

Summary.
We consider modeling and Bayesian analysis for panel-count data when the termination time for each subject may depend on its history of the recurrent events. We propose a fully specified semiparametric model for the joint distribution of the recurrent events and the termination time. For this model, we provide a natural motivation, derive several novel properties, and develop a Bayesian analysis based on a Markov chain Monte Carlo algorithm. Comparisons are made to other existing models and methods for panel-count data. We demonstrate the usefulness of our new models and methodologies through the reanalysis of a data set from a clinical trial.
Keywords: Frailty, Gibbs sampling, Posterior distribution, Recurrent events
Résumé
Nous considérons la modélisation et l’analyse bayésienne pour des données de dénombrements groupés quand le temps de terminaison de chaque sujet peut dépendre de l’historique des événements récurrents. Nous proposons un modèle semi-paramétrique complètement spécifié pour la distribution jointe des événements récurrents et le temps de terminaison. Pour ce modèle, nous fournissons une motivation naturelle, dérivons plusieurs propriétés nouvelles et développons une analyse bayésienne basée sur un algorithme de Monte-Carlo par chaînes de Markov. Des comparaisons sont effectuées avec d’autres modèles et méthodes pour des dénombrements d’événements groupés. Nous montrons l’utilité de nos nouveaux modèles et méthodologies par la ré-analyse des données d’un essai clinique.
1. Introduction
Grouped recurrent event times data, popularly called panel-count data, occur when a subject, inspected over time only at scheduled time points 0 = a0 < a1 < ⋯ < aJ, may experience repeated occurrences of an important but possibly nonfatal event. Suppose Ni(t) denotes the number of nonfatal events experienced by subject i up to time t. The number of new recurrent events Nij = Ni(aj) – Ni(aj−1) to subject i occuring in the inspection interval Ij = (aj−1, aj] is recorded until the termination time Ti, when the subject is removed from observation either due to an adverse response such as detection of a relapse to malignancy or due to a positive response such as cure. We are assuming that the inspection times are the same for all subjects and Ti is discrete and take values only a1, … , aJ. Let xi be the explanatory variable for subject i. We use the notation D = {Nij : j = 1, … , Ti; xi; Ti : i = 1, … , n} to denote the observed panel-count data.
See Gail, Santner, and Brown (1980), Oakes (1982), Sinha (1993), Scheike and Zhang (1998), Sun and Wei (2000), and the references therein for different examples of panel-count data from various clinical trials and animal experiments. Earlier works on modeling panel-count data often assumed that given the covariate x of the subject, the risk of termination at any time point t does not depend on the past history of nonfatal events . But, this assumption will not be valid when, say, the termination is caused by either a significant positive response (say, cure or discharge from the treatment) or a negative response (such as relapse to malignancy). This risk of termination is often associated with the history of nonfatal events. For the bladder cancer data example (Sun and Wei, 2000) from the Veterans Administration Cooperative Urological Research Group (VACURG), the time-independent covariates available for each patient are a binary variable indicating treatment (1 for treatment thiotepa and 0 for placebo), number of tumors at entry, and the size of the largest tumor at entry. After the surgery to remove his/her primary tumor, each patient was monitored every month during scheduled clinic visit for number of new (since the previous clinic visit) superficial bladder tumors. A patient was terminated from further observation during a particular clinic visit probably due to clinically significant improvement of his/her disease symptoms. As the treatment (thiotepa and placebo) had to be instilled in the bladder, we can expect that a patient who is very prone to superficial bladder tumors is also expected to take longer than usual time to termination. A previous analysis of this data ignored the possible association between the risk of termination and the history of superficial tumors. Unlike the previous analysis, we treat both the event of termination (cure) and the recurrent events of superficial tumors as important responses.
