

Abstract
Christianson syndrome (CS), an X-linked neurological disorder characterized by postnatal attenuation of brain growth (postnatal microcephaly), is caused by mutations in SLC9A6 (also termed NHE6), the gene encoding endosomal Na+/H+ exchanger 6 (NHE6). To hasten treatment development, we established CS patient-derived induced pluripotent stem cell (iPSC) lines representing a mutational spectrum, as well as biologically related and isogenic control lines. We demonstrated that pathogenic mutations lead to loss of protein function by a variety of mechanisms: the majority of mutations caused loss of mRNA due to nonsense-mediated mRNA decay; however, a recurrent, missense mutation (the G383D mutation) had both loss-of-function and dominant-negative activities. Regardless of mutation, all patient-derived neurons demonstrated reduced neurite growth and arborization, likely underlying diminished postnatal brain growth in patients. Phenotype rescue strategies showed mutation-specific responses: a gene transfer strategy was effective in nonsense mutations, but not in the G383D mutation, wherein residual protein appeared to interfere with rescue. In contrast, application of exogenous trophic factors (BDNF or IGF-1) rescued arborization phenotypes across all mutations. These results may guide treatment development in CS, including gene therapy strategies wherein our data suggest that response to treatment may be dictated by the class of mutation.
One Sentence Summary:
Neurons from patient-derived iPSCs demonstrate mutation-specific responses to potential therapeutic strategies for Christianson syndrome.
Introduction
Christianson syndrome (CS) is an X-linked neurological disorder characterized by impaired cognitive development and epilepsy, with a distinctive social communication phenotype (1, 2). CS is caused by diverse mutations in SLC9A6 (also termed NHE6), the gene encoding the endosomal Na+/H+ exchanger 6 (NHE6) (2, 3). Given the social phenotype, which includes a happy demeanor and autistic features, and the presentation of seizures and postnatal microcephaly, CS was originally termed X-linked Angelman syndrome (AS) (3). Postnatal microcephaly (as opposed to primary microcephaly) is diagnosed when children are born with typical head circumference yet undergo subsequent attenuation of the expected brain growth in the first years of life (4). The human brain increases in size by approximately two- to three-fold in the first years of life, coincident with the emergence of experience-dependent acquisition of brain function and behavior. Thereby, study of human genetic disorders with postnatal microcephaly, such as CS or AS, may allow for the identification of mechanisms involved in early childhood brain development, including those governing cognitive, motor, and social development (4). As neurogenesis is largely complete in human brain prior to birth, postnatal microcephaly genes often regulate later neurodevelopmental processes involved in neuronal connectivity such as neuronal axonogenesis, axonal and dendritic arborization, and synaptic development.
Endosomes play an important role in the development of neuronal connectivity via a range of mechanisms, including by their involvement in the trafficking, recycling, and degradation of cargo, as well as by serving in endosomal signaling (5, 6). In particular, neurotrophin signaling, such as through the brain-derived neurotrophic factor (BDNF)/tropomyosin receptor kinase B (TrkB) pathway, represents the classically studied endosomal signaling mechanism essential for neurite outgrowth and arborization (5). Tight regulation of intra-endosomal proton concentration serves an essential role in endosome maturation and function (7). The vacuolar H+-ATPase (V-ATPase) is a pump that mediates acidification of endosomes and lysosomes. The endosomal NHEs (particularly NHE6 and NHE9) allow proton efflux and counter the V-ATPase by regulating relative alkalization of the lumen (8). Loss of NHE6 in primary mouse neurons revealed a unique endosomal phenotype, namely, over-acidification of the endosome lumen (9). Reduced endosomal pH in developing NHE6-null neurons was associated with attenuation of BDNF signaling via TrkB and diminished neuronal arborization in vitro and in vivo. Exogenous addition of BDNF to NHE6-null murine cultures rescued the arborization defect, consistent with the previously shown pH-dependent decrease in ligand-receptor binding (10).
Given the disease burden experienced by people with CS, we sought to develop an induced pluripotent stem cell (iPSC) system that could be utilized to study the cellular mechanisms of CS in human neurons, as well as serve for therapeutic development. In CS, a majority of mutations are nonsense mutations or strong splice mutations; however, additional mutations representing missense or in-frame amino acid deletions have also been reported (2, 3). Patient-derived iPSCs offer the opportunity to dissect mechanisms mediated by specific human mutations from the endogenous genomic locus. As has been well demonstrated recently for therapeutic development for cystic fibrosis, specific patient mutations show evidence of mutation-specific responses to treatments (11). Given our prior data on BDNF, one rescue strategy examined here is the exogenous, or cell-non-autonomous, addition of trophic factors. Such factors might include not only BDNF but also insulin-like growth factor-1 (IGF-1). BDNF and IGF-1 have demonstrated activity on neurite growth and arborization (12), and further still, IGF-1 is in clinical trials for other neurodevelopmental disorders with postnatal microcephaly and/or autistic features (for example, NCT01970345, NCT01525901; clinicaltrials.gov). We also examined a rescue strategy involving re-expression of NHE6 via transfection of human NHE6 cDNA into mutant neurons. This therapeutic strategy may be pursued through gene therapy or perhaps through upregulation of the related endosomal NHE9 in patients with CS.
In this study, we present a substantial iPSC resource that we have developed to study CS disease mechanisms and therapeutic responses. This resource includes a series of pathogenic mutations, as well as genetically related controls and isogenic controls. We utilized this resource to determine specific action of mutations in patient-derived neurons, as well as distinct mutation-specific responses to rescue strategies. Whereas exogenous addition of BDNF or IGF-1 to patient neurons rescued neuronal phenotypes regardless of patient mutation, gene transfer strategies showed successful rescue in a fashion that is dependent on the nature of the mutation.
Results
A majority of CS mutations lead to loss of protein through nonsense-mediated decay (NMD) mechanisms
In order to establish a patient-derived cellular model for translational research in CS, we reprogrammed peripheral blood mononuclear cells (PBMCs) from five families into iPSCs. The cells from the affected child each contained a distinct mutation in NHE6 (13) (Table 1). A majority of NHE6 mutations in CS appear to be loss-of-function, such as putatively protein-truncating due to early frameshift, nonsense, or splicing mutations, whereas several missense or in-frame deletions have also been reported, likely with residual protein (2, 3). Our iPSC collection included a mutational series of four distinct frameshift/nonsense mutations and one recurrent missense mutation in NHE6, the latter of which we hypothesized could also affect mRNA splicing. These mutations are as follows: Family 1, a single base pair duplication in exon 11 leading to a frameshift and premature stop codon between predicted transmembrane domains (TM) 10 and 11 (c.1414dupA, p.R472fsX4); Family 2, a nonsense mutation in exon 12 in predicted TM12 (c.1568G>A, p.W523X); Family 3, a missense mutation at the first base pair of exon 9 in predicted TM8 (c.1148G>A, p.G383D); Family 4, an eight base pair duplication in exon 3 leading to a frameshift and premature stop codon in predicted TM4 (c.540_547dupAGAAGTAT, p.F183fsX1); and Family 5, a nonsense mutation in exon 14 located in the cytoplasmic tail (c.1710G>A, p.W570X) (2) (Fig. 1A; Table 1; and figs. S1 and S2). iPSCs were also derived from each proband’s genetically related, non-carrier male sibling for use as a paired control. Control and CS iPSCs had similar morphologies to human embryonic stem cells (hESCs), expressed endogenous pluripotency markers, had normal karyotypes, formed embryoid bodies in vitro, and formed teratomas in vivo, thereby demonstrating successful reprogramming (table S1 and figs. S3 to S10).
Table 1. iPSC resources for study of CS.
Resources include: 1) iPSCs derived from five families with distinct mutations in NHE6, including paired patient and biologically related, non-carrier, unaffected male siblings (Family 1–5); 2) isogenic CS and genome-corrected iPSC lines (Isogenic Pair 1); and 3) isogenic control and NHE6 induced-mutation iPSC lines (Isogenic Pair 2). Mutations in NHE6 are listed based on Ensembl transcript ENST00000370695. TM were predicted using TMHMM based on ENSP00000359729.
| Patient-derived iPSCs | |||
| Family | Disease status | NHE6 mutation | Location of mutation |
| 1 | |||
| Unaffected brother | Y/+ | - | |
| 2 | |||
| Unaffected brother | Y/+ | - | |
| 3 | |||
| Unaffected brother | Y/+ | - | |
| 4 | |||
| Unaffected brother | Y/+ | - | |
| 5 | |||
| Unaffected brother | Y/+ | - | |
| Genome-edited iPSCs | |||
| Pair | Genotype | NHE6 mutation | Location of mutation |
| 1 | |||
| Isogenic corrected | c.[1404T>A; 1407T>C; 1408T>C; 1410G>A; 1413T>A], p.(L468=; N469=; L470=; G471=) | Mutation corrected (+ silent mutations) | |
| 2 | |||
| NHE6 induced mutations | |||
| c.345_424del, p.L116TfsX5 | Exon 2, TM3 | ||
Fig. 1. Analysis of NHE6 gene expression in iPSCs from patients with NHE6 mutations and from related controls.
