

Abstract
Efficiently carrying out the oxygen reduction reaction (ORR) is critical for many applications in biology and chemistry, such as bioenergetics and fuel cells, respectively. In biology, this reaction is carried out by large, transmembrane oxidases such as heme-copper oxidases (HCOs) and cytochrome bd oxidases. Common to these oxidases is the presence of a glutamate residue next to the active site, but its precise role in regulating the oxidase activity remains to be understood. In order to gain insight into the role, we herein report that incorporation of glutamate next to a designed heme-copper center in two biosynthetic models of HCOs improves O2-binding affinity, facilitates protonation of reaction intermediates, and eliminates release of reactive oxygen species. High-resolution crystal structures of the models revealed extended, water-mediated hydrogen bonding networks involving the glutamate. Electron paramagnetic resonance of the cryoreduced oxy-ferrous centers at cryogenic temperature followed by thermal annealing allowed observation of the key hydroperoxo intermediate that can be attributed to the hydrogen bonding network. By demonstrating these important roles of glutamate in oxygen reduction biochemistry, this work offers deeper insights into its role in native oxidases, may guide the design of more efficient artificial ORR enzymes or catalysts for applications such as fuel cell cathodes.
Graphical Abstract
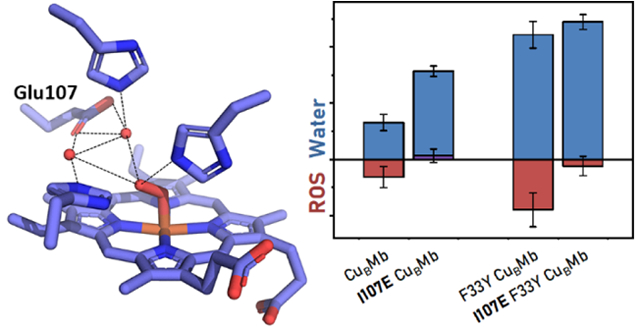
The oxygen reduction reaction (ORR) catalyzed by terminal oxidases, such as heme-copper oxidases (HCOs)1,2 and cytochrome bd oxidases,3 plays an important role in bioenergetic processes such as respiration. Both HCOs and cytochrome bd oxidases are heme-containing transmembrane proteins that efficiently deliver electrons to oxygen and use the energy of reduction to generate a proton gradient that drives energy-intensive processes such as the synthesis of ATP. In HCOs, the ORR is catalyzed by a heme-copper site, while the reaction is proposed to be carried out by a single heme d center in cytochrome bd oxidases.
In addition to electron delivery, efficient reduction of oxygen by terminal oxidases requires coordinated delivery of protons during the catalytical cycle (Scheme 1). Protons make their way into the active sites by proton pathways that are extended, hydrogen-bonded chains of amino acids and water molecules.4 One example is the D-pathway in HCOs which delivers the majority of the protons used in the catalytic cycle and ends at a glutamate (Glu) residue near the active site.5 Similarly, crystal and cryo-electron microscopy structures of cytochrome bd oxidase and mutational studies suggest that a Glu near the heme d is critical in oxidase activity.6–11
Scheme 1.
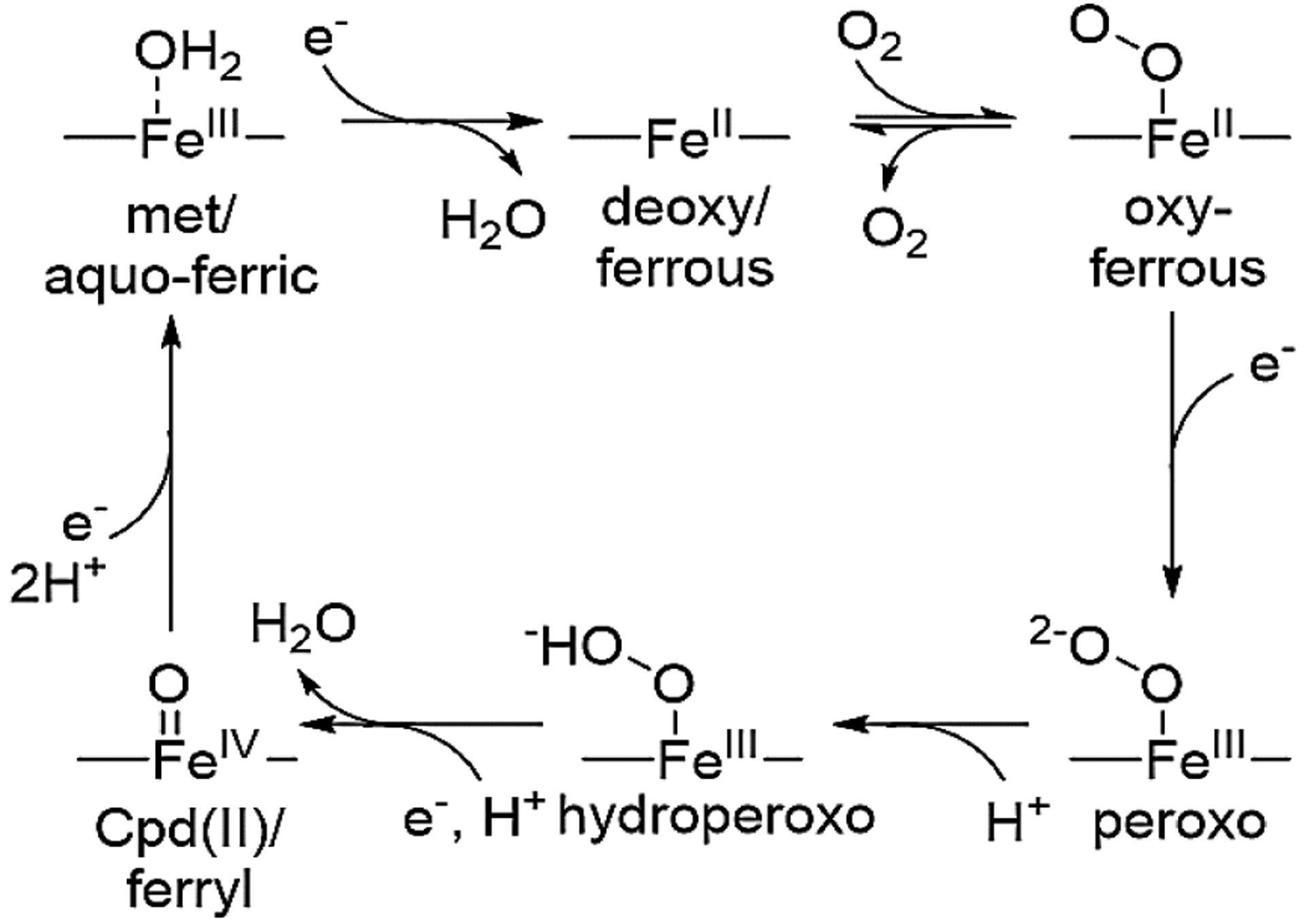
Proposed HCO reaction mechanism
While biology has evolved through millions of years to produce these oxidases for such important function in bioenergetics, the precise role of the Glu in regulating the oxidase activity remains to be understood. It has been difficult to pinpoint subtle structural features around the Glu, including the weak hydrogen bonding interactions in these large membrane enzymes, for which high-resolution crystal structures are not easily obtained. To overcome this limitation, synthetic models of HCOs using organic molecules have been obtained,12 but it has been difficult to incorporate residues such as Glu that often play a role in weak, non-covalent interactions at precise location in the models. To address these issues, we have been using small stable proteins with rigid scaffolds, such as sperm whale myoglobin (swMb), to prepare biosynthetic models of HCOs, allowing both active site structures such as the heme-copper center and residues that are involved in weak and non-covalent interactions to be readily incorporated and studied. We have successfully engineered several variants of swMb to be structural and functional mimics of terminal oxidases.13–18 The initial variant, L29H/F43H-swMb, called CuBMb, displayed relatively slow ORR.19,20 A subsequent improvement was achieved by introducing a tyrosine to yield either F33Y-CuBMb or G65YCuBMb, which achieved higher rates and over 1000 turnovers of oxygen reduction.20 When combined with an efficient redox partner for electron transfer, one of the variants displayed ORR rate that rivals that of native HCOs.21,22 Although our previous computer modeling suggests that G65YCuBMb is a closer structural model than F33YCuBMb, for the positioning of Tyr in a similar position as Tyr244 of bovine HCO, and G65YCuBMb is slightly more active, we choose to focus our efforts on F33YCuBMb in this study, because it is much more stable and crystallizable than G65YCuBMb, while still displaying HCO activity.
While this success of biosynthetic models is exciting, they still release 7–32% of reactive oxygen species (ROS) during turnover, in comparison with almost no ROS in native oxidases.20,21 Release of such ROS is an indicator of subpar efficiency of the enzyme. Successful rational improvement of the enzyme function to eliminate this ROS release would not only be a true test of our understanding of native enzymes, but also allow application of this knowledge to the design of more efficient artificial ORR enzymes or catalysts for use in fuel cell cathodes.