Various methods of panel-count data and recurrent event times data, published so far, have put high emphasis on partially specified models. In such work, the model and associated inferential procedure depend on the particular underlying inferential objective, which can be of the following four types: (1) inference about N(t) (e.g., Sun and Wei, 2000), under the assumption that given x, the process N(t) is independent of T; (2) marginal inference about N(t) unconditional on the past history of events (e.g., Wang, Qin, and Chang, 2001); (3) inference about T given the history of N(·); and (4) inference about the termination modulated process N(t)l[T≤t] (e.g., Ghosh and Lin, 2000), where 1A is the indicator function.
In many applications, it may be desirable to have a fully specified model which can be used for addressing all four objectives together within a unified statistical analysis and also perform prediction, model diagnostic, simultaneous inferences on multiple parameters, etc. Especially when both the nonfatal events and the termination are deemed as important responses, and the termination depends on the history of nonfatal events, addressing each objective through separate analysis may be misleading. The results of the analysis of different partially specified models focusing on different objectives may conflict with each other and may be difficult to unify. Our aim is to develop and analyze a fully specified stochastic model of the joint distribution of the nonfatal events and the termination time. This new model also allows us to understand the prognostic values of the history of nonfatal events for evaluating the risk of termination.
A fully parametric model by Lancaster and Intrator (1998), here onward referred to as the LI model, allows only positive dependence between a subject’s history of nonfatal events and risk of termination. Our model represents a much broader class than the LI model and we can incorporate a negative association between N(t) and T. The data example discussed in this article may require a negative association between N(t) and T. The details of the comparison between our model and the LI model are given later. Recently introduced class of models for panel-count data in animal carcinogenicity experiments (e.g., Dunson, 2000) also do not incorporate the association between T and N(t).
In Section 2, we present a fully specified stochastic model for panel-count data with a dependent termination process. We present novel properties of our model and relationship of our model to other existing models for recurrent events data. Existing Bayesian literature on panel-count data is very limited. See Ibrahim, Chen, and Sinha (2001) for a current literature survey on Bayesian survival analysis. One of our goals is to demonstrate the use and advantages of the Bayesian paradigm for such problems. In Section 3, we present the likelihood of our model and associated Bayesian methods for analyzing such data. In Section 4, our methodology is illustrated with the analysis of panel-count data from the bladder cancer study from Sun and Wei (2000). In Section 5, we conclude our article with some closing remarks.
2. Model for Panel-Count Data
We assume that each Ni(t) is a realization of a compound nonhomogeneous Poisson process (Karlin and Taylor, 1981). Given the subject-specific unobservable frailty wi, the cumulative number of nonfatal events to the subject i, under observation at time t (that is, Ti ≥ t) is given by
| (1) |
where μ0(t) is the baseline cumulative intensity and β1 is the regression parameter of the covariate effect on the conditional intensity in (1). Here w1, … , wn are assumed to be independent Ga(η, η) with common density g(w | η) = ηηwη−1e−ηw/Γ(η). The mean of wi is taken to be unity to assure the identifiability of the model parameters and the variance of wi is κ = η−1. We assume that κ > 0 to exclude the oversimplified situation when, within the same subject, the increments of N(t) in disjoint intervals are independent. A finite mean frailty distribution is warranted here to assure a finite expected number of events within any finite time interval.
The model further assumes that the hazard of Ti at time t depends on the history of point process via the unobservable frailty wi. We use a discretized version of the Cox model (Cox, 1972),
| (2) |
where β2 is the regression parameter and is the discretized baseline conditional hazard rate.
Here, −∞ < α < + ∞ quantifies the nature of dependence between and the hazard of Ti. When α = 0, we have free of and we get the model of Sinha (1993), where the hazard of Ti at time t is assumed independent of . When α > 0, there is positive association between Ni and Ti, that is, given the same value of xi, a subject with a higher rate of nonfatal events has a higher risk of termination than another with a lower rate of nonfatal events. Similarly, α < 0 indicates a negative relationship between Ti and Ni. When α > 1, the variability of termination times are more than the heterogeneity of subjects in terms of their rates of nonfatal events and 0 < α < 1 implies the opposite. Again, α = 1 implies that the heterogeneity among subjects in terms of rates of nonfatal events is the same as the heterogeneity in terms of their termination times. When α = 1 and πj = exp[−ρ{μ0(aj) – μ0(aj−1)}], we get the LI model (Lancaster and Intrator, 1998). This shows that the LI model allows only positive association and has a restrictive assumption of a very precise algebraic relationship between πj and μ0. We investigate some further useful and desirable properties of our model.
