

Abstract
Three-dimensional (3D) bioprinting is a promising technology to produce tissue-like structures, but a lack of diversity in bioinks is a major limitation. Ideally each cell type would be printed in its own customizable bioink. To fulfill this need for a universally applicable bioink strategy, we developed a versatile, bioorthogonal bioink crosslinking mechanism that is cell compatible and works with a range of polymers. We term this family of materials UNIversal, Orthogonal Network (UNION) bioinks. As demonstration of UNION bioink versatility, gelatin, hyaluronic acid (HA), recombinant elastin-like protein (ELP), and polyethylene glycol (PEG) were each used as backbone polymers to create inks with storage moduli spanning 200 to 10,000 Pa. Because UNION bioinks are crosslinked by a common chemistry, multiple materials can be printed together to form a unified, cohesive structure. This approach is compatible with any support bath that enables diffusion of UNION crosslinkers. Both matrix-adherent human corneal mesenchymal stromal cells and non-matrix-adherent human induced pluripotent stem cell-derived neural progenitor spheroids were printed with UNION bioinks. The cells retained high viability and expressed characteristic phenotypic markers after printing. Thus, UNION bioinks are a versatile strategy to expand the toolkit of customizable materials available for 3D bioprinting.
Keywords: 3D bioprinting, bioink, biomaterials, bioorthogonal chemistry
Graphical Abstract
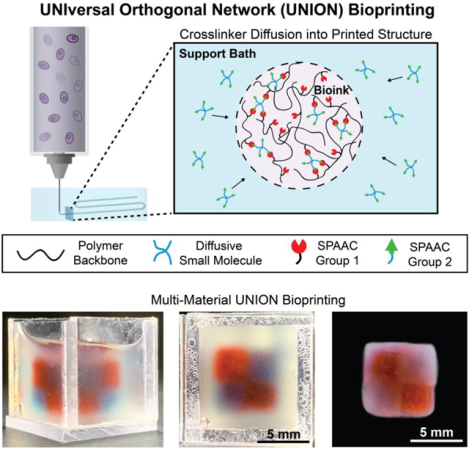
A family of materials, termed UNIversal, Orthogonal Network (UNION) bioinks, is developed for 3D bioprinting of multi-material structures using a cell-compatible, bioorthogonal crosslinking mechanism. UNION bioinks can be created from a wide range of polymers, work with a variety of cell types, and offer biochemical and mechanical tunability, thus expanding the toolkit of customizable materials available for 3D bioprinting.
1. Introduction
Three-dimensional (3D) bioprinting has emerged as a promising technology for producing complex, functional tissue constructs containing precisely patterned cells.[1–5] However, bioprinting remains limited by the number of materials that can be used as bioinks, especially in comparison to the vast array of biomaterials developed for non-printed tissue engineering scaffolds. Here, a bioink is defined as a printable composite that includes both cells and polymer.[6] Since a cell’s phenotype is exquisitely sensitive to the biochemical and mechanical properties of its surroundings, the matrix cues presented by the bioink will become increasingly important as bioengineers attempt to fabricate more cellularly diverse engineered tissues.[7,8] Ideally, each cell type would be printed in its own customizable bioink matrix, such that the bioink is tailored to fit the cellular and structural needs of the desired tissue application. With the limited number of bioinks available today, there is a clear need for a universal bioink strategy that can be easily customized to support any type of cell.[9]
One factor prohibiting the use of many previously developed biomaterials as bioinks is the intrinsic difficulty of printing soft materials without support into air. To overcome this limitation, freeform printing into gel-based baths was developed to support extrusion printing of soft biological materials into complex structures. Freeform reversible embedding of suspended hydrogels (FRESH) printing involves extruding a bioink into a reversible gel support bath that provides physical reinforcement to structures like arches and overhangs, even when printing weak biomaterials.[10–14] In addition to enabling the fabrication of constructs with overhang features, the FRESH bioprinting technique also enables printing of inks with a broad variety of rheological properties.[10,15] While in the support bath, the bioink is crosslinked to stabilize the printed construct, a process also known as “curing”. The support bath is then liquified, typically through a change in temperature, and the bioprinted structure is removed for downstream applications. While this technique offers great improvements in fidelity and structural complexity compared to open-air printing, demonstrations have been limited to only a few materials and primarily have been used to print acellular structures. This is partly due to the limited number of effective crosslinking strategies, which contributes to the overall lack of bioink diversity.
To achieve greater bioink diversity, new crosslinking strategies are required. Many previously reported strategies have used crosslinking reactions with off-target chemical or biological reactivity that may hinder cell function and compromise cell viability. For example, light-curable inks require cytotoxic initiators, alginate inks commonly use super-physiological levels of Ca2+, and collagen inks often use large pH shifts that preclude cell encapsulation.[15–18] Importantly, many of these bioink crosslinking strategies are specific to a particular polymer (e.g. Ca2+ crosslinks alginate but not collagen inks). Therefore, it is difficult to print multiple materials into a single, integrated structure. Thus, to realize the immense potential of bioprinting, it is necessary to develop a universal family of bioinks that (1) use a cell-compatible crosslinking method that works with a variety of polymers, (2) can integrate together into coherent structures, (3) are versatile for use with different support baths, and (4) are biochemically and mechanically customizable for multiple cell types.
Here, we report the development of a universal bioink strategy that uses a bioorthogonal crosslinking mechanism to enable freeform bioprinting of various cell types with a range of polymers. We term this family of materials UNIversal, Orthogonal Network (UNION) bioinks. This strategy provides a toolkit of bioinks that can be customized for specific biological applications without redesigning the crosslinking mechanism for each bioink used. This bioprinting approach is compatible with polymers that are extrudable and able to be chemically modified with multiple bioorthogonal functional groups. Furthermore, because each individual bioink uses a common bioorthogonal crosslinking chemistry, multiple inks can be printed together into a single, cohesive construct. This is in contrast to previous demonstrations of multi-material printing in which the individual materials are typically crosslinked using distinct crosslinking mechanisms specific to each polymer.[3,19,20] Bioorthogonal chemistries enable the rapid formation of a covalent bond between two distinct, complementary chemical functional groups.[21–26] These reactions are ideally suited for bioprinting since they are chemically specific, produce no toxic side products, proceed rapidly under ambient conditions, and can be designed to proceed without external catalysts or triggers.[27–29] Importantly, bioorthogonally crosslinked bioinks have no cross-reactivity with other biomolecules, including those present on the surface of cells, in the culture medium, or in the ink itself, making this a universal strategy.
2. Results and Discussion
UNION bioinks are prepared by grafting one of the bioorthogonal chemical groups onto the backbone of a polymer prior to mixing with cells (Figure 1A). The complementary biorthogonal chemical group is presented on a crosslinking molecule that is added into the support bath prior to printing (Figure 1B). After the UNION bioink is extruded into the bath, the crosslinkers passively diffuse into the printed structure and spontaneously react with the bioink, covalently crosslinking the polymer to stiffen and stabilize the final structure. Once the gel support bath has been liquified, the final printed structure can be removed.
Figure 1. UNIversal Orthogonal Network (UNION) bioprinting in a support bath uses diffusable crosslinkers to enable bioorthogonal stabilization of UNION bioinks.
