

Abstract
Signal transducer and activator of transcription 3 (STAT3) has been recognized for its key role in the progression of cancer, where it is frequently upregulated or constitutively hyperactivated, contributing to tumor cell proliferation, survival, and migration, as well as angiogenesis and suppression of anti-tumor immunity. Given the ubiquity of dysregulated STAT3 activity in cancer, it has long been considered a highly attractive target for the development of anti-cancer therapies. Efforts to target STAT3, however, have proven to be especially challenging, perhaps owing to the fact that transcription factors lack targetable enzymatic activity and have historically been considered “undruggable”. Small molecule inhibitors targeting STAT3 have been limited by insufficient selectivity and potency. More recently, therapeutic approaches that selectively target STAT3 protein for degradation have been developed, offering novel strategies that do not rely on inhibition of upstream pathways or direct competitive inhibition of the STAT3 protein. Here, we review these emerging approaches, including the development of STAT3 proteolysis targeting chimera (PROTAC) agents, as well as preclinical and clinical studies of chemically stabilized antisense molecules, such as the clinical agent AZD9150. These therapeutic strategies may robustly reduce the cellular activity of oncogenic STAT3 and overcome the historical limitations of less selective small molecules.
Keywords: STAT3, PROTACs, ASOs, cancer, therapeutics
Introduction
Signal transducer and activator of transcription (STAT) proteins comprise a family of transcription factors involved in cell growth, survival, and differentiation.1 Highly conserved in structure, the seven members of the STAT family (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, STAT6) are critical mediators of intracellular signaling, relaying and integrating extracellular stimuli from cytokines and growth factors into responses that alter levels of gene transcription in accordance with their respective functions.2 Perhaps the most well-studied STAT, STAT3 is frequently dysregulated and implicated in cancer development and progression. Numerous studies have provided evidence linking overexpression and/or hyperactivation of STAT3 to tumor growth, invasion, and angiogenesis via transcription of target genes encoding positive regulators of the cell cycle and negative regulators of apoptosis signaling.3–7 Additionally, STAT3 promotes tumor cell production of immunosuppressive cytokines, and STAT3 activation in infiltrating immune cells acts, in general, to suppress anti-tumor immunity.8 Analyses of clinical specimens from many solid tumor cancers have demonstrated an association between aberrant STAT3 activation in the tumor microenvironment and poor patient prognosis.9 Collectively, these factors identify STAT3 as a promising therapeutic target in cancer.
Canonical activation of STAT3 involves phosphorylation of a specific carboxyl-terminal tyrosine, Y705, by upstream kinases, including Janus kinases (JAKs), epidermal growth factor receptor (EGFR), c-Src, and BCR/ABL. In the well-characterized IL-6/JAK/STAT3 signaling pathway, IL-6 binds to its cognate cell surface receptor, forming a heterohexameric IL-6/IL-6R/gp130 complex. Formation of this complex leads to activation of associated members of the JAK family of tyrosine kinases, which directly phosphorylate STAT3.8 Receptor tyrosine kinases such as EGFR undergo autophosphorylation following ligand binding, and these phosphorylation sites then serve as docking sites for the Src-homology 2 (SH2) domain of STAT3, facilitating EGFR-mediated phosphorylation of STAT3.10,11 EGFR, and other receptor tyrosine kinases, also induce activation of c-Src, leading to phosphorylation of STAT3 by this non-receptor tyrosine kinase.12 Phosphorylation of STAT3 Y705 leads to homodimerization that is facilitated by the SH2 domain of each monomer interacting with the phosphorylated Y705 residue on the other partner. Once dimerized, STAT3 translocates into the nucleus and binds to a response element in the promoter regions of target genes, inducing the transcription of these genes. Serine phosphorylation of STAT3, most frequently on S727, also modulates STAT3 transcriptional activity.13
More recently, studies of non-canonical pathways of STAT3 activation and signaling have identified the potential of these alternate mechanisms to contribute to cancer development.14 Regulation of mitochondrial function is one area in which STAT3 has been discovered to play a role. Translocation of STAT3 to the mitochondria is dependent on S727 phosphorylation, and leads to interaction with the electron transport chain to regulate cellular respiration. Mitochondrial STAT3 is observed in non-transformed cells, but dysfunction of its normal role in the mitochondria, resulting in increased respiration, has been shown to contribute to RAS-mediated cellular transformation.14,15
Additional studies of non-canonical STAT3 signaling have indicated that dimerization can occur in the absence of Y705 phosphorylation, and that unphosphorylated STAT3 (uSTAT3) dimers and monomers can interact with interferon gamma-activated sequence (GAS) DNA-binding sites.16–18 Interestingly, uSTAT3 has been shown to regulate the transcription of a distinct set of genes, including cyclin B1, E2F1, c-MET and M-RAS, and also interacts with unphosphorylated NF-κB to activate a set of NF-κB-dependent genes responsive to the uSTAT3/NF-κB complex.14,16,17,19,20 Due to the oncogenic nature of the uSTAT3 regulated proteins c-MET and M-RAS, and the aberrant co-expression of both uSTAT3 and phosphorylated STAT3 in cancer cells, emerging evidence indicates that unphosphorylated and phosphorylated STAT3 work in concert through canonical and non-canonical pathways to promote cancer development.14 Hence, therapeutic approaches that target both the phosphorylated and unphosphorylated forms of STAT3 may be necessary to efficiently reduce STAT3-mediated oncogenic signaling in cancer cells. In this regard, antisense and PROTAC approaches, which target the expression of STAT3, may be particularly advantageous compared with small molecule competitive inhibitors.
Proteolysis targeting chimeras (PROTACs)
Proteolysis targeting chimera (PROTAC) technology has emerged as a novel approach that enables modulation of protein expression levels via targeted degradation.21–23 PROTAC drugs show particular promise in that their mode of action is rapid and catalytic, and they degrade rather than inhibit the targeted protein of interest (POI) to effectively and holistically reduce the activity of the protein in the cell.21–23 Structurally, PROTAC drugs are heterobifunctional molecules comprised of a “warhead” ligand that recruits the POI, connected by a linker to a ligand that recruits an E3 ubiquitin ligase (Figure 1A). The technology exploits the eukaryotic ubiquitin-proteasome system, such that the PROTAC molecule brings the POI and the E3 ubiquitin ligase moiety into close proximity, whereupon the E3 ligase recruits an E2 ubiquitin-conjugating enzyme that subsequently transfers ubiquitin to the target POI. The ubiquitinated POI is then targeted for degradation by the 26S proteasome (Figure 1B).
Figure 1. The structure and mechanism of PROTACs.