Theorem 1.
The conditional risk of termination, , depends on the events history only through the cumulative number of events Ni(aj). For the special case α = 1,
| (3) |
where .
The proof of Theorem 1 is given in the Appendix. Please note that Theorem 1 is very different from Implications 1–3 in Lancaster and Intrator (1998). When α ≠ 1, we can numerically evaluate the quantities and P[Ti = aj | xi, Ti > aj−1], but no closed form expressions of these quantities are available. Unlike the model of Wang et al. (2001), P(Ti = aj | xi, Ti, > aj−1) in our model does not follow a discretized Cox structure. We present the following theorem about the conditional distribution of nonfatal events in any interval given past history of events.
Theorem 2.
The conditional distribution of Nij | xi, , Ti > aj−1 depends on only through the Ni(aj−1). For the case α = 1, this distribution is a generalized negative binomial with conditional expectation
| (4) |
The proof is given in the Appendix. The result in (4) is similar to the property of the model proposed by Oakes (1982). These two theorems also demonstrate that our model is very different from the modulated renewal process of Oakes and Cui (1994). For α ≠ 1, no such simple expression exists for , but, these can be numerically computed given the parameter values. Following the steps similar to the derivation of (4), we can also numerically evaluate E[Nij | xi, Ti > aj−1]. The details are omitted here. These demonstrate that through our fully specified model along with Bayesian analysis, we can estimate different important model quantities of interest using a single unified inference procedure.
3. Likelihood and Posterior
It follows from (1) that, given wi, Nij for j = 1, … , J are independent Poi{wiexp(β1xi)/μj}, where μj = μ0(aj) – μ0(aj−1) and Poi(γ) denotes the Poisson distribution with mean γ. The likelihood based on the sampling distributions of the Nij is
| (5) |
where Mi, is the set of inspection intervals when the subject i is under observation. Using (2), we obtain the likelihood based on the sampling distributions of the T1, … , Tn as
| (6) |
where ti = a(i) ∈ {a1, … , aJ} is the termination time of the subject i and . In (2), we use a discretized Weibull baseline hazard rate , with unknown parameter vector ϕ = (λ, ν). This model on πj allows for a very wide class for the hazard. In principle, it is possible to use a more flexible model with completely unspecified πj.
The observed data likelihood L0(β1, β2, α, ϕ, η | D) is obtained via the multiple integration , where g(w | η) is the joint density of w = (w1, … , wn) given η. We present a semiparametric Bayes survival analysis approach (e.g., Sinha and Dey, 1997; Ibrahim et al., 2001) using a prior process to summarize the prior information on μ0(·) and a prior distribution for the rest of the parameters. We assume that the prior information about μ0(t) is independent of the prior information about the rest of the parameters, π(μ, β, η, ϕ, α) = π(μ) × π(β, η, ϕ, α). As (5) involves μ0 only through (μ1, … , μJ), we only need the joint prior density of the increments (μ1, … , μJ). Using an independent increment gamma process prior (Kalbfleisch, 1978) on μ0(t) we have the prior distribution μj ~ Ga(cγj, c) with independence for j = 1, … , J. The joint prior density of the increments is given by
| (7) |
The hyperparameters, c and γj’s, assumed to be known here, are determined by the prior expectation (mean) and the precision of the prior expectation of μ0(t). But, it is possible to put another level of hierarchy in this model using a hyperprior for (c, γ).
