

There are significant neurogenic and inflammatory influences on blood pressure, yet the role played by each of these processes in the development of hypertension is unclear. Tumor necrosis factor α (TNFα) has emerged as a critical modulator of blood pressure and neural plasticity; however, the mechanism by which TNFα signaling contributes to the development of hypertension is uncertain.
Keywords: blood pressure, cytokine, excitatory neural transmission, inflammation, neural plasticity, NMDA receptor
Abstract
There are significant neurogenic and inflammatory influences on blood pressure, yet the role played by each of these processes in the development of hypertension is unclear. Tumor necrosis factor α (TNFα) has emerged as a critical modulator of blood pressure and neural plasticity; however, the mechanism by which TNFα signaling contributes to the development of hypertension is uncertain. We present evidence that following angiotensin II (AngII) infusion the TNFα type 1 receptor (TNFR1) plays a key role in heightened glutamate signaling in the hypothalamic paraventricular nucleus (PVN), a key central coordinator of blood pressure control. Fourteen day administration of a slow-pressor dose of AngII in male mice was associated with transcriptional and post-transcriptional (increased plasma membrane affiliation) regulation of TNFR1 in the PVN. Further, TNFR1 was shown to be critical for elevated NMDA-mediated excitatory currents in sympathoexcitatory PVN neurons following AngII infusion. Finally, silencing PVN TNFR1 prevented the increase in systolic blood pressure induced by AngII. These findings indicate that TNFR1 modulates a cellular pathway involving an increase in NMDA-mediated currents in the PVN following AngII infusion, suggesting a mechanism whereby TNFR1 activation contributes to hypertension via heightened hypothalamic glutamate-dependent signaling.
SIGNIFICANCE STATEMENT Inflammation is critical for the emergence of hypertension, yet the mechanisms by which inflammatory mediators contribute to this dysfunction are not clearly defined. We show that tumor necrosis factor α receptor 1 (TNFR1) in the paraventricular hypothalamic nucleus (PVN), a critical neuroregulator of cardiovascular function, plays an important role in the development of hypertension in mice. In the PVN, TNFR1 expression and plasma membrane localization are upregulated during hypertension induced by angiotensin II (AngII). Further, TNFR1 activation was essential for NMDA signaling and the heightening NMDA currents during hypertension. Finally, TNFR1 silencing in the PVN inhibits elevated blood pressure induced by AngII. These results point to a critical role for hypothalamic TNFR1 signaling in hypertension.
Introduction
A significant risk factor for diseases of the brain, vasculature, and heart (Nadar et al., 2006), hypertension is currently recognized as a leading contributor to the global burden of disease (Olsen et al., 2016). Angiotensin II (AngII) is an important humoral factor implicated in hypertension that is elevated in a subpopulation of hypertensive individuals (Laragh, 2001). Although the hypertensive actions of circulating AngII may be mediated by distinct mechanisms, experimental hypertension models implicate the brain as a critical target (Ferguson, 2009). Specifically, there is evidence that AngII is capable of elevating blood pressure by acting on circumventricular organs (CVOs), which in turn have direct neural projections to hypothalamic circuits (Mangiapane and Simpson, 1980a,b; Lind et al., 1983; Ferguson, 2009). One well characterized CVO–hypothalamic pathway implicated in AngII-mediated hypertension involves an excitatory pathway between the subfornical organ (SFO) and the paraventricular nucleus (PVN) of the hypothalamus (Bains and Ferguson, 1995; Llewellyn et al., 2012). In particular, the SFO–PVN pathway is implicated in hypertension (Young et al., 2012) achieved by continuous systemic administration of a subpressor dose of AngII (i.e., “slow-pressor”; Dickinson and Lawrence, 1963) that may model certain features of the most common form of hypertension, essential or primary hypertension, including a gradual rise in blood pressure and an increase in sympathetic activation (Grassi and Ram, 2016; Lerman et al., 2019). However, the signaling mechanisms in this circuit that contribute to hypertension are not well characterized.
Significantly, there is evidence that the hypertension mediated by AngII is associated with glutamate signaling in the PVN (Basting et al., 2018; Zhou et al., 2019). The NMDA-type glutamate receptor, an established molecular substrate of synaptic plasticity (Baez et al., 2018), is active in the PVN during acutely elevated blood pressure (Li et al., 2001; Martins-Pinge et al., 2013) as well as during hypertension associated with AngII (Glass et al., 2015) or other hypertension models (Li et al., 2003; Coleman et al., 2010; Li et al., 2017). Additionally, convergent gene targeting, ultrastructural, and neurophysiological evidence supports a critical role for NMDA receptor signaling in the PVN during AngII-dependent hypertension (Wang et al., 2013; Glass et al., 2015); however, the mechanisms mediating this are unclear.
Cytokines are critical contributors to both glutamate-dependent plasticity (Rizzo et al., 2018) and hypertension (Norlander et al., 2018). Thus, cytokines may play an important role in modulating excitatory signaling pathways in the hypothalamus contributing to AngII-mediated hypertension. In particular, the proinflammatory mediator tumor necrosis factor α (TNFα) has been implicated in autonomic processes, including the development of hypertension in species as diverse as mice (Sriramula et al., 2008) and humans (Puszkarska et al., 2019).
Tumor necrosis factor α has been implicated in hypertension by acting on various target organs including the heart (Sriramula and Francis, 2015) and kidney (Mehaffey and Majid, 2017). Additionally TNFα also plays a role in hypertension by acting centrally, particularly in brain cardiovascular regulatory circuits (Zhu and Ho, 1998) including the PVN. In the PVN, TNFα is basally expressed (Breder et al., 1993; Shi et al., 2010; Du et al., 2015) and is elevated in experimental hypertension models (Sriramula et al., 2013; Dai et al., 2015; Dange et al., 2015). In addition, exogenous administration of TNFα in the PVN influences sympathetic activity and blood pressure (Bardgett et al., 2014; Shi et al., 2014). In the brain, TNFα has been shown to potentiate NMDA receptor-mediated signaling (Han and Whelan, 2010; Zhang et al., 2011). Importantly, TNFα receptor 1 (TNFR1) is the major mediator of the actions of TNFα and is implicated in glutamate-dependent neural communication (Del Rivero et al., 2019; Valentinova et al., 2019). However, within the PVN, the role of TNFR1 in NMDA-mediated signaling in the context of AngII-dependent hypertension is unknown.
In the present study, a combination of in situ hybridization, immunoelectron microscopy, patch-clamp recording, and viral-mediated gene silencing in male mice was used to assess the role of PVN TNFR1 in NMDA receptor signaling and AngII-dependent hypertension.
Materials and Methods
Subjects
The experimental subjects were adult (postnatal >60 d) male wild-type C57BL/6 mice bred and maintained in a colony at Weill Cornell Medicine. Mice weighing 23–28 grams were housed in groups of at least two animals per cage maintained on a 12 h light/dark cycle (lights out at 6:00 P.M.) with unlimited access to water and rodent chow in their home cages. In addition, similarly housed and maintained male TNFR1 knock-out (KO) mice and their wild-type littermates on the C57BL/6 background (The Jackson Laboratory) were used to functionally characterize TNFR1 in specific neurophysiological studies. All experiments were approved by the Institutional Animal Care and Use Committees at Weill Cornell Medicine in accordance with guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize the number of animals used and their suffering.
Slow-pressor AngII infusion and blood pressure measurement
As described previously (Capone et al., 2012; Coleman et al., 2013), mice were anesthetized with isoflurane and implanted subcutaneously with osmotic mini-pumps (ALZET) loaded with vehicle [0.1% bovine serum albumin (BSA)/saline] or AngII dissolved in vehicle (600 ng/kg/min) for delivery over 14 d. All mice were habituated to the blood pressure measurement conditions including handling and exposure to the apparatus beginning 7–10 d before induction of hypertension. Mice were handled by the same investigator or investigators for each experimental procedure. Mice were then given at least two prebaseline blood pressure test trials before the start of baseline testing. Blood pressure measurements were recorded during the light period (3:00–4:00 P.M.). A tail-cuff blood pressure system (MC-4000, Hatteras) was used to measure systolic blood pressure in mice treated with vehicle or AngII. In each blood pressure assessment session, 10–20 blood pressure measurements were recorded over a 10 min period. Blood pressure measurements were averaged for each mouse, which were then combined to generate group mean systolic blood pressure values across treatments. Measurements were made at baseline and post-treatment.
In situ hybridization
An in situ hybridization approach using the RNAscope 2.5 HD Brown Chromogenic Reagent Kit (ACD) was used to assess TNFR1 mRNA in the PVN as previously described (Wang et al., 2012). Following experimental treatment, mice were deeply anesthetized with sodium pentobarbital (150 mg/kg, i.p.), and their brains were rapidly fixed via aortic arch perfusion at a flow rate of 20 ml/min sequentially with 5 ml of 1000 U/ml heparin in 0.9% saline immediately followed by 30 ml of 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.4. After dissection from the cranium, each brain was postfixed in 4% PFA in PB/15% sucrose overnight and then cryoprotected in 30% sucrose/PB solution for 1–2 d. Then brains were frozen, cryosectioned (30 µm), collected in cryoprotectant solution, and stored at −20°C.
To process tissue with probes under identical labeling conditions and reduce variability in labeling between sections because of various experimental conditions (e.g., differences in probe concentration, processing from experimental runs), brain sections from each treatment group were mounted on a single glass slide and were run in tandem. Two levels of the PVN (rostral, ∼0.6 mm posterior to bregma; caudal, ∼1.0 mm posterior to bregma; Paxinos and Franklin, 2000) were mounted on separate sets of slides (1 slide/region) for processing and analysis. After mounting, slides were dehydrated overnight and baked for 1 h at 60°C immediately before processing. After peroxidase quenching and incubation in target retrieval solution and protease, sections were then hybridized with proprietary probes (mouse TNFR1 target region 331–1345; probe Mm-Tnfrsf1a, catalog #426541) for 2 h at 40°C. Some sections were run with the positive housekeeping control probe Ppib (catalog #313911) and the bacterial negative control probe DapB (catalog #310043). The hybridization signal was detected using the chromogen diaminobenzidine (DAB) followed by blue hematoxylin counterstaining.
Color images of the PVN were captured at 40× using an Eclipse Nikon 80i microscope interfaced to a digital camera and adjusted for contrast. The number of brown particles overlying each blue Nissl-stained cell were counted manually (Wang et al., 2014) and were verified using a grain counting feature of MCID Image Analysis software (InterFocus Imaging) according to manufacturer guidelines, as previously described (Glass et al., 2008). Particles were counted from a single plane of section which was selected randomly. Four fields containing the PVN for each subregion for each subject were analyzed. The number of particles/cell (i.e., particle density), and the total number of particle-containing cells was assessed in each condition.