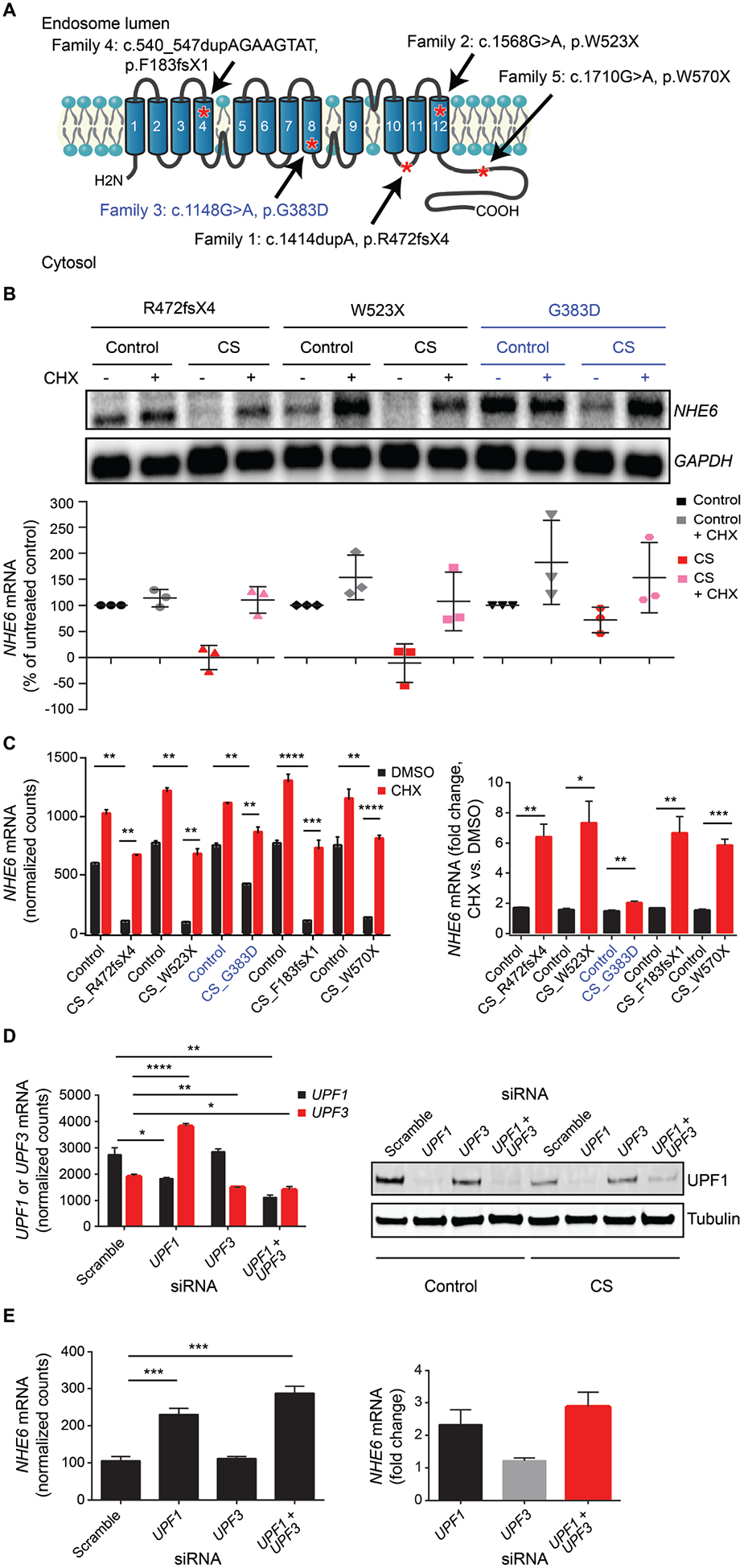
(A) iPSCs were derived from five patients with CS and from their genetically related, unaffected brothers without NHE6 mutation as controls. Shown is a schematic of the NHE6 protein with the predicted locations of the proband mutations, based on NHE6 transcript NM_001042537. NHE6 TM were predicted by the transmembrane hidden Markov model (TMHMM) (30) for Ensembl transcript ENST00000370695 and protein ENSP00000359729 (31). (B) NHE6 mRNA expression in control and CS iPSCs was quantified based on northern blots. Northern blotting was performed using samples from CS and control iPSCs treated (+) or untreated (−) with CHX. Expression of NHE6 mRNA was normalized to GAPDH and plotted as a percentage of untreated control cells. n = 3 experiments. (See fig. S11 for northern blots of Families 4 and 5, and data file S7 for mRNA expression quantification data.) (C) NHE6 mRNA expression in control and CS iPSCs was quantified based on NanoString data. RNA samples collected from CS and paired control iPSCs treated (+) or untreated (−) with CHX were analyzed using a customized NanoString platform. Expression of NHE6 mRNA was normalized to hybridization controls and to a set of housekeeping genes: B2M, GAPDH, GUSB, HPRT1, RPL13a, RPL27, POLR2A (left panel). Fold change in NHE6 mRNA expression, in the presence or absence of CHX treatment, was quantified for each CS and paired control iPSC (right panel). n = 15 samples per group (3 replicate wells from each of 5 control lines with or without CHX treatment and 3 replicate wells from each of 5 CS lines with or without CHX treatment). (D) UPF1 and UPF3 mRNA expression in CS iPSCs from Family 1 treated with a control set of scrambled siRNA oligonucleotides or siRNA oligonucleotides targeting UPF genes was quantified. Following siRNA treatment, expression of UPF1 and UPF3 mRNA was determined using a customized NanoString platform (left panel). Western blotting was performed using antibodies against UPF1 (top) to test for knockdown of UPF1 and against tubulin (bottom) as a loading control (right panel). Shown is a representative blot using control and CS samples from Family 2. (E) NHE6 mRNA expression in CS iPSCs from Family 1 treated with a control set of scrambled siRNA oligonucleotides or siRNA oligonucleotides targeting UPF genes was quantified. Following siRNA treatment, expression of NHE6 mRNA was determined using a customized NanoString platform (left panel). NHE6 mRNA was normalized to hybridization controls and to a set of housekeeping genes: B2M, GAPDH, HPRT1, RPL13a, POLR2A. Fold change in NHE6 mRNA expression after knockdown of UPF1, UPF3, or UPF1 + UPF3 was quantified. Shown are results for samples from Family 1 CS iPSCs (right panel). n = 4 replicates. Data represent means ± SEM. Unpaired Student’s t tests were used. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
To determine the effects of the pathogenic mutations on NHE6 gene and protein expression, we conducted mRNA and western blot analyses of representative mutations and subclonal lines. In all four of the putative protein-truncating nonsense mutations, we found NHE6 mRNA to be absent. These mRNA results were corroborated using two distinct methods: northern blot (Fig. 1B and fig. S11) and NanoString nCounter (Fig. 1C). These results are most consistent with a loss of mRNA by nonsense-mediated decay (NMD).
To test if NHE6 mRNA is degraded by NMD, we treated iPSCs with cycloheximide (CHX), which prevents NMD by inhibiting translation and thereby interfering with recognition of a premature stop codon. Upon CHX treatment, the NHE6 transcript was recovered in CS lines with these nonsense mutations (Fig. 1, B and C). Notably, the NHE6 missense mutation G383D behaved differently than the nonsense mutations. The NHE6 mRNA transcript in cells with the missense mutation was not absent but instead was reduced to 58% ± 18.4% of control amounts by northern blot analysis and to 56.4% ± 4.7% by NanoString nCounter analysis. However, the amount of transcript was recoverable with CHX treatment to 77.9% ± 6.4% of control amounts, suggesting some component of NMD (Fig. 1, B and C). Because CHX may have broad effects beyond blocking NMD, we conducted additional controls. To further implicate NMD as the cause for loss of NHE6 mRNA in mutant lines, we targeted NMD using specific siRNA targeting of NMD factors UPF1 and UPF3 (14, 15) (Fig. 1D). Again, we observed a strong recovery of NHE6 mRNA after siRNA-mediated reduction of NMD factors. Alone, reduction of UPF1 showed the strongest result, whereas combination siRNA against UPF1 and UPF3 showed additive effects on recovery of NHE6 mRNA (Fig. 1E).
Loss of mRNA in the nonsense/frameshift mutations was associated with an apparent complete loss of protein. Using western blot on NHE6 immunoprecipitates, NHE6 protein appeared to be absent in all CS lines carrying nonsense/frameshift mutations (Fig. 2, A and B). Therefore, these results establish that the majority of CS mutations, namely those leading to nonsense mutations, are likely to cause disease by a loss-of-function mechanism involving NMD. The results further indicate that truncated NHE6 proteins are unlikely to be translated in large quantities.
Fig. 2. Analysis of protein expression of pathogenic NHE6 mutations and additional study of the complex alternative splicing and missense mutation G383D.
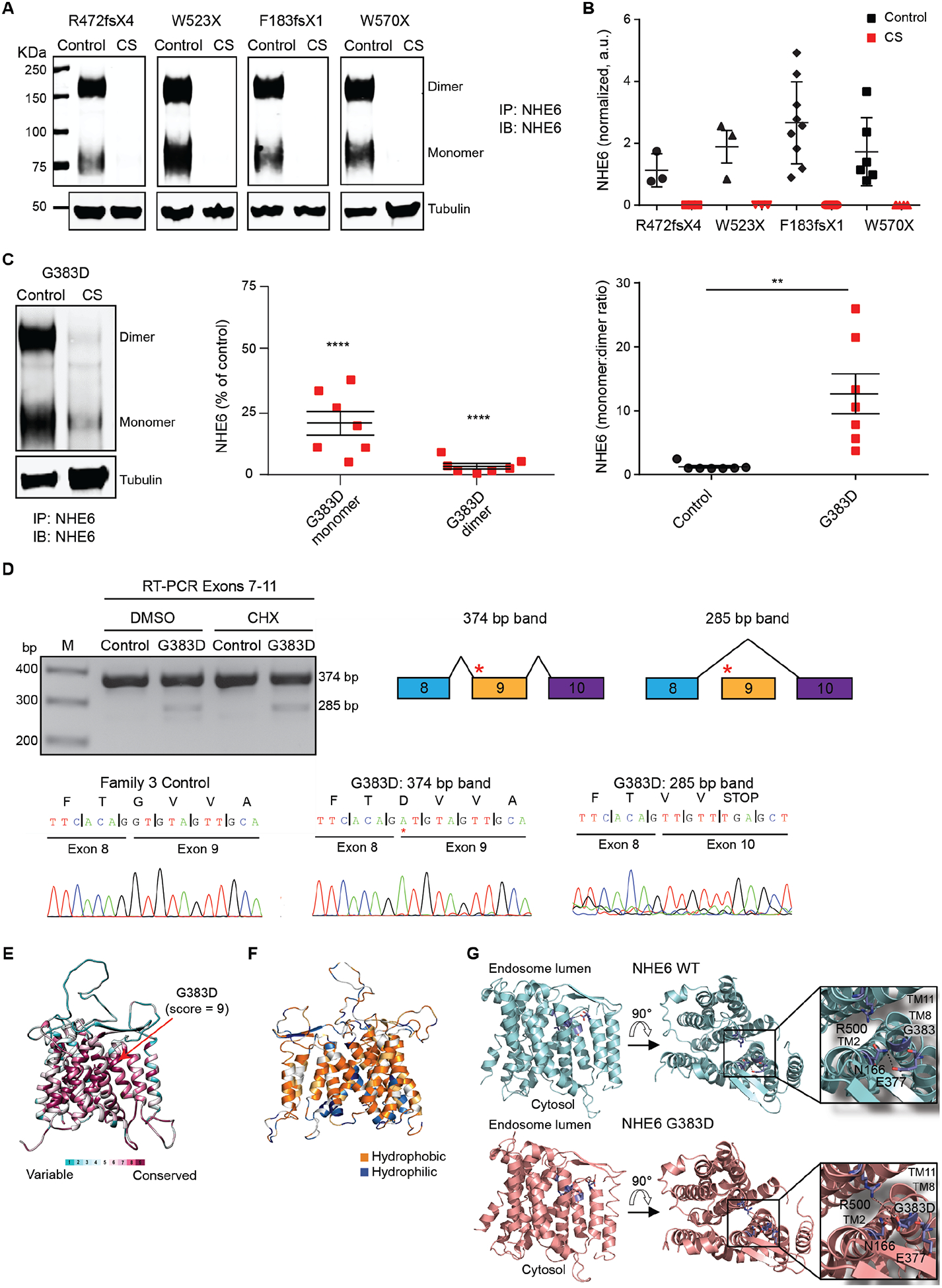
(A) Western blotting was performed to determine NHE6 protein expression in control and CS iPSCs. Lysates of CS and paired control iPSCs from Families 1, 2, 4, and 5 were immunoprecipitated using a custom-made anti-NHE6 antibody and subsequently analyzed by western blot using the same antibody. Blotting against tubulin was used as a loading control. (B) NHE6 protein expression, based on western blots such as those presented in (A), was quantified. The signal from immunoprecipitated NHE6 was normalized to the respective signal from tubulin and the average signal intensity for the control of Family 1 was set to a value of 1. n ≥ 3 experiments. (C) Western blotting was performed to determine NHE6 protein expression specifically in control and CS iPSCs from Family 3 (left panel). Samples were prepared and analyzed as described for panel (A). Amounts of NHE6 protein monomer and dimer in CS and paired control samples from Family 3 were quantified and the monomer:dimer ratios determined (middle and right panels). The signal from immunoprecipitated NHE6 was normalized to the respective signal from tubulin; the data are plotted as a percent of control. n = 7 experiments. (D) The G383D mutation generates an alternative transcript by exon skipping. RT-PCR was performed on cDNA collected from iPSCs treated or untreated with CHX. The resulting bands were sequenced and revealed the presence of an alternative splicing event in which exon 9 was skipped. The red asterisk in the diagram indicates the approximate location of the c.1148G>A/p.G383D mutation. (E) Conservation of NHE6 protein was analyzed using ConSurf. Shown are variable and conserved regions (turquoise through maroon) mapped onto the NHE6 structural model. (F) Hydrophobicity analysis of the NHE6 structural model was performed. Hydrophobic (orange) residues are located in TM, whereas hydrophilic (blue) residues are located in loop regions. (G) The protein structure of wild-type (WT) and G383D-mutant NHE6 was modeled. The model of NHE6 predicts that the G383D mutation disrupts helix packing between TM8 and TM2, possibly due to hydrogen bonding of D383 with N166 and loss of an interaction between N166 and E377 (insets, dotted lines). Alternatively, the G383D variant could form a salt-bridge with R500 on TM11 (lower inset, dotted line). Data represent means ± SEM. Unpaired Student’s t tests were used (B and C). ** P < 0.01, **** P < 0.0001.