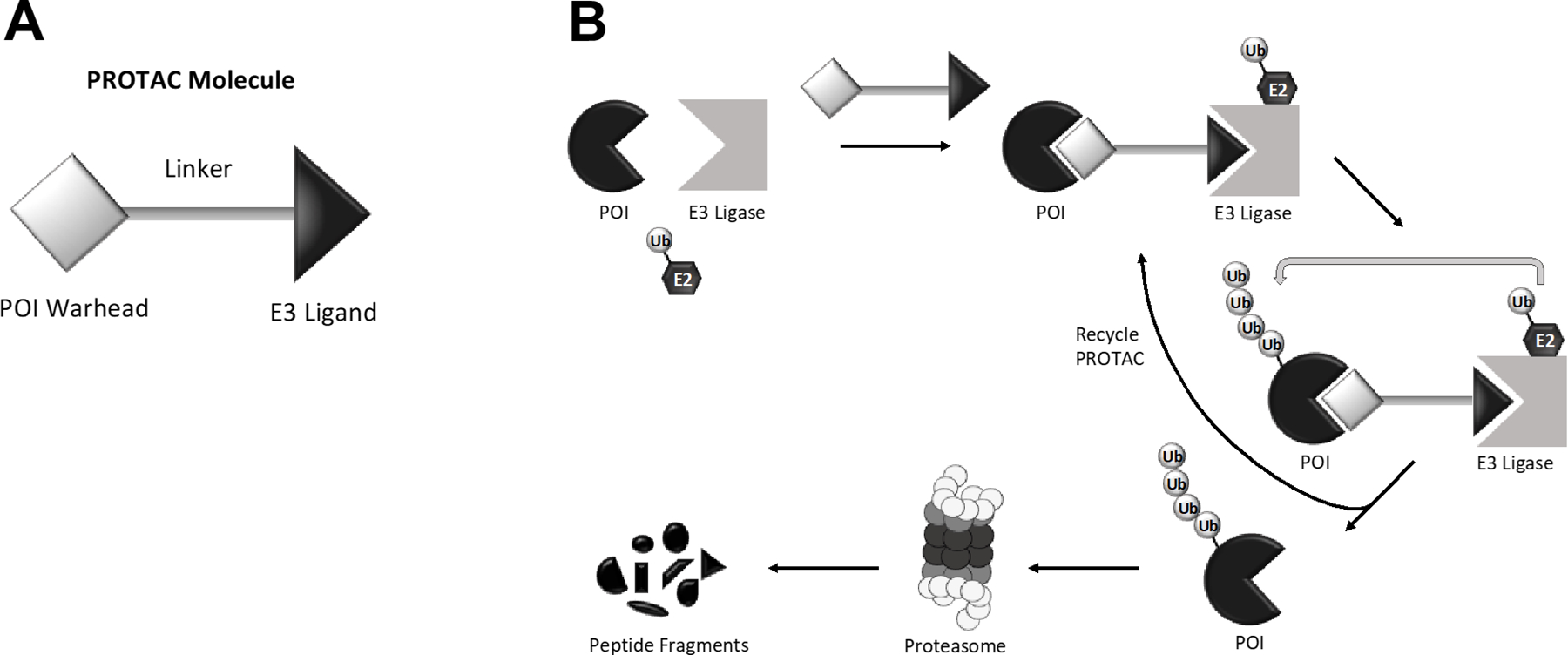
A) The general structure of a PROTAC molecule consists of a “warhead” ligand that recruits the protein of interest (POI), a linker, and an E3 ligand that recruits an E3 ubiquitin ligase. B) The “warhead” ligand and the E3 ligand bring into close proximity the protein of interest (POI) and an E3 ubiquitin ligase, respectively. The E3 ubiquitin ligase, in concert with an E2 ubiquitin-conjugating enzyme, promotes polyubiquitination of the POI. Polyubiquitination of the POI results in degradation by the 26S proteasome. This process is catalytic, meaning that the freed PROTAC molecule can participate in another reaction. Figure adapted from Pei et al. and Sun et al.21,22
The concept of PROTACs was introduced in 2001 by Deshaies, Crews, Sakamoto and colleagues who created the first PROTAC molecule, consisting of an angiogenesis inhibitor, ovalicin, targeting the enzyme methionine aminopeptidase-2 (MetAP-2) linked to a phosphopeptide that recruits the SCFβ-TRCP E3 ubiquitin ligase.22,24 While studies with this first-generation PROTAC faced limitations in cell permeability, they provided proof-of-concept evidence that spurred the development of PROTACs incorporating non-peptidic ligands for E3 ligases. The use of small molecules (as opposed to peptides) as warheads or E3 ligase ligands for PROTAC drugs is advantageous due to their low molecular weight, lack of labile peptide bonds, and potential for greater cell permeability and potency.23 A key advancement occurred when Bradner and colleagues successfully employed the small molecule phthalimide as a ligand recruiter for cereblon E3 ubiquitin ligase.25 PROTAC drugs have now been designed to recruit an expanding repertoire of E3 ligases, including the E3 ligases MDM2, CRBN, SCFβ-TRCP, VHL, and IAPs, representing only a fraction of the vast family of E3 ubiquitin ligases found in the human genome.21,22
A key strength of PROTAC approaches lies in the rapid and catalytic nature of the process. The binding of a PROTAC drug to a target POI is a transient interaction, and once a successful ubiquitin transfer has been catalyzed, leading to destruction of the POI, the PROTAC molecule can dissociate to participate in another reaction (Figure 1B).22 The catalytic nature of PROTAC drugs means, in principle, that efficient potency can be achieved using lower drug concentrations and shorter exposure times than would be needed for a typical competitive inhibitor. The ability to utilize low doses of PROTAC drugs may limit nonspecific adverse toxicities. Studies have shown that PROTACs can achieve near complete depletion of the target protein in as little as several hours after treatment. PROTACs targeting the estrogen receptor alpha (ERα) demonstrated efficient depletion of the protein one hour after treatment of cells in culture at nanomolar concentrations.26–28 Remarkably, not only is depletion of target proteins generally sustained after only a single treatment, but downstream signaling is also reduced. Depletion of ERα was sustained for up to 48 hours after treatment and downstream estrogen signaling was inhibited, while PROTAC degraders of anaplastic lymphoma kinase (ALK) showed persistent inhibition of ALK downstream signaling for at least 24 hours following a 2-hour treatment with 30 nM of drug.26,27,29
Currently, a number of PROTAC-based cancer therapeutics have been developed by groups in both academia and industry. These efforts include, but are not limited to, PROTACs targeting ALK (implicated in lymphomas), ERα (implicated in estrogen receptor-positive breast cancer), androgen receptor (AR; implicated in prostate cancer), eukaryotic translation initiation factor 4E (eIF4E; a critical regulator of cell proliferation), and BCR-ABL (a prominent driver of chronic myelogenous lymphoma).21,27,29,30 Efforts by the pharmaceutical company Arvinas, Inc. have propelled two PROTACs into Phase I clinical studies, ARV-110 and ARV-471, which act to target AR and ER, respectively (NCT03888612; NCT04072952).31,32 Preliminary results indicate a generally positive safety profile and tolerability. Although challenges remain in the development of PROTAC drugs (see below), they are emerging as promising therapeutic agents for targets that have proven difficult to treat by conventional methods.
PROTACs targeting STAT3
Recently, Bai et al. reported the development of a small-molecule PROTAC targeting STAT3.28 Known as SD-36, this PROTAC potently and selectively degraded STAT3 protein in acute myeloid leukemia (AML) and anaplastic large-cell lymphoma (ALCL) cells, while also mediating complete tumor regression in AML and ALCL mouse xenograft models.
The warhead for SD-36 was the small molecule SI-109 (Figure 2A), a STAT3 SH2 domain inhibitor reported to bind to STAT3 with high affinity (Ki = 9 nM).28 To generate the SD-36 PROTAC, SI-109 was attached via a six carbon linker to a ligand analog of lenalidomide, which acts to recruit the CRBN E3 ligase (Figure 2B). A modified version, SD-36Me, containing a methyl group on the lenalidomide analog moiety, was also generated; SD-36Me lacked the ability to recruit CRBN and was used as a negative control (Figure 2C).
Figure 2. Chemical structures of STAT3 inhibitor, PROTAC, and PROTAC control.
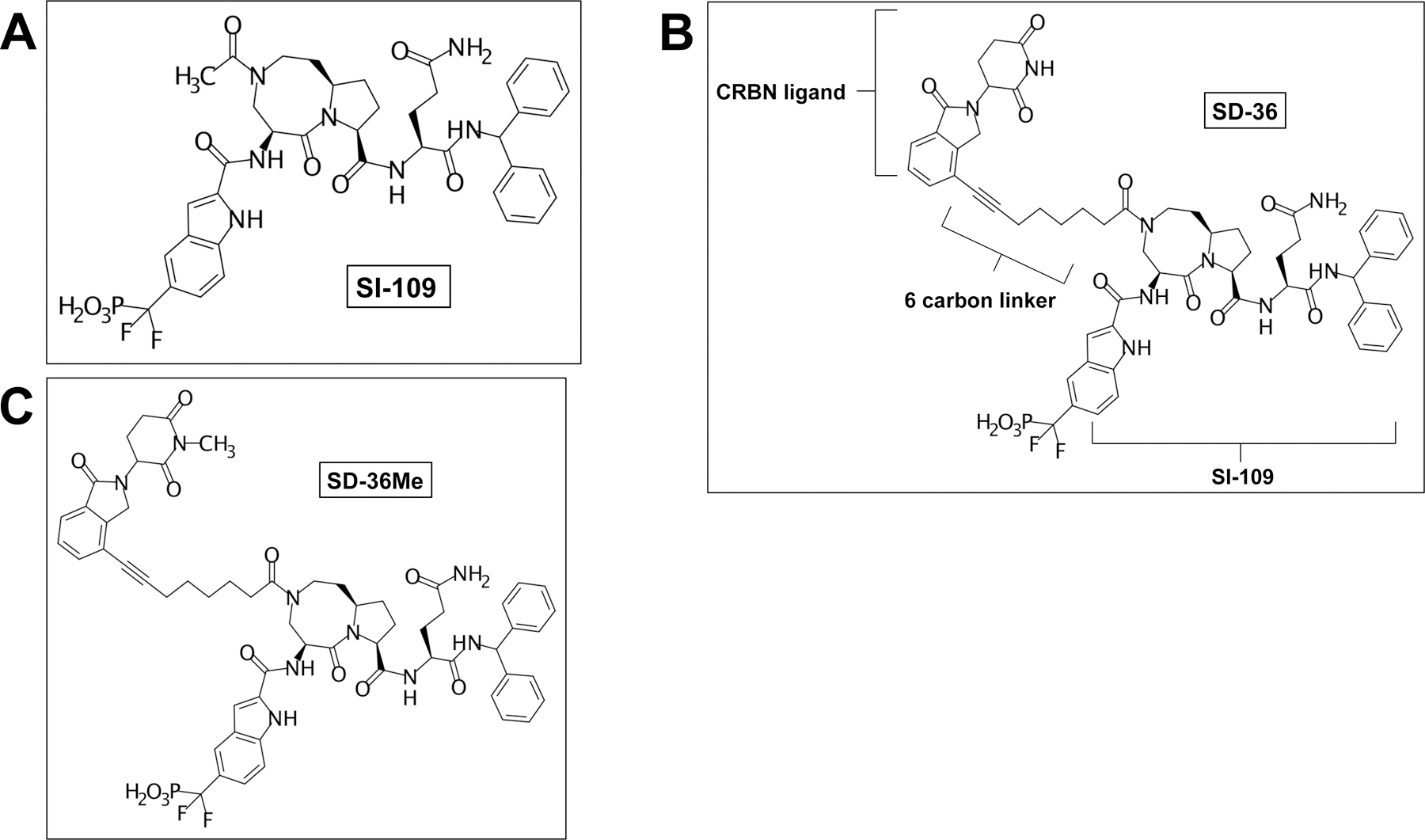
A) Chemical structure of the STAT3 SH2 domain inhibitor SI-109. B) Chemical structure of SD-36. C) Chemical structure of SD-36Me, which contains an additional methyl group on the lenalidomide analog moiety. SD-36Me is deficient in recruiting the E3 ligase and acts as a negative control.
