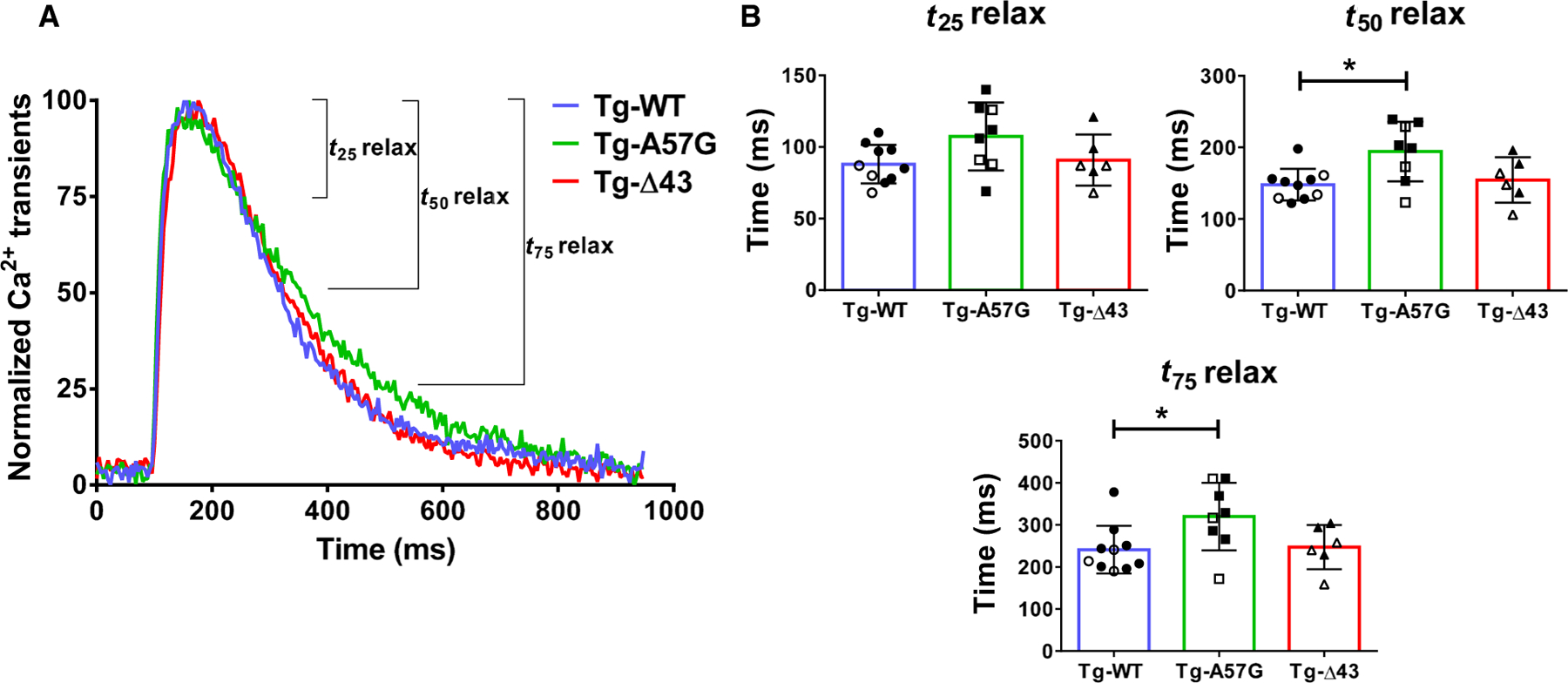
(A) Calcium transients in intact PM fibers from Tg-A57G and Tg-Δ43 compared with Tg-WT (3F and 7M) mice. Open symbols depict F, while closed symbols depict M mice. (B) T25 relax, T50 relax, and T75 relax depict the time (in ms) from peak [Ca2+] to 25%, 50%, and 75% toward baseline. Note delayed relaxation times for Tg-A57G compared to Tg-WT and Tg-Δ43 mice. Values are mean of n = 8 (3F and 5M) Tg-A57G, n = 6 (3F and 3M) Tg-Δ43, and n = 10 (3F and 7M) Tg-WT animals T SD with *P < 0.05 calculated by one-way ANOVA with Tukey’s multiple comparison test.
In vivo assessments of Tg-ELC mice show LV hypertrophy and abnormal cardiac function in Tg-A57G animals and LV hypertrophy without cardiac dysfunction in Tg-Δ43 mice
To gain insight into A57G-versus Δ43-related heart remodeling in vivo, cardiac morphology and function were assessed in all experimental groups of mice using B- and M-mode echocardiography, and pulse-wave and tissue Doppler analysis, as described in Yadav et al. [28]. Heart rate (HR) and body temperature (37.0 ± 1 °C) were carefully monitored and controlled during data acquisition to avoid any effects on cardiac function and subsequent data analysis. The average HR was 459 ± 61 and 444 ± 58 for Tg-A57G and Tg-Δ43 mice, respectively, compared to 477 ± 45 for WT (Table 3). No differences in ejection fraction (EF) or fractional shortening (FS) were observed among Tg-A57G, Tg-Δ43, and Tg-WT animals (Table 3). M-mode echocardiographic measurements revealed a significant increase in LV anterior wall thickness in diastole and systole (LVAW;d,s) and in posterior wall thickness (LVPW;d,s) for Tg-A57G compared with Tg-WT (Fig. 5A; Table 3). Likewise, Tg-Δ43 hearts demonstrated significant increases in LVPW;d,s and LVAW;d, indicating cardiac hypertrophy in N terminus ELC-truncated Tg-Δ43 mice (Table 3). Consistently, LV mass (in mg) obtained from M-mode measurements was increased in Tg-Δ43 (169 ± 52) compared with Tg-WT (123 ± 28) mice and in Tg-A57G (168 ± 43) versus Tg-WT animals, but the differences did not reach statistical significance (Fig. 5A; Table 3). No differences were observed in LV inner diameter (LVID) in systole and diastole among all mouse models.
Table 3.
