

Abstract
Glioblastoma multiforme (GBM) is the most prevalent and aggressive form of glioma, with poor prognosis and high mortality rates. As GBM is a highly vascularized cancer, antiangiogenic therapies to halt or minimize the rate of tumor growth are critical to improving treatment. In this review, antiangiogenic therapies, including small-molecule drugs, nucleic acids and proteins and peptides, are discussed. The authors further explore biomaterials that have been utilized to increase the bioavailability and bioactivity of antiangiogenic factors for better antitumor responses in GBM. Finally, the authors summarize the current status of biomaterial-based targeting moieties that target endothelial cells in GBM to more efficiently deliver therapeutics to these cells and avoid off-target cell or organ side effects.
Keywords: : antiangiogenesis, biomaterials, cancer, glioblastoma, targeting agents
Background
Glioblastoma
Glioblastoma multiforme (GBM), or glioblastoma, is the most common, aggressive malignant brain tumor, accounting for 57.3% of all gliomas and 14.6% of all primary brain and other CNS tumors [1]. Because of its infiltrative nature, response to treatment and survival rate, GBM is classified as a grade IV CNS tumor according to the WHO classification [2]. The unique characteristics of GBM render it largely incurable and deadly despite scientific and technological advancements. GBM has a low relative survival estimate, with only 6.8% of patients surviving 5 years post-diagnosis [1]. Risk factors for glioblastoma development have been poorly defined except for age [1]. Annual age-specific incidence rates of GBM vary widely but increase with age from 0.18 per 100,000 in children and adolescents aged 0–19 years to a peak of 15.29 per 100,000 in adults aged 75–84 years [1]. The ratio of GBM incidence is higher in males compared with females (1.58:1), and whites have a 1.95 higher risk compared with blacks [1].
GBM is a diffuse astrocytic tumor mostly composed of poorly differentiated neoplastic astrocytes [3]. Ninety-five percent of cases appear in the supratentorial region, and less commonly in the brainstem, cerebellum and spinal cord [1,4]. Morphologically, GBM is known to have an infiltrative growth pattern that makes it difficult to distinguish from normal tissue, high amounts of vascularization and/or necrosis, and this accounts for the poor prognosis associated with the disease [5–7]. In addition to the WHO grade IV CNS tumor classification, GBM is often classified in two different clinical forms: primary and secondary [8]. Primary GBM, which makes up ∼90% of cases of GBM, has a de novo origin, manifesting rapidly, without a prior less malignant precursor lesion [9]. Secondary GBM arises from a slowly progressive, lower grade astrocytoma (WHO grade II or III). Understanding the distinctive molecular mechanisms of GBM is integral to bettering GBM outcomes, as it may respond differently to various treatments [8].
Standard care for glioblastoma
The current standard of treatment for GBM is maximal surgical resection, followed by extensive radiation therapy (RT) and oral temozolomide (TMZ), a chemotherapeutic agent [10,11]. Surgery is a vital component in the treatment of GBM, as it is used as both a diagnostic and a cytoreduction tool [12]. A goal of surgery is to reduce the number of tumor cells; thus, less RT or chemotherapy is needed to kill the remaining cells [13]. Because of the infiltrative nature of GBM tumors, they are difficult to resect without damaging areas of the brain that control speech, motor function and the senses [14].
Ultimately, without follow-up treatment, most glioblastomas recur because it is challenging to resect all tumor cells during resection surgery. Thus, following surgery, a combination of RT and chemotherapy is used to prolong survival. TMZ is an oral, alkylating chemotherapeutic agent that is the first-line drug used to treat GBM [15]. It functions by causing DNA damage that leads to DNA double-strand breaks and cell death [15,16]. Its popularity arose because of its ability to penetrate the CNS and cross the blood–brain barrier (BBB) as well as its relatively low toxicity and bioavailability [17]. Previous studies have shown that RT and TMZ prolong median survival more than RT alone, with minimal increase in toxicity [18,19]. In a clinical trial by Stupp et al., published in 2005, newly diagnosed patients were given RT (2 Gy 5 days per week for 6 weeks) in combination with daily TMZ (75 mg/m2 body surface area) (known as the Stupp protocol) [18]. Stupp et al. [18] and Clarke et al. [11] both stated, however, that combining RT with TMZ leads to a median progression-free survival of only approximately 7 months, and thus new therapies for GBM are still much needed.
An ongoing challenge with regard to the current treatment of GBM involves administering drugs at therapeutic levels to the target site, an issue that is partially due to glioblastomas causing dysfunction of the BBB. This barrier has a unique feature that tightly regulates the movement of ions, molecules and foreign materials between the blood and the brain, ensuring an optimal environment for the brain to function correctly [20]. Although the dysfunction caused by glioblastomas promotes blood vessel leakiness because of the downregulation of tight junctions, such as claudin and occludin, there is an upregulation of transporter proteins; namely, efflux proteins [21]. This paradoxical behavior allows for extravasation of tumor-promoting substances while limiting the entry of antitumorigenic compounds [21].
As previously mentioned, GBM possesses a high degree of infiltration, making it nearly impossible to remove the entire tumor, which leads to recurrence [22] and the need for multiple rounds of varying therapeutic agents and RT. The heterogeneity found in glioblastomas often presents a barrier to the efficacious treatment of these aggressive tumors, which carry stem cell-like characteristics that promote resistance to RT and chemotherapy through various complex mechanisms [21,23]. For example, GBMs can gain resistance to the chemotherapeutic agent TMZ because of varying expression levels of DNA alkylating proteins and DNA repair enzymes such as O6-methylguanine-DNA-methyltransferase [24]. These GBM characteristics often influence treatment and make targeted treatment difficult to develop.
Despite all the existing methods for improving the delivery of systemic drugs and enhancing the delivery across the BBB, the clinical outcome of GBM treatment has still been modest. Thus, investigation of new antiangiogenic drugs and their combinations with other therapies as well as the utilization of biomaterials and targeting agents to increase their efficacy to treat and manage GBM effectively is needed.
Angiogenesis in glioblastoma
Angiogenesis is a normal body process in which new blood vessels are formed from pre-existing ones. However, during tumor growth, angiogenesis aids in the development of an extensive vascular unit to provide the tumor with necessary nutrients and oxygen supply. As a tumor grows beyond normal size, there is a higher demand for nutrients and oxygen, which is rectified by the creation of new vessels [25]. The increase in tumor-associated blood vessels contributes to vessel density, which can be seen at both the site of the tumor and the metastasized location [26]. In addition, the creation of new blood vessels can transport tumor cells to other parts of the body, thereby aiding in the metastasis of a malignant tumor [27]. With an increased network of blood vessels, tumor and cancer cells can easily metastasize [28]; however, metastasis is actually rare in GBM, and thus this is not as relevant for this type of tumor [29]. Tumor endothelial cells that line the blood vessels surrounding and supporting the tumor site also play an essential part in tumor progression. The tumor site induces proliferation of these surrounding endothelial cells, which leads to the growth of the tumor alongside a more extensive vasculature [30]. GBM is characterized as highly vascularized, and GBM tumors have shown the ability to sprout vessels from pre-existing blood vessels to create a neurovascular unit that, in part, consists of endothelial cells [31]. Since GBM tumor cells as well as peritumoral cells encourage endothelial proliferation [32], targeting proliferating endothelial cells in charge of angiogenesis during GBM growth with an antiangiogenic agent may halt or minimize the rate at which the tumor grows and develops. In doing so, this can provide a more effective treatment for GBM. The cells that support tumor growth do not develop drug resistance as easily compared with cancer cells, as they are genetically stable and less likely to mutate [33].
In this review, the authors discuss antiangiogenic therapies that have been explored for the treatment of GBM. The current standard of treatment for GBM is maximal surgical resection, followed by extensive RT with TMZ; however, outcomes remain modest. The authors explore the range of antiangiogenic therapies, including small-molecule drugs, nucleic acids such as plasmid DNA, siRNA and shRNA and peptides and polymers that have been used for the treatment of GBM. The authors also evaluate applications that combine antiangiogenic agents with standard chemotherapy and RT regimens as a strategy to sensitize tumors to these therapies. The authors further explore how the local or systemic delivery of antiangiogenic agents can be enhanced with the use of biomaterials that enable sustained release and protection of antiangiogenic therapeutics, which can in turn increase the bioavailability and bioactivity of these therapeutics for better antitumor responses in GBM. The authors' findings highlight the importance of antiangiogenic treatment approaches that utilize biomaterials, as tumors in the brain are often localized near vessels and nervous tissue, and there are several barrier systems in the brain that interfere with the delivery of drugs to these areas. Finally, the authors summarize the current status of biomaterial-based moieties that target endothelial cells in GBM to more efficiently deliver therapeutics to these cells and avoid off-target cell or organ side effects.
Several articles have reviewed biomaterial-based therapies for glioblastoma but have not focused on antiangiogenic therapies [34,35]. Clavreul et al. discussed convection-enhanced delivery devices, implantable polymer devices, nanocarriers and cellular vehicles used to deliver antiangiogenic factors in intracranial animal models of GBM but not in subcutaneous models [36]. In this review, the authors assess antiangiogenic therapies for GBM that are delivered systemically by themselves (i.e., orally in animal feed), locally using implantable biomaterials (i.e., wafers, disks, nanofibers) or systemically or locally using injectable biomaterials (i.e., polymeric nanoparticles [NPs] and microparticles, lipid-based NPs/liposomes and nonviral gene-delivery vectors) and were evaluated in either a subcutaneous or intracranial model. Several articles have reviewed targeting moieties for glioma but were focused on those targeting glial cells [37,38]. Sakurai et al. reviewed targeting ligands to endothelial cells for different cancers, including glioma [39]. To the authors' knowledge, this is the first review that provides a focused and in-depth evaluation of the application of targeting moieties/ligands for endothelial cells specifically for the treatment or assessment of GBM.
Table 1 lists the antiangiogenic agents that have been evaluated for the treatment of glioblastoma. Figure 1 is a schematic that summarizes the different applications that have utilized antiangiogenic agents, combination therapies that have been investigated, biomaterials that have been applied and tumor models that have been utilized to test these therapies.
Table 1. . Antiangiogenic therapies that have been investigated for the treatment of glioblastoma.
| Antiangiogenic factor | Combined with | Biomaterials | In vivo model | Outcome | Ref. |
|---|---|---|---|---|---|
| Small-molecule drugs | |||||
| Minocycline (local) | Radiotherapy or TMZ (chemotherapy, systemic) | pCPP:SA | Rat intracranial 9L GBM model | Minocycline delivered locally potentiates the effects of both radiotherapy and oral TMZ in increasing median survival | [40] |
| Minocycline (local) | BCNU (chemotherapy, systemic) | pCPP:SA | Rat intracranial 9L GBM model | The combination of intracranial minocycline and systemic BCNU extended median survival compared with systemic BCNU alone | [41] |
| Minocycline (local or systemic) |
BCNU (chemotherapy, systemic) | EVAc disk | Rat intracranial C6 GBM model | Treatment with a combination of minocycline delivered locally in a controlled-release polymer and systemic BCNU 5 days after tumor implantation resulted in an extension of median survival time compared with BCNU alone | [42] |
| Docetaxel (systemic) | PEG-PCL nanoparticles with IL-13 peptide and RGD targeting agents (IRNPs) | Mouse intracranial C6 GBM model | Docetaxel-IRNPs displayed best antitumor effect, with a median survival time of 35 days, which was significantly longer than that seen with mono-modified and unmodified nanoparticles | [43] | |
| Sorafenib (local) |
LNCs made from Lipoid S75-3 (soybean lecithin with 70% phosphatidylcholine and 10% phosphatidylethanolamine) and Kolliphor HS15 (mixture of free PEG 660 and PEG 660 hydroxystearate) | Mouse intracranial U87MG GBM model | SFN-LNCs or free SFN decreased the proportion of proliferating cells in the tumor relative to control groups. SFN-LNCs were more effective than free SFN in inducing early tumor vascular normalization | [36] | |
| Nelfinavir or amprenavir (systemic) | Oral with feed | Mouse subcutaneous U87MG Matrigel plugs GBM model (nelfinavir only) |
In vitro: nelfinavir and amprenavir downregulate VEGF and HIF-1 expression In vivo: nelfinavir decreased angiogenesis |
[44] | |
| Combretastatin (local) | BIC (chemotherapy, local) | PLGA nanofibrous membrane | Mouse intracranial C6 GBM model | BICC/PLGA nanofibrous membranes (i.e. BIC combined with combretastatin) decreased malignancy, retarded tumor growth and prolonged survival compared with BIC/PLGA nanofibrous membranes | [45] |
| Nucleic acids | |||||
| Plasmid-encoding secreted HGFK1 gene (pHGFK1) (local) | Radiotherapy (IR) | H1, a cationic co-polymer consisting of low-molecular-weight (600 Da) PEI linked by β-cyclodextrin and conjugated with folic acid Form H1/pHGFK1 nanoparticles (peritumoural injection) |
Mouse subcutaneous U87MG GBM model Mouse intracranial U118 cell model |
H1/pHGFK1 nanoparticles significantly inhibited tumor growth and prolonged survival in tumor-bearing mice and enhanced the antitumoral efficacy of IR | [46] |
| Plasmid-encoding non-collagenous C-terminal globular NC1 domain of type VIII collagen a1 chain, Vastatin (local) | TMZ (chemotherapy, systemic) | PEI600-CyD-Folate (H1), a cationic co-polymer consisting of low-molecular-weight (600 Da) PEI linked by β-cyclodextrin and conjugated with folic acid | Mouse intracranial U8MG or U87-ATR (chemoresistant) GBM model | Enhancing Vastatin expression by intracerebral injection of H1-Vastatin significantly prolonged animal survival, comparable to the effect of endostatin, the most studied endogenous antiangiogenic polypeptide. Synergistic effect in extending survival was detected when H1-Vastatin was administered with TMZ in GBM chemoresistant murine models |
[47] |
| RNAi plasmid targeting MMP-2 (siRNA MMP-2) (local) | PTX (local) | PEI nonviral vector PLGA nanofibrous membrane |
Mouse intracranial U87MG GBM model | PTX/siRNA dual implant significantly enhanced tumor growth inhibition relative to PTX implant only | [48] |
| Plasmid expressing interfering RNA targeting VEGF (shVEGF) (systemic) | Dox (chemotherapy, systemic) |
dtACPP-modified nanoparticles | Mouse U87MG intracranial GBM model | Utilizing dtACPP-modified nanoparticles to co-deliver plasmid expressing interfering RNA targeting VEGF (shVEGF) and Dox (designated dtACPPD/shVEGFDOX) resulted in effective shutdown of blood vessels and cell apoptosis within the tumor | [49] |
| Soluble VEGFR (sFlt-1) and an angiostatin–endostatin fusion gene (statin-AE) with Sleeping Beauty transposon (local) | PEI nonviral vector | Mouse subcutaneous U373 or U87MG GBM model Mouse intracranial U87MG GBM model |
Co-injection of transgenes sFlt-1 and statin-AE showed marked antitumor activity, as demonstrated by reduction of tumor vessel density, inhibition or termination of glioma growth and increase in animal survival Sustained tumor regression of intracranial gliomas was achieved only when statin-AE and sFlt-1 transposons were co-administered with SB-transposase-encoding DNA to facilitate long-term expression |
[50] | |
| Peptides | |||||
| PF-4/CTF, a 23 amino acid CTF of PF-4 (local) | PLGA microspheres | Mouse subcutaneous U87MG GBM model Mouse intracranial U87MG GBM model |
A single injection of microspheres containing PF-4/CTF resulted in reduction in tumor volume, with a significant decrease in angiogenesis and an increase in apoptosis | [51] | |
| Fc-endostatin (local implantation or CED or systemic injection) | TMZ (chemotherapy, systemic) | pCPP:SA wafer | Rat intracranial 9L GBM model | Systemically or locally delivered mFc-endostatin prolonged the survival of rats implanted with intracranial 9L gliosarcoma. This benefit was further enhanced when mFc-endostatin was combined with the oral chemotherapeutic agent TMZ | [52] |
| Bevacizumab (systemic) | PLGA nanoparticles | Mouse intracranial U87MG GBM model | Bevacizumab-loaded PLGA nanoparticles were able to increase the penetration and residence time of bevacizumab in the brain and demonstrated a reduction in tumor growth accompanied by a higher antiangiogenic effect compared with free bevacizumab | [53] | |
| Biomaterials as standalone therapeutics | |||||
| Carbon nanoparticles | Nanoparticles of carbon allotropes (UDD and MW/RF) | In ovo: chorioallantoic membrane chicken embryo U87MG model | UDD and MW/RF nanoparticles reduced tumor mass and volume and inhibited new blood vessel development in cultured GBM tumors | [54] | |
BCNU: Bis-chloroethylnitrosourea; BIC: Bis-chloroethylnitrosourea, irinotecan and cisplatin; BICC: Bis-chloroethylnitrosourea, irinotecan, cisplatin and combretastatin; CED: Convection-enhanced delivery; pCPP:SA: Poly(1,3-bis-[p-carboxyphenoxy propane]-co-[sebacic anhydride]); CTF: C-terminal fragment; Dox: Doxorubicin; dtACPP: Dual-triggered nanoparticle system decorated with an activatable cell-penetrating peptide; GBM: Glioblastoma multiforme; EVAc: Ethylene vinyl acetate copolymer; IRNPs: IL-13 RGD nanoparticles; IR: Ionizing radiation; LNC: Lipid nanocapsule; MW/RF: Microwave/radiofrequency; pCPP:SA: Polyanhydride poly[bis(p-carboxyphenoxy)propane-sebacic acid]; PEG: Polyethylene glycol; PEG-PCL: Polyethylene glycol–poly(ε-caprolactone); PEI: Polyethylenimine; PLGA: Poly[(d,l)-lactide-co-glycolide]; PTX: Paclitaxel; RGD: Arg-Gly-Asp; SFN: Sorafenib; TMZ: Temozolomide; UDD: Ultradispersed detonation diamond.
