

Abstract
The controlled release of drugs using nanoparticle-based delivery vehicles is a promising strategy to improve the safety and efficacy of chemotherapy. We have developed a simple, scalable, and reproducible strategy to synthesize a drug delivery system by loading the prodrug 6-maleimidocaproyl-hydrazone doxorubicin (DOX-EMCH) into the empty core of virus-like particles (VLPs) derived from Physalis mottle virus (PhMV) via a combination of chemical conjugation to cysteine residues and π–π stacking interactions with the anchored doxorubicin molecule. The DOX-EMCH prodrug features an acid-sensitive hydrazine linker that triggers the release of doxorubicin in the slightly acidic extracellular tumor microenvironment or acidic endosomal or lysosomal compartments following cellular uptake. The VLP external surface was coated with polyethylene glycol (PEG) to prevent non-specific uptake and improve biocompatibility. The DOX-PhMV-PEG particles were stable in vitro and showed significantly greater efficacy in vivo compared to free doxorubicin in a breast tumor mouse model (using MDA-MB-231 cells and nude mice): 92% of the tumor-bearing mice treated with DOX-PhMV-PEG were completely cured compared to 27% of those treated with free doxorubicin under the same conditions, representing a 3.4-fold improvement. These results lay a foundation for the further development of our biological drug delivery system for a new generation of chemotherapy products.
Keywords: nanomedicine, virus-like particle, pH sensitivity, doxorubicin, tumor microenvironment
Graphical Abstract
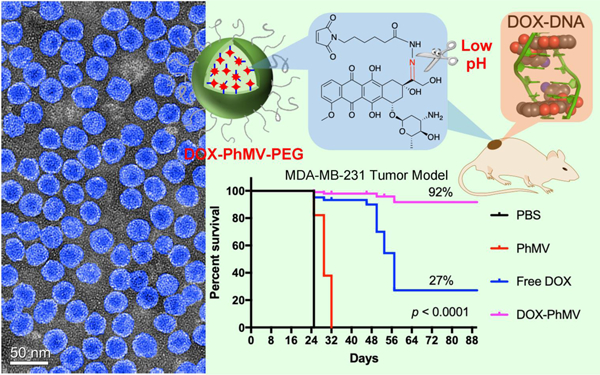
The authors developed a pH-responsive drug delivery system (DDS) based on virus-like particles derived from Physalis mottle virus (PhMV) loaded with 6-maleimidocaproyl-hydrazone doxorubicin (DOX-PhMV-PEG). The DDS demonstrated significantly greater efficacy in vivo compared to free doxorubicin in a breast tumor mouse model: 92% of the tumor-bearing mice treated with DOX-PhMV-PEG were completely cured compared to 27% of those treated with free doxorubicin under the same conditions, representing a 3.4-fold improvement.
1. Introduction
Chemotherapy plays a vital role in the fight against cancer but conventional chemotherapy is far from satisfactory due to its limited efficacy and often severe off-target effects.[1] For example, more than 40% of drugs in development are only sparingly soluble in water.[2] The direct administration of such hydrophobic drugs promotes aggregation that can trigger embolism in capillaries.[3] Systemic administration also results in an undesirable biodistribution, off-target effects against healthy cells and limited accumulation in the tumor, encouraging the emergence of drug-resistant cancer cells.[4] To overcome these limitations, various controlled release strategies have been developed, many of which involve a drug delivery system (DDS) based on nanotechnology.[4] Successful DDS approaches prevent the release of drugs into the circulation and achieve delivery to tumors, followed by drug release induced by the unique properties of the tumor microenvironment or by external stimuli (e.g. light, ultrasound, heat or a magnetic field).[4, 5a, b] Such delivery systems may allow the re-evaluation of drugs that were formerly considered too toxic for systemic administration.
The realization of DDS requires nanocarriers to sequester the drug molecules and release them on demand.[3] Nanocarriers must satisfy multiple criteria to be effective delivery vehicles: (1) they must be biocompatible to avoid toxicity and prolong their half-life in circulation; (2) they must possess functional groups that enable them to carry large drug payloads; (3) there must be a mechanism that allows targeting to disease sites, such as tumors; and (4) they must be sensitive to stimuli that allow controlled drug release.[3, 4, 5a] Various nanocarriers have been developed to address these requirements, including polymeric, magnetic and silica-based nanoparticles, liposomes, carbon nanotubes, and engineered nanoparticles based on viruses.[4, 5b] Proof of concept has been demonstrated in vitro and in vivo. However, few of these vehicles have reached clinical development and only a handful been approved by the United States Food and Drug Administration (FDA).[5b] Despite these advances, current DDS are still inefficient due to their rapid clearance from circulation, low targeting specificity, and inability to achieve controlled release.[4] DDS strategies must therefore be optimized to address these challenges.
Plant viruses have recently emerged as promising biomimetic nanomaterials for biomedical applications.[6a–c] Among their many advantages, plant viruses are inherently stable, they come in a variety of shapes and sizes, they are biocompatible and biodegradable, they are noninfectious in humans, and the presence of particular amino acid side chains at defined sites ensures they are easy to modify with targeting ligands, cloaking molecules and drug payloads.[6a–c] Furthermore, the ability of plant virus capsids to assemble in the absence of nucleic acid allows the production of empty virus-like particles (VLPs) that can be loaded with drugs.[6a] We have previously generated DDS based on Tobacco mosaic virus (TMV),[7a, b] Cowpea chlorotic mottle virus (CCMV)[8a, b] and Cowpea mosaic virus (CPMV)[9] for the delivery of drugs including chemotherapy, immunotherapy, nucleic acid therapy, as well as protein drugs. Most recently we have investigated the suitability of VLPs based on Physalis mottle virus (PhMV).[10]
PhMV (Tymovirus, Tymoviridae) forms a ~30-nm icosahedral capsid comprised of 180 identical coat protein subunits (21 kDa) with lysine residues exposed on the external surface and cysteine residues on the internal surface, providing handles that allow the chemical modification of both surfaces independently with spatial control.[10] We previously reported that PhMV-based VLPs are highly biocompatible, with long circulation times in vivo and a preference to accumulate in tumors.[11a, b] PhMV-based VLPs could therefore provide a highly suitable platform for the development of new DDS formulations.