The missense G383D mutation demonstrates complex effects on mRNA splicing and on protein coding
We have previously reported a patient with a de novo c.1148G>A (p.G383D) mutation (2). Through international ascertainment of patients, we have identified a second unrelated pedigree with this same mutation presented here (see Supplementary Materials for details on patients); therefore, we have now determined this mutation to be recurrent in patients with CS. The NHE6 gene and protein in cells with the missense mutation G383D behaved differently than the nonsense mutations. By contrast to nonsense mutations, the amount of NHE6 protein in the G383D cells was decreased, but NHE6 protein was not entirely absent. Relative to control, NHE6 G383D protein in cells was 20.7% ± 4.7% (NHE6 monomer) and 3.1% ± 1.1% (NHE6 dimer) (Fig. 2C). The reduction in amount of detectable NHE6 protein was more prominent for the dimer as compared to the monomer, such that the proportion of available monomer (reflected by monomer to dimer ratio) was substantially elevated in the G383D cells compared to control cells (Fig. 2C).
In addition to creating a missense coding change, the G383D mutation occurs at the first base pair of exon 9 and therefore directly adjacent to the 3’ splice acceptor site (fig. S1). We predicted that this mutation may disrupt splicing, namely, skipping of exon 9 and splicing of exon 8 to exon 10, thereby leading to a frameshift and premature stop codon two codons downstream of the exon 8/10 junction. RT-PCR and sequencing of cDNA from iPSCs with the G383D mutation revealed the presence of an alternative splicing event generated by skipping of exon 9 (Fig. 2D). Therefore, two mRNAs are produced: one with exon skipping leading to a nonsense mutation (subject to NMD, as supported by transcript recovery in the presence of CHX, which reduces the amount of mRNA in these cells); and a second mRNA encoding for a protein product with the G383D missense mutation.
With regard to the G383D mutation, through use of structural modeling of the protein, we investigated whether this missense mutation was predicted to affect protein structure. We constructed a homology model of NHE6 based on the crystal structure of the Escherichia coli Na+/H+ exchanger NhaA (16), which was previously used to model other members of the SLC9 family (17). Consurf analysis, which calculates the rate of evolution at each amino acid based on phylogenetic relations between homologous sequences, showed the highest conservation score for the G383 residue (Fig. 2E). (See also fig. S12 for sequence comparisons demonstrating conservation of the G383 residue across NHE homologs.) The NHE6 structural model showed that, similar to other membrane proteins, hydrophobic residues are located in the TM (transmembrane domains), whereas polar residues are located in loops (Fig. 2F); these results increased our confidence in the model. The model predicted that the G383 residue is positioned toward the endosomal end of NHE6 facing the core of the transporter on TM8. This domain has been proposed to function in the regulation of ion transport mediated by NHE family members (18). Mutation of this nonpolar glycine residue to an acidic aspartate residue is therefore likely to impact the transporter’s structure by disrupting helix packing between TM7 and TM8. Modeling of the G383D variant suggested that this residue may also disrupt the transporter structure by forming a hydrogen bond with N166 on TM2 (Fig. 2G). Alternatively, the G383D variant could form a salt-bridge with R500 on TM11, thereby stabilizing the inward open NHE6 conformation (Fig. 2G). Taken together, structural modeling suggests that the G383D mutation hampers the structure and dynamics of TM of NHE6, negatively affecting ion transport. In summary, the G383D mutation is predicted to be loss-of-function by complex mechanisms, including defects in splicing and a protein missense mutation. The residual mutant protein product is predicted to have impaired transport based on structural analyses.
Postnatal microcephaly is associated with defects in neuronal arborization in patient-derived neurons and is not attributable to differences in proliferation or cell death
Postnatal microcephaly, such as observed in patients with CS, may result from deficiencies of neuronal arborization in the neocortex, as opposed to differences in progenitor cell proliferation; in contrast, primary microcephaly generally occurs as a result of defects in cortical progenitor neurogenesis, cell fate determination, or cell death pathways (4). To investigate the potential mechanisms of postnatal microcephaly in CS, we differentiated CS and control iPSCs to an excitatory cortical neuronal fate using a monolayer differentiation protocol involving dual SMAD inhibition (19) (fig. S13). Our differentiation protocol demonstrated robust downregulation of stem cell genes and upregulation of neuronal genes in iPSC-derived neurons (figs. S14 and S15).
In order to examine the possibility that differences in neuronal progenitor proliferation, cell fate determination, or cell death could contribute to CS-associated microcephaly, CS and control neuronal cultures were examined using immunocytochemistry and RNA expression profiling. By immunocytochemistry, CS and control neuronal cultures had comparable percentages of cells expressing markers for neuronal progenitors (PAX6), neurons (MAP2), and deep-layer cortical projection neurons (TBR1, CTIP2) (fig. S14). We further examined the cell fate of CS and control neuronal cultures using our NanoString-based panel of mRNAs related to neuronal cell fate determination (data file S1 and fig. S15). Upon neuronal differentiation, iPSC-derived neurons upregulated neuronal genes and downregulated stem cell fate genes (data files S2 to S4). Further, in differential gene expression studies, which were performed using iPSCs or iPSC-derived neurons, we found neurons from both genotypes to express comparable and high amounts of neuronal progenitor, neuronal, and cortical markers, and very low amounts of mesodermal, endodermal, glial, and midbrain markers (figs. S15 to S21, table S2, and data files S2 to S5). Therefore, these analyses demonstrate that cortical cell fate determination in these cultures does not appear to be affected by NHE6 mutations in patient-derived cells. We also ruled out differences in neuronal cell proliferation or cell death between CS and control iPSC-derived neurons. The number of mitotically cycling cells positive for Ki67 was not statistically different between CS and control cultures at day 35 after neuronal induction (fig. S22). Similarly, there was no difference in the number of apoptotic cells between CS and control neuronal cultures (fig. S22). We conclude that mechanisms frequently involved in primary microcephaly such as alterations in cell fate determination, progenitor cell proliferation, or cell death are unlikely to contribute to CS-associated secondary or postnatal microcephaly.
Reduced neuronal arborization may contribute to postnatal microcephaly in CS, particularly in light of our data above showing no differences in cell proliferation or death. In prior studies in NHE6-null mice, we identified abnormalities in neuronal arborization both in vitro and in vivo (9). Therefore, to expand on our prior findings in mouse but now in neurons from patients, we examined neurite outgrowth and arborization in neurons differentiated from iPSCs from patients with CS. Across all CS mutations examined, using a variety of different approaches, we saw prominent defects reflecting abnormal neurite growth and arborization in CS. We conducted time-lapse live-imaging analysis of CS neurons plated on laminin stripes (Fig. 3A). The neurons adhered along the laminin stripes and extended and retracted their neurites. We found decreases in the elongation rates and retraction rates, in comparison to their respective controls, for specific CS lines (Fig. 3B). Additionally, using Sholl analysis on GFP-transfected neurons to assess arborization, we observed strong defects in arborization in CS neurons regardless of the CS mutation studied (Fig. 3, C and D, and fig. S23). Taken together, these data from live imaging and Sholl analysis suggest that defects in arborization might underlie phenotypes of patients with CS such as postnatal microcephaly.
Fig. 3. Rescue of deficits in neuronal arborization by re-expression of NHE6 in CS lines with frameshift or nonsense mutations but not in the CS missense mutation line.