Figure 1. . Anti-angiogenic therapies that have been investigated for the treatment of glioblastomas.
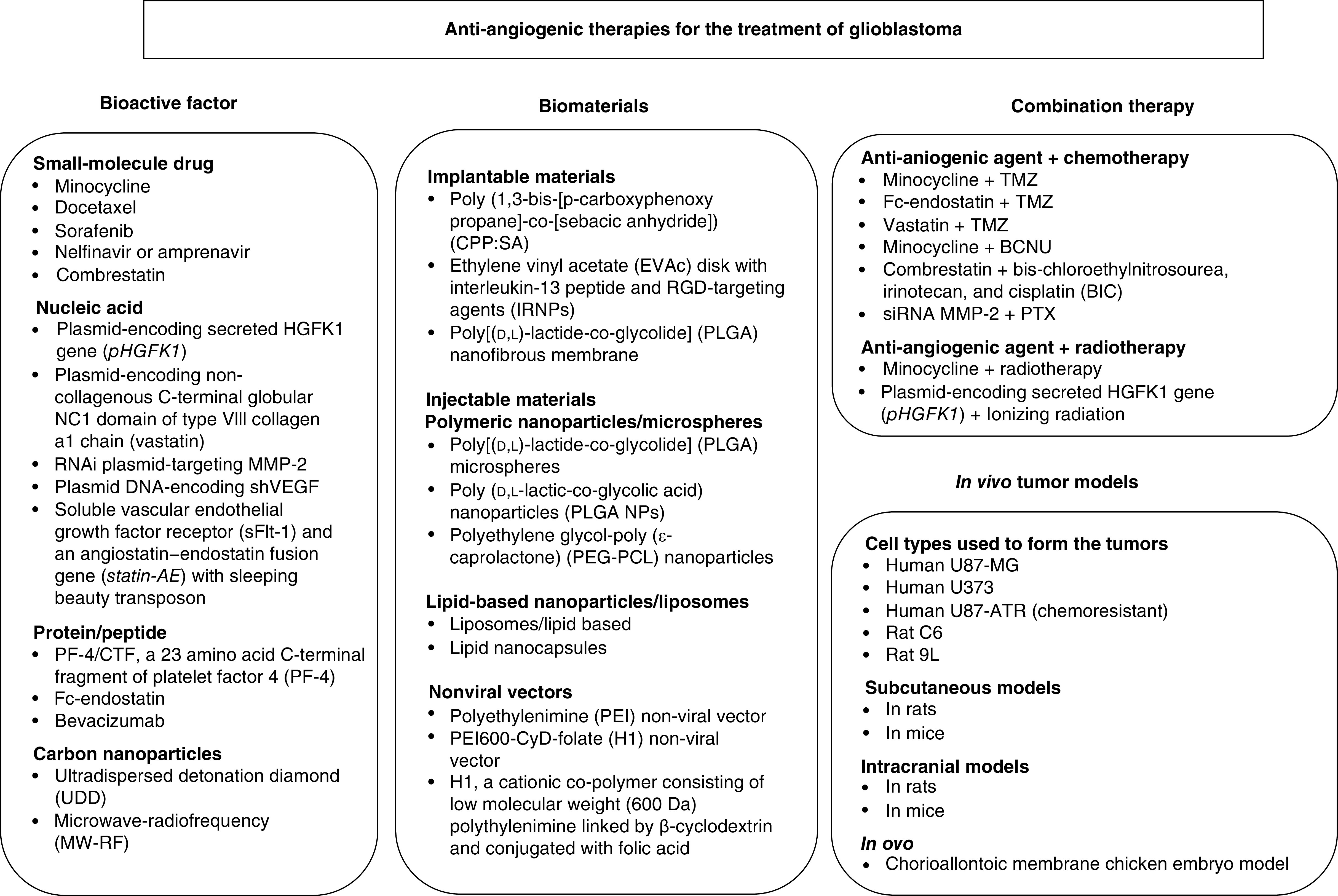
BCNU: Carmustine; PTX: Paclitaxel; Statin-AE: Angiostatin-endostatin fusion protein; TMZ: Temozolomide.
Antiangiogenic bioactive factors for the treatment of glioblastoma
Small-molecule drugs
Small-molecule drugs are small organic compounds that are typically less than 1 kDa compared with biologics such as proteins and nucleic acids, which range from a few kDa to 1000 kDa [55]. Thus, the size of these drugs allows them to more easily cross the plasma membrane of a cell. Small-molecule drugs are, unlike biologics, non-immunogenic, which can increase clearance [55]. The size and non-immunogenicity of small-molecule drugs can increase their availability and distribution in vivo.
Minocycline
Minocycline, a second-generation tetracycline, is a commonly prescribed antibiotic for bacterial infections [40,56]. Minocycline is a bacteriostatic antibiotic that binds to the bacterial 30 S ribosomal subunit, preventing protein synthesis from occurring [57]. Aside from its antibacterial properties, studies have shown that minocycline has antiangiogenic effects on tumors [58] by inhibiting matrix metalloproteinases (MMPs), specifically collagenase and type IV collagenase/gelatinase [59,60]. MMPs are enzymes with proteolytic activity that are vital for angiogenesis, as they provide room for endothelial cell migration and tube formation [61]. MMPs function by degrading and remodeling the extracellular matrix, detaching pericytes from vessels, and through various other mechanisms that contribute to angiogenesis [62]. Thus, the properties of MMPs make them an attractive target for angiogenic inhibition. In addition to its antiangiogenic effects, minocycline is a highly lipophilic drug that is capable of transportation across hydrophobic barriers, and it also has high bioavailability and great tissue penetration [63–65]. The lipophilicity of minocycline allows it to easily traverse the BBB and accumulate in the CNS and cerebrospinal fluid, making it an effective agent for treating neurological disorders [66].
Docetaxel
Docetaxel (DTX), a second-generation taxane agent, is an antineoplastic chemotherapy drug that can induce cell cycle arrest and apoptosis of not only tumor cells but also endothelial cells [43,67]. DTX's main mechanism is binding to β-tubulin in microtubule spindles during the G2/M phase of the cell cycle [68,69]. By binding to the spindles, it stabilizes their structure, preventing their depolymerization and ultimately causing cell death [70]. Moreover, DTX can induce apoptosis by phosphorylating Bcl-2, thus inactivating its antiapoptotic activity in tumor cells [71]. Sweeney et al. showed that this antineoplastic agent possesses the ability to inhibit angiogenesis in vitro and in vivo and that these effects can be fully enhanced via a synergistic combination with anti-VEGF antibodies [72]. One drawback of DTX, however, is its poor aqueous solubility and poor tissue selectiveness, which increases the chance of toxicity and failure to significantly treat gliomas [73–76]. However, studies have used nanomaterials, such as NPs, scaffolds and liposomes, to deliver DTX and overcome the obstacles associated with the utilization of DTX, ensuring a more effective and precise treatment [43,77,78].
Sorafenib
Sorafenib (SFN), a multikinase inhibitor, is an anticancer agent commonly used for the treatment of unresectable hepatocellular carcinoma and advanced renal cell carcinoma [79,80]. It functions by targeting and blocking critical components of tumor growth signaling as well as angiogenesis [81]. Specifically, SFN inhibits the activity of Raf proteins, serine/threonine kinases, which are integral to the Ras/Raf/MEK/ERK signaling cascade [82]. The Ras/Raf/MEK/ERK cascade is key in activating transcription factors and regulating gene expression relating to growth and apoptosis [83,84]. Often, this pathway is mutated or overexpressed in oncogenesis [85,86]; thus, by using SFN to block Raf activity, growth signaling and cell proliferation can be inhibited [87]. Additionally, SFN has demonstrated antiangiogenic activity by inhibiting the proangiogenic receptors VEGFR-1, VEGFR-2 and VEGFR-3 as well as PDGFR [88,89]. Joensuu et al. showed that VEGFR is highly expressed in GBM, making SFN a potential clinical candidate for treatment of the disease [90]. Nonetheless, past studies have shown that SFN has a modest effect, whether as monotherapy or combination therapy, on the treatment of progressive and recurrent GBM [91–95]. Limited results may be due to SFN's poor solubility and oral route of administration, which may reduce its ability to traverse the BBB [36]. Because of SFN's dual mechanism of action, it harbors the potential for treatment of GBM; however, new systems of delivery are needed to improve treatment of GBM.
Nelfinavir/amprenavir
Nelfinavir and amprenavir, both protease inhibitors, are commonly used to treat HIV [44]. Normally, these protease inhibitors block the activity of HIV proteases, preventing the cleavage of viral polyproteins and, hence, production of immature and noninfectious viral particles [96,97]. In 1994, Gupta and Singh demonstrated that protease inhibitors such as amprenavir and nelfinavir can inhibit PI3K/Akt signaling, increasing radiosensitivity of tumor cells [98]. Previous studies closely linked PI3K/Akt signaling pathway to VEGF and HIF-1α expression levels [99,100], which prompted Pore et al. to demonstrate that these HIV protease inhibitors can indeed decrease VEGF and HIF-1α expression and angiogenesis in GBMs [44]. Therefore, nelfinavir and amprenavir could potentially be used as antiangiogenic therapeutic agents for GBM.
Combretastatin
Combretastatins are a group of small stilbenoid molecules that have the ability to downregulate various signaling systems, leading to the inhibition of cell proliferation and angiogenesis [101]. They function by binding at or near the colchicine-binding site, causing the destabilization of the tubulin polymers of the cytoskeleton, leading to morphological changes in endothelial cells and apoptosis [101,102]. Additionally, Su et al. showed that administration of combretastatin A-4 decreases expression of two potent mediators of angiogenesis, VEGF and VEGFR-2, in tumor tissue, thus inhibiting angiogenesis [103]. With this in mind, Tseng et al. investigated the effects of combretastatin A-4 on GBM in combination with three chemotherapeutic agents, bis-chloroethylnitrosourea (BCNU), irinotecan and cisplatin (BIC), from a poly[(d,l)-lactide-co-glycolide] (PLGA) nanofibrous membrane [45]. Together, these agents were able to attenuate the malignancy, retard tumor growth and ultimately prolong survival.
Nucleic acids
Gene therapy is designed to introduce genetic materials (nucleic acids) that have the capacity to correct altered or mutated genes or cause site-specific modifications, such as restoring, increasing or halting gene expression in cells [104]. These genetic materials include plasmid DNA encoding a protein of interest, siRNA, shRNA or microRNA.
Plasmid DNA encoding HGFK1
Plasmid DNA molecules are closed, double-stranded, circular DNA molecules that have become important in gene therapy [105] because of their ability to have therapeutic genes inserted directly into them [106]. Zhang et al. utilized polyethylenimine (PEI)600-CyD (H1) NPs to encapsulate plasmid-encoding secreted HGFK1 [46], which is known to be a potent antiangiogenic factor [107]. Furthermore, HGFK1 has been shown to have a high affinity to MET, a proto-oncogene that stimulates proliferation, mobility and angiogenesis and is frequently upregulated or overactivated in GBM [108–110]. By introducing the NP encapsulated plasmid, Zhang et al. were able to block MET and show its potential to reduce angiogenesis, inhibit proliferation and induce apoptosis in GBMs [46].
Plasmid DNA encoding non-collagenous C-terminal globular NC1 domain of type VIII collagen a1 chain (Vastatin)
Recently, using plasmid DNA technology, Li et al. were able to successfully deliver the non-collagenous C-terminal globular NC1 domain of type VIII collagen a1 chain, Vastatin [47]. Vastatin is an endogenous polypeptide that is part of a family known as collagen-derived antiangiogenic factors [111]. This family is composed of well-known antiangiogenic factors, such as endostatin, arresten and canstatin, that arise from various parent collagen molecules containing NC1 and play various roles in angiogenesis inhibition [112,113]. Previously, Vastatin has been shown to inhibit proliferation and migration of bovine aortic endothelial cells as well as tumor growth in hepatocellular carcinoma and have antiangiogenic activity in ocular neovascularization [111,114,115]. Since Vastatin is derived from type VIII collagen, Li et al. were interested in determining whether Vastatin was effective as a treatment for GBM since it is known that collagen VIII expression is increased in brain tumors [47,116]. This study effectively indicated that plasmid DNA encoding Vastatin induces antiangiogenesis and prolongs survival in GBM [47].