Previously, we have investigated the structural features of the oxy- and peroxo- intermediates of F33Y-CuBMb using EPR and crystallographic studies.21 Based on the results, we proposed that oxygen activation for complete reduction to water could be attributed to engineered protic residues that organize an extended hydrogen bonding network of water molecules in the active-site. While rational protein design has been extremely successful, it has largely focused on design of interactions directly to the substrate or cofactor. On the other hand, extended hydrogen bonding networks, particularly those mediated by water molecules are well known in natural enzymes;23–29 however, reports of rationally incorporating such water mediated interactions have been limited.30–32 Building on our previous results, we herein report incorporation of a Glu residue, which is known to be important in both HCOs and bd oxidases, into CuBMb and F33Y-CuBMb through rational extension of the water-mediated hydrogen bonding network in its active site, and show that the introduced Glu plays a key role in eliminating ROS formation. In addition, we have obtained insights into the role of Glu using high-resolution crystal structures and EPR of cryoreduced oxy-I107E-CuBMb.
MATERIALS AND METHODS
Protein mutagenesis, expression, and purification.
Site directed mutagenesis, and protein expression and purification were carried out as previously reported,20 with minor changes to improve yield and purity. The source of the cloned gene is described earlier;33 the sequence corresponds to Uniprot P02185–1. For consistency, sequence numbering used here and in the accompanying manuscript omits the initiator methionine, which has been the convention in the literature since the initial sequencing of myoglobin obtained directly from sperm whale tissue revealed that the protein sequence begins with the subsequent valine.34
Expression was carried out with E. coli BL-21 DE3 Star strain (Life Technologies); other strains produced inferior yield and purity. Dialysis of protein after refolding was carried out against 10 mM tris sulfate pH 8.0. Dialysis was limited to two exchanges - first for 4 hours, second overnight. Protein was loaded onto the size exclusion column within 12 hours of completing dialysis. Protein batch activity was validated by oxygen reduction activity assays.17,20,35
Oxygen reduction assays.
The oxygen reduction rate assays were carried out as previously reported, by first determining the rates of O2 consumption in the absence and presence of the ROS scavengers, catalase and superoxide dismutase, and then calculate the rates of water and ROS formation using equations described in a previous publication,17,20,35 using a Clark type oxygen electrode, ascorbate and TMPD as reductant and mediator, respectively, and ROS scavengers.
Oxygen affinity assays.
Measurements of O2 affinity were performed as described earlier.15
Crystallization, I107E-CuBMb.
Resting state (aquo-met-ferric) crystals of I107E-CuBMb were grown under modified conditions previously reported.20 Sitting drop vapor diffusion was utilized to increase drop size and allow for larger crystals. Protein was concentrated to 1.5–2.0 mM in 20 mM tris sulfate pH 8.0 buffer. Crystallization solution was composed of 100 mM tris sulfate pH 8.6, 200 mM sodium acetate, and 30% w/v PEG 10,000. Solutions were mixed 1:1 to yield a final drop volume of 150–200 μL. This was equilibrated by vapor diffusion against 30 mL of crystallization solution, at 4°C, in dark. Crystals formed within 1 week. Two crystal forms were observed - long needles which diverged into fan shapes (P21) and thicker rectangular crystals (P212121). The orthorombic crystal form was propagated by seeding new crystallization drops.
Crystallization, I107E-F33Y-CuBMb.
Resting state (aquo-met-ferric) crystals of I107E-CuBMb were grown under modified conditions previously reported.20 Sitting drop vapor diffusion was utilized to increase drop size and allow for larger crystals. Protein was concentrated to 1.5–2.0 mM in 20 mM tris sulfate pH 8.0 buffer. Crystallization solution was composed of 100 mM tris sulfate pH 8.6, 200 mM sodium acetate, and 30% w/v PEG 10,000. Solutions were mixed 1:1 to yield a final drop volume of 150–200 μL. This was equilibrated by vapor diffusion against 30 mL of crystallization solution, at 4°C, in dark. Crystals formed within 1 week.
Crystal preparation, oxy-I107E-CuBMb.
Oxy-I107E CuBMb crystals were prepared by anaerobic reduction of aquo-met-ferric I107ECuBMb and exposure to oxygen, based on previously reported methods.36,37 Several large and robust orthorhombic crystals were harvested into a small container. This was brought into an anaerobic atmosphere chamber (Coy Labs) by 27 headspace purges, down to 8 in. Hg of vacuum each, in the antechamber. Subsequently, crystals were transferred to a small finely-fritted filter reservoir, filled with degassed crystallization solution containing 20 mM dithionite. These crystals were left to soak for 10 minutes to reduce protein and eliminate residual oxygen. Soaking solution was gently removed from the reservoir through the frit using a syringe, driven by a syringe pump and connected with plastic tubing to the bottom of the filter reservoir. Solution was replaced by manually adding fresh degassed solution into the filter reservoir. This washing was repeated four times total. Deoxy crystals were harvested into small (~2 μL) drops on slides and sealed in a high pressure bomb. The bomb was removed from the chamber and immediately pressurized to ~1500 psi O2 for 1–10 minutes. The bomb was carefully vented over 30s and the crystal harvested after brief soak in O2 supersaturated cryoprotectant (50% PEG 400 supersaturated with O2 under similar pressurization conditions).
Crystallography, general methods.
Unless otherwise noted, X-ray diffraction data were collected under cryogenic conditions, data reduction and scaling were carried out with HKL2000, refinement was carried out in Phenix, and model building was carried out using Coot. Graphics were generated using VMD with Tachyon rendering. Omit map images generated using PyMol.
Crystallography, met-I107E-CuBMb.
X-ray diffraction data were collected on beamline x29 at NSLS. The structure was solved using molecular replacement using the structure of CuBMb (PDB ID: 4FWZ) as a model. Sidechains and water molecules were rebuilt manually and iteratively with refinement.
Crystallography, met-I107E-F33Y-CuBMb.
X-ray diffraction data were collected on beamline 21-ID-G at APS. The structure was solved by isomorphous replacement using the backbone and heme coordinates from the crystal structure of F33Y-CuBMb (PDB ID: 4FWX). R-free flags were copied from the 4FWX dataset and extended to the resolution of this dataset with random selection. Sidechains and water molecules were rebuilt manually and iteratively with refinement.
Crystallography, 1.18Å oxy-I107E-CuBMb.
Single crystal spectroscopic data and X-ray diffraction data were collected at SSRL beamline 11–1. The oxygen-bound state of the protein was confirmed using the microspectrophotometer;38,39 high attenuation of the X-ray beam was used to minimize radiolytic reduction. Data reduction and scaling was carried out with XDS, pointless, aimless, and truncate, automated using the script ‘autoxds.’ The structure was solved by isomorphous replacement using the backbone and heme coordinates from the crystal structure of F33Y-CuBMb (PDB ID: 4FWX). R-free flags were copied from the 4FWX dataset and extended to the resolution of this dataset with random selection. Refinement was performed with Phenix and REFMAC. Sidechains and water molecules were rebuilt manually and iteratively with refinement. The O-O group was modeled as peroxide (PER), which would be consistent with the final state in the crystal, due to reduction by the X-ray beam.
Cryoreduced oxy-ferrous EPR sample preparation.
Each protein was concentrated to ~1–3 mM concentration and reduced with excess dithionite anaerobically under nitrogen atmosphere. Dithionite was removed under anaerobic conditions by a small GE Sephadex G-25 desalting column. The protein was reconcentrated anaerobically. The protein solution was removed from the anaerobic chamber and immediately, slowly, and gently sparged with pure oxygen gas from a syringe. The protein was briefly centrifuged to remove bubbles and mixed with pure glycerol to yield 15% final glycerol concentration. This solution was quickly transferred to an EPR tube and slowly frozen with liquid nitrogen.
Samples were transferred to a glass dewar filled with liquid nitrogen and irradiated with ~3 Mrad of gamma radiation over 440 minutes using a Gammacell 220 irradiator containing a 60Co source. Irradiated samples were stored and transported in liquid nitrogen at all times.
EPR spectroscopy.
X-band CW EPR spectroscopy was performed using a Bruker ESP-300 spectrometer equipped with an Oxford Instruments ESR-900 helium flow cryostat. EPR spectra were recorded at 20K, a modulation frequency of 100 kHz, a modulation amplitude of 5 G, microwave power of 2 mW and with a sweep rate of 10 G/s. Spectra were recorded at microwave frequencies of approximately 9.364 GHz, with the precise microwave frequencies recorded for individual spectra using a Hewlett-Packard Microwave Frequency Counter (HP5352B).
Safety Statement.
No unexpected or unusually high safety hazards were encountered with the reported work.
RESULTS AND DISCUSSION
To engineer a Glu into the water mediated hydrogen bonding network of CuBMb and F33Y-CuBMb, we start with their crystal structures (Figure 1, left).20 In our previous study, we proposed that the presence of W1 in the active site of both proteins and its interactions with active site residues are crucial for imparting complete reduction of O2 to H2O, instead of partially reduced ROS such as superoxide or peroxide.36 The location and choice of introducing a Glu around the active site were based on visual analysis and basic rotamer modeling in PyMol. The I107E mutation showed the most optimal H-bonding interaction with W1, with minimal perturbation of the active site (Figure 1, center). This choice has been corroborated by a previous study showing that the I107E mutation improved NO reduction activity of a related model, presumably due to improved protonation.40
Figure 1.