Tissue preparation and immunohistochemistry for electron microscopy
Following deep anesthesia with sodium pentobarbital (150 mg/kg, i.p.), mouse brains were rapidly fixed via aortic arch perfusion at a flow rate of 20 ml/min sequentially with: (1) 15 ml of 1000 U/ml heparin in 0.9% saline; and (2) 40 ml of a mixture of 3.75% acrolein/2% PFA in 0.1 m PB. After dissection from the cranium, each brain was postfixed in 2% PFA in PB for 60 min. For each animal, sections extending through the rostrocaudal extent of the PVN were coronally sectioned (40 µm) using a vibratome. For single labeling of TNFR1, tissue sections were processed for immunocytochemical detection of TNFR1 using a previously described immunogold-silver (IGS) labeling method (Milner et al., 2011). Sections were punch coded and pooled into single containers to ensure that tissue sections were identically exposed to reagents. To remove excess aldehydes, brain tissue was incubated in 1.0% sodium borohydride in PB, followed by washing in PB. After this, brain sections were washed in 0.1 m Tris-buffered saline (TBS) followed by a 30 min incubation in 0.5% BSA to lessen nonspecific labeling. After rinsing in TBS, brain sections were then incubated for 48 h in a primary rabbit anti-TNFR1 antiserum (1:100) diluted in 0.1% BSA. Following primary antiserum incubation, sections were washed in TBS. In preparation for IGS labeling, brain tissue was first rinsed in 0.01 m PBS, pH 7.4. Then, to reduce nonspecific binding of gold particles, brain sections were incubated for 10 min in a blocking solution consisting of 0.8% BSA and 0.1% gelatin in PBS. After this blocking step, sections were incubated for 2 h in anti-rabbit 1 nm gold particle-conjugated IgG [1:50; Electron Microscopy Sciences (EMS)] diluted in the blocking solution. Following this, tissue was rinsed in the blocking solution followed by washing in PBS. Brain sections were then incubated in 2% glutaraldehyde in PBS for 10 min followed by rinsing in PBS. Next, the nano-gold particles were enlarged using an IntenSE M silver enhancement kit for 5–6 min (GE Healthcare).
Other experiments assessed dual labeling of TNFR1 by IGS and fluorogold (FG) by the avidin-biotin-peroxidase complex (ABC) DAB method as described previously (Milner et al., 2011). For this, forebrain sections were incubated in an antisera cocktail of rabbit anti-TNFR1 (1:100) and guinea pig anti-FG (1:1000). Following this incubation, sections were first processed for FG by ABC/DAB. Sections were washed in TBS, incubated in anti-guinea pig IgG conjugated to biotin, rinsed in TBS, then incubated for 30 min in in ABC (1:100; VECTASTAIN Elite ABC-HRP Kit, Vector Laboratories) in TBS. The bound peroxidase was visualized by reaction for 5–6 min in DAB (Sigma-Aldrich) and 0.003% hydrogen peroxide in TBS, and then washed in TBS. Sections were then processed for TNFR1 IGS labeling using an anti-rabbit nano-gold-conjugated IgG, as described above.
Electron microscopy
For electron microscopic analysis, tissue was postfixed for 1 h in a solution of 2% osmium tetroxide (EMS) in PB. Brain sections were then embedded in electron microscopy (EM) BED 812 between two sheets of ACLAR plastic.
The surface of each flat-embedded forebrain section containing the PVN was cut in 60–80 nm sections with a diamond knife using an ultramicrotome (Ultratome, NOVA, LKB). The ultrathin sections from the caudal PVN were collected on 400-mesh, thin-bar copper grids (EMS) and counterstained with uranyl acetate and Reynold's lead citrate (Milner et al., 2011). Labeling for TNFR1 in the PVN was analyzed using a transmission electron microscope (Tecnai 12 BioTwin, FEI) interfaced to a digital camera (Advantage HR/HR-B CCD Camera System, Advanced Microscopy Techniques) that was used to collect digital images from the sampled tissue.
Ultrastructural analysis
To ensure that tissue was sampled from regions of even reagent penetrance, electron micrographs were captured from the embedding media–tissue transition zone (Glass et al., 2015). The classification of profiles was based on well established guidelines for the ultrastructural identification of neuronal and glial elements (Peters et al., 1991). Somata were distinguished by the presence of nuclei, Golgi bodies, as well as rough endoplasmic reticula. Profiles were defined as dendritic if they contained regular microtubule arrays, endomembranous organelles, and/or postsynaptic densities. Structures that were at least 0.2 µm in diameter and that also contained numerous small synaptic vesicles were characterized as axon terminals, whereas profiles <0.2 µm and lacking small synaptic vesicles were designated as unmyelinated axons. Irregularly shaped profiles devoid of cytoplasmic organelles or containing arrays of filaments were considered to be glia. Profiles were determined to be IGS labeled if they contained at least one particle per small profile, or two particles for larger profiles, provided that structures not expected to be labeled for the primary antisera, such as myelin, were devoid of gold-silver deposits (Hara and Pickel, 2008).
An established procedure for the apportionment of particulate IGS labeling within subcellular compartments was used to estimate the distribution of TNFR1 in cytoplasmic and plasma membrane compartments of dendritic and terminal profiles (Glass et al., 2015; Marques-Lopes et al., 2017). A total of 17,340 µm2 of tissue was sampled from the PVN of vehicle-treated (100 fields at 86.7 µm2/field) and AngII-treated (100 fields at 86.7 µm2/field) animals. The subcellular localization of TNFR1-immunogold silver particles was defined as either on the plasma membrane (i.e., directly touching the membrane), near the plasma membrane (within 70 nm of the plasma membrane), affiliated with mitochondria or cytoplasmic (>70 nm from the plasma membrane).
For dual labeling, the dense uniform black IGS particles are easily distinguished from the diffuse brown/black immunoperoxidase precipitate. The numbers of postsynaptic (e.g., dendritic) profiles singly labeled for TNFR1, FG, or both TNFR1 and FG were counted according to established procedures (Beckerman et al., 2013).
Primary antisera
A rabbit polyclonal antiserum (Abnova) was used to label TNFR1 (Glass et al., 2017). A guinea pig polyclonal antibody (Protos Biotech) was used to identify FG (Glass et al., 2015). A chicken polyclonal antiserum (Aves Labs) was used to label green fluorescent protein (GFP; Marques-Lopes et al., 2015). A mouse antibody (Chemicon) was used to label NeuN (Glass et al., 2008). The specificity of these reagents has been reported in the respective citations.
Retrograde labeling of spinally projecting PVN neurons
As previously described (Wang et al., 2013), mice were anesthetized (87.5 mg/kg ketamine/12.5 mg/kg xylazine, i.p., or isoflurane), and their spinal cords were exposed at the T2–T4 level through dorsal laminectomy. Under a surgical microscope, the tip of a glass pipette filled with rhodamine-labeled fluorescent microspheres (0.04 μm; FluoSpheres, Thermo Fisher Scientific) or FG (4%; Fluorochrome) was pressure ejected (50 nl) bilaterally into the intermediolateral nucleus (IML) region of the spinal cord, and the incision was sutured after the injection.
Whole-cell voltage and current clamp
Whole-cell configuration of visually identified neurons was established as previously described (Wang et al., 2013). Mice were anesthetized with 4% isoflurane, and their brains were removed and immersed in sucrose-artificial CSF (aCSF). Coronal brain slices (200 µm in thickness) containing the PVN were cut using a Leica VT1000s vibratome and stored in a custom-designed chamber containing sucrose-aCSF with 95% O2 and 5% CO2 at 32°C for 1 h. The aCSF was composed of the following (in mmol/L): 125 sucrose, 26 NaHCO3, 5 KCl, 1 NaH2PO4, 5 MgSO4, 1 CaCl2, 10 glucose, and 4.5 lactic acid, at pH 7.4. The aCSF with lactic acid (l-aCSF) was composed of the following (in mmol/L): 124 NaCl, 26 NaHCO3, 5 KCl, 1 NaH2PO4, 2 MgSO4, 2 CaCl2, 10 glucose, and 4.5 lactic acid, with 95% O2 and 5% CO2, at pH 7.4. The PVN was identified using the lateral ventricle, fornix, and optic chiasm as landmarks. The coronal slices containing the PVN were then transferred to a glass-bottom recording chamber and continuously perfused with a modified Mg2+-free l-aCSF (in mmol/L): 121 NaCl, 5 KCl, 1.8 CaCl2, 0.01 glycine, 1 Na-pyruvate, 20 glucose, 26 NaHCO3, 1 NaH2PO4, and 4.5 lactic acid, with 95% O2 and 5% CO2, at pH 7.4 at a rate of 2 ml/min. Rhodamine-labeled spinal PVN neurons in brain slices were briefly identified under an epifluorescence microscope (E600, Nikon) with a combination of FITC filter and differential interference contrast optics. Neurons located in the medial one-third of the PVN area between the third ventricle and the fornix were patched for whole-cell current and voltage-clamps separately (Li et al., 2002) using an Axopatch 200B amplifier (Molecular Devices). Because current flow through NMDA receptors is largely blocked by Mg2+ ions at resting membrane potentials (Mayer et al., 1984) to elicit NMDA receptor-mediated spontaneous firing and inward ionic currents maximally, before the recording, cells were superfused with the modified Mg2+-free l-aCSF and then NMDA (30 μmol/L)-containing buffers. To block voltage-gated Na+ channels and non-NMDA receptor-mediated cation channels, 1 μmol/L TTX and 5 μmol/L CNQX were added to the Mg2+-free l-aCSF buffer. The holding potential was at −60 mV and NMDA (30 μmol/L)-containing Mg2+-free extracellular solution was perfused toward the patched neuron for 30 s (Coleman et al., 2010; Suh et al., 2010). Recording pipette tip resistances were 5–8 MΩ as filled with the following intracellular solution (in mmol/L): 130 K-gluconate; 10 NaCl, 1.6 MgCl2, 1 EGTA, 10 HEPES, and 2 Mg-ATP, adjusted to pH 7.3. After formation of a gigaohm seal, the electrode capacitance was nullified. After breaking the plasma membrane, the cell membrane capacitance (Cm) was read directly from Membrane Test of Window pClamp 8.2 (Molecular Devices). Cm and series resistance were monitored throughout the recording, with series resistance generally compensated >80%. Signals were low-pass filtered at 2 kHz and acquired at a sampling rate of 5–10 kHz.
Spatial–temporal gene silencing
Knockdown of TNFR1 was achieved by viral-mediated transfer of a TNFR1 short hairpin RNA (shRNA; Test vector). The test vector was a neurotropic serotype 2 recombinant adeno-associated virus (rAAV; ∼4.7 kb) expressing an enhanced GFP reporter (rAAV-TNFR1) produced by Vector Biolabs. A vector expressing enhanced GFP was used as a control (Control vector)
Virus administration
Viral vectors were unilaterally or bilaterally microinjected into the PVN via stereotaxic surgical procedures as previously described (Glass et al., 2015). Under deep isoflurane anesthesia, rAAV-TNFR1 shRNA or rAAV-GFP (∼100 nl/hemisphere, 3.2 × 1013 genome copies/ml) were injected into the PVN ∼1.0 mm posterior and 0.2 mm lateral to bregma, at a depth of 4.8 mm (Paxinos and Franklin, 2000). Microinjections were made by interfacing a picospritzer (Picospritzer II, General Valve) to a glass pipette (World Precision Instruments) with the tip pulled to a diameter of ∼50 µm, via a pipette holder and plastic tubing. Injections were made over a 10 min interval. To prevent leakage, the pipette was left in place for an additional 10 min. Bone wax was used to cover the bore hole, and the mice were allowed to recover in their home cages. Mice were allowed to recover for 14 d to allow for maximal gene knockdown.