RNAi plasmid targeting MMP-2
Another promising approach in gene therapy pertains to the biological process of RNAi. This process uses siRNAs to block the expression of specific genes, resulting in decreased production of the respective protein [117]. The siRNAs are dsRNAs of approximately 19–22 bp with an overhang of two to three nucleotides on the 3′ end [118,119]. Lei et al. applied this technology to provide new insights into the treatment of gliomas [48]. The researchers utilized a designer RNAi plasmid targeting MMP-2, a vital proteinase needed for both tumor invasion and angiogenesis. This study revealed that inhibition of MMP-2 mRNA and protein expression impeded tumor invasion and tumor-induced angiogenesis [48].
Plasmid-encoding interfering RNA targeting VEGF (shVEGF)
Besides siRNAs, short hairpin RNAs can be used in RNAi applications to inhibit gene expression and protein production. Huang et al. combined RNAi and plasmids to selectively inhibit the expression of VEGF [49]. The researchers successfully showed that the delivery of plasmid expressing shVEGF, in combination with the chemotherapeutic agent doxorubicin (Dox), decreased blood vessel numbers and killed cells within the tumor, thereby reducing tumor growth and increasing median survival.
Sleeping Beauty transposon with soluble VEGFR (sFlt-1) & angiostatin–endostatin fusion protein (statin-AE)
Ohlfest et al. utilized the Sleeping Beauty (SB) transposon to deliver two potent antiangiogenic genes: a soluble VEGFR (sFlt-1) and an angiostatin–endostatin fusion protein (statin-AE) [50]. The SB transposon system is made out of a transposon carrying genes of interest and a transposase enzyme that has the ability to recognize, excise and integrate the gene of interest into genomic DNA in a ‘copy-and-paste' manner [120,121]. SB has been shown to provide long-term gene expression, making it a promising therapeutics delivery system [122,123]. The sFlt-1 is a soluble variant of VEGFR with antagonistic activity toward VEGF [124,125]. Angiostatin and endostatin express antitumor activity individually but work as effectively or even greater together as statin-AR, a fusion gene [126–131]. Ohlfest et al. showed that this combinatorial antiangiogenic gene therapy can sustain tumor regression, eliminate new tumor growth and increase survival significantly.
Proteins or peptides
The market for peptide and protein therapeutics has been growing much faster than the market for small-molecule drugs [132]. Compared with small-molecule drugs, proteins and peptides can bind to their target with multiple points of contact, which makes them highly selective [132]. The basic building blocks of proteins and peptides are amino acids. Compared with proteins, peptides tend to be smaller in size, which makes them great candidates for penetrating tissues and overcoming barriers [133].
C-terminal fragment of PF-4 peptide
Benny et al. designed microspheres loaded with the C-terminal fragment of PF-4 (PF-4/CTF) to investigate its effects on glioblastomas [51]. PF-4 is a potent chemokine that inhibits endothelial cell proliferation, migration and angiogenesis through receptor-dependent and -independent mechanisms [98,134,135]. PF-4/CTF, also known as PF447-70, is a modified fragment from the C-terminus of its parent peptide, PF-4 [136]. Although smaller, this domain retains the antiangiogenic effects of the parent peptide and is able to inhibit angiogenesis induced in vitro by either FGF-2 or VEGF [137–139]. Benny et al. delivered PF-4/CTF-loaded microspheres to glioblastoma, resulting in a 65% reduction in tumor volume, significant decrease in angiogenesis and increase in apoptosis. These results add to previous data provided by Bello et al. that PF-4/CTF administration can delay malignant glioma growth [140].
Endostatin peptide
Endostatin, a 20-kDa CTF of collagen XVIII, is a well-known antiangiogenic factor first isolated in 1997 that successfully stopped the growth of Lewis lung cancer metastases and blocked angiogenesis [141]. It has also proven to be effective in other types of tumors, such as renal cell carcinoma and gliomas [131,142]. Although promising as an antiangiogenic therapeutic agent, its mechanism of action and effects are complex and have yet to be clearly defined [143–145]. A shortcoming is that the previously used endostatin had a short half-life and lacked four amino acids at the NH2 termini, which are important for antitumor activity [146–148]. To improve these deficiencies, Lee et al. linked an antibody (IgG) Fc domain to a molecule of endostatin, constructing ‘Fc-endostatin’, which resulted in improved half-life and efficacy [149]. Grossman et al. utilized this Fc-endostatin molecule to explore its efficacy in treating gliomas using systemic or local delivery [52]. Their study showed that administration of Fc-endostatin, whether by systemic or local delivery, prolonged survival. Hence, using this fused molecule opens a new door to treating such aggressive tumors.
Bevacizumab
In 2009, bevacizumab (Avastin®) was approved by the US FDA as a single agent for second-line treatment of progressive GBM in patients who do not response to standard treatment such as TMZ [53,150]. It is a humanized anti-VEGF monoclonal antibody aimed at preventing the activation of VEGFR tyrosine kinases [13,151]. Bevacizumab exerts its effect by blocking the interaction between VEGF-A and the VEGFRs VEGFR-1 and VEGFR-2 on endothelial cells [152]. This reduces angiogenesis, which subsequently reduces the growth of tumor cells expressing VEGFRs and disrupts the vasculature niche, causing 'starvation' of the tumor [22]. As demonstrated by Sousa et al., there is a reduction in GBM tumor growth, accompanied by higher antiangiogenic effect, when delivering bevacizumab [53]. These effects have been shown to improve patient outcomes. Studies have also shown that bevacizumab can be started either early or late in recurrent GBM without a decrease in efficacy [153]. The current treatment regimen for bevacizumab is intravenous administration every 2 weeks [53,154]. However, this administration regimen has made it difficult for patients to adhere to therapy, emphasizing the need for alternative antiangiogenic treatments [53]. In addition, GBM tumors often have the ability to continue to progress during bevacizumab treatment, as the tumors can become resistant to bevacizumab due to being able to adapt by using other proangiogenic pathways besides VEGF-A [154]. To overcome bevacizumab resistance, other angiogenic factors and signaling pathways need to be targeted in combination with bevacizumab treatment [155]. Thus, complementary approaches, such as the combination of these inhibitors with agents targeting alternative mechanisms of blood vessel formation, are urgently needed.
Biomaterials as standalone therapeutics
Besides small-molecule drugs, nucleic acids, proteins and peptides, biomaterials themselves can be bioactive and have antiangiogenic properties. Past studies have indicated that NPs of carbon allotropes are highly bioactive, influence gene expression and exert antiangiogenic properties on blood vessels [54,156,157]. Grodzik et al. utilized two NPs of carbon allotropes, ultradispersed detonation diamond (UDD) and microwave/radiofrequency (MW/RF), to observe their effects on GBM [54]. Results indicated that UDD NPs significantly decreased VEGF and FGF-2 expression, whereas MW/RF NPs reduced only VEGF expression. The difference between these two types of carbon NPs could be due to the presence of different functional groups on their surfaces, which affects binding to domains of the factors or their receptors. It is known that both VEGF and FGF-2 are proangiogenic factors that play a role in new blood vessel formation, so by reducing these factors, angiogenesis is inhibited [158–160]. Ultimately, Grodzik et al. determined that UDD and MW/RF carbon NPs indeed inhibited angiogenesis and decreased tumor mass and volume in GBMs. The mechanism of action of these NPs remains unclear; however, it is believed that these NPs function by inhibiting angiogenesis by binding to VEGF and FGF-2 or their receptors when levels are elevated [54,157], inhibiting their actions.
Combination therapies
Antiangiogenic therapy & chemotherapy
As previously mentioned, angiogenesis is a hallmark of glioblastoma, making it an excellent target for clinical treatment through its inhibition. However, generally, antiangiogenic drugs have only modestly improved survivability. Thus, significant efforts and investments have been made to develop more effective ways to treat GBM, such as combining antiangiogenic drugs with chemotherapy and RT.
A frequent chemotherapeutic agent used in the standard treatment of GBM is TMZ. It is concomitantly used with RT and can penetrate the CNS with relatively low toxicity [17]. Because of TMZ's promising outcomes and properties, multiple studies have combined it with antiangiogenic agents to observe the potential of dual therapy. Bow et al. demonstrated that the delivery of minocycline potentiated effects of oral TMZ and increased median survival (29 days) when compared with single-agent TMZ (21 days), minocycline (19 days) and control groups (14 days) [40]. Two other common antiangiogenic agents that have been used in combination with TMZ are Fc-endostatin and Vastatin. Grossman et al. showed that Fc-endostatin itself enhanced survival to 15 days compared with control animals (13 days), but when used with TMZ, survival was increased to 28 days [52]. Li et al. demonstrated that local delivery of Vastatin synergized with TMZ and restored the sensitivity of chemoresistant tumors [47]. Alone, Vastatin significantly increased median survival to 34 days relative to untreated mice (23 days), whereas the combination of Vastatin and TMZ further increased it to 54 days.
BCNU (carmustine) is another chemotherapeutic agent that is FDA-approved for the treatment of newly diagnosed high-grade glioma and recurrent glioblastoma [161]. Several groups have utilized BCNU in conjunction with antiangiogenic agents to demonstrate efficacy in the treatment of glioblastoma [41,42,162]. Weingart et al. demonstrated that the delivery of minocycline locally or systematically did not have a positive effect on survival [42]. However, when local delivery of minocycline from a polymer was combined with systemic BCNU, median survival was extended by 93% compared with systemic BCNU alone. Frazier et al. also used minocycline, but with a pCCP:SA wafer for delivery [41]. In this study, minocycline implantation prolonged survival (19 days) when compared with the control group (14 days), with even more favorable effects when minocycline was combined with BCNU (42 days). Sonabend et al. utilized a unique combination consisting of the IL-12 gene and BCNU [162]. This gene/drug combination proved to be potent, enhancing survival in 100% of the mice to 95 days after treatment compared with IL-12 (25% long-term survival) and BCNU (75% long-term survival) alone [162]. Other chemotherapeutic agents, such as BIC, Dox and paclitaxel (PTX), have also demonstrated promising effects when combined with various antiangiogenic agents. Tseng et al. loaded antiangiogenic combretastatin with BIC into PLGA nanofibers and surgically implanted it onto the brain surfaces of glioma-bearing rats [45]. BIC and combretastatin improved survival time (86.50 days) when compared with BIC alone (60.00 days) and the control groups (22.87 days) [45]. Additionally, BIC and combretastatin attenuated malignancy and retarded tumor growth. In another study, an RNAi plasmid targeting MMP-2 was combined with PTX and concurrently given to mice. This gene/drug dual-delivery system significantly enhanced tumor growth inhibition by a factor of about 30-fold relative to the drug implant only [48]. Similarly, a plasmid expressing interfering RNA targeting VEGF was used with the agent Dox, and the combined treatment yielded an effective shutdown of blood vessels and cell apoptosis within the tumor [49]. These studies strongly suggest that synergistic therapy can achieve significantly better results than monotherapy. Thus, combining antiangiogenic agents with chemotherapeutic drugs provides a potential strategy and regimen for treating GBM.
Antiangiogenic therapy & RT
RT is the current, standard adjuvant approach for post-resection glioblastoma and primary treatment of unresectable tumors [163], demonstrating great efficacy during post-resection [164–166]. Although RT has been proven to increase survival, novel techniques to further enhance RT, such as the concurrent delivery of antiangiogenic agents with RT, need to be developed.
Aside from combining minocycline with TMZ, Bow et al. also observed the effects of minocycline with RT [40]. The researchers showed that minocycline potentiated the effects of RT, just as it did with TMZ. The median survival with RT alone was 31 days, whereas those treated with the dual therapy of minocycline and RT had an increased median survival of 74 days. Another approach delivered a plasmid-encoding secreted HGFK1 gene (pHGFK1) locally with ionizing radiation (IR) [46]. The median survival time of mice treated with IR, pHGK1 and IR + pHGFK1 was 65, 64 and 78 days, respectively. When compared with other groups, the IR + pHGFK1 group had significantly prolonged survival time. The pHGFK1 sensitized GBM to IR treatment, thus improving the outcome. The therapeutic benefits of RT have significantly improved throughout the decades; however, to further improve its efficacy, dual therapy with RT and antiangiogenic agents needs to be explored to maximize effectiveness.
Biomaterials utilized in antiangiogenic therapies
Synthetic and natural biomaterials can be tailored to the application of interest by altering the biomaterials' properties. This makes biomaterials a versatile component of glioblastoma therapies that can be utilized to prevent off-target site effects, increase local bioavailability, control drug release and enhance bioactivity of the drug by protecting it from degradation.
Implantable biomaterials
Compared with systemic delivery, local delivery of therapeutics decreases systemic toxicity and increases the bioavailability of the drug. By delivering drugs locally, the drug concentration at the site of the tumor can be maximized, and nontarget systemic exposure and organ toxicity can be minimized. Furthermore, local delivery can increase the efficacy of the drug by bypassing the harsh environment encountered in the long journey the drug has to take to reach the targeted site of interest. Local delivery of drugs can be achieved by implantable biomaterials such as scaffolds, wafers, disks and membranes.
Poly(1,3-bis-[p-carboxyphenoxy propane]-co-[sebacic anhydride]) wafer
Polyanhydrides such as poly(1,3-bis-[p-carboxyphenoxy propane]-co-[sebacic anhydride]) (pCPP:SA) can degrade into nontoxic metabolites [167]. This type of polymer degrades by surface erosion, and thus drug release is based on a zero-order kinetic rule [168,169]. Hydrolysis rates can be controlled by the content of the CCP monomer (i.e., more CPP results in slower degradation) of the polymer, which can be varied from a few days to several years, making this a versatile polymer for drug delivery [170].
The most extensively used polyanhydride is pCPP:SA [170]. Among the implantable devices made from pCPP:SA that have been investigated thus far, Gliadel®, a commercial implant for the delivery of the chemotherapy drug BCNU, is the most advanced biomaterial studied for the treatment of brain gliomas [171]. It has been approved by the FDA for the treatment of newly diagnosed high-grade malignant glioma with surgery and RT as well as recurrent GBM with surgery. The pCPP:SA biodegradable polymers are made by melt condensation [172]. Frazier et al. used pCPP:SA polymer with 50% minocycline in combination with oral BCNU [41]. To make the biomaterial polymer, the drug and polymer were dissolved in methylene chloride and then dried in a vacuum desiccator. The polymer was then molded by compression molding into a 3 × 3 × 1-mm disk weighing 10 mg. The polymer released 26% of the drug on day 1 and 1.7% of the drug per day after day 1. Bow et al. also used this polymer loaded with 50% minocycline and tested it in combination with oral TMZ or RT [40]. Grossman et al. employed the polymer with 40% (w/w) mFC-endostatin, using a similar protocol, to produce 14-mg wafers [52]. These wafers were tested alone or in combination with oral TMZ. Interestingly, the researchers found that the delivery of mFC-endostatin either systemically or locally with the polymer had the same effect on prolonging survival; thus, local delivery of the bioactive factor with the wafers was no more efficient in the treatment of glioblastoma.