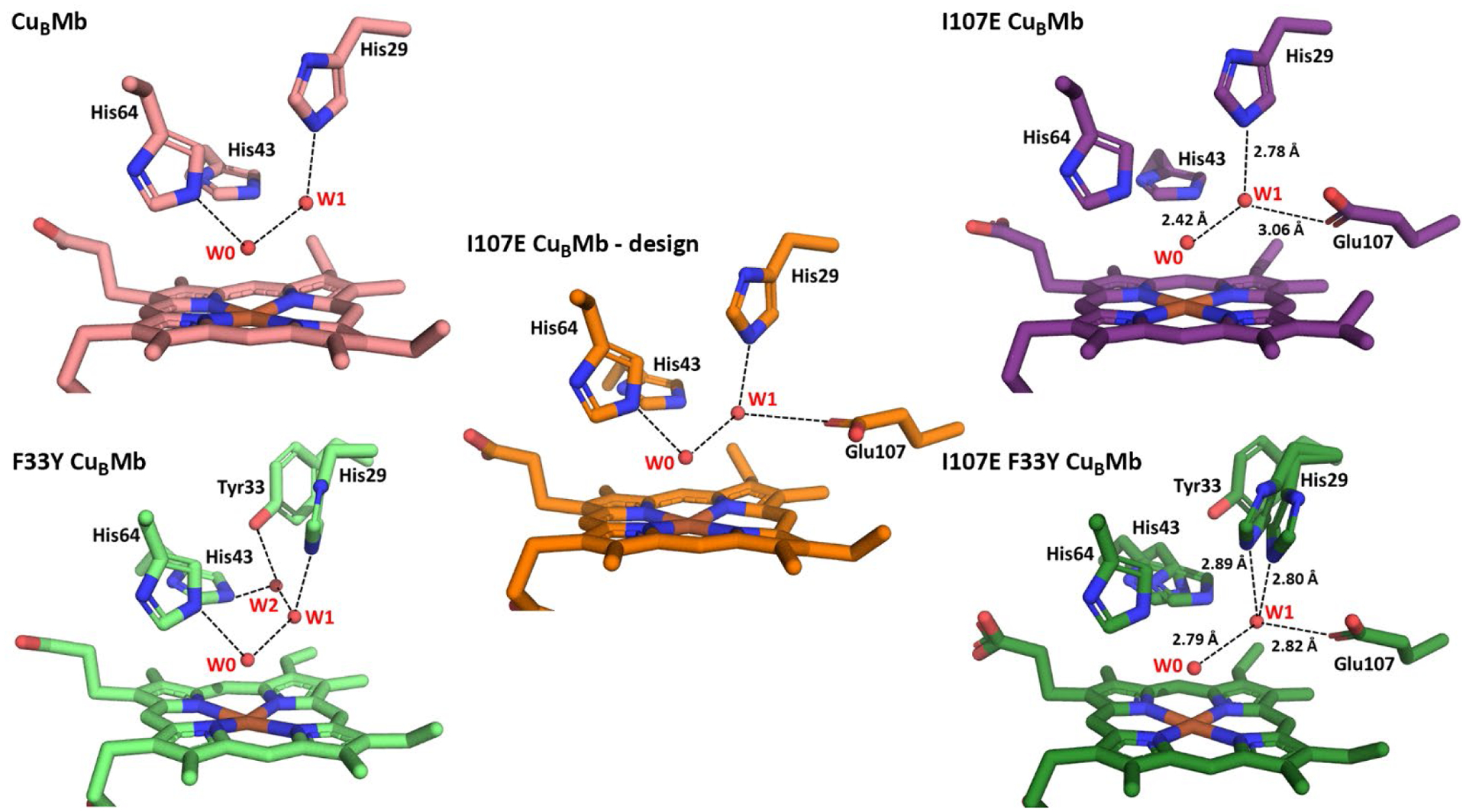
Structures of CuBMb variants. (left) Crystal structures of CuBMb and F33Y-CuBMb, highlighting water-mediated hydrogen-bonding network in the active site. (center) Computational model of I107E-CuBMb based on crystal structure of CuBMb and I107E containing Mb variant from PDB 3M38. (right) Crystal structures of I107E-CuBMb and I107E/F33Y-CuBMb.
Construction, expression, and purification of I107E-CuBMb and I107E/F33Y-CuBMb was carried out using a protocol reported previously,20 and their identities were confirmed by sequencing of the DNA and mass spectrometry of the purified proteins. As purified, the proteins displayed a Soret peak at 408 nm and a visible spectrum indicative of a high spin ferric heme (Figure S1), consistent with the aquo-ferric (met) resting state of Mb.41 The extinction coefficients of the Soret peak in the resting states of I107E-CuBMb and I107E/F33Y-CuBMb were determined to be 181 mM−1 and 175 mM−1, respectively.
The successful incorporation of a carboxylate group to interact with the water-mediated H-bonding network of both proteins was confirmed by obtaining crystal structures of the resting states of I107E-CuBMb and I107E/F33Y-CuBMb determined at 1.70 Å and 1.47 Å resolution, respectively (Figure 1, right). In both structures, the active site contains a water molecule (W0) coordinating the heme and forming a H-bond with His64 and a second water molecule (W1) near His29 and engaging in a H-bond with W0 (Figure S2), similar to those found in previous structures of CuBMb variants, suggesting that introducing the I107E mutation caused almost no perturbation of the active structure, including the delicate H-bonding network. More importantly, the structure revealed that the W1 near His29 also engages in a H-bond with Glu107, as predicted in our initial computational design of the mutants.
Having confirmed the presence of a new H-bond in both mutants, we proceeded to investigate its effect on the functional properties of the enzymes.20 As shown in Figure 2, CuBMb reduced O2 at a total rate of 0.49 s−1, producing H2O at 0.33 s−1 and ROS at 0.16 s−1, which amounts to 32% selectivity for ROS as reported previously.20 Introducing I107E almost completely eliminated the ROS formation. Similarly, F33YCuBMb reduced O2 at a total rate of 1.56 s−1, producing water at 1.1 s−1 and ROS at 0.5 s−1, which equates to 29% selectivity for ROS.20 Introducing the I107E mutation into F33Y-CuBMb reduced the ROS formation to <5% in I107E/F33Y-CuBMb. Therefore, the I107E mutation has resulted in effective elimination of ROS release in both proteins. The rate of water formation of CuBMb was roughly doubled by the I107E mutation. The I107E mutation did not increase the rate of water production of F33Y-CuBMb.
Figure 2.
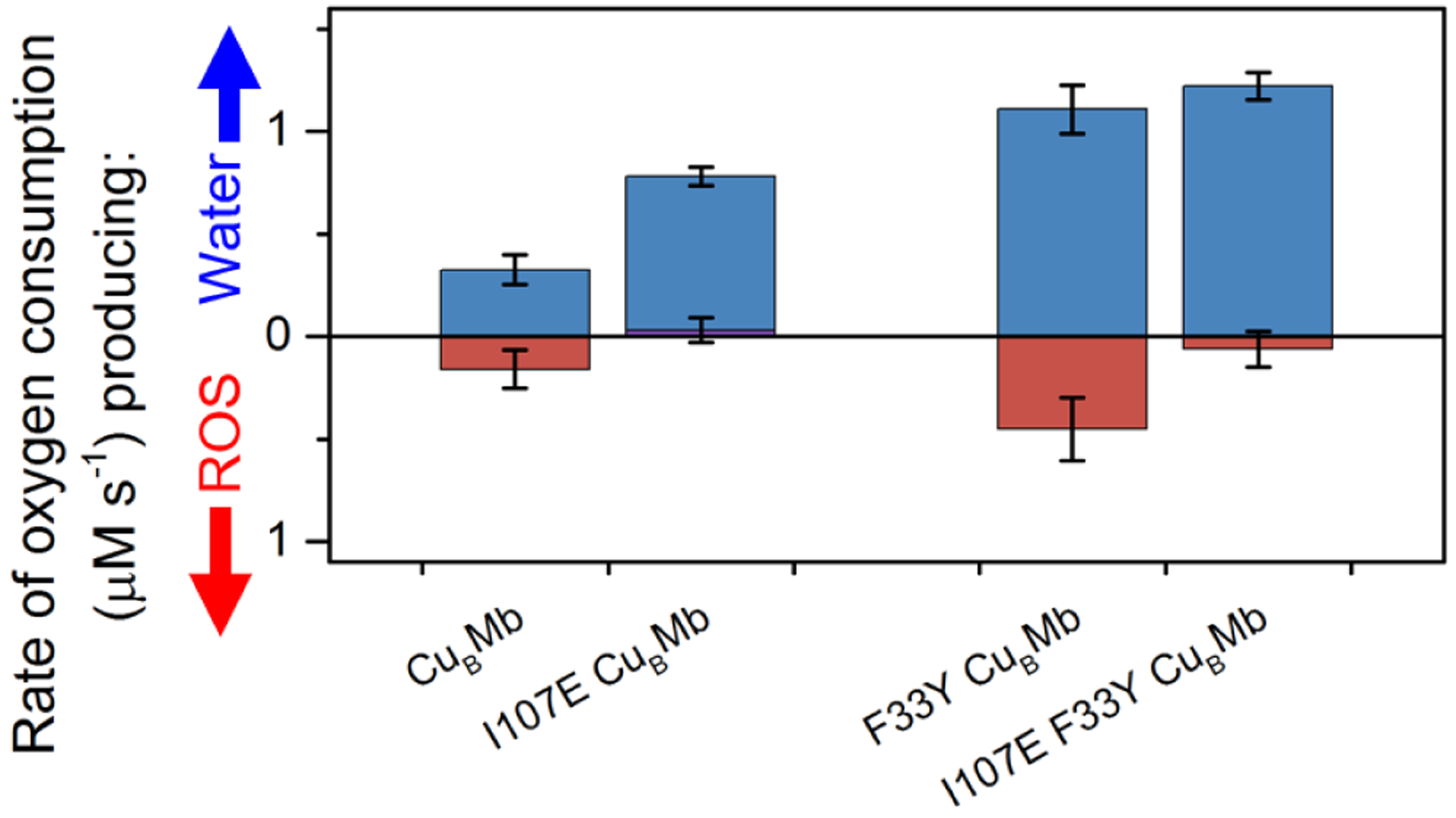
Rates of O2 consumption yielding water (blue) or reactive oxygen species (ROS) (red), measured and calculated as reported previously.20,17,35 Rates of CuBMb, F33Y-CuBMb, have been reported previously20 and are included here for visual comparison.