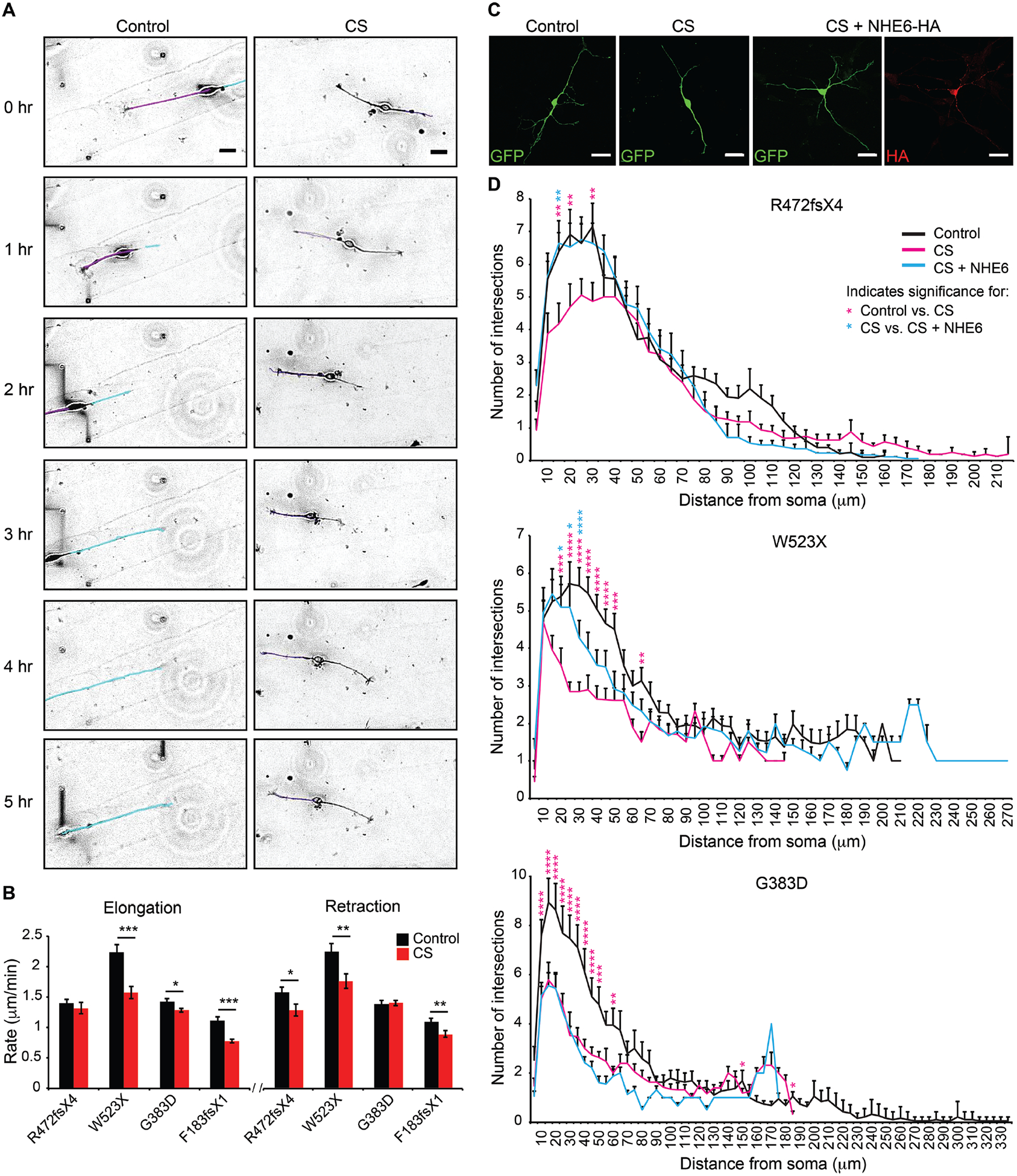
(A) Neurite outgrowth dynamics were monitored in iPSC-derived neurons. Time-lapse images of iPSC-derived neurons plated on laminin-striped coverslips were acquired using Volocity software at four XY positions every 5 min for 12 hr. Representative images are shown from the CS and paired control lines for Family 3, G383D. Tracings of neurites are shown in purple and cyan for the control line to demonstrate tracking of different neurites. Scale bars, 20 μm. (B) The rates of neurite elongation and retraction were measured using ImageJ software for the first 2 hr. Control Family 1: n = 17 cells/ 32 neurites/ 841 measurements; Family 1, R472fsX4: n = 13 cells/ 25 neurites/ 875 measurements; Control Family 2: n = 12 cells/ 15 neurites/ 635 measurements; Family 2, W523X: n = 8 cells/ 9 neurites/ 724 measurements; Control Family 3: n = 28 cells/ 55 neurites/ 827 measurements; Family 3, G383D: n = 52 cells/ 126 neurites/ 2,163 measurements; Control Family 4: n = 8 cells/ 17 neurites/ 622 measurements; and Family 4, F183fsX1: n = 9 cells/ 13 neurites/ 708 measurements. (C) Representative images reflective of each condition of gene transfection experiments are shown from Family 1, R472fsX4. iPSC-derived neurons were transfected with a construct encoding for GFP alone or with two constructs together encoding for GFP + full-length human NHE6-HA and analyzed after 5 days. Scale bars, 20 μm. (D) Sholl analysis of control iPSC-derived neurons (black lines), CS iPSC-derived neurons (magenta lines), and CS iPSC-derived neurons transfected with a construct encoding for NHE6-HA (cyan lines) was performed. The average number of neurite intersections with Sholl radii was plotted as a function of distance from the soma for each condition. n = 20–25 cells per condition. Data represent means ± SEM. Welch’s t tests (B) and two-way ANOVA followed by Bonferroni correction for multiple comparisons (D) were used. * P < 0.045, ** P < 0.008, *** P < 0.00008 (B). * P < 0.05, ** P < 0.01, *** P < 0.0001, **** P < 0.00001 (D). Magenta asterisks denote significance when comparing CS to control cells. Cyan asterisks denote significance when comparing CS to CS cells transfected with a construct encoding for NHE6-HA.
Gene transfer leads to cell-autonomous rescue of neuronal arborization in patient neurons with nonsense mutations but not in neurons with the missense mutation
Using Sholl analysis to assess arborization, we observed strong reductions in neuronal arborization regardless of the CS mutation studied (Fig. 3D). However, with regard to rescue of these defects using cDNA transfection in patient-derived neurons, cells with nonsense mutations behaved alike to each other, but differently from the cells with the G383D missense/splice mutation. For these studies, we transfected CS and control neurons with a single construct encoding for GFP or with a construct encoding for GFP together with a construct encoding for full-length NHE6 tagged with HA (Fig. 3C). Expression of NHE6-HA by gene transfer led to prominent, cell-autonomous rescue of arbor complexity (by Sholl analysis) in neurons with nonsense mutations; however, rescue completely failed in neurons from the patient with the complex missense/splice mutation, G383D (Fig. 3D and fig. S23).
G383D missense mutant protein has both loss-of-function and dominant-negative properties
The ability to rescue patient-derived neurons with nonsense mutations by way of expression of wild-type NHE6, but the failure to rescue arborization in G383D-mutant neurons with the same strategy suggests that the residual endogenously expressed NHE6 G383D protein may exert a dominant-negative function. We hypothesized that the mechanism may be potentially through formation of non-functional heterodimers with exogenously expressed NHE6. This is an important question with regard to therapy because if residual, non-functioning protein is active as a dominant-negative in this class of CS mutations, then gene therapy strategies will probably not be a feasible approach in these patients.
To assess the functional properties of G383D protein, we next tested the ability of G383D mutant protein to alkalinize endosomes, an established function of endosomal NHEs. For these studies, we generated a CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 genome-edited HEK293T cell line wherein deleterious mutations were induced in NHE6 and in which we observed a decrease in endosomal pH relative to control (fig. S24). Exogenous expression of wild-type NHE6 in NHE6-null HEK293T cells resulted in an alkalization of endosomes, as compared to untransfected NHE6-null HEK293T cells. However, exogenous expression of NHE6 G383D in NHE6-null HEK293T cells did not alkalinize endosomes (Fig. 4A). These results indicate that the G383D missense protein fails to function appropriately in cation exchange, consistent with loss-of-function predictions by the structural modeling (Fig. 2G). Co-expression of NHE6 G383D together with wild-type NHE6 inhibited the ability of exogenously expressed wild-type NHE6 to alkalinize endosomes (Fig. 4A).
Fig. 4. Loss-of-function and dominant-negative activity of NHE6 G383D mutation in regulation of intra-endosomal pH, and mislocalization in cells.
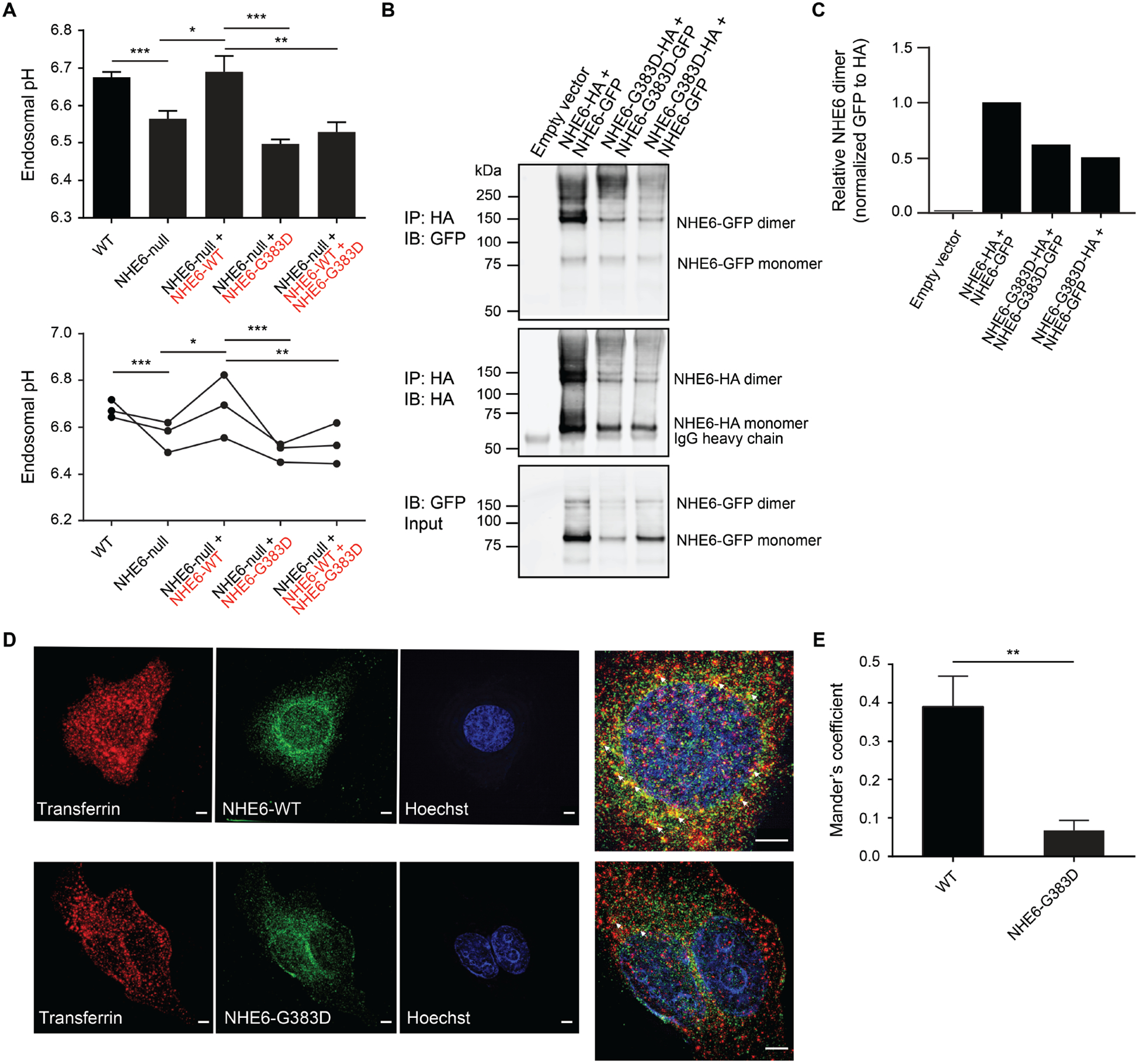
(A) The ability of NHE6 G383D-mutant protein to rescue endosomal pH defects in NHE6-null HEK293T cells was determined. Endosomal pH was assayed using a transferrin-based method in which cells were incubated with pH-sensitive and pH-insensitive fluorescent conjugates of transferrin and then analyzed by flow cytometry. Endosomal pH was measured in wild-type HEK293T cells (WT) (black font), HEK293T cells lacking NHE6 by way of gene editing (NHE6-null) (black font), and HEK293T cells lacking NHE6 but expressing mCherry-tagged WT (red font) and/or G383D-mutant NHE6 (red font). Data are presented as a bar graph (top) and as individual data points wherein each dot represents the average of three replicates from one independent experiment (bottom). n = 3 experiments. (B) Lysates from HEK293T cells expressing HA- or GFP-tagged forms of NHE6, as indicated, were immunoprecipitated using an anti-HA antibody and subsequently analyzed by western blot using antibodies against GFP (top panel) and HA (middle panel). Protein input was determined by western blot of lysates that had not undergone immunoprecipitation for HA-tagged protein using an antibody against GFP (bottom panel). (C) The relative amount of immunoprecipitated NHE6 dimer was quantified based on results shown in panel (B). (D) HeLa cells expressing HA-tagged WT or G383D-mutant NHE6 were incubated with Alexa Fluor 568-conjugated transferrin (red) for 10 min and subsequently immunostained for HA (green) and imaged using srSIM. Arrows (white) in the magnified images indicate areas of co-localization between transferrin and NHE6 in endosomes. Scale bars, 5 μm. (E) Co-localization of NHE6 WT protein vs. NHE6 G383D-mutant protein with transferrin was quantified using ImageJ, and a Mander’s coefficient was determined. Data represent means ± SEM. Unpaired Student’s t tests were used. * P < 0.05, ** P < 0.01, *** P < 0.001.