PLGA nanofibrous membrane
The copolymer PLGA is a polyester often used as a biomaterial for the delivery of bioactive factors for many applications, including cancer, because of its inert properties, great biocompatibility, biodegradability and versatility [173,174]. Furthermore, the degradation rate of PLGA can be optimized by altering the lactic-to-glycolic acid ratio, molecular weight and end cap groups. PLGA is a polymer that has been used extensively for the delivery of bioactive factors, including drugs, proteins and genes. Lei et al. utilized electrospinning to fabricate a PLGA nanofibrous membrane to deliver RNAi plasmids targeting MMP-2 [48]. The PEI/DNA NPs in phosphate-buffered saline were incorporated into the PLGA solution by forming an emulsion using a sonicator. The emulsion was then electrospun, and 3 × 1-mm fiber disks were obtained by punching the fiber mats obtained. The researchers demonstrated that the release of PEI/DNA NPs was mainly controlled by pore formation in the electrospun fiber matrix that resulted from the polyethylene glycol (PEG) (incorporated as porogen) leakage. The constant leakage rate of PEG resulted in a near constant release rate of the DNA. The disk released less than 20% DNA in 6 weeks, indicating that the disk was able to sustain the release for a prolonged time frame.
Ethylene vinyl acetate copolymer disk
Ethylene vinyl acetate copolymer is a non-biodegradable polymer that has been clinically used for birth control applications [175] and the treatment of glaucoma [176]. Ethylene vinyl acetate copolymer disks or cylinders are usually fabricated by solvent casting and then allowing the polymer to dry. Weingart et al. used ethylene vinyl acetate copolymer 3 × 3 × 1-mm disks to administer minocycline with or without systemic delivery of BCNU [42]. The material was first washed in absolute ethyl alcohol to extract the inflammatory antioxidant butylhydroxytoluene [177]. The solution of drug in methylene chloride was poured into glass cylindrical molds. The polymer contained 50% minocycline by weight. The polymer was cut into a 3 × 3 × 1-mm disk for the intracranial study. The polymer was able to release minocycline in vitro for 90 days. The researchers demonstrated that the local delivery of minocycline with the polymer in combination with systemic BCNU improved median survival by 93% compared with systemic BCNU alone; however, systemic minocycline in combination with systemic BCNU was no different compared with systemic BCNU alone, indicating the importance of local delivery of the antiangiogenic factor with the biomaterial.
Injectable biomaterials
Compared with implantable devices, smaller biomaterials, such as NPs, microparticles and liposomes, can be delivered by stereotactic injection to the tumor site because of their size and shape. Stereotactic injection still requires a craniotomy, but it is not as invasive as craniotomy to implant devices, which may damage surrounding brain tissue. These injectable biomaterials include polymeric NPs and microparticles, lipid-based NPs/liposomes and nonviral-based polyplexes for gene delivery.
Polymeric NPs and microparticles
Besides being applied to make scaffolds, PLGA has been extensively used to make microparticles and NPs. Sousa et al. fabricated bevacizumab-loaded PLGA microparticles using a W/O/W emulsion solvent evaporation method [53]. The researchers demonstrated that ∼14.0% of bevacizumab released after 7 days. The intranasal delivery of the PLGA microsphere allowed for direct administration of the drug to the CNS, bypassing the BBB. This prevented off-target organ toxicity and increased bioavailability at the targeted side. Furthermore, Sousa et al. concluded that the increase in resident time of the drug at the brain could be attributed to the protection of the antibody by the biomaterial. Benny et al. utilized PLGA microspheres to deliver the 23 amino acid CTF of PF-4, PF-4/CTF [51]. The microspheres were again fabricated using the W/O/W emulsion solvent evaporation method. The PLGA microspheres were able to maintain morphological integrity and release the protein continuously for 30 days. This demonstrated that local delivery of the peptide with the PLGA microspheres was much more effective than systemic delivery of the drug. Furthermore, the microsphere prolonged biological activity by acting as a protective system for the peptide.
Gao et al. utilized PEG–poly(ε-caprolactone) (PEG-PCL) NPs loaded with DTX that had IL-13 peptides to target GBM cells and Arg-Gly-Asp (RGD) (IL-13 RGD nanoparticles, IRNPs) to target the neovasculature [43]. The particles were prepared by an emulsion/solvent evaporation method, using a sonicator to make the emulsion. PCL is a biodegradable polymer approved for human use for drug delivery applications by the FDA and widely used in drug delivery applications [178,179,180]. PEG is a hydrophilic polymer that is often conjugated to other polymers or biomaterials to increase hydrophilicity. Xin et al. showed that there was a higher accumulation of methoxy PEG-PCL than conventional NPs at the intracranial tumor [181]. This occurs as a result of the enhanced permeability and retention effect, where NPs spontaneously accumulate in the pathological area because of the hydrophilic surface and ability to stay longer in the circulation. Furthermore, the hydrophilic PEG shell prevents aggregation of NPs and binding between particles and plasma protein, thereby avoiding recognition and removal by the mononuclear phagocyte system by preventing opsonization. Thus, PEG-PCL NPs are a promising biomaterial for the treatment of glioblastoma.
NPs of carbon allotropes have been used in antiangiogenic therapy. Grodzik et al. investigated two NPs of carbon allotropes, UDD and MW/RF, to inhibit angiogenesis in GBM. The material itself is the angiogenic factor and thus was not loaded with any drugs [54]. The authors discussed the mechanism earlier.
Lipid-based NPs/liposomes
Clavreul et al. utilized lipid nanocapsules (LNCs), as they are associated with several advantages, including fabrication by a phase inversion process using generally safe excipients without the use of organic solvents; high stability, payload and encapsulation efficiency; ease of scaling up production; and ability to modify the surface with targeting moieties [82,182]. The researchers loaded SFN into LNCs made from Lipoid S75-3 (soybean lecithin with 70% phosphatidylcholine and 10% phosphatidylethanolamine) and Kolliphor HS15 (mixture of free PEG 660 and PEG 660 hydroxystearate). Nanocarriers can help overcome the poor solubility of SFN in water as well as unwanted side effects [183]. Most of the SFN (i.e., 80% after 5 days) remains associated with the LNCs, which allows time for cells to capture the loaded nanocapsules before the drug is released [183]. The SFN-loaded LNCs were more effective in inducing early tumor vascular normalization than free SFN, which could be due to the ability of LNCs to improve drug retention within the tumor [184].
Huang et al. developed a dual-triggered nanoparticle system decorated with an activatable cell-penetrating peptide (dtACPP) [49]. This peptide can be triggered by both lowered tumor extracellular pH and MMP-2. The particles co-delivered plasmid expressing interfering RNA targeting VEGF (shVEGF) and Dox simultaneously. The dtACPP was conjugated to the surface of the nonviral vector dendrigraft poly-L-lysine to form the NPs to deliver shVEGF-DOX. This dual-delivery system with an activatable cell-penetrating peptide was able to effectively shut down blood vessels and result in cell apoptosis in the tumor.
Nonviral gene-delivery vectors
To successfully deliver nucleic acids such as plasmid DNA, viral or nonviral vectors are needed. All gene delivery vectors used in Table 1 were nonviral vectors. Although nonviral vectors are not as efficient as viral vectors, they are often used because of their lower immunogenicity and lack of limitations in the gene insert size [185].
PEI is a widely used polymeric, nonviral gene delivery vector because its density of amines provides positive charges to bind to DNA. PEI can be branched, consisting of primary, secondary and tertiary amines, or linear, consisting of secondary and primary amines [186]. Linear PEI and branched PEI are solid and liquid, respectively [187]. Linear PEI has a greater transfection efficiency compared with branched PEI because of the inherent kinetic instability under salt conditions, which results in aggregation of the polyplexes into larger size particles [175]. These larger particles result in increased interaction with the cell membrane, cytoplasm and nucleus. Lei et al. used 25-kDA linear PEI to deliver an RNAi plasmid targeting MMP-2 [48]. The polyplexes were embedded in PLGA nanofibrous membrane. Ohlfest et al. utilized 22-kDa linear PEI to co-deliver soluble VEGFR (sFlt-1) and an angiostatin–endostatin fusion gene (statin-AE) using the SB transposon by intratumoral injection [50].
PEI has been modified in many different ways to improve its ability as a nonviral transfection agent. Degradable linkages can be incorporated into high-molecular-weight PEI to improve its clearance [188]. Furthermore, biocompatible membrane-permeating moieties can be incorporated into low-molecular-weight polymers to improve its transfection activity [189,190]. Zhang et al. utilized a polymer, H1, which consists of low-molecular-weight (600 Da) PEI linked by β-cyclodextrin and conjugated with folic acid [46]. Cyclodextrin has low toxicity and does not elicit an immune response [183,191] and thus can reduce the toxicity of polycations [192]. This nonviral vector is the first reported nonviral vector that can achieve similar levels of transgene expression compared with adenovirus [46]. Li et al. also used the H1 polymer to deliver non-collagenous C-terminal globular NC1 domain of type VIII collagen a1 chain, Vastatin [47]. H1 is designed to target tumor and atypical cells by binding to the folate receptors that are enriched on cancer cell surfaces.
In vivo models
In vivo models are a key component of drug discovery and evaluating the efficiency, safety and effectiveness of therapeutics.
Subcutaneous/heterotopic/ectopic models
Research often starts with ectopic tumor models by implanting the scaffolds subcutaneously, outside of their original anatomical location [193–195]. Subcutaneous implantations are the easiest to perform and are often done in rodents because of their low cost and lax skin that provides versatility for large implants and their availability as immunodeficient animals for xenografts [196]. Pore et al. [44], Zhang et al. [46], Ohlfest et al. [50] and Benny et al. [51] utilized the subcutaneous model. Tumor cells were subcutaneously implanted on the dorsal part of the mice and/or rats, where the cells proliferated and formed quantifiable glioma tumors under the skin. Subcutaneous tumors facilitate close monitoring but are not the most representative of the actual growth and behavior of human cancer [197].
Intracranial/orthotopic
Although easy to create and monitor, subcutaneous models are associated with several limitations. The treatment is not tested in the brain microenvironment, the actual tumor site, so the antiangiogenic agent does not take into account the constraints that may exist, including having to cross the BBB [184], and thus overinterpretation of results can often occur [198]. The location poses a limitation because of the differences in angiogenic activity and the microenvironment's not providing the required conditions to portray a true malignant brain tumor.
By contrast, implantation of tumor cells in the flank of an animal or in intracranial mouse or rat tumor models is more challenging but is able to provide more clinically relevant results. Bow et al. [40], Frazier et al. [41], Weingart et al. [42] and Grossman et al. [52] utilized the rat intracranial model, whereas Gao et al. [43], Clavreul et al. [36], Tseng et al. [45], Zhang et al. [46], Li et al. [78], Lei et al. [48], Huang et al. [49], Ohlfest et al. [50], Benny et al. [51] and Sousa et al. [53] used the mouse intracranial model. Rat brains are much larger (∼1200 mg) compared with mouse brains (∼400 mg), and thus the tumor cell implantation process is more precise, and relatively larger volumes (∼20 μl) of cells can be injected into the area [199].
The orthotopic procedure consists of creating a scalp incision, cutting through the skull and placing a piece of tumor in the brain, where it begins to gradually grow near or in the brain tissue into larger masses [40]. Intracranial tumors are favored over subcutaneous tumors because the drug's testing site will be very similar to the original environment of a human malignant glioma. When administering drugs for the treatment of intracranial tumors, intracranial tumor models pose true challenges, such as getting across the BBB and avoiding systemic toxicity, in addition to the unique environments of crucial cell processes that help tumors survive, proliferate, differentiate and migrate [40,51].
Ohlfest et al., like some other groups, utilized the subcutaneous model first because of the ease of establishing and monitoring the tumor and then utilized the intracranial model to more accurately study the designed therapy [50]. The researchers observed that there was a decrease in efficacy in the intracranial model compared with the subcutaneous model and attributed this to the sixfold decrease in plasmid dose (adjusted for volume of the mouse brain).
Cells used to establish glioma models
As mentioned earlier, drug and treatment advancements are made possible through xenograft models, where human-derived tissues or cells from a patient's tumor are implanted, in this case intracranially, into immunodeficient mice and/or rats [200]. Xenografts allow for research to be completed without working on a living human being, but rather an animal that represents a human. Furthermore, they allow for the understanding of tumors and drug efficacy and act as a way to increase the number of preclinical subjects available to test with when conducting research.
Xenograft models can utilize various types of cell lines to establish the tumor. The most common one used in the applications listed in Table 1 is U87MG, which is one of the two most studied glioma cell lines [198]. After U87MG, the next two most common cell lines used to create tumors to test antiangiogenic agents for GBM are the C6 and 9L rat glioma cell lines. The C6 and 9L gliomas were induced in adult rats by repeated injections of methylnitrosourea [199]. Besides xenograft tumor models, cell lines can be implanted as allograft tumors in syngeneic animals [198].
In addition to the three common cell lines that were used in the applications discussed, Li et al. utilized a unique version of U87MG that acquired TMZ resistance, designated U87-ATR, to form xenograft gliomas [47]. The researchers utilized this chemoresistant model to examine the effect of the chemotherapy drug TMZ and its combination with the antiangiogenic agent Vastatin on the intrinsic and acquired chemoresistance that is often acquired with TMZ treatment.
Another GBM cell line that is used is U373. Ohlfest et al. used both U87MG and U373 to establish a mouse subcutaneous tumor model [50]. Compared with U87MG cells, which have more neuronal morphology, U373 cells have more mesenchymal morphology. U373 also differs from U87MG in terms of proliferation rate (i.e., less proliferative compared with U87MG) [201]. Interestingly, U373 cells can protect U87MG cells from TMZ-induced cell death, and, conversely, U87MG cells appear to increase the sensitivity of U373 cells to TMZ [201]. The effectiveness of antiangiogenic agents on in vivo tumor models may depend on the growth potential of the cell line used to form the tumor. The choice of which animal model (mouse vs rat cells used to form the tumor) can often depend on the preference for or experience and expertise of the research lab with different rodent types and cell lines and the nature of the study that will be conducted and therapy to be tested.
Targeting agents for endothelial cells
All cells in the human body possess surface marker molecules that indicate and give identity to a specific cell and can be implicated in different cellular functions including growth, differentiation and apoptosis. Thus, the development of targeting agents allows for specified recognition of cells of interest [202]. The importance of identifying and targeting cell-specific surface markers is highly desirable in developing effective drug delivery and treatment methods and potentially less harm to healthy cells [203]. The building blocks of blood vessels are endothelial cells, and thus antiangiogenic factors should be targeted to cell surface receptors of these cells. Furthermore, the surrounding vital endothelial cells near tumors tend to express some specific cell surface receptors and are therefore distinguishable from normal endothelial cells and can also be targeted [204]. Table 2 lists applications that utilize ligands bound to biomaterials to better target endothelial cells and improve the efficacy of the delivered bioactive factor being investigated for the treatment or study of glioma.