Interestingly, we noted that the raw O2 consumption traces as a function of O2 concentration for the I107E mutants showed rate beginning to saturate at lower O2 concentrations than in the variants without the mutation (Figure S3), suggesting that the I107E mutation imparts enhanced oxygen affinity. We confirmed this suggestion by measuring the oxygen affinities of these variants using laser-flash photolysis (Table S1 and Figure S4), which indeed showed that the I107E mutation resulted in substantial increases in O2 affinity for both proteins. Previous studies at tuning the O2-binding affinity of Mb have shown that mutations of Leu29 and Ile107 are critical for tuning the affinity.42–44 An increased O2 affinity has been typically associated with larger hydrophobic sidechains and water exclusion in this region. The observation of increase in affinity with the introduction of a charged sidechain is noteworthy and has not been previously reported, to our knowledge. Furthermore, it is known that increased O2 affinity slows auto-oxidation and thus release of ROS from myoglobin,45–49 which may contribute to the effective elimination of ROS species formation during the O2 reduction by the I017E-CuBMb and I107E/F33Y-CuBMb.
In order to gain further understanding of how the introduced Glu residue tunes the reactivity of CuBMb and F33Y-CuBMb, we sought to probe the interactions of the oxygen-bound intermediate. We and others have previously shown that EPR is a valuable tool for probing the interactions within the active sites of heme and non-heme iron proteins.50–68 In particular, cryoreduction of oxy-ferrous heme proteins to yield EPR-active peroxo- and hydroperoxo-ferric heme states, which retain the conformation of the oxy heme precursor, has been extremely successful in probing the interactions of oxygen with its active-site environment in oxygen-activating proteins.50–58,65 As mentioned earlier, this technique was used to understand the structural features responsible for F33Y-CuBMb reactivity.36 Here, we apply this technique to compare the structural features and interactions of the newly designed I107E-CuBMb and I107E/F33Y-CuBMb.
The EPR spectra of 77K cryoreduced oxy-CuBMb and oxy-F33Y-CuBMb, and their I107E variants are shown in Figure 3 and their g-values are summarized in Table 1. Both cryoreduced CuBMb and F33Y-CuBMb spectra collected at 20K exhibit two new species. The EPR parameters of the first species, with g1 = 2.24 in both proteins, suggest that it is a ferric peroxo state in which the peroxo ligand engages in a hydrogen bond with the distal His64.50–53,61 The somewhat broadened g1 feature in F33Y-CuBMb indicates a slight conformational distribution. The second species with g1 = 2.29 in both proteins is in the range characteristic of ferric-hydroperoxo intermediates.
Figure 3.
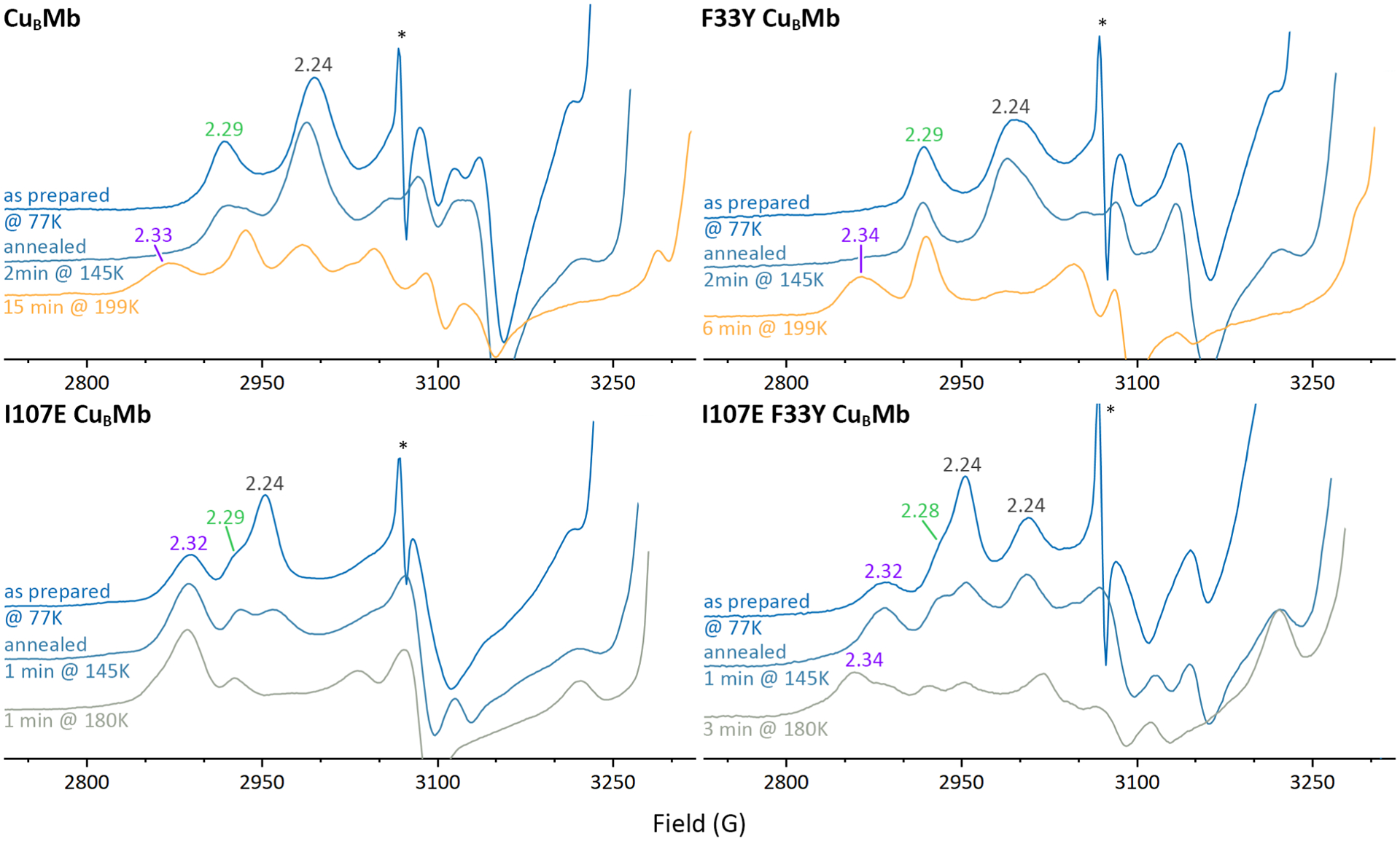
Representative 20 K EPR spectra of oxy-CuBMb proteins after 77K radiolytic reduction with a ~3Mrad dose of γ radiation from 60Co, and after subsequent stepwise annealing as indicated (sharp signal marked by asterisk is due to hydrogen atoms radiolytically generated at 77K in quartz EPR tube).
Table 1.
g-Values of Cryoreduced Oxy-ferrous Myoglobins
| Protein | g1 | g2 | g3 |
|---|---|---|---|
| CuBMb | |||
| peroxo | 2.24 | 2.13 | 1.96 |
| hydroperoxo1 | 2.29 | 2.16 | 1.94 |
| hydroperoxo2 | 2.34 | 2.18 | 1.93 |
| F33Y-CuBMb | |||
| peroxo | 2.24 | 2.13 | 1.96 |
| hydroperoxo1 | 2.29 | 2.17 | 1.95 |
| hydroperoxo2 | 2.34 | 2.19 | 1.94 |
| I107E-CuBMb | |||
| peroxo | 2.27 | 2.16 | nd |
| hydroperoxo A1 | 2.29 | 2.20 | nd |
| hydroperoxo B1 | 2.32 | 2.17 | 1.94 |
| I107E/F33Y-CuBMb | |||
| peroxo A | 2.23 | 2.12 | nd |
| peroxo B | 2.27 | 2.16 | nd |
| hydroperoxo A1 | 2.28 | 2.17 | nd |
| hydroperoxo B1 | 2.32 | 2.17 | 1.95 |
| hydroperoxo C2 | 2.34 | 2.19 | 1.91 |
Primary hydroperoxo product formed at 77K.
Hydroperoxo product formed upon annealing.
Annealing of cryoradiolytically generated peroxo-heme species has been shown to provide further insights into the local environment. Therefore, we performed EPR of the frozen samples after sequential annealing at progressively higher temperatures. Spectroscopic traces of the samples at key points are shown in Figure 3, and the full series of spectra is shown in Figure S5. Annealing of CuBMb and F33Y-CuBMb to 145K causes no change in speciation and only slight changes in lineshapes reflecting conformational changes. Annealing the CuBMb peroxo species for 15 minutes at 199K generates a hydroperoxo state with g1 = 2.34, which can be assigned as arising from protonation of some g1 = 2.24 peroxo centers and some rearrangement of the conformation of the g1 = 2.29 hydroperoxo state into a more rhombic hydroperoxo state. In contrast, annealing of cryoreduced F33Y-CuBMb for only 6 minutes at 199K causes essentially complete protonation of the peroxo form with conversion to the g1 = 2.34 hydroperoxo state, consistent with facilitated proton transfer by this mutation.