Light microscopic immunohistochemistry
Animals were deeply anesthetized with sodium pentobarbital (150 mg/kg, i.p.), and their brains were perfusion fixed with 4% PFA in PB. After dissection from the cranium, each brain was postfixed in 4% PFA in PB overnight, and the forebrain containing the PVN was sectioned (40 µm) using a vibratome (model VT1000X, Leica Microsystems). Sections were punch coded and pooled into single containers to ensure that tissue sections were identically exposed to reagents (Milner et al., 2011). Next, sections were incubated for 30 min in 0.5% BSA to minimize nonspecific labeling, followed by 24 or 48 h incubation in primary rabbit anti-GFP (1:1000; Thermo Fisher Scientific), rabbit anti-TNFR1 (1:250), or mouse anti-NeuN (1:1000) antisera. Sections were then washed in 0.1 m TBS, incubated in anti-rabbit IgG conjugated to biotin, rinsed in TBS, then incubated for 30 min with ABC in TBS. The bound peroxidase was visualized by reaction for 5–6 min in DAB and 0.003% hydrogen peroxide in TBS. Sections were mounted on glass slides in 0.05 m PB, dehydrated through an ascending series of alcohol through xylene, and coverslipped with DPX (Sigma-Aldrich). These sections were examined using a Nikon light microscope.
Light microscopic cell counting
Cell counting was performed using relative optical density measurements via Microcomputer Imaging Device software (MCID, Imaging Research), as previously described (Glass et al., 2008). Briefly, mounted sections were viewed with a Nikon Microphot-FX microscope (Nikon) equipped with a digital CoolSNAP camera (Photometrics). The light microscopic images were acquired through an interface between the camera and a Macintosh computer. Pixel intensity thresholding procedures were performed as per manufacturer guidelines. Electronic images were imported into MCID, which calculates a relative threshold level for each image, then adjusted using an object enhancement filter that maximizes the contrast between large objects and background. Cell counts were also performed manually to verify the consistency of automated tallies. For bilateral microinjections, labeled cells in each hemisphere were pooled for analysis. For unilateral microinjections, labeling was calculated as the ratio of the number of labeled cells in the injected hemisphere (i.e., ipsilateral) to the number in the noninjected hemisphere (i.e., contralateral).
In situ microfluorography
Production of reactive oxygen species (ROS) in PVN neurons was determined using dihydroethidium (DHE; Thermo Fisher Scientific). Superoxide oxidizes the cell-permanent DHE to 2-hydroxyethidium and other oxidation products (Wang et al., 2013), which interact with DNA and are detectable using an ethidium (ETH) bromide filter (Chroma Technology) on an Eclipse C-CU microscope (Nikon). Methods for ROS detection in dissociated cells using DHE have been described previously (Coleman et al., 2013). Briefly, slices containing the PVN were incubated with 0.01% thermolysin, 0.01% Pronase and DHE (2 µmol/L) containing l-ACSF for 30 min and then PVN cells were obtained by mechanical stirring. Time-resolved fluorescence using IPLab software (Scanalytics) was measured at 1 min intervals with an exposure time of 150 ms for 40 min using a Nikon Diaphot 300 inverted microscope equipped with a CCD digital camera (Princeton Instruments). Bath application of TNFα was performed after a stable baseline measurement for 10 min. The increase in ROS signal induced by TNFα was expressed as the ratio of Ft/Fo, where Ft is fluorescence after the application of TNFα, and Fo is the baseline fluorescence in the same cell (Coleman et al., 2013). For analysis of ROS signals, the baseline background intensity was subtracted from the detected ETH signals.
Drugs and reagents
AngII, TNFα, the TNFR1 transduction inhibitor R-7050, NMDA, TTX, CNQX, Pronase, and thermolysin were obtained from Sigma-Aldrich.
Statistical analyses
Blood pressure results were analyzed by repeated-measures ANOVA followed by post hoc testing (Fisher's PLSD). In situ hybridization, immunoelectron microscopic, and electrophysiological data were analyzed by t tests or factorial ANOVA followed by post hoc testing (Fisher's PLSD). In cases where distributions deviated from equality of variance, data were analyzed by Welch's test, Welch's ANOVA, or the Mann–Whitney U test, as indicated in the text. Statistical analyses were conducted using Prism 6 (GraphPad) or StatView 5.0 software.
Image preparation
Light and electron micrographs were prepared by adjusting images for contrast, sharpness, and/or brightness using Photoshop 11 software. These images were then imported into PowerPoint to add lettering and symbols. Prism 6 software was used to produce the graphical figures (GraphPad Software).
Results
The slow-pressor response to AngII is associated with an increase in TNFR1 mRNA in the caudal PVN
Functionally, the major effects of TNFα are transduced by binding and activation of TNFR1 (Nadeau and Rivest, 1999; Bette et al., 2003), which has been implicated in diverse hypertension models (Yu et al., 2017; Wei et al., 2018). In addition, TNFR1 is prominently expressed in the PVN (Nadeau and Rivest, 1999; Rizk et al., 2001), where it is frequently present in somata and dendritic compartments (Glass et al., 2017). However, the relationship between AngII-dependent hypertension and TNFR1 transcription in the PVN is unknown.
To investigate the relationship between the slow-pressor response to AngII and TNFR1 gene expression, mice were infused with vehicle (n = 7) or AngII (n = 7) for 14 d. There was a significant effect of treatment (F(1,12) = 12.4, p < 0.005, ANOVA), session (F(4,48) = 3.3, p < 0.03, repeated-measures ANOVA), and a session by treatment interaction (F(4,48) = 4.4, p = 0.004, repeated-measures ANOVA) on systolic arterial blood pressure (Fig. 1A). Systolic blood pressure was elevated in AngII-infused mice compared with vehicle-administered animals at day 9 (p < 0.05), day 11 (p < 0.001), and day 13 (p < 0.002).
Figure 1.

Slow-pressor AngII infusion is associated with both elevated blood pressure and increased TNFR1 transcription in the caudal PVN. A, Mice infused systemically with AngII showed a progressive increase in systolic blood pressure (SBP) compared with mice given vehicle, reaching a peak at day 13 [*p < 0.05, **p < 0.002 vehicle (Veh) vs AngII days 9–13; #p < 0.05, ##p < 0.002, ###p < 0.0005, AngII Day 0 vs Days 9–13). B, In both groups, TNFR1 mRNA was quantified in the caudal PVN (cPVN; ∼1.0 mm posterior to bregma) at the level shown in the schematic adapted from Paxinos and Franklin (2000). C, Mice infused with AngII and killed at day 14 showed a significantly higher particle density of TNFR1 mRNA compared with vehicle-infused mice (*p < 0.03). D, F, Light micrographs of the dorsal medial hypothalamus showing the PVN (dashed) from sections processed for TNFR1 in situ hybridization (arrows) in mice infused with Veh (D) or AngII (F). E, G, The area bounded by the dashed boxes shows the PVN in Veh (E) and AngII-treated (G) mice at a higher magnification. 3v, Third ventricle; ot, optic tract. Scale bars: D, F, 1 mm; E, G, 0.05 mm.
The caudal PVN contains the major population of spinally projecting sympathoexcitatory neurons that are critical for blood pressure regulation (Sawchenko and Swanson, 1983). Therefore, we investigated TNFR1 transcription in the caudal PVN (Fig. 1B, approximate location) in vehicle- and AngII-infused mice by in situ hybridization. Mice infused with AngII had a higher density of TNFR1 in the caudal PVN compared with vehicle-infused animals (t(12) = 2.2, p < 0.05, unpaired t test; Fig. 1C–G). There was no significant difference with respect to the number of cells showing TNFR1 transcript in vehicle- and AngII-treated mice (t(12) = 1.2, >0.2; data not shown).
The rostral PVN is populated by neuroendocrine neurons that contribute to blood pressure (Capone et al., 2012). We therefore next measured TNFR1 in the rostral PVN (∼0.6 mm posterior to bregma) in vehicle- and AngII-infused mice. It was found that there was no significant difference in PVN TNFR1 transcript in vehicle- and AngII-treated animals in the rostral PVN (t(12) = 1.3, p > 0.2, unpaired t test; data not shown). There was also no between-group difference with respect to the number of cells showing TNFR1 transcript (t(12) = 0.3, >0.7; data not shown).
These results demonstrate that the slow-pressor response to AngII is associated with an increase in TNFR1 gene expression in the caudal PVN, a region that contains the major population of spinally projecting sympathoexcitatory neurons that are important in blood pressure regulation (Sawchenko and Swanson, 1983).
The slow-pressor response to AngII is associated with an increase in both the density of TNFR1-labeled dendritic profiles and plasma membrane-affiliated TNFR1 in the PVN
Post-translational modifications that contribute to protein transport from intracellular to potentially functional sites on the plasma membrane play an important role in the signaling properties of cytokine receptors (Moraga et al., 2014). There is, however, no evidence that hypertension is associated with alterations in the subcellular location of TNFR1 in PVN neurons.
The effect of hypertension on the subcellular localization of TNFR1 in caudal PVN neurons was investigated by immunoelectron microscopy in mice treated with vehicle (n = 3) or AngII (n = 3) for 14 d. Although there were no between-group differences in baseline blood pressure (Veh: 104.4 ± 3.1 mmHg vs AngII: 106.1 ± 1.7 mmHg), AngII-treated mice had significantly higher blood pressure compared with vehicle-infused animals by day 13 of treatment (Veh: 102.2 ± 3.1 mmHg vs AngII: 136.3 ± 3.4 mmHg; F(1,4) = 38.0, p < 0.004, one-way ANOVA).
A total of 248 dendritic profiles were counted in PVN samples from vehicle-treated mice, and 330 were counted in the PVN in AngII-treated animals. Examples of TNFR1 IGS-labeled dendritic profiles are shown in Figure 2, A and B. The total number of IGS particles for TNFR1 in dendritic profiles was significantly higher in AngII-treated mice compared with vehicle-treated mice (t(4) = 3.2, p < 0.04, unpaired t test; Fig. 2C). Additionally, significantly more TNFR1-labeled dendritic profiles were counted in AngII-treated mice compared with vehicle-treated mice (U = 0, p < 0.05 Mann–Whitney U test; Fig. 2D).
Figure 2.

Chronic infusion of slow-pressor AngII is associated with increased TNFR1 labeling and elevated plasma membrane-affiliated TNFR1 in PVN neurons. A, B, Electron micrographs illustrating TNFR1 IGS labeling in dendrites (TNFR1-d) of PVN neurons from vehicle (Veh)-infused (A) and AngII-infused (B) mice. Immunoreactivity for TNFR1 is seen in the cytoplasm (black arrows) in both Veh-infused (A) and AngII-infused (B) mice. In addition, TNFR1 labeling is also present on the plasma membrane (gray arrows) of dendrites in mice receiving AngII. Some TNFR1 dendritic profiles receive asymmetric-type excitatory synapses (arrowheads) from unlabeled axon terminals (ut). C, Quantitatively, compared with Veh-treated mice, animals receiving AngII had a significantly greater number of IGS particles for TNFR1 in dendritic profiles. D, In addition, the number of TNFR1-labeled dendritic profiles was higher in AngII-infused mice compared with Veh-treated animals. There were also differences in the subcellular distribution of IGS particles for TNFR1 in AngII- and Veh-administered mice. E, In particular, the density of TNFR1 on the dendritic plasma membrane was greater in mice infused with AngII compared with Veh. F–H, There were no significant differences in the densities of TNFR1 labeling near the plasma membrane (F), in the cytoplasm (G), or affiliated with mitochondria (H) in either treatment group. *p < 0.05 AngII compared to Veh. Scale bars, 500 nm.