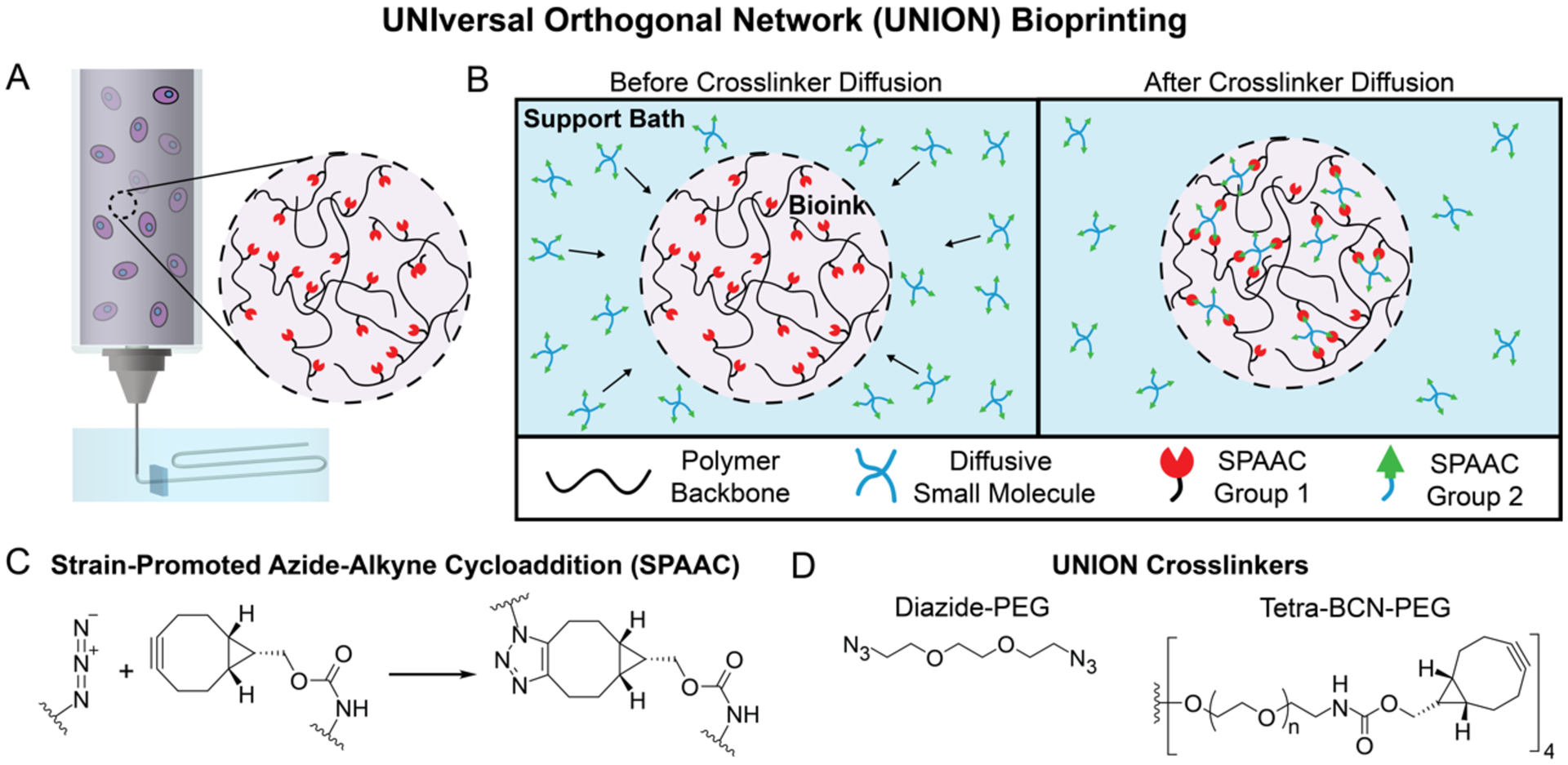
A. UNION inks are mixed with cells in a syringe and extruded into a gel support bath. B. UNION crosslinkers with complementary chemical groups diffuse into printed UNION bioinks and covalently crosslink the polymer into a network. C. Strain-promoted azide-alkyne cycloaddition (SPAAC) copper-free, click-chemistry is a specific and cytocompatible, bioorthogonal chemistry for crosslinking between azides and bicyclononynes in UNION bioprinting. D. UNION crosslinkers include a small diazide-PEG (MW 200 Da), and a larger tetra-BCN-PEG (MW 20,000 Da).
To produce a universal crosslinking scheme, we chose to use the bioorthogonal strain-promoted azide-alkyne cycloaddition (SPAAC) reaction between azides and bicyclononynes (BCN) (Figure 1C). This is a water-stable form of copper-free, click-chemistry with reasonable reaction kinetics for homogeneous encapsulation of cells within hydrogels.[24,30–32] SPAAC chemistries are only one of several classes of bioorthogonal chemistries currently under investigation for tissue engineering applications, and others may also be suitable for UNION bioinks.[28] The selectivity of SPAAC reactions ensures that once the bioorthogonal groups are grafted onto a polymer, they will only be crosslinked by the corresponding bioorthogonal partner group and will not cross-react with unmodified polymers or biochemical groups present on the cell surface or in the cell culture medium.
The UNION bioink strategy is compatible with any polymer that is water soluble, amenable to conjugation chemistry, non-cytotoxic, and extrudable into a support bath. To demonstrate the versatility of this crosslinking strategy, gelatin and polyethylene glycol (PEG) were chosen as examples of natural and synthetic polymers, respectively, for BCN grafting. Both polymers were functionalized under anhydrous conditions using N-hydroxysuccinimide (NHS) ester amidation of primary amines. In a parallel demonstration, hyaluronic acid (HA) and a recombinant elastin-like protein (ELP) were functionalized with azide functional groups, again as examples of a natural polymer and an engineered polymer, respectively. For the four polymers selected here, each was successfully conjugated with between 2 to 22 SPAAC functional groups per polymer chain (Figure S1). Thus, four ink materials in total were prepared: gelatin-BCN, PEG-BCN, HA-Azide, and ELP-Azide.
Crosslinkers for UNION bioinks must be (i) multifunctional (i.e. have 2 or more reactive groups per molecule) in order to crosslink the printed polymer into a stable network, and (ii) soluble to enable diffusion through the gel support bath. For the BCN-functionalized inks, a small molecule diazide-PEG (MW 200 Da, Figure 1D) was purchased for use as a crosslinker. In comparison to the highly hydrophilic azide groups, BCN functional groups are hydrophobic, and typically need to be grafted to large hydrophilic polymers to remain soluble at a usable crosslinking concentration. We determined that functionalization of a multi-arm PEG at a ratio of one BCN functional group per ~5 kDa of PEG was sufficient for solubility up to at least 10 mg mL−1. Therefore, a tetra-BCN-PEG (MW 20 kDa, Figure 1D) was synthesized for use as a crosslinker for the azide-modified inks.
When mixed with their corresponding UNION crosslinker (either diazide-PEG or tetra-BCN-PEG), gelatin-BCN, PEG-BCN, HA-Azide, and ELP-Azide were all found to produce hydrogels, as demonstrated by oscillatory shear rheology (Figure 2 and S2). As cells are known to respond to mechanical cues present in their microenvironment,[33–35] we explored the tunability of UNION ink mechanics. Soft tissues typically have shear storage moduli spanning from ~100 Pa to ~10 kPa;[36] thus, bioinks with mechanical properties within this range may be useful for tissue engineering applications. Here, the final hydrogel storage moduli varied from 200 Pa to 10,000 Pa and could be tuned by at least one order of magnitude for each polymer by changing its weight percentage (Figure 2A–D).
Figure 2. UNION inks can be formulated from a range of polymers.

A-D. UNION inks fabricated from (A) gelatin-BCN, (B) PEG-BCN, (C) HA-Azide, and (D) ELP-Azide with their corresponding UNION crosslinker (A-B, diazide-PEG; C-D, tetra-BCN-PEG) form hydrogels (G’ > G”, where filled markers are the storage moduli, G’, and open markers are the loss moduli, G”) with a range of mechanical stiffness depending on the polymer concentration (wt% shown in each legend, n = 3). UNION inks can be printed and crosslinked into logpile structures in a gel support bath and maintain shape fidelity after being released, as shown in representative top-down (left) and side-view (right) photographs for each ink.
Using freeform embedded printing techniques, UNION inks can be printed into complex shapes in a gel support bath. As a first demonstration, we printed logpile structures using each of our functionalized ink materials into a commercially available gel support bath for FRESH printing. The LifeSupport bath is a slurry of gelatin microparticles that provides support during printing and can then be melted away at 37 °C.[15] The appropriate crosslinker (either diazide-PEG or tetra-BCN-PEG) is added to the support bath during hydration of the gelatin microparticles to achieve a final concentration of 1 mg mL−1 or 5 mg mL−1, respectively. The total quantity of crosslinker added was estimated to be at least a two-fold excess relative to the theoretical amount needed to fully crosslink all printed components. Importantly, the addition of a crosslinker does not alter the shear-thinning behavior of the bath (Figure S3), and thus does not affect the support function of the gel bath, which still allows features such as windows and overhangs to be printed. Using a custom syringe-based extruder, 8-mm logpiles were printed using each of the UNION inks into a LifeSupport bath containing the appropriate crosslinkers at room temperature (Figure S4, CAD target structure shown in Figure S5A). The printed structures were allowed to cure for at least 1 hour in the gel support bath to facilitate complete crosslinking before being heated to 37 °C to melt and remove the gelatin support bath. For all four ink formulations, the hydrogel modulus reached 90% of its final value within 30 minutes of reaction time (Figure S2). This suggests a post-printing diffusion time of 1 hour should be sufficient for the construct to crosslink into a self-supporting structure. Finally, to assess post-printing gelation, the elastic moduli of printed disks were measured by unconfined compressive testing (Figure S6) following crosslinking and release from the support bath. Elastic moduli of released prints ranged between 800 – 8,000 Pa. A table detailing the ink concentration and crosslinking conditions for all structures is presented in the Supporting Information (Table S1).
As with other bioprinting strategies, the print resolution is a function of these bioink parameters (Table S1), support bath parameters, and printer parameters such as nozzle diameter and print speed.[15,37,38] Here, the diameter of individual UNION ink filaments was altered from 100 μm with a 30-gauge needle to 400 μm with a 22-gauge needle (Figure S7). As reported previously by Lee et al, micron-sized surface roughness is observed on individual filaments printed in LifeSupport baths due to the colloidal bath suspension.[15] Once printed, the individual ink filaments have limited flow and can crosslink together to produce a cohesive structure with multiple layers. In the logpile structures, visible void spaces remain after printing, and shape fidelity is maintained after release from the support bath (Figure 2A–D, bottom and Figure S4).