To dissect the biochemical mechanism of the dominant-negative activity exerted by the G383D protein, we investigated if the G383D protein could form heterodimers with wild-type NHE6 protein in HEK293T cells. We found that exogenously expressed G383D protein could form homodimers with itself, as well as heterodimers with exogenously expressed wild-type NHE6 protein (Fig. 4, B and C). Using super-resolution structured illumination microscopy (srSIM) in cells expressing exogenous wild-type NHE6 or G383D-mutant NHE6, the cellular localization of G383D protein was altered relative to control. Control protein showed a strong co-localization with fluorescent-conjugated transferrin in early endosomes, whereas G383D-mutant NHE6 protein demonstrated minimal co-localization with transferrin-positive early endosomes (Fig. 4, D and E). In summary, we find that the NHE6 G383D mutant does not function in the alkalization of endosomes, reflecting the loss-of-function nature of this mutation. In addition, our dimerization and pH studies also demonstrate that the G383D-mutant protein functions as a dominant-negative, potentially by inhibiting the wild-type NHE6 protein function upon formation of (non-functional) heterodimers.
Patient neurons exhibit rescue of neuronal arborization by exogenous trophic factor treatment (BDNF or IGF-1) regardless of mutation
Herein, we have established a substantial cellular resource that may be used in cell-based assays for pre-clinical development of therapeutics. In addition to a gene therapy strategy, we considered two alternative strategies for cell rescue that might serve as potential CS treatments: 1) agents that alkalinize the early endosome, such as quinolines (such as, chloroquine), weak bases, or V-ATPase inhibitors; and 2) growth factors such as BDNF, which we have shown to have efficacy in NHE6-null primary mouse cultures (9). To assess the premise for studying agents that alkalinize the endosome, we conducted extensive studies of intra-endosomal pH across all available subclonal lines derived from patients with CS using two methods: ratiometric analysis of cells following endocytosis of pH-sensitive and pH-insensitive fluorescent conjugates of transferrin; and ratiometric analysis of cells expressing vesicle-associated membrane protein 3 (VAMP3)-pHluorin2. In the context of either method, we identified decreases in intra-endosomal pH that were recoverable by correction of the mutation using CRISPR/Cas9 (Fig. 5A–C and figs. S25 to S27). (See Supplementary Materials and Methods and figs. S25 to S31 for detailed methods and additional results on intra-endosomal pH studies, including studies in patient-derived lines, and in lines in which NHE6 mutations have been induced in a founder cell line.) However, we did observe variability in the strength of the effect of over-acidification of the endosome across different families. Some families showed strong effects, whereas other families did not show a phenotype of endosome over-acidification (figs. S27 to S29). Given this variability, we did not pursue the alkalinizing agents further as a rescue strategy.
Fig. 5. Correction of endosomal pH defects in genome-edited CS iPSCs.
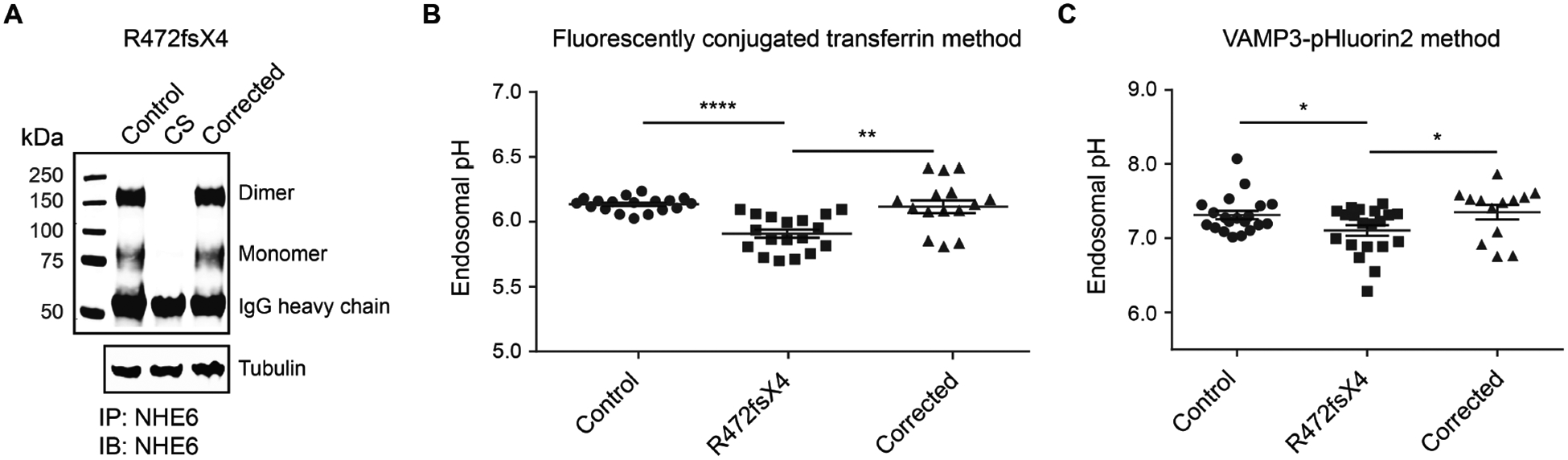
(A) Western blotting was performed to confirm expression of NHE6 protein in CS iPSCs that had undergone genome editing to correct the R472fsX4 mutation. Lysates of paired control, CS, and genome-corrected iPSCs from Family 1 were immunoprecipitated using a custom-made anti-NHE6 antibody and subsequently analyzed by western blot using the same antibody. Blotting against tubulin was used as a loading control. (B and C) Whether correction of the NHE6 R472fsX4 mutation might rescue endosomal pH defects present in mutant iPSCs was determined. Endosomal pH was assayed using two different ratiometric methods, a transferrin-based method (B) and a VAMP3-pHluorin2-based method (C). For the transferrin-based method, cells were incubated with pH-sensitive and pH-insensitive fluorescent conjugates of transferrin and then analyzed by flow cytometry. For the VAMP3-pHluorin2-based method, cells were transfected with a construct allowing for expression of VAMP3-pHluorin2 and then analyzed by flow cytometry. Endosomal pH was measured in paired control, R472fsX4-mutant, and genome-corrected iPSCs. Data are presented as individual data points wherein each dot represents a technical replicate. n ≥ 3 experiments. Data represent means ± SEM. Unpaired Student’s t tests were used. * P < 0.05, ** P < 0.01, **** P < 0.0001.
We next tested the extent to which neuronal arborization could be restored in patient-derived neurons pharmacologically through treatment with exogenous BDNF or IGF-1. We tested these two growth factors as proof-of-principle molecules, as we have previously shown that BDNF signaling via TrkB is diminished and the related arborization defects may be rescued by exogenous application of excess BDNF (9). Further still, IGF-1 is also a trophic factor that augments neurite growth, and this agent is currently in clinical trials for a number of neurodevelopmental disorders (20, 21).
When CS and control neurons were treated for three days with exogenous addition of either BDNF or IGF-1, we observed substantial rescue of the arborization defect in all family lines tested, regardless of mutation (Fig. 6, A and B). Treatment with BDNF or IGF-1 increased the mean length per neurite as well as the number of branchpoints per neurite in all CS lines tested, as compared to untreated CS neurons (Fig. 6B). Therefore, neurite morphology defects in CS cultures may be restored by exogenous trophic factor treatment. Together, these results suggest that independent exposure to BDNF or IGF-1 can rescue the neuronal arborization phenotype in CS neurons and that these growth factor treatments warrant further exploration in existing animal models and other pre-clinical studies. Unlike the gene transfer experiments above, these exogenous trophic factors demonstrate efficacy in vitro across patient-derived neurons with diverse mutations.
Fig. 6. Rescue of deficiencies in neuronal arborization, regardless of mutation type, by BDNF or IGF-1 treatment.