In addition to distinctions in protein levels, there were also differences between vehicle- and AngII-treated mice with respect to the subcellular localization of TNFR1. Mice receiving AngII had a higher density of IGS labeling for TNFR1 on the plasma membrane of dendritic profiles of PVN neurons compared with vehicle-treated mice (U = 0, p < 0.05, Mann–Whitney U test; Fig. 2E). However, there were no significant between-treatment differences in TNFR1 IGS labeling near (<70 nm) the plasma membrane (U = 4, p > 0.8, Mann–Whitney U test; Fig. 2F), in the cytoplasm (U = 3, p > 0.5, Mann–Whitney U test; Fig. 2G), or affiliated with mitochondria (U = 3, p > 0.5, Mann–Whitney U test; Fig. 2H). There were also no significant differences in either the surface (vehicle, 4.4 ± 0.19 µm; vs AngII, 4.9 ± 0.24 µm; t(4) = 1.6, p > 0.17, unpaired t test; data not shown) or cross-sectional (vehicle, 1.4 ± 0.19 µm2; vs AngII, 1.6 ± 0.09 µm2; t(4) = 0.97, p > 0.38, unpaired t test; data not shown) areas of dendritic profiles between the two treatment groups. These results demonstrate an increase in the plasma membrane affiliation of TNFR1 in presumably functional compartments of PVN neurons following AngII administration.
Slow-pressor AngII is associated with an increase in TNFR1 labeling in somatodendritic profiles of PVN neurons projecting to the spinal cord
Among the various neuronal phenotypes populating the PVN, spinally projecting sympathoexcitatory neurons have been prominently implicated in hypertension (Li and Pan, 2017). However, it is not known whether TNFR1 is expressed in PVN neurons projecting to the spinal cord.
Immunohistochemistry was combined with retrograde neuronal tracing to investigate the extent of TNFR1 labeling in PVN–spinal cord projection neurons. Following spinal microinjection of FG, light microscopic analysis revealed retrogradely labeled neurons in the PVN (n = 3; Fig. 3A) at the caudal level of the nucleus. Adjacent sections were processed for immunoperoxidase labeling of FG and IGS labeling of TNFR1 by dual labeling EM. Ultrathin sections were taken from an area of the PVN retrogradely labeled from the IML and a total of 85 fields (1225 nm2/field) were sampled for the presence of neuronal cell bodies and dendrites expressing immunolabeling for FG, TNFR1, or both FG and TNFR1. Dual-labeled somatodendritic profiles were found in the PVN (Fig. 3B,C). A total of 77 labeled profiles were sampled from the PVN, the majority of which were dually labeled (TNFR1: 28 profiles; FG: 12 profiles; TNFR1 + FG: 37 profiles; Fig. 3D).
Figure 3.

TNFR1 is expressed in PVN neurons projecting to the IML. A, Light micrograph showing FG-labeled neurons in the PVN. B, An electron micrograph showing a FG-labeled somatodendritic profile (FG+TNFR1-s) adjacent to the lumen (lu) of a blood vessel. C, The area in the bounded box is shown at a higher magnification. Diffuse immunoperoxidase reaction product for FG is present in the cytoplasm. In addition, IGS particles for TNFR1 (arrows) are also present throughout the dendritic profile. D, Neurons in the PVN labeled for both FG and TNFR1 were more numerous than those exclusively labeled for either TNFR1 or FG. Scale bars: A, 0.25 mm; B, 5 µm; inset, 200 nm.
It was shown earlier that TNFR1 IGS labeling was elevated in dendritic profiles of PVN neurons in AngII-infused mice (Fig. 2C). To investigate the effect of AngII on TNFR1 SIG labeling in PVN–spinal cord projection neurons, mice were microinjected with FG in the IML and infused with either vehicle (n = 3) or AngII (n = 3) for 14 d. Mice receiving AngII showed an increase in blood pressure at day 14 when compared with mice treated with vehicle (baseline: vehicle, 107.4 ± 0.7 mmHg; vs AngII, 103.1 ± 3.7 mmHg; day 13 treatment: vehicle, 103.1 ± 1.9 mmHg; vs AngII, 137.1 ± 3.3 mmHg; F(3,6) = 20.9, p < 0.006, repeated-measures ANOVA). A total of 180 somatodendritic profiles were counted (30/animal) across both treatment groups, examples of which are shown in Figure 4, A and B. Total IGS particles for TNFR1 were higher in PVN FG-labeled profiles in AngII-treated versus vehicle-infused mice (t(4) = 5.4, p < 0.006, unpaired t test; Fig. 4C). Given the increase in plasma membrane TNFR1 found in PVN dendrites (Fig. 2E) following AngII, the extent of plasma membrane-affiliated TNFR1 in PVN–spinal cord projection neurons was also assessed in vehicle- and AngII-infused mice. There was an increase in plasma membrane-associated TNFR1 in dual-labeled profiles from AngII-infused versus vehicle-infused mice, although this just failed to reach statistical significance (t(4) = 2.7, p = 0.053, unpaired t test; data not shown). These results show an increase in total TNFR1 SIG labeling in spinally projecting PVN neurons following AngII hypertension.
Figure 4.
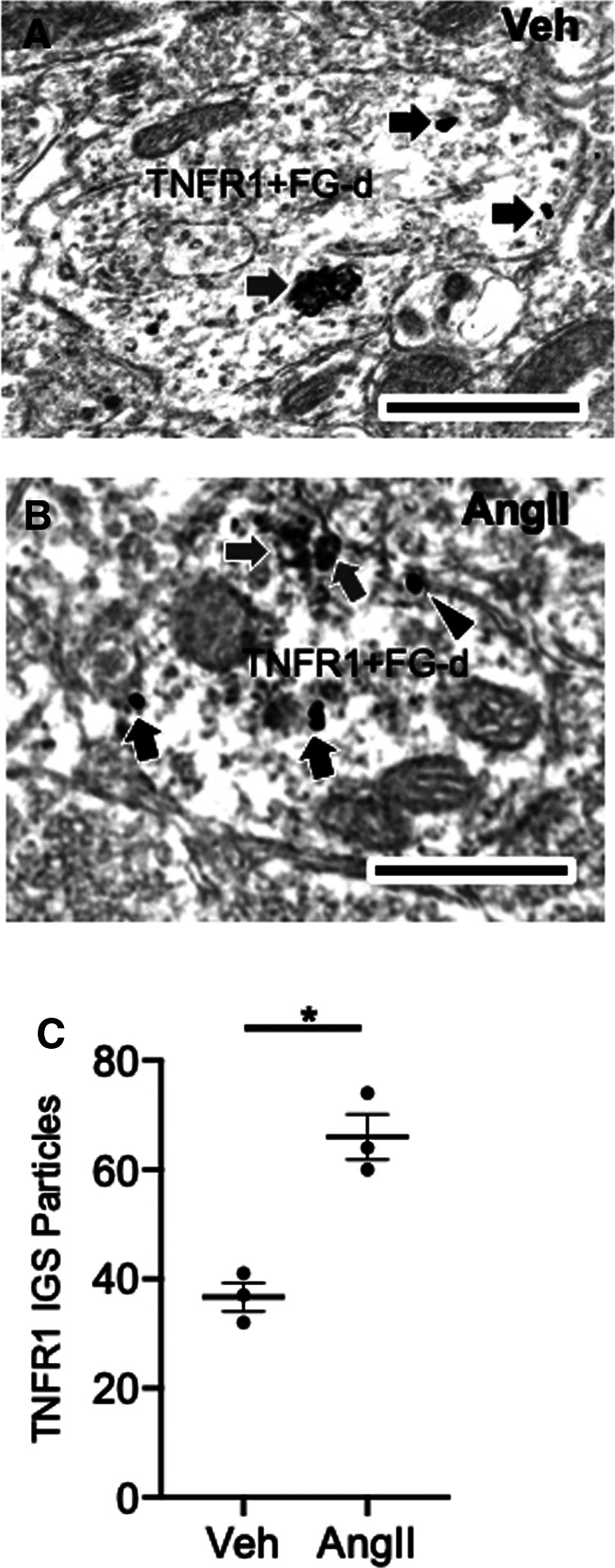
Elevated TNFR1 IGS labeling in somatodendritic profiles of PVN neurons retrogradely labeled from the spinal cord in AngII-infused mice. A, B, Electron micrographs showing dual immunoperoxidase and IGS labeling for FG and TNFR1, respectively, in dendritic profiles of PVN neurons from vehicle-infused (A) and AngII-infused (B) mice. Dendritic profiles (TNF+FG-d) show aggregates of peroxidase reaction product for FG (gray arrows) and TNFR1 SIG-labeling (black arrows) in intracellular and plasmalemmal (arrowhead) locations. C, A scatter plot showing total TNFR1 labeling in dual TNFR1+FG-labeled profiles in the PVN. There was an increase in total TNFR1 SIG labeling in dual-labeled profiles of mice treated with AngII relative to Veh(*p < 0.05). Scale bars: A, B, 500 nm.
TNFα increases spontaneous firing rates and NMDA currents in PVN–IML projection neurons in naive mice
It has been shown that TNFα influences excitatory neural signaling in several brain areas (Santello and Volterra, 2012); however, the effect of TNFα on the activity of PVN neurons projecting to the spinal cord is uncertain. To examine the influence of TNFα on the spontaneous firing of PVN projection neurons, we tested the effect of TNFα application on PVN-spinal cord neurons by whole-cell current-clamp in PVN slices of naive mice. Recordings were made in visually identified PVN neurons retrogradely labeled by rhodamine microspheres microinjected into the IML. Spontaneous postsynaptic currents were recorded in neurons following the application of vehicle or TNFα (Fig. 5A,B). There was an increase in the firing rate of neurons (n = 5; three mice) after TNFα application compared with vehicle (t(4) = 5.8, p < 0.005, paired t test; Fig. 5B). In addition, we also evaluated the effect of TNFα on excitatory currents elicited by NMDA. It was found that, compared with treatment with vehicle, treatment with TNFα resulted in an increase in NMDA currents (Fig. 5C,D) in PVN projection neurons (t(14) = 2.49, p < 0.03, paired t test; n = 8, three mice). These results demonstrate that TNFα stimulation results in an increase in both spontaneous firing rates and NMDA-elicited currents in PVN–spinal cord projection neurons in experimentally untreated mice.
Figure 5.

TNFα increases spontaneous firing and NMDA currents in spinally projecting PVN neurons in experimentally untreated animals. A, Representative traces of whole-cell current-clamp recordings of spontaneous firing after vehicle (top trace) and TNFα (0.1 nm; bottom trace). B, There is a significant increase in the spontaneous firing rate in spinally labeled PVN neurons after TNFα application compared with vehicle (**p < 0.005). C, Representative current traces from PVN neurons after NMDA (30 μm) application and pretreatment with vehicle (left trace) or TNFα (right trace). D, There is a significant increase in NMDA currents following TNFα compared with vehicle (*p < 0.03).