The joint posterior for our model is given by
| (8) |
where is the product of gamma frailty densities and π(β, ϕ, η, α) is given below. Instead of dealing with complicated L0((β1, β2, α, ϕ, η | D), we use popular MCMC tools (e.g., Chen, Ibrahim, and Shao, 2000) to sample from (8) and then use those samples to do the posterior inference. For practical convenience, we further assume that
| (9) |
This assumption is reasonable, as β1 and β2 are the parameters associated with effects of covariate x, respectively, on N(t) and on T. In many practical applications, the priors on β1 and β2 will be very noninformative to balance both the skeptical and the enthusiastic views about the effects of covariates such as assigned treatments. It is also a common practice to assume priors on baseline functions μ and π to be independent of the rest of the parameters. As we can expect to have independent expert opinions about two different types of events N(·) and T, we have the assumption of mutual independence between the set of parameters associated with N(·) and the set associated with T. The prior belief on α, the parameter quantifying the association between the two types of events, is expected to be obtained independently from others. The parameter π(α) is assumed to be N(α0, σ3) with α0 = 0 as we do not know a priori whether the association between the two classes of events should be either positive or negative. For further convenience we use N(β10, σ1) for π(β1), N(β20, σ2) for π(β2), and Ga(λ | A1, B1) × Ga(ν | A2, B2) for the density of π(ϕ). We are choosing the forms of the priors that are flexible, yet facilitate easy-to-sample conditional posteriors at each iteration of the MCMC. We would like to emphasize that if needed, MCMC is very capable of handling more complicated prior structures such as a multivariate π(β, η, ϕ, α) inducing prior dependencies among the parameters.
The full conditional distributions for the MCMC steps are given as follows using the notation [θ | rest] to denote the conditional distribution of ϕ given all other parameters:
.
Here Vij is the indicator variable of whether subject i is examined at time aj. Except for μj and λ, all other conditional densities are not in standard form. Fortunately, the conditional densities for β1, β2, η, v, and α are log concave. So, a readily available software program for the adaptive rejection sampling method (Gilks and Wild, 1992) has been used to sample from these log-concave densities. To avoid lengthening the article, the proofs of their log concavities are omitted. Notice that the conditional density of each wi is proportional to f(w) × h(w), where f(w) is proportional to gamma density function and h(w) is a bounded function. To sample wi’s, we have used a Metropolis-Hastings algorithm with acceptance probability r(wold, wnew) = min[{h(wnew)/h(wold)}, 1]. Here, wnew is the candidate value simulated from f(w) and wold is the current value of wi at this iteration. In particular we have used 10 Gibbs chains with 20,000 iterations in each chain. To monitor the convergence of MCMC, we have checked that the estimated scale reduction factor of Gelman and Rubin (1992) is less than 1.2 for each of the parameters. This is a very popular and easy method to monitor the convergence of the MCMC algorithm. We have also checked several other convergence diagnostics of the MCMC using the publicly available software package CODA (Best, Cowles, and Vines, 1995).
4. Analysis of the Bladder Cancer Data
We reanalyze the bladder cancer data from the VACURG (Sun and Wei, 2000), allowing a possible association between the history of superficial tumor occurrences and the risk of termination. The data set is available at the website www.blackwellpublishers.co.uk/rss. We use only the binary regression variable treatment in our model. For each patient, some of the monthly clinic visits (before termination) have missing observations as the patients have missed some of their clinic visits. We assume here that patterns of missed clinic visits are noninformative in the sense that the probability of a patient missing a particular clinic visit does not depend on the unobserved number of tumors in that month. This assumption of missing at random (Little and Rubin, 1987) may not be valid for this data example, but the patterns of missing data and methodologies dealing with them are beyond the scope of this article. To accommodate missing counts in some of the months, the set Mi in (5) should contain only those months when subject i has been observed to have his/her scheduled clinic visits.
Hyperprior specification:
We have used zero mean and high variance (1000) for the regression parameters β1 and β2 to reflect our prior opinions of no covariate effects on the rate of nonfatal events as well as on the risk of termination. Because we do not have prior input from any expert, we are using diffuse priors for β1 and β2 and similar diffuse priors later for other parameters. We have noticed that our analysis is not sensitive to the choice of the values of the hyperparameters as long as the priors remain sufficiently diffuse.