UNION printing extends beyond compositional variation to complex shapes and multi-material printing. Windows can be effectively printed in a LifeSupport bath without crowning or deformation, demonstrating controlled FRESH printing of UNION bioinks in a support bath containing crosslinkers (Figure 3A, left; CAD target structure shown in Figure S5B). The open structure remains and does not collapse following removal of the construct from the support bath (Figure 3A, right). Introducing a UNION crosslinker also allows for increased complexity of the printed structure by enabling multiple bioinks to be printed and crosslinked together using the same bioorthogonal chemistry. As a demonstration, gelatin-BCN and PEG-BCN polymers were printed simultaneously in the same support bath containing diazide-PEG crosslinkers (Video S1) to form a single integrated structure (Figure 3B; CAD target structure shown in Figure S5C). Similarly, HA-Azide and ELP-Azide bioinks can printed simultaneously in a support bath containing the tetra-BCN-PEG crosslinker. The integrity of the interface between the two UNION inks was tested by printing dual-material dogbone structures for tensile testing (CAD target structure shown in Figure S5D). Elongation of the dual-material dogbones was similar to the least extensible bioink component for both printed structures, illustrating that adhesion between the two bioinks is comparable to single bioink cohesion (Figure 3C). These results also suggest that multiple polymers conjugated with the same SPAAC reactive group could be blended to create an infinite library of bioinks with different polymer compositions for further customization. Furthermore, because UNION inks rely on a biorthogonal reaction, they are also compatible with other bioink curing strategies (e.g. enzymatic, ionic, or photo-crosslinking) that could be used alongside this technique in the future. Thus, the UNION strategy enables the potential for compositional flexibility not previously found in other bioink designs.
Figure 3. UNION inks can be utilized in fabrication of windows and multi-material printing.
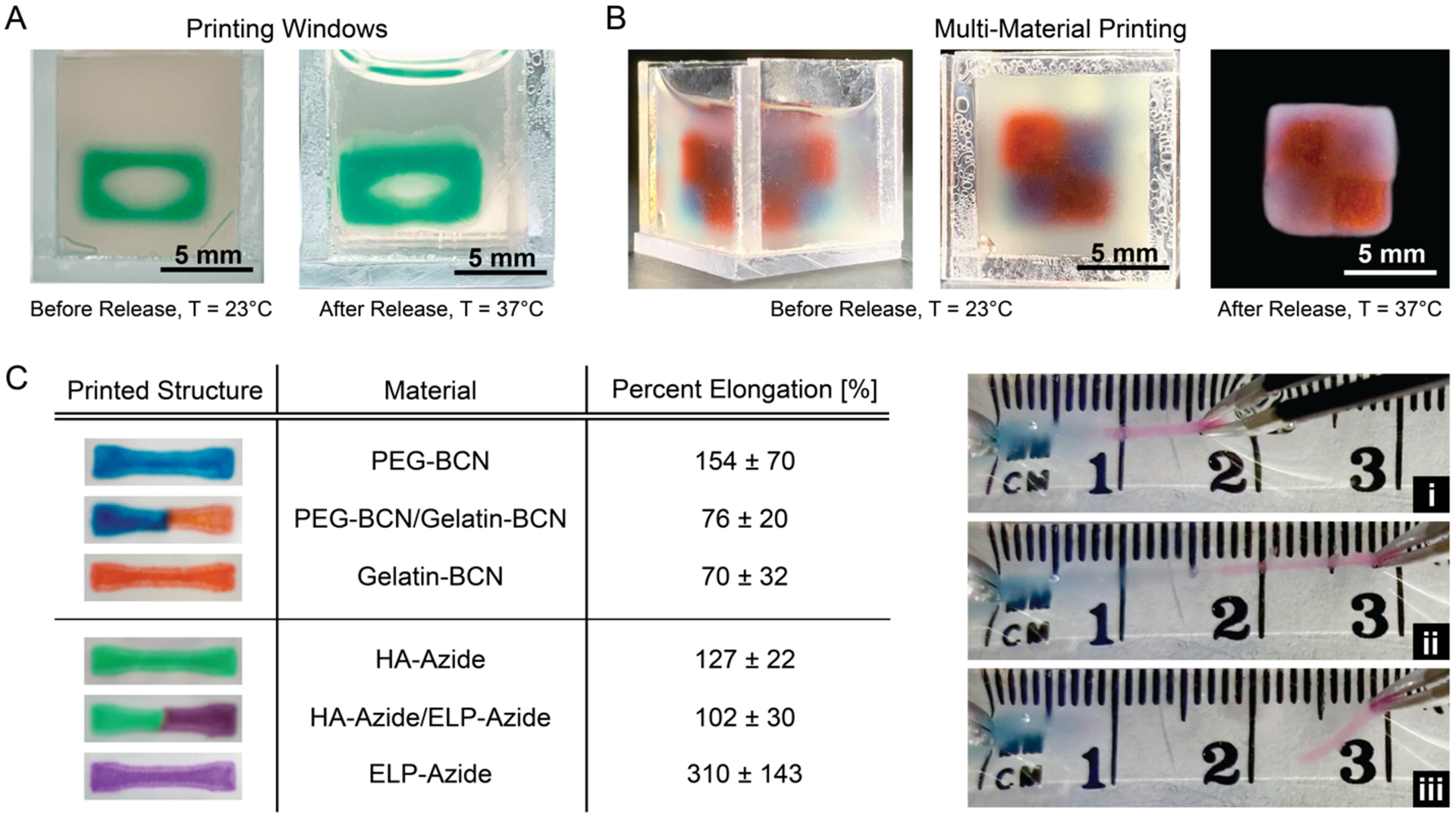
A. HA-Azide inks are printed into a structure with an elliptical window in a LifeSupport bath with tetra-BCN-PEG crosslinkers (left) and retain their open structure after release from the support bath (right). B. Gelatin-BCN (red) and PEG-BCN (blue) inks are printed side-by-side into a cohesive 3D checkerboard structure in LifeSupport with diazide-PEG crosslinker. C. Dogbone structures were printed using either single-material or dual-material (PEG-BCN/gelatin-BCN and HA-Azide/ELP-Azide) inks. Percent elongation before fracture was manually determined for each construct and is presented in the table. Data are averages ± standard deviation, n ≥ 3.
Next, we showed that the versatility of UNION inks also extends to the choice of support bath. UNION inks should be compatible with other commonly available aqueous support baths without concern for chemical cross-reactivity due to the bioorthogonal nature of the crosslinking chemistry; however, care must be taken to ensure that the support bath allows diffusion of the crosslinkers into the printed ink. Here, both a 26 wt% (w/v) Pluronic F-127 support bath (Figure 4A, right) and the previously described gelatin microparticle support bath (LifeSupport, Figure 4A, left) were both found to be compatible with UNION inks and crosslinkers. To achieve homogeneous crosslinking and stabilization of the printed structure, the UNION crosslinkers must be able to diffuse through the support bath and through the crosslinked ink. Diffusion coefficients were determined experimentally for both support baths, using either fluorescently-tagged diffusants and a custom dialysis chamber (for the Pluronic bath) or fluorescence recovery after photobleaching (for the LifeSupport bath) (Figure S8). The diazide-PEG crosslinker had a comparable diffusion coefficient in the Pluronic support bath (90 μm2 s−1) as in the LifeSupport bath (87 μm2 s−1) (Figure 4B and Figure S8). As expected, this is less than the theoretical Stokes-Einstein diffusion coefficient in water (~250 μm2 s−1) since the support baths are both gel phase. In comparison, larger diffusants (20-kDa and 40-kDa dextran with hydrodynamic radii (RH) about three times and five times larger than diazide-PEG, respectively) had smaller diffusion coefficients in LifeSupport (33 μm2 s−1 and 18 μm2 s−1, respectively), which again were less than the theoretical diffusion coefficient in water (~70 μm2 s−1 and ~50 μm2 s−1, respectively). As expected, the diffusion coefficients (D) for these different diffusants scale approximately with the inverse of their hydrodynamic radii (i.e. D ~ 1/RH).
Figure 4. UNION crosslinkers diffuse through gel support baths and UNION inks.
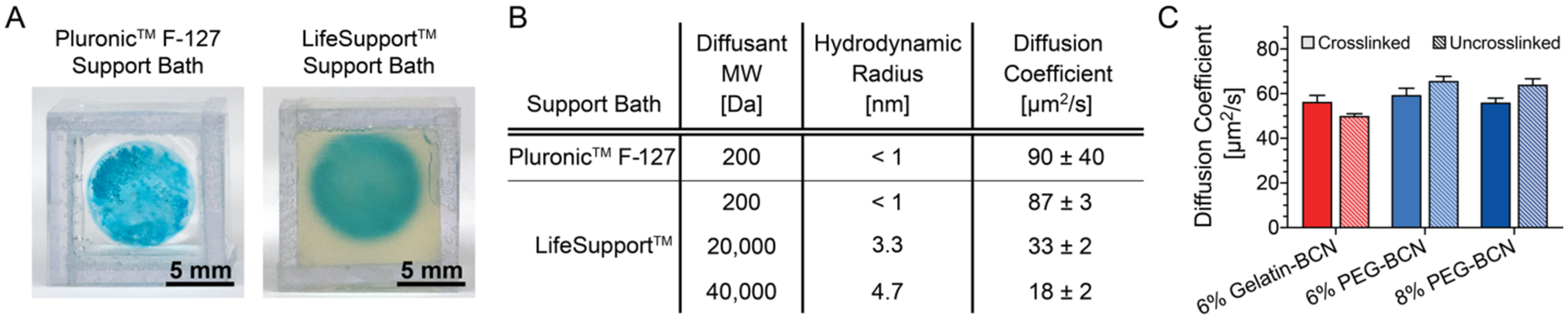
A. PEG-BCN inks successfully print disks in both 26% (w/v) Pluronic F-127 (left) and LifeSupport (right) baths. B. Small (fluorescently-tagged, 200-Da diazide-PEG) and large (fluorescently-tagged, 20,000-Da or 40,000-Da dextran) molecules diffuse through Pluronic and LifeSupport baths, n ≥ 3, data are averages ± standard deviation. C. Diffusion of fluorescently-tagged dextran (MW 10,000 Da) is similar in crosslinked and uncrosslinked gelatin-BCN (6 wt%) and PEG-BCN UNION (6 and 8 wt%) inks, n = 3, data are averages ± standard deviation.