TNFR1 signaling contributes to NMDA-mediated currents in PVN projection neurons of naive mice and mice treated with 14 d AngII
TNFα has been shown to modulate excitatory currents mediated by ionotropic glutamate receptor activation (Santello and Volterra, 2012), a process implicated in hypertension (Wang et al., 2013). However, the role of TNFR1 signaling on NMDA currents in PVN neurons basally and following AngII hypertension is unknown.
We investigated the role of TNFR1 on NMDA-induced currents by whole-cell voltage clamp in PVN slices of mice. Recordings were made in visually identified PVN neurons retrogradely labeled by rhodamine microspheres microinjected in the IML. There was a significant effect of treatment on inward currents induced by NMDA (F(2,14) = 5.3, p < 0.02, one-way ANOVA). First, in wild-type mice (n = 3) pretreatment with the TNFR1 transduction inhibitor R-7050 (Gururaja et al., 2007) significantly inhibited the inward current induced by NMDA application in PVN neurons (NMDA, n = 8 neurons; NMDA + R-7050, n = 5 neurons; p < 0.02; Fig. 6A,B). In addition, in TNFR1 KO mice (n = 3) there was a reduced NMDA-mediated inward current generated in PVN neurons (n = 4 neurons; p = 0.02; Fig. 6B). These results demonstrate that TNFR1 signaling plays an important role in NMDA-mediated currents in PVN-spinal projection neurons.
Figure 6.
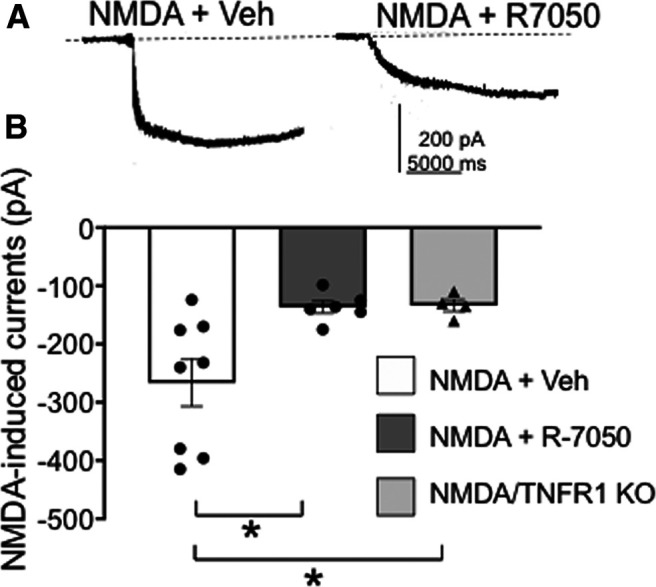
TNFR1 activation plays a role in NMDA-induced currents in PVN–spinal cord projection neurons in experimentally naive mice. A, Whole-cell patch-current recordings of PVN-spinal cord projecting neurons in brain slices following NMDA and vehicle (30 μm; left trace) as well as NMDA after TNFR1 blockade by 5 μm R-7050 (right trace). B, Scatter plot illustrates that NMDA-mediated currents are inhibited following the TNFR1 antagonist and also in TNFR1 knock-out mice [*p < 0.01 NMDA + vehicle (Veh) vs NMDA + R-7050; *p < 0.01 NMDA + Veh vs NMDA/TNFR1 KO].
Whole-cell voltage clamp was also used to assess the role of TNFR1 on NMDA-induced currents following hypertension in PVN neurons of mice infused with 14 d vehicle or AngII. Systolic blood pressure in AngII-treated mice was higher at day 14 compared with vehicle-infused mice (vehicle, n = 4: 100.9 ± 6.6; vs AngII, n = 6: 141.8 ± 5.3; p < 0.009). There was a significant effect of treatment on NMDA receptor currents in PVN neurons from mice treated with vehicle or AngII with or without pretreatment with R-7050 (Fig. 7A–E). There was a main effect of R-7050 (F(1,25) = 2.4, p < 0.0001, two-way ANOVA) and an interaction between R-7050 and AngII infusion (F(1,25) = 49.9, p = 0.009, two-way ANOVA) but no effect of AngII (F(1,25) = 2.4, p > 0.1, two-way ANOVA) on NMDA receptor currents. NMDA currents were greater in PVN neurons (n = 8) from AngII-infused mice versus neurons (n = 6) from mice infused with vehicle (t(11.8) = 2.2, p < 0.05, Welch's t test). In addition, pretreatment with R-7050 suppressed NMDA-mediated currents in PVN neurons (n = 8) from AngII-infused mice compared with neurons (n = 8) pretreated with vehicle (t(7.2) = 6.9, p = 0.0002, Welch's t test). Although R-7050 reduced NMDA currents in PVN neurons (n = 7) from vehicle-infused mice (60 ± 7%), the magnitude of NMDA-mediated current suppression was significantly greater (90 ± 2%) in neurons from Ang-infused mice (t(6.5) = 3.4, p < 0.02; unpaired t test; Fig. 7F). These results demonstrate that the inhibition of TNFR1 signaling reduces NMDA-mediated currents in the nonhypertensive state, while TNFR1 signaling blockade also potently inhibits NMDA-dependent currents in the context of AngII-dependent hypertension.
Figure 7.
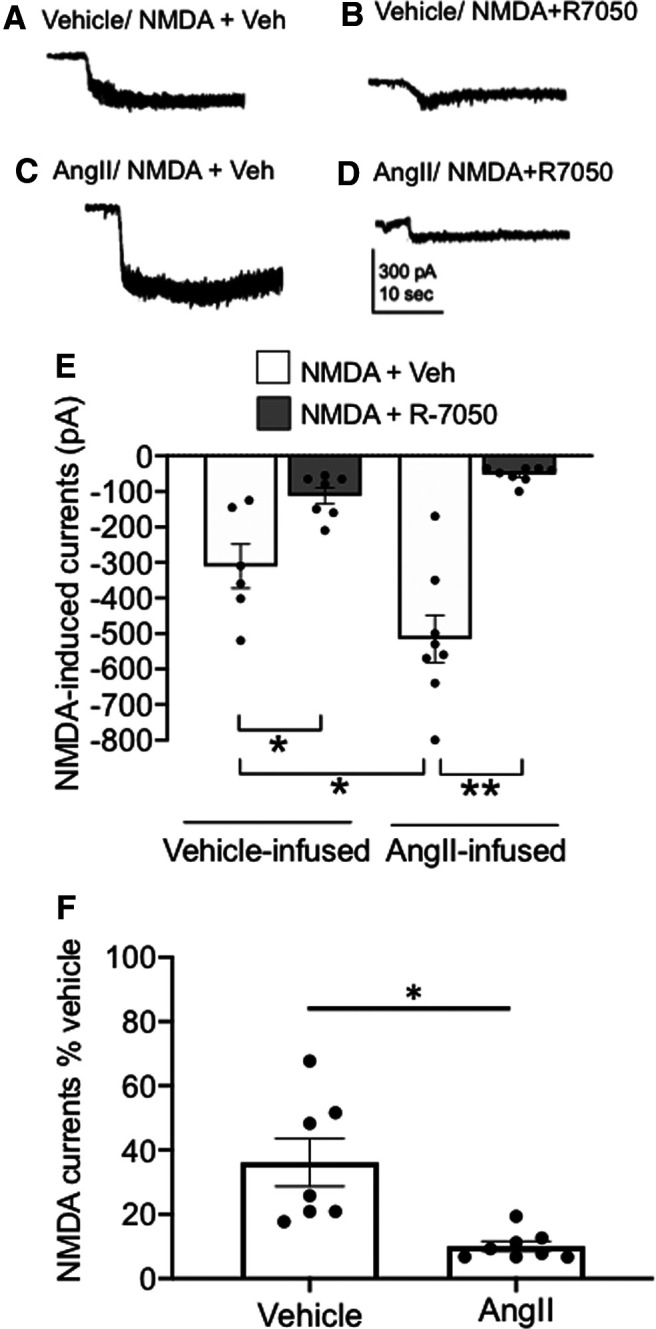
TNFR1 activity contributes to elevated NMDA-induced currents in PVN neurons following slow-pressor AngII. A, B, Representative current traces of whole-cell patch recordings of inward currents from mice infused with vehicle for 14 d and superfused with NMDA (30 μm) and vehicle (A) or NMDA and 5 μm R-7050 (B). C, D, Current traces are also shown in PVN slices from mice infused with AngII for 14 d and superfused with NMDA and vehicle (C) or NMDA and R-7050 (D). E, Scatter plots illustrating that the increase in NMDA-elicited currents in mice treated with AngII compared with saline is attenuated by TNFR1 inhibition (*p < 0.02, Veh-infused/NMDA + Veh vs Veh-infused/NMDA + R-7050; *p < 0.05, Veh-infused/NMDA + Veh vs AngII-infused/NMDA + Veh; **p < 0.05, Ang-infused/NMDA + Veh vs AngII-infused/NMDA + R-7050). F, The suppression of NMDA receptor currents by R-7050 is significantly greater in AngII-infused versus vehicle-infused mice (*p < 0.02).
Knockdown of TNFR1 in the PVN is associated with a suppression of the slow-pressor response to AngII
If TNFR1 signaling has functional relevance for hypertension, then silencing TNFR1 in PVN neurons would be expected to impair the increase in blood pressure following slow-pressor AngII. Traditionally, inhibitors of TNFR1 signaling have been limited to monoclonal antibodies that paradoxically increase receptor activity by antibody-induced receptor cross-linking (Engelmann et al., 1990) or genetic models lacking spatial or tissue specificity (Pfeffer et al., 1993; Peschon et al., 1998). Gene silencing via shRNA is an alternative method to inhibit TNFR1 function without these limitations (Yu et al., 2017), yet has never been used in the PVN in the context of hypertension. To assess the role of PVN TNFR1 signaling in blood pressure, the TNFR1 gene was silenced by local viral-mediated shRNA delivery in mice followed by 14 d administration of a slow-pressor dose of AngII (Glass et al., 2015).
We first investigated the effect of PVN rAAV-shTNFR1 microinjection on local TNFR1 expression. Control and test vectors were unilaterally microinjected into the PVN, resulting in regional expression of the GFP reporter in the hemisphere of microinjection (Fig. 8A,D) 3 weeks later. Compared with control vector-injected mice (n = 3; Fig. 8B), there was a decrease in TNFR1 in the injected hemisphere of mice receiving TNFR1 shRNA (n = 4; Fig. 8E) as assessed by immunohistochemistry (Fig. 8G ; t(5) = 5.7, p < 0.003, unpaired t test). Compared with control vector-injected animals (n = 3) mice receiving the TNFR1 shRNA vector (n = 3) also had lower TNFR1 mRNA in the PVN (t(4) = 5.1, p < 0.007, unpaired t test). There was no difference in NeuN labeling in the PVN of mice microinjected with either vector (Fig. 8C,F,H; t(4) = −1.9, p > 0.1, unpaired t test), indicating that silencing of TNFR1 in the PVN was not associated with neuron loss. The production of ROS is a major effect of TNFα stimulation (Sriramula and Francis, 2015). To investigate the functional consequences of TNFR1 silencing, TNFα-induced ROS signal was measured in isolated PVN cells of mice receiving control or test vectors. In isolated GFP-expressing cells (n = 18, three mice) from control injected mice, there was an increase in ROS signal following TNFα (t(17) = 4.2, p < 0.007, paired t test; Fig. 8I); however, TNFα did not affect ROS production in isolated GFP-expressing cells (n = 26, three mice) from TNFR1 shRNA-injected mice (t(25) = 1.6, p > 0.1, paired t test; Fig. 8J).