For hyperparameters τ = 1/c and γ1, … , γJ associated with the gamma process in (7), we take τ = 5.0 and . These choices imply that the prior guess for μj is and the precision of the prior guess is very low, c = 0.2 (or variability of the prior guess is 5.0). Our choice of the hyperparameters reflects our prior belief, although a very weak one, that the overall rate of superficial tumors is slightly higher than 0.01 per unit time. The selection of such a diffuse prior process allows the conclusion to be driven by the likelihood.
Table 1 presents the results of our data analysis. The posterior estimate of β2 is 0.0427, indicating a slight increase in risk of drop-out due to cure (positive response). There is not much evidence for the treatment effect on termination time because the 95% credible interval for β2 contains zero. There is, however, good evidence that thiotepa reduces the rate of superficial tumors as the 95% credible interval of β1 is (0.538, 2.77). Please note that the posterior standard deviation (0.507) of β1 is around 10% larger than the standard error of the corresponding estimate of Sun and Wei (2000). But, unlike the previous analysis, the standard deviation of the marginal posterior does take care of the additional uncertainty in the posterior estimate of a parameter due to the presence of the rest of the parameters of the model.
Table 1.
Summary of posterior analysis
| Parameter | Mean | SE | 95% Credible interval |
|---|---|---|---|
| β1 | −1.491 | 0.507 | (−0.538, −2.77) |
| β2 | 0.0427 | 0.229 | (−0.401, 0.580) |
| α | −0.0137 | 0.0295 | (−0.081, 0.037) |
| η | 0.1313 | 0.0298 | (0.0817, 0.1968) |
| ν | 2.192 | 0.1849 | (1.778, 2.546) |
| λ | 0.0006 | 0.0005 | (0.0001, 0.0019) |
The posterior mean of α is negative, indicating some evidence of negative association between {N(t)} and T. So, the patients who are more prone to superficial tumors are likely to be terminated later (longer follow-up times). But, the evidence for negative α is not strong as the 95% credible interval for α contains zero. There is also strong evidence that the baseline hazard rate for the termination is increasing over time (that is, ν is positive). The results of our Bayesian analysis are in general agreement with the findings of Sun and Wei (2000).
Model validation:
Model validation diagnostics are very important in Bayesian survival analysis (Sinha and Dey, 1997; Ibrahim et al., 2001). We developed a method for model validation for panel-count data via a Bayesian cross-validated residual called the conditional predictive ordinate (CPO). The cross-validated posterior predictive probability evaluated at the observed data from patient i is given as
| (10) |
where D(−i) is the observed data with the patient i removed, θ = (μ, β1, β2, η, α, λ, ν) is the set of parameters of the model, and . The final expression of the CPO in (10) follows from a result of Gelfand, Dey, and Chang (1992). This expression actually enables us to use the samples from the posterior p(θ | D) to compute the CPO using the following Monte Carlo integration algorithm:
Step 1: Simulate from from p(θ | D) MCMC;
Step 2: For each θl, simulate from p(wi | θl, D) for i = 1, … , n;
Step 3: For l = 1, … , M, compute P(Ni = Ni,obs, Ti = ti | θl) using ;
Step 4: Finally, compute .
As they are probabilities, the CPOs lie between 0 and 1 and a higher value of the CPO implies a better agreement between the model and the observation. Typically, CPOs are plotted against their covariate values to check whether the CPO values have any relationship with the covariates. Figure 1 presents the plot of the CPO of each subject versus the corresponding termination time. Figure 1 also uses separate symbols for 47 subjects corresponding to the placebo group and the rest corresponding to the treatment group. From this plot, there appears to be neither evidence of any possible outlier nor evidence of a treatment-dependent pattern. The pattern of CPOs does not appear to depend on the termination times. This plot suggests that our model fits the data adequately.