Similar to the necessity for crosslinker transport through the support bath, the crosslinker must also diffuse through the printed bioink to achieve homogeneous crosslinking throughout the hydrogel network. Polymer crosslinking to form a hydrogel network is known to potentially reduce the diffusion coefficient depending on the diffusant size relative to the hydrogel network mesh size.[39,40] To determine if the mesh size of the crosslinked ink hinders further diffusion of the crosslinker, we compared the diffusivity of a fluorescently-labeled molecule (10 kDa dextran, RH ~ 2.3 nm) in uncrosslinked vs. fully crosslinked inks. This fluorescent diffusant was of a size similar to that of the larger UNION crosslinker, but lacked any reactive groups, and thus would not alter the network mesh size while diffusing through the hydrogel. The diffusion coefficient was determined to be similar (~60 μm2 s−1) in both gelatin-BCN and PEG-BCN networks, regardless of the whether the ink was uncrosslinked or fully crosslinked (Figure 4C). Similarly, the diffusion coefficients in both 6 wt% and 8 wt% PEG-BCN networks were similar, within experimental error of the measurement (Figure 4C). Together, these data suggest that even when fully crosslinked, the network mesh size of UNION bioinks is sufficiently large to enable the diffusion of UNION crosslinkers.
With this experimentally determined diffusion coefficient, we estimated that the characteristic diffusion length (L ~ (D·t)1/2) of these crosslinking reagents in the bioinks is on the order of 500 μm for a 1-hour crosslinking duration (t). As 1 hour is a realistic upper-bound for cell-processing time, this characteristic diffusion length represents a theoretical upper limit for printed filament size using diffusive UNION bioinks. Printing filaments larger than this size may result in incomplete crosslinking at the center of the filament unless longer crosslinking times are employed, which may be detrimental to encapsulated cells. On the other hand, a lower feature size limit is reached when the UNION bioink material diffuses into the support bath before crosslinking can begin (~5 minutes, Figure S2). For a free, 40-kDa polymer in LifeSupport, this gives a theoretical filament resolution limit on the order of 50 μm, which is on the same length-scale as the smallest feature that can be practically extruded using microextrusion syringe bioprinting in LifeSupport.[15] Optimization of the support bath for these specific bioinks could further improve feature resolution, as has been demonstrated for other bioink/support bath systems.[41,42]
Key to the adaptability of a universal bioink system is its ability to be customized to match the desired bioprinting application. Several decades of biomaterials research have demonstrated that different cell types have different matrix requirements and that cell-matrix interactions influence cell phenotype.[43–45] To demonstrate the versatility of UNION bioinks, we selected two cell types with distinctly different matrix requirements: human corneal mesenchymal stromal cells (c-MSCs) and human induced-pluripotent stem cell-derived neural progenitor cells (hiPSC-NPCs). First, we confirmed that the presence of cells would not interfere with bioink crosslinking and found that inclusion of c-MSCs did not significantly alter the crosslinking kinetics or final modulus of gelatin-BCN UNION bioinks (Figure 5A). Next, we printed c-MSCs separately in a 6 wt% gelatin-BCN bioink, a 3 wt% HA-Azide bioink, and a 4 wt% ELP-Azide bioink into disks (8 mm × 0.5 mm) in a LifeSupport bath with either diazide-PEG crosslinker (for gelatin-BCN) or tetra-BCN-PEG (for HA-Azide and ELP-Azide) to demonstrate a clinically relevant, mesenchymal cell type in cell-adhesive matrices. Cells remained highly viable (>85%) after exposure to bioprinting and crosslinking in all of the cell-adhesive UNION bioinks: gelatin-BCN (Figure 5B), HA-Azide, and ELP-Azide (Figure S9). These data suggest that c-MSCs can tolerate the 1–4 hrs of crosslinking time that was used for these different inks (Table S1). Because c-MSCs are an adherent-dependent cell type, they were not tested in the PEG-BCN bioink, which lacks cell-adhesive domains that are required to maintain viability of mesenchymal-type cells.[46,47]
Figure 5. Human corneal mesenchymal stromal cells and neural progenitor cell spheroids are supported in gelatin-BCN and PEG-BCN UNION bioinks, respectively.
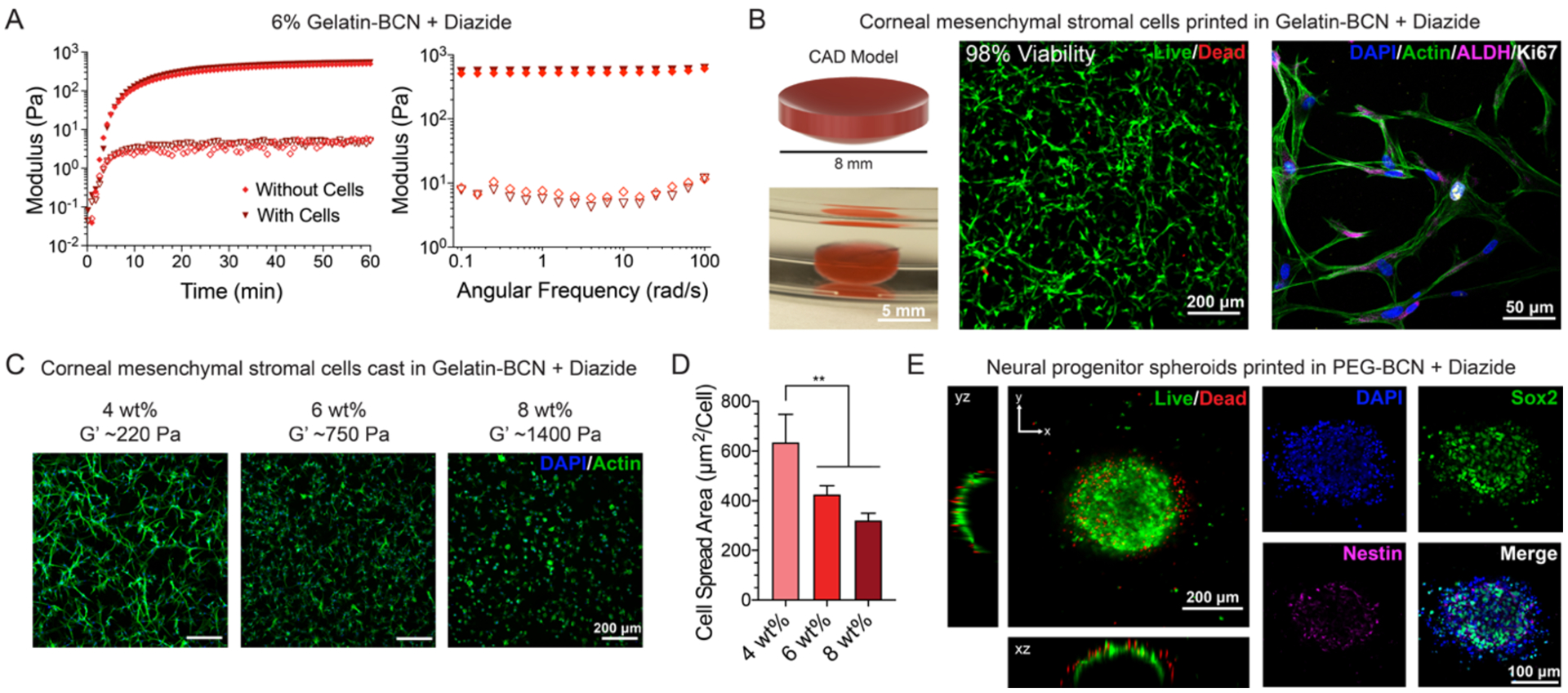
A. The presence of human corneal mesenchymal stromal cells (c-MSCs) does not affect UNION bioink gelation kinetics or final stiffness (filled markers are the storage moduli, G’, and open markers are the loss moduli, G”). B. (Left) Human c-MSCs are printed in gelatin-BCN in the shape of a corneal dome (top) and retain this shape after release from the LifeSupport bath (bottom). (Middle) c-MSCs in gelatin-BCN UNION bioink retain high viability (98%) 24 hours after printing as tested by a Live/Dead cytotoxicity assay, n = 3. (Right) Cytoskeletal staining reveals well spread c-MSCs that stain positive for corneal stromal cell marker aldehyde dehydrogenase 3A1 (ALDH3A1) and proliferation marker Ki-67 after 7 days in printed gelatin-BCN UNION bioinks. C. Representative images of c-MSCs in cast gelatin-BCN hydrogels with varying weight percent, crosslinked with diazide-PEG, after 3 days. D. Cell spread area for c-MSCs in gelatin-BCN hydrogels with varying weight percent, quantified from phalloidin staining. **p<0.01. Error bars are ± standard deviation, n ≥ 3. E. (Left) Human induced pluripotent stem cell-derived neural progenitor cell (hiPSC-NPC) spheroids printed in PEG-BCN bioinks maintain a highly viable core after 24 hours. (Right) NPC spheroids maintain a stem-cell phenotype as demonstrated by positive staining for the neural stem cell markers nestin and Sox2 after 3 days in culture in printed PEG-BCN UNION bioinks.