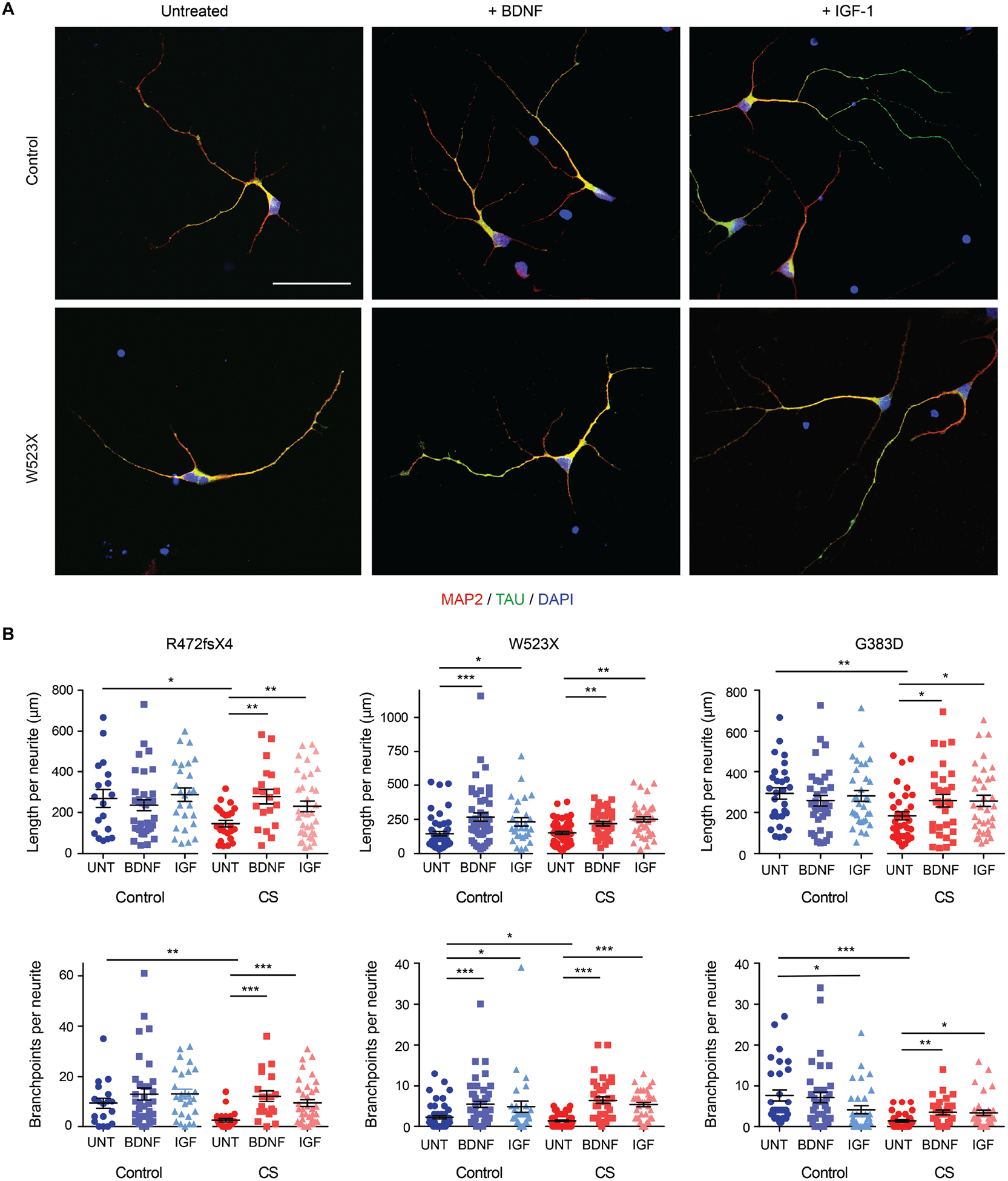
(A) Immunostaining of control and CS iPSC-derived neurons left untreated or treated with BDNF or IGF-1 was performed for MAP2 (red), a marker of dendrites, and TAU (green), a marker of axons. Nuclei were labelled using DAPI (blue). Representative images are shown from the CS and paired control lines for Family 2, W523X. Scale bar, 50 μm. (B) The recovery of deficiencies in neuronal arborization present in CS iPSC-derived neurons by way of growth factor treatment was quantified. Average length (top panels) and number of branchpoints per neurite (bottom panels) were measured using Neurolucida tracing software for control and CS iPSC-derived neurons left untreated (UNT) or treated with BDNF or IGF-1 (IGF). Neurites stained with either MAP2 or TAU were analyzed; no distinction was made between dendrites or axons during measurements. Control Family 1: n = 18 (UNT), 35 (BDNF), 26 (IGF) neurites; Family 1, R472fsX4: n = 24 (UNT),19 (BDNF), 36 (IGF) neurites; Control Family 2: n = 54 (UNT), 48 (BDNF), 28 (IGF) neurites; Family 2, W523X: n = 53 (UNT), 37 (BDNF), 37 (IGF) neurites; Control Family 3: n = 30 (UNT), 35 (BDNF), 32 (IGF) neurites; and Family 3, G383D: n = 32 (UNT), 31 (BDNF), 35 (IGF) neurites. Data represent means ± SEM. Welch’s t tests were used. * P < 0.05, ** P < 0.005, *** P < 0.0005.
Discussion
Despite increasing CS diagnoses in the clinic, much remains to be understood concerning the pathogenesis of CS, and there are currently no specific treatments. The generation of iPSCs derived from patients with CS presents a new opportunity to study the pathogenesis of CS and to develop treatment strategies. In this study, we examined the properties of patient-derived neurons that underlie deficits in circuit development as well as two rescue strategies. We show that human neurons derived from patients with CS have reduced complexity of neuronal arbors regardless of the nature of the mutation. These deficits in neuronal growth and arborization may underlie the postnatal microcephaly reported in CS (2). We also demonstrate that the neuronal arborization defect could be rescued by expression of wild-type NHE6 in cells with an NHE6 nonsense mutation and loss of protein due to NMD; however, cells from a patient with residual, non-functional NHE6 protein, namely cells from a patient with the recurrent G383D missense mutation, could not be rescued. We further provide biochemical evidence that residual, non-functional G383D protein can act as a dominant-negative through heterodimerization with the exogenously added wild-type protein. By contrast, the neuronal arborization defect could be rescued in vitro by exogenous addition of BDNF or IGF-1 regardless of mutation.
Our analyses of iPSCs from patients with CS spanning a series of mutations demonstrate at least two mechanistic classes of mutations. The majority of known mutations in patients with CS (Class 1 mutations) are predicted to be loss-of-function based on disruption of coding through introduction of nonsense mutations (2). Loss-of-function mutations by way of splicing mutations are also common and may act similarly, yielding complete loss of protein. We show here through measurement of mRNA expression in patient-derived cells, including in human neurons, that the mRNA is absent in CS cells, consistent with nonsense-mediated mRNA decay. We implicate NMD directly through recovery of mRNA after inhibition of NMD by CHX or siRNA-mediated reduction in expression of different components of the NMD machinery.
Our studies also examine the G383D mutation in depth as a model of a second class of mutations wherein residual, non-functional protein remains (Class 2 mutations). We demonstrate here that the G383D mutation is recurrent in CS, as we herein report two families with this mutation (Supplementary Materials). In patient cells, we show that this mutation has complex effects on mRNA and protein coding: First, we find that the G to A mutation at the first base of exon 9 leads to exon skipping and production of an mRNA with a nonsense mutation that is recoverable with CHX treatment. We estimate that this mechanism reduces mRNA amounts by approximately 50% compared to control. Second, importantly, this mutation leads to residual protein with the G383D mutation, which is predicted to perturb exchanger function based on our structural predictions. We also find that this residual protein appears to alter the cellular distribution of NHE6, with mutant NHE6 exhibiting reduced co-localization to transferrin-positive endosomes. Finally, we find that the G383D protein can heterodimerize with wild-type NHE6 protein. The G383D protein cannot function in the alkalization of endosomes, and when co-expressed with wild-type NHE6 protein, the G383D protein can interfere with wild-type NHE6 function in the alkalization of endosomes. Thereby, this G383D mutation represents a distinct class of NHE6 mutations wherein the mutation has both loss-of-function properties and also dominant-negative properties through residual expression of a mutant protein. At this time there are several published examples of mutations in patients with CS that might fit this second class of NHE6 mutations wherein residual dominant-interfering protein is likely (22).
The fact that CS has distinct mechanistic classes of pathogenic mutations has critical therapeutic importance. Our data show mutation-specific responses to treatments with regard to gene transfer using DNA transfection. Neurons with nonsense mutations in NHE6 exhibit clear rescue of arborization phenotypes via cDNA transfer; however, cells expressing the G383D missense mutation fail to be rescued by this strategy. This result indicates that gene therapy based in gene transfer strategies is unlikely to be effective in patients with mutations of this class with residual protein. Mutation-specific mechanisms of treatment response may well be more the rule than the exception in genetic disease, as has been studied in depth in cystic fibrosis (11). However, exogenous treatments of CS neurons with growth factors such as BDNF or IGF-1 rescue arborization defects regardless of the mutation class. BDNF agonists have been subject to efforts in pre-clinical studies of neurological disease with mixed results (23). Alternatively, IGF-1 has been shown recently to be a potential therapy for the autism-associated syndromes Rett syndrome and Phelan-McDermid syndrome. Pre-clinical studies in Rett syndrome showed that treatment of iPSC-derived neurons with IGF-1 increased numbers of glutamatergic synapses (24). Furthermore, treatment of a Rett syndrome mouse model with IGF-1 rescued brain size, synapse density, and excitatory synapse transmission to control values (25). In a phase I clinical trial for IGF-1 in Rett syndrome, patients showed improvement in symptoms of anxiety and breathing difficulty (20). In addition, IGF-1 treatment of iPSC-derived neurons from Phelan-McDermid syndrome patients improved deficits in mature excitatory synapses, as compared to controls (26). A preliminary clinical trial of IGF-1 in Phelan-McDermid syndrome showed improvement of social withdrawal and restricted behaviors (21). Additionally, a phase II clinical trial for IGF-1 is in progress with respect to a potential treatment for autism spectrum disorder (NCT01970345), and phase II trials have been completed with respect to the use of an IGF-1 analog (NNZ-2566) as a potential treatment for Fragile X syndrome (NCT01894958) and Rett syndrome (NCT02715115). Careful attention assessing the efficacy of IGF-1 in these other disorders may have relevance to therapeutic development in CS. Additionally, further research into efficacy of IGF-1 treatment may also be warranted in CS mouse models in vivo.
Results of our study of intra-endosomal pH across a diverse range of patient-derived and induced (through genome editing) mutations also provide new insights into the function of NHE6 as an endosomal proton/cation exchanger. Prior data from our laboratory and others have demonstrated reductions in organellar luminal pH in the context of NHE6 mutation or deficiency in inbred, laboratory strains in mouse (9) and in immortalized, mammalian cell lines (27, 28). A similar phenotype is observed upon Nhx1 mutation in yeast (17, 29). Here we observed the predictable responses, that is, elevation of endosomal pH with correction of the NHE6 mutation using CRISPR/Cas9; however, using patient-derived lines, compared to biologically related controls from human populations, we observed a range in the strength of the effect on intra-endosomal pH. There may be several explanations for this, among them are that cells in culture undergo genetic changes that compensate for the effect of the NHE6 mutation. In addition, it is possible that there is more redundancy or induction of compensatory mechanisms in some lines as compared to others, given the wide range in background genomic variation in these human lines. Our transcriptome data support a link between loss of NHE6 and potential compensatory changes in expression of V-ATPase genes (figs. S19 to S21, table S2, and data files S2 to S5), thereby further implicating a role for NHE6 in cross-membrane proton transport.
Although it is likely that mutation of NHE6 affects proton leak, the defective mechanisms may also involve protein functions that are not best exemplified by measurable changes in proton concentration within the endosome lumen. At the same time, the defects observed in neuronal development, such as in arborization, do not appear to clearly correlate with the strength of the effects on acidification of the endosome lumen. Overall, the weight of the data here suggests that NHE6 also functions in a process yet to be determined (not apparently attributable solely to alterations in intra-endosomal pH) that perturbs neuronal development. These results also might have therapeutic relevance, as they raise the question as to whether alkalinizing agents (such as V-ATPase inhibitors or weak bases) will be effective in treating CS. In developing potential alternative treatments, in this description of this new iPSC resource, we demonstrated that there are distinct mutational classes in CS and, further, that these classes need to be considered carefully as we embark on studies of treatment development. Our data indicate that CS mutations with residual protein are unlikely to respond to gene transfer strategies, whereas growth factor treatments may represent a more fruitful approach to treatments.
