

Abstract

Mixed-halide perovskites offer bandgap tunability essential for multijunction solar cells; however, a detrimental halide segregation under light is often observed. Here we combine simultaneous in situ photoluminescence and X-ray diffraction measurements to demonstrate clear differences in compositional and optoelectronic changes associated with halide segregation in MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 films. We report evidence for low-barrier ionic pathways in MAPb(Br0.5I0.5)3, which allow for the rearrangement of halide ions in localized volumes of perovskite without significant compositional changes to the bulk material. In contrast, FA0.83Cs0.17Pb(Br0.4I0.6)3 lacks such low-barrier ionic pathways and is, consequently, more stable against halide segregation. However, under prolonged illumination, it exhibits a considerable ionic rearrangement throughout the bulk material, which may be triggered by an initial demixing of A-site cations, altering the composition of the bulk perovskite and reducing its stability against halide segregation. Our work elucidates links between composition, ionic pathways, and halide segregation, and it facilitates the future engineering of phase-stable mixed-halide perovskites.
The success of metal halide perovskites (MHPs) is often attributed to their excellent optoelectronic properties, coupled with the relatively facile processing techniques needed to fabricate corresponding photovoltaic devices.1,2 In addition, the large tolerance of the perovskite crystal lattice to structural distortions allows for the mixing of different ions across similar lattice sites in the perovskite structure, an important factor for the stoichiometric tuning of MHPs.3−7 Such stoichiometric engineering of the perovskite composition grants a relatively straightforward control method over a range of basic optoelectronic and material properties, such as the bandgap energy,3−6,8,9 crystallinity,8,10 and chemical stability.6,10,11 For photovoltaic applications, bandgap control is often achieved in lead-based MHPs by mixing iodide and bromide ions across X-anion sites in the ABX3 perovskite structure,3,4,6,8 yielding APb(BrxI(1–x))3 compositions, where x indicates the halide content of the perovskite and the A-cation sites are usually occupied by MA+ (methylammonium, CH3NH3+), FA+ (formamidinium, CH(NH2)2+), Cs+, or a mixture thereof. Control of the material bandgap is particularly important for tandem photovoltaic applications, for which the total efficiency of the tandem stack is strongly dependent on the bandgaps of the composite layers.12,13 Because of the bandgap tunability of MHPs, research into perovskite-perovskite14−17 and perovskite-silicon18,19 tandem solar cells has been greatly successful, opening a major commercial opportunity for perovskite device technologies.
While mixing iodide and bromide ions in the perovskite structure conveniently allows for bandgap tuning, it unfortunately also facilitates a compositional instability problem known as halide segregation.3 During the halide segregation process, excited charge carriers generated either through photon absorption3,20−23 or current injection24−28 drive the separation of the halide species, resulting in the formation of perovskite regions with differing halide composition and hence different bandgap energies. In particular, the formation of iodide-rich regions causes low-bandgap domains within the bulk perovskite material into which excited charge carriers funnel and collect, resulting in a red-shifted emission. In addition, such low-bandgap regions can compromise the charge-carrier extraction efficiency23,24 and the open-circuit voltage25,29 of MHP photovoltaic devices, lowering their performance. Interestingly, ceasing the generation of excited charge carriers allows the halide ions to remix, and eventually the perovskite material returns to its presegregated state.3,20,23,30 While the remixing dynamic is thought to occur for entropic reasons,21,31 the cause of the initial segregation is still much debated, with several potential mechanisms having been proposed.32 Empirically, many factors have been found to affect the halide segregation mechanism, such as crystallinity,10,24,33−35 trap state density,20,23,36−43 and the average stoichiometry of the perovskite.6,10,11,44,45 Interestingly, the composition of the A-cation site appears to have a particularly strong effect on the extent to which halide segregation occurs. MAPb(BrxI(1–x))3 materials are known to segregate under relatively low photon doses for intermediate iodide-bromide ratios (0.2 < x < 1).3,11,21,22 In contrast, mixed A-cation FA(1–y)CsyPb(BrxI(1–x))3 perovskites of comparable bromide content x are known to be much more stable and possess a much lower probability for ionic separation within the perovskite structure, with much higher photon doses required to induce halide separation6,10,24 provided the Cs content y remains within the range of ∼10–30%.10 However, the underlying reasons behind the influence of these factors on the segregation dynamics, and how they may be interconnected, is still largely unclear.11
Exploration of the causes of halide segregation in mixed-halide perovskites has been further complicated by the separate use of two measurement techniques, namely, photoluminescence (PL)3,20−22 and X-ray diffraction (XRD).3,10,46,47 While each technique offers valuable insights, they have mostly been used in isolation, even though each tends to probe different regions of the perovskite. PL techniques reflect the electronic energy levels at sites where charge carriers recombine, meaning they mostly probe the iodide-rich domains formed through the segregation process, since charge carriers will quickly diffuse to such low-energy regions.3,20−22 XRD measurements are more sensitive to the volume average of the crystalline perovskite and, as such, give a more comprehensive picture of the compositional variations within the whole material. While PL measurements of mixed-halide perovskites consistently exhibit red-shifted emission following halide segregation, XRD measurements of a segregated material tend to vary more substantially and, additionally, depend on the perovskite composition.48 These considerations suggest that an understanding of the halide segregation process would hugely benefit from in situ, combined measurements of changes in PL and XRD patterns under illumination. Such in situ XRD/PL measurements could grant an understanding of both the underlying ionic movement and the electronic changes that occur during the halide segregation process, elucidating, for example, links between material composition and phase stability.
In this study, we implemented in situ, combined XRD/PL measurements to elucidate why MA-cation mixed-halide perovskites exhibit a substantially different propensity toward halide segregation compared with certain mixed formamidinium-cesium (FACs) cation counterparts. We explore two specific compositions, MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3, under long-term illumination and subsequent periods of darkness. While MA-cation mixed-halide perovskites are the prototypical compositions often investigated in halide-segregation studies,3,11,21,22 the vastly more stable FA0.83Cs0.17Pb(Br0.4I0.6)36,10,24 is a much more relevant material for high-performance photovoltaic devices,6 especially as the top (sun-facing) absorber layer in perovskite-perovskite tandem solar cells.14,17,49 Selecting perovskite compositions with a roughly even bromide/iodide ratio ensures that the halide segregation process will occur over a reasonable time frame in our experiments.11 We note that differences in the halide compositions and manufacturing processes for the two examined compositions may have had a slight influence over the resulting halide segregation dynamics in the perovskite films. However, FACs lead halide perovskites have been observed to possess a much greater stability against halide segregation as compared to MA-based perovskites over a wide range of manufacturing methods and halide contents.11 Therefore, the A-site cation choice will be the dominant factor in the phase stability of lead mixed-halide perovskites of similar iodide and bromide content.
We further note that links between the film crystallinity and the A-site cation composition6,10,24 highlight that FACs perovskites likely experience more ideal film formation dynamics than MA-based perovskites and that the stability of FACs perovskites may be partly associated with their increased crystallinity. In Section 2.1 of the Supporting Information we show that the width of the XRD peaks recorded from our FA0.83Cs0.17Pb(Br0.4I0.6)3 thin films are indeed slightly smaller than from our MAPb(Br0.5I0.5)3 films, in agreement with these literature reports.11 Further discussion regarding such slight material differences and how they may affect the halide segregation dynamics is provided in Section 3 of the Supporting Information.
Our combined XRD/PL measurements allow for a detailed understanding of the halide segregation process in each perovskite material, derived from the observation of both incipient low-bandgap regions in PL measurements and the analysis of the bulk material gained from XRD measurements. We conclude that, for MAPb(Br0.5I0.5)3, halide segregation initiates in minority regions of the perovskite material, rather than throughout the bulk. Such regions must hence be particularly susceptible to ionic rearrangements, for example, through the existence of fast ionic pathways near grain boundaries.50−54 We find that the growth and accumulation of these halide-segregated minority regions gradually reduces the volume of the remaining well-mixed perovskite phase. However, the composition (as inferred from the lattice parameters) of the remnant mixed-halide majority phase itself remains unchanged throughout the halide segregation dynamics. These observations demonstrate that, for MAPb(Br0.5I0.5)3, the segregation of halide ions occurs within and spreads from specific locations inside the material. Conversely, we find that in order for halide segregation to occur in FA0.83Cs0.17Pb(Br0.4I0.6)3, large amounts of ionic rearrangement are induced throughout the perovskite in unison, indicating a lack of regions that are particularly susceptible to halide segregation in this material and explaining the typically high photostability of FACs A-cation mixed-halide perovskites,6,10,24 for Cs content of ∼10–30%. We propose that, for such FACs perovskites, halide segregation may instead be triggered by the initial segregation of the A-site cations, which enriches the bulk perovskite in either FA or Cs and, in turn, reduces the stability of the perovskite against halide segregation.