The 77K EPR spectra of the cryoreduced oxy-I107E-CuBMb variant are distinctly different from those of the oxy-CuBMb and its F33Y variant. The major signal produced at 77K, with g1 = 2.27 is assigned as a peroxo ferric species significantly more rhombic than the peroxo species in the other two proteins. This species is also observed in the 77K of the double mutant. The higher rhombicity of the ferric peroxo signals of these I107E-containing mutants compared to those without the mutation is an indicator of a stronger H-bond interaction between the cryogenerated peroxo ligand, and thus the parent O2 ligand in the ferrous-oxy complex, and a nearby hydrogen bond donor. Furthermore, prior to annealing, a highly rhombic hydroperoxo signal with g1 = 2.32 is observed in the I107E mutants which is not observed in CuBMb and F33Y-CuBMb until they were annealed at 173K (Figure S5). The lower rhombicity hydroperoxo ferric signal, g1 = 2.28-2.29, manifests as a shoulder of the major peroxo peak in both I107E variants. The spectroscopic distinction between the detected hydroperoxo intermediates with g1 = 2.29 and 2.32-4 may be explained by different geometry of Fe-O-O-H unit.57
In the case of cryoreduced I107E mutant we observed the nearly complete decay of the major peroxo signals in the I107E mutant upon annealing at 180K for one minute (Figure 3 and S5). The decay of the peroxo signal is concomitant with the increase of the g1 = 2.32-2.34 hydroperoxo signal, indicative that protonation of the peroxo species has produced the hydroperoxo state. This observation of rapid protonation of the major peroxo species in 1 minute annealing at 180K to yield hydroperoxo in the protein containing the I107E mutation is in stark contrast with the results of annealing of CuBMb, without the I107E mutation, where residual peroxo state remains after three minutes of 180K annealing, and strongly supports the generation of a very mobile proton in the active site by this mutation. Intriguingly, in the double mutant, the presence of the F33Y change slightly stabilize the peroxo state and slowed its protonation at 180K (Figure 3 and S5).
Further analysis of the time-dependence of the final annealing spectra in Figure S5 is shown in Figure 4. While all samples eventually decay to an EPR silent state, the proteins lacking the I107E mutation require longer times and higher temperatures (199K) than the proteins containing the mutation (180K). Consistent with previous results on myoglobin, subsequent cryoreduction of this EPR-silent state in I107E/F33Y-CuBMb produces a g1 = 2.54 signal consistent with reductive generation of a ferric hydroxo (Fe(III)-OH; Figure S6). These results confirm that (1) the EPR-silent state is the ferryl compound II (Fe(IV)=O), and furthermore that (2) the active site environment of I107E variants of CuBMb facilitates protonation of the Fe ligand.
Figure 4.
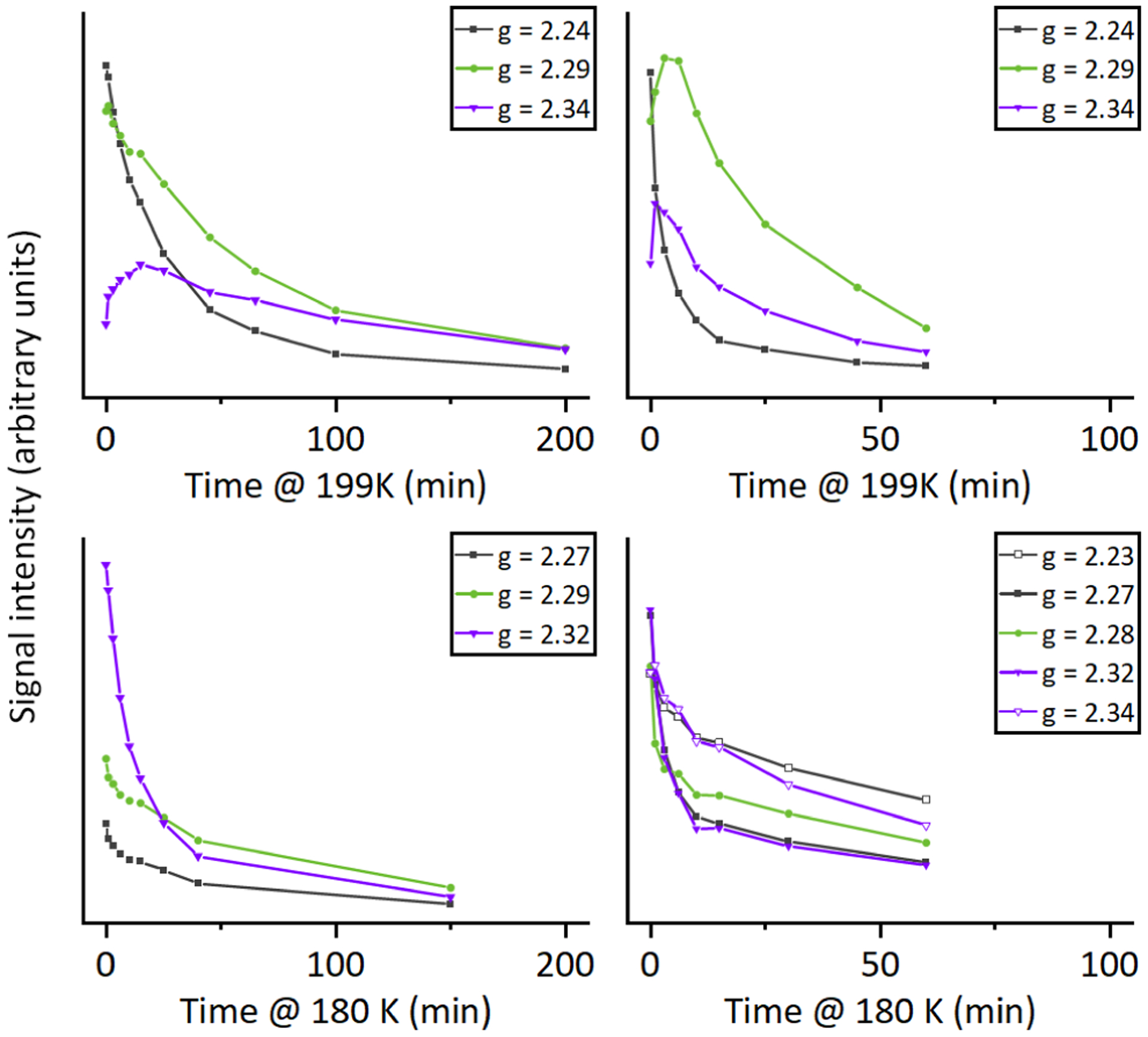
Temperature dependence of the g1 peak intensity (in arbitrary units) of the peroxo (gray ■, □) and hydroperoxo (green ● and purple ▽, ▼) states, showing faster decay at lower temperatures in I107E mutants.
This contrast between the decay kinetics of the (hydro)peroxo species of I107E and parent enzymes at cryogenic temperatures further supports the design hypothesis, that Glu107 stabilizes an additional proton in the active site, enabling the hydrogen bonding network to efficiently activate oxygen by quickly delivering two protons to promote heterolytic O-O bond cleavage during turnover. This study provides structural and spectroscopic basis for previously observed D2O/H2O isotopic effects in electro-chemical studies on the role of I107E mutation on the rate-determining step of O2 reduction in the CuBMb variant,13 and demonstrated the dominant role of proton transfer residue in HCO activity.
In order to gain further insight into the structural basis for oxygen activation of these efficient enzymes, we obtained the crystal structure of the dioxygen-bound intermediate. Oxy-I107E-CuBMb crystals were obtained as previously reported, by reducing ferric crystals, followed by rapid exposure to oxygen (details in SI). The active site structure of oxy-I107E-CuBMb, solved at 1.18 Å resolution, is shown in Figure 5. While the occupancy of the dioxygen molecule is incomplete (see Figure S7 for alternative occupancies), likely due to large crystals and short soak times to prevent autooxidation, the structure still provides a structural basis for understanding the role of Glu107. As predicted from design, a water molecule is present within hydrogen bonding distance of the bound dioxygen, and furthermore, this water molecule engages in H-bonds with Glu107 and His29. The geometry of the H-bonding interactions with oxygen is close to ideal, suggesting that the interactions likely polarize the O2 bond and could facilitate reduction and O-O bond cleavage.
Figure 5.