Table 2. . Biomaterial-based targeting agents that have been utilized to target endothelial cells in the treatment of glioblastoma.
| Target on biomaterial | Target on cell | Endothelial cells used in vitro and in animal models | Biomaterial | Bioactive factor | Outcome | Ref. |
|---|---|---|---|---|---|---|
| Monoclonal antibody | ||||||
| Anti-CD105 antibodies | Endoglin/CD105 |
In vitro: mouse endothelial cell line MS1 and mouse breast cancer cell line 4T1 In vivo: mouse subcutaneous U87MG GBM model |
Perfluorocarbon-containing lipid-shelled MBs for ultrasound contrast agents | None, contrast agent |
In vitro: attachment numbers of the CD105-targeted MBs significantly correlated with the CD105 expression levels of the cells in the parallel flow chamber test In vivo: there was a good correlation between the in vivo molecular ultrasound signals with the CD105-targeted MBs and the ex vivo expression levels of CD105 |
[205] |
| Peptides | ||||||
| cRGD and IL-13 peptide |
αvβ3 integrin on neovasculature |
In vitro: HUVECs In vivo: mouse intracranial C6 GBM model |
PEG-PCL NPs | DTX |
In vitro: IRNPs had high uptake into HUVEC cells In vivo: IRNPs had higher localization in the GBM site than mono-modified NPs |
[43] |
| cRGDyC | αvβ3 | In vitro: HUVECs | Liposomes formed with DPPG, cholesterol, DSPE-mPEG2000 and DSPE-PEG2000-MAL in chloroform and methanol | BSH | In vitro: cRGDyC liposome uptake was significantly higher than plain liposomes in integrin αvβ3-positive HUVEC and U87MG cells | [206] |
| iNGR (CRNGRGPDC sequence) | APN/CD13 |
In vitro: HUVECs In vivo: mouse intracranial U87MG GBM model |
Liposomes made with HSPC, cholesterol and PEG (2000)-DSPE | Dox |
In vitro: Uptake of iNGR-SSL by U87MG cells and HUVECs was significantly greater than that of unmodified liposomes In vivo: accumulation of iNGR-SSL/DiR was significantly increased compared with unmodified liposomes |
[207] |
| iNGR | APN/CD13 |
In vitro: HUVECs In vivo: mouse intracranial U87MG GBM model |
Poly (ethyleneglycol)-poly (L-lactic-co-glycolic acid) NPs | PTX |
In vitro: iNGR-NP-PTX inhibited tube formation more efficiently than unconjugated NPs In vivo: iNGR-NP exhibited the highest fluorescence intensity in the brain compared with other NPs tested |
[208] |
| NGR ligand | APN/CD13 |
In vitro: primary rat BCECs In vivo: mouse intracranial C6 GBM model |
PDA-coated mesoporous silica NPs (MSNs) | Dox |
In vitro: MSN-DOX-PDA-NGR had higher intracellular accumulation in primary BCECs and C6 cells than nontargeting NPs In vivo: MSN-DOX-PDA-NGR had higher accumulation in intracranial tumor tissue than undecorated NPs |
[209] |
| CGKRK (Cys-Gly-Lys-Arg-Lys) | Heparan sulfate on neovascular endothelial cells |
In vitro: HUVECs In vivo: mouse intracranial C6 GBM model |
PEG-PLGA NPs | PTX |
In vitro: cellular fluorescence intensities of CGKRK-NP and PC-NP were significantly higher at various concentrations and all experiment time points compared with normal NPs In vivo: PC-NP (CGKRK and Pep 1) exhibited the most accumulation in glioma |
[210] |
| CGKRK peptide | Heparan sulfate |
In vitro: HUVECs and U87MG cells In vivo: mouse subcutaneous U87MG GBM model |
PEG-co-PCL NPs | PTX |
In vitro: fluorescence intensity of CGKRK-NP on both HUVEC and U87MG cells was significantly enhanced when compared with that of NPs In vivo: following CGKRK-NP-DiR injection, mice exhibited a much stronger fluorescence intensity at the tumor site at every imaging time when compared with that of the NP group |
[211] |
| Tf and PFVYLI (PFV) | TfR | In vitro: bEnd.3 brain endothelial cells | Liposomal NPs made from DOTAP, DOPE, NHS-PEG(2000)-DSPE and cholesterol |
Dox and Erlo | In vitro: Tf-PFV liposomes showed significantly higher cellular uptake compared with single ligand or plain liposomes | [212] |
APN: Aminopeptidase N; BCEC: Brain capillary endothelial cell; BSH: Sodium borocaptate; cRGD: Cyclic Arg-Gly-Asp; cRGDyC: Cyclic Arg-Gly-Asp-Tyr-Cys; DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane chloride; DPPG: 1,2-dipalmitoyl-sn-glycero-3-phospho-rac-(1-glycerol); Dox: Doxorubicin; DTX: Docetaxel; Erlo: Erlotinib; GBM: Glioblastoma multiforme; HSPC: Hydrogenated soy phosphatidylcholine; HUVEC: Human umbilical vein endothelial cell; MB: Microbubble; NGR: Asp-Gly-Arg; NHS-PEG(2000)-DSPE: 3-(N-succinimidyloxyglutaryl) aminopropyl, polyethyleneglycol-carbamyl distearoylphosphatidyl-ethanolamine; NP: Nanoparticle; PDA: Polydopamine; PEG-PCL: Polyethylene glycol–poly(ε-caprolactone); PEG-PLGA: Polyethylene glycol–poly[(d,l)-lactide-co-glycolide]; PTX: Paclitaxel; Tf: Transferrin; TfR: Transferrin receptor.
Monoclonal antibodies
Monoclonal antibodies are monovalent antibodies produced by B-lymphocytes that bind to specific target antigens [213]. Monoclonal antibodies have exquisite specificity that decreases off-target effects and, hence, decreases toxicity [214]. These antibodies are widely used in the treatment of cancer and other diseases. As of 2019, 79 monoclonal antibodies have been approved by the FDA for therapeutic use, 30 of which have been approved for the treatment of cancer [213]. Besides being therapeutic agents for cancer, these antibodies or their fragments can be used as targeting agents to direct the homing of drugs to endothelial cells at tumor sites [215].
Endoglin (CD105) antibody
Endoglin (CD105) is a promising target because it is overexpressed in proliferating endothelial cells, such as those found in tumors [216], as it is an alternative proangiogenic growth factor [205]. Liu et al. used a CD105 antibody (anti-CD105) to target this overexpressed biomarker [205]. This study used perfluorocarbon-containing lipid-shelled microbubbles (MBs) for ultrasound contrast agents combined with anti-CD105 antibodies. The MBs were tested in vitro in the mouse endothelial cell line MS1 and the mouse breast cancer cell line 4T1 as well as in vivo in a mouse subcutaneous U87MG tumor model. The results of the in vitro and in vivo studies showed higher cellular uptake in the CD105-targeted MBs than the untargeted MBs. Liu et al. utilized these MBs for molecular ultrasound imaging to investigate the expression level of the biomarker CD105 during glioblastoma progression. However, anti-CD105 may be a promising target for the delivery of antiangiogenic therapy, as it can target overexpressed endoglin on proliferating endothelial cells.
Peptides
Another effective method of targeting cells is through the use of peptide targeting agents. The advantage of these peptides compared with antibodies is their low molecular weight (<10 kDa), which allows for better penetration of solid tumors [217].
αvβ3 integrin
The receptor αvβ3 integrin is an overexpressed receptor found on both proliferating endothelial cells and GBM tumor cells themselves [218]. Gao et al. examined the ability of NPs conjugated with IL-13 peptide and RGD to target IL13Rα2 in GBM cells and αvβ3 integrin in neurovascular endothelial cells [43]. These PEG-PCL NPs loaded with DTX display high uptake by human umbilical vein endothelial cells (HUVECs) in in vitro studies. When tested in vivo using a mouse intracranial C6 GBM model, the NPs had higher localization in GBM than in other groups of free drugs or unconjugated NPs. In addition to endothelial cells, U87MG glioblastoma cells have high expression of αvβ3 compared with other cell types [206]. Kang et al. utilized cyclic Arg-Gly-Asp-Tyr-Cys peptide to target αvβ3 integrin on both HUVEC and U87MG cells [206]. The researchers conjugated liposomes, formed with 1,2-dipalmitoyl-sn-glycero-3-phospho-rac-(1-glycerol), cholesterol, 1,2-Distearoyl-ras-glycerol-3-Phosphoethanolamine-N-Polyethyleneglycol-2000 (DSPE-mPEG2000) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-MAL), with the cyclic Arg-Gly-Asp-Tyr-Cys peptide. The results showed higher uptake of the cyclic Arg-Gly-Asp-Tyr-Cys peptide liposomes in both cell lines compared with unconjugated liposomes.
Aminopeptidase N (CD13)
Aminopeptidase N (also known as CD13) is a receptor for the Asp-Gly-Arg (NGR) peptides found on the endothelial cells and tumor blood vessels of some cancers, including glioblastoma [219]. It has also been shown to target glioblastoma tumor cells (i.e., U87MG and C6 cell lines) [207–209]. Zhou et al. [207] and Kang et al. [208] performed studies to target aminopeptidase N/CD13 by conjugating liposomes and NPs with the iNGR peptide (cyclized peptide sequence CRNGRGPDC) to deliver chemotherapy drugs [207,208]. The researchers performed in vitro studies utilizing HUVEC and U87MG cells. Both cell lines demonstrated higher uptake and better overall efficiency of the biomaterials conjugated with iNGR compared with those left unconjugated. The results were further supported by in vivo studies performed in mouse U87MG intracranial models. Both iNGR-conjugated biomaterials demonstrated higher accumulation in the tumor site and proved to be more effective than unconjugated biomaterials. Furthermore, iNGR can be cut by an enzyme to remove the GPDC sequence and result in the CRNGR sequence, which can bind NRP-1, which is overexpressed on tumor vessels and glioblastoma cells [220]. Hu et al. also targeted aminopeptidase N/CD13 by modifying polydopamine-coated mesoporous silica NPs with the NGR ligand to deliver Dox [209]. In vitro studies performed on primary rat brain capillary endothelial cells and C6 cells have demonstrated higher accumulation of NPs for both cell lines compared with nontargeted NPs. In vivo studies in a mouse C6 intracranial tumor model further supported the efficiency of these targeted NPs, as they showed higher accumulation at the tumor site than unmodified NPs [209].
Heparan sulfate
Heparan sulfate proteoglycans play a crucial role in the process of angiogenesis in glioblastomas and can be found on neovascular endothelial cells [221]. Therefore, various studies have been done to target heparan sulfate receptors on tumor endothelial cells with the use of CGKRK (Cys-Gly-Lys-Arg-Lys) peptide. Lv et al. conjugated PTX-loaded PEG-PLGA NPs with CGKRK peptide to target endothelial cells for more efficient drug delivery [210]. In vitro studies performed on HUVECs demonstrated higher cellular fluorescence intensity of CGKRK-NPs, indicating higher uptake, compared with unconjugated NPs. An in vivo study was also done using a mouse intracranial C6 glioma model [210]. The results demonstrated that CGKRK-NPs had much higher accumulation in gliomas compared with the control. In addition to endothelial cells, Hu et al. demonstrated that CRGKRK peptide can be used to target tumor cells [211]. The researchers used polyethylene glycol–poly(ε-caprolactone) (PEG-co-PCL) NPs to deliver PTX, and in vitro studies demonstrated higher fluorescence intensity of NPs conjugated with CGKRK on HUVEC and U87MG cells than plain NPs. When tested on subcutaneous U87MG tumor-bearing mice, the fluorescence intensity was much higher in the tumor site in mice given the CGKRK-NP injection compared with other NPs administered.
Transferrin
Transferrin receptors are found on the surface of GBM cells [222] as well as brain endothelial cells [212]. Lakkadwala et al. added transferrin and PFVTLI (PFV) ligands (a synthetic peptide with only six amino acids which represents the c-terminal portion of C105Y) to the surface of liposomal NPs made from 1,2-dioleoyl-3-trimethylammonium-propane chloride, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, 3-(N-succinimidyloxyglutaryl) aminopropyl, polyethyleneglycol-carbamyl distearoylphosphatidyl-ethanolamine and cholesterol [212]. The transferrin ligand was chosen to target transferrin receptors present on U87MG, bEnd.3, and primary glial cell lines, whereas the addition of PFVTLI was used to aid in the diffusion of the drug across the BBB. In vitro studies performed in U87MG, bEnd.3, and primary glial cells demonstrated that the transferrin-PFV (Tf-PFV) liposomes showed a higher cellular uptake than unmodified liposomes or liposomes modified with only a single ligand.
The results of the studies that evaluated endothelial-targeting ligands demonstrated higher efficacy of the drug being delivered in vivo to a tumor model because of its ability to target endothelial cells. Many of these targeting agents can also target GBM cells, thus further increasing their utility. Based on the results, conjugating a biomaterial with a ligand according to the target receptor provides a promising method for more effective delivery of a bioactive factor, such as an antiangiogenic drug, to endothelial cells involved in glioblastoma growth.
Conclusion
GBM is an aggressive and highly vascularized brain tumor with multiple limitations and challenges with regard to its therapeutic management. With the advent of antiangiogenic strategies, the outlook for the future management of GBM has the potential to be significantly improved, primarily due to the combination of these agents with chemoradiation, which often augments their effects and prolongs survival. Currently, most approaches have paired the local delivery of an antiangiogenic factor with the systemic delivery of chemotherapy drugs. Future work should consider designing treatments that deliver both drug types locally to enhance their therapeutic potential. Biomaterials can be used to overcome the off-target effects that occur because of systemic delivery of antiangiogenic agents and hence reduce the occurrence of adverse events. Furthermore, biomaterials can be used to increase bioavailability of drugs by enhancing the penetration of the BBB and preserving their pharmacological activity. Targeting agents can be covalently linked to the biomaterials to further increase targeting to the site of interest. Improvements in targeted delivery of these therapies is important for efficient and safe destruction of the tumor. Angiogenesis is a normal physiological process in certain organs in the body and a natural response to injury. Thus, off-target effects that occur because of systemic delivery may be detrimental to these organs or the body's healing process. The application of biomaterials and targeting moieties is key to avoiding these negative effects.
Future perspective
One critical area that requires greater depth of research is the identification of receptors or markers that are unique to endothelial cells in GBM, primarily because many of the receptors on endothelial cells are also expressed in other cell types. Additional research is also needed to identify other proangiogenic pathways that might be active in GBM, as many of these pathways might underlie the mechanisms of resistance to current therapeutic modalities. Finally, as the scientific community acquires a deeper understanding of the therapeutic approaches that work for GBM patients, it is important to identify new combination strategies to better treat GBM.
Executive summary.
Glioblastoma & angiogenesis
Glioblastoma multiforme (GBM), or glioblastoma, is the most prevalent and aggressive form of glioma, with poor prognosis and high mortality rates.