As c-MSCs showed the highest cell viability in gelatin-BCN, this material was used for subsequent c-MSC printing demonstrations. Additionally, gelatin is the partially hydrolyzed form of collagen, the primary extracellular component of the human cornea, and contains peptide sequences known to promote cell adhesion and spreading; thus, gelatin-BCN may be well-suited for applications in corneal tissue engineering.[48,49] The native human cornea has a convex lens shape and is typically about 500 microns in thickness.[50] To create a bioprinted mimic of the cornea, c-MSCs were printed in a 6 wt% gelatin-BCN bioink in the shape of a 500 micron-thick corneal dome in a LifeSupport bath with diazide-PEG crosslinker (Figure 5B). After 7 days in culture, the c-MSCs were well-spread within the UNION gelatin bioink and stained positive for aldehyde dehydrogenase 3A1 (ALDH3A1), a corneal crystallin and a common marker of corneal stromal cells,[51] and the proliferation marker Ki67 (Figure 5B), suggesting that c-MSCs can proliferate in UNION bioinks. Cell proliferation rates assessed by positive Ki67 staining in printed samples were observed to be similar to those in cast gelatin-BCN and collagen control cultures (Figure S10). The cMSCs printed within the gelatin-BCN bioink also maintained a morphology similar to that in 3D collagen control cultures at day 7 (Figure S10). Finally, to demonstrate how mechanical tunability of our UNION bioinks might influence cell phenotype, c-MSCs were grown in 4, 6, and 8 wt% gelatin-BCN scaffolds for 3 days. c-MSC spreading was greatest (~600 μm2/cell) in the weakest gelatin-BCN bioink (4 wt%, G’ ~ 200 Pa) when compared to the stiffer bioinks (~400 μm2/cell in 6 wt%, G’ ~ 900 Pa; ~300 μm2/cell in 8 wt%, G’ ~ 1,000 Pa) (Figure 5C). These results are consistent with reports of greater cell spreading in more compliant 3D hydrogel environments.[52–54] Together, these data demonstrate that UNION bioinks can be formulated and tuned to support the viability, spreading, and proliferation of an adhesion-dependent cell type.
In a separate demonstration, spheroids (i.e. spherical clusters of cells) of hiPSC-NPCs were printed and cultured in a PEG-BCN bioink. NPCs are not matrix-adhesion-dependent and do not require matrix signaling to maintain viability, and hence are particularly well-suited to be grown within a PEG bioink lacking cell-adhesive domains. This cell type is being investigated in a number of different potential regenerative medicine therapies and also has promise in the fabrication of in vitro models of the blood-brain-barrier.[55,56] We previously demonstrated that this cell type can maintain its phenotype when cultured within a bioink with a shear storage modulus of ~1,000 Pa[16]; therefore, we selected a PEG-BCN formulation of 6 wt%, which had similar mechanical properties. The spheroids were mixed with PEG-BCN, printed into disks (8 mm × 0.5 mm) in LifeSupport containing diazide-PEG crosslinkers, and incubated for 1 hour prior to removal from the support bath.
One day after printing, the interior of the spheroids contained mostly living cells, as confirmed by Live/Dead staining (Figure 5E). In contrast, the periphery of the spheroid had a thin layer that contained isolated dead cells. This is similar to observations of decreased cell viability in printed spheroids of breast cancer cells,[57] and is likely due to the increased size of a spheroid (~400 microns) compared to the size of a single cell (~10 microns), leading to increased fluid stresses as the bioink passes through the nozzle (d = 838 microns). Here, spheroids were printed using an 18-gauge needle to better accommodate the large size of the cell clusters. As UNION bioinks are liquids in the syringe, some cell sedimentation was observed prior to printing, especially with the larger neural spheroids. In the future, viscosity modifiers could be added to the ink to reduce cell sedimentation, as is commonly done with other liquid bioinks.[58–60] In our previous work with this cell type, a culture timepoint of 3 days was validated as appropriate to evaluate potential biomaterial-induced changes in stemness maintenance.[16,61,62] Thus, we selected this same culture timepoint for analysis of NPCs in UNION bioinks. After 3 days of culture within the PEG bioinks, the hiPSC-NPC spheroids retained their stem-like phenotype, as demonstrated by positive staining for the neural stem cell markers Sox2 and nestin (Figure 4B).[63,64] As expected, the spheroids remained spherical in shape within the non-biodegradable, non-cell-adhesive PEG bioink. These data demonstrate that UNION bioinks can be formulated to support the viability and phenotype maintenance of non-matrix-adhesion-dependent cell types.
3. Conclusion
In summary, the UNION bioink strategy offers a versatile method to create bioinks from a wide range of polymers. This strategy is also compatible with a choice of support bath materials that allow for crosslinker diffusion. Thus, this universal strategy could be useful for a wide variety of potential biological and multi-material applications, such as in vitro tumor models, human organoid models of tissue development, and multi-cell-type constructs for regenerative medicine. UNION bioinks can produce cohesive bioprinted structures from distinct bioinks by introducing a common crosslinking chemistry, removing the need for separate curing strategies for each ink. The bioorthogonal nature of the crosslinking chemistry also prevents off-target cross-reactivity with biomolecules present on the cell surface or in the cell culture medium, expanding the possibility of different tissue types that can be fabricated. We demonstrate that UNION bioinks can be formulated to achieve a range of mechanical and biochemical properties, enabling the bespoke customization of each bioink to achieve successful printing of diverse cells, including clinically relevant cell types that are both matrix-adhesion-dependent and adhesion-independent. Altogether, these findings demonstrate the UNION bioink strategy as a universal bioink platform. We envision that this strategy will enable multi-material and multi-cellular bioprinting of complex mimics of in vivo architectures, and may enhance the translational and therapeutic potential of 3D bioprinting technology.
4. Methods and Materials
Bioink and Crosslinker Synthesis and Characterization:
The four materials used as UNION bioinks (gelatin-BCN, PEG-BCN, HA-Azide, and ELP-Azide) and the tetra-BCN-PEG crosslinker were all synthesized using carbodiimide chemistry. Briefly, to synthesize gelatin-BCN and PEG-BCN, either gelatin (Type A, 200 Bloom, MP Biomedicals) or PEG-amine (4-arm 20 kDa for crosslinker, 8-arm 40 kDa for bioink, Creative PEGworks) was dissolved at 10 mg mL−1 in anhydrous dimethyl sulfoxide (DMSO, Fisher). (1R, 8S, 9S)-bicyclo[6.1.0]-non-4-yn-9ylmethyl N-succinimidyl carbonate (BCN-NHS, Sigma) was then added dropwise to the polymer solution to achieve a concentration of 1 molar equivalent (NHS relative to amine groups) for PEG-amine or 0.5 molar equivalent for gelatin. Triethylamine (Fisher) was added as a basic catalyst at 1.5 molar equivalents relative to NHS. The reaction was purged with nitrogen and allowed to proceed overnight at room temperature with constant stirring. Successful conjugation and estimated percent modification was determined by fluorescence measurements. Briefly, gelatin-BCN and PEG-BCN were reacted with fluorescein (FAM)-Azide overnight and then dialyzed to remove any unbound dye. The concentration of conjugated FAM-Azide was estimated by comparing against a standard curve relating fluorescence intensity and FAM-Azide concentration. Then the degree of modification was determined based on the theoretical maximum amount of dye that could have been conjugated given complete conversion of primary amines to BCN.
To prepare ELP-Azide, first the recombinant ELP (37 kDa, includes 13 primary amines and a cell-adhesive Arg-Gly-Asp peptide sequence) was expressed in Escherichia coli and purified as described previously.[65] ELP was dissolved at 10 mg mL−1 in anhydrous DMSO (Fisher) to which was added 1 molar equivalent of azido-PEG4-succinimydyl ester (Azido-PEG4-NHS, BroadPharm), along with 1.5 molar equivalents of triethylamine (Fisher). The reaction was purged with nitrogen and allowed to proceed overnight at room temperature with constant stirring. To produce HA-Azide, a tetrabutylammonium (TBA) salt of HA (40 kDa, Life-Core) was prepared from its sodium salt form by counterion exchange to increase solubility. To synthesize HA-Azide, the HA-TBA salt was dissolved at 10 mg mL−1 in anhydrous DMSO. Then 1.5 molar equivalents of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, Thermo Fisher), 1.5 molar equivalents of N-hydroxysuccinimide (NHS, Thermo Fisher), 1.5 molar equivalents of 3-azido-propyl-amine (Click Chemistry Tools), and 3 molar equivalents of the basic catalyst 4-methylmorpholine (Sigma) were added to the HA solution. The reaction was allowed to proceed overnight at room temperature with constant stirring. The degree of azide substitution for ELP-Azide and HA-Azide was determined using 1H-NMR.
For all BCN- and azide-functionalized polymers, once the reaction was complete, the functionalized polymers were dialyzed against double deionized water, sterile filtered through a 0.22-μm filter, and lyophilized to produce white powders. PEG-BCN polymers were flash frozen in liquid nitrogen prior to lyophilization to prevent cryogelation during freezing. Polymers were stored at −20 °C (gelatin-BCN, ELP-Azide, HA-Azide) or −80 °C (PEG-BCN). Azide-PEG3-azide (diazide-PEG, MW 200 Da, Lumiprobe) was purchased and used as received.