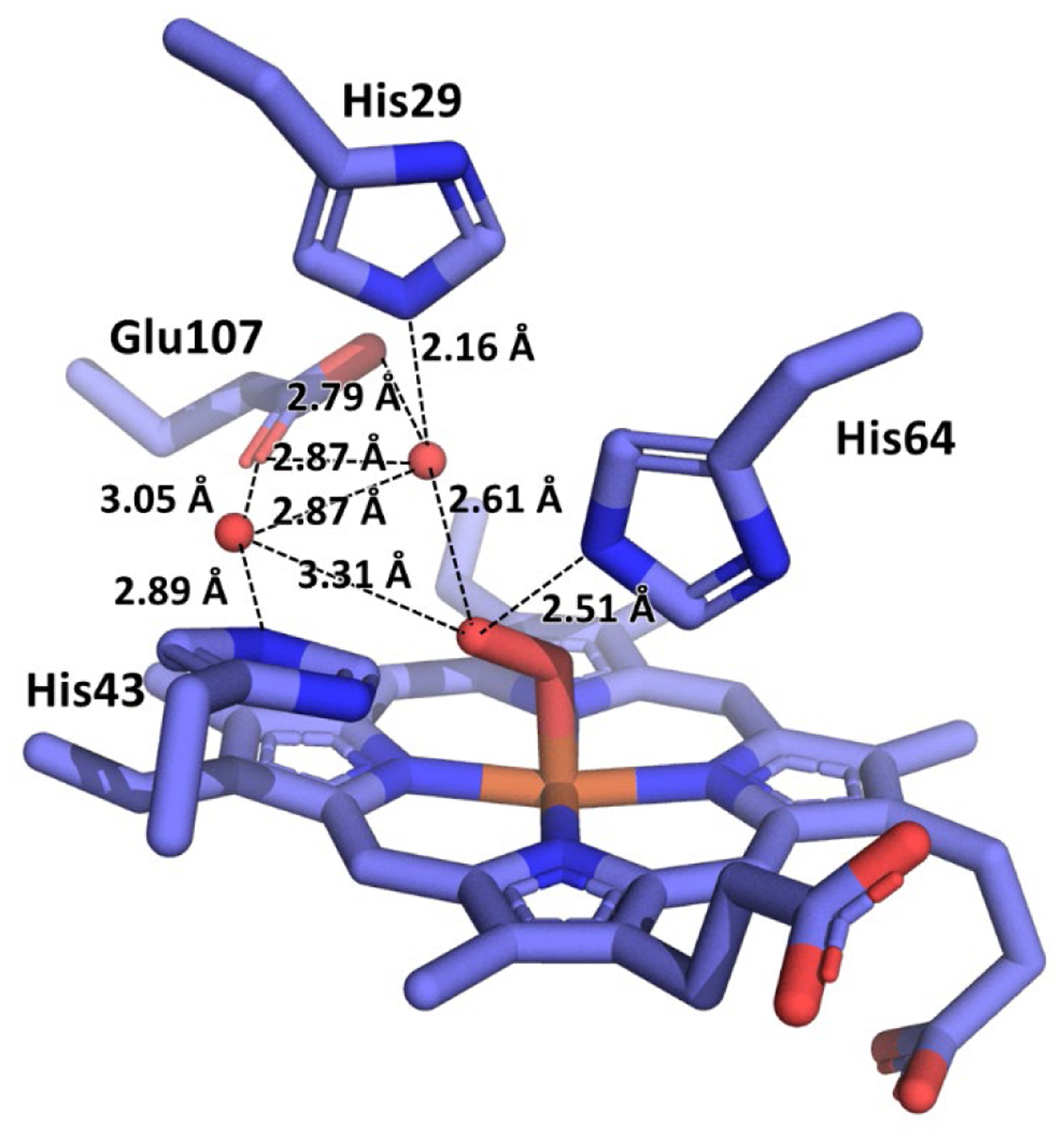
Structure of the active site of oxy-I107E-CuBMb at 1.18 Å resolution, with pertinent interatomic distances labeled. Distal pocket omit map shown in Figure S2.
CONCLUSION
Taken together, the results above demonstrate that the extension of the hydrogen bonding network within the active site of our designed biosynthetic models of HCO by introducing Glu107 improves O2-binding affinity, facilitates protonation of reaction intermediates and eliminates ROS release, resulting in cleaner oxygen reductase chemistry. We have provided X-ray structural and EPR spectroscopic evidence that the enhanced catalytic performance is a result of Glu107 engaging in strong hydrogen bonds with the active site water network, stabilizing protons in the active site (Scheme 2), which are then available for efficient oxygen polarization, activation, and protonation of the formed peroxo- or hydroperoxo intermediates during turnover. These results support the role of the D-pathway-terminating Glu residue, and potentially the Glu residue near the heme d in bd oxidases, in providing protons to the bound dioxygen during the first steps of oxygen reduction. Moreover, the results presented here, extensively characterizing the hydrogen bonding networks of 4 different variants of Mb, clearly demonstrate that designing long-range, water-mediated hydrogen-bonding networks - a feature that has seen some but, minimal pursuit in either synthetic models using organic molecules as ligand or rational enzyme design strategies30–32 - can play a key role in conferring and fine-tuning enzymatic function, and should be pursued more actively in biomimetic modeling of native enzymes.
Scheme 2.
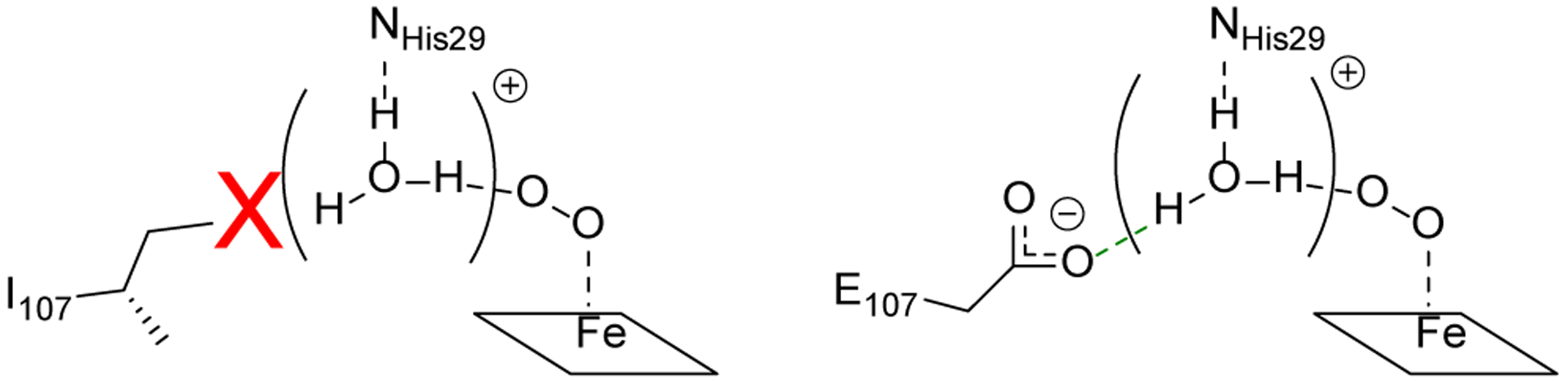
Proposed interaction of Glu107
Supplementary Material
ACKNOWLEDGMENT
The authors wish to thank Dr. Julian Reed for lending a helping hand in various experiments, Dr. Stoyan Toshkov for assistance with γ-irradiation, Dr. Howard Robinson for assistance with data collection at NSLS, Dr. Vukica Srajer and Dr. Robert Henning for beamline support at BioCARS for spectroscopic screening of oxy crystals, Dr. Vivian Stojanoff for beamline support at SSRL, and Drs. Parisa Hosseinzadeh, Shiliang Tian, and Ilya Denisov for helpful discussion.
Funding Sources
Research reported in this publication was supported by National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM062211 (YL) and R01GM111097 (BMH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This research used beamline x29 of the National Synchrotron Light Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-AC02-98CH10886.
This research used the Life Sciences Collaborative Access Team beamline 21-ID-G at the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
ABBREVIATIONS
- Mb
myoglobin
- swMb
sperm whale Mb
- WTMb
wild type swMb
- CuBMb
L29H F43H swMb
- HCO
heme-copper oxidase
- EPR
electron paramagnetic resonance
- ENDOR
electron-nuclear double resonance
Footnotes
This material is available free of charge via the Internet at http://pubs.acs.org.
Supporting Info
Oxygen affinities, X-ray diffraction data collection and refinement statistics and omit maps, resting state UV-Vis spectra, oxygen consumption rate curves as a function of oxygen concentration, full EPR spectral sets of annealing experiments, active-site crystal structures.