SD-36 demonstrated efficient reduction of STAT3 levels in human hematopoietic cell lines, promoting ≥90% degradation in the AML cell line MOLM-16 after 4-hour treatment at 250 nM and ≥50% in the ALCL cell lines SU-DHL, DEL, and KI-JK after 7-hour treatment at the same concentration. The small molecule inhibitors SI-109 and SD-36Me, on the other hand, failed to promote STAT3 degradation.
Evaluation of the SD-36 selectivity revealed that treatment of MOLM-16 cells for 4 hours, or SU-DHL cells for 18 hours, with 0.3 µM SD-36 resulted in nearly complete degradation of STAT3.33 By contrast, treatment with concentrations as high as 10 µM had little impact on other expression levels of other STAT family members, including STAT1, STAT2, STAT4, STAT5A/B, and STAT6. Interestingly, binding analyses determined that SD-36 exhibits a Kd of 44 µM for STAT3, and also binds with moderately high affinities to STAT1 and STAT4 (Kds ~ 1 µM).33 Hence, while SD-36 binds to all three of these STAT proteins, it is exquisitely selective for degradation of STAT3. This unique selectivity underscores a promising, yet complex, aspect to the development of selective PROTAC degraders. Specifically, the degradation selectivity of a PROTAC will be determined by the precise physical structure of the ternary complex. The promise lies in the fact that highly selective degraders can be generated, even when limited by a warhead ligand that is not entirely specific for the POI, as in the case of SD-36. The complexity, or difficulty, lies in how to design the PROTAC molecule to generate a ternary complex with selective degradation capability. Currently, a great deal of effort is being expended to empirically optimize linker length and composition, as well as the ligands for E3 ligases, although this can be a daunting task in the process of developing PROTACs selective for different POIs.
Additional studies determined that SD-36 efficiently degraded mutated STAT3 proteins containing the D661Y and K658R mutations that are seen in hematologic cancers such as T-cell large granular lymphocytic leukemia as well as an engineered Y705F mutant.34,35 The ability of SD-36 to stimulate degradation of the Y705F mutant indicates the capacity of this PROTAC to degrade STAT3 that may be involved in non-canonical signaling pathways. Indeed, SD-36 induced reduction of STAT3 levels in the mitochondria. Inhibition of leukemia and lymphoma cell growth by SD-36 was associated with G1 arrest and downregulation of c-Myc.
Treatment of mice harboring MOLM-16 or SU-DHL-1 tumors with a single intravenous dose of SD-36 (25 mg/kg) resulted in nearly complete depletion of tumoral STAT3 after 6 hours, and this maximal reduction was sustained for up to 24 hours. Treatment with a maximum dose of 100 mg/kg up to three times per week for up to 4 weeks led to complete and long-term tumor regression in MOLM-16, SU-DHL-1, and SUP-M2 xenograft models. Tumor regression after the last dosing persisted for at least 2 weeks for MOLM-16 and >100 days for SU-DHL-1 and SUP-M2 xenograft models. Evaluation of spleen, liver, heart, and kidney in immunocompetent mice showed no substantial toxicity from treatment with 100 mg/kg SD-36, and red and white blood cell counts remained relatively constant. Thus, SD-36 was well tolerated in immunocompetent mice.
Collectively, these results have identified PROTAC approaches, including treatment with SD-36, as advantageous and promising therapeutic strategies for eliminating the cellular activity of STAT3, a transcription factor that has historically evaded efficacious molecular targeting. Importantly, SD-36 appears to be capable of degrading STAT3 that is activated via non-canonical pathways, where phosphorylation of Y705 is not required. As emerging evidence indicates that these alternate, non-canonical pathways contribute alongside canonical STAT3 signaling to promote tumor progression, PROTACs offer a unique advantage by robustly inhibiting the total pool of cellular STAT3 protein. In view of the important role that STAT3 plays in the development of both solid and hematologic malignancies it is likely that considerable effort will be invested in generating and optimizing new STAT3-targeting PROTAC molecules in the near future. It is also worth considering, however, given the complex position STAT3 holds within the immune system, if deleterious effects may arise from complete elimination of STAT3. An immunodeficiency disorder leading to increased plasma IgE levels and recurring staphylococcal abscesses and pneumonia, hyperimmunoglobulin E syndrome (HIES), results from expression of dominant-negative mutations in STAT3, emphasizing the need for meticulous evaluations of toxicity.36,37
Current Challenges to Development of PROTAC Drugs
Current challenges in the development of PROTAC drugs include minimization of nonspecific binding and off-target effects, optimization of warhead-linker-E3 ligand combinations, and efficient delivery to tumor cells. The elaborate process of employing the cellular ubiquitin-proteasome system for selective degradation necessitates the need for precise and specific drug design conducive to the formation of a productive ternary complex; structural design of PROTAC molecules will thus require intensive studies to optimize drug function and pharmacological properties for their specific target proteins.38
The linker is crucial to the biological function of the PROTAC. In addition to properly situating the POI and E3 ubiquitin ligase for the formation of a ternary complex, it also affects physicochemical properties including propensity for cellular uptake, stability, and aqueous solubility.38 However, optimization of the linker and linkage site is often a difficult and demanding process.39 Ongoing efforts to identify small-molecule ligands for E3 ligases that will enable recruitment of the more than 600 E3 ligases present in the human genome represents another area that requires extensive development and optimization. The differential expression of E3 ubiquitin ligases in tissues and variation in subcellular localization and regulation may present the potential for tissue-specific as well as spatially and temporally confined protein degradation.40,41 Moreover, the use of different E3 ligases will modify the range of proteins that can be targeted for degradation.42 These considerations highlight the complex nature of PROTAC design, which due to its modularity, presents both challenges and potential opportunities for the development of efficacious and highly specific PROTACs.
An inherent consequence of the design of current PROTAC molecules is their relatively large molecular size, which adversely impacts a number of parameters, including metabolism, stability, and uptake.38 Optimization of linker design and length, as well as optimization of other components contributing to the large size, should help to improve the physicochemical properties of PROTACs and efforts to optimize pharmacokinetic parameters are intensively ongoing.40 Interestingly, due to their catalytic mode of action and ability to initiate several cycles of degradation, PROTACs show sub-stoichiometric activity.41 For example, in vivo studies of PROTACs targeting RIPK2 demonstrated lasting pharmacodynamic efficacy at low doses even after levels of the PROTAC were no longer measurable.43 Evidence of the lower and infrequent dosages required by PROTACs may prove to be advantageous despite somewhat unfavorable physicochemical properties, although the therapeutic benefit of PROTACs needs to be determined through comprehensive clinical investigations.
Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are synthetic oligonucleotides that selectively target the expression of mRNAs. ASOs typically consist of short, single-stranded DNA or RNA sequences approximately 20 base pairs in length that bind to single-stranded RNA sequences in a complementary fashion.44 Contingent on their specific design, ASOs are capable of altering protein expression by reducing mRNA levels or by modulating alternative splicing patterns.45 ASOs can operate through several mechanisms, including targeting mRNA for degradation by the cellular endonuclease ribonuclease H (RNase H) or by binding and blocking pre-mRNA splice sites to alter splicing and generation of the final mRNA product. Hybridization of ASOs to AUG translation start sites can also prevent the recruitment of ribosome translational machinery and, hence, subsequent protein expression.44,45
A multitude of chemical modifications aimed at enhancing intracellular delivery and stability have advanced the development of ASOs as therapeutic agents. Prominent limitations to ASOs include rapid degradation by cellular nucleases and potential toxicities due to their immunostimulatory properties. The poor ability of negatively charged oligonucleotides to cross cell membranes also poses a problem to clinical application. One modification that increases the resistance of ASOs to degradation is the replacement of a non-bridging oxygen atom of the phosphodiester linkage by a sulfur atom to produce a phosphorothioate backbone (Figure 3). This modification reduces the sensitivity of ASOs to nucleolytic degradation, stabilizes the longevity of ASOs in tissues, and improves binding to plasma proteins.45,46 Non-specific binding to proteins can, however, induce toxicity, while immune stimulation, likely due to CpG motifs in phosphorothioate oligonucleotides, has also been reported.47,48 ASOs containing modifications to the phosphate backbone that are aimed at increasing resistance to nucleases are generally referred to as first-generation ASO therapeutics. The toxicity of first-generation ASOs containing phosphorothioate backbones led to the development of second-generation ASOs containing modifications to the 2’ position of the ribose sugar. Common additions produce 2’-O-methyl (2’-OME) and 2’-O-methoxyethel (2’-O-MOE) nucleotides (Figure 3) that have been shown to increase nuclease resistance and binding affinity to target RNAs while lowering toxicity by reducing non-specific binding.46,49 As modifications to the 2’ position of the ribose sugar reduce the activity of RNase H, these modifications may be particularly valuable in instances where altering of mRNA splicing, as opposed to degradation of the target mRNA, is the goal. In cases where degradation is the primary goal, the strategic use of a gapmer design comprising a stretch of 2’ modified oligoribonucleotides flanking either side of an internal DNA segment has been shown to restore RNase H-mediated degradation.49 It is critical that the internal DNA segment, modified or not, is capable of being recognized by RNase H when hybridized.
Figure 3. Common structural modifications of ASOs.
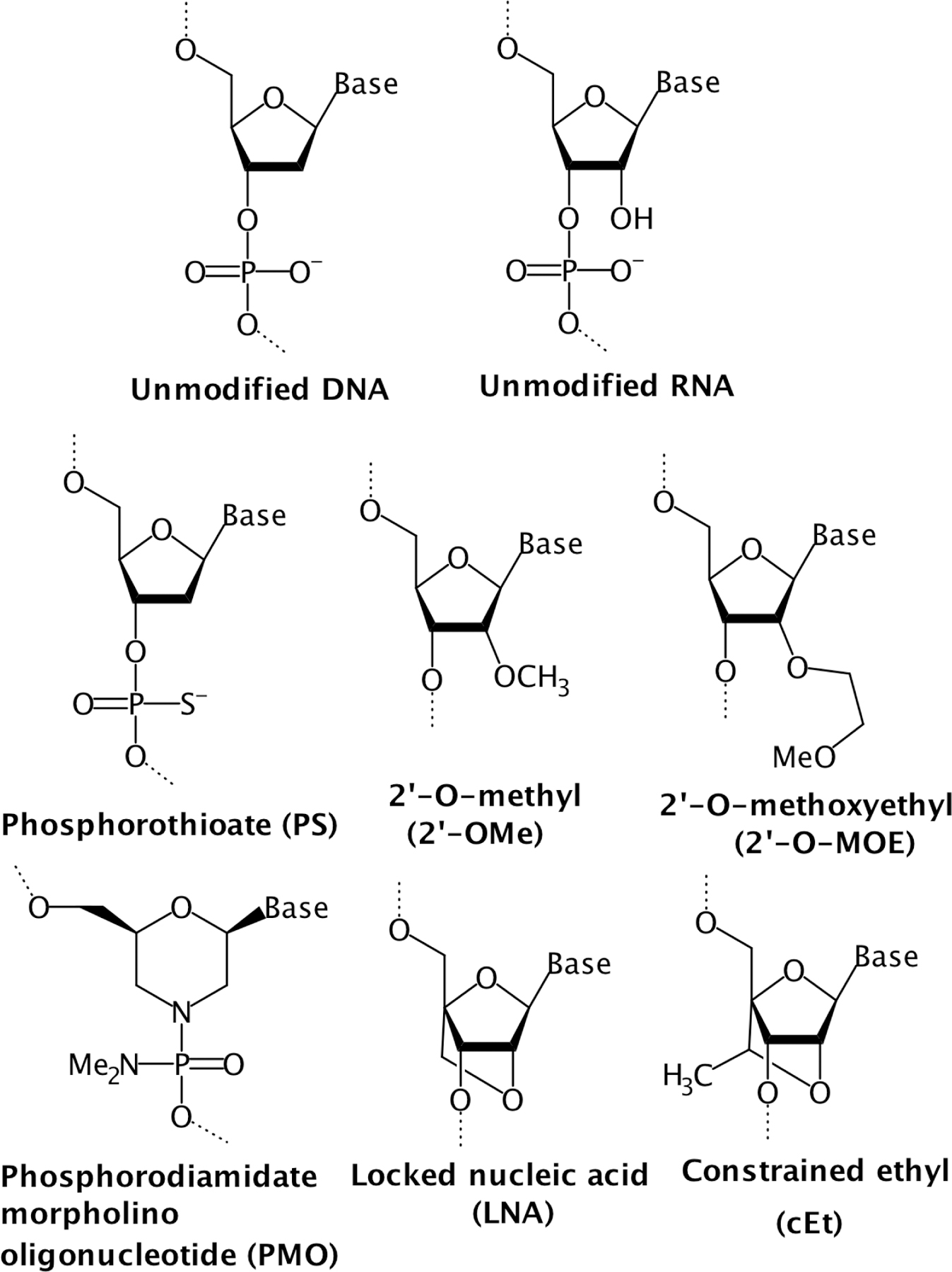
First-generation ASOs incorporating modifications of the phosphate backbone include phosphorothioate (PS) oligonucleotides, while second-generation ASOs harboring 2’ ribose modifications include 2’-O-methyl (2’-OMe) and 2’-O-methoxyethyl (2’-O-MOE) residues. Additional modifications incorporated in constrained ASOs include locked nucleic acid (LNA) and constrained ethyl (cEt) modifications.
Finally, various other modifications have been implemented to further enhance the stability and binding affinity of first- and second-generation ASOs. These include the substitution of a morpholine ring in place of the deoxyribose in the sugar-phosphate backbone to form neutrally charged phosphorodiamidate morpholino oligonucleotides (PMO) composed of phosphorodiamidate linkages (Figure 3).50 ASOs containing this modification do not facilitate RNase H recruitment and are thereby primarily used to inhibit translation of the target mRNA or alter splicing events through steric blocking at a splice junction sequence.46 A conformational constraint introduced by bridging the 2’ oxygen and 4’ carbon of the ribose moiety in RNA, referred to as locked nucleic acid (LNA), considerably increases binding affinity while also augmenting nuclease resistance (Figure 3).51 A constrained ethyl (cEt) nucleotide formed from the addition of a methyl group to LNA residues has also demonstrated improved binding affinity and resistance to nucleases (Figure 3). Despite auspicious improvement in hybridization, LNA- and cEt-based ASOs have been observed to induce liver toxicity due to partial complementary binding to off-target transcripts, emphasizing the importance of ASO selection and design.50,52,53 In many cases, chimeric approaches utilizing a combination of modified residues may be employed to refine the activity of ASOs for their intended use, as well as help to compensate for detrimental effects caused by the use of a particular modification.