Figure 1.
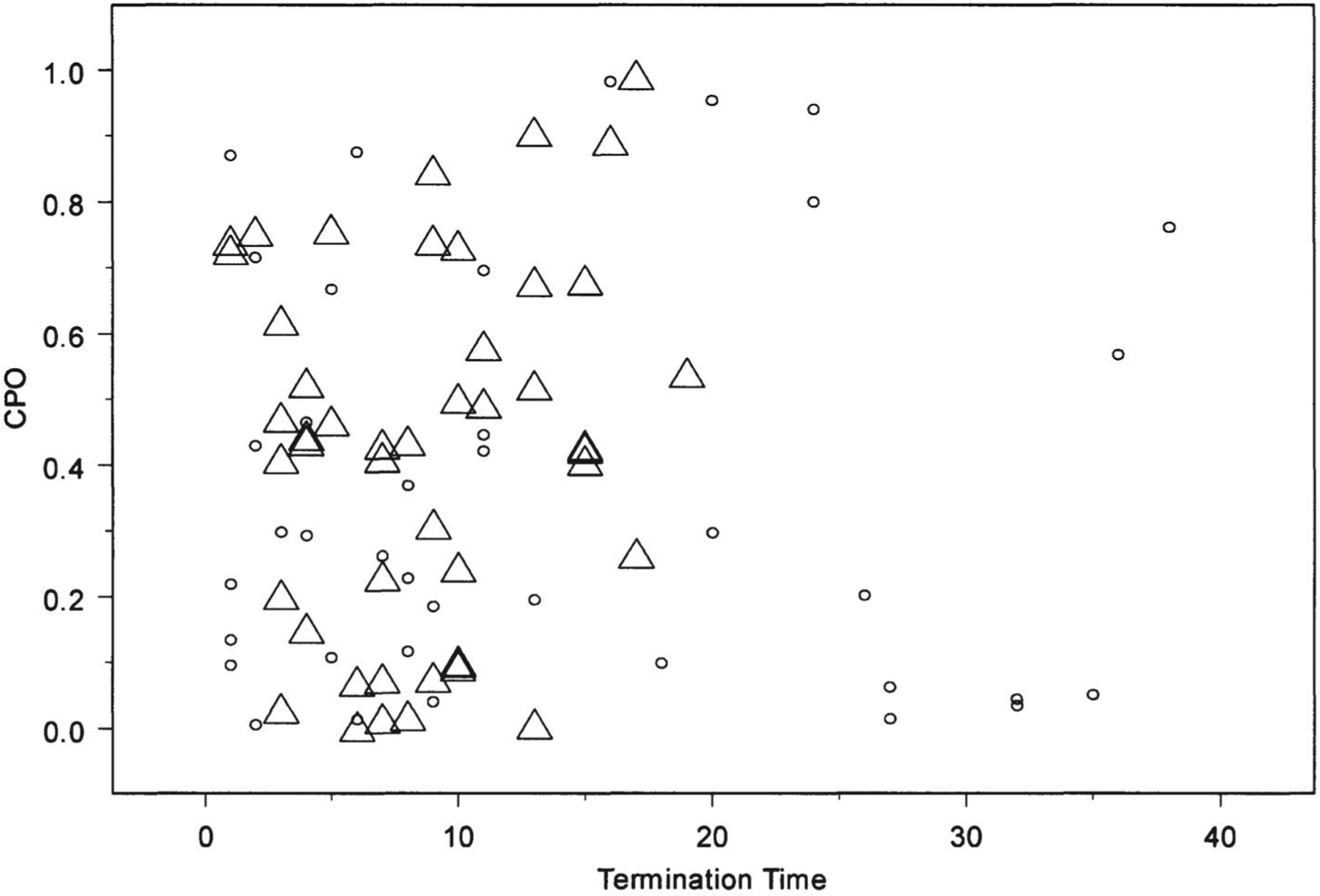
Plot of termination time versus CPO for each patient: ○ for treatment and Δ for placebo.