Through the results of our studies described here, we present merits and potentially some limitations in using iPSC systems. Our studies have examined highly penetrant pathogenic mutations, yet there remains variation in the prominence of some cellular phenotypes likely due to modulation by background genetic variation. Other limitations of this study include that our experiments are in vitro, and that the iPSC-derived neurons represent early embryonic differentiating neurons. Further still, we have examined only one neuron type, excitatory neurons, in monolayer cultures. In the future, cellular phenotypes in distinct neural cell types, as well as in more complex tissue models such as organoids, may be examined. Also, moving forward, successful rescue strategies presented here would be ideally corroborated in animal studies in vivo.
Finally, a major strength of our study is the establishment and validation of these iPSC resources for the study of cellular mechanisms of disease and pre-clinical drug development in CS. Our resource, which will be widely shared, in part through the National Institute of Mental Health (NIMH) Repository and Genomics Resource (Study 200), has established patient-derived cells with both related (unaffected brother) and isogenic (from mutation correction) controls, as well as induced mutations in a normal male founder iPSC line and in HEK293T cells. An important strength of these iPSC systems is the access to multiple human mutations, which are often hard to model in the genome of experimental animals. As we have done here, this sort of patient cell resource permits in depth study of the consequences of disease-causing mutations and potential rescue strategies in this cellular context.
Materials and Methods
Study design
The objective of the current study was to identify the underlying cellular and molecular mechanisms associated with mutations in NHE6 (encoded by SLC9A6) in patients with CS and to establish rescue of cellular phenotypes. To generate iPSCs, peripheral blood was obtained from five different patients with CS and from unaffected genetically related brothers. CS status was defined as the presence of a pathogenic NHE6 mutation, and families were recruited sequentially, wherein a patient with CS and a non-carrier brother were present, from our CS study described previously (2). Commercially available human iPSCs derived from fibroblasts of a healthy male were also obtained, and NHE6 mutations were induced in these cells using CRISPR/Cas9-based genome editing. A similar approach was also used to induce NHE6 mutations in the HEK293T cell line. The Institutional Review Boards (IRBs) at Brown University and Lifespan Healthcare approved all human subjects research protocols. The study involved controlled laboratory experiments and bioinformatics-based analyses. We determined cellular (neuronal arborization, endosomal acidification, cell death, and cell fate) and molecular (changes in gene expression) phenotypes and tested potential rescue strategies using patient-derived iPSCs or iPSC-derived neurons. Some assays were also performed in cells (iPSCs or cultured cells) that had undergone CRISPR/Cas9-based genome editing to induce or correct NHE6 mutations. Samples were chosen and allocated based on genotype. Statistical design and analyses were guided by a study statistician (R.N.J.) (see Statistics section). Experiments were replicated at minimum three times across multiple subclonal lines from the various patient-derived and control iPSC lines. Different numbers of replicates were used for experiments as specified in the figure legends. Multiple distinct family mutations and different subclonal lines were used, and orthogonal experimental approaches were taken. For studies in which analysis of cellular phenotype using microscopy images was performed, such as neuronal arborization and neurite outgrowth studies, the analyzer was blinded as to the genotype. See the Materials and Methods section in the Supplementary Materials for more detailed methods. Also, see data files S6 and S7 for primary data for main text and supplemental figures, respectively, in which experiments had an n < 20.
Statistics
Statistical tests used in comparing data have been indicated in the legends to figures. Unless otherwise indicated, unpaired Student’s t tests were used to compare data. Cell proliferation and cell death, neurite elongation and retraction, and neurite length and branching were compared using two-tailed Welch’s t tests. Analysis of Sholl data was performed using two-way ANOVA followed by Bonferroni correction for multiple comparisons. NanoString gene expression differences were tested for significance with two-tailed Welch’s t tests followed by Benjamini-Yekutieli FDR adjustment. Fold changes with an adjusted P-value < 0.05 were considered significant.
Supplementary Material
Materials and Methods
Fig. S1. Family 1 to Family 5 CS-related mutations in NHE6 cDNA.
Fig. S2. Sanger sequencing chromatograms of paired control and CS iPSCs.
Fig. S3. Morphological characterization of paired control and CS iPSCs.
Fig. S4. Karyotype analysis of paired control and CS iPSC lines.
Fig. S5. Staining for alkaline phosphatase activity in paired control and CS iPSC lines.
Fig. S6. Immunostaining of paired control and CS iPSC colonies for pluripotency markers.
Fig. S7. FACS analysis of paired control and CS iPSCs for stem cell-specific markers.
Fig. S8. qRT-PCR analyses of pluripotency markers in undifferentiated iPSCs (UD) as compared to hESC-derived embryoid bodies (EB).
Fig. S9. In vitro differentiation potential of paired control and CS iPSCs.
Fig. S10. In vivo differentiation potential of paired control and CS iPSCs.
Fig. S11. Northern blot analysis of Family 4 and Family 5 paired control and CS iPSCs.
Fig. S12. Conservation of the G383 residue across NHE homologs.
Fig. S13. Protocol for neuronal differentiation from iPSCs.
Fig. S14. Neuronal and cortical marker expression in iPSC-derived neurons.
Fig. S15. Gene expression of cell fate markers in iPSC-derived neurons.
Fig. S16. Unsupervised hierarchical clustering of iPSCs and iPSC-derived neurons by gene expression.
Fig. S17. Volcano plot of gene expression changes in iPSCs as compared to iPSC-derived neurons.
Fig. S18. Volcano plots of gene expression changes in CS vs. control iPSCs and iPSC-derived neurons.
Fig. S19. NHE family member gene expression changes in CS iPSCs and iPSC-derived neurons.
Fig. S20. V-ATPase gene expression changes in CS iPSCs and iPSC-derived neurons.
Fig. S21. WGCNA-based network construction and module detection in CS iPSCs.
Fig. S22. Analysis of iPSC-derived neuronal cultures for proliferation and apoptosis.
Fig. S23. Rescue of deficits in neuronal arborization complexity by re-expression of NHE6 in CS lines with frameshift or nonsense mutations but not in the CS missense mutation line.
Fig. S24. Validation of control and CRISPR/Cas9-induced NHE6-null HEK293T cell lines and characterization of endosomal pH.
Fig. S25. Establishment of isogenic, genome-corrected iPSCs for Family 1 using genome editing.
Fig. S26. Calibration curve and flow cytometer profiles for ratiometric analysis of endosomal pH using pH-sensitive and pH-insensitive fluorescent conjugates of transferrin.
Fig. S27. Analysis of endosomal pH in paired control and CS iPSCs from Family 1 to Family 5 using the transferrin method.
Fig. S28. Analyses of endosomal pH in paired control and CS iPSCs from Family 1 to Family 5 using the VAMP3-pHluorin2 method.
Fig. S29. Analysis of endosomal pH in paired control and CS iPSC-derived neurons.
Fig. S30. Establishment of iPSCs with NHE6 mutations and isogenic controls using genome editing of a normal founder line.
Fig. S31. Analysis of endosomal pH in paired isogenic control iPSCs and iPSCs with induced NHE6 mutations using the transferrin method.
Table S1. Characterization of PBMC-derived iPSC lines.
Table S2. Statistical analysis of genes included in the blue module using a Hypergeometric test.
Data file S1. NanoString customized probe set.
Data file S2. Average normalized NanoString count data of gene regulation in iPSCs and iPSC-derived neurons from patients with CS and controls.
Data file S3. By-sample normalized NanoString count data of gene regulation in iPSCs and iPSC-derived neurons from patients with CS and controls.
Data file S4. By-sample raw NanoString count data of gene regulation in iPSCs and iPSC-derived neurons from patients with CS and controls.
Data file S5. Genes in each gene module from the WGCNA-based analyses.
Data file S6. Primary data for main text figures.
Data file S7. Primary data for supplementary figures.
Acknowledgements:
We thank the families who participated in this study.
Funding: The study was supported by the following: Aspire I Junior Faculty Award (to S.B.L.), Center of Biomedical Excellence Dietary Supplements and Inflammation-NIGMS P20GM103641 Pilot Project Award (to S.B.L.), and SC INBRE NIGMS P20GM103499 Pilot Award (to S.B.L.); NIGMS Postbaccalaureate Research Education Program (PREP) R25GM064118 (to S.A.); Brown Institute for Brain Science Pilot Award and Brown University Seed Award (to D.H.-K.); NIGMS R01GM108911 (to A.S.); and Burroughs Wellcome Fund Career Award for Medical Scientists 1006815.01 (to E.M.M.), NIMH R01MH105442 (to E.M.M.), NIMH R01MH102418 (to E.M.M.), NIMH R21MH115392 (to E.M.M.), NINDS/NIA R01NS113141 (to E.M.M.), and Angelman Syndrome Foundation General Research Grant (to E.M.M.). This study was also supported by the Hassenfeld Child Health Innovation Institute at Brown University, the Brown University Flow Cytometry and Sorting Facility, and the Cincinnati Children’s Hospital Medical Center Pluripotent Stem Cell Facility.
Competing interests: A.S. is co-founder of AIchemy, and Scientific Advisory Board Member of Trilogy Sciences and Parthenon Therapeutics.
Footnotes
Data and materials availability: The CS and paired control iPSC lines are available through the NIMH Repository and Genomics Resource (Study 200). Constructs and other cell lines newly described in this study are available from the corresponding author upon reasonable request and can be shared upon implementation of a Uniform Biological Material Transfer Agreement. All data are included in this published article and the accompanying Supplementary Materials files. See data files S6 and S7 for primary data for main text and supplemental figures, respectively, in which experiments had an n < 20.