ASOs target mRNA for degradation through binding to target RNA and forming a RNA-DNA duplex. These hybrid duplexes are recognized by RNase H, which subsequently cleaves and degrades the RNA strand.50 Around 6–7 nucleotides of an RNA-DNA complex are generally required to hybridize for recognition by RNase H, and successful targeting by the ASO causes downregulation of the target gene protein product.44 This approach has been employed in diseases associated with hyperactivated or mutated proteins. One such example is Inotersen, a second-generation RNase H-recruiting ASO that decreases the production of transthyretin, a protein that can accumulate in deposits in peripheral nerves, kidney, and gastrointestinal tract, causing transthyretin amyloidosis. Inotersen has advanced to Phase III clinical trials, where it has been shown to reduce TTR levels and improve quality of life.54
ASOs that modulate splicing to alter the RNA product, and subsequently the protein product, have been applied to diseases that manifest from aberrant splicing events. Strategic design of ASOs targeting splice sites can produce modifications of mRNA that include exon skipping or regulation of the population of mRNA variants. Eteplirsen, developed as a therapy for Duchenne muscular dystrophy, is a PMO-modified ASO that facilitates skipping of exon 51 in dystrophin pre-mRNA to form a splice variant that encodes a protein which retains partial activity. Following a Phase II clinical trial and follow-up study, Eteplirsen was provisionally approved by the Food and Drug Administration (FDA). Another FDA-approved ASO that alters splicing is Nusinersen, developed as a therapeutic for spinal muscular atrophy. Nusinersen induces inclusion of exon 7 during splicing of the survival motor neuron 2 pre-mRNA to restore a functional protein.55
ASOs targeting STAT3
Early antisense approaches against STAT3 utilized unmodified oligonucleotide sequences designed and selected for low overall free energy between the ASO and STAT3 mRNA.56 The more effective of these unmodified STAT3 ASOs, AS10, when transfected into A549 non-small cell lung cancer (NSCLC) cells downregulated expression of total STAT3, phospho(Y705)-STAT3 (p-STAT3), and the STAT3 target gene encoding BCL-XL. Furthermore, cell proliferation was inhibited in a dose-dependent manner and induction of apoptosis was observed at a rate significantly higher than the control group.56 STAT3 AS-ON, another early STAT3 ASO, was shown to inhibit expression of STAT3 protein and cell proliferation in HEp-2 human laryngeal carcinoma cells.57 In an in vivo study evaluating the role of STAT3 in trinitrobenzene sulfonic acid-induced colitis, intracolonically administered STAT3 ASO lowered the levels of inflammatory cytokines produced and increased apoptosis of lamina propria mononuclear cells.58 Limitations of these unmodified STAT3 ASOs, however, included modest potencies and nonspecific toxicities.56
Extensive studies on modifications to ASOs resulting in improved potency led to the development of AZD9150, a chemically modified ASO designed to target STAT3 mRNA.59 Based on second-generation 2’-O-methoxyethel (2’-O-MOE)-based ASOs, AZD9150 is a generation 2.5 (Gen 2.5) ASO containing 2’ and 4’ constrained ethyl (cEt) residues. AZD9150 employs a gapmer approach where a central segment of 8–10 deoxynucleotides containing phosphorothioate backbones is flanked by a few cEt residues on either side. The mechanism of action involves recognition by RNase H and cleavage of the target RNA within the resulting RNA-DNA hybrid, effecting a downregulation of STAT3 expression.59
AZD9150 demonstrated 10 to 20 times greater potency than its second generation ASO counterpart in human epidermoid cancer A431 cells in the absence of transfection agent, achieving IC50 values for knockdown of STAT3 mRNA in the nanomolar range and selective and near-complete reduction of STAT3 protein over STAT1 and STAT5.59 In human ALCL and AML cell lines exhibiting hyperactivation of STAT3, AZD9150 selectively depleted STAT3, inhibited proliferation, and induced apoptosis. Notably, downstream STAT3 target genes such as MCL-1, c-MYC, BCL-6, and cyclin D1 were downregulated in lymphoma cells while MCL-1 and other oncogenes were downregulated in leukemia cells.59,60 Leukemia and myelodysplastic stem cells treated with AZD9150 also demonstrated uptake of the ASO and increased erythoid and myleloid differentiation.60 Inhibition of STAT3 expression in neuroblastoma cells impaired growth in colony formation assays and decreased cell migration.61
In in vivo mouse studies, AZD9150 was successful at reducing STAT3 expression and inhibiting the growth of lung and lymphoma xenograft tumors, although no change was observed in established neuroblastoma xenograft tumors.59,61 However, treatment with AZD9150 did decrease STAT3 expression in mice harboring neuroblastoma xenograft tumors and strongly inhibited the formation of secondary tumors.61 Furthermore, while AZD9150 alone did not impair growth of established neuroblastoma tumors, the addition of AZD9150 to cisplatin treatment significantly sensitized the tumors to cisplatin and significantly lengthened survival time in comparison to mice treated with either agent alone. This suggests a potential clinical benefit of AZD9150 in patients who have developed cisplatin resistance, as commonly occurs in lung, ovarian, colorectal, and head and neck cancers.61,62 Similarly, in studies investigating radioresistance, a considerable clinical problem, mice bearing HNSCC tumors showed increased survival and impaired tumor growth in response to the combination of radiation and AZD9150.63
Preclinical studies assessing the safety profiling of AZD9150 in mice and cynomolgus monkeys showed toxicity levels comparable to previous second-generation ASOs, likely due to the increased protein binding generated by phosphorothioate-modified residues.64 However, no new harmful effects were observed, and AZD9150 entered Phase Ib clinical testing in patients with relapsed or treatment refractory lymphoma (NCT01563302).64,65 Of the 30 patients enrolled with non-Hodgkin’s lymphoma, all had undergone previous systemic treatments and the majority had diffuse large B-cell lymphoma (DLBCL), for which activated STAT3 expression has been observed to accompany poor overall survival.66 The initial cycle began with a loading dose of thrice weekly treatments of 2 mg/kg administered via intravenous infusion, with subsequent weekly doses escalating to 3 mg/kg if disease progression occurred. Two complete responses, two partial responses, and one stable disease response were observed among the patients with DLBCL, amounting to a 13% clinical benefit. AZD9150 was found to be well-tolerated with no adverse events sufficiently serious to be cause for premature discontinuation. Common grade 1 and 2 adverse events consisted of transaminitis, fatigue, nausea, anemia, and thrombocytopenia, while some grade 3 and 4 events involving thrombocytopenia also occurred.65 It is probable that the reduction of platelets causing thrombocytopenia is an on-target effect resulting from inhibition of STAT3, requiring the recommended Phase II dose to remain at 3 mg/kg.59,65
Studies evaluating pharmacokinetic properties showed AZD9150 to be comparable to previous second-generation ASOs.59 In contrast to dosing based on ideal body weight, more recently, population pharmacokinetic analysis of AZD9150 proposed pharmacokinetic exposure to be minimally affected by a change to flat dosing at 200 mg weekly.67 This analysis was congruent with preliminary results from an ongoing Phase Ib/II clinical trial assessing AZD9150 in patients with advanced solid tumors receiving a flat dose of 200 mg (D5660C00016, Study 16).67
Overall, in the clinic, AZD9150 appears to be well-tolerated and provides a therapeutic benefit for a subset of patients with treatment refractory DLBCL. Further investigations to understand the basis for the differential responses among patients is merited. Moreover, concerns remain over dose-limiting toxicities of AZD9150, such as the thrombocytopenia observed in treated subjects.65 Compelling evidence for AZD9150 as an advantageous therapy lies in its superior potency over previous ASO iterations in both preclinical and clinical settings and demonstrable efficacy in inhibiting STAT3 in multiple preclinical models. In vivo studies involving combination therapies of AZD9150 with cisplatin and radiation have also proven promising. Furthermore, AZD9150 targets STAT3 directly at the level of expression, theoretically extending its effects to non-canonical STAT3 signaling, including oncogenic signaling via uSTAT3.
Small molecule competitive inhibitors of STAT3
Efforts are also ongoing to develop competitive inhibitors of STAT3, a few of which have advanced to the stage of clinical testing, although limited efficacy remains a significant obstacle. The small molecule inhibitors OPB-31121 and OPB-111077, developed by Otsuka Pharmaceutical Company, bind with high affinity to the STAT3 SH2 domain to directly inhibit STAT3 activity and have been tested in Phase I trials in patients with solid tumors.68,69 OPB-31121 was found to have dose-limiting toxicities of vomiting and diarrhea in addition to observation of disease progression (NCT00955812).70 OPB-111077 was found to be better tolerated; however, only modest anti-tumor activity was observed (NCT01711034).68 C188–9, another small molecule inhibitor, binds to the STAT3 SH2 domain with a Kd of ~5 nM and is undergoing Phase I testing in patients with advanced cancers (NCT03195699).71,72
Napabucasin (BBI608) is a small molecule inhibitor of STAT3 that was demonstrated to be well-tolerated in a Phase I trial in Japanese patients with advanced gastric cancer when administered in combination with paclitaxel.73 A Phase III trial evaluating the efficacy of napabucasin plus best supportive care versus placebo plus best supportive care in advanced colorectal cancer revealed no difference in overall survival between treatment groups (NCT01830621).74 A Phase III study of napabucasin in addition to nab-paclitaxel with gemcitabine is currently ongoing in patients with pancreatic adenocarcinoma (NCT02993731).75
A STAT3 decoy oligonucleotide consisting of a double-stranded 15-base pair sequence corresponding to the STAT3 genomic response element, competitively inhibits binding of activated STAT3 to target gene promoters, abrogating expression of these genes, and demonstrated pharmacodynamic activity following intratumoral delivery in a Phase 0 trial.76,77 A chemically stabilized version of the decoy (cyclic STAT3 decoy) exhibited anti-tumor effects when delivered systemically to tumor-bearing mice and was well tolerated.77,78 Future studies with the cyclic STAT3 decoy will address pharmacology and toxicology, prior to evaluation in human clinical trials.