Figure 1 presents PL spectra and XRD patterns recorded for MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 perovskite thin films under illumination, collected in situ utilizing our custom-built setup that allows for simultaneous, time-resolved XRD and PL measurements. During the measurements, MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 films were exposed to 15 min and 6 h of illumination, respectively, from a 470 nm wavelength, continuous-wave laser. The intensity of illumination was set to 190 mW cm–2 for both compositions. This intensity, while somewhat above 1 sun and the level required to induce halide segregation in the MAPb(Br0.5I0.5)3 perovskite film, was needed to induce halide segregation in the more stable FA0.83Cs0.17Pb(Br0.4I0.6)3 film within a reasonable time frame. All perovskite films investigated in this study were overcoated with a layer of poly(methyl methacrylate) (PMMA) to provide protection against unwanted interactions between the perovskite and the surrounding air.20 Full details of the experimental apparatus and parameters are provided in Section 5 of the Supporting Information.
Figure 1.
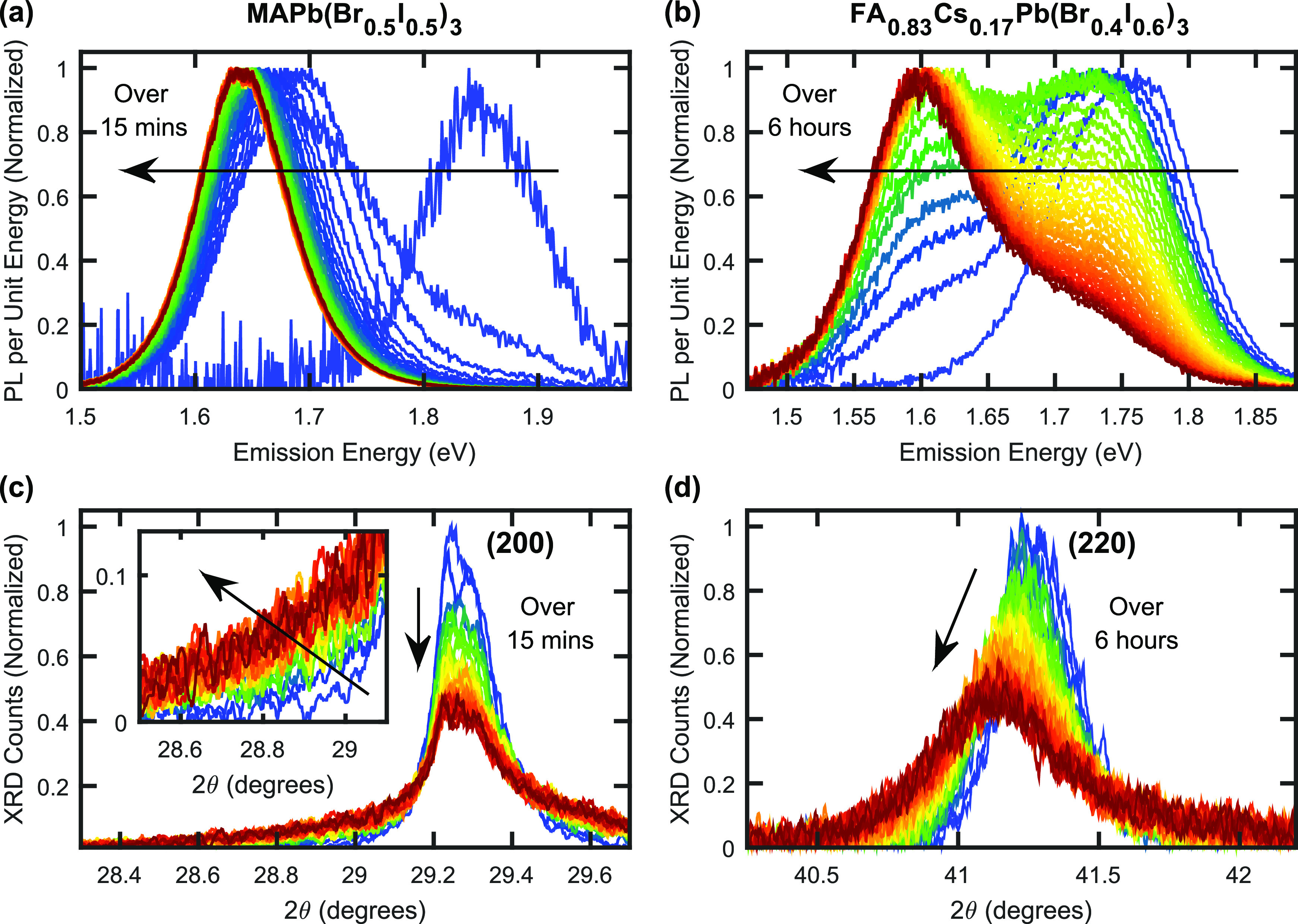
(a) Normalized PL spectra for an MAPb(Br0.5I0.5)3 thin film coated with PMMA, recorded over 15 min of illumination under a light of intensity of 190 mW cm–2, wavelength 470 nm. (b) Normalized PL spectra for an FA0.83Cs0.17Pb(Br0.4I0.6)3 thin film coated with PMMA, recorded over 6 h of illumination under a light of intensity of 190 mW cm–2, wavelength 470 nm. Red-shifts in (a, b) are caused by halide segregation. (c) XRD patterns recorded in situ at the same time and on the same MAPb(Br0.5I0.5)3 thin film as the data shown in (a). The angle axis is scaled to focus on the region around the cubic (200) peak in the recorded XRD data. (inset) Enlarged region of the low angle tail of the (200) peak. (d) XRD patterns recorded in situ at the same time and on the same FA0.83Cs0.17Pb(Br0.4I0.6)3 thin film as the data shown in (b). The angle axis is scaled to show the cubic (220) peak in the recorded XRD data. (c, d) The Cu Kα1 line was used as incident radiation.
The normalized PL spectra of MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 thin films develop in a qualitatively similar manner, with the growth and eventual dominance of a low-energy PL peak—ascribed to iodide-rich regions—clearly visible in Figure 1a,b, respectively. However, as expected, the time scales for halide segregation to occur in the two compositions were significantly different, with the PL spectrum of MAPb(Br0.5I0.5)3 reaching equilibrium on the order of tens of minutes (Figure 1a), while changes still occurred in the PL spectrum of FA0.83Cs0.17Pb(Br0.4I0.6)3 after several hours (Figure 1b). Despite the large difference in time and photon doses required to induce halide segregation in the two different perovskite thin films, the observed red-shifts of the corresponding PL spectra appear comparable.
In contrast, XRD patterns for MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 films, recorded in situ at the same time as the PL spectra discussed above and under the same illumination conditions, show markedly different dynamics (Figure 1c,d, respectively). On the one hand, for MAPb(Br0.5I0.5)3, Figure 1c shows that, over the course of 15 min of illumination, the (200) diffraction peak in the XRD pattern decreased in amplitude but remained centered at a fixed 2θ value of ∼29.25°. For FA0.83Cs0.17Pb(Br0.4I0.6)3, on the other hand, Figure 1d illustrates that the (220) diffraction peak shifted to lower angles and widened significantly during the 6 h of illumination. In addition, substantial differences can be observed in the tail regions of these peaks, farther away from their center positions. For MAPb(Br0.5I0.5)3, a significant growth of XRD intensity in the high- and low-angle tails of the (200) peak can be observed, as highlighted in the inset to Figure 1c for the low-angle tail. For FA0.83Cs0.17Pb(Br0.4I0.6)3, such growth in the low-angle tail amplitude to the (220) peak is negligible (Figure 1d); however, a slight amount of high-angle tail growth is evident (see Figures S3 and S4 for an analysis of this high-angle region). The specific XRD peaks discussed here were selected based on their prominence in the corresponding XRD patterns (see Section 5 in the Supporting Information for a discussion), with XRD patterns over a wider range of scattering angles provided in Figures S1 and S2. Figures S10 and S11 in the Supporting Information show that the behavior of the (200) and (220) XRD peaks of the MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 films, respectively, is similar to that observed for additional XRD peaks for these films.