Prediction:
Please note that one of the major advantages of a Bayesian analysis is that one can compute the predictive distributions of nonfatal events and termination time of a future patient given the values of his/her covariate x. The steps for simulating termination time T and number of events until termination N(T) of a future patient are very similar to the computation of CPO in the last paragraph:
Step 1: Simulate from p(θ | D) using MCMC;
Step 2: For each θl, simulate wl from Ga(w | ηl);
Step 3: For l = 1, … , M, simulate Tl using ;
Step 4: Finally, simulate N(Tl) from Poi{μ0l(Tl)exp(β1lx)wlx}.
These {T1, N(T1)}, … , {TM, N(TM)} are MCMC samples from the bivariate predictive distribution of {T, N(T)} of a future patient with covariate value x. Now these MCMC samples can be used to compute different quantities (e.g., mean, median, standard deviation, prediction interval, etc.) related to the predictive distributions of T and N(T).
5. Remarks
5.1. Censoring of the Termination
In many data examples encountered in practice, the termination time Ti may be subjected to right-censoring. Typically, the censoring time Ci is assumed independent of Ti and we further assume that risk of Ci does not depend on events history. To allow possible right-censoring, we need a straight-forward modification of L2 in (6) as
| (11) |
where the observed ti = a(i) ∈ {a1, … , aJ} is the minimum of termination time Ti and censoring time Ci, and is the indicator of censoring of termination for subject i. Our methodology can accommodate this case with this minor modification of the likelihood and without any major modification of the MCMC tool.
A potentially serious censoring problem may arise when Ti is actually continuous, but only observed as interval censored within the grid intervals a1 < a2 < ⋯ < aJ. Treating Ti as discrete when Ti is in reality only interval censored may give us biased inference, and the level and direction of bias in the inference are, at this point, unknown.
5.2. Comparison to LI Model
The LI model (Lancaster and Intrator, 1998), developed in the context of modeling the joint distribution of hospitalization rate and death in AIDS patients, assumes that the counting process N(t) (hospitalizations) occurs as a compound Poisson process and each hospitalization has a fixed (but unknown) probability of causing the termination (death). Due to the structure of the LI model, only positive association between rate of N(·) and risk of T is allowed. The assumption of discrete T is not very credible when the termination event is death. In Lancaster and Intrator (1998), T was assumed to be equal to one of a1, … , aJ. For a patient with T ∈ Ij = (aj−1, aj], the increment Nj = N(aj) – N(aj−1) is informative about T – aj–1 (length of interval-censored part of T) and ignoring the difference aj – T in the likelihood may bias the inference. Neither the LI model nor our model can describe accurately a situation with continuous termination time, as we need to use an approximation where the termination time is interval censored and the number of hospitalizations in the last observation interval can be informative about the exact termination time within the interval. For many data examples including the VACURG data, the termination event is not necessarily a random consequence of a nonfatal event, and there are cases when a history of higher rates of nonfatal events implies an expected delay in the favorable termination event such as the cure or discharge from hospital. The LI cannot accommodate this kind of negative relationship between recurrent events history and risk of termination.
Acknowledgements
The authors would like to thank the referee and the associate editor for helpful comments that greatly improved the presentation. Dr Sinha’s research was partially supported by NCI grant R01-CA69222. Dr Maiti’s research was partially supported by NSF grant SES-0221857.
Appendix
Proof of Theorem 1.
The conditional discrete hazard of the termination event given the nonfatal events history is given by
where g(w | η) is the Ga(η, η) density of the frailty w. The above integral expressions are derived from the sampling distribution of in (1) and the conditional hazard in (2). The rest of the proof follows from here.
Proof of Theorem 2.
Let us define the vector (N1, … , Nj)—the vector of number of nonfatal events to a subject in first j intervals. Also, note that N(aj−1) = N1 + ⋯ + Nj−1. Then,
Note that the above probability needs to be evaluated only up to the normalizing constant as a function of m. The rest of the proof follows from here.