Indirect targeting of STAT3
The challenge of directly targeting STAT3 has resulted in substantial attention being paid to indirect targeting of STAT3 via inhibition of the upstream activators of STATs, Janus kinases (JAKs). Several JAK inhibitors have been developed and tested in clinical trials in patients with solid tumors, with two inhibitors currently approved by the FDA for other indications.79,80 Ruxolitinib, a JAK1/2-selective inhibitor, is one such FDA approved agent, and clinical studies of ruxolitinib in solid tumors have been reported.80 Although a Phase II study of ruxolitinib in combination with capecitabine in patients with metastatic pancreatic cancer showed greater overall survival in a subset of patients with elevated C-reactive protein (CRP) levels (NCT01423604), the overwhelming majority of clinical trials with ruxolitinib have been terminated due to lack of efficacy.81 Phase III trials studying ruxolitinib in patients with pancreatic cancer and in patients with elevated CRP levels were terminated due to a lack of difference observed in overall or progression-free survival between treatment groups (NCT02117479) (NCT02119663).82 In a Phase II study of ruxolitinib in triple-negative breast cancer, no objective responses despite demonstration of inhibitory activity against STAT3 led to termination of the trial (NCT01562873). A Phase I/II trial of ruxolitinib and the EGFR inhibitor erlotinib demonstrated that this combination was well-tolerated but lacked clinical activity in patients with lung adenocarcinoma (NCT02155465).83,84
Other JAK inhibitors have also suffered from inadequate anti-tumor activity when advanced to clinical trials in solid tumors. INCB052793, which selectively inhibits JAK1, entered a Phase I/II trial in patients with advanced malignancies that was prematurely terminated as the drug was shown to be ineffective (NCT02265510).85 Similarly, despite promising preclinical results, a Phase I clinical trial of AZD1480, an ATP-competitive inhibitor selective for JAK1/2, was halted due to lack of efficacy in addition to dose-limiting neurotoxicities.86 This dose-limiting toxicity exemplifies another limitation exhibited by JAK inhibitors; a Phase Ib study of the JAK1/2 inhibitor, momelotinib, when administered in combination with erlotinib in patients with EGFR-mutated metastatic NSCLC, was terminated due to dose-limiting toxicities of diarrhea and neutropenia (NCT02206763)87.
Thus, despite extensive efforts, efficacious indirect targeting of STAT3 in clinical studies using JAK inhibitors has proven challenging, with disappointing results stemming from lack of clinical activity and observation of adverse toxicities. Hence, direct targeting with agents such as SD-36 and AZD9150, or small molecule competitive inhibitors, may be advantageous in their capacity to selectively and more completely eliminate expression or function of STAT3 protein, increasing the specificity of effects and decreasing toxicity due to off-target activity. The PROTAC approach of SD-36, in particular, may be favorable due to its catalytic mode of action that may, in theory, lower the amount of drug required and potentially reduce risk of toxicities.
Targeting STAT3 in the tumor microenvironment
Although tightly regulated in normal cells, STAT3 signaling in cancer cells is often hyperactivated due to aberrantly active receptor tyrosine kinases (eg. EGFR, c-MET) or non-receptor tyrosine kinases (c-Src and BCR-ABL), or inactivation or downregulation of critical negative regulators such as protein inhibitor of activated STAT (PIAS), suppressor of cytokine signaling (SOCs) proteins, protein tyrosine phosphatases (PTPases), and members of the protein tyrosine phosphatase receptor (PTPR) family.8,88 Hyperactivation of STAT3 in tumor cells leads to production and secretion of immunosuppressive cytokines and growth factors, including VEGF, IL-10, and IL-6.8,88 These factors can act in a paracrine fashion to further propagate STAT3 activation in the tumor microenvironment. Activation of STAT3 in tumor infiltrating immune cells acts to suppress anti-tumor immunity by inhibiting the differentiation of dendritic cells (DCs), downregulating T helper (Th1 and Th2) cell activity, and contributing to the polarization of macrophages to an M2 immunosuppressive phenotype.89,90 Furthermore, T regulatory cells (Tregs) are positively modulated by STAT3 and have been observed to accumulate in tumors. The levels of myeloid-derived suppressor cells (MDSCs), associated with a number of immunosuppressive properties including impairment of CD4+ and CD8+ T cell activation, are also enhanced by STAT3. Accordingly, STAT3 mediates a number of immune evading and suppressing events that contribute to cancer progression.89–91
It is thus important to bear in mind the immunosuppressive role of STAT3 activation within the tumor microenvironment in order to better understand the anti-cancer activity of STAT3 inhibitors. The reduction of immunosuppressive Tregs levels following treatment with STAT3 inhibitor has been shown to augment the efficacy of radiation therapy in head and neck cancer.63 Given the role of STAT3 in the tumor microenvironment, it is possible that downregulation of STAT3-induced pro-inflammatory cytokines such as IL-6 and immunosuppressive MDSCs may generate immunostimulatory responses that enhance clinical benefit. Thus, identifying relationships between STAT3 inhibition and responses within the tumor microenvironment may prove valuable to designing targeted therapeutic strategies with optimal efficacy.92 A Phase Ib/II clinical trial evaluating the efficacy of AZD9150 in combination with durvalumab, an FDA-approved antibody that inhibits binding of PD-L1 to PD-1, and AZD9150 in combination with durvalumab plus chemotherapy is currently ongoing (NCT03421353).93
Conclusions
The development of PROTAC drugs and ASOs targeting STAT3 presents promising therapeutic strategies for inhibiting STAT3 expression, a transcription factor that has long been a challenging and highly pursued target. Notably, the mechanisms of action of these two therapies result in elimination of STAT3 at the level of expression, offering a more holistic approach to targeting STAT3, particularly in light of emerging evidence implicating non-canonical STAT3 signaling in the progression of cancer. PROTAC and ASO therapeutics may thus be advantageous over small molecule inhibitors operating through inhibition of upstream pathways or competitive inhibition of STAT3 protein, which are often limited by poor potency and the development of resistance.
SD-36, a novel PROTAC targeting STAT3 protein, has demonstrated robust and selective depletion of STAT3 protein in leukemia and lymphoma cell lines as well as complete and persisting tumor regression in mouse xenograft models. Encouragingly, SD-36 was well-tolerated in immunocompetent mice, although rigorous studies to further assess its safety profile will be important before clinical application is possible. Furthermore, although these initial studies show promising evidence of tumor regression in hematopoietic malignancies, it will be imperative to examine the activity of SD-36 in solid tumors and also investigate the potential deleterious effects that may arise from potently eliminating STAT3 expression in normal tissues. The ASO AZD9150 is generally well-tolerated and has shown potency at inhibiting STAT3 expression in a number of preclinical models as well as clinical benefit in a subset of patients with treatment refractory lymphoma. Phase II/III trials further evaluating the efficacy of AZD9150 in a range of tumor types are ongoing or pending. The potential for combining novel PROTAC and ASO agents targeting STAT3 with conventional chemotherapy and radiation represents a promising avenue for tumor reduction or eradication in the clinic.
Acknowledgements
This work was supported by the following grants: NIH R35CA231998 (J.R.G.) and R01DE028289 (D.E.J. and J.R.G.).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Clevenger CV. Roles and regulation of stat family transcription factors in human breast cancer. Am J Pathol 2004;165(5):1449–1460. doi: 10.1016/S0002-9440(10)63403-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamran MZ, Patil P, Gude RP. Role of STAT3 in Cancer Metastasis and Translational Advances. Buijs JT, ed. BioMed Research International. 2013;2013:421821. doi: 10.1155/2013/421821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an Oncogene. Cell. 1999;98(3):295–303. doi: 10.1016/S0092-8674(00)81959-5 [DOI] [PubMed] [Google Scholar]
- 4.Darnell JE. Validating Stat3 in cancer therapy. Nature Medicine. 2005;11(6):595–596. doi: 10.1038/nm0605-595 [DOI] [PubMed] [Google Scholar]
- 5.Timofeeva OA, Tarasova NI, Zhang X, et al. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. PNAS 2013;110(4):1267–1272. doi: 10.1073/pnas.1211805110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu H, Jove R. The STATs of cancer — new molecular targets come of age. Nature Reviews Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275 [DOI] [PubMed] [Google Scholar]
- 7.Turkson J STAT proteins as novel targets for cancer drug discovery. Expert Opinion on Therapeutic Targets. 2004;8(5):409–422. doi: 10.1517/14728222.8.5.409 [DOI] [PubMed] [Google Scholar]