Echocardiography assessment of heart morphology and function of Tg-WT, Tg-A57G, and Tg-Δ43 mice. Values are mean ± SD for n = # animals. d, diastole; e’, early diastolic mitral annulus velocity; EF%, ejection fraction; FS%, fractional shortening; HR, heart rate in beats per minute (bpm); IVCT, isovolumetric contraction time; IVRT, isovolumetric relaxation time; LVAW, left ventricular anterior wall; LVID, LV inner diameter; LVPW, LV posterior wall; MPI, myocardial performance index; MV E/A, mitral valve inflow—peak E (early)-wave-to-A (late)-wave velocity ratio; s, systole.
| Tg-WT | Tg-A57G | Tg-Δ43 | |
|---|---|---|---|
| n = # animals (# M and # F) | 9 (6M, 3F) | 9 (3M, 6F) | 9 (6M, 3F) |
| B- and M-modes | |||
| HR (bpm) | 477 ± 45 | 459 ± 61 | 444 ± 58 |
| EF (%) | 54 ± 3 | 55 ± 6 | 52 ± 3 |
| FS (%) | 31 ± 7 | 40 ± 14 | 42 ± 8 |
| LVAW;d (mm) | 0.96 ± 0.07 | 1.17 ± 0.11* | 1.10 ± 0.12* |
| LVAW;s (mm) | 1.43 ± 0.16 | 1.70 ± 0.22* | 1.59 ± 0.31 |
| LVID;d (mm) | 3.94 ± 0.56 | 3.66 ± 0.36 | 3.60 ± 0.44 |
| LVID;s (mm) | 2.74 ± 0.62 | 2.20 ± 0.66 | 2.27 ± 0.64 |
| LVPW;d (mm) | 1.00 ± 0.18 | 1.43 ± 0.40* | 1.51 ± 0.38* |
| LVPW;s (mm) | 1.32 ± 0.23 | 1.94 ± 0.47* | 1.88 ± 0.42* |
| LV mass/corr (mg) | 123 ± 28 | 168 ± 43 | 169 ± 52 |
| PW-Doppler and TDI | |||
| IVCT (ms) | 12.8 ± 2.33 | 14.2 ± 2.88 | 13.93 ± 3.70 |
| IVRT (ms) | 16.4 ± 2.73 | 22.9 ± 1.34*,# | 17.2 ± 1.15 |
| MV A (mm·s−1) | 449 ± 120 | 359 ± 180 | 431 ± 104 |
| MV E (mm·s−1) | 697 ± 160 | 571 ± 144 | 676 ± 153 |
| MV E/A | 1.44 ± 0.25 | 1.88 ± 0.75 | 1.57 ± 0.58 |
| MPI | 0.65 ± 0.13 | 0.80 ± 0.13*,# | 0.65 ± 0.11 |
| E/e’ | −24.2 ± 5.55 | −22.2 ± 6.65 | −27.1 ± 6.68 |
| e’ (mm·s−1) | −26.2 ± 6.65 | −28.0 ± 9.52 | −25.8 ± 6.14 |
P < 0.05 Tg-A57G/Tg-Δ43 versus Tg-WT.;
P < 0.05 Tg-A57G versus Tg-Δ43, one-way ANOVA with Tukey’s multiple comparison test.
Fig. 5.
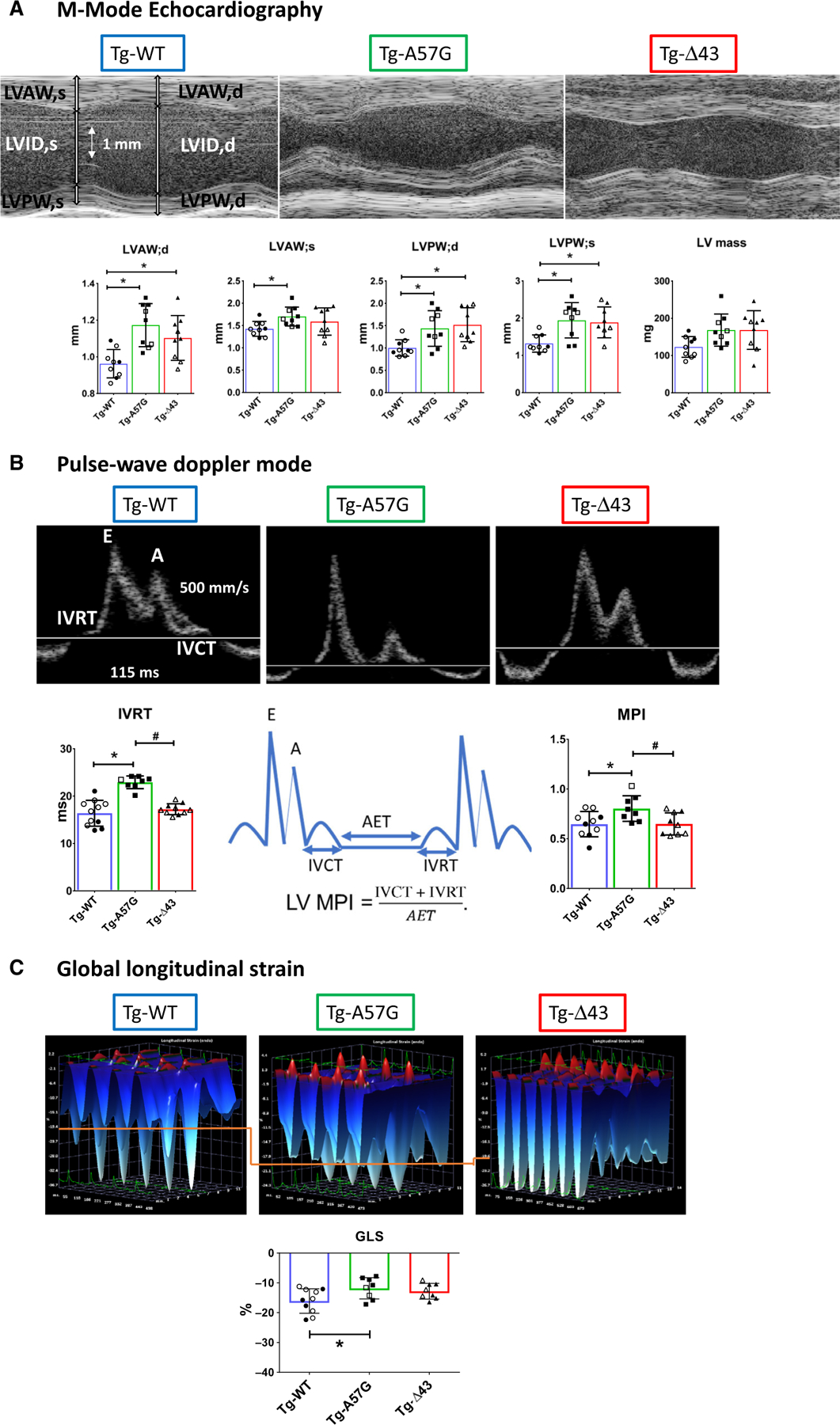
Echocardiography measurements in Tg-WT, Tg-A57G, and Tg-Δ43 mice. (A) Upper panel: Representative echocardiographic images of mouse hearts in M-mode. Lower panel: The assessment of heart morphology and function in Tg-A57G (n = 9 mice) and Tg-Δ43 (n = 9 mice) compared to Tg-WT (n = 9 mice). Open symbols depict F, while closed symbols depict M mice. Note increased wall thickness in Tg-A57G and Tg-Δ43 compared to Tg-WT. LVAW;d/s, left ventricular anterior wall thickness in diastole/systole; LVPW, LV posterior wall thickness. (B) Upper panel: Representative pulse-wave and tissue Doppler images of transgenic hearts. Lower panel: Assessment of IVRT (isovolumetric relaxation time) and MPI (myocardial performance index) in mice. Note increased IVRT and MPI in Tg-A57G compared to Tg-WT and Tg-Δ43. (C) Upper panel: Representative three-dimensional longitudinal strain deformation images for Tg-WT, Tg-A57G, and Tg-Δ43 hearts showing contraction (red, positive) and relaxation (blue, negative) over six LV segments during four consecutive cardiac cycles. The orange line has been added to aid comparison of the images’ scale. Lower panel: Speckle tracking-based strain analysis (Global longitudinal strain) on Tg-WT, Tg-A57G, and Tg-Δ43. Lower (less negative values) in Tg-A57G versus Tg-WT, indicative of poorer global cardiac function in HCM Tg-A57G mouse model. Data are mean of n = 9 animals/per group T SD with *P < 0.05 depicting significance between Tg-WT and Tg-A57G and #P < 0.05 between Tg-A57G and Tg-Δ43, one-way ANOVA with Tukey’s multiple comparison test.