Figure 8.

Local delivery of rAAV expressing TNFR1 shRNA in PVN results in a reduction of TNFR1 levels. Light micrographs showing expression of the GFP reporter protein (A) and TNFR1 immunoreactivity (B) following unilateral microinjection of the control vector in the PVN (white dashed lines). Similar light micrographs of GFP labeling (D), as well as TNFR1 immunolabeling (E), after unilateral microinjection of the TNFR1 shRNA expressing test vector. After TNFR1 shRNA injection, TNFR1-labeled cells were reduced in the ipsilateral hemisphere as a percentage of the contralateral hemisphere (G; **p < 0.006 control vs shRNA). In PVN sections processed for NeuN, there was no difference in labeling in the ipsilateral and contralateral hemispheres of control-injected or shRNA-injected mice (C, F, H). In control-injected mice, ROS signal was increased in isolated PVN cells in response to TNFα (I; **p < 0.007), whereas ROS was not elevated by TNFα in mice injected with TNFR1 shRNA (J). 3v, Third ventricle. Scale bars, 0.5 mm.
To assess the role of PVN TNFR1 in hypertension, mice received local bilateral microinjections of vectors. Neither mice injected with rAAV expressing TNFR1 shRNA (n = 6) nor those administered the control vector (n = 6) differed in blood pressure before or 21 d following bilateral PVN microinjections (Fig. 9A; GFP: baseline, 100.3 mmHg ± 2.5 mmHg; vs day 21, 100.8 mmHg ± 3.9 mmHg; TNFR1: baseline, 102.3 mmHg ± 2.2 mmHg; vs day 21, 101.7 mmHg ± 5.0 mmHg; F(1,10) = 0.14, p > 0.7, repeated-measures ANOVA). Mice were then infused with vehicle or AngII. There was a significant effect of treatment (F(1,10) = 8.3, p < 0.02, ANOVA), session (F(4,40) = 2.6, p < 0.05, repeated-measures ANOVA) and a session by vector interaction (F(4,40) = 3.9, p < 0.01, repeated-measures ANOVA) with respect to systolic blood pressure. Following AngII administration, mice receiving the control vector showed an increase in systolic blood pressure over the course of treatment (Fig. 9B) reaching significant elevations at day 12 (p < 0.05) and day 14 (p < 0.05) compared with baseline and TNFR1 knock-down mice. Unlike control animals, however, mice given the TNFR1 shRNA did not respond with an increase in systolic blood pressure at any time point (Fig. 9B). Body weight increased in both control mice (Fig. 9C) and test vector-injected mice (Fig. 9D) during the 3 week postinjection period (F(2,20) = 43.6, p < 0.001, repeated-measures ANOVA), but there was no difference in body weight between the two treatment groups (F(2,20) = 0.53, p > 0.5, repeated-measures ANOVA). There was a significant decrease in TNFR1 labeling in the PVN (Fig. 9E; F(1,10) = 7.9, p < 0.02, one-way ANOVA) of mice receiving bilateral rAAV-TNFR1 shRNA (Fig. 9H,I) compared with those receiving the control vector (Fig. 9F,G). In sum, these results demonstrate that functional TNFR1 expression in the PVN is critical for the elevated systolic blood pressure in response to slow-pressor AngII administration.
Figure 9.

Spatial-temporal knockdown of TNFR1 in PVN neurons is associated with an inhibition of the hypertensive response to slow-pressor AngII infusion. A, Mice receiving bilateral microinjection of control or shRNA vectors did not differ in systemic systolic blood pressure 21 d postinjection versus preinjection. B, There was a significant difference in the hypertensive response to AngII in the control and test vector-treated groups (*p < 0.02, control vs shRNA on day 12 and day 14; #p < 0.02, ##p < 0.005, control day 0 vs days 12 and 14). Mice microinjected with the control vector showed a significant increase in blood pressure, whereas mice receiving the shRNA vector did not demonstrate any treatment-dependent elevation in blood pressure over the 14 d infusion period. C, D, There were no significant differences in body weight gain postinjection (*p < 0.05, control weeks 1–3; #p < 0.05, shRNA weeks 1–3). E, There was a significant reduction in TNFR1 expression in mice microinjected with the TNFR1 shRNA (*p < 0.05). F, G, Micrographs illustrating bilateral expression of the reporter protein GFP (F) and TNFR1 (G) in the PVN are shown after bilateral microinjection of control vector. H, I, Bilateral expression of GFP (H) and TNFR1 (I) is also shown after TNFR1 shRNA microinjection. Scale bar, 0.5 mm.
Discussion
Our results provide novel evidence that the slow-pressor response to AngII is associated with an increase in TNFR1 signaling in the PVN characterized by increased TNFR1 gene transcription and subcellular protein localization. In addition, AngII hypertension is also accompanied by an increase in TNFR1 levels and TNFR1-associated NMDA receptor currents in spinally projecting neurons in the PVN. The functional importance of TNFR1 in hypertension is further supported by our finding that silencing TNFR1 in the PVN inhibits the hypertension induced by slow-pressor AngII. These results point to an important role for TNFR1 signaling in PVN sympathoexcitatory neurons during the emergence of the slow-pressor response to AngII.
Within the PVN, we found that hypertension was associated with alterations in TNFR1 at distinct stages of its life cycle, including gene transcription and protein transport. First, it was shown that TNFR1 mRNA was increased in the PVN in mice infused for 14 d with AngII. The increase in TNFR1 transcript was found in the caudal region, but not the rostral region, of the PVN. Second, it was also shown that there was an elevation in plasma membrane-affiliated TNFR1 in PVN neurons of mice chronically infused with AngII. The latter findings are significant since following gene transcription and protein synthesis TNFR1 is transported to the plasma membrame, where it is positioned for the binding of extracellular TNFα (Brenner et al., 2015). We also found that TNFR1 immunoreactivity and plasma membrane affiliation were elevated in PVN neurons projecting to the IML. Thus, these results demonstrate that there is an increase in the pool of TNFR1 potentially available for TNFα binding and transduction of intracellular signaling pathways in PVN neurons projecting to the spinal cord during AngII-dependent hypertension.
Prior evidence for both elevated TNFα (Kang et al., 2008; Yu et al., 2015) and NMDA-type glutamate receptor signaling in the PVN during hypertension (Glass et al., 2015) suggests that TNFα may couple to a heightening of NMDA receptor signaling in the PVN during hypertension, although there has been little prior direct evidence for this. We presently report that TNFR1 signaling is critical for the expression of NMDA receptor-mediated currents in PVN neurons under basal and hypertensive states. Specifically, in single-cell patch-clamped PVN neurons, we show that NMDA receptor-dependent currents are inhibited by either constitutive knockout of the TNFR1 gene or pharmacological inhibition of the TNF receptor-associated death domain (TRADD)/receptor interacting protein 1 (RIP1) transduction pathway immediately downstream of TNFR1 activation. Importantly, the enhancement of NMDA receptor currents is shown to occur in PVN neurons that project to the IML, demonstrating that sympathoexcitatory neurons are targets of TNFR1-dependent modulation of NMDA receptor signaling. Moreover, these actions appear to have significant relevance for blood pressure regulation, given that NMDA receptor currents are even further potentiated in PVN neurons from mice following AngII-dependent hypertension and are blocked by inhibiting the TNFR1-coupled TRADD/RIP1 transduction pathway.
Functional TNFR1 may influence NMDA receptor signaling and hypertension by two modes of action involving either transcriptional/translational processes or rapid signaling events. TNFα is known to regulate gene expression via its coupling to the activity of the transcription factor nuclear factor (NF)-κB, a pivotal interface among synaptic activity, gene expression, and neuronal plasticity (Albensi and Mattson, 2000; Meffert et al., 2003). Genes regulated by NF-κB include NMDA receptor subunit genes (Tai et al., 2009) and NMDA receptor-interacting molecules [e.g., PSD-95, SAP97 (synapse-associated protein 97)] known to modulate receptor function (Schmeisser et al., 2012). In addition, other targets include kinases (e.g., PKA) known to regulate plasma membrane availability of NMDA receptors (Kaltschmidt et al., 2006) or molecules like the catalytic gp91phox subunit (Anrather et al., 2006), whose activity is known to modulate NMDA receptor signaling (Wang et al., 2013).
In addition to transcriptional effects, TNFR1 stimulation may also have rapid effects through its ability to modulate intraneuronal signaling cascades. In this context, we report that in the case of the NMDA receptor, signaling was inhibited in slices by acute (30 min) blockade of the TRADD/RIP1 signaling pathway, a time frame that is likely too short for new gene expression. Although the TRADD/RIP1 pathway has been classically linked to several signaling pathways involving transcriptional events via the regulation of NF-κB, other evidence also suggests a role in rapid nontranscriptional signaling processes including nonexcitotoxic effects of glutamate (Butler et al., 2002). Additional evidence from invertebrate models indicates that constitutive NF-κB itself can play a signaling role outside of the nucleus by contributing to the regulation of local events, including basal levels of glutamate activity (Meffert and Baltimore, 2005). Significantly, one postulated mechanism of local nontranscriptional NF-κB activity includes the regulation of postsynaptic clustering of glutamate receptors (Heckscher et al., 2007). Additionally, the TRADD/RIP1 pathway also engages other signaling molecules, including MAP kinase p38 (Schneider-Brachert et al., 2013), which is known to rapidly impact NMDA receptor signaling and neural plasticity (Weng et al., 2016). Current data cannot distinguish whether transcriptional, nontranscriptional, or a combination of these processes contribute to NMDA receptor signaling during hypertension, but this can be investigated in the future.
The relevant source of TNFα targeting TNFR1 in the PVN may originate from the circulation, infiltrating immune cells, or local release by resident brain cells (Vezzani and Viviani, 2015). Within the PVN, an intrinsic TNFα system involving glia (Shi et al., 2010; Du et al., 2015), and possibly neurons (Breder et al., 1993), has been previously described. Significantly, evidence suggests that the hypertension-associated increase in TNFα may originate from a microglial source (Shi et al., 2010; Sriramula et al., 2011; Cardinale et al., 2012; Yu et al., 2015). This is particularly noteworthy in that glial-derived TNFα has been implicated in models of neuronal plasticity by influencing the coordination of plasma membrane glutamate receptors and increasing neuronal excitability (Beattie et al., 2002; Stellwagen and Malenka, 2006).
In conclusion, the present report shows that TNFR1 activity in the PVN is an important contributor to slow-onset AngII-dependent hypertension, possibly acting via modulation of NMDA receptor signaling in sympathoexcitatory neurons.