As GBM is a highly vascularized cancer, antiangiogenic therapies to halt or minimize the rate of tumor growth are critical to improving treatment.
Antiangiogenic bioactive factors for the treatment of GBM
Antiangiogenic therapies can be small-molecule drugs, nucleic acids or proteins/peptides.
The combination of antiangiogenic therapy with chemotherapy or radiation can augment the effects of the antiangiogenic agents and prolong survival.
Biomaterials utilized in antiangiogenic therapies
Biomaterials can be utilized to increase bioavailability of the drug at the site of interest while preventing off-target organ toxicity and enhancing the bioactivity of the drug by protecting the therapeutic agents.
Biomaterials have been utilized to deliver antiangiogenic agents systemically using injectable materials (i.e., polymeric nano- and microparticles, lipid-based nanoparticles/liposomes and nonviral gene-delivery vectors) or locally using injectable or implantable materials (i.e., wafers, disks, nanofibers).
Targeting agents for endothelial cells
Targeting agents can be covalently linked to biomaterials to further increase targeting to the site of interest.
The biomaterial-based targeting agents that have been used to target endothelial cells for GBM treatment include monoclonal antibodies and peptides.
In addition to targeting endothelial cells at the tumor site, these targeting moieties usually can target GBM tumor cells, as the target receptors are expressed on both cell types.
Footnotes
Disclaimer
The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Financial & competing interests disclosure
The authors acknowledge support by the National Institute of General Medical Sciences of the NIH under award number 1SC3GM135138-01. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Ostrom QT, Cioffi G, Gittleman H et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 21(Suppl. 5), v1–v100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Provides information on glioblastoma multiforme (GBM) tumors.
- 2.Urbanska K, Sokolowska J, Szmidt M, Sysa P Glioblastoma multiforme – an overview. Contemp. Oncol. (Pozn.) 18(5), 307–312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakada M, Kita D, Watanabe T et al. Aberrant signaling pathways in glioma. Cancers (Basel) 3(3), 3242–3278 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirsching HG, Galanis E, Weller M Glioblastoma. Handb. Clin. Neurol. 134, 381–397 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Mansouri A, Karamchandani J, Das S Molecular genetics of secondary glioblastoma. : Glioblastoma. De Vleeschouwer S, Brisbane, Australia: (2017). [PubMed] [Google Scholar]
- 6.Liu S, Wang Y, Xu K et al. Relationship between necrotic patterns in glioblastoma and patient survival: fractal dimension and lacunarity analyses using magnetic resonance imaging. Sci. Rep. 7(1), 8302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aldape K, Zadeh G, Mansouri S, Reifenberger G, Von Deimling A Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 129(6), 829–848 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Ohgaki H, Kleihues P Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 170(5), 1445–1453 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohgaki H, Kleihues P The definition of primary and secondary glioblastoma. Clin. Cancer Res. 19(4), 764–772 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Alifieris C, Trafalis DT Glioblastoma multiforme: pathogenesis and treatment. Pharmacol. Ther. 152, 63–82 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Clarke J, Butowski N, Chang S Recent advances in therapy for glioblastoma. Arch. Neurol. 67(3), 279–283 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Wilson TA, Karajannis MA, Harter DH Glioblastoma multiforme: state of the art and future therapeutics. Surg. Neurol. Int. 5, 64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perry J, Okamoto M, Guiou M, Shirai K, Errett A, Chakravarti A Novel therapies in glioblastoma. Neurol. Res. Int. 2012, 428565 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis ME Glioblastoma: overview of disease and treatment. Clin. J. Oncol. Nurs. 20(Suppl. 5), S2–S8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aasland D, Gotzinger L, Hauck L et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR-CHK1, p21, and NF-kappaB. Cancer Res. 79(1), 99–113 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Day SE, Waziri A Clinical trials of small molecule inhibitors in high-grade glioma. Neurosurg. Clin. N. Am. 23(3), 407–416 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Friedman HS, Kerby T, Calvert H Temozolomide and treatment of malignant glioma. Clin. Cancer Res. 6(7), 2585–2597 (2000). [PubMed] [Google Scholar]
- 18.Stupp R, Mason WP, Van Den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352(10), 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Parisi S, Corsa P, Raguso A et al. Temozolomide and radiotherapy versus radiotherapy alone in high grade gliomas: a very long term comparative study and literature review. Biomed. Res. Int. 2015, 620643 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daneman R, Prat A The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 7(1), a020412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noch EK, Ramakrishna R, Magge R Challenges in the treatment of glioblastoma: multisystem mechanisms of therapeutic resistance. World Neurosurg. 116, 505–517 (2018). [DOI] [PubMed] [Google Scholar]; • Discusses challenges in treating glioblastoma.
- 22.Wainwright DA, Nigam P, Thaci B, Dey M, Lesniak MS Recent developments on immunotherapy for brain cancer. Expert Opin. Emerg. Drugs 17(2), 181–202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shergalis A, Bankhead A, Luesakul U 3rd, Muangsin N, Neamati N Current challenges and opportunities in treating glioblastoma. Pharmacol. Rev. 70(3), 412–445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee SY Temozolomide resistance in glioblastoma multiforme. Genes Dis. 3(3), 198–210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondo M, Asai T, Katanasaka Y et al. Anti-neovascular therapy by liposomal drug targeted to membrane type-1 matrix metalloproteinase. Int. J. Cancer 108(2), 301–306 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Yang M, Baranov E, Li XM et al. Whole-body and intravital optical imaging of angiogenesis in orthotopically implanted tumors. Proc. Natl Acad. Sci. USA 98(5), 2616–2621 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zuazo-Gaztelu I, Casanovas O Unraveling the role of angiogenesis in cancer ecosystems. Front. Oncol. 8, 248 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skobe M, Rockwell P, Goldstein N, Vosseler S, Fusenig NE Halting angiogenesis suppresses carcinoma cell invasion. Nat. Med. 3(11), 1222–1227 (1997). [DOI] [PubMed] [Google Scholar]; • Discusses the importance of halting angiogenesis.
- 29.Rosen J, Blau T, Grau SJ, Barbe MT, Fink GR, Galldiks N Extracranial metastases of a cerebral glioblastoma: a case report and review of the literature. Case Rep. Oncol. 11(2), 591–600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Folkman J, Watson K, Ingber D, Hanahan D Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 339(6219), 58–61 (1989). [DOI] [PubMed] [Google Scholar]
- 31.Das S, Marsden PA Angiogenesis in glioblastoma. N. Engl. J. Med. 369(16), 1561–1563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'alessio A, Proietti G, Lama G et al. Analysis of angiogenesis related factors in glioblastoma, peritumoral tissue and their derived cancer stem cells. Oncotarget 7(48), 78541–78556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerbel R, Folkman J Clinical translation of angiogenesis inhibitors. Nat. Rev. Cancer 2(10), 727–739 (2002). [DOI] [PubMed] [Google Scholar]; •• Discusses the successful translation of angiogenesis inhibitors to clinical application.
- 34.Miranda A, Blanco-Prieto MJ, Sousa J, Pais A, Vitorino C Breaching barriers in glioblastoma. Part II: targeted drug delivery and lipid nanoparticles. Int. J. Pharm. 531(1), 389–410 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Nam L, Coll C, Erthal LCS et al. Drug delivery nanosystems for the localized treatment of glioblastoma multiforme. Materials (Basel) 11(5), 779–807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clavreul A, Roger E, Pourbaghi-Masouleh M, Lemaire L, Tetaud C, Menei P Development and characterization of sorafenib-loaded lipid nanocapsules for the treatment of glioblastoma. Drug Deliv. 25(1), 1756–1765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma P, Debinski W Receptor-targeted glial brain tumor therapies. Int. J. Mol. Sci. 19(11), 3326–3363 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, Meng Y, Li C, Qian M, Huang R Receptor-mediated drug delivery systems targeting to glioma. Nanomaterials (Basel) 6(1), 3–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakurai Y, Akita H, Harashima H Targeting tumor endothelial cells with nanoparticles. Int. J. Mol. Sci. 20(23), 5819–5833 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bow H, Hwang LS, Schildhaus N et al. Local delivery of angiogenesis-inhibitor minocycline combined with radiotherapy and oral temozolomide chemotherapy in 9L glioma. J. Neurosurg. 120(3), 662–669 (2014). [DOI] [PubMed] [Google Scholar]; • Discusses minocycline and temozolomide combination therapy.
- 41.Frazier JL, Wang PP, Case D et al. Local delivery of minocycline and systemic BCNU have synergistic activity in the treatment of intracranial glioma. J. Neurooncol. 64(3), 203–209 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Weingart JD, Sipos EP, Brem H The role of minocycline in the treatment of intracranial 9L glioma. J. Neurosurg. 82(4), 635–640 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Gao H, Yang Z, Cao S et al. Tumor cells and neovasculature dual targeting delivery for glioblastoma treatment. Biomaterials 35(7), 2374–2382 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Pore N, Gupta AK, Cerniglia GJ, Maity A HIV protease inhibitors decrease VEGF/HIF-1alpha expression and angiogenesis in glioblastoma cells. Neoplasia 8(11), 889–895 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tseng YY, Su CH, Yang ST et al. Advanced interstitial chemotherapy combined with targeted treatment of malignant glioma in rats by using drug-loaded nanofibrous membranes. Oncotarget 7(37), 59902–59916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang W, Duan R, Zhang J et al. H1/pHGFK1 nanoparticles exert anti-tumoural and radiosensitising effects by inhibition of MET in glioblastoma. Br. J. Cancer 118(4), 522–533 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Y, Li J, Woo YM et al. Enhanced expression of Vastatin inhibits angiogenesis and prolongs survival in murine orthotopic glioblastoma model. BMC Cancer 17(1), 126 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lei C, Cui Y, Zheng L, Chow PK, Wang CH Development of a gene/drug dual delivery system for brain tumor therapy: potent inhibition via RNA interference and synergistic effects. Biomaterials 34(30), 7483–7494 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Huang S, Shao K, Liu Y et al. Tumor-targeting and microenvironment-responsive smart nanoparticles for combination therapy of antiangiogenesis and apoptosis. ACS Nano. 7(3), 2860–2871 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Ohlfest JR, Demorest ZL, Motooka Y et al. Combinatorial antiangiogenic gene therapy by nonviral gene transfer using the Sleeping Beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Mol. Ther. 12(5), 778–788 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Benny O, Kim SK, Gvili K et al. In vivo fate and therapeutic efficacy of PF-4/CTF microspheres in an orthotopic human glioblastoma model. FASEB J. 22(2), 488–499 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Grossman R, Tyler B, Hwang L et al. Improvement in the standard treatment for experimental glioma by fusing antibody Fc domain to endostatin. J. Neurosurg. 115(6), 1139–1146 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Sousa F, Dhaliwal HK, Gattacceca F, Sarmento B, Amiji MM Enhanced anti-angiogenic effects of bevacizumab in glioblastoma treatment upon intranasal administration in polymeric nanoparticles. J. Control Release 309, 37–47 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Grodzik M, Sawosz E, Wierzbicki M et al. Nanoparticles of carbon allotropes inhibit glioblastoma multiforme angiogenesis in ovo. Int. J. Nanomedicine 6, 3041–3048 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao L, Ren TH, Wang DD Clinical pharmacology considerations in biologics development. Acta Pharmacol. Sin. 33(11), 1339–1347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nazarian S, Akhondi H Minocycline. : StatPearls. StatPearls Publishing LLC, FL, USA: (2020). [PubMed] [Google Scholar]
- 57.Nagarakanti S, Bishburg E Is minocycline an antiviral agent? A review of current literature. Basic Clin. Pharmacol. Toxicol. 118(1), 4–8 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Tamargo RJ, Bok RA, Brem H Angiogenesis inhibition by minocycline. Cancer Res. 51(2), 672–675 (1991). [PubMed] [Google Scholar]
- 59.Guerin C, Laterra J, Masnyk T, Golub LM, Brem H Selective endothelial growth inhibition by tetracyclines that inhibit collagenase. Biochem. Biophys. Res. Commun. 188(2), 740–745 (1992). [DOI] [PubMed] [Google Scholar]
- 60.Greenwald RA, Moak SA, Ramamurthy NS, Golub LM Tetracyclines suppress matrix metalloproteinase activity in adjuvant arthritis and in combination with flurbiprofen, ameliorate bone damage. J. Rheumatol. 19(6), 927–938 (1992). [PubMed] [Google Scholar]
- 61.Nguyen M, Arkell J, Jackson CJ Human endothelial gelatinases and angiogenesis. Int. J. Biochem. Cell Biol. 33(10), 960–970 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Rundhaug JE Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 9(2), 267–285 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brogden RN, Speight TM, Avery GS Minocycline: a review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs 9(4), 251–291 (1975). [DOI] [PubMed] [Google Scholar]
- 64.Agwuh KN, Macgowan A Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J. Antimicrob. Chemother. 58(2), 256–265 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Klein NC, Cunha BA Tetracyclines. Med. Clin. North Am. 79(4), 789–801 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Garrido-Mesa N, Zarzuelo A, Galvez J Minocycline: far beyond an antibiotic. Br. J. Pharmacol. 169(2), 337–352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farha NG, Kasi A Docetaxel. : StatPearls. StatPearls Publishing LLC, FL, USA: (2020). [PubMed] [Google Scholar]
- 68.Antonarakis ES, Armstrong AJ Evolving standards in the treatment of docetaxel-refractory castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 14(3), 192–205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kavallaris M Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 10(3), 194–204 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Fulton B, Spencer CM Docetaxel. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic efficacy in the management of metastatic breast cancer. Drugs 51(6), 1075–1092 (1996). [DOI] [PubMed] [Google Scholar]
- 71.Haldar S, Basu A, Croce CM Bcl2 is the guardian of microtubule integrity. Cancer Res. 57(2), 229–233 (1997). [PubMed] [Google Scholar]
- 72.Sweeney CJ, Miller KD, Sissons SE et al. The antiangiogenic property of docetaxel is synergistic with a recombinant humanized monoclonal antibody against vascular endothelial growth factor or 2-methoxyestradiol but antagonized by endothelial growth factors. Cancer Res. 61(8), 3369–3372 (2001). [PubMed] [Google Scholar]