Pulse-wave and tissue Doppler imaging (TDI) showed significantly increased isovolumetric relaxation time (IVRT, in ms) in Tg-A57G (22.9 ± 1.34) compared with WT (16.4 ± 2.73) mice, but no differences were observed in Tg-Δ43 (17.2 ± 1.15) versus WT animals (Fig. 5B, Table 3). This result was consistent with delayed Ca2+ transients only observed in Tg-A57G mice, suggesting HCM-related diastolic dysfunction in this mouse model, as observed previously [7]. The myocardial performance index (MPI), widely accepted as an indicator of cardiac function and independent from HR and LV shape [29], was significantly higher in Tg-A57G mice: MPI = 0.80 ± 0.13 compared with Tg-WT: MPI = 0.65 ± 0.13 and Tg-Δ43: MPI = 0.65 ± 0.11, indicating signs of HCM-related pathology only in Tg-A57G mice (Fig. 5B; Table 3).
We also performed speckle tracking-based echocardiography (STE) (Fig. 5C), shown to be a valuable tool in assessing myocardial function in clinical and animal studies [30]. It represents the degree of myocardial lengthening or shortening and provides an accurate representation of global and regional LV function. One important parameter we can derive from these analyses is global longitudinal strain (GLS), defined as the change in length of myocardial tissue compared to the original length and used as a marker of myocardial deformation and LV impairment [30]. A greater degree of deformation or increased strain is associated with numerically lower (more negative) GLS values. As shown in Fig. 5C, STE analysis showed a reduced LV deformation/shortening in Tg-A57G mice, manifested by a lesser negative value of GLS = −11.8 ± 3.56% compared to Tg-WT (GLS = −16.0 ± 4.09%), while no significant changes were observed in Tg-Δ43 myocardium. Representative three-dimensional LV longitudinal strain deformation images obtained from Tg-WT, Tg-A57G, and Tg-Δ43 animals show contraction/thickening (red/positive values) or relaxation/thinning (blue/negative values) over six LV segments during four consecutive cardiac cycles (Fig. 5C). Note that LV longitudinal strain deformation has a different scale for Tg-WT compared to Tg-A57G and Tg-Δ43, and an orange line linking the scales between the graphs has been drawn to aid with comparison.
Interstitial fibrosis in HCM-A57G, and not Δ43 hearts, underlies pathological heart remodeling in Tg-A57G mice compared with the physiologic-like cardiac hypertrophy observed in Tg-Δ43 animals
Fibrosis is a common pathology in HCM, causing mechanical and electrical cardiac dysfunction [31]. Pathological cardiac remodeling in hypertrophic cardiomyopathy disease is often characterized by elevated appearance of fibrotic lesions that can be quantitatively assessed by myocardial hydroxyproline (HOP) concentration [32]. HOP is a major component of collagen produced from hydroxylation of proline amino acid and is used as a diagnostic marker for fibrosis. The amount of HOP was measured in LV and intra-ventricular septum (IVS), and both regions were shown to express high amount of fibrotic tissues in HCM disease (Fig. 6). For HOP assessments, n = 10–14 heart samples from Tg-WT (10 mice), Tg-A57G (8 mice), and Tg-Δ43 (8 mice) were used. Tg-A57G hearts showed a significantly higher HOP content (in nmoles HOP per mg tissue) that was equal to 5.5 ± 1.6 compared with Tg-WT (3.9 ± 1.4) and Tg-Δ43 (3.7 ± 1.0) mice (Fig. 6). These results are in accord with our previous findings on occurrences of fibrosis observed in Tg-A57G mouse model compared with Tg-WT and assessed by Masson’s trichrome staining [7]. Results of Fig. 6 are also consistent with histopathological findings in Tg-A57G versus Tg-Δ43 hearts showing positive Masson’s staining only in Tg-A57G myocardium [8]. The HOP data quantitatively differentiate the pathological type of remodeling associated with HCM-A57G mutation compared with the N-ELC deletion mutation that leads to physiologic-like cardiac hypertrophy in Tg-Δ43 mice. Altogether, our in vivo and in vitro studies clearly demonstrate that both ELC mutants recapitulate LV cardiac hypertrophy, but only the A57G mutation leads to a pathological cardiac remodeling with hypercontractile cross-bridge behavior and subsequent diastolic dysfunction.
Fig. 6.