The observed differences in the evolution of the XRD diffraction patterns for MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 under illumination suggest that fundamentally different ionic movements accompany and drive halide segregation in these two materials. To explore the causes of such differences further, we examine the changes in XRD peak amplitudes and the peak integrals, as a function of time under illumination and during the subsequent recovery in the dark. Figure 2a presents time-resolved XRD peak amplitudes for the (200) diffraction peak of MAPb(Br0.5I0.5)3 films and the (220) peak of an FA0.83Cs0.17Pb(Br0.4I0.6)3 film, exposed to different periods of 190 mW cm–2 intensity illumination and in subsequent darkness. In general, the XRD peak amplitudes of both perovskites decrease under illumination and increase again as they recover in the periods of postillumination darkness. However, the MAPb(Br0.5I0.5)3 films experienced faster dynamics than the FA0.83Cs0.17Pb(Br0.4I0.6)3 film under both illumination and darkness, highlighting the higher susceptibility to ionic movement for MAPb(Br0.5I0.5)3. In addition, the initial decrease of XRD peak amplitude under illumination is significantly faster than the subsequent recovery of amplitude under darkness for all perovskite films (see also Figure S5), in agreement with previous studies demonstrating that the initial halide segregation proceeds much more rapidly than the subsequent remixing of halide ions under darkness.3 We further confirmed that the 18 h of X-ray exposure themselves did not significantly affect the perovskite material, by placing an MAPb(Br0.5I0.5)3 perovskite thin film in darkness but under X-ray examination for 18 h (black plot in Figure 2a), which resulted in negligible loss in XRD peak amplitude over the course of the experiment. Figures S6–S9 further provide PL spectra and XRD patterns taken before and after the periods of illumination and after corresponding periods of subsequent darkness, for each of the examined perovskite films.
Figure 2.

(a) Maximum amplitude of and (b) integral over the (200) peak (MAPb(Br0.5I0.5)3 thin-film samples) and the (220) peak (FA0.83Cs0.17Pb(Br0.4I0.6)3 thin-film sample) in the XRD patterns of metal halide perovskite films coated with PMMA. The thin-film samples were either illuminated for 15 min under 190 mW cm–2 intensity light of 470 nm wavelength (highlighted by the shaded orange region at early times), illuminated for 6 h under the same illumination parameters (highlighted by the shaded yellow region) or not illuminated, after which all thin-film samples were left in darkness until 18 h of total experimental time had passed. In (b), the integral was taken from 27.0° to 31.5° 2θ for the (200) diffraction peak of the MAPb(Br0.5I0.5)3 perovskite thin films and from 39.5° to 42.75° 2θ for the (220) peak of the FA0.83Cs0.17Pb(Br0.4I0.6)3 thin film. The data plots in (a) are normalized to their initial value, and the data plots in (b) are normalized to an average of the data within the first 2 min of the corresponding experiment.
Interestingly, we find that, unlike the XRD peak amplitudes, the integrals over the same XRD peaks (Figure 2b) remain remarkably constant during illumination and subsequent recovery, indicating a lack of film degradation into nonperovskite products during the experiments. The only exception to this observation is when the less-stable MAPb(Br0.5I0.5)3 film is illuminated for the prolonged period of 6 h, during which the integrated XRD signal eventually declines, indicating a small amount of material degradation, and we therefore exclude these data from the following analysis. Any potential emergence of nonperovskite material would be unlikely to produce XRD diffraction signal exclusively within the chosen integral limits used for the respective (200) and (220) peaks of the MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 films, and so a significant formation of these products would have reduced the overall integral over these XRD peaks, which is not observed. In particular, we can rule out the generation of two specific nonperovskite degradation products during the periods of illumination. First, little or no material converted from a perovskite crystalline structure into more amorphous material, given that a coherent reflection of X-rays may only occur from regions exhibiting a well-ordered structure.55 If a significant volume of the perovskite material had converted from an ordered structure into a nonperovskite, random arrangement then a significant decrease in total XRD diffraction signal would be evident in Figure 2b. Second, little or no perovskite material converted into PbI2—or its mixed-halide equivalents—over the course of the experiment. PbI2 is a very common degradation product for many perovskites, but it contributes insignificant XRD diffraction amplitude in the 2θ regions over which these second-order peaks were integrated.5,56−58 Besides, we confirmed that neither of the two perovskites featured a growth in the prominent peak between 12° and 13° 2θ (for Cu Kα1 incident radiation) typically associated with PbI256,58,59 in their recorded XRD pattern as a result of the prolonged illumination (see Figures S12 and S13).
Given the absence of significant breakdown into nonperovskite structures or amorphous material for both compositions during the conducted experiments, we conclude that the changes occurring in the XRD patterns (Figures 1c,d) principally result from two effects. First, the red-shifts in the PL emission spectra (Figures 1a,b) are indicative of halide segregation and halide ion movement, and we conclude that such ionic movement is likely primarily responsible for the concomitant changes in the associated XRD patterns (Figures 1c,d). Iodide-rich regions of perovskite will generate XRD peak positions at lower values of 2θ than bromide-rich regions, given that these peaks are linked with the lattice spacing of a given structure.3−5,55 The increased spread of XRD diffraction amplitude across a wide range of 2θ values as illumination progresses (Figures 1c,d) may then be primarily explained by a widening of the compositional range present across different perovskite regions. Second, as a less significant effect, a breakdown of crystalline volumes into smaller domains may contribute to the broadening of XRD peaks60 and result in an associated growth in their tails,61,62 as described in detail in Supporting Information Section 8. This scenario may apply to the MAPb(Br0.5I0.5)3 film, for which Figure 1c indicates a symmetric signal growth in both high- and low-angle tails of the (200) XRD peak, which could to some extent derive from a breakdown of crystalline domains. In contrast, Figure 1d demonstrates an absence of such symmetric tail growth for FA0.83Cs0.17Pb(Br0.4I0.6)3, suggesting domain breakdown to be insignificant here. Additionally, we note that a minor extent of XRD peak broadening may result from strain being introduced into the perovskite crystal by halide ion movement and the fracturing of crystalline domains.
In order for us to examine the compositional evolution within the MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 films, we proceed by converting both PL emission energies and XRD 2θ angles to an estimate of the bromide content of the emitting/diffracting perovskite region. Section 7 of the Supporting Information provides full details on how these conversions were performed, based on known dependencies of average PL emission and XRD peak positions on bromide content x for a range of corresponding perovskite stoichiometries across the iodide-bromide series. By remapping the PL and XRD measurements displayed in Figure 1 onto the same horizontal axis (axis of abscissa), now given in terms of bromide content, we are thus able to directly correlate the optoelectronic changes with the local compositional changes occurring upon halide segregation. We note that, because this approach does not capture any potential effects arising from crystal domain fracturing, these bromide content axes should be viewed as estimates, rather than precise measurements. Through this approach, we are able to elucidate how the associated underlying ionic rearrangement differs between MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 perovskites films.
We first focus our analysis on MAPb(Br0.5I0.5)3, for which Figure 3a,b presents in situ PL and XRD data taken simultaneously over 15 min of 190 mW cm–2 intensity illumination. A comparison of PL spectra and XRD patterns, now both plotted against an estimated bromide content axis, strikingly highlights how little of the XRD diffraction signal in Figure 3b correlates with the bromide content associated with the low-energy PL peak in Figure 3a. These observations confirm that iodide-rich regions of perovskite form only a tiny fraction of the total perovskite volume,29 while contributing to the vast majority of the PL emission because charge-carrier diffusion to these regions is highly effective. The XRD spectra displayed in Figure 3b also demonstrate that halide segregation results in complex compositional changes, most of which—because of the sensitivity of PL techniques to only the most iodide-rich, lowest bandgap perovskite domains—cannot be detected with PL measurements. Our work here therefore highlights the importance of examining the halide segregation process through techniques that are sensitive to the bulk of the perovskite material, such as XRD measurements.