Accession Codes
Crystal structure of I107E CuB myoglobin: PDB ID 7KYR. Crystal structure of I107E F33Y CuB myoglobin: PDB ID 7L3U. Crystal structure of oxy-I107E CuB myoglobin: PDB ID 7L3Y. Sperm whale myoglobin: UNP Accession P02185–1
REFERENCES
- (1).Ferguson-Miller S, and Babcock GT (1996) Heme/Copper terminal oxidases. Chem. Rev. Wash. DC U. S 96, 2889–2907. [DOI] [PubMed] [Google Scholar]
- (2).Namslauer A, and Brzezinski P (2004) Structural elements involved in electron-coupled proton transfer in cytochrome c oxidase. FEBS Lett. 567, 103–110. [DOI] [PubMed] [Google Scholar]
- (3).Borisov VB, Gennis RB, Hemp J, and Verkhovsky MI (2011) The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta-Bioenerg 1807, 1398–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brzezinski P, and Gennis RB (2008) Cytochrome c oxidase: exciting progress and remaining mysteries. J. Bioenerg. Biomembr 40, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kaila VRI, Verkhovsky MI, and Wikström M (2010) Proton-coupled electron transfer in cytochrome Oxidase. Chem. Rev. Wash. DC U. S 110, 7062–7081. [DOI] [PubMed] [Google Scholar]
- (6).Murali R, and Gennis RB (2018) Functional importance of Glutamate-445 and Glutamate-99 in proton-coupled electron transfer during oxygen reduction by cytochrome bd from Escherichia coli. Biochim. Biophys. Acta BBA - Bioenerg 1859, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Safarian S, Rajendran C, Müller H, Preu J, Langer JD, Ovchinnikov S, Hirose T, Kusumoto T, Sakamoto J, and Michel H (2016) Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 352, 583–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rappaport F, Zhang J, Vos MH, Gennis RB, and Borisov VB (2010) Heme–heme and heme-ligand interactions in the di-heme oxygen-reducing site of cytochrome bd from Escherichia coli revealed by nanosecond absorption spectroscopy. Biochim. Biophys. Acta BBA - Bioenerg 1797, 1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Belevich I, Borisov VB, Zhang J, Yang K, Konstantinov AA, Gennis RB, and Verkhovsky MI (2005) Time-resolved electrometric and optical studies on cytochrome bd suggest a mechanism of electron-proton coupling in the di-heme active site. Proc. Natl. Acad. Sci. U. S. A 102, 3657–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Theßeling A, Rasmussen T, Burschel S, Wohlwend D, Kägi J, Müller R, Böttcher B, and Friedrich T (2019) Homologous bd oxidases share the same architecture but differ in mechanism. Nat. Commun 10, 5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yang K, Zhang J, Vakkasoglu AS, Hielscher R, Osborne JP, Hemp J, Miyoshi H, Hellwig P, and Gennis RB (2007) Glutamate 107 in Subunit I of the Cytochrome bd Quinol Oxidase from Escherichia coli Is Protonated and near the Heme d/Heme b595 Binuclear Center. Biochemistry 46, 3270–3278. [DOI] [PubMed] [Google Scholar]
- (12).Adam SM, Wijeratne GB, Rogler PJ, Diaz DE, Quist DA, Liu JJ, and Karlin KD (2018) Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function. Chem. Rev 118, 10840–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mukherjee S, Mukherjee M, Mukherjee A, Bhagi-Damodaran A, Lu Y, and Dey A (2018) O2 Reduction by Biosynthetic Models of Cytochrome c Oxidase: Insights into Role of Proton Transfer Residues from Perturbed Active Sites Models of CcO. ACS Catal. 8, 8915–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Reed JH, Shi Y, Zhu Q, Chakraborty S, Mirts EN, Petrik ID, Bhagi-Damodaran A, Ross M, Moënne-Loccoz P, Zhang Y, and Lu Y (2017) Manganese and Cobalt in the Nonheme-Metal-Binding Site of a Biosynthetic Model of Heme-Copper Oxidase Superfamily Confer Oxidase Activity through Redox-Inactive Mechanism. J. Am. Chem. Soc 139, 12209–12218. NIHMS ID: NIHMS916523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bhagi-Damodaran A, Kahle M, Shi Y, Zhang Y, Ädelroth P, and Lu Y (2017) Insights Into How Heme Reduction Potentials Modulate Enzymatic Activities of a Myoglobin-based Functional Oxidase. Angew. Chem. Int. Ed 56, 6622–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bhagi-Damodaran A, Michael MA, Zhu Q, Reed J, Sandoval BA, Mirts EN, Chakraborty S, Moënne-Loccoz P, Zhang Y, and Lu Y (2016) Why copper is preferred over iron for oxygen activation and reduction in haem-copper oxidases. Nat. Chem 9, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bhagi-Damodaran A, Petrik ID, Marshall NM, Robinson H, and Lu Y (2014) Systematic Tuning of Heme Redox Potentials and Its Effects on O2 Reduction Rates in a Designed Oxidase in Myoglobin. J. Am. Chem. Soc 136, 11882–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sigman JA, Kwok BC, and Lu Y (2000) From myoglobin to heme-copper oxidase: Design and engineering of a CuB center into sperm whale myoglobin. J Am Chem Soc 122, 8192. [Google Scholar]
- (19).Sigman JA, Kim HK, Zhao X, Carey JR, and Lu Y (2003) The role of copper and protons in heme-copper oxidases: Kinetic study of an engineered heme-copper center in myoglobin. Proc. Natl. Acad. Sci. U. S. A 100, 3629–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Miner KD, Mukherjee A, Gao Y-G, Null EL, Petrik ID, Zhao X, Yeung N, Robinson H, and Lu Y (2012) A designed functional metalloenzyme that reduces O2 to H2O with over one thousand turnovers. Angew. Chem. Int. Ed 51, 5589–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yu Y, Cui C, Liu X, Petrik ID, Wang J, and Lu Y (2015) A Designed Metalloenzyme Achieving the Catalytic Rate of a Native Enzyme. J. Am. Chem. Soc 137, 11570–11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mukherjee S, Mukherjee A, Bhagi-Damodaran A, Mukherjee M, Lu Y, and Dey A (2015) A biosynthetic model of cytochrome c oxidase as an electrocatalyst for oxygen reduction. Nat. Commun 6, 8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kwon H, Basran J, Devos JM, Suardíaz R, Kamp M. W. van der, Mulholland AJ, Schrader TE, Ostermann A, Blakeley MP, Moody PCE, and Raven EL (2020) Visualizing the protons in a metalloenzyme electron proton transfer pathway. Proc. Natl. Acad. Sci 117, 6484–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Efimov I, Badyal SK, Metcalfe CL, Macdonald I, Gumiero A, Raven EL, and Moody PCE (2011) Proton Delivery to Ferryl Heme in a Heme Peroxidase: Enzymatic Use of the Grotthuss Mechanism. J. Am. Chem. Soc 133, 15376–15383. [DOI] [PubMed] [Google Scholar]
- (25).Amaya JA, Batabyal D, and Poulos TL (2020) Proton Relay Network in the Bacterial P450s: CYP101A1 and CYP101D1. Biochemistry 59, 2896–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Batabyal D, and Poulos TL (2013) Crystal Structures and Functional Characterization of Wild-Type CYP101D1 and Its Active Site Mutants. Biochemistry 52, 8898–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Stok JE, Yamada S, Farlow AJ, Slessor KE, and De Voss JJ (2013) Cytochrome P450cin (CYP176A1) D241N: Investigating the role of the conserved acid in the active site of cytochrome P450s. Biochim. Biophys. Acta BBA - Proteins Proteomics 1834, 688–696. [DOI] [PubMed] [Google Scholar]
- (28).Madrona Y, Hollingsworth SA, Khan B, and Poulos TL (2013) P450cin Active Site Water: Implications for Substrate Binding and Solvent Accessibility. Biochemistry 52, 5039–5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chreifi G, Baxter EL, Doukov T, Cohen AE, McPhillips SE, Song J, Meharenna YT, Soltis SM, and Poulos TL (2016) Crystal structure of the pristine peroxidase ferryl center and its relevance to proton-coupled electron transfer. Proc. Natl. Acad. Sci 113, 1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Huang X, Yang J, and Zhu Y (2013) A solvated ligand rotamer approach and its application in computational protein design. J. Mol. Model 19, 1355–1367. [DOI] [PubMed] [Google Scholar]
- (31).Jiang L, Althoff EA, Clemente FR, Doyle L, Röthlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, Hilvert D, Houk KN, Stoddard BL, and Baker D (2008) De Novo Computational Design of Retro-Aldol Enzymes. Science 319, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Jiang L, Kuhlman B, Kortemme T, and Baker D (2005) A “solvated rotamer” approach to modeling water-mediated hydrogen bonds at protein–protein interfaces. Proteins Struct. Funct. Bioinforma 58, 893–904. [DOI] [PubMed] [Google Scholar]
- (33).Springer BA, and Sligar SG (1987) High-level expression of sperm whale myoglobin in Escherichia coli. Proc. Natl. Acad. Sci 84, 8961–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Edmundson AB, and Hirs CHW (1961) The Amino-Acid Sequence of Sperm Whale Myoglobin: Chemical Studies. Nature 190, 663–665. [DOI] [PubMed] [Google Scholar]