Bioink and Support Bath Preparation:
Lyophilized gelatin-BCN, PEG-BCN, HA-Azide, and ELP-Azide were dissolved to the appropriate concentration (typically 60 mg mL−1, 60 mg mL−1, 30 mg mL−1, and 30 mg mL−1, respectively, see Table S1) in phosphate buffered saline (PBS) and added to a 2.5 mL Hamilton syringe for printing. Either food coloring, Alcian Blue (ThermoFisher Scientific), or Coomassie Blue (ThermoFisher Scientific) were used for acellular printing to aid in visualization of printed structures. Pluronic support baths were prepared by dissolving 7.8 g Pluronic F-127 (Sigma) in 30 mL of sterile, cold PBS (26% w/v) and stirring overnight at 4 °C. UNION crosslinkers were added to the Pluronic to a final concentration of either 1 mg mL−1 (diazide-PEG) or 5 mg mL−1 (tetra-BCN-PEG); the solution was thoroughly mixed and then centrifuged to remove any bubbles. LifeSupport (FluidForm Inc.) is the commercialized support bath produced using the FRESH 2.0 process as previously described,[15] and baths were prepared following the manufacturer’s recommendations. Briefly, lyophilized LifeSupport was hydrated using sterile, cold PBS or media containing UNION crosslinker (1 mg mL−1 diazide-PEG or 5 mg mL−1 tetra-BCN-PEG). The hydrated slurry was centrifuged, and the supernatant was removed. Support baths were added to custom-made polycarbonate containers, centrifuged to remove any bubbles, and kept on ice prior to use. Printing was performed with a MakerGear M2 modified into a 3D dual-extruder bioprinter[15] using 27-gauge needles (unless indicated otherwise) at a print speed of 23 mm s−1, extrusion width of 0.21 mm, and layer height of 0.084 mm. 3D printed models were sliced using either Repetier Host (Hot-World GmbH & Co. KG) or Simplify3D. Printed disks were 8 mm × 0.5 mm (Figures 4A, 5E), the printed elliptical window (Figure 3A) was an open-source calibration standard for 3D bioprinting (NIH 3D Print Exchange, Model ID 3DPX-011749),[66] the dual material checkerboard (Figure 3B) was a 2×2×2 cube of alternating 3-mm cubes of gelatin-BCN and PEG-BCN, and the dogbones were 16 mm × 3.2 mm ×. 0.8 mm. All printed structures were cured for at least 1 hour (37 °C for Pluronic and room temperature or 4 °C for LifeSupport baths, see Table S1) before the support baths were melted for 15 minutes (4°C for Pluronic and 37 °C for LifeSupport baths). Printed structures were removed from melted support baths and rinsed with PBS for further analysis or culture.
Multi-material Bioprinter Modification and Printing:
A MakerGear M2 Rev E plastic 3D printer was modified into a 3D bioprinter. The entire plastic extrusion apparatus was removed and replaced with a mount capable of holding two custom designed Replistruder 4 syringe pumps. Additionally, the control board of the printer was replaced with a Duet 2 WiFi board with a PanelDue 5i touch screen controller (Duet3D, UK). The Duet WiFi board utilizes a preinstalled RepRapFirmware and is compatible with all open-source 3D printing software. This board, in conjunction with the Replistruder 4 syringe pumps, allows for automated two material printing. Prior to printing, two Hamilton gastight syringes are filled with their respective bioinks. These syringes are loaded into the Replistruder 4 syringe pumps and the appropriate dispense tips are attached (typically 27-gauge needles). The physical separation of the two dispense tips are measured using a precision square reference block, and the offsets are recorded using the tool offset G-Code commands (G10) built into the Duet WiFi’s RepRapFirmware. When the material is switched during multi-material printing, a G-Code script is automatically executed that removes the active dispense tip from the support bath, switches the active tool to the next material, moves that tool to its origin (which is the measured offset to the other tool), and then resumes printing with the new material. To prevent the inactive dispense tip from drying out, the nozzle is submerged in DI water when not actively printing.
Bioink Mechanical Characterization:
Mechanical testing of UNION bioinks was performed using an ARG2 stress-controlled rheometer (TA Instruments). Bioinks were mixed to the final polymer concentration with stochiometric quantities of the appropriate UNION crosslinker (5:1 Azide:BCN for gelatin-BCN + Diazide, 10:1 Azide:BCN for PEG-BCN + Diazide, 3:1 Azide:BCN for ELP-Azide + Tetra-BCN, and 1:1 Azide:BCN for ELP-Azide + Tetra-BCN) and added to the rheometer stage using a pipette (45 μL of bioink solution). Rheological measurements were performed using a 20-mm cone and plate geometry. Gelation time sweeps were performed at a frequency of 1 rad s−1 with a strain of 1%. Frequency sweeps were performed between 0.1 and 100 rad s−1 at a strain of 1%. All measurements were confirmed to be within the linear viscoelastic regime of the bioinks. Compression tests were performed with an 8-mm parallel plate geometry at a constant linear rate of 10 μm s−1, and the elastic modulus was fitted to the linear portion of the stress/strain curve. Viscosity tests of the support bath material were performed with a 40-mm parallel plate geometry at a shear rate ranging from 0 to 100 s−1 over the course of 120 s.
Diffusion Characterization:
For diffusion testing, diazide-PEG was modified with fluorescein-dibenzocyclooctyne (FITC-DBCO, BroadPharm). Diazide-PEG was dissolved in anhydrous DMSO to a concentration of 3 mg mL−1, and 1 molar equivalent of FITC-DBCO was added. The reaction was allowed to proceed overnight to produce diazide-PEG-FITC. Diffusional testing for the Pluronic support baths was performed using custom-made dialysis chambers over the course of 24 hours. Briefly, 5 mm holes were punched into rubber septums that were affixed with a cyanoacrylate adhesive (Locktite 401) to one side of a 0.1–0.5 mL dialysis cassette (3.5K MWCO, Thermo Scientific) to produce a chamber (Figure S8). The other side of the cassette was sealed to prevent evaporation. Approximately 500 μL of cold 26% Pluronic support bath loaded with 10 μg mL−1 diazide-PEG-FITC was introduced into the dialysis cassette using a syringe. 5 mL of double deionized water was added to the chamber, and then 100 μL samples were collected from the chamber after 15 minutes and 1, 2, 4, and 8 hours. Total released diazide-PEG-FITC at each time point was determined by comparing fluorescence to a standard curve. A diffusion coefficient was estimated using a simple, short-time, one-dimensional diffusion model fit to the cumulative released diffusant over time.[67]
Diffusivity within the LifeSupport bath was assessed by fluorescence recovery after photobleaching (FRAP) experiments. Briefly, LifeSupport was hydrated with PBS containing FITC-labelled diffusant (either 1 μg mL−1 diazide-PEG-FITC, 10 μg mL−1 20 kDa FITC-dextran, or 10 μg mL−1 40 kDa FITC-dextran, Sigma). Approximately 150 μL of diffusant-loaded LifeSupport was placed in a clear bottom, 96-well plate and centrifuged to remove any bubbles. FRAP experiments were performed using a confocal microscope (Leica SPE) with 30 seconds of photobleaching (100 μm × 100 μm area, 488 nm laser, 100% intensity) and 90 seconds of capture time. Similarly, to quantify the diffusivity within the bioinks, BCN-functionalized UNION bioinks were loaded with 2 mg mL−1 of nonreactive, 10-kDa FITC-dextran (Sigma) and then crosslinked with diazide-PEG using bioink to crosslinker reactive group ratios of 1:0 (i.e. uncrosslinked) or 1:1 (i.e. fully crosslinked). Crosslinked bioinks were cured at room temperature for 1 hour to ensure full crosslinking prior to FRAP experiments. Diffusion coefficients for each condition were calculated using the open source MATLAB code ‘frap_analysis’ based on the Hankel transform method.[68]
Cell Culture and Analysis:
All UNION bioinks for cell studies were dissolved in the appropriate fresh cell culture medium to their final concentration, and cell-containing bioinks were printed as either 8 mm × 0.5 mm disks or 8 mm × 0.5 mm cornea-shaped domes into sterile LifeSupport baths hydrated with medium and either 1 mg mL−1 diazide-PEG (for gelatin-BCN and PEG-BCN inks) or 5 mg mL−1 tetra-BCN (for HA-Azide and ELP-Azide inks). Cell-laden bioinks were incubated for at least 1 hour at room temperature or 4 °C before melting and removal of the support bath (Table S1).