Figure 3.

(a) PL spectra for an MAPb(Br0.5I0.5)3 thin film coated with PMMA, recorded over 15 min of 190 mW cm–2, 470 nm wavelength illumination. Spectra are plotted against the estimated bromide content of the emitting perovskite, which was determined from the dependence of the perovskite bandgap on halide composition reported in the literature, as described in Section 7 of the Supporting Information. (b) XRD patterns for the same MAPb(Br0.5I0.5)3 thin film as in (a), recorded in situ over the same 15 min period of 190 mW cm–2 illumination, also plotted against the estimated bromide content of the reflecting perovskite determined from literature reports (see Section 7 of the Supporting Information). (c) The blue line plots the integral over the PL spectra in (a), and the orange circles plot the integral over the XRD patterns in (b), both taken over the bromide-content interval indicated by the shaded red regions in (a, b), which centers around the bromide composition matching the final position of the PL peak.
We confirm that the process of compositional change primarily causes the observed changes to both the PL and XRD data by examining the temporal evolution of the respective signals around the low-bromide (iodide-rich) region of the PL spectra and XRD patterns. Figure 3c presents such time-resolved integrals over the PL and XRD signals presented in Figure 3a,b, between the estimated bromide contents of x = 0.07 and x = 0.16, as indicated by the shaded red columns. Conversion of both horizontal axes to values of estimated bromide content thus allows a direct comparison to be made between the evolution of PL and XRD data in the same corresponding region. Figure 3c clearly illustrates the time-resolved formation of iodide-rich regions within MAPb(Br0.5I0.5)3 via both PL and XRD measurements, with the integral of both signal types showing the same rise and plateau over time commonly associated with PL measurements of halide segregation in the literature.20,21Figure 3c therefore confirms that the rise in low-angle XRD tail and low-energy PL signals predominantly results from halide ion rearrangement, with any potential breakdown of crystalline domains being a secondary effect.
Our analysis of these observations allows us to elucidate the halide ion dynamics within the MAPb(Br0.5I0.5)3 perovskite under illumination. The growth of the low- and high-angle tails surrounding the main XRD (200) peak (evident in Figure 3b) is ascribed to the buildup of iodide- and bromide-enriched material as the halide segregation process proceeds. Interestingly, the evolution of the core (200) diffraction peak over time can be explained by the accumulated effect of continuous ionic movements that are initially restricted to specific regions of the perovskite, before these specific regions increase in number due to crystalline domain breakdown and grow in volume, thereby spreading throughout the bulk perovskite material. We propose that, during any given time interval, a subfraction of the bulk material converts into iodide-rich and bromide-rich material, with the nature of the remaining bulk perovskite phase remaining unchanged. As time progresses, such changes become visible in Figure 3b as a rise in the high- and low-angle tails of the (200) peak and a significant decrease of the (200) peak itself, while the overall 2θ position of the (200) peak remains constant. If instead, significant, concurrent ionic rearrangements occurred in large volumes of the perovskite simultaneously, this would be recorded as a shift or change in shape of the main (200) XRD peak, neither of which are strongly apparent in Figure 3b. We therefore conclude that halide segregation in MAPb(Br0.5I0.5)3 is initiated in specific volumes of the perovskite, with the remaining bulk remaining compositionally stable, as indicated by the unchanged 2θ central position of the XRD peak. Under continued illumination, the regions in which such segregation occurs gradually grow to consume much of the overall volume.
For the halide segregation pathways we propose for MAPb(Br0.5I0.5)3, the associated ionic movement will be relatively facile, as only the volumes of perovskite most prone to halide segregation will initially participate. Therefore, such segregation pathways help to explain the relative ease by which MA-cation mixed-halide perovskites are observed to initiate halide segregation. We propose that ionic movement might commence in regions such as grain boundaries that may offer enhanced ion mobilities,50−54 which could facilitate the segregation of halide ions in certain, small regions of the perovskite material with initially minor effects on the bulk material. The evolution of the XRD pattern under illumination of MAPb(Br0.5I0.5)3 further demonstrates that these specific regions gradually grow, eventually engulfing most of the material. Such a growth of halide-segregated material could result from an extension of these domains away from grain boundaries and into the bulk, as charge-carrier funneling and lattice distortion progresses with time under illumination. However, we suggest that an alternative process, deriving from the fracturing of crystalline domains under illumination, may also be in operation. As discussed in detail in Section 8 of the Supporting Information, the observed increase in the full width at half-maximum of the (200) peak is commensurate with a significant contraction in crystalline domain size under illumination, if ionic redistribution is neglected. Therefore, halide segregation in MAPb(Br0.5I0.5)3 may partly progress through the gradual fracturing of crystalline domains under illumination, which increases the volume of grain boundaries, generating more regions where halide segregation may easily occur. This proposed picture of the halide segregation mechanism in certain mixed-halide perovskites is congruent with literature reports of halide segregation often being associated with perovskite regions surrounding grain boundaries in the material.42,43,63
We proceed by examining the halide segregation mechanism for the much more compositionally stable FA0.83Cs0.17Pb(Br0.4I0.6)3 perovskite and find it to be markedly different from that described above for MAPb(Br0.5I0.5)3. As already discussed above, the evolution of XRD patterns of FA0.83Cs0.17Pb(Br0.4I0.6)3 under illumination, as shown in Figure 1d, highlights a different underlying type of ionic motion. Here, the actual (220) diffraction peak of FA0.83Cs0.17Pb(Br0.4I0.6)3 perovskite is found to shift and widen over 6 h of illumination, with a tail appearing at higher angles. Such pronounced changes to the core diffraction peak suggest that large amounts of ionic rearrangement occur throughout the perovskite volume, with a significant change to the average composition of the majority phase. To examine such changes to the bulk of the material, we therefore compare shifts in the main (220) diffraction peak with those in the PL emission peak associated with only the initially well-mixed, bulk phase of the FA0.83Cs0.17Pb(Br0.4I0.6)3, as shown in Figure 4a,b for the first 6 h of 190 mW cm–2 intensity illumination. To obtain the PL spectral component associated with only the bulk phase, the low-energy PL peak deriving from the iodide-rich minority phase was captured with a Gaussian fit and removed from the PL spectra. A direct comparison of the PL emission peaks and XRD patterns of the bulk perovskite phase (Figures 4a,b), plotted against a common axis of bromide content, shows clear parallel dynamics. Both PL and XRD peaks of the bulk perovskite phase shift to lower bromide content and broaden significantly with illumination time. In Figure 4c, the peak position of the PL spectra displayed in Figure 4a is compared with that of the (220) XRD peaks in Figure 4b, in terms of bromide content at which the maximum signal occurs, highlighting the similarity in time scales of the changes to the PL spectra and XRD patterns. Overall, these observations suggest that, for FA0.83Cs0.17Pb(Br0.4I0.6)3, prolonged illumination primarily affects the optoelectronic properties and the ionic distribution across the whole bulk material, rather than just within a minority phase exhibiting high ion mobility, as for MAPb(Br0.5I0.5)3.
Figure 4.

(a) PL spectra showing the mixed-phase perovskite emission from an FA0.83Cs0.17Pb(Br0.4I0.6)3 thin film coated with a PMMA layer, recorded over 6 h under light of intensity 190 mW cm–2, wavelength 470 nm. The spectra are plotted on a compositional axis determined from PL measurements, as described in Section 7 of the Supporting Information. The low-energy, iodide-rich PL peak has been captured by a Gaussian fit and removed from the PL spectra. (b) XRD patterns recorded in situ at the same time and on the same FA0.83Cs0.17Pb(Br0.4I0.6)3 film as the data shown in (a). The patterns are plotted on an estimated bromide compositional axis determined from data in literature reports, which may be subjected to errors pertaining to A-site cation movement, as described in Section 7 of the Supporting Information. See Figure S3 for the behavior of the maximum and mean values of the XRD signal, which shift and remain relatively constant over time, respectively. The differently colored data plots shown in (a, b) were taken at 1 h intervals, and similarly colored plots in (a, b) correspond to measurements made at approximately the same time. (c) The blue dots plot the peak position of the PL signal shown in (a), and the orange circles plot the peak position of the XRD signal shown in (b), both over 6 h of illumination.