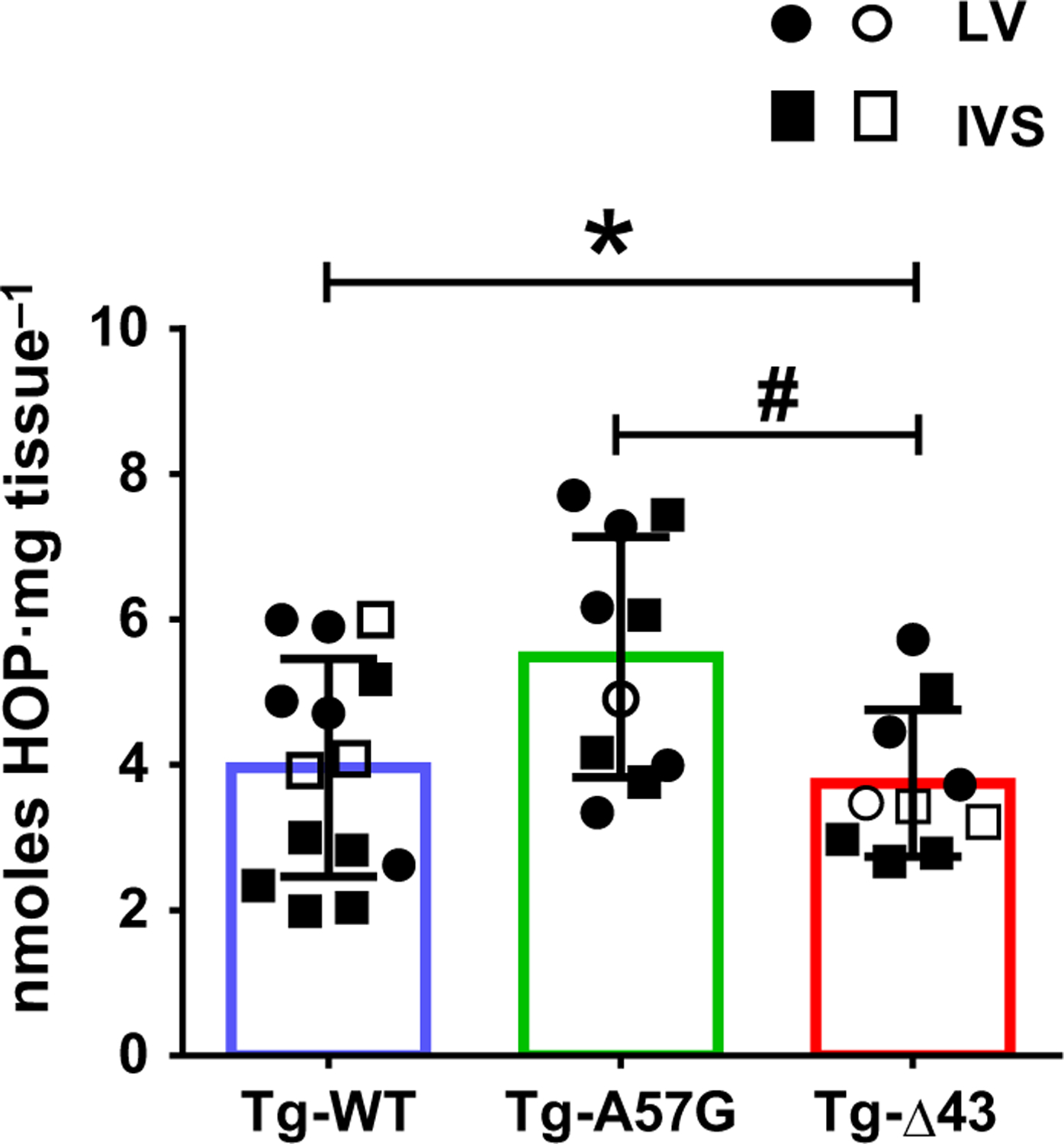
Hydroxyproline (HOP) content in the hearts of Tg-WT, Tg-A57G, and Tg-Δ43 mice. Ten to fourteen samples from the hearts of eight to ten mice were used: 10 hearts for Tg-WT (n = 14 samples) and eight hearts for Tg-A57G (n = 10 samples) and Tg-Δ43 (n = 10 samples). Open symbols depict preparations from F, while closed symbols depict preparations from M mice. Note increased HOP only in the hearts of Tg-A57G mice. Data are mean of n samples ± SD with *P < 0.05 depicting significance between Tg-WT and Tg-A57G and #P < 0.05 between Tg-A57G and Tg-Δ43, one-way ANOVA with Tukey’s multiple comparison test.
Discussion
Cardiac muscle contraction consists of ATP-dependent cyclic interactions of myosin cross-bridges with Ca2+/tropomyosin/troponin—regulated actin filaments [33,34]. Considering the importance of cardiac essential light chains for myosin function and the structural stability of the myosin lever arm, it is not surprising that similar to other genes (MYH7, MBPC3, MYL2), hereditary mutations in MYL3, residing in human ventricular ELC, can lead to HCM [3,35–39]. In this study, we continue our efforts in unraveling the mechanisms associated with the ELC-A57G mutation found to cause HCM in humans [3] and in mice [7]. Specifically, we aim to understand the role the unique N terminus ELC extension plays in the actin–myosin interaction and generation of contractile force in healthy Tg-WT hearts as well as how it works in HCM, recapitulated in Tg-A57G mouse model [6–8]. The question is whether N-ELC, by virtue of its direct contacts with actin [5,8,15–18,20,21], can regulate the proportion of myosin heads that are available for their interaction with actin and force production in muscle, and whether it can regulate the myosin ATP turnover rate and the energetic state of the sarcomere. Our previous study revealed that ablation of the N-ELC in Tg-Δ43 mice led to cardiac hypertrophy, but the hearts still maintained normal cardiac performance [5]. The development of two different forms of cardiac hypertrophy is assessed at the level of myosin cross-bridges and confirmed in vivo by echocardiography using two ELC-humanized transgenic mice: Tg-A57G, as a model of HCM, and Tg-Δ43, as a model of physiologic-like cardiac growth.
Using a human β-cardiac myosin interactive head motif (IHM) quasi-atomic model, the Padron Group defined important interactions sites between adjacent myosin heads and associated protein partners, including myosin ELC, and how the IHM structure, considered to be the ‘hot spot’ for HCM mutations, may change in the presence of cardiomyopathy mutations [13]. Proposed hypotheses suggested that impaired IHM formation and stability, and attenuation of the super-relaxed state and increased energy consumption may be the mechanisms underlying the altered contractility and diastolic relaxation in HCM [40]. Like many of MHC mutations, A57G-ELC is located near the MHC converter and lever arm domains; the structural regions in the myosin head that are part of IHM, and similar to other disease-associated mutations positioned in IHM, are thought to be likely pathogenic [13,41]. It has been postulated that mutations that were seen to destabilize the myosin-sequestered state, in which the heads are inactive and unable to interact with actin, were considered to have the major negative impact on heart remodeling in HCM [14]. The inhibition of ATP turnover by myosin heads in the SRX state is caused by binding of the myosin heads to the core of the thick filament forming the IHM [13]. We used the SRX paradigm to analyze the effect of ELC modifications in the hearts of Tg-A57G and Tg-Δ43 mice on the sequestered myosin state and the proportion of heads that are available to interact with actin, using mantATP chase assays developed by Cooke et al. [10,11]. We show that cardiac N-ELC directly contributes to the regulation of actin–myosin interactions by controlling the proportion of myosin heads that are functionally available to interact with actin and produce force (Table 1). The loss of the N-ELC in Tg-Δ43 mice promotes the SRX state with low ATP turnover (Fig. 1) and shows reduced actin-activated myosin ATPase activity (Fig. 2). This inhibited ATP turnover by Δ43-ELC myosin could eventually result in a low metabolic rate of Tg-Δ43 hearts, leading to cardiac hypertrophic growth, as actually observed in aging Tg-Δ43 mice [5,8].
The question still remains by what mechanisms a single mutation in the ELC protein leads to decreased SRX cross-bridge population, while a large deletion Δ43 leads to increased SRX population? Since the A57G ELC mutation is part of IHM [13,41], it is expected to result in the pathological phenotype, which was actually confirmed in vivo in Tg-A57G mice [6–8]. The hypercontractility phenotype associated with A57G was manifested by increased calcium sensitivity of contraction in skinned papillary muscle fibers and increased cardiac output in vivo by echocardiography and invasive hemodynamics [7]. Using small-angle X-ray diffraction, we showed that A57G mutation led to decreased interfilament lattice spacing and increased I1,1/I1,0 ratio in rigor when all cross-bridges are attached to actin, both measures suggestive of hypercontractile cross-bridge behavior [8]. In agreement with our previously published structural data, Figs 1 and 2 of the current report support the A57G-mediated hypercontractile activity manifested by inhibited myosin’s sequestered state and enhanced transition from the SRX to DRX state as well as increased actin-activated myosin ATPase activity of A57G-mutated cardiac preparations. Findings for A57G are consistent with other studies linking HCM mutations in cardiac sarcomere with a disrupted SRX [22,25], but are contradictory to other reports showing HCM-associated R58Q mutation in myosin RLC promoting the SRX state [24,42]. Structural work from the Houdusse laboratory reconciles these models and explains how distinct HCM mutations can have disparate effects on the motor mechanochemical parameters and yet lead to the same disease [41].