Human corneal mesenchymal stromal cells (c-MSCs) were isolated from donor corneas (Lions Eye Institute for Transplant and Research) according to established protocols[69,70] and expanded in growth medium (MEM-Alpha (Corning), 10% fetal bovine serum (Gibco), GlutaMax (Gibco), non-essential amino acids (Gibco), and Antibiotic-Antimycotic, which contains penicillin, streptomycin, and Amphotericin B (Gibco)). Prior to printing, c-MSCs were trypsinized, counted, pelleted, resuspended in 6% w/v gelatin-BCN at a cell density of 3×106 mL−1, and placed in a 2.5 mL Hamilton syringe fitted with a 27-gauge needle for printing. Following printing, c-MSC-laden constructs were maintained in growth medium, and the medium was changed daily. For cast gel experiments, c-MSCs were trypsinized, counted, pelleted, resuspended in either 4%, 6%, or 8% w/v gelatin-BCN at a cell density of 3×106 mL−1, and then mixed with a stoichiometric (5:1 Azide:BCN) amount of diazide-PEG crosslinker. The cell suspensions were mixed thoroughly, and then 10 μl of the mixture was pipetted into 4-mm diameter × 0.5-mm deep silicone molds. The gels were allowed to crosslink for 30 minutes, and then enough c-MSC growth medium was added to cover the gels. The medium was changed daily for the duration of the culture period (3 or 7 days). For collagen control cultures, c-MSCs were trypsinized, counted, pelleted, and resuspended at a cell density of 3×106 mL−1 in 3 mg mL−1 neutralized bovine Collagen I (Gibco). 10 μL of the resulting mixture was pipetted into circular 4-mm diameter × 0.5-mm deep silicone molds. The gels were allowed to crosslink for 30 minutes at 37 °C, and then c-MSC growth medium was added to cover the gels and was changed daily.
Human induced pluripotent stem cells (hiPSCs, line number 511.3) were graciously provided by Dr. Theo Palmer and Julien Roth. hiPSC-neural progenitor cell (hIPSC-NPCs) were differentiated and formed spheroids in accordance with previously described protocols.[71,72] To generate cell clusters of equal sizes (herein defined as approximately 5,000 cells per cluster) for differentiation, hiPSCs were dissociated into single cells with Accutase (Corning), centrifuged in AggreWell plates (Stemcell Technologies), and incubated within the AggreWell plate for 48 hr in Essential 8 (E8) medium (ThermoFisher Scientific) supplemented with the RHO/ROCK inhibitor Y-27632 (Stemcell Technologies). To initiate neural differentiation, hiPSC clusters were lifted from the Aggrewell plate and cultured within ultra-low-attachment plastic dishes (Corning) in Essential 6 (E6) medium (ThermoFisher Scientific) supplemented with two SMAD pathway inhibitors: 100 nM LDN-193189 (Cayman Chemical Company) and 10 μM SB-431542 (Tocris Bioscience). Neural spheroids were used at day 7 following treatment with the two SMAD pathway inhibitors. The spheroids were removed from their culture dishes and resuspended in 6% w/v PEG-BCN, and the bioink was then loaded into a 2.5 mL Hamilton syringe fitted with an 18-gauge needle and printed. Following printing, spheroid-laden constructs were maintained in neural medium consisting of Neurobasal-A (1x, Thermo Fisher Scientific), 1% N-2 Supplement (ThermoFisher Scientific), 2% B-27 Supplement minus vitamin A (ThermoFisher Scientific), GlutaMax (Gibco) and Non-Essential Amino Acids (MEM NEAA, Gibco).
Cell viability was assessed using Live/Dead staining (Life Technologies) following the manufacturer’s instructions. For immunofluorescence imaging, individual printed disk samples were fixed in 4% paraformaldehyde and permeabilized with a 0.25% Triton X-100 in PBS solution. Permeabilized samples were blocked with 5% bovine serum albumin (BSA, Roche) and 5% goat serum (Gibco) and then rinsed thoroughly. Each sample was treated with the appropriate primary antibody overnight at 4 °C: for c-MSCs, Ki-67 (mouse, Cell Signaling, 9449, 1:400 dilution) and aldehyde dehydrogenase 3a1 (rabbit, Abcam, ab76976, 1:200); for neural spheroids, nestin (mouse, BD Pharmingen, 556309, 1:400) and Sox2 (rabbit, Millipore, AB5603, 1:400). Samples were then washed and stained with the appropriate secondary antibodies overnight at 4 °C: AF488 goat anti-rabbit (Invitrogen, A11034, 1:500) and AF647 goat anti-mouse (Invitrogen, A21242, 1:500). Samples were washed again and then incubated with DAPI (Molecular Probes, 1:1000) and/or TRITC-phalloidin (Sigma Aldrich, 1:100). Samples were mounted with antifade reagent (Cell Signaling Technologies) on coverslips and imaged using a Leica SPE confocal microscope.
Statistical Analysis:
Two-tailed Student’s t-tests were used when comparing two experimental groups, and one-way analysis of variance (ANOVA) with Tukey post-hoc testing was used to compare more than two experimental groups.
Supplementary Material
Acknowledgements
We thank T. Palmer for providing hiPCSs, O. Kuzmenko and J. Goldberg for providing donor human corneal stromal tissue, G. Fernandes-Cunha for help with c-MSC culture, P. Cai for help with bioink synthesis, C. Madl for discussions and expertise on bioorthogonal chemistries, and the Feinberg group at Carnegie Mellon University for hosting the 2019 3D Bioprinting Open-Source Workshop. S.C.H. acknowledges support by the National Science Foundation (DMR-1508006 and DMR-1808415) and the National Institutes of Health (U19-AI116484, R01-EB027171, and R01-HL142718). S.M.H. acknowledges support from a NIH NRSA pre-doctoral fellowship (F31-EY030731) and a Stanford Bio-X Interdisciplinary Graduate Fellowship. C.D.L. acknowledges support from a Kodak Fellowship. L.G. B. acknowledges support from an NSF Graduate Research Fellowship. D.M. acknowledges support from the National Eye Institute (K08-EY028176 and P30-EY026877), Research to Prevent Blindness career development and department core grant, the Matilda Ziegler Foundation, and the Department of Veterans Affairs (I21 RX003179). A.W.F. acknowledges support from the Food and Drug Administration (R01-FD006582) and the National Science Foundation (CMMI 1454248). D.J.S. acknowledges support from a NIH NRSA postdoctoral fellowship (F32-HL142229). J.G.R. acknowledges support from an NSF Graduate Research Fellowship and a Stanford Graduate Fellowship.
Footnotes
Disclosures
The authors declare the following competing interests: A.W.F. is CTO and co-founder of FluidForm Inc., manufacturer of LifeSupport.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author and includes the following data: degree of chemical modification of polymers for UNION inks, kinetics of UNION bioink crosslinking, uniaxial unconfined compression of printed discs, viscosity of LifeSupport with and without UNION crosslinkers, printed logpiles in LifeSupport before release from the support bath, CAD models of the printed structures, characterization of printing resolution, characterization of UNION crosslinker diffusion coefficients, human corneal mesenchymal stromal cell (c-MSCs) viability in UNION bioinks, proliferation and morphology of c-MSCs in UNION bioinks and control materials, table of ink formulations and crosslinking conditions for all printed structures, and video of dual UNION bioink FRESH printing in a LifeSupport bath.
Contributor Information
Sarah M. Hull, Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA.
Dr. Christopher D. Lindsay, Department of Materials Science & Engineering, Stanford University, Stanford, CA 94305, USA.
Lucia G. Brunel, Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA
Julien G. Roth, Institute for Stem Cell Biology and Regenerative Medicine, Stanford University, Stanford, CA 94305, USA
Dr. David Myung, Department of Ophthalmology, Stanford University, Stanford, CA 94305, USA Division of Ophthalmology, VA Palo Alto Health Care System, Stanford, CA 94305, USA.
Dr. Adam W. Feinberg, Department of Biomedical Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA Department of Materials Science & Engineering, Carnegie Mellon University, Pittsburgh, PA 15213, USA.