Footnotes
This research was supported by National Institutes of Health Grants HL-135498 (to M.J.G.) and HL-136520 (to M.J.G., T.A.M.).
The authors declare no competing financial interests.
References
- Albensi BC, Mattson MP (2000) Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse 35:151–159. [DOI] [PubMed] [Google Scholar]
- Anrather J, Racchumi G, Iadecola C (2006) NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem 281:5657–5667. 10.1074/jbc.M506172200 [DOI] [PubMed] [Google Scholar]
- Baez MV, Cercato MC, Jerusalinsky DA (2018) NMDA receptor subunits change after synaptic plasticity induction and learning and memory acquisition. Neural Plast 2018:5093048 10.1155/2018/5093048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains JS, Ferguson AV (1995) Paraventricular nucleus neurons projecting to the spinal cord receive excitatory input from the subfornical organ. Am J Physiol 268:R625–R633. 10.1152/ajpregu.1995.268.3.R625 [DOI] [PubMed] [Google Scholar]
- Bardgett ME, Holbein WW, Herrera-Rosales M, Toney GM (2014) Ang II-salt hypertension depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-α. Hypertension 63:527–534. 10.1161/HYPERTENSIONAHA.113.02429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basting T, Xu J, Mukerjee S, Epling J, Fuchs R, Sriramula S, Lazartigues E (2018) Glutamatergic neurons of the paraventricular nucleus are critical contributors to the development of neurogenic hypertension. J Physiol 596:6235–6248. 10.1113/JP276229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science 295:2282–2285. 10.1126/science.1067859 [DOI] [PubMed] [Google Scholar]
- Beckerman MA, Van Kempen TA, Justice NJ, Milner TA, Glass MJ (2013) Corticotropin-releasing factor in the mouse central nucleus of the amygdala: ultrastructural distribution in NMDA-NR1 receptor subunit expressing neurons as well as projection neurons to the bed nucleus of the stria terminalis. Exp Neurol 239:120–132. 10.1016/j.expneurol.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bette M, Kaut O, Schäfer MK, Weihe E (2003) Constitutive expression of p55TNFR mRNA and mitogen-specific up-regulation of TNF alpha and p75TNFR mRNA in mouse brain. J Comp Neurol 465:417–430. 10.1002/cne.10877 [DOI] [PubMed] [Google Scholar]
- Breder CD, Tsujimoto M, Terano Y, Scott DW, Saper CB (1993) Distribution and characterization of tumor necrosis factor-alpha-like immunoreactivity in the murine central nervous system. J Comp Neurol 337:543–567. 10.1002/cne.903370403 [DOI] [PubMed] [Google Scholar]
- Brenner D, Blaser H, Mak TW (2015) Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15:362–374. 10.1038/nri3834 [DOI] [PubMed] [Google Scholar]
- Butler MP, Moynagh PN, O'Connor JJ (2002) Methods of detection of the transcription factor NF-κB in rat hippocampal slices. J Neurosci Methods 119:185–190. 10.1016/S0165-0270(02)00171-1 [DOI] [PubMed] [Google Scholar]
- Capone C, Faraco G, Peterson JR, Coleman C, Anrather J, Milner TA, Pickel VM, Davisson RL, Iadecola C (2012) Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci 32:4878–4886. 10.1523/JNEUROSCI.6262-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale JP, Sriramula S, Mariappan N, Agarwal D, Francis J (2012) Angiotensin II-induced hypertension is modulated by nuclear factor-Bin the paraventricular nucleus. Hypertension 59:113–121. 10.1161/HYPERTENSIONAHA.111.182154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman CG, Wang G, Park L, Anrather J, Delagrammatikas GJ, Chan J, Zhou J, Iadecola C, Pickel VM (2010) Chronic intermittent hypoxia induces NMDA receptor-dependent plasticity and suppresses nitric oxide signaling in the mouse hypothalamic paraventricular nucleus. J Neurosci 30:12103–12112. 10.1523/JNEUROSCI.3367-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman CG, Wang G, Faraco G, Marques Lopes J, Waters EM, Milner TA, Iadecola C, Pickel VM (2013) Membrane trafficking of NADPH oxidase p47(phox) in paraventricular hypothalamic neurons parallels local free radical production in angiotensin II slow-pressor hypertension. J Neurosci 33:4308–4316. 10.1523/JNEUROSCI.3061-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai SY, Peng W, Zhang YP, Li JD, Shen Y, Sun XF (2015) Brain endogenous angiotensin II receptor type 2 (AT2-R) protects against DOCA/salt-induced hypertension in female rats. J Neuroinflammation 12:47. 10.1186/s12974-015-0261-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dange RB, Agarwal D, Teruyama R, Francis J (2015) Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. J Neuroinflammation 12:31. 10.1186/s12974-015-0242-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rivero T, Fischer R, Yang F, Swanson KA, Bethea JR (2019) Tumor necrosis factor receptor 1 inhibition is therapeutic for neuropathic pain in males but not in females. Pain 160:922–931. [DOI] [PubMed] [Google Scholar]
- Dickinson CJ, Lawrence JR (1963) A slowly developing pressor response to small concentrations of angiotensin. Its bearing on the pathogenesis of chronic renal hypertension. Lancet 1:1354–1356. 10.1016/s0140-6736(63)91929-9 [DOI] [PubMed] [Google Scholar]
- Du D, Jiang M, Liu M, Wang J, Xia C, Guan R, Shen L, Ji Y, Zhu D (2015) Microglial P2X7 receptor in the hypothalamic paraventricular nuclei contributes to sympathoexcitatory responses in acute myocardial infarction rat. Neurosci Lett 587:22–28. 10.1016/j.neulet.2014.12.026 [DOI] [PubMed] [Google Scholar]
- Engelmann H, Holtmann H, Brakebusch C, Avni Y, Sarov I, Nophar Y, Hadas E, Leitner O, Wallach D (1990) Antibodies to a soluble form of a tumor necrosis factor (TNF) receptor have TNF-like activity. J Biol Chem 265:14497–14504. [PubMed] [Google Scholar]
- Ferguson AV (2009) Angiotensinergic regulation of autonomic and neuroendocrine outputs: critical roles for the subfornical organ and paraventricular nucleus. Neuroendocrinol 89:370–376. 10.1159/000211202 [DOI] [PubMed] [Google Scholar]
- Glass MJ, Hegarty DM, Oselkin M, Quimson L, South SM, Xu Q, Pickel VM, Inturrisi CE (2008) Conditional deletion of the NMDA-NR1 receptor subunit gene in the central nucleus of the amygdala inhibits naloxone-induced conditioned place aversion in morphine dependent mice. Exp Neurol 213:57–70. 10.1016/j.expneurol.2008.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Wang G, Coleman CG, Chan J, Ogorodnik E, Van Kempen TA, Milner TA, Butler SD, Young CN, Davisson RL, Iadecola C, Pickel VM (2015) NMDA receptor plasticity in the hypothalamic paraventricular nucleus contributes to the elevated blood pressure produced by angiotensin II. J Neurosci 35:9558–9567. 10.1523/JNEUROSCI.2301-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Chan J, Pickel VM (2017) Ultrastructural characterization of tumor necrosis factor alpha receptor type 1 distribution in the hypothalamic paraventricular nucleus of the mouse. Neuroscience 352:262–272. 10.1016/j.neuroscience.2017.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Ram VS (2016) Evidence for a critical role of the sympathetic nervous system in hypertension. J Am Soc Hypertens 10:457–466. 10.1016/j.jash.2016.02.015 [DOI] [PubMed] [Google Scholar]
- Gururaja TL, Yung S, Ding R, Huang J, Zhou X, McLaughlin J, Daniel-Issakani S, Singh R, Cooper RD, Payan DG, Masuda ES, Kinoshita T (2007) A class of small molecules that inhibit TNFalpha-induced survival and death pathways via prevention of interactions between TNFalphaRI, TRADD, and RIP1. Chem Biol 14:1105–1118. 10.1016/j.chembiol.2007.08.012 [DOI] [PubMed] [Google Scholar]
- Han P, Whelan PJ (2010) Tumor necrosis factor alpha enhances glutamatergic transmission onto spinal motoneurons. J Neurotrauma 27:287–292. 10.1089/neu.2009.1016 [DOI] [PubMed] [Google Scholar]
- Hara Y, Pickel VM (2008) Preferential relocation of the N-methyl-d-aspartate receptor NR1 subunit in nucleus accumbens neurons that contain dopamine D1 receptors in rats showing an apomorphine-induced sensorimotor gating deficit. Neuroscience 154:965–977. 10.1016/j.neuroscience.2008.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckscher ES, Fetter RD, Marek KW, Albin SD, Davis GW (2007) NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ. Neuron 55:859–873. 10.1016/j.neuron.2007.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prüllage M, Pfeiffer J, Lindecke A, Staiger V, Israël A, Kaltschmidt C, Mémet S (2006) NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol Cell Biol 26:2936–2946. 10.1128/MCB.26.8.2936-2946.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YM, Zhang ZH, Xue B, Weiss RM, Felder RB (2008) Inhibition of brain proinflammatory cytokine synthesis reduces hypothalamic excitation in rats with ischemia-induced heart failure. Am J Physiol 295:H227–H236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laragh J (2001) Laragh's lessons in pathophysiology and clinical pearls for treating hypertension. Am J Hypertens 14:186–194. 10.1016/S0895-7061(00)01317-0 [DOI] [PubMed] [Google Scholar]
- Lerman LO, Kurtz TW, Touyz RM, Ellison DH, Chade AR, Crowley SD, Mattson DL, Mullins JJ, Osborn J, Eirin A, Reckelhoff JF, Iadecola C, Coffman TM (2019) Animal models of hypertension: a scientific statement from the American Heart Association. Hypertension 73:e87–e120. 10.1161/HYP.0000000000000090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Pan HL (2017) Glutamatergic regulation of hypothalamic presympathetic neurons in hypertension. Curr Hypertens Rep 19:78. 10.1007/s11906-017-0776-4 [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL (2002) Nitric oxide inhibits spinally projecting paraventricular neurons through potentiation of presynaptic GABA release. J Neurophysiol 88:2664–2674. 10.1152/jn.00540.2002 [DOI] [PubMed] [Google Scholar]
- Li DP, Zhou JJ, Zhang J, Pan HL (2017) CaMKII regulates synaptic NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension. J Neurosci 37:10690–10699. 10.1523/JNEUROSCI.2141-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, Mayhan WG, Patel KP (2001) NMDA-mediated increase in renal sympathetic nerve discharge within the PVN: role of nitric oxide. Am J Physiol Heart Circ Physiol 281:H2328–H2336. 10.1152/ajpheart.2001.281.6.H2328 [DOI] [PubMed] [Google Scholar]
- Li YF, Cornish KG, Patel KP (2003) Alteration of NMDA NR1 receptors within the paraventricular nucleus of hypothalamus in rats with heart failure. Circ Res 93:990–997. 10.1161/01.RES.0000102865.60437.55 [DOI] [PubMed] [Google Scholar]
- Lind RW, Ohman LE, Lansing MB, Johnson AK (1983) Transection of subfornical organ neural connections diminishes the pressor response to intravenously infused angiotensin II. Brain Res 275:361–364. 10.1016/0006-8993(83)90998-8 [DOI] [PubMed] [Google Scholar]
- Llewellyn T, Zheng H, Liu X, Xu B, Patel KP (2012) Median preoptic nucleus and subfornical organ drive renal sympathetic nerve activity via a glutamatergic mechanism within the paraventricular nucleus. Am J Physiol 302:R424–432. 10.1152/ajpregu.00403.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiapane ML, Simpson JB (1980a) Subfornical organ lesions reduce the pressor effect of systemic angiotensin II. Neuroendocrinology 31:380–384. 10.1159/000123107 [DOI] [PubMed] [Google Scholar]