Here we describe a simple, scalable, and reproducible DDS produced by loading PhMV-based VLPs with the prodrug 6-maleimidocaproyl-hydrazone doxorubicin (DOX-EMCH) via specific thiol-maleimide conjugation to cysteine residues and π–π stacking interaction with the anchored doxorubicin moieties. The external surface of the DOX-PhMV VLPs was coated with polyethylene glycol (PEG) to avoid nonspecific uptake and improve biocompatibility. Doxorubicin is one of the most widely used antineoplastic drugs,[12] but its high toxicity limits dosing.[13] A DDS could eliminate these adverse effects by ensuring that doxorubicin is delivered to the tumor, while avoiding or decreasing its accumulation in healthy tissue. To evaluate the PhMV DDS, we investigated the physicochemical characteristics of the VLPs, the doxorubicin release profiles, in vitro cell uptake and cytotoxicity, and finally their efficacy in vivo in a preclinical MDA-MB-231 xenograft breast tumor model.
2. Materials and methods
2.1. Preparation of VLPs:
The VLPs were prepared by expressing PhMV coat protein in E. coli as previously described.[11a, 14] DOX-EMCH (MedChemExpress, Monmouth Junction, NJ, USA) was conjugated to the internal surface of PhMV by mixing 5 molar equivalents per coat protein (Eq/CP) of the prodrug with 2 mg/mL PhMV particles in 50 mM PBS (pH 7.0) overnight at room temperature. The loaded VLPs were purified by sucrose cushion ultracentrifugation (0.1 mL 30% sucrose cushion, 116,525 g, 1 h), then re-dissolved in 50 mM PBS (pH 7.4), mixed with 10 Eq/CP 2000 Da mPEG-NHS (Nanocs, New York, NY, USA) for 3 h at room temperature and processed twice by ultracentrifugation as described above to purify the DOX-PhMV-PEG particles. The VLPs were stored in 50 mM PBS (pH 8.0) and the concertation was determined using the Pierce BCA assay kit (Thermo Fisher Scientific, Waltham, MA, USA). The concentration of doxorubicin was determined by measuring the absorption at 488 nm (ε= 11,500 L mol−1 cm−1). The structural integrity of the VLPs was confirmed by transmission electron microscopy (TEM), size-exclusion chromatography (SEC), denaturing SDS-PAGE and native agarose gel electrophoresis as described in the Supporting Information). UV/vis absorption and fluorescence were determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
2.2. Doxorubicin release profile:
To measure the pH-triggered release of doxorubicin we dialyzed 200 µL 2.5 mg/mL DOX-PhMV-PEG against 12 mL of PBS (pH 7.4 or 6.4) or citrate buffer (pH 5.2) in 10 kDa Slide-A-Lyzer MINI dialysis tubes (Thermo Fisher Scientific) at 37 °C with gentle shaking. We collected 100-µL fractions every few hours for 192 h and measured the fluorescence intensity in a Tecan Infinite M200 plate reader against a doxorubicin standard curve (Figure S1).
2.3. Cell uptake:
MDA-MB-231 (breast cancer), PC-3 (prostate cancer), SKOV-3 (ovarian cancer) and A2780 cells (ovarian cancer) were grown in tissue culture media (as per manufacturer’s [ATCC, Manassas, VA, USA] recommendation) and then incubated with 10 µg/mL doxorubicin or an equivalent dose of DOX-PhMV-PEG particles for 3 h at 37 °C. The cells were then observed by laser confocal scanning microscopy (LCSM) to determine the intracellular localization of doxorubicin (for detailed methods see Supporting Information) and quantitative data were collected by flow cytometry using an Accuri C6 instrument (BD Biosciences, Franklin Lakes, NJ, USA) as previously described.[11a]
2.4. Cytotoxicity and DNA damage assays:
The cytotoxicity of DOX-PhMV-PEG particles and the free drug was compared in MDA-MB-231 (breast cancer), SKOV-3 (ovarian cancer), A2780 (ovarian cancer) and PC-3 (prostate cancer) cell lines using an MTT assay (ATCC). Cells were treated with each sample normalized to doxorubicin concentrations in the range 0.01−50 μM in triplicate for 48 h, and absorbance at 570 nm was measured in a Tecan Infinite M200 plate reader. Each assay was performed in triplicate and the normalized cell proliferation values were averaged for each treatment concentration to calculate IC50 values.
DNA damage was detected using an eBioscience phosphohistone H2A.X assay (Thermo Fisher Scientific). Briefly, MDA-MB-231 cell lines were plated at a density of 2.5 × 104 cells/well in triplicate over 1.5-mm coverslips in a 12-well plate 24 h before the experiment. The cells were then exposed to 2 μM doxorubicin or an equivalent dose of DOX-PhMV-PEG particles (or PBS as a control) at 37 °C for 24 h. The cells were then fixed in 4% (v/v) paraformaldehyde, permeabilized with