To understand the reasons for the observed shift of the (220) XRD diffraction peak of FA0.83Cs0.17Pb(Br0.4I0.6)3 under prolonged illumination, we consider the possibilities of ionic rearrangements and of the introduction of lattice strain,64 which could shift crystal plane spacings. Interestingly, when the XRD signal is evaluated across its full spread, we find that the mean bromide content value (as opposed to just the peak position) is largely unchanged with illumination time (see Figure S3 for a comparison of XRD peak and mean values over illumination time). This observation essentially results from the shift of the XRD peak maximum to lower 2θ angles (lower bromide contents) being largely counterbalanced by a growth of XRD signal in the high-angle (high-bromide) tail of the (220) peak (see Figures 1d and S3). As a result, the mean 2θ position of the XRD (220) peak shifts only by a tiny amount, 0.07°, over the 6 h illumination period, which would correspond to only an ∼0.2% increase in the average lattice parameter of the perovskite but could also be explained through other effects, such as changes in XRD reflectivity for regions of different halide content (see the associated discussion in Section 4 of the Supporting Information). We therefore conclude that the evolution in the shape of the XRD (220) peak of FA0.83Cs0.17Pb(Br0.4I0.6)3 is primarily caused by ionic rearrangement in the perovskite crystal, rather than large-scale lattice strain.
To elucidate the specific ionic rearrangement occurring in FA0.83Cs0.17Pb(Br0.4I0.6)3 under illumination we first analyze the reasons for the growth of signal in the high-angle (high-bromide) tail of the (220) XRD peak (evident in Figure 1d and highlighted in Figure S3a). This increase of XRD diffraction amplitude in the range of ∼41.5°–42.75° 2θ corresponds to the formation of perovskite material with a range of pseudocubic lattice parameters between 5.98 and 6.15 Å. Given the variation of ionic radii in the participating FA, Cs, Br, and I ions, it is of course possible for perovskite regions to be generated with a range of different compositions yet similar lattice spacings and hence similar XRD patterns. In Section 4.1 of the Supporting Information we discuss possible candidates for the origin of this XRD signal, which range from FAPbBr3 to CsPb(Br0.4I0.6)3 perovskite compositions. While we are unable to determine the exact composition of the perovskite that generates the high-angle tail of the (220) XRD peak in Figure 1d, we find it to be compatible with a phase enriched in either FA or Cs as a result of an A-site cation rearrangement, comparable to that observed in similar perovskite materials.64
Therefore, we suggest that the increase of high-angle XRD diffraction amplitude for FA0.83Cs0.17Pb(Br0.4I0.6)3 under illumination may partially derive from the initial demixing of A-site cations in the perovskite material. The formation of a phase with either an FA- or Cs-enrichment would result in the depletion of FA or Cs, respectively, from the remaining bulk perovskite, which—alongside corresponding halide ion rearrangements—can increase the average lattice spacing of the bulk perovskite. As shown in Figure 4b, the shift in lattice spacing of the bulk perovskite is reflected by the associated XRD signal shifting to lower values of estimated bromide content. As discussed in Section 4.1 of the Supporting Information, the newly formed FA- or Cs-enriched perovskite phase apparent in the high-angle tail of the (220) XRD peak is expected to have a higher bandgap than that of the original FA0.83Cs0.17Pb(Br0.4I0.6)3 material. Therefore, over the course of the illumination, the bandgap of the remaining FA- or Cs-depleted bulk perovskite would be expected to red-shift. Accordingly, Figure 4a highlights this red-shift of the bulk phase of the FA0.83Cs0.17Pb(Br0.4I0.6)3 perovskite over the course of the illumination period. Interestingly, it has previously been shown10 that FA(1–y)CsyPb(Br0.4I0.6)3 films only exhibit a stable phase space substantially free from halide segregation for cesium contents y approximately between 0.1 and 0.3. Similarly, FAPb(BrxI(1–x))3 (y = 0)3,8 and CsPb(BrxI(1–x))3 (y = 1)7 perovskites have been found to be highly susceptible to halide segregation. Rehman et al.10 have attributed such effects to a lowering of material crystallinity (as derived from charge-carrier lifetimes and XRD peak widths) outside the range of 0.1 < y < 0.3, which in turn reduces the stability of the perovskite against halide segregation, as expected from the strong correlation between crystallinity and photostability reported in the literature.11,33 Therefore, we propose that, for FA0.83Cs0.17Pb(Br0.4I0.6)3, illumination could first result in the formation of a minority FA- or Cs-enriched phase and the FA- or Cs-depletion of the majority bulk phase, as a result of A-site cation separation. The composition of the bulk perovskite would be shifted from the maximally stable cesium composition of y ≈ 0.2, which in turn would reduce the overall stability of the material against halide segregation, hastening the formation of iodide-rich domains in the bulk that lead to the ultimately observed low-energy PL emission. As such, halide segregation in the FACs A-cation mixed-halide perovskite would therefore proceed via a two-step process initiated by A-cation segregation that ultimately enables halide segregation. This multistep process appears to be applicable to the whole bulk of the material and to present a larger hurdle against halide segregation than that operational in an equivalent MA-based perovskite.
In conclusion, by comparing simultaneous, in situ PL and XRD measurements, we have elucidated differences in the halide segregation mechanism between MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 perovskites. Overall, the different structural and optoelectronic changes we observe following illumination of the two types of mixed-halide perovskite explain some of the strong variations in phase stability for different mixed-halide perovskites reported in the literature.11 For MAPb(Br0.5I0.5)3, we conclude that the presence of fast ionic pathways initially results in facile halide segregation in specific subvolumes of material under illumination, with the remaining volume of bulk phase maintaining its original mixed-halide composition. Such specific, halide-segregated volumes ultimately grow at the expense of the well-mixed bulk phase as halide segregation proceeds, for example, by expanding away from grain boundaries further into the bulk or by the fracturing of crystalline domains, which enlarges the interface areas with crystalline imperfections. In contrast, FA0.83Cs0.17Pb(Br0.4I0.6)3 exhibits large, concurrent changes occurring across the whole bulk material, but only after a much longer illumination period of comparable intensity. In particular, the change in shape and peak position of the main (220) XRD diffraction peak of FA0.83Cs0.17Pb(Br0.4I0.6)3 indicates a concurrent ionic rearrangement throughout the whole of the perovskite volume; that is, unlike in the MAPb(Br0.5I0.5)3 perovskite, there appear to be few fast ionic pathways that facilitate halide segregation in localized regions. The lack of such fast ionic pathways in FA0.83Cs0.17Pb(Br0.4I0.6)3 means that any phase segregation must instead occur across the whole of the more resistant bulk perovskite material, raising hurdles to these effects. Such an absence of ionic pathways is likely caused by the FA0.83Cs0.17Pb(Br0.4I0.6)3 material possessing superior crystallinity properties over the MAPb(Br0.5I0.5)3 perovskite. For materials such as FA0.83Cs0.17Pb(Br0.4I0.6)3, their mixed A-cation nature, while allowing for a high crystallinity that suppresses halide segregation in the first instance, may ultimately still be their Achilles’ heel, given that A-cation segregation may subsequently trigger halide segregation in turn. For such materials, further stabilization against halide segregation may thus result if processing protocols can be developed that further stabilize A-cation mixing. In addition, we note that other factors may also contribute to the different time scales required to induce halide segregation in MAPb(Br0.5I0.5)3 and FA0.83Cs0.17Pb(Br0.4I0.6)3 thin films, such as trap state density.20 However, we conclude that the ionic pathways available in a perovskite material are a key indicator of stability against halide segregation. Our results therefore also highlight the importance of bulk-sensitive measurements in determining the susceptibility of a mixed-halide perovskite to halide segregation and indicate that ionic mobilities, grain boundaries, and crystallinity are key areas of research if completely phase-stable materials are to be manufactured for photovoltaic applications.