Contributor Information
Debajyoti Sinha, Department of Biometry, Medical University of South Carolina, Charleston, South Carolina 29425, U.S.A..
Tapabrata Maiti, Department of Statistics, Iowa State University, Ames, Iowa 50011, U.S.A..
References
- Best NG, Cowles MK, and Vines SK (1995). CODA Manual Version 0.30, MRC Biostatistics Unit, Cambridge, U.K. [Google Scholar]
- Chen M-H, Ibrahim JG, and Shao Q-M (2000). Monte-Carlo Methods in Bayesian Computation. New York: Springer-Verlag. [Google Scholar]
- Cox DR (1972). Regression models and life tables. Journal of the Royal Statistical Society, Series B 34, 187–220. [Google Scholar]
- Dunson DB (2000). Models for papilloma multiplicity and regression: Application to transgenic mouse studies. Applied Statistics 49, 19–30. [Google Scholar]
- Gail MH, Santner TJ, and Brown CC (1980). An analysis of comparative carcinogenesis experiments with multiple times to tumor. Biometrics 36, 255–266. [PubMed] [Google Scholar]
- Gelfand AE, Dey DK, and Chang H (1992). Model determination using predictive distributions with implementation via sampling-based methods In Bayesian Statistics, Volume 4, Bernardo JM, Berger JO, Dawid AP, and Smith AFM (eds), 147–167. Oxford: Oxford University Press. [Google Scholar]
- Gelman A and Rubin DB (1992). Inference from iterative simulation using multiple sequences. Statistical Science 7, 457–741. [Google Scholar]
- Ghosh D and Lin DY (2000). Nonparametric analysis of recurrent events and death. Biometrics 56, 554–562. [DOI] [PubMed] [Google Scholar]
- Gilks WR and Wild P (1992). Adaptive rejection sampling for gibbs sampling. Applied Statistics 41, 337–348. [Google Scholar]
- Ibrahim JG, Chen M-H, and Sinha D (2001). Bayesian Survival Analysis. New York: Springer-Verlag. [Google Scholar]
- Kalbfleisch JD (1978). Nonparametric Bayesian analysis of survival time data. Journal of the Royal Statistical Society, Series B 40, 214–221. [Google Scholar]
- Karlin S and Taylor HM (1981). A First Course in Stochastic Processes. London: Academic Press. [Google Scholar]
- Lancaster T and Intrator O (1998). Panel data with survival: Hospitalization of HIV-positive patients. Journal of the American Statistical Association 93, 46–51. [Google Scholar]
- Little Roderick JA and Rubin DB (1987). Statistical Analysis with Missing Data. New York: Wiley. [Google Scholar]
- Oakes D (1982). Frailty models for multiple event times In Survival Analysis: State of the Art, Klein JP and Goel PK (eds), 371–379. Netherlands: Kluwer Academic Publisher. [Google Scholar]
- Oakes D and Cui L (1994). On semiparametric inference for modulated renewal processes. Biometrika 81, 83–90. [Google Scholar]
- Scheike TH and Zhang M-J (1998). Cumulative regression function tests for regression models for longitudinal data. The Annals of Statistics 26, 1328–1355. [Google Scholar]
- Sinha D (1993). Semiparametric Bayesian analysis of multiple event time data. Journal of the American Statistical Association 88, 979–983. [Google Scholar]
- Sinha D and Dey DK (1997). Semiparametric Bayesian analysis of survival data. Journal of the American Statistical Association 92, 1195–1212. [Google Scholar]
- Sun J and Wei LJ (2000). Regression analysis of panel-count data with covariate-dependent observation and censoring times. Journal of the Royal Statistical Society, Series B 62, 293–302. [Google Scholar]
- Wang M-C, Qin J, and Chang C-T (2001). Analyzing recurrent event data with informative censoring. Journal of the American Statistical Association 96, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]