- Mangiapane ML, Simpson JB (1980b) Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. Am J Physiol 239:R382–R389. 10.1152/ajpregu.1980.239.5.R382 [DOI] [PubMed] [Google Scholar]
- Marques-Lopes J, Lynch MK, Van Kempen TA, Waters EM, Wang G, Iadecola C, Pickel VM, Milner TA (2015) Female protection from slow-pressor effects of angiotensin II involves prevention of ROS production independent of NMDA receptor trafficking in hypothalamic neurons expressing angiotensin 1A receptors. Synapse 69:148–165. 10.1002/syn.21800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Lopes J, Tesfaye E, Israilov S, Van Kempen TA, Wang G, Glass MJ, Pickel VM, Iadecola C, Waters EM, Milner TA (2017) Redistribution of NMDA receptors in estrogen receptor β-containing paraventricular hypothalamic neurons following slow-pressor angiotensin II hypertension in female mice with accelerated ovarian failure. Neuroendocrinology 104:239–256. 10.1159/000446073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins-Pinge MC, Mueller PJ, Foley CM, Heesch CM, Hasser EM (2013) Regulation of arterial pressure by the paraventricular nucleus in conscious rats: interactions among glutamate, GABA, and nitric oxide. Front Physiol 3:490. 10.3389/fphys.2012.00490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB (1984) Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309:261–263. 10.1038/309261a0 [DOI] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D (2005) Physiological functions for brain NF-kappaB. Trends Neurosci 28:37–43. 10.1016/j.tins.2004.11.002 [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (2003) NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci 6:1072–1078. 10.1038/nn1110 [DOI] [PubMed] [Google Scholar]
- Mehaffey E, Majid DSA (2017) Tumor necrosis factor-α, kidney function, and hypertension. Am J Physiol Renal Physiol 313:F1005–F1008. 10.1152/ajprenal.00535.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner TA, Waters EM, Robinson DC, Pierce JP (2011) Degenerating processes identified by electron microscpic immunocytochemical methods. In: Neurodegeneration, methods and protocols (Manfredi G, Kawamata H, eds), pp 23–59. New York: Springer. [DOI] [PubMed] [Google Scholar]
- Moraga I, Spangler J, Mendoza JL, Garcia KC (2014) Multifarious determinants of cytokine receptor signaling specificity. Adv Immunol 121:1–39. 10.1016/B978-0-12-800100-4.00001-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadar SK, Tayebjee MH, Messerli F, Lip GY (2006) Target organ damage in hypertension: pathophysiology and implications for drug therapy. Curr Pharm Des 12:1581–1592. 10.2174/138161206776843368 [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S (1999) Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience 93:1449–1464. 10.1016/s0306-4522(99)00225-0 [DOI] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS, Harrison DG (2018) The immunology of hypertension. J Exp Med 215:21–33. 10.1084/jem.20171773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen MH, Angell SY, Asma S, Boutouyrie P, Burger D, Chirinos JA, Damasceno A, Delles C, Gimenez-Roqueplo AP, Hering D, López-Jaramillo P, Martinez F, Perkovic V, Rietzschel ER, Schillaci G, Schutte AE, Scuteri A, Sharman JE, Wachtell K, Wang JG (2016) A call to action and a lifecourse strategy to address the global burden of raised blood pressure on current and future generations: the Lancet Commission on hypertension. Lancet 388:2665–2712. 10.1016/S0140-6736(16)31134-5 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB (2000) The mouse brain in stereotaxic coordinates. San Diego: Academic. [Google Scholar]
- Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM (1998) TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol 160:943–952. [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster H (1991) The fine structure of the nervous system. New York: Oxford UP. [Google Scholar]
- Pfeffer K, Matsuyama T, Kündig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Krönke M, Mak TW (1993) Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457–467. 10.1016/0092-8674(93)90134-C [DOI] [PubMed] [Google Scholar]
- Puszkarska A, Niklas A, Głuszek J, Lipski D, Niklas K (2019) The concentration of tumor necrosis factor in the blood serum and in the urine and selected early organ damages in patients with primary systemic arterial hypertension. Medicine (Baltimore) 98:e15773. 10.1097/md.0000000000015773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizk NM, Joost HG, Eckel J (2001) Increased hypothalamic expression of the p75 tumor necrosis factor receptor in New Zealand obese mice. Horm Metab Res 33:520–524. 10.1055/s-2001-17209 [DOI] [PubMed] [Google Scholar]
- Rizzo FR, Musella A, De Vito F, Fresegna D, Bullitta S, Vanni V, Guadalupi L, Stampanoni Bassi M, Buttari F, Mandolesi G, Centonze D, Gentile A (2018) Tumor necrosis factor and interleukin-1β modulate synaptic plasticity during neuroinflammation. Neural Plast 2018:8430123. 10.1155/2018/8430123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Volterra A (2012) TNFα in synaptic function: switching gears. Trends Neurosci 35:638–647. 10.1016/j.tins.2012.06.001 [DOI] [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW (1983) The organization of forebrain afferents to the paraventricular and supraoptic nuclei of the rat. J Comp Neurol 218:121–144. 10.1002/cne.902180202 [DOI] [PubMed] [Google Scholar]
- Schmeisser MJ, Baumann B, Johannsen S, Vindedal GF, Jensen V, Hvalby ØC, Sprengel R, Seither J, Maqbool A, Magnutzki A, Lattke M, Oswald F, Boeckers TM, Wirth T (2012) IκB kinase/nuclear factor κB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J Neurosci 32:5688–5703. 10.1523/JNEUROSCI.0111-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Brachert W, Heigl U, Ehrenschwender M (2013) Membrane trafficking of death receptors: implications on signalling. Int J Mol Sci 14:14475–14503. 10.3390/ijms140714475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK (2010) Brain microglial cytokines in neurogenic hypertension. Hypertension 56:297–303. 10.1161/HYPERTENSIONAHA.110.150409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Jiang SJ, Wang GH, Xu AL, Guo L (2014) Pro-inflammatory cytokines in paraventricular nucleus mediate the cardiac sympathetic afferent reflex in hypertension. Auton Neurosci 186:54–61. 10.1016/j.autneu.2014.10.001 [DOI] [PubMed] [Google Scholar]
- Sriramula S, Haque M, Majid DS, Francis J (2008) Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51:1345–1351. 10.1161/hypertensionaha.107.102152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula S, Francis J (2015) Tumor necrosis factor - alpha is essential for angiotensin II-induced ventricular remodeling: role for oxidative stress. PLoS One 10:e0138372 10.1371/journal.pone.0138372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula S, Cardinale JP, Lazartigues E, Francis J (2011) ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res 92:401–408. 10.1093/cvr/cvr242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula S, Cardinale JP, Francis J (2013) Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II-induced hypertension. PLoS One 8:e63847. 10.1371/journal.pone.0063847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059. 10.1038/nature04671 [DOI] [PubMed] [Google Scholar]
- Suh YH, Terashima A, Petralia RS, Wenthold RJ, Isaac JT, Roche KW, Roche PA (2010) A neuronal role for SNAP-23 in postsynaptic glutamate receptor trafficking. Nat Neurosci 13:338–343. 10.1038/nn.2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai DJ, Su CC, Ma YL, Lee EH (2009) SGK1 phosphorylation of IkappaB Kinase alpha and p300 Up-regulates NF-kappaB activity and increases N-Methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem 284:4073–4089. 10.1074/jbc.M805055200 [DOI] [PubMed] [Google Scholar]
- Valentinova K, Tchenio A, Trusel M, Clerke JA, Lalive AL, Tzanoulinou S, Matera A, Moutkine I, Maroteaux L, Paolicelli RC, Volterra A, Bellone C, Mameli M (2019) Morphine withdrawal recruits lateral habenula cytokine signaling to reduce synaptic excitation and sociability. Nat Neurosci 22:1053–1056. 10.1038/s41593-019-0421-4 [DOI] [PubMed] [Google Scholar]
- Vezzani A, Viviani B (2015) Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacol 96:70–82. 10.1016/j.neuropharm.2014.10.027 [DOI] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, Luo Y (2012) RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14:22–29. 10.1016/j.jmoldx.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Coleman CG, Chan J, Faraco G, Marques-Lopes J, Milner TA, Guruju MR, Anrather J, Davisson RL, Iadecola C, Pickel VM (2013) Angiotensin II slow-pressor hypertension enhances NMDA currents and NOX2-dependent superoxide production in hypothalamic paraventricular neurons. Am J Physiol 304:R1096–1106. 10.1152/ajpregu.00367.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Su N, Wang LC, Wu X, Bui S, Nielsen A, Vo HT, Luo Y, Ma XJ (2014) Quantitative ultrasensitive bright-field RNA in situ hybridization with RNAscope. Methods Mol Biol 1211:201–212. 10.1007/978-1-4939-1459-3_16 [DOI] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Felder RB (2018) Blood-borne interleukin-1β acts on the subfornical organ to upregulate the sympathoexcitatory milieu of the hypothalamic paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol 314:R447–R458. 10.1152/ajpregu.00211.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng W, Chen Y, Wang M, Zhuang Y, Behnisch T (2016) Potentiation of Schaffer-collateral CA1 synaptic transmission by eEF2K and p38 MAPK mediated mechanisms. Front Cell Neurosci 10:247. 10.3389/fncel.2016.00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN, Cao X, Guruju MR, Pierce JP, Morgan DA, Wang G, Iadecola C, Mark AL, Davisson RL (2012) ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest 122:3960–3964. 10.1172/JCI64583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Xue BJ, Wei SG, Zhang ZH, Beltz TG, Guo F, Johnson AK, Felder RB (2015) Activation of central PPAR-γ attenuates angiotensin II-induced hypertension. Hypertension 66:403–411. 10.1161/HYPERTENSIONAHA.115.05726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wei SG, Weiss RM, Felder RB (2017) TNF-α receptor 1 knockdown in the subfornical organ ameliorates sympathetic excitation and cardiac hemodynamics in heart failure rats. Am J Physiol Heart Circ Physiol 313:H744–H756. 10.1152/ajpheart.00280.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR (2011) TNF-α contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain 152:419–427. 10.1016/j.pain.2010.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JJ, Ma HJ, Shao JY, Pan HL, Li DP (2019) Impaired Hypothalamic Regulation of Sympathetic Outflow in Primary Hypertension. Neurosci Bull 35:124–132. 10.1007/s12264-018-0316-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Ho IK (1998) NMDA-R1 antisense oligonucleotide attenuates withdrawal signs from morphine. Eur J Pharmacol 352:151–156. 10.1016/s0014-2999(98)00367-7 [DOI] [PubMed] [Google Scholar]