Contrary to the observations in HCM-A57G, the deletion of the N-ELC in Tg-Δ43 mice led to stabilization of the SRX (Fig. 1), decreased myosin ATPase activity (Fig. 2), and longer relaxation times of force transients (Fig. 3). These data are in agreement with the findings of Miller and coworkers who suggested a new role for the N-ELC to inhibit the reversal of the power stroke, thereby increasing the number of myosin heads available for myosin–actin interaction [21]. Based on our data, we can conclude that cardiac N-ELC works as a molecular linker and/or energetic switch of the actin–myosin interaction by regulating the transition of myosin cross-bridges from SRX to DRX state and altering the proportion of myosin heads that are available for the interaction with actin. The lack of N-ELC in Tg-Δ43 mice promotes the SRX and counteracts the hypercontractile HCM cross-bridge behavior. Interestingly, results of our previous investigation demonstrated that the cross-bridge kinetics in Tg-Δ43 skinned PM preparations were accelerated owing to an acceleration of the cross-bridge detachment step and that the number of strongly attached cross-bridges was decreased because of a reduction of the equilibrium constant of the force generation step [19]. Quantum dot-labeled actin motility assays performed on myosin purified from Tg-Δ43 and Tg-A57G hearts led us to the conclusion that the N-ELC uses the novel mechanism of step-size and step-frequency modulation to affect power output, providing basis for regulating cardiac myosin power generation in vivo, and that the disease-causing A57G mutation located in the vicinity of the N-ELC may work through direct alterations of N-ELC–actin interactions and force production in muscle [15,18]. These earlier data are fully supported by the current investigation and highlight the importance of the N-ELC in maintaining the myosin motor function during force generation and optimal cardiac performance.
In vivo results, from echocardiography examination of Tg-A57G and Tg-Δ43 mice and from intact papillary muscle fiber studies, confirmed an A57G-mediated pathological type of heart remodeling compared to physiologic-like hypertrophy observed in transgenic ELC-Δ43 mice (Tables 2 and 3). In accord with A57G-induced increase in the Ca2+ sensitivity of force [8], delayed Ca2+ transients were observed in Tg-A57G mice (Fig. 4), suggesting diastolic dysfunction in HCM-A57G model. MPI and GLS indices and increased fibrosis, quantified by the amount of HOP/mg of LV and IVS samples (Fig. 6), supported decompensated function in Tg-A57G mice and no abnormalities in function for Tg-Δ43 hearts. While LV fibrosis is an important feature of HCM [43–45], it is not present in physiologic cardiac hypertrophy [46], consistent with our findings. It is worth mentioning that the findings of the current report are in agreement with our previous work showing that passive tension/stiffness is only increased in Tg-A57G, indicating that fibrosis and resting stiffness contribute to the pathological HCM remodeling [8].
Conclusions
We have proposed a regulatory mechanism for the development of different forms of hypertrophy phenotypes in mice expressing mutations in cardiac myosin essential light chain. Cardiac myosin ELC is a sarcomeric protein that contains a unique N terminus (N-ELC) capable of regulating cardiac contractility and the proportion of myosin heads that are available for the interaction with actin. The A57G HCM-causing mutation is positioned in the vicinity of N-ELC and is shown to promote the shift from the SRX to DRX state that mechanistically underlies the clinical phenotype of hypercontractility associated with HCM. The N-ELC truncation in Tg-Δ43 mice leads to physiologic-like hypertrophy by promoting the SRX-sequestered state of cardiac myosin. Importantly, deletion of N-ELC counteracts the A57G-mediated hypercontractile cross-bridge behavior. Our data suggest that cardiac N-ELC may work as a molecular linker and/or energetic switch of the actin–myosin interaction, and as such could be targeted therapeutically to modify the proportion of myosin heads readily available for the interaction with actin, power stroke execution, and force production in cardiac muscle. To date, this is the first study showing that alterations of the SRX state in ELC mouse models of different cardiac remodeling may result in pathological or physiologic-like cardiac hypertrophy in mice.
Materials and methods
Transgenic mice
All animal procedures and experiments were performed in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ (NIH Publication 85–23, revised 2011). All the protocols were approved by the Institutional Animal Care and Use Committee at the University of Miami Miller School of Medicine, which has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare (OLAW), NIH. The assurance number is #A-3224–01, approved through November 30, 2019. We are registered with USDA APHIS, registration # 58-R-007, approved through December 3, 2020. We have full accreditation with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC International), site 001069, latest effective date, November 8, 2016. Mice were euthanized through CO2 inhalation followed by cervical dislocation. IACUC protocol # is 18–110-LF.
The generation of Tg mice, expressing human ventricular ELC-WT or human ELC mutants, used in this study was previously described in Kazmierczak et al. [5] for Tg-Δ43 and Tg-WT-ELC mice, and in Muthu et al. [8] for Tg-A57G. Tg-A57G lines: L1 and L5 expressing 80% and 74% of A57G mutant, respectively, and Tg-Δ43 L8 and L9 expressing 34–39% and 35–40% of Δ43-ELC were used in this study. Tg-WT L1 and L4 expressing 76% and 71% of WT-ELC, respectively, were used as controls. The percentage of protein expression indicates the amount of replacement of the endogenous mouse ventricular ELC by the human ventricular WT or mutants. All experiments were performed on 8- to 11-month-old male (M) and female (F) mice.
Preparation of skinned papillary muscle (PM) fibers
The mouse LV PM were isolated from the hearts of F and M mice from all groups, dissected into small bundles, and chemically skinned for 24 h at 4 °C. The skinning solution contained 50% glycerol and 50% pCa 8 solution [10−8 m (Ca2+), 1 mm free (Mg2+), total MgPr (propionate) =3.88 mm, 7 mm EGTA, 2.5 mm (Mg-ATP2−), 20 mm MOPS pH 7.0, 15 mm creatine phosphate, 15 U·mL−1 of phosphocreatine kinase, ionic strength = 150 mm (adjusted with KPr), and 1% Triton X-100] [24,28]. Muscles were then transferred to the fresh solution without Triton X-100 and stored at −20 °C until used (5–10 days).