- (35).Liu X, Yu Y, Hu C, Zhang W, Lu Y, and Wang J (2012) Significant increase of oxidase activity through the genetic incorporation of a tyrosine-histidine cross-link in a myoglobin model of heme-copper oxidase. Angew. Chem. Int. Ed Engl 51, 4312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Petrik ID, Davydov R, Ross M, Zhao X, Hoffman B, and Lu Y (2016) Spectroscopic and Crystallographic Evidence for the Role of a Water-Containing H-Bond Network in Oxidase Activity of an Engineered Myoglobin. J. Am. Chem. Soc 138, 1134–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Unno M, Chen H, Kusama S, Shaik S, and Ikeda-Saito M (2007) Structural Characterization of the Fleeting Ferric Peroxo Species in Myoglobin: Experiment and Theory. J. Am. Chem. Soc 129, 13394–13395. [DOI] [PubMed] [Google Scholar]
- (38).Meharenna YT, Doukov T, Li H, Soltis SM, and Poulos TL (2010) Crystallographic and Single-Crystal Spectral Analysis of the Peroxidase Ferryl Intermediate. Biochemistry 49, 2984–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cohen AE, Doukov T, and Soltis MS (2016) UV-Visible Absorption Spectroscopy Enhanced X-ray Crystallography at Synchrotron and X-ray Free Electron Laser Sources. Protein Pept. Lett 23, 283–290. [DOI] [PubMed] [Google Scholar]
- (40).Lin Y-W, Yeung N, Gao Y-G, Miner KD, Tian S, Robinson H, and Lu Y (2010) Roles of glutamates and metal ions in a rationally designed nitric oxide reductase based on myoglobin. Proc. Natl. Acad. Sci 107, 8581–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Antonini E, and Brunori M (1971) Hemoglobin and Myoglobin in their Reactions with Ligands (Frontiers of Biology, Vol. 21). North-Holland. [Google Scholar]
- (42).Carver TE, Brantley RE, Singleton EW, Arduini RM, Quillin ML, Phillips GN, and Olson JS (1992) A novel site-directed mutant of myoglobin with an unusually high O2 affinity and low autooxidation rate. J. Biol. Chem 267, 14443–14450. [PubMed] [Google Scholar]
- (43).Draghi F, Miele AE, Travaglini-Allocatelli C, Vallone B, Brunori M, Gibson QH, and Olson JS (2002) Controlling Ligand Binding in Myoglobin by Mutagenesis. J. Biol. Chem 277, 7509–7519. [DOI] [PubMed] [Google Scholar]
- (44).Smerdon SJ, Krzywda S, Brzozowski AM, Davies GJ, Wilkinson AJ, Brancaccio A, Cutruzzola F, Allocatelli CT, Brunori M, Brantley RE Jr., Carver TE, Eich RF, Singleton E, and Olson JS (1995) Interactions among residues CD3, E7, E10, and E11 in myoglobins: Attempts to simulate the ligand-binding properties of Aplysia myoglobin. Biochemistry 34, 8715–8725. [DOI] [PubMed] [Google Scholar]
- (45).Brantley RE, Smerdon SJ, Wilkinson AJ, Singleton EW, and Olson JS (1993) The mechanism of autooxidation of myoglobin. J. Biol. Chem 268, 6995–7010. [PubMed] [Google Scholar]
- (46).Arcon JP, Rosi P, Petruk AA, Marti MA, and Estrin DA (2015) Molecular Mechanism of Myoglobin Autoxidation: Insights from Computer Simulations. J. Phys. Chem. B 119, 1802–1813. [DOI] [PubMed] [Google Scholar]
- (47).Springer BA, Egeberg KD, Sligar SG, Rohlfs RJ, Mathews AJ, and Olson JS (1989) Discrimination between oxygen and carbon monoxide and inhibition of autooxidation by myoglobin. Site-directed mutagenesis of the distal histidine. J. Biol. Chem 264, 3057–3060. [PubMed] [Google Scholar]
- (48).Zhao X, Vyas K, Nguyen BD, Rajarathnam K, Mar GNL, Li T, Phillips GN, Eich RF, Olson JS, Ling J, and Bocian DF (1995) A Double Mutant of Sperm Whale Myoglobin Mimics the Structure and Function of Elephant Myoglobin. J. Biol. Chem 270, 20763–20774. [DOI] [PubMed] [Google Scholar]
- (49).Shibata T, Matsumoto D, Nishimura R, Tai H, Matsuoka A, Nagao S, Matsuo T, Hirota S, Imai K, Neya S, Suzuki A, and Yamamoto Y (2012) Relationship between Oxygen Affinity and Autoxidation of Myoglobin. Inorg. Chem 51, 11955–11960. [DOI] [PubMed] [Google Scholar]
- (50).Symons MCR, Petersen RL, and Perutz MF (1978) Electron capture by oxyhaemoglobin: an e. s. r. study. Proc. R. Soc. Lond. B Biol. Sci 201, 285–300. [DOI] [PubMed] [Google Scholar]
- (51).Davydov RM (1980) [Optical and ESR-spectroscopic study of electronic adducts of oxymyoglobin and oxyhemoglobin]. Biofizika 25, 203–207. [PubMed] [Google Scholar]
- (52).Kappl R, Höhn-Berlage M, Hüttermann J, Bartlett N, and Symons MCR (1985) Electron spin and electron nuclear double resonance of the [FeO2]− centre from irradiated oxy-hemo- and oxymyoglobin. Biochim. Biophys. Acta BBA - Protein Struct. Mol. Enzymol 827, 327–343. [Google Scholar]
- (53).Davydov R, Laryukhin M, Ledbetter-Rogers A, Sono M, Dawson JH, and Hoffman BM (2014) Electron Paramagnetic Resonance and Electron-Nuclear Double Resonance Studies of the Reactions of Cryogenerated Hydroperoxoferric-Hemoprotein Intermediates. Biochemistry 53, 4894–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Davydov R, Kofman V, Nocek JM, Noble RW, Hui H, and Hoffman BM (2004) Conformational Substates of the Oxyheme Centers in α and β Subunits of Hemoglobin As Disclosed by EPR and ENDOR Studies of Cryoreduced Protein. Biochemistry 43, 6330–6338. [DOI] [PubMed] [Google Scholar]
- (55).Davydov R, and Hoffman BM (2011) Active intermediates in heme mono reactions as revealed by cryoreduction/annealing, EPR/ENDOR studies. Arch. Biochem. Biophys 507, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, and Hoffman BM (2001) Hydroxylation of camphor by reduced oxy-cytochrome P450cam: mechanistic implications of EPR and ENDOR studies of catalytic intermediates in native and mutant enzymes. J. Am. Chem. Soc 123, 1403–1415. [DOI] [PubMed] [Google Scholar]
- (57).Davydov R, Kofman V, Fujii H, Yoshida T, Ikeda-Saito M, and Hoffman BM (2002) Catalytic Mechanism of Heme Oxygenase through EPR and ENDOR of Cryoreduced Oxy-Heme Oxygenase and Its Asp 140 Mutants. J. Am. Chem. Soc 124, 1798–1808. [DOI] [PubMed] [Google Scholar]
- (58).Davydov R, Sudhamsu J, Lees NS, Crane BR, and Hoffman BM (2009) EPR and ENDOR Characterization of the Reactive Intermediates in the Generation of NO by Cryoreduced Oxy-Nitric Oxide Synthase from Geobacillus stearothermophilus. J. Am. Chem. Soc 131, 14493–14507. [DOI] [PubMed] [Google Scholar]
- (59).Davydov RM, Chauhan N, Thackray SJ, Anderson JLR, Papadopoulou ND, Mowat CG, Chapman SK, Raven EL, and Hoffman BM (2010) Probing the ternary complexes of indoleamine and tryptophan 2,3-dioxygenases by cryoreduction EPR and ENDOR spectroscopy. J. Am. Chem. Soc 132, 5494–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Davydov R, Macdonald IDG, Makris TM, Sligar SG, and Hoffman BM (1999) EPR and ENDOR of catalytic intermediates in cryoreduced native and mutant oxy-cytochromes P450cam: Mutation-induced changes in the proton delivery system. J. Am. Chem. Soc 121, 10654–10655. [Google Scholar]
- (61).Davydov R, Gilep AA, Strushkevich NV, Usanov SA, and Hoffman BM (2012) Compound I is the reactive intermediate in the first monooxygenation step during conversion of cholesterol to pregnenolone by cytochrome P450scc: EPR/ENDOR/cryoreduction/annealing studies. J. Am. Chem. Soc 134, 17149–17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Davydov R, Dawson JH, Perera R, and Hoffman BM (2013) The use of deuterated camphor as a substrate in (1)H ENDOR studies of hydroxylation by cryoreduced oxy P450cam provides new evidence of the involvement of Compound I. Biochemistry 52, 667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Davydov R, Osborne RL, Kim SH, Dawson JH, and Hoffman BM (2008) EPR and ENDOR Studies of Cryoreduced Compounds II of Peroxidases and Myoglobin. Proton-Coupled Electron Transfer and Protonation Status of Ferryl Hemes. Biochemistry 47, 5147–5155. [DOI] [PubMed] [Google Scholar]
- (64).Garcia-Serres R, Davydov RM, Matsui T, Ikeda-Saito M, Hoffman BM, and Huynh BH (2007) Distinct Reaction Pathways Followed upon Reduction of Oxy-Heme Oxygenase and Oxy-Myoglobin as Characterized by Mössbauer Spectroscopy. J. Am. Chem. Soc 129, 1402–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Denisov IG (2010) Cryoradiolysis as a method for mechanistic studies in inorganic biochemistry, pp 109–142. John Wiley & Sons, Inc. [Google Scholar]
- (66).Davydov R, Matsui T, Fujii H, Ikeda-Saito M, and Hoffman BM (2003) Kinetic Isotope Effects on the Rate-Limiting Step of Heme Oxygenase Catalysis Indicate Concerted Proton Transfer/Heme Hydroxylation. J. Am. Chem. Soc 125, 16208–16209. [DOI] [PubMed] [Google Scholar]
- (67).Davydov R, Perera R, Jin S, Yang T-C, Bryson TA, Sono M, Dawson JH, and Hoffman BM (2005) Substrate Modulation of the Properties and Reactivity of the Oxy-Ferrous and Hydroperoxo-Ferric Intermediates of Cytochrome P450cam As Shown by Cryoreduction-EPR/ENDOR Spectroscopy. J. Am. Chem. Soc 127, 1403–1413. [DOI] [PubMed] [Google Scholar]
- (68).Davydov R, Razeghifard R, Im S-C, Waskell L, and Hoffman BM (2008) Characterization of the Microsomal Cytochrome P450 2B4 O2 Activation Intermediates by Cryoreduction and Electron Paramagnetic Resonance. Biochemistry 47, 9661–9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.