- 73.Kushwah V, Jain DK, Agrawal AK, Jain S Improved antitumor efficacy and reduced toxicity of docetaxel using anacardic acid functionalized stealth liposomes. Colloids Surf. B Biointerfaces 172, 213–223 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Van Der Veldt AA, Hendrikse NH, Smit EF et al. Biodistribution and radiation dosimetry of 11C-labelled docetaxel in cancer patients. Eur. J. Nucl. Med. Mol. Imaging 37(10), 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Forsyth P, Cairncross G, Stewart D, Goodyear M, Wainman N, Eisenhauer E Phase II trial of docetaxel in patients with recurrent malignant glioma: a study of the National Cancer Institute of Canada Clinical Trials Group. Invest. New Drugs 14(2), 203–206 (1996). [DOI] [PubMed] [Google Scholar]
- 76.Sanson M, Napolitano M, Yaya R et al. Second line chemotherapy with docetaxel in patients with recurrent malignant glioma: a phase II study. J. Neurooncol. 50(3), 245–249 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Kadari A, Pooja D, Gora RH et al. Design of multifunctional peptide collaborated and docetaxel loaded lipid nanoparticles for antiglioma therapy. Eur. J. Pharm. Biopharm. 132, 168–179 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Li AJ, Zheng YH, Liu GD, Liu WS, Cao PC, Bu ZF Efficient delivery of docetaxel for the treatment of brain tumors by cyclic RGD-tagged polymeric micelles. Mol. Med. Rep. 11(4), 3078–3086 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Abdelgalil AA, Alkahtani HM, Al-Jenoobi FI Sorafenib. Profiles Drug Subst. Excip. Relat. Methodol. 44, 239–266 (2019). [DOI] [PubMed] [Google Scholar]
- 80.Wilhelm S, Carter C, Lynch M et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 5(10), 835–844 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Ben Mousa A Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J. Gastroenterol. 14(1), 40–42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cervello M, Bachvarov D, Lampiasi N et al. Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle 11(15), 2843–2855 (2012). [DOI] [PubMed] [Google Scholar]
- 83.Li L, Zhao GD, Shi Z, Qi LL, Zhou LY, Fu ZX The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 12(5), 3045–3050 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang F, Steelman LS, Lee JT et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia 17(7), 1263–1293 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Mccubrey JA, Steelman LS, Chappell WH et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 1773(8), 1263–1284 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roberts PJ, Der CJ Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26(22), 3291–3310 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Gauthier A, Ho M Role of sorafenib in the treatment of advanced hepatocellular carcinoma: an update. Hepatol. Res. 43(2), 147–154 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Adnane L, Trail PA, Taylor I, Wilhelm SM Sorafenib (BAY 43–9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 407, 597–612 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol. Cancer Ther. 7(10), 3129–3140 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Joensuu H, Puputti M, Sihto H, Tynninen O, Nupponen NN Amplification of genes encoding KIT, PDGFRalpha and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J. Pathol. 207(2), 224–231 (2005). [DOI] [PubMed] [Google Scholar]
- 91.Schiff D, Jaeckle KA, Anderson SK et al. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North Central Cancer Treatment Group study/Alliance N0572. Cancer 124(7), 1455–1463 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hottinger AF, Ben Aissa A, Espeli V et al. Phase I study of sorafenib combined with radiation therapy and temozolomide as first-line treatment of high-grade glioma. Br. J. Cancer 110(11), 2655–2661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reardon DA, Vredenburgh JJ, Desjardins A et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J. Neurooncol. 101(1), 57–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Galanis E, Anderson SK, Lafky JM et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): a North Central Cancer Treatment Group trial. Clin. Cancer Res. 19(17), 4816–4823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jo Y, Kim EH, Sai S et al. Functional biological activity of sorafenib as a tumor-treating field sensitizer for glioblastoma therapy. Int. J. Mol. Sci. 19(11), 3684–3701 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fung HB, Kirschenbaum HL, Hameed R Amprenavir: a new human immunodeficiency virus type 1 protease inhibitor. Clin. Ther. 22(5), 549–572 (2000). [DOI] [PubMed] [Google Scholar]
- 97.Bardsley-Elliot A, Plosker GL Nelfinavir: an update on its use in HIV infection. Drugs 59(3), 581–620 (2000). [DOI] [PubMed] [Google Scholar]
- 98.Gupta SK, Singh JP Inhibition of endothelial cell proliferation by platelet factor-4 involves a unique action on S phase progression. J. Cell Biol. 127(4), 1121–1127 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pore N, Jiang Z, Shu HK, Bernhard E, Kao GD, Maity A Akt1 activation can augment hypoxia-inducible factor-1alpha expression by increasing protein translation through a mammalian target of rapamycin-independent pathway. Mol. Cancer Res. 4(7), 471–479 (2006). [DOI] [PubMed] [Google Scholar]
- 100.Zhong H, Chiles K, Feldser D et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 60(6), 1541–1545 (2000). [PubMed] [Google Scholar]
- 101.Sherbet GV Suppression of angiogenesis and tumour progression by combretastatin and derivatives. Cancer Lett. 403, 289–295 (2017). [DOI] [PubMed] [Google Scholar]
- 102.Nainwal LM, Alam MM, Shaquiquzzaman M, Marella A, Kamal A Combretastatin-based compounds with therapeutic characteristics: a patent review. Expert Opin. Ther. Pat. 29(9), 703–731 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Su M, Huang J, Liu S et al. The anti-angiogenic effect and novel mechanisms of action of combretastatin A-4. Sci. Rep. 6, 28139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chew SA, Moscato S, George S, Azimi B, Danti S Liver cancer: current and future trends using biomaterials. Cancers (Basel) 11(12), 2026–2066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schmeer M, Buchholz T, Schleef M Plasmid DNA manufacturing for indirect and direct clinical applications. Hum. Gene Ther. 28(10), 856–861 (2017). [DOI] [PubMed] [Google Scholar]
- 106.Mali S Delivery systems for gene therapy. Indian J. Hum. Genet. 19(1), 3–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xin L, Xu R, Zhang Q, Li TP, Gan RB Kringle 1 of human hepatocyte growth factor inhibits bovine aortic endothelial cell proliferation stimulated by basic fibroblast growth factor and causes cell apoptosis. Biochem. Biophys. Res. Commun. 277(1), 186–190 (2000). [DOI] [PubMed] [Google Scholar]
- 108.Tulasne D, Deheuninck J, Lourenco FC et al. Proapoptotic function of the MET tyrosine kinase receptor through caspase cleavage. Mol. Cell Biol. 24(23), 10328–10339 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cruickshanks N, Zhang Y, Yuan F, Pahuski M, Gibert M, Abounader R Role and therapeutic targeting of the HGF/MET pathway in glioblastoma. Cancers (Basel) 9(7), 87–102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cruickshanks N, Zhang Y, Hine S et al. Discovery and therapeutic exploitation of mechanisms of resistance to MET inhibitors in glioblastoma. Clin. Cancer Res. 25(2), 663–673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shen Z, Yao C, Wang Z et al. Vastatin, an endogenous antiangiogenesis polypeptide that is lost in hepatocellular carcinoma, effectively inhibits tumor metastasis. Mol. Ther. 24(8), 1358–1368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Monboisse JC, Oudart JB, Ramont L, Brassart-Pasco S, Maquart FX Matrikines from basement membrane collagens: a new anti-cancer strategy. Biochim. Biophys. Acta 1840(8), 2589–2598 (2014). [DOI] [PubMed] [Google Scholar]
- 113.Assadian S, Teodoro JG Regulation of collagen-derived antiangiogenic factors by p53. Expert Opin. Biol. Ther. 8(7), 941–950 (2008). [DOI] [PubMed] [Google Scholar]
- 114.Xu R, Yao ZY, Xin L, Zhang Q, Li TP, Gan RB NC1 domain of human type VIII collagen (alpha 1) inhibits bovine aortic endothelial cell proliferation and causes cell apoptosis. Biochem. Biophys. Res. Commun. 289(1), 264–268 (2001). [DOI] [PubMed] [Google Scholar]
- 115.Zhang L, Shen X, Lu Q et al. A potential therapeutic strategy for inhibition of ocular neovascularization with a new endogenous protein: rhEDI-8t. Graefes Arch. Clin. Exp. Ophthalmol. 250(5), 731–739 (2012). [DOI] [PubMed] [Google Scholar]
- 116.Paulus W, Sage EH, Liszka U, Iruela-Arispe ML, Jellinger K Increased levels of type VIII collagen in human brain tumours compared to normal brain tissue and non-neoplastic cerebral disorders. Br. J. Cancer 63(3), 367–371 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Izquierdo M Short interfering RNAs as a tool for cancer gene therapy. Cancer Gene Ther. 12(3), 217–227 (2005). [DOI] [PubMed] [Google Scholar]
- 118.Kim DH, Rossi JJ Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 8(3), 173–184 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Li CX, Parker A, Menocal E, Xiang S, Borodyansky L, Fruehauf JH Delivery of RNA interference. Cell Cycle 5(18), 2103–2109 (2006). [DOI] [PubMed] [Google Scholar]
- 120.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91(4), 501–510 (1997). [DOI] [PubMed] [Google Scholar]
- 121.Ma K, Wang DD, Lin Y, Wang J, Petrenko V, Mao C Synergetic targeted delivery of Sleeping-Beauty transposon system to mesenchymal stem cells using LPD nanoparticles modified with a phage-displayed targeting peptide. Adv. Funct. Mater. 23(9), 1172–1181 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Grabundzija I, Irgang M, Mates L et al. Comparative analysis of transposable element vector systems in human cells. Mol. Ther. 18(6), 1200–1209 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat. Genet. 25(1), 35–41 (2000). [DOI] [PubMed] [Google Scholar]
- 124.Kendall RL, Wang G, Thomas KA Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem. Biophys. Res. Commun. 226(2), 324–328 (1996). [DOI] [PubMed] [Google Scholar]
- 125.Jinjun W, Zhaowei W, Qiang L et al. sFLT-1 inhibits proliferation, migration, and invasion of colorectal cancer SW480 cells through vascular mimicry formation suppression. Tumour Biol 39(5), 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 126.Scappaticci FA, Contreras A, Smith R et al. Statin-AE: a novel angiostatin-endostatin fusion protein with enhanced antiangiogenic and antitumor activity. Angiogenesis 4(4), 263–268 (2001). [DOI] [PubMed] [Google Scholar]
- 127.Scappaticci FA, Smith R, Pathak A et al. Combination angiostatin and endostatin gene transfer induces synergistic antiangiogenic activity in vitro and antitumor efficacy in leukemia and solid tumors in mice. Mol. Ther. 3(2), 186–196 (2001). [DOI] [PubMed] [Google Scholar]
- 128.Kirsch M, Strasser J, Allende R, Bello L, Zhang J, Black PM Angiostatin suppresses malignant glioma growth in vivo. Cancer Res. 58(20), 4654–4659 (1998). [PubMed] [Google Scholar]
- 129.O'reilly MS, Holmgren L, Shing Y et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79(2), 315–328 (1994). [DOI] [PubMed] [Google Scholar]
- 130.Read TA, Sorensen DR, Mahesparan R et al. Local endostatin treatment of gliomas administered by microencapsulated producer cells. Nat. Biotechnol. 19(1), 29–34 (2001). [DOI] [PubMed] [Google Scholar]
- 131.Dhanabal M, Ramchandran R, Volk R et al. Endostatin: yeast production, mutants, and antitumor effect in renal cell carcinoma. Cancer Res. 59(1), 189–197 (1999). [PubMed] [Google Scholar]
- 132.Craik DJ, Fairlie DP, Liras S, Price D The future of peptide-based drugs. Chem. Biol. Drug Des. 81(1), 136–147 (2013). [DOI] [PubMed] [Google Scholar]
- 133.Bhutia SK, Maiti TK Targeting tumors with peptides from natural sources. Trends Biotechnol. 26(4), 210–217 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Maione TE, Gray GS, Petro J et al. Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 247(4938), 77–79 (1990). [DOI] [PubMed] [Google Scholar]
- 135.Ruegg C, Hasmim M, Lejeune FJ, Alghisi GC Antiangiogenic peptides and proteins: from experimental tools to clinical drugs. Biochim. Biophys. Acta 1765(2), 155–177 (2006). [DOI] [PubMed] [Google Scholar]
- 136.Bikfalvi A Platelet factor 4: an inhibitor of angiogenesis. Semin. Thromb. Hemost. 30(3), 379–385 (2004). [DOI] [PubMed] [Google Scholar]
- 137.Hagedorn M, Zilberberg L, Lozano RM et al. A short peptide domain of platelet factor 4 blocks angiogenic key events induced by FGF-2. FASEB J. 15(3), 550–552 (2001). [DOI] [PubMed] [Google Scholar]
- 138.Jouan V, Canron X, Alemany M et al. Inhibition of in vitro angiogenesis by platelet factor-4-derived peptides and mechanism of action. Blood 94(3), 984–993 (1999). [PubMed] [Google Scholar]
- 139.Lozano RM, Redondo-Horcajo M, Jimenez MA et al. Solution structure and interaction with basic and acidic fibroblast growth factor of a 3-kDa human platelet factor-4 fragment with antiangiogenic activity. J. Biol. Chem. 276(38), 35723–35734 (2001). [DOI] [PubMed] [Google Scholar]
- 140.Bello L, Giussani C, Carrabba G et al. Suppression of malignant glioma recurrence in a newly developed animal model by endogenous inhibitors. Clin. Cancer Res. 8(11), 3539–3548 (2002). [PubMed] [Google Scholar]
- 141.O'Reilly MS, Boehm T, Shing Y et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88(2), 277–285 (1997). [DOI] [PubMed] [Google Scholar]
- 142.Peroulis I, Jonas N, Saleh M Antiangiogenic activity of endostatin inhibits C6 glioma growth. Int. J. Cancer 97(6), 839–845 (2002). [DOI] [PubMed] [Google Scholar]
- 143.Walia A, Yang JF, Huang YH, Rosenblatt MI, Chang JH, Azar DT Endostatin's emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim. Biophys. Acta 1850(12), 2422–2438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Folkman J Antiangiogenesis in cancer therapy – endostatin and its mechanisms of action. Exp. Cell Res. 312(5), 594–607 (2006). [DOI] [PubMed] [Google Scholar]; •• Discusses antiangiogenesis in cancer therapy and endostatin as an antiangiogenic treatment.
- 145.Ramchandran R, Karumanchi SA, Hanai J, Alper SL, Sukhatme VP Cellular actions and signaling by endostatin. Crit. Rev. Eukaryot. Gene Expr. 12(3), 175–191 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thomas JP, Arzoomanian RZ, Alberti D et al. Phase I pharmacokinetic and pharmacodynamic study of recombinant human endostatin in patients with advanced solid tumors. J. Clin. Oncol. 21(2), 223–231 (2003). [DOI] [PubMed] [Google Scholar]
- 147.Sim BK, Fogler WE, Zhou XH et al. Zinc ligand-disrupted recombinant human endostatin: potent inhibition of tumor growth, safety and pharmacokinetic profile. Angiogenesis 3(1), 41–51 (1999). [DOI] [PubMed] [Google Scholar]
- 148.Tjin Tham Sjin RM, Satchi-Fainaro R, Birsner AE, Ramanujam VM, Folkman J, Javaherian K A 27-amino-acid synthetic peptide corresponding to the NH2-terminal zinc-binding domain of endostatin is responsible for its antitumor activity. Cancer Res. 65(9), 3656–3663 (2005). [DOI] [PubMed] [Google Scholar]
- 149.Lee TY, Tjin Tham Sjin RM, Movahedi S et al. Linking antibody Fc domain to endostatin significantly improves endostatin half-life and efficacy. Clin. Cancer Res. 14(5), 1487–1493 (2008). [DOI] [PubMed] [Google Scholar]
- 150.Kazazi-Hyseni F, Beijnen JH, Schellens JH Bevacizumab. Oncologist 15(8), 819–825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Seystahl K, Gramatzki D, Roth P, Weller M Pharmacotherapies for the treatment of glioblastoma – current evidence and perspectives. Expert Opin. Pharmacother. 17(9), 1259–1270 (2016). [DOI] [PubMed] [Google Scholar]
- 152.Gerriets V, Kasi A Bevacizumab. : StatPearls. FL, USA: (2020). [PubMed] [Google Scholar]
- 153.Piccioni DE, Selfridge J, Mody RR et al. Deferred use of bevacizumab for recurrent glioblastoma is not associated with diminished efficacy. Neuro. Oncol. 16(6), 815–822 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Castro BA, Aghi MK Bevacizumab for glioblastoma: current indications, surgical implications, and future directions. Neurosurg. Focus 37(6), E9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Discusses the use of bevacizumab for glioblastoma.