- 8.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nature Reviews Clinical Oncology. 2018;15(4):234–248. doi: 10.1038/nrclinonc.2018.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu P, Wu D, Zhao L, et al. Prognostic role of STAT3 in solid tumors: a systematic review and meta-analysis. Oncotarget. 2016;7(15):19863–19883. doi: 10.18632/oncotarget.7887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quesnelle KM, Boehm AL, Grandis JR. STAT-mediated EGFR signaling in cancer. Journal of Cellular Biochemistry. 2007;102(2):311–319. doi: 10.1002/jcb.21475 [DOI] [PubMed] [Google Scholar]
- 11.Grandis JR, Drenning SD, Zeng Q, et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. PNAS 2000;97(8):4227–4232. doi: 10.1073/pnas.97.8.4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 2004;23(48):8017–8023. doi: 10.1038/sj.onc.1208159 [DOI] [PubMed] [Google Scholar]
- 13.Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interv 2011;11(1):18–26. doi: 10.1124/mi.11.1.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava J, DiGiovanni J. Non-canonical Stat3 signaling in cancer. Molecular Carcinogenesis. 2016;55(12):1889–1898. doi: 10.1002/mc.22438 [DOI] [PubMed] [Google Scholar]
- 15.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 Supports Ras-Dependent Oncogenic Transformation. Science. 2009;324(5935):1713–1716. doi: 10.1126/science.1171721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nkansah E, Shah R, Collie GW, et al. Observation of unphosphorylated STAT3 core protein binding to target dsDNA by PEMSA and X-ray crystallography. FEBS Letters. 2013;587(7):833–839. doi: 10.1016/j.febslet.2013.01.065 [DOI] [PubMed] [Google Scholar]
- 17.Braunstein J, Brutsaert S, Olson R, Schindler C. STATs Dimerize in the Absence of Phosphorylation. J Biol Chem 2003;278(36):34133–34140. doi: 10.1074/jbc.M304531200 [DOI] [PubMed] [Google Scholar]
- 18.Timofeeva OA, Chasovskikh S, Lonskaya I, et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J Biol Chem Published online February 29, 2012:jbc.M111.323899. doi: 10.1074/jbc.M111.323899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFκB. Genes Dev 2007;21(11):1396–1408. doi: 10.1101/gad.1553707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J, Chatterjee-Kishore M, Staugaitis SM, et al. Novel Roles of Unphosphorylated STAT3 in Oncogenesis and Transcriptional Regulation. Cancer Res 2005;65(3):939–947. [PubMed] [Google Scholar]
- 21.Sun X, Gao H, Yang Y, et al. PROTACs: great opportunities for academia and industry. Signal Transduction and Targeted Therapy. 2019;4(1):1–33. doi: 10.1038/s41392-019-0101-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pei H, Peng Y, Zhao Q, Chen Y. Small molecule PROTACs: an emerging technology for targeted therapy in drug discovery. RSC Adv 2019;9(30):16967–16976. doi: 10.1039/C9RA03423D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.An S, Fu L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553–562. doi: 10.1016/j.ebiom.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. PNAS 2001;98(15):8554–8559. doi: 10.1073/pnas.141230798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winter GE, Buckley DL, Paulk J, et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241):1376–1381. doi: 10.1126/science.aab1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bondeson DP, Mares A, Smith IED, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nature Chemical Biology. 2015;11(8):611–617. doi: 10.1038/nchembio.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohoka N, Okuhira K, Ito M, et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-dependent Protein Erasers (SNIPERs). J Biol Chem 2017;292(11):4556–4570. doi: 10.1074/jbc.M116.768853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai L, Zhou H, Xu R, et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell. 2019;36(5):498–511.e17. doi: 10.1016/j.ccell.2019.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang C, Han X-R, Yang X, et al. Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). European Journal of Medicinal Chemistry. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naito M, Ohoka N, Shibata N, Tsukumo Y. Targeted Protein Degradation by Chimeric Small Molecules, PROTACs and SNIPERs. Front Chem 2019;7. doi: 10.3389/fchem.2019.00849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arvinas Inc. A Phase 1, Open-Label, Dose Escalation Clinical Trial to Evaluate the Safety, Tolerability and Pharmacokinetics of ARV-471 in Patients With Estrogen Receptor Positive/Human Epidermal Growth Factor Receptor 2 Negative (ER+/HER2-) Locally Advanced or Metastatic Breast Cancer, Who Have Received Prior Hormonal Therapy and Chemotherapy in the Locally Advanced/Metastatic Setting. clinicaltrials.gov; 2020. Accessed June 18, 2020 https://clinicaltrials.gov/ct2/show/NCT04072952
- 32.Arvinas Inc. A Phase 1, Open-Label, Dose Escalation Clinical Trial to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ARV-110 in Patients With Metastatic Castration-Resistant Prostate Cancer Who Have Progressed on at Least Two Prior Systemic Therapies. clinicaltrials.gov; 2019. Accessed June 18, 2020 https://clinicaltrials.gov/ct2/show/NCT03888612
- 33.Zhou H, Bai L, Xu R, et al. Structure-Based Discovery of SD-36 as a Potent, Selective, and Efficacious PROTAC Degrader of STAT3 Protein. J Med Chem 2019;62(24):11280–11300. doi: 10.1021/acs.jmedchem.9b01530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu Z-Y, Fan L, Wang L, et al. STAT3 mutations are frequent in T-cell large granular lymphocytic leukemia with pure red cell aplasia. Journal of Hematology & Oncology. 2013;6(1):82. doi: 10.1186/1756-8722-6-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Teramo A, Barila G, Calabretto G, et al. STAT3 mutation impacts biological and clinical features of T-LGL leukemia. Oncotarget. 2017;8(37):61876–61889. doi: 10.18632/oncotarget.18711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee H, Pal SK, Reckamp K, Figlin RA, Yu H. STAT3: A Target to Enhance Antitumor Immune Response. In: Dranoff G, ed. Cancer Immunology and Immunotherapy. Current Topics in Microbiology and Immunology. Springer; 2011:41–59. doi: 10.1007/82_2010_51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minegishi Y, Saito M, Tsuchiya S, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062. doi: 10.1038/nature06096 [DOI] [PubMed] [Google Scholar]
- 38.Churcher I Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? J Med Chem 2018;61(2):444–452. doi: 10.1021/acs.jmedchem.7b01272 [DOI] [PubMed] [Google Scholar]
- 39.Li X, Song Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. Journal of Hematology & Oncology. 2020;13(1):50. doi: 10.1186/s13045-020-00885-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chamberlain PP, Hamann LG. Development of targeted protein degradation therapeutics. Nature Chemical Biology. 2019;15(10):937–944. doi: 10.1038/s41589-019-0362-y [DOI] [PubMed] [Google Scholar]
- 41.Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 2017;16(2):101–114. doi: 10.1038/nrd.2016.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai AC, Toure M, Hellerschmied D, et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl 2016;55(2):807–810. doi: 10.1002/anie.201507634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mares A, Miah AH, Smith IED, et al. Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Commun Biol 2020;3. doi: 10.1038/s42003-020-0868-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Fusco D, Dinallo V, Marafini I, Figliuzzi MM, Romano B, Monteleone G. Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease. Front Pharmacol 2019;10. doi: 10.3389/fphar.2019.00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nature Reviews Neurology. 2018;14(1):9–21. doi: 10.1038/nrneurol.2017.148 [DOI] [PubMed] [Google Scholar]
- 46.Bennett CF, Baker BF, Pham N, Swayze E, Geary RS. Pharmacology of Antisense Drugs. Annu Rev Pharmacol Toxicol 2017;57(1):81–105. doi: 10.1146/annurev-pharmtox-010716-104846 [DOI] [PubMed] [Google Scholar]
- 47.Henry SP, Templin MV, Gillett N, Rojko J, Levin AA. Correlation of Toxicity and Pharmacokinetic Properties of a Phosphorothioate Oligonucleotide Designed to Inhibit ICAM-1: Toxicologic Pathology. Published online July 2, 2016. doi: 10.1177/019262339902700117 [DOI] [PubMed] [Google Scholar]
- 48.Stein CA. The experimental use of antisense oligonucleotides: a guide for the perplexed. J Clin Invest 2001;108(5):641–644. doi: 10.1172/JCI13885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quemener AM, Bachelot L, Forestier A, Donnou‐Fournet E, Gilot D, Galibert M-D. The powerful world of antisense oligonucleotides: From bench to bedside. WIREs RNA n/a(n/a):e1594. doi: 10.1002/wrna.1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nature Biotechnology. 2017;35(3):238–248. doi: 10.1038/nbt.3765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh SK, Koshkin AA, Wengel J, Nielsen P. LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem Commun 1998;(4):455–456. doi: 10.1039/A708608C [DOI] [Google Scholar]
- 52.Swayze EE, Siwkowski AM, Wancewicz EV, et al. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res 2007;35(2):687–700. doi: 10.1093/nar/gkl1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burel SA, Hart CE, Cauntay P, et al. Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res 2016;44(5):2093–2109. doi: 10.1093/nar/gkv1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mathew V, Wang AK. Inotersen: new promise for the treatment of hereditary transthyretin amyloidosis. Drug Des Devel Ther 2019;13:1515–1525. doi: 10.2147/DDDT.S162913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bennett CF. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annual Review of Medicine. 2019;70(1):307–321. doi: 10.1146/annurev-med-041217-010829 [DOI] [PubMed] [Google Scholar]