For muscle mechanics experiments, small muscle strips composed of approximately four to six single muscle fibers were isolated from a batch of glycerinated PM bundles and attached to the force transducer of the Guth Muscle Research System (Heidelberg, Germany). Then, the fibers were placed in a 1-mL cuvette and skinned in 1% Triton X-100 (in pCa 8 buffer) for 30 min, washed three times for 5 min in pCa 8 buffer, and tested for steady-state force development in pCa 4 solution [composition is the same as pCa 8 buffer except the (Ca2+) = 10−4 m]. After the initial steady-state force determined in pCa 4 (in kN·m−2), muscle strips were relaxed in pCa 8 and exposed to solutions of increasing Ca2+ concentrations to establish the force-pCa dependence and myofilament Ca2+ sensitivity (pCa50) [6,7]. Muscle strips that passed the quality control of reaching reproducible values of maximal force and pCa50 were used to perform the SRX experiments.
ATP chase experiments
To measure the super-relaxed state of myosin in skinned PM fibers from all groups of mice, we used fluorescent mantATP, an analogue of ATP that binds strongly to myosin and shows ~ 2.5-fold increase in fluorescence emission compared to its dissociated form when exchanged for dark ATP [12]. The mantATP/ATP chase experiments were performed using IonOptix Calcium and Contractility Recording System as described in detail in Yadav et al. [24]. PM fibers of about 100 µm in diameter were dissected from glycerinated PM bundles, rinsed in pCa 8 solution followed by several washes with rigor buffer containing 120 mm KPr, 5 mm MgPr, 2.5 mm K2HPO4, 2.5 mm KH2PO4, and 50 mm 3-(n-morpholino) propanesulfonic acid, pH 6.8, and fresh 2 mm DTT. Subsequently, the fibers were attached to a glass slide and covered with a coverslip to form a flow chamber for nucleotide exchange. The experiment was initiated with fibers’ incubation with 250 µM mantATP (in rigor buffer) until maximum fluorescence was monitored. Then, mantATP was chased with 4 mm unlabeled ATP (in rigor buffer) that resulted in fluorescence decay as hydrolyzed mantATP was exchanged for dark ATP. The sarcomere length of fibers varied from 1.9 to 2.2 µm and was found to cause no changes in fluorescence decay curves [24]. Fluorescence intensity as a function of time was fit to a two-exponential decay function using a nonlinear least-squares algorithm in GRAPHPAD PRISM version 7 (GraphPad Software, San Diego, CA, USA). P1 and P2 depict the fractions of fluorescence amplitude, characterized by the fast T1 and slow T2 fluorescence lifetimes, and indicate the proportion of myosin heads in the DRX and SRX states, respectively. 1/T1 and 1/T2 represent the rates of ATP turnover per myosin head in the DRX and SRX states.
Actin-activated myosin ATPase activity assays
For myosin ATPase experiments, myosin was purified from left and right ventricles of mouse hearts and actin was extracted from rabbit skeletal acetone powder and purified as described previously [5,24]. Briefly, 0.6 µM myosin was complexed with increasing actin concentrations (0–15 µM) in the reaction solution containing 25 mm imidazole pH 7.0, 4 mm MgCl2, 1 mm EGTA, and 1 mm DTT (final KCl salt concentration was 40 mm). The reaction was initiated with the addition of 2.5 mm ATP, allowed to proceed for 7 min at 22 °C, and terminated by addition of 4% ice-cold trichloroacetic acid. Precipitated proteins were cleared by centrifugation, and the inorganic phosphate was determined according to the Fiske and Subbarrow method. The Vmax and Km values were established using the Michaelis–Menten equation as described previously [5].
Intact papillary muscles studies
Intact PM fiber studies were performed according to protocols described in detail in [26]. Briefly, after euthanasia, the hearts of mice were excised, rinsed in Krebs-Ringer solution containing 30 mm 2,3-butanedione monoxime (BDM). Then, the PM were carefully dissected from the LV in preoxygenated (95% O2–5% CO2) BDM–Krebs–Ringer solution consisting of (in mm): Na+ 152, K+ 3.6, Cl− 135, , Mg2+ 0.6, H2PO4− 1.3, , Ca2+ 2.5, and glucose 5.6. Subsequently, PM were incubated in BDM–Krebs–Ringer buffer containing 50 µm Fura-2 AM (Life Technologies Corporation, Eugene, OR, USA), dissolved in DMSO and 0.1% pluronic acid at room temperature for 1.5 h in dark, followed by 3 washes in BDM–Krebs–Ringer solution to remove excess Fura-2 AM [26]. Physiologic cross-sectional area (in mm2), used for normalizing peak force, was calculated as muscle mass (mg) divided by the product of muscle density (~ 1.05 mg·mm−3) and muscle fiber length (~ 0.5 mm—optimal muscle length). The final contractile force data were presented as force/area in mN·mm−2. An inverted fluorescence microscope equipped with a calcium photometry objective and MyoCam-S™ digital variable field rate CCD video system was used to detect and record [Ca2+] transients of fibers loaded with Fura-2 AM. Fura-2 AM fluorescence was collected by a CCD camera from 340 and 380 nm channels, followed by background subtraction. Up to 1000 length recordings per second were collected, and the data points were averaged from ~ 20 force and [Ca2+] transient peaks. The data were analyzed using the IONWIZARD software [26].
Echocardiography
We monitored cardiac function and morphology of Tg-A57G, and Tg-Δ43 versus Tg-WT hearts using a VEVO 2100 and VEVO 1.7.1 software (Visual Sonics Inc.) image system as described previously [26,47]. Heart images were recorded from mice under isoflurane inhalation anesthesia (1–2%) with monitored heart rates (HR) (>400 beats per min) and body temperature (37 ± 1 °C). M-mode and B-mode images were saved for analysis of cardiac morphology and function. The following parameters were determined: LV end-diastolic and end-systolic dimensions, LV posterior and anterior wall thickness, EF, and FS. End-diastolic and end-systolic endocardial volumes were calculated from the bidimensional long-axis parasternal views [26,47].
Speckle tracking-based echocardiographic (STE) strain analysis
As recently described [28], high-resolution long-axis B-mode images were used to perform STE analyses using the vevo strain software (vevo lab 2.1.0). Changes in length (in the radial axis from the center of the ventricular cavity outward and in the longitudinal axis from the apex to the base of the heart) relative to initial length (final length/initial length) and strain rate, describing the rate of change of deformation over time, were measured and analyzed. Global LV longitudinal and radial strain and strain rates were evaluated in both epicardial and endocardial regions. In addition, B-mode images were recorded, with vector diagrams depicting magnitude and direction of endocardial deformation [28]. The LV global longitudinal strain (GLS) assessments, depicting the maximal shortening of myocardial longitudinal length during systole, were compared to those in diastole and the resting myocardial length. A greater degree of deformation or increased strain/sarcomere shortening was expressed as a numerically lower (more negative) GLS value [28].
Pulse-wave (PW) and Tissue Doppler Imaging (TDI)
A Vevo-2100 imaging system, equipped with MS 400 transducer, was used to assess the LV diastolic function of mice that was analyzed using the values derived from the mitral inflow velocity curves obtained by pulse-wave Doppler method, as described previously [26,28]. Doppler indexes included the ratio of peak velocity of early to late diastolic filling of mitral inflow (E/A), isovolumetric contraction time (IVCT), isovolumetric relaxation time (IVRT), and ejection time (ET). Myocardial performance index (MPI) was calculated using the following equation [28]:
Tissue Doppler imaging was used for visualization of tissue movement in which the negative waves represent the early diastolic mitral annulus velocity or e’.