References and Notes
- 1.Christianson AL, Stevenson RE, van der Meyden CH, Pelser J, Theron FW, van Rensburg PL, Chandler M, Schwartz CE, X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J Med Genet 36, 759–766 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pescosolido MF, Stein DM, Schmidt M, El Achkar CM, Sabbagh M, Rogg JM, Tantravahi U, McLean RL, Liu JS, Poduri A, Morrow EM, Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Ann Neurol 76, 581–593 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, Kroken M, Mattingsdal M, Egeland T, Stenmark H, Sjoholm H, Server A, Samuelsson L, Christianson A, Tarpey P, Whibley A, Stratton MR, Futreal PA, Teague J, Edkins S, Gecz J, Turner G, Raymond FL, Schwartz C, Stevenson RE, Undlien DE, Stromme P, SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet 82, 1003–1010 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Dyck LI, Morrow EM, Genetic control of postnatal human brain growth. Curr Opin Neurol 30, 114–124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barford K, Deppmann C, Winckler B, The neurotrophin receptor signaling endosome: Where trafficking meets signaling. Dev Neurobiol 77, 405–418 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yap CC, Winckler B, Harnessing the power of the endosome to regulate neural development. Neuron 74, 440–451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casey JR, Grinstein S, Orlowski J, Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 11, 50–61 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Ohgaki R, van IJzendoorn SC, Matsushita M, Hoekstra D, Kanazawa H, Organellar Na+/H+ exchangers: novel players in organelle pH regulation and their emerging functions. Biochemistry 50, 443–450 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Ouyang Q, Lizarraga SB, Schmidt M, Yang U, Gong J, Ellisor D, Kauer JA, Morrow EM, Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 80, 97–112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrington AW, St Hillaire C, Zweifel LS, Glebova NO, Philippidou P, Halegoua S, Ginty DD, Recruitment of actin modifiers to TrkA endosomes governs retrograde NGF signaling and survival. Cell 146, 421–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brodlie M, Haq IJ, Roberts K, Elborn JS, Targeted therapies to improve CFTR function in cystic fibrosis. Genome Med 7, 101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozdinler PH, Macklis JD, IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat Neurosci 9, 1371–1381 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Kunisato A, Wakatsuki M, Shinba H, Ota T, Ishida I, Nagao K, Direct generation of induced pluripotent stem cells from human nonmobilized blood. Stem Cells Dev 20, 159–168 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Gupta P, Li YR, Upf proteins: highly conserved factors involved in nonsense mRNA mediated decay. Mol Biol Rep 45, 39–55 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Kim YK, Furic L, Desgroseillers L, Maquat LE, Mammalian Staufen1 recruits Upf1 to specific mRNA 3’UTRs so as to elicit mRNA decay. Cell 120, 195–208 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Lee C, Yashiro S, Dotson DL, Uzdavinys P, Iwata S, Sansom MS, von Ballmoos C, Beckstein O, Drew D, Cameron AD, Crystal structure of the sodium-proton antiporter NhaA dimer and new mechanistic insights. J Gen Physiol 144, 529–544 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kondapalli KC, Hack A, Schushan M, Landau M, Ben-Tal N, Rao R, Functional evaluation of autism-associated mutations in NHE9. Nat Commun 4, 2510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendus-Altenburger R, Kragelund BB, Pedersen SF, Structural dynamics and regulation of the mammalian SLC9A family of Na(+)/H(+) exchangers. Curr Top Membr 73, 69–148 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, Kirwan P, Livesey FJ, Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc 7, 1836–1846 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Khwaja OS, Ho E, Barnes KV, O’Leary HM, Pereira LM, Finkelstein Y, Nelson CA 3rd, Vogel-Farley V, DeGregorio G, Holm IA, Khatwa U, Kapur K, Alexander ME, Finnegan DM, Cantwell NG, Walco AC, Rappaport L, Gregas M, Fichorova RN, Shannon MW, Sur M, Kaufmann WE, Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci U S A 111, 4596–4601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolevzon A, Bush L, Wang AT, Halpern D, Frank Y, Grodberg D, Rapaport R, Tavassoli T, Chaplin W, Soorya L, Buxbaum JD, A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol Autism 5, 54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondapalli KC, Prasad H, Rao R, An inside job: how endosomal Na(+)/H(+) exchangers link to autism and neurological disease. Front Cell Neurosci 8, 172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagahara AH, Tuszynski MH, Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 10, 209–219 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR, A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M, Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A 106, 2029–2034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, Dolmetsch RE, SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ilie A, Gao AY, Reid J, Boucher A, McEwan C, Barriere H, Lukacs GL, McKinney RA, Orlowski J, A Christianson syndrome-linked deletion mutation ((287)ES(288)) in SLC9A6 disrupts recycling endosomal function and elicits neurodegeneration and cell death. Mol Neurodegener 11, 63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prasad H, Rao R, The Na+/H+ exchanger NHE6 modulates endosomal pH to control processing of amyloid precursor protein in a cell culture model of Alzheimer disease. J Biol Chem 290, 5311–5327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brett CL, Tukaye DN, Mukherjee S, Rao R, The yeast endosomal Na+K+/H+ exchanger Nhx1 regulates cellular pH to control vesicle trafficking. Mol Biol Cell 16, 1396–1405 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krogh A, Larsson B, von Heijne G, Sonnhammer EL, Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305, 567–580 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Giron CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Keenan S, Lavidas I, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Nuhn M, Parker A, Patricio M, Pignatelli M, Rahtz M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Birney E, Harrow J, Muffato M, Perry E, Ruffier M, Spudich G, Trevanion SJ, Cunningham F, Aken BL, Zerbino DR, Flicek P, Ensembl 2016. Nucleic Acids Res 44, D710–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warlich E, Kuehle J, Cantz T, Brugman MH, Maetzig T, Galla M, Filipczyk AA, Halle S, Klump H, Scholer HR, Baum C, Schroeder T, Schambach A, Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol Ther 19, 782–789 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, Wells JM, Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516, 400–404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiser A, Sali A, Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol 374, 461–491 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Hunte C, Screpanti E, Venturi M, Rimon A, Padan E, Michel H, Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature 435, 1197–1202 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Shen MY, Sali A, Statistical potential for assessment and prediction of protein structures. Protein Sci 15, 2507–2524 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schushan M, Barkan Y, Haliloglu T, Ben-Tal N, C(alpha)-trace model of the transmembrane domain of human copper transporter 1, motion and functional implications. Proc Natl Acad Sci U S A 107, 10908–10913 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, Ben-Tal N, ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Wolfenden R, Experimental measures of amino acid hydrophobicity and the topology of transmembrane and globular proteins. J Gen Physiol 129, 357–362 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, van der Spoel D, Hess B, Lindahl E, GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlessinger A, Wittwer MB, Dahlin A, Khuri N, Bonomi M, Fan H, Giacomini KM, Sali A, High selectivity of the gamma-aminobutyric acid transporter 2 (GAT-2, SLC6A13) revealed by structure-based approach. J Biol Chem 287, 37745–37756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schlessinger A, Sun NN, Colas C, Pajor AM, Determinants of substrate and cation transport in the human Na+/dicarboxylate cotransporter NaDC3. J Biol Chem 289, 16998–17008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langfelder P, Horvath S, WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang B, Horvath S, A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Langfelder P, Zhang B, Horvath S, Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24, 719–720 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Bruder JM, Monu NC, Harrison MW, Hoffman-Kim D, Fabrication of polymeric replicas of cell surfaces with nanoscale resolution. Langmuir 22, 8266–8270 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Ma L, Ouyang Q, Werthmann GC, Thompson HM, Morrow EM, Live-cell Microscopy and Fluorescence-based Measurement of Luminal pH in Intracellular Organelles. Front Cell Dev Biol 5, 71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galli T, Chilcote T, Mundigl O, Binz T, Niemann H, De Camilli P, Tetanus toxin-mediated cleavage of cellubrevin impairs exocytosis of transferrin receptor-containing vesicles in CHO cells. J Cell Biol 125, 1015–1024 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li D, Herault K, Zylbersztejn K, Lauterbach MA, Guillon M, Oheim M, Ropert N, Astrocyte VAMP3 vesicles undergo Ca2+ -independent cycling and modulate glutamate transporter trafficking. J Physiol 593, 2807–2832 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahon MJ, pHluorin2: an enhanced, ratiometric, pH-sensitive green florescent protein. Adv Biosci Biotechnol 2, 132–137 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. Family 1 to Family 5 CS-related mutations in NHE6 cDNA.
Fig. S2. Sanger sequencing chromatograms of paired control and CS iPSCs.
Fig. S3. Morphological characterization of paired control and CS iPSCs.
Fig. S4. Karyotype analysis of paired control and CS iPSC lines.
Fig. S5. Staining for alkaline phosphatase activity in paired control and CS iPSC lines.
Fig. S6. Immunostaining of paired control and CS iPSC colonies for pluripotency markers.
Fig. S7. FACS analysis of paired control and CS iPSCs for stem cell-specific markers.
Fig. S8. qRT-PCR analyses of pluripotency markers in undifferentiated iPSCs (UD) as compared to hESC-derived embryoid bodies (EB).
Fig. S9. In vitro differentiation potential of paired control and CS iPSCs.
Fig. S10. In vivo differentiation potential of paired control and CS iPSCs.
Fig. S11. Northern blot analysis of Family 4 and Family 5 paired control and CS iPSCs.
Fig. S12. Conservation of the G383 residue across NHE homologs.
Fig. S13. Protocol for neuronal differentiation from iPSCs.
Fig. S14. Neuronal and cortical marker expression in iPSC-derived neurons.
Fig. S15. Gene expression of cell fate markers in iPSC-derived neurons.
Fig. S16. Unsupervised hierarchical clustering of iPSCs and iPSC-derived neurons by gene expression.
Fig. S17. Volcano plot of gene expression changes in iPSCs as compared to iPSC-derived neurons.
Fig. S18. Volcano plots of gene expression changes in CS vs. control iPSCs and iPSC-derived neurons.
Fig. S19. NHE family member gene expression changes in CS iPSCs and iPSC-derived neurons.
Fig. S20. V-ATPase gene expression changes in CS iPSCs and iPSC-derived neurons.
Fig. S21. WGCNA-based network construction and module detection in CS iPSCs.
Fig. S22. Analysis of iPSC-derived neuronal cultures for proliferation and apoptosis.
Fig. S23. Rescue of deficits in neuronal arborization complexity by re-expression of NHE6 in CS lines with frameshift or nonsense mutations but not in the CS missense mutation line.
Fig. S24. Validation of control and CRISPR/Cas9-induced NHE6-null HEK293T cell lines and characterization of endosomal pH.
Fig. S25. Establishment of isogenic, genome-corrected iPSCs for Family 1 using genome editing.
Fig. S26. Calibration curve and flow cytometer profiles for ratiometric analysis of endosomal pH using pH-sensitive and pH-insensitive fluorescent conjugates of transferrin.
Fig. S27. Analysis of endosomal pH in paired control and CS iPSCs from Family 1 to Family 5 using the transferrin method.
Fig. S28. Analyses of endosomal pH in paired control and CS iPSCs from Family 1 to Family 5 using the VAMP3-pHluorin2 method.
Fig. S29. Analysis of endosomal pH in paired control and CS iPSC-derived neurons.
Fig. S30. Establishment of iPSCs with NHE6 mutations and isogenic controls using genome editing of a normal founder line.
Fig. S31. Analysis of endosomal pH in paired isogenic control iPSCs and iPSCs with induced NHE6 mutations using the transferrin method.
Table S1. Characterization of PBMC-derived iPSC lines.
Table S2. Statistical analysis of genes included in the blue module using a Hypergeometric test.
Data file S1. NanoString customized probe set.
Data file S2. Average normalized NanoString count data of gene regulation in iPSCs and iPSC-derived neurons from patients with CS and controls.
Data file S3. By-sample normalized NanoString count data of gene regulation in iPSCs and iPSC-derived neurons from patients with CS and controls.
Data file S4. By-sample raw NanoString count data of gene regulation in iPSCs and iPSC-derived neurons from patients with CS and controls.
Data file S5. Genes in each gene module from the WGCNA-based analyses.
Data file S6. Primary data for main text figures.
Data file S7. Primary data for supplementary figures.