Acknowledgments
The authors gratefully acknowledge the financial support from the Engineering and Physical Sciences Research Council (U.K.). A.J.K. thanks the University College Oxford for graduate scholarship support from the Oxford-Radcliffe endowment. R.D.J.O. expresses his gratitude to the Penrose Scholarship for funding his studentship. L.M.H. thanks the Alexander von Humboldt Foundation for support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenergylett.0c02475.
Sample fabrication methods, basic XRD measurements, additional sample discussion, additional XRD analysis, concurrent XRD/PL measurement overview, method of estimating halide contents from PL and XRD data, crystallite size estimates (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Snaith H. J. Present Status and Future Prospects of Perovskite Photovoltaics. Nat. Mater. 2018, 17, 372. 10.1038/s41563-018-0071-z. [DOI] [PubMed] [Google Scholar]
- Jena A. K.; Kulkarni A.; Miyasaka T. Halide Perovskite Photovoltaics: Background, Status, and Future Prospects. Chem. Rev. 2019, 119, 3036–3103. 10.1021/acs.chemrev.8b00539. [DOI] [PubMed] [Google Scholar]
- Hoke E. T.; Slotcavage D. J.; Dohner E. R.; Bowring A. R.; Karunadasa H. I.; McGehee M. D. Reversible Photo-Induced Trap Formation in Mixed-Halide Hybrid Perovskites for Photovoltaics. Chem. Sci. 2015, 6, 613–617. 10.1039/C4SC03141E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh J. H.; Im S. H.; Heo J. H.; Mandal T. N.; Seok S. I. Chemical Management for Colorful, Efficient, and Stable Inorganic–Organic Hybrid Nanostructured Solar Cells. Nano Lett. 2013, 13, 1764–1769. 10.1021/nl400349b. [DOI] [PubMed] [Google Scholar]
- Jesper Jacobsson T.; Correa-Baena J.-P.; Pazoki M.; Saliba M.; Schenk K.; Gratzel M.; Hagfeldt A. Exploration of the Compositional Space for Mixed Lead Halogen Perovskites for High Efficiency Solar Cells. Energy Environ. Sci. 2016, 9, 1706–1724. 10.1039/C6EE00030D. [DOI] [Google Scholar]
- McMeekin D. P.; Sadoughi G.; Rehman W.; Eperon G. E.; Saliba M.; Hörantner M. T.; Haghighirad A.; Sakai N.; Korte L.; Rech B.; et al. A Mixed-Cation Lead Mixed-Halide Perovskite Absorber for Tandem Solar Cells. Science 2016, 351, 151–155. 10.1126/science.aad5845. [DOI] [PubMed] [Google Scholar]
- Beal R. E.; Slotcavage D. J.; Leijtens T.; Bowring A. R.; Belisle R. A.; Nguyen W. H.; Burkhard G. F.; Hoke E. T.; McGehee M. D. Cesium Lead Halide Perovskites with Improved Stability for Tandem Solar Cells. J. Phys. Chem. Lett. 2016, 7, 746–751. 10.1021/acs.jpclett.6b00002. [DOI] [PubMed] [Google Scholar]
- Rehman W.; Milot R. L.; Eperon G. E.; Wehrenfennig C.; Boland J. L.; Snaith H. J.; Johnston M. B.; Herz L. M. Charge-Carrier Dynamics and Mobilities in Formamidinium Lead Mixed-Halide Perovskites. Adv. Mater. 2015, 27, 7938–7944. 10.1002/adma.201502969. [DOI] [PubMed] [Google Scholar]
- Parrott E. S.; Green T.; Milot R. L.; Johnston M. B.; Snaith H. J.; Herz L. M. Interplay of Structural and Optoelectronic Properties in Formamidinium Mixed Tin–Lead Triiodide Perovskites. Adv. Funct. Mater. 2018, 28, 1802803. 10.1002/adfm.201802803. [DOI] [Google Scholar]
- Rehman W.; McMeekin D. P.; Patel J. B.; Milot R. L.; Johnston M. B.; Snaith H. J.; Herz L. M. Photovoltaic Mixed-Cation Lead Mixed-Halide Perovskites: Links between Crystallinity, Photo-Stability and Electronic Properties. Energy Environ. Sci. 2017, 10, 361–369. 10.1039/C6EE03014A. [DOI] [Google Scholar]
- Knight A.; Herz L. M. Preventing Phase Segregation in Mixed-Halide Perovskites: A Perspective. Energy Environ. Sci. 2020, 13, 2024–2046. 10.1039/D0EE00788A. [DOI] [Google Scholar]
- Shockley W.; Queisser H. J. Detailed Balance Limit of Efficiency of p-n Junction Solar Cells. J. Appl. Phys. 1961, 32, 510–519. 10.1063/1.1736034. [DOI] [Google Scholar]
- Vos A. D. Detailed Balance Limit of the Efficiency of Tandem Solar Cells. J. Phys. D: Appl. Phys. 1980, 13, 839. 10.1088/0022-3727/13/5/018. [DOI] [Google Scholar]
- Eperon G. E.; Leijtens T.; Bush K. A.; Prasanna R.; Green T.; Wang J. T.-W.; McMeekin D. P.; Volonakis G.; Milot R. L.; May R.; et al. Perovskite-Perovskite Tandem Photovoltaics with Optimized Band Gaps. Science 2016, 354, 861–865. 10.1126/science.aaf9717. [DOI] [PubMed] [Google Scholar]
- Rajagopal A.; Yang Z.; Jo S. B.; Braly I. L.; Liang P.-W.; Hillhouse H. W.; Jen A. K.-Y. Highly Efficient Perovskite–Perovskite Tandem Solar Cells Reaching 80% of the Theoretical Limit in Photovoltage. Adv. Mater. 2017, 29, 1702140. 10.1002/adma.201702140. [DOI] [PubMed] [Google Scholar]
- Forgács D.; Gil-Escrig L.; Pérez-Del-Rey D.; Momblona C.; Werner J.; Niesen B.; Ballif C.; Sessolo M.; Bolink H. J. Efficient Monolithic Perovskite/Perovskite Tandem Solar Cells. Adv. Energy Mater. 2017, 7, 1602121. 10.1002/aenm.201602121. [DOI] [Google Scholar]
- Lin R.; Xiao K.; Qin Z.; Han Q.; Zhang C.; Wei M.; Saidaminov M. I.; Gao Y.; Xu J.; Xiao M.; et al. Monolithic All-Perovskite Tandem Solar Cells with 24.8% Efficiency Exploiting Comproportionation to Suppress Sn (II) Oxidation in Precursor Ink. Nat. Energy 2019, 4, 864–873. 10.1038/s41560-019-0466-3. [DOI] [Google Scholar]
- Bush K. A.; Palmstrom A. F.; Yu Z. J.; Boccard M.; Cheacharoen R.; Mailoa J. P.; McMeekin D. P.; Hoye R. L.; Bailie C. D.; Leijtens T.; et al. 23.6%-Efficient Monolithic Perovskite/Silicon Tandem Solar Cells with Improved Stability. Nat. Energy 2017, 2, 17009. 10.1038/nenergy.2017.9. [DOI] [Google Scholar]
- Duong T.; Wu Y.; Shen H.; Peng J.; Fu X.; Jacobs D.; Wang E.-C.; Kho T. C.; Fong K. C.; Stocks M.; et al. Rubidium Multication Perovskite with Optimized Bandgap for Perovskite-Silicon Tandem with over 26% Efficiency. Adv. Energy Mater. 2017, 7, 1700228. 10.1002/aenm.201700228. [DOI] [Google Scholar]
- Knight A. J.; Wright A. D.; Patel J. B.; McMeekin D. P.; Snaith H. J.; Johnston M. B.; Herz L. M. Electronic Traps and Phase Segregation in Lead Mixed-Halide Perovskite. ACS Energy Lett. 2019, 4, 75–84. 10.1021/acsenergylett.8b02002. [DOI] [Google Scholar]