Assessment of fibrosis by hydroxyproline (HOP) assay
Fibrosis and collagen content was assessed in the hearts of 8- to 11-month-old M and F Tg-A57G, Tg-Δ43, and Tg-WT using HOP assay, as described previously [26,47]. Briefly, 15–20 mg of flash-frozen tissue from LV and IVS of mice hearts were first boiled in 200 µL of 6 m HCl at 110 °C overnight, and then, 5 µL aliquots of hydrolyzed tissue were added to 80 µL of 100 % isopropanol and allowed to react with 40 µL of 7% chloramine-T solution mixed at a 1 : 4 ratio with acetate citrate buffer [0.695 m sodium acetate, 0.174 m citric acid, 0.435 m NaOH, and 38.5% (vol/vol) isopropanol] for 5 min at room temperature [26]. 0.5 mL of Ehrlich reagent was then added to the mixture at a 3 : 13 ratio with isopropanol and incubated at 55 °C for 30 min. The mixture was subsequently placed on ice for 5 min and centrifuged at 5000 g for 1 min at 4 °C. 200 µL aliquots were then placed in a 96-well plate, and the absorbance was measured at 558 nm. The total amount of HOP was calculated using the standard curve of trans-4-hydroxy-L-proline (0–500 µm). Assay was performed in triplicate on n = 10–14 heart samples from eight to ten Tg-A57G, Tg-Δ43, and Tg-WT mice.
Statistical analysis
Unless otherwise stated, all values are shown as means ± SD (standard deviation). Statistically significant differences between multiple groups were assessed using one-way ANOVA and Tukey’s multiple comparison test using GRAPHPAD PRISM software version 7.0 for Windows. Significance was defined as P < 0.05.
Acknowledgements
This work was supported by the National Institutes of Health [Grant number HL123255 (DSC)]. We would like to thank Nicole Fernandez and Jacob Rowe for providing assistance with the ATP chase experiments.
Abbreviations
- A wave
mitral valve inflow (late)
- B-mode
brightness mode (ultrasound)
- DRX
disordered relaxed state
- E
mitral valve inflow (early)
- e’
early diastolic mitral annulus velocity
- ELC
essential light chain
- GLS
global longitudinal strain
- HCM
hypertrophic cardiomyopathy
- HOP
hydroxyproline
- IHM
interactive head motif
- LV
left ventricle
- M-mode
time-motion display of the ultrasound wave
- N-ELC
N terminus essential light chain
- P1
fraction of myosin cross-bridges in DRX
- P2
fraction of myosin cross-bridges in SRX
- PM
papillary muscles
- PW-Doppler
pulse-wave Doppler mode
- SL
sarcomere length
- SRX
super-relaxed state
- STE
speckle tracking-based echocardiography
- T1
nucleotide turnover lifetime in DRX
- T2
nucleotide turnover lifetime in SRX
- TDI
tissue Doppler imaging
- Tg
transgenic
- WT
wild-type
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Hernandez OM, Jones M, Guzman G & Szczesna-Cordary D (2007) Myosin essential light chain in health and disease. Am J Physiol Heart Circ Physiol 292, H1643–H1654. [DOI] [PubMed] [Google Scholar]
- 2.Buckingham M, Kelly R, Tajbakhsh S & Zammit P (1998) The formation and maturation of skeletal muscle in the mouse: the myosin MLC1F/3F gene as a molecular model. Acta Physiol Scand 163, S3–S5. [DOI] [PubMed] [Google Scholar]
- 3.Lee W, Hwang TH, Kimura A, Park SW, Satoh M, Nishi H, Harada H, Toyama J & Park JE (2001) Different expressivity of a ventricular essential myosin light chain gene Ala57Gly mutation in familial hypertrophic cardiomyopathy. Am Heart J 141, 184–189. [DOI] [PubMed] [Google Scholar]
- 4.Aydt EM, Wolff G & Morano I (2007) Molecular modeling of the myosin-S1(A1) isoform. J Struct Biol 159, 158–163. [DOI] [PubMed] [Google Scholar]
- 5.Kazmierczak K, Xu Y, Jones M, Guzman G, Hernandez OM, Kerrick WGL & Szczesna-Cordary D (2009) The role of the N-terminus of the myosin essential light chain in cardiac muscle contraction. J Mol Biol 387, 706–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kazmierczak K, Yuan C-C, Liang J, Huang W, Rojas AI & Szczesna-Cordary D (2014) Remodeling of the heart in hypertrophy in animal models with myosin essential light chain mutations. Front Physiol 5, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kazmierczak K, Paulino EC, Huang W, Muthu P, Liang J, Yuan CC, Rojas AI, Hare JM & Szczesna-Cordary D (2013) Discrete effects of A57G-myosin essential light chain mutation associated with familial hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol 305, H575–H589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muthu P, Wang L, Yuan CC, Kazmierczak K, Huang W, Hernandez OM, Kawai M, Irving TC & Szczesna-Cordary D (2011) Structural and functional aspects of the myosin essential light chain in cardiac muscle contraction. FASEB J 25, 4394–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matusovsky OS, Mayans O & Szczesna-Cordary D (2015) Molecular mechanism of muscle contraction: new perspectives and ideas. Biomed Res Int 2015, 694345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hooijman P, Stewart MA & Cooke R (2011) A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J 100, 1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart MA, Franks-Skiba K, Chen S & Cooke R (2010) Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci USA 107, 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNamara JW, Li A, Dos Remedios CG & Cooke R (2015) The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys Rev 7, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE & Padron R (2017) Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife, 6, pii: e24634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trivedi DV, Adhikari AS, Sarkar SS, Ruppel KM & Spudich JA (2018) Hypertrophic cardiomyopathy and the myosin mesa: viewing an old disease in a new light. Biophys Rev 10, 27–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Yuan CC, Kazmierczak K, Szczesna-Cordary D & Burghardt TP (2018) Single cardiac ventricular myosins are autonomous motors. Open Biol 8, pii: 170240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guhathakurta P, Prochniewicz E, Roopnarine O, Rohde JA & Thomas DD (2017) A cardiomyopathy mutation in the myosin essential light chain alters actomyosin structure. Biophys J 113, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guhathakurta P, Prochniewicz E & Thomas DD (2015) Amplitude of the actomyosin power stroke depends strongly on the isoform of the myosin essential light chain. Proc Natl Acad Sci USA 112, 4660–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Ajtai K, Kazmierczak K, Szczesna-Cordary D & Burghardt TP (2016) N-terminus of cardiac myosin essential light chain modulates myosin step-size. Biochemistry 55, 186–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Muthu P, Szczesna-Cordary D & Kawai M (2013) Characterizations of myosin essential light chain’s N-terminal truncation mutant Delta43 in transgenic mouse papillary muscles by using tension transients in response to sinusoidal length alterations. J Muscle Res Cell Motil 34, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowey S, Saraswat LD, Liu H, Volkmann N & Hanein D (2007) Evidence for an interaction between the SH3 domain and the N-terminal extension of the essential light chain in class II myosins. J Mol Biol 371, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller MS, Palmer BM, Ruch S, Martin LA, Farman GP, Wang Y, Robbins J, Irving TC & Maughan DW (2005) The essential light chain N-terminal extension alters force and fiber kinetics in mouse cardiac muscle. J Biol Chem 280, 34427–34434. [DOI] [PubMed] [Google Scholar]