Dr. Sarah C. Heilshorn, Department of Materials Science & Engineering, Stanford University, Stanford, CA 94305, USA
References
- [1].Murphy SV, Atala A, Nat. Biotechnol 2014, 32, 773. [DOI] [PubMed] [Google Scholar]
- [2].Ozbolat IT, Hospodiuk M, Biomaterials 2016, 76, 321. [DOI] [PubMed] [Google Scholar]
- [3].Liu W, Zhang YS, Heinrich MA, De Ferrari F, Jang HL, Bakht SM, Alvarez MM, Yang J, Li YC, Trujillo-de Santiago G, Miri AK, Zhu K, Khoshakhlagh P, Prakash G, Cheng H, Guan X, Zhong Z, Ju J, Zhu GH, Jin X, Shin SR, Dokmeci MR, Khademhosseini A, Adv. Mater 2017, 29, 1604630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kang HW, Lee SJ, Ko IK, Kengla C, Yoo JJ, Atala A, Nat. Biotechnol 2016, 34, 312. [DOI] [PubMed] [Google Scholar]
- [5].Ma X, Qu X, Zhu W, Li YS, Yuan S, Zhang H, Liu J, Wang P, Lai CSE, Zanella F, Feng GS, Sheikh F, Chien S, Chen S, Proc. Natl. Acad. Sci. U. S. A 2016, 113, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Groll J, Burdick JA, Cho D-W, Derby B, Gelinsky M, Heilshorn SC, Jüngst T, Malda J, Mironov VA, Nakayama K, Ovsianikov A, Sun W, Takeuchi S, Yoo JJ, Woodfield TBF, Biofabrication 2018, 11, 13001. [DOI] [PubMed] [Google Scholar]
- [7].Chen KG, Mallon BS, McKay RDG, Robey PG, Cell Stem Cell 2014, 14, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gu Q, Tomaskovic-Crook E, Wallace GG, Crook JM, Adv. Healthc. Mater 2017, 6, 1700175. [DOI] [PubMed] [Google Scholar]
- [9].Skardal A, Bioprinting 2018, 10, e00026. [Google Scholar]
- [10].Hinton TJ, Jallerat Q, Palchesko RN, Park JH, Grodzicki MS, Shue HJ, Ramadan MH, Hudson AR, Feinberg AW, Sci. Adv 2015, 1, e1500758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bhattacharjee T, Zehnder SM, Rowe KG, Jain S, Nixon RM, Sawyer WG, Angelini TE, Sci. Adv 2015, 1, e1500655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Highley CB, Rodell CB, Burdick JA, Adv. Mater 2015, 27, 5075. [DOI] [PubMed] [Google Scholar]
- [13].Compaan AM, Song K, Huang Y, ACS Appl. Mater. Interfaces 2019, DOI 10.1021/acsami.8b13792. [DOI] [PubMed] [Google Scholar]
- [14].Noor N, Shapira A, Edri R, Gal I, Wertheim L, Dvir T, Adv. Sci 2019, 6, 1900344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee A, Hudson AR, Shiwarski DJ, Tashman JW, Hinton TJ, Yerneni S, Bliley JM, Campbell PG, Feinberg AW, Science (80-.) 2019, 365, 482. [DOI] [PubMed] [Google Scholar]
- [16].Lindsay CD, Roth JG, LeSavage BL, Heilshorn SC, Acta Biomater. 2019, 95, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ouyang L, Highley CB, Sun W, Burdick JA, Adv. Mater 2017, 29, DOI 10.1002/adma.201604983. [DOI] [PubMed] [Google Scholar]
- [18].Isaacson A, Swioklo S, Connon CJ, Exp. Eye Res 2018, 173, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dubbin K, Hori Y, Lewis KK, Heilshorn SC, Adv. Healthc. Mater 2016, 5, 2488. [DOI] [PubMed] [Google Scholar]
- [20].Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA, Lewis JA, Adv. Mater 2014, 26, 3124. [DOI] [PubMed] [Google Scholar]
- [21].Baskin JM, Bertozzi CR, QSAR Comb. Sci 2007, 26, 1211. [Google Scholar]
- [22].Ossipov DA, Hilborn J, Macromolecules 2006, 39, 1709. [Google Scholar]
- [23].Malkoch M, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsbury K, Hawker CJ, Chem. Commun 2006, 2774. [DOI] [PubMed] [Google Scholar]
- [24].Madl CM, Katz LM, Heilshorn SC, Adv. Funct. Mater 2016, 26, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang H, Dicker KT, Xu X, Jia X, Fox JM, ACS Macro Lett. 2014, 3, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dicker KT, Song J, Moore AC, Zhang H, Li Y, Burris DL, Jia X, Fox JM, Chem. Sci 2018, 9, 5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Azagarsamy MA, Anseth KS, ACS Macro Lett. 2013, 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Madl CM, Heilshorn SC, Adv. Funct. Mater 2018, 28, 1706046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gopinathan J, Noh I, Tissue Eng. Regen. Med 2018, 15, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Deforest CA, Polizzotti BD, Anseth KS, Nat. Mater 2009, 8, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hermann CD, Wilson DS, Lawrence KA, Ning X, Olivares-Navarrete R, Williams JK, Guldberg RE, Murthy N, Schwartz Z, Boyan BD, Biomaterials 2014, 35, 9698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee HJ, Fernandes-Cunha GM, Na KS, Hull SM, Myung D, Adv. Healthc. Mater 2018, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Baker BM, Chen CS, J.Cell Sci 2012, 125, 3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Boontheekul T, Hill EE, Kong HJ, Mooney DJ, Tissue Eng 2007, 13, 1431. [DOI] [PubMed] [Google Scholar]
- [35].Chaudhuri O, Koshy ST, Branco Da Cunha C, Shin JW, Verbeke CS, Allison KH, Mooney DJ, Nat. Mater 2014, 13, 970. [DOI] [PubMed] [Google Scholar]
- [36].Guimarães CF, Gasperini L, Marques AP, Reis RL, Nat. Rev. Mater 2020, DOI 10.1038/s41578-019-0169-1. [DOI] [Google Scholar]
- [37].Miri AK, Mirzaee I, Hassan S, Mesbah Oskui S, Nieto D, Khademhosseini A, Zhang YS, Lab Chip 2019, 19, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee JM, Ng WL, Yeong WY, Appl. Phys. Rev 2019, 6, 11307. [Google Scholar]
- [39].Lustig SR, Peppas NA, J. Appl. Polym. Sci 1988, 36, 735. [Google Scholar]
- [40].Amsden B, Macromolecules 1998, 31, 8382. [Google Scholar]
- [41].Shin S, Hyun J, ACS Appl. Mater. Interfaces 2017, 9, 26438. [DOI] [PubMed] [Google Scholar]
- [42].O’Bryan CS, Bhattacharjee T, Hart S, Kabb CP, Schulze KD, Chilakala I, Sumerlin BS, Sawyer WG, Angelini TE, Sci. Adv 2017, 3, e1602800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- [44].Saha K, Keung AJ, Irwin EF, Li Y, Little L, Schaffer DV, Healy KE, Biophys. J 2008, 95, 4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee HP, Lippens E, Duda GN, Mooney DJ, Nat. Mater 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hern DL, Hubbell JA, J. Biomed. Mater. Res 1998, 39, 266. [DOI] [PubMed] [Google Scholar]
- [47].Wu J, Rnjak-Kovacina J, Du Y, Funderburgh ML, Kaplan DL, Funderburgh JL, Biomaterials 2014, 35, 3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ma Z, He W, Yong T, Ramakrishna S, Tissue Eng. 2005, 11, 1149. [DOI] [PubMed] [Google Scholar]
- [49].Huang Y, Onyeri S, Siewe M, Moshfeghian A, Madihally SV, Biomaterials 2005, 26, 7616. [DOI] [PubMed] [Google Scholar]
- [50].Doughty MJ, Zaman ML, Surv. Ophthalmol 2000, 44, 367. [DOI] [PubMed] [Google Scholar]
- [51].Pei Y, Reins RY, McDermott AM, Exp. Eye Res 2006, 83, 1063. [DOI] [PubMed] [Google Scholar]
- [52].Stowers RS, Allen SC, Suggs LJ, Proc. Natl. Acad. Sci 2015, 112, 1953 LP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dikovsky D, Bianco-Peled H, Seliktar D, Biophys. J 2008, 94, 2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cavo M, Fato M, Peñuela L, Beltrame F, Raiteri R, Scaglione S, Sci. Rep 2016, 6, 35367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Goldman S, Nat. Biotechnol 2005, 23, 862. [DOI] [PubMed] [Google Scholar]
- [56].Lippmann ES, Al-Ahmad A, Palecek SP, V Shusta E, Fluids Barriers CNS 2013, 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Duarte Campos DF, Lindsay CD, Roth JG, Lesavage BL, Seymour AJ, Krajina BA, Ribeiro R, Costa P, Blaeser A, Heilshorn SC, Front. Bioeng. Biotechnol 2020, 8, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Dubbin K, Tabet A, Heilshorn SC, Biofabrication 2017, 9, 044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Na K, Shin S, Lee H, Shin D, Baek J, Kwak H, Park M, Shin J, Hyun J, J. Ind. Eng. Chem 2018, 61, 340. [Google Scholar]
- [60].Chimene D, Lennox KK, Kaunas RR, Gaharwar AK, Ann. Biomed. Eng 2016, 44, 2090. [DOI] [PubMed] [Google Scholar]
- [61].Madl CM, Lesavage BL, Dewi RE, Dinh CB, Stowers RS, Khariton M, Lampe KJ, Nguyen D, Chaudhuri O, Enejder A, Heilshorn SC, Nat. Mater 2017, 16, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Madl CM, LeSavage BL, Dewi RE, Lampe KJ, Heilshorn SC, Adv. Sci 2019, 6, 1801716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Mckay R, Science (80-.) 1997, 276, 66. [DOI] [PubMed] [Google Scholar]
- [64].Ahmed S, J. Cell. Biochem 2009, 106, 1. [DOI] [PubMed] [Google Scholar]
- [65].Straley KS, Heilshorn SC, Soft Matter 2009, 5, 114. [Google Scholar]
- [66].Pusch K, Hinton TJ, Feinberg AW, HardwareX 2018, 3, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wilkinson DS, in Mass Transp. Solids Fluids, Cambridge University Press, Cambridge, 2000, pp. 64–68. [Google Scholar]
- [68].Jönsson P, Jonsson MP, Tegenfeldt JO, Höök F, Biophys. J 2008, 95, 5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Du Y, Funderburgh ML, Mann MM, SundarRaj N, Funderburgh JL, Stem Cells 2005, 23, 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Eslani M, Putra I, Shen X, Hamouie J, Tadepalli A, Anwar KN, Kink JA, Ghassemi S, Agnihotri G, Reshetylo S, Mashaghi A, Dana R, Hematti P, Djalilian AR, Stem Cells 2018, 36, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Yoon SJ, Elahi LS, Pașca AM, Marton RM, Gordon A, Revah O, Miura Y, Walczak EM, Holdgate GM, Fan HC, Huguenard JR, Geschwind DH, Pașca SP, Nat. Methods 2019, 16, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Pasca S, Yoon S-J, Protoc. Exch 2018, 1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.