- 155.Haibe Y, Kreidieh M, El Hajj H et al. Resistance mechanisms to anti-angiogenic therapies in cancer. Front. Oncol. 10, 221 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bakowicz-Mitura K, Bartosz G, Mitura S Influence of diamond powder particles on human gene expression. Surf. Coat. Technol. 201(13), 6131–6135 (2007). [Google Scholar]
- 157.Murugesan S, Mousa SA, O'Connor LJ, Lincoln DW, Linhardt RJ 2nd Carbon inhibits vascular endothelial growth factor- and fibroblast growth factor-promoted angiogenesis. FEBS Lett. 581(6), 1157–1160 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hillen F, Griffioen AW Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev. 26(3–4), 489–502 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brekken RA, Thorpe PE Vascular endothelial growth factor and vascular targeting of solid tumors. Anticancer Res. 21(6B), 4221–4229 (2001). [PubMed] [Google Scholar]
- 160.Reijneveld JC, Voest EE, Taphoorn MJ Angiogenesis in malignant primary and metastatic brain tumors. J. Neurol. 247(8), 597–608 (2000). [DOI] [PubMed] [Google Scholar]
- 161.Reithmeier T, Graf E, Piroth T, Trippel M, Pinsker MO, Nikkhah G BCNU for recurrent glioblastoma multiforme: efficacy, toxicity and prognostic factors. BMC Cancer 10, 30 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sonabend AM, Velicu S, Ulasov IV et al. A safety and efficacy study of local delivery of interleukin-12 transgene by PPC polymer in a model of experimental glioma. Anticancer Drugs 19(2), 133–142 (2008). [DOI] [PubMed] [Google Scholar]
- 163.Barani IJ, Larson DA Radiation therapy of glioblastoma. Cancer Treat. Res. 163, 49–73 (2015). [DOI] [PubMed] [Google Scholar]
- 164.Walker MD, Alexander E Jr, Hunt WE et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J. Neurosurg. 49(3), 333–343 (1978). [DOI] [PubMed] [Google Scholar]
- 165.Walker MD, Green SB, Byar DP et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N. Engl. J. Med. 303(23), 1323–1329 (1980). [DOI] [PubMed] [Google Scholar]
- 166.Katsigiannis S, Krischek B, Barleanu S et al. Impact of time to initiation of radiotherapy on survival after resection of newly diagnosed glioblastoma. Radiat. Oncol. 14(1), 73 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kumar N, Langer RS, Domb AJ Polyanhydrides: an overview. Adv. Drug Deliv. Rev. 54(7), 889–910 (2002). [DOI] [PubMed] [Google Scholar]
- 168.Torres MP, Determan AS, Anderson GL, Mallapragada SK, Narasimhan B Amphiphilic polyanhydrides for protein stabilization and release. Biomaterials 28(1), 108–116 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jain JP, Modi S, Domb AJ, Kumar N Role of polyanhydrides as localized drug carriers. J. Control Release 103(3), 541–563 (2005). [DOI] [PubMed] [Google Scholar]
- 170.Sun L, Zhou S, Wang W, Su Q, Li X, Weng J Preparation and characterization of protein-loaded polyanhydride microspheres. J. Mater. Sci. Mater. Med. 20(10), 2035–2042 (2009). [DOI] [PubMed] [Google Scholar]
- 171.Brem H, Lawson HC The development of new brain tumor therapy utilizing the local and sustained delivery of chemotherapeutic agents from biodegradable polymers. Cancer 86(2), 197–199 (1999). [DOI] [PubMed] [Google Scholar]
- 172.Tamargo RJ, Myseros JS, Epstein JI, Yang MB, Chasin M, Brem H Interstitial chemotherapy of the 9L gliosarcoma: controlled release polymers for drug delivery in the brain. Cancer Res. 53(2), 329–333 (1993). [PubMed] [Google Scholar]
- 173.Kou JH, Emmett C, Shen P et al. Bioerosion and biocompatibility of poly(D,L-lactic-co-glycolic acid) implants in brain. J. Control. Release 43(2), 123–130 (1997). [Google Scholar]
- 174.Menei P, Daniel V, Montero-Menei C, Brouillard M, Pouplard-Barthelaix A, Benoit JP Biodegradation and brain tissue reaction to poly(D,L-lactide-co-glycolide) microspheres. Biomaterials 14(6), 470–478 (1993). [DOI] [PubMed] [Google Scholar]
- 175.Wightman L, Kircheis R, Rossler V et al. Different behavior of branched and linear polyethylenimine for gene delivery in vitro and in vivo. J. Gene Med. 3(4), 362–372 (2001). [DOI] [PubMed] [Google Scholar]
- 176.Langer R, Folkman J Polymers for the sustained release of proteins and other macromolecules. Nature 263, 797–800 (1976). [DOI] [PubMed] [Google Scholar]
- 177.Langer R, Brem H, Tapper D Biocompatibility of polymeric delivery systems for macromolecules. J. Biomed. Mater. Res. 15(2), 267–277 (1981). [DOI] [PubMed] [Google Scholar]
- 178.Pitt CG Poly(ε-caprolactone) and its copolymers. : Biodegradable Polymers as Drug Delivery Systems. Chasin M, Langer R (). Marcel Dekker Inc., NY, USA: (1990). [Google Scholar]
- 179.Langer R MCE. : Biodegradable Polymers as Drug Delivery Systems. Marcel Decker Inc., NY, USA: (1990). [Google Scholar]
- 180.Chawla JS, Amiji MM Biodegradable poly(epsilon-caprolactone) nanoparticles for tumor-targeted delivery of tamoxifen. Int. J. Pharm. 249(1–2), 127–138 (2002). [DOI] [PubMed] [Google Scholar]
- 181.Xin H, Chen L, Gu J et al. Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(varepsilon-caprolactone) nanoparticles: in vitro and in vivo evaluation. Int. J. Pharm. 402(1–2), 238–247 (2010). [DOI] [PubMed] [Google Scholar]
- 182.Huynh NT, Passirani C, Saulnier P, Benoit JP Lipid nanocapsules: a new platform for nanomedicine. Int. J. Pharm. 379(2), 201–209 (2009). [DOI] [PubMed] [Google Scholar]
- 183.Saliou B, Thomas O, Lautram N et al. Development and in vitro evaluation of a novel lipid nanocapsule formulation of etoposide. Eur. J. Pharm. Sci. 50(2), 172–180 (2013). [DOI] [PubMed] [Google Scholar]
- 184.Clavreul A, Pourbaghi-Masouleh M, Roger E, Menei P Nanocarriers and nonviral methods for delivering antiangiogenic factors for glioblastoma therapy: the story so far. Int. J. Nanomedicine 14, 2497–2513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Furgeson DY, Chan WS, Yockman JW, Kim SW Modified linear polyethylenimine-cholesterol conjugates for DNA complexation. Bioconjug. Chem. 14(4), 840–847 (2003). [DOI] [PubMed] [Google Scholar]
- 186.Gonzalez H, Hwang SJ, Davis ME New class of polymers for the delivery of macromolecular therapeutics. Bioconjug. Chem. 10(6), 1068–1074 (1999). [DOI] [PubMed] [Google Scholar]
- 187.Lungu CN, Diudea MV, Putz MV, Grudzinski IP Linear and branched PEIs (polyethylenimines) and their property space. Int. J. Mol. Sci. 17(4), 555 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Langer R, Folkman J Polymers for the sustained release of proteins and other macromolecules. Nature 263(5580), 797–800 (1976). [DOI] [PubMed] [Google Scholar]
- 189.Petersen H, Merdan T, Kunath K, Fischer D, Kissel T Poly(ethylenimine-co-L-lactamide-co-succinamide): a biodegradable polyethylenimine derivative with an advantageous pH-dependent hydrolytic degradation for gene delivery. Bioconjug. Chem. 13(4), 812–821 (2002). [DOI] [PubMed] [Google Scholar]
- 190.Han S, Mahato RI, Kim SW Water-soluble lipopolymer for gene delivery. Bioconjug. Chem. 12(3), 337–345 (2001). [DOI] [PubMed] [Google Scholar]
- 191.Pack DW, Hoffman AS, Pun S, Stayton PS Design and development of polymers for gene delivery. Nat. Rev. Drug Discov. 4(7), 581–593 (2005). [DOI] [PubMed] [Google Scholar]
- 192.Davis ME, Brewster ME Cyclodextrin-based pharmaceutics: past, present and future. Nat. Rev. Drug Discov. 3(12), 1023–1035 (2004). [DOI] [PubMed] [Google Scholar]
- 193.Eskildsen T, Taipaleenmaki H, Stenvang J et al. MicroRNA-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc. Natl Acad. Sci. USA 108(15), 6139–6144 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Li Y, Fan L, Liu S et al. The promotion of bone regeneration through positive regulation of angiogenic–osteogenic coupling using microRNA-26a. Biomaterials 34(21), 5048–5058 (2013). [DOI] [PubMed] [Google Scholar]
- 195.Chen L, Holmstrom K, Qiu W et al. MicroRNA-34a inhibits osteoblast differentiation and in vivo bone formation of human stromal stem cells. Stem Cells 32(4), 902–912 (2014). [DOI] [PubMed] [Google Scholar]
- 196.Scott MA, Levi B, Askarinam A et al. Brief review of models of ectopic bone formation. Stem Cells Dev. 21(5), 655–667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lwin TM, Hoffman RM, Bouvet M Advantages of patient-derived orthotopic mouse models and genetic reporters for developing fluorescence-guided surgery. J. Surg. Oncol. 118(2), 253–264 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W Glioma: experimental models and reality. Acta Neuropathol. 133(2), 263–282 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Discusses animal models for glioma.
- 199.Barth RF, Kaur B Rat brain tumor models in experimental neuro-oncology: the C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J. Neurooncol. 94(3), 299–312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Richmond A, Su Y Mouse xenograft models vs GEM models for human cancer therapeutics. Dis. Model Mech. 1(2–3), 78–82 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Motaln H, Koren A, Gruden K, Ramsak Z, Schichor C, Lah TT Heterogeneous glioblastoma cell cross-talk promotes phenotype alterations and enhanced drug resistance. Oncotarget 6(38), 40998–41017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zhang X, Lan Y, Xu J et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 47(D1), D721–D728 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bode AM, Dong Z Molecular and cellular targets. Mol. Carcinog. 45(6), 422–430 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C, St Croix B Genes that distinguish physiological and pathological angiogenesis. Cancer Cell 11(6), 539–554 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Liu C, Yan F, Xu Y, Zheng H, Sun L InVivo molecular ultrasound assessment of glioblastoma neovasculature with endoglin-targeted microbubbles. Contrast Media Mol. Imaging 2018, 8425495 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kang W, Svirskis D, Sarojini V, Mcgregor AL, Bevitt J, Wu Z Cyclic-RGDyC functionalized liposomes for dual-targeting of tumor vasculature and cancer cells in glioblastoma: an in vitro boron neutron capture therapy study. Oncotarget 8(22), 36614–36627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhou JE, Yu J, Gao L et al. iNGR-modified liposomes for tumor vascular targeting and tumor tissue penetrating delivery in the treatment of glioblastoma. Mol. Pharm. 14(5), 1811–1820 (2017). [DOI] [PubMed] [Google Scholar]
- 208.Kang T, Gao X, Hu Q et al. iNGR-modified PEG-PLGA nanoparticles that recognize tumor vasculature and penetrate gliomas. Biomaterials 35(14), 4319–4332 (2014). [DOI] [PubMed] [Google Scholar]
- 209.Hu J, Zhang X, Wen Z et al. Asn-Gly-Arg-modified polydopamine-coated nanoparticles for dual-targeting therapy of brain glioma in rats. Oncotarget 7(45), 73681–73696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lv L, Jiang Y, Liu X et al. Enhanced antiglioblastoma efficacy of neovasculature and glioma cells dual targeted nanoparticles. Mol. Pharm. 13(10), 3506–3517 (2016). [DOI] [PubMed] [Google Scholar]
- 211.Hu Q, Gao X, Kang T et al. CGKRK-modified nanoparticles for dual-targeting drug delivery to tumor cells and angiogenic blood vessels. Biomaterials 34(37), 9496–9508 (2013). [DOI] [PubMed] [Google Scholar]
- 212.Lakkadwala S, Singh J Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surf. B Biointerfaces 173, 27–35 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lu RM, Hwang YC, Liu IJ et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 27(1), 1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shepard HM, Phillips GL, Thanos CD, Feldmann M Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. (Lond.) 17(3), 220–232 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Weiner GJ Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 15(6), 361–370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fonsatti E, Jekunen AP, Kairemo KJ et al. Endoglin is a suitable target for efficient imaging of solid tumors: in vivo evidence in a canine mammary carcinoma model. Clin. Cancer Res. 6(5), 2037–2043 (2000). [PubMed] [Google Scholar]
- 217.Scodeller P, Asciutto EK Targeting tumors using peptides. Molecules 25(4), 808–831 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Schnell O, Krebs B, Wagner E et al. Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol. 18(3), 378–386 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Pasqualini R, Koivunen E, Kain R et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 60(3), 722–727 (2000). [PMC free article] [PubMed] [Google Scholar]
- 220.Ahmad A, Mondal SK, Mukhopadhyay D, Banerjee R, Alkharfy KM Development of liposomal formulation for delivering anticancer drug to breast cancer stem-cell-like cells and its pharmacokinetics in an animal model. Mol. Pharm. 13(3), 1081–1088 (2016). [DOI] [PubMed] [Google Scholar]
- 221.Qiao D, Meyer K, Mundhenke C, Drew SA, Friedl A Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 signaling in brain endothelial cells. Specific role for glypican-1 in glioma angiogenesis. J. Biol. Chem. 278(18), 16045–16053 (2003). [DOI] [PubMed] [Google Scholar]
- 222.Prabhakar K, Afzal SM, Kumar PU, Rajanna A, Kishan V Brain delivery of transferrin coupled indinavir submicron lipid emulsions – pharmacokinetics and tissue distribution. Colloids Surf. B Biointerfaces 86(2), 305–313 (2011). [DOI] [PubMed] [Google Scholar]