- Draguta S.; Sharia O.; Yoon S. J.; Brennan M. C.; Morozov Y. V.; Manser J. S.; Kamat P. V.; Schneider W. F.; Kuno M. Rationalizing the Light-Induced Phase Separation of Mixed Halide Organic–Inorganic Perovskites. Nat. Commun. 2017, 8, 200. 10.1038/s41467-017-00284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker A. J.; Sadhanala A.; Deschler F.; Gandini M.; Senanayak S. P.; Pearce P. M.; Mosconi E.; Pearson A. J.; Wu Y.; Srimath Kandada A. R.; et al. Defect-Assisted Photoinduced Halide Segregation in Mixed-Halide Perovskite Thin Films. ACS Energy Lett. 2017, 2, 1416–1424. 10.1021/acsenergylett.7b00282. [DOI] [Google Scholar]
- Knight A. J.; Patel J. B.; Snaith H. J.; Johnston M. B.; Herz L. M. Trap States, Electric Fields, and Phase Segregation in Mixed-Halide Perovskite Photovoltaic Devices. Adv. Energy Mater. 2020, 10, 1903488. 10.1002/aenm.201903488. [DOI] [Google Scholar]
- Braly I. L.; Stoddard R. J.; Rajagopal A.; Uhl A. R.; Katahara J. K.; Jen A. K.-Y.; Hillhouse H. W. Current-Induced Phase Segregation in Mixed Halide Hybrid Perovskites and its Impact on Two-Terminal Tandem Solar Cell Design. ACS Energy Lett. 2017, 2, 1841–1847. 10.1021/acsenergylett.7b00525. [DOI] [Google Scholar]
- Duong T.; Mulmudi H. K.; Wu Y.; Fu X.; Shen H.; Peng J.; Wu N.; Nguyen H. T.; Macdonald D.; Lockrey M.; et al. Light and Electrically Induced Phase Segregation and its Impact on the Stability of Quadruple Cation High Bandgap Perovskite Solar Cells. ACS Appl. Mater. Interfaces 2017, 9, 26859–26866. 10.1021/acsami.7b06816. [DOI] [PubMed] [Google Scholar]
- Li G.; Rivarola F. W. R.; Davis N. J.; Bai S.; Jellicoe T. C.; de la Peña F.; Hou S.; Ducati C.; Gao F.; Friend R. H.; et al. Highly Efficient Perovskite Nanocrystal Light-Emitting Diodes Enabled by a Universal Crosslinking Method. Adv. Mater. 2016, 28, 3528–3534. 10.1002/adma.201600064. [DOI] [PubMed] [Google Scholar]
- Vashishtha P.; Halpert J. E. Field-Driven Ion Migration and Color Instability in Red-Emitting Mixed Halide Perovskite Nanocrystal Light-Emitting Diodes. Chem. Mater. 2017, 29, 5965–5973. 10.1021/acs.chemmater.7b01609. [DOI] [Google Scholar]
- Shynkarenko Y.; Bodnarchuk M. I.; Bernasconi C.; Berezovska Y.; Verteletskyi V.; Ochsenbein S.; Kovalenko M. V. Direct Synthesis of Quaternary Alkylammonium Capped Perovskite Nanocrystals for Efficient Blue and Green Light-Emitting Diodes. ACS Energy Lett. 2019, 4, 2703–2711. 10.1021/acsenergylett.9b01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahesh S.; Ball J. M.; Oliver R. D.; McMeekin D. P.; Nayak P.; Johnston M. B.; Snaith H. Revealing the Origin of Voltage Loss in Mixed-Halide Perovskite Solar Cells. Energy Environ. Sci. 2020, 13, 258–267. 10.1039/C9EE02162K. [DOI] [Google Scholar]
- Yoon S. J.; Draguta S.; Manser J. S.; Sharia O.; Schneider W. F.; Kuno M.; Kamat P. V. Tracking Iodide and Bromide Ion Segregation in Mixed Halide Lead Perovskites During Photoirradiation. ACS Energy Lett. 2016, 1, 290–296. 10.1021/acsenergylett.6b00158. [DOI] [Google Scholar]
- Brivio F.; Caetano C.; Walsh A. Thermodynamic Origin of Photoinstability in the CH3NH3Pb(I(1–x)Brx)3 Hybrid Halide Perovskite Alloy. J. Phys. Chem. Lett. 2016, 7, 1083–1087. 10.1021/acs.jpclett.6b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan M. C.; Ruth A.; Kamat P. V.; Kuno M. Photoinduced Anion Segregation in Mixed Halide Perovskites. Trends Chem. 2020, 2, 282–301. 10.1016/j.trechm.2020.01.010. [DOI] [Google Scholar]
- Hu M.; Bi C.; Yuan Y.; Bai Y.; Huang J. Stabilized Wide Bandgap MAPbBrxI3–x Perovskite by Enhanced Grain Size and Improved Crystallinity. Adv. Sci. 2016, 3, 1500301. 10.1002/advs.201500301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Jia Y.-H.; Fang H.-H.; Loi M. A.; Xie F.-Y.; Gong L.; Qin M.-C.; Lu X.-H.; Wong C.-P.; Zhao N. Composition-Tuned Wide Bandgap Perovskites: From Grain Engineering to Stability and Performance Improvement. Adv. Funct. Mater. 2018, 28, 1803130. 10.1002/adfm.201803130. [DOI] [Google Scholar]
- Xie Y.-M.; Zeng Z.; Xu X.; Ma C.; Ma Y.; Li M.; Lee C.-S.; Tsang S.-W. FA-Assistant Iodide Coordination in Organic–Inorganic Wide-Bandgap Perovskite with Mixed Halides. Small 2020, 16, 1907226. 10.1002/smll.201907226. [DOI] [PubMed] [Google Scholar]
- Belisle R. A.; Bush K. A.; Bertoluzzi L.; Gold-Parker A.; Toney M. F.; McGehee M. D. Impact of Surfaces on Photoinduced Halide Segregation in Mixed-Halide Perovskites. ACS Energy Lett. 2018, 3, 2694–2700. 10.1021/acsenergylett.8b01562. [DOI] [Google Scholar]
- Abdi-Jalebi M.; Andaji-Garmaroudi Z.; Cacovich S.; Stavrakas C.; Philippe B.; Richter J. M.; Alsari M.; Booker E. P.; Hutter E. M.; Pearson A. J.; et al. Maximizing and Stabilizing Luminescence From Halide Perovskites with Potassium Passivation. Nature 2018, 555, 497. 10.1038/nature25989. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wang Y.; Nie Z.; Ren Y.; Zeng H. Laser Induced Ion Migration in All-Inorganic Mixed Halide Perovskite Micro-Platelets. Nanoscale Advances 2019, 1, 4459–4465. 10.1039/C9NA00565J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishna R. G.; Kobosko S. M.; Kamat P. V. Mixed Halide Perovskite Solar Cells. Consequence of Iodide Treatment on Phase Segregation Recovery. ACS Energy Lett. 2018, 3, 2267–2272. 10.1021/acsenergylett.8b01450. [DOI] [Google Scholar]
- Zhou Y.; Wang F.; Cao Y.; Wang J.-P.; Fang H.-H.; Loi M. A.; Zhao N.; Wong C.-P. Benzylamine-Treated Wide-Bandgap Perovskite with High Thermal-Photostability and Photovoltaic Performance. Adv. Energy Mater. 2017, 7, 1701048. 10.1002/aenm.201701048. [DOI] [Google Scholar]
- Yang J.-N.; Song Y.; Yao J.-S.; Wang K.-H.; Wang J.-J.; Zhu B.-S.; Yao M.-M.; Rahman S. U.; Lan Y.-F.; Fan F.-J.; et al. Potassium-Bromide Surface Passivation on CsPbI3–xBrx Nanocrystals for Efficient and Stable Pure Red Perovskite Light Emitting Diodes. J. Am. Chem. Soc. 2020, 142, 2956–2967. 10.1021/jacs.9b11719. [DOI] [PubMed] [Google Scholar]