- 22.Anderson RL, Trivedi DV, Sarkar SS, Henze M, Ma W, Gong H, Rogers CS, Gorham JM, Wong FL, Morck MM et al. (2018) Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci USA 115, E8143–E8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashrafian H, Redwood C, Blair E & Watkins H (2003) Hypertrophic cardiomyopathy: a paradigm for myocardial energy depletion. Trends Genet 19, 263–268. [DOI] [PubMed] [Google Scholar]
- 24.Yadav S, Kazmierczak K, Liang J, Sitbon YH & Szczesna-Cordary D (2019) Phosphomimetic-mediated in vitro rescue of hypertrophic cardiomyopathy linked to R58Q mutation in myosin regulatory light chain. FEBS J 286, 151–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNamara JW, Li A, Smith NJ, Lal S, Graham RM, Kooiker KB, van Dijk SJ, Remedios CGD, Harris SP & Cooke R (2016) Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J Mol Cell Cardiol 94, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan CC, Kazmierczak K, Liang J, Zhou Z, Yadav S, Gomes AV, Irving TC & Szczesna-Cordary D (2018) Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc Natl Acad Sci USA 115, E2338–E2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mast F & Elzinga G (1990) Heat released during relaxation equals force-length area in isometric contractions of rabbit papillary muscle. Circ Res. 67, 893–901. [DOI] [PubMed] [Google Scholar]
- 28.Yadav S, Yuan CC, Kazmierczak K, Liang J, Huang W, Takeuchi LM, Kanashiro-Takeuchi RM & Szczesna-Cordary D (2019) Therapeutic potential of AAV9-S15D-RLC gene delivery in humanized MYL2 mouse model of HCM. J Mol Med (Berl) 97, 1033–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaefer A, Meyer GP, Hilfiker-Kleiner D, Brand B, Drexler H & Klein G (2005) Evaluation of Tissue Doppler Tei index for global left ventricular function in mice after myocardial infarction: comparison with Pulsed Doppler Tei index. Eur J Echocardiogr 6, 367–375. [DOI] [PubMed] [Google Scholar]
- 30.Bauer M, Cheng S, Jain M, Ngoy S, Theodoropoulos C, Trujillo A, Lin FC & Liao R (2011) Echocardiographic speckle-tracking based strain imaging for rapid cardiovascular phenotyping in mice. Circ Res 108, 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rockey DC, Bell PD & Hill JA (2015) Fibrosis–a common pathway to organ injury and failure. N Engl J Med 372, 1138–1149. [DOI] [PubMed] [Google Scholar]
- 32.Wang B, Omar A, Angelovska T, Drobic V, Rattan SG, Jones SC & Dixon IM (2007) Regulation of collagen synthesis by inhibitory Smad7 in cardiac myofibroblasts. Am J Physiol Heart Circ Physiol 293, H1282–H1290. [DOI] [PubMed] [Google Scholar]
- 33.Geeves MA & Holmes KC (2005) The molecular mechanism of muscle contraction. Adv Protein Chem 71, 161–193. [DOI] [PubMed] [Google Scholar]
- 34.Lin BL, Song T & Sadayappan S (2017) Myofilaments: movers and rulers of the sarcomere. Compr Physiol 7, 675–692. [DOI] [PubMed] [Google Scholar]
- 35.Caleshu C, Sakhuja R, Nussbaum RL, Schiller NB, Ursell PC, Eng C, De Marco T, McGlothlin D, Burchard EG & Rame JE (2011) Furthering the link between the sarcomere and primary cardiomyopathies: restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am J Med Genet A 155, 2229–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ & Elliott PM (2009) Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2, 436–441. [DOI] [PubMed] [Google Scholar]
- 37.Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG & Seidman CE (2008) Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 358, 1899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olson TM, Karst ML, Whitby FG & Driscoll DJ (2002) Myosin light chain mutation causes autosomal recessive cardiomyopathy with mid-cavitary hypertrophy and restrictive physiology. Circulation 105, 2337–2340. [DOI] [PubMed] [Google Scholar]
- 39.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L & Epstein ND (1996) Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 13, 63–69. [DOI] [PubMed] [Google Scholar]
- 40.Spudich JA (2015) The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem Soc Trans 43, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robert-Paganin J, Auguin D & Houdusse A (2018) Hypertrophic cardiomyopathy disease results from disparate impairments of cardiac myosin function and auto-inhibition. Nat Commun 9, 4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kampourakis T, Ponnam S & Irving M (2018) Hypertrophic cardiomyopathy mutation R58Q in the myosin regulatory light chain perturbs thick filament-based regulation in cardiac muscle. J Mol Cell Cardiol 117, 72–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konhilas JP, Watson PA, Maass A, Boucek DM, Horn T, Stauffer BL, Luckey SW, Rosenberg P & Leinwand LA (2006) Exercise can prevent and reverse the severity of hypertrophic cardiomyopathy. Circ Res 98, 540–548. [DOI] [PubMed] [Google Scholar]
- 44.Yuan CC, Muthu P, Kazmierczak K, Liang J, Huang W, Irving TC, Kanashiro-Takeuchi RM, Hare JM & Szczesna-Cordary D (2015) Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc Natl Acad Sci USA 112, E4138–E4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Xu Y, Kerrick WGL, Wang Y, Guzman G, Diaz-Perez Z & Szczesna-Cordary D (2006) Prolonged Ca2+ and force transients in myosin RLC transgenic mouse fibers expressing malignant and benign FHC mutations. J Mol Biol 361, 286–299. [DOI] [PubMed] [Google Scholar]
- 46.Olah A, Nemeth BT, Matyas C, Hidi L, Lux A, Ruppert M, Kellermayer D, Sayour AA, Szabo L, Torok M et al. (2016) Physiological and pathological left ventricular hypertrophy of comparable degree is associated with characteristic differences of in vivo hemodynamics. Am J Physiol Heart Circ Physiol 310, H587–H597. [DOI] [PubMed] [Google Scholar]
- 47.Yuan CC, Kazmierczak K, Liang J, Kanashiro-Takeuchi R, Irving TC, Gomes AV, Wang Y, Burghardt TP & Szczesna-Cordary D (2017) Hypercontractile mutant of ventricular myo sin essential light chain leads to disruption of sarcomeric structure and function and results in restrictive cardiomyopathy in mice. Cardiovasc Res 113, 1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]