- 56.Zhu B-R, Cai J-M, Tang G-S, et al. [Effects of STAT3 antisense oligonucleotide on proliferation and apoptosis of non-small cell lung cancer cell line A549]. Ai Zheng 2007;26(8):820–827. [PubMed] [Google Scholar]
- 57.Wang J-G, Li X-M, Chen Y-H, Lu X-Y. [Molecular mechanisms involved in regulation of proliferation and apoptosis by STAT3 antisense oligonucleotide and chemotherapy in laryngeal cancer cells]. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 2007;42(3):222–226. [PubMed] [Google Scholar]
- 58.Bai A, Hu P, Chen J, et al. Blockade of STAT3 by antisense oligonucleotide in TNBS-induced murine colitis. Int J Colorectal Dis 2007;22(6):625–635. doi: 10.1007/s00384-006-0229-z [DOI] [PubMed] [Google Scholar]
- 59.Hong D, Kurzrock R, Kim Y, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Science Translational Medicine. 2015;7(314):314ra185–314ra185. doi: 10.1126/scitranslmed.aac5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shastri A, Choudhary G, Teixeira M, et al. Antisense STAT3 inhibitor decreases viability of myelodysplastic and leukemic stem cells. J Clin Invest 2018;128(12):5479–5488. doi: 10.1172/JCI120156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Odate S, Veschi V, Yan S, Lam N, Woessner R, Thiele CJ. Inhibition of STAT3 with the Generation 2.5 Antisense Oligonucleotide, AZD9150, Decreases Neuroblastoma Tumorigenicity and Increases Chemosensitivity. Clin Cancer Res 2017;23(7):1771–1784. doi: 10.1158/1078-0432.CCR-16-1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun C-Y, Nie J, Huang J-P, Zheng G-J, Feng B. Targeting STAT3 inhibition to reverse cisplatin resistance. Biomedicine & Pharmacotherapy. 2019;117:109135. doi: 10.1016/j.biopha.2019.109135 [DOI] [PubMed] [Google Scholar]
- 63.Oweida AJ, Darragh L, Phan A, et al. STAT3 Modulation of Regulatory T Cells in Response to Radiation Therapy in Head and Neck Cancer. J Natl Cancer Inst 2019;111(12):1339–1349. doi: 10.1093/jnci/djz036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burel SA, Han S-R, Lee H-S, et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther 2013;23(3):213–227. doi: 10.1089/nat.2013.0422 [DOI] [PubMed] [Google Scholar]
- 65.Reilley MJ, McCoon P, Cook C, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6(1):119. doi: 10.1186/s40425-018-0436-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ok CY, Chen J, Xu-Monette ZY, et al. Clinical Implications of Phosphorylated STAT3 Expression in de novo Diffuse Large B-cell Lymphoma. Clin Cancer Res 2014;20(19):5113–5123. doi: 10.1158/1078-0432.CCR-14-0683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu H, Tong X, Mugundu G, et al. Population pharmacokinetic analysis of danvatirsen supporting flat dosing switch. J Pharmacokinet Pharmacodyn. 2019;46(1):65–74. doi: 10.1007/s10928-019-09619-6 [DOI] [PubMed] [Google Scholar]
- 68.Tolcher A, Flaherty K, Shapiro GI, et al. A First-in-Human Phase I Study of OPB-111077, a Small-Molecule STAT3 and Oxidative Phosphorylation Inhibitor, in Patients with Advanced Cancers. The Oncologist. 2018;23(6):658–e72. doi: 10.1634/theoncologist.2017-0325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brambilla L, Genini D, Laurini E, et al. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of the Signal Transducer and Activator of Transcription 3 (STAT3). Molecular Oncology. 2015;9(6):1194–1206. doi: 10.1016/j.molonc.2015.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bendell JC, Hong DS, Burris HA, et al. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother Pharmacol 2014;74(1):125–130. doi: 10.1007/s00280-014-2480-2 [DOI] [PubMed] [Google Scholar]
- 71.Bharadwaj U, Eckols TK, Xu X, et al. Small-molecule inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma. Oncotarget. 2016;7(18):26307–26330. doi: 10.18632/oncotarget.8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oral STAT3 Inhibitor, TTI-101, in Patients With Advanced Cancers - Full Text View - ClinicalTrials.gov. Accessed June 22, 2020 https://clinicaltrials.gov/ct2/show/NCT03195699
- 73.SHITARA K, YODO Y, IINO S. A Phase I Study of Napabucasin Plus Paclitaxel for Japanese Patients With Advanced/Recurrent Gastric Cancer. In Vivo. 2019;33(3):933–937. doi: 10.21873/invivo.11561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jonker DJ, Nott L, Yoshino T, et al. Napabucasin versus placebo in refractory advanced colorectal cancer: a randomised phase 3 trial. The Lancet Gastroenterology & Hepatology. 2018;3(4):263–270. doi: 10.1016/S2468-1253(18)30009-8 [DOI] [PubMed] [Google Scholar]
- 75.Sumitomo Dainippon Pharma Oncology, Inc. A Phase III Study of BBI-608 Plus Nab-Paclitaxel With Gemcitabine in Adult Patients With Metastatic Pancreatic Adenocarcinoma. clinicaltrials.gov; 2020. Accessed August 21, 2020 https://clinicaltrials.gov/ct2/show/NCT02993731
- 76.Lee DS, O’Keefe RA, Ha PK, Grandis JR, Johnson DE. Biochemical Properties of a Decoy Oligodeoxynucleotide Inhibitor of STAT3 Transcription Factor. International Journal of Molecular Sciences. 2018;19(6):1608. doi: 10.3390/ijms19061608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sen M, Thomas SufiM, Kim S, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov 2012;2(8):694–705. doi: 10.1158/2159-8290.CD-12-0191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sen M, Paul K, Freilino ML, et al. Systemic Administration of a Cyclic Signal Transducer and Activator of Transcription 3 (STAT3) Decoy Oligonucleotide Inhibits Tumor Growth without Inducing Toxicological Effects. Mol Med 2013;20(1):46–56. doi: 10.2119/molmed.2013.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buchert M, Burns CJ, Ernst M. Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene. 2016;35(8):939–951. doi: 10.1038/onc.2015.150 [DOI] [PubMed] [Google Scholar]
- 80.Kontzias A, Kotlyar A, Laurence A, Changelian P, O’Shea JJ. Jakinibs: A New Class of Kinase Inhibitors in Cancer and Autoimmune Disease. Curr Opin Pharmacol 2012;12(4):464–470. doi: 10.1016/j.coph.2012.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hurwitz HI, Uppal N, Wagner SA, et al. Randomized, Double-Blind, Phase II Study of Ruxolitinib or Placebo in Combination With Capecitabine in Patients With Metastatic Pancreatic Cancer for Whom Therapy With Gemcitabine Has Failed. J Clin Oncol 2015;33(34):4039–4047. doi: 10.1200/JCO.2015.61.4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hurwitz H, Van Cutsem E, Bendell J, et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Invest New Drugs. 2018;36(4):683–695. doi: 10.1007/s10637-018-0580-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stover DG, Gil Del Alcazar CR, Brock J, et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer. 2018;4:10. doi: 10.1038/s41523-018-0060-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu HA, Perez L, Chang Q, et al. A phase 1/2 trial of ruxolitinib and erlotinib in patients with EGFR-mutant lung adenocarcinomas with acquired resistance to erlotinib. J Thorac Oncol 2017;12(1):102–109. doi: 10.1016/j.jtho.2016.08.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Incyte Corporation. A Phase 1/2, Open-Label, Dose-Escalation, Safety and Tolerability Study of INCB052793 in Subjects With Advanced Malignancies. clinicaltrials.gov; 2020. Accessed August 21, 2020 https://clinicaltrials.gov/ct2/show/results/NCT02265510
- 86.Plimack ER, LoRusso PM, McCoon P, et al. AZD1480: A Phase I Study of a Novel JAK2 Inhibitor in Solid Tumors. Oncologist 2013;18(7):819–820. doi: 10.1634/theoncologist.2013-0198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Padda S, Reckamp K, Koczywas M, et al. P2.03–043 A Phase 1b Study of Erlotinib and Momelotinib for TKI-Naïve EGFR-Mutated Metastatic Non-Small Cell Lung Cancer. Journal of Thoracic Oncology. 2017;12(11):S2143–S2144. doi: 10.1016/j.jtho.2017.09.1294 [DOI] [Google Scholar]
- 88.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews Immunology. 2007;7(1):41–51. doi: 10.1038/nri1995 [DOI] [PubMed] [Google Scholar]
- 89.Wang Y, Shen Y, Wang S, Shen Q, Zhou X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Letters. 2018;415:117–128. doi: 10.1016/j.canlet.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferguson SD, Srinivasan VM, Heimberger AB. The role of STAT3 in tumor-mediated immune suppression. J Neurooncol 2015;123(3):385–394. doi: 10.1007/s11060-015-1731-3 [DOI] [PubMed] [Google Scholar]
- 91.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology. 2009;9(3):162–174. doi: 10.1038/nri2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kroemer G, Galluzzi L, Zitvogel L. STAT3 inhibition for cancer therapy: Cell-autonomous effects only? OncoImmunology. 2016;5(5):e1126063. doi: 10.1080/2162402X.2015.1126063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.AZD9150 Plus Durvalumab Alone or in Combination With Chemotherapy in Patients With Advanced, Solid Tumours and in Patients With Non-Small-Cell Lung Cancer - Tabular View - ClinicalTrials.gov Accessed June 22, 2020 https://clinicaltrials.gov/ct2/show/record/NCT03421353