- Tang X.; van den Berg M.; Gu E.; Horneber A.; Matt G. J.; Osvet A.; Meixner A. J.; Zhang D.; Brabec C. J. Local Observation of Phase Segregation in Mixed-Halide Perovskite. Nano Lett. 2018, 18, 2172–2178. 10.1021/acs.nanolett.8b00505. [DOI] [PubMed] [Google Scholar]
- Li W.; Rothmann M. U.; Liu A.; Wang Z.; Zhang Y.; Pascoe A. R.; Lu J.; Jiang L.; Chen Y.; Huang F.; et al. Phase Segregation Enhanced Ion Movement in Efficient Inorganic CsPbIBr2 Solar Cells. Adv. Energy Mater. 2017, 7, 1700946. 10.1002/aenm.201700946. [DOI] [Google Scholar]
- Yang Z.; Rajagopal A.; Jo S. B.; Chueh C.-C.; Williams S.; Huang C.-C.; Katahara J. K.; Hillhouse H. W.; Jen A. K.-Y. Stabilized Wide Bandgap Perovskite Solar Cells by Tin Substitution. Nano Lett. 2016, 16, 7739–7747. 10.1021/acs.nanolett.6b03857. [DOI] [PubMed] [Google Scholar]
- Li N.; Zhu Z.; Li J.; Jen A. K.-Y.; Wang L. Inorganic CsPb1–xSnxIBr2 for Efficient Wide-Bandgap Perovskite Solar Cells. Adv. Energy Mater. 2018, 8, 1800525. 10.1002/aenm.201800525. [DOI] [Google Scholar]
- Sutter-Fella C. M.; Ngo Q. P.; Cefarin N.; Gardner K. L.; Tamura N.; Stan C. V.; Drisdell W. S.; Javey A.; Toma F. M.; Sharp I. D. Cation-Dependent Light-Induced Halide Demixing in Hybrid Organic–Inorganic Perovskites. Nano Lett. 2018, 18, 3473–3480. 10.1021/acs.nanolett.8b00541. [DOI] [PubMed] [Google Scholar]
- Beal R. E.; Hagström N. Z.; Barrier J.; Gold-Parker A.; Prasanna R.; Bush K. A.; Passarello D.; Schelhas L. T.; Brüning K.; Tassone C. J.; et al. Structural Origins of Light-Induced Phase Segregation in Organic-Inorganic Halide Perovskite Photovoltaic Materials. Matter 2020, 2, 207–219. 10.1016/j.matt.2019.11.001. [DOI] [Google Scholar]
- Brennan M. C.; Draguta S.; Kamat P. V.; Kuno M. Light-Induced Anion Phase Segregation in Mixed Halide Perovskites. ACS Energy Lett. 2018, 3, 204–213. 10.1021/acsenergylett.7b01151. [DOI] [Google Scholar]
- McMeekin D. P.; Mahesh S.; Noel N. K.; Klug M. T.; Lim J.; Warby J. H.; Ball J. M.; Herz L. M.; Johnston M. B.; Snaith H. J. Solution-Processed All-Perovskite Multi-Junction Solar Cells. Joule 2019, 3, 387–401. 10.1016/j.joule.2019.01.007. [DOI] [Google Scholar]
- Yang B.; Brown C. C.; Huang J.; Collins L.; Sang X.; Unocic R. R.; Jesse S.; Kalinin S. V.; Belianinov A.; Jakowski J.; et al. Enhancing Ion Migration in Grain Boundaries of Hybrid Organic–Inorganic Perovskites by Chlorine. Adv. Funct. Mater. 2017, 27, 1700749. 10.1002/adfm.201700749. [DOI] [Google Scholar]
- Shao Y.; Fang Y.; Li T.; Wang Q.; Dong Q.; Deng Y.; Yuan Y.; Wei H.; Wang M.; Gruverman A.; et al. Grain Boundary Dominated Ion Migration in Polycrystalline Organic–Inorganic Halide Perovskite Films. Energy Environ. Sci. 2016, 9, 1752–1759. 10.1039/C6EE00413J. [DOI] [Google Scholar]
- Meggiolaro D.; Mosconi E.; De Angelis F. Formation of Surface Defects Dominates Ion Migration in Lead-Halide Perovskites. ACS Energy Lett. 2019, 4, 779–785. 10.1021/acsenergylett.9b00247. [DOI] [Google Scholar]
- Khassaf H.; Yadavalli S. K.; Zhou Y.; Padture N. P.; Kingon A. I. Effect of Grain Boundaries on Charge Transport in Methylammonium Lead Iodide Perovskite Thin Films. J. Phys. Chem. C 2019, 123, 5321–5325. 10.1021/acs.jpcc.9b00538. [DOI] [Google Scholar]
- Yun J. S.; Seidel J.; Kim J.; Soufiani A. M.; Huang S.; Lau J.; Jeon N. J.; Seok S. I.; Green M. A.; Ho-Baillie A. Critical Role of Grain Boundaries for Ion Migration in Formamidinium and Methylammonium Lead Halide Perovskite Solar Cells. Adv. Energy Mater. 2016, 6, 1600330. 10.1002/aenm.201600330. [DOI] [Google Scholar]
- Simon S. H.The Oxford Solid State Basics ;OUP Oxford, 2013. [Google Scholar]
- Habisreutinger S. N.; Leijtens T.; Eperon G. E.; Stranks S. D.; Nicholas R. J.; Snaith H. J. Carbon Nanotube/Polymer Composites as a Highly Stable Hole Collection Layer in Perovskite Solar Cells. Nano Lett. 2014, 14, 5561–5568. 10.1021/nl501982b. [DOI] [PubMed] [Google Scholar]
- Dualeh A.; Tétreault N.; Moehl T.; Gao P.; Nazeeruddin M. K.; Grätzel M. Effect of Annealing Temperature on Film Morphology of Organic–Inorganic Hybrid Pervoskite Solid-State Solar Cells. Adv. Funct. Mater. 2014, 24, 3250–3258. 10.1002/adfm.201304022. [DOI] [Google Scholar]
- Gujar T. P.; Unger T.; Schönleber A.; Fried M.; Panzer F.; van Smaalen S.; Köhler A.; Thelakkat M. The Role of PbI2 in CH3NH3PbI3 Perovskite Stability, Solar Cell Parameters and Device Degradation. Phys. Chem. Chem. Phys. 2018, 20, 605–614. 10.1039/C7CP04749E. [DOI] [PubMed] [Google Scholar]
- Buin A.; Comin R.; Xu J.; Ip A. H.; Sargent E. H. Halide-Dependent Electronic Structure of Organolead Perovskite Materials. Chem. Mater. 2015, 27, 4405–4412. 10.1021/acs.chemmater.5b01909. [DOI] [Google Scholar]
- Scherrer P. Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen. Mathematisch-Physikalische Klasse 1918, 2, 98–100. [Google Scholar]
- De Keijser T. H.; Langford J. I.; Mittemeijer E. J.; Vogels A. B. P. Use of the Voigt Function in a Single-Line Method for the Analysis of X-ray Diffraction Line Broadening. J. Appl. Crystallogr. 1982, 15, 308–314. 10.1107/S0021889882012035. [DOI] [Google Scholar]
- Nandi R. K.; Sen Gupta S. P. The Analysis of X-ray Diffraction Profiles From Imperfect Solids by an Application of Convolution Relations. J. Appl. Crystallogr. 1978, 11, 6–9. 10.1107/S0021889878012595. [DOI] [Google Scholar]
- Bischak C. G.; Hetherington C. L.; Wu H.; Aloni S.; Ogletree D. F.; Limmer D. T.; Ginsberg N. S. Origin of Reversible Photoinduced Phase Separation in Hybrid Perovskites. Nano Lett. 2017, 17, 1028–1033. 10.1021/acs.nanolett.6b04453. [DOI] [PubMed] [Google Scholar]
- Lin Y.-H.; Sakai N.; Da P.; Wu J.; Sansom H. C.; Ramadan A. J.; Mahesh S.; Liu J.; Oliver R. D. J.; Lim J.; et al. A Piperidinium Salt Stabilizes Efficient Metal-Halide Perovskite Solar Cells. Science 2020, 369, 96–102. 10.1126/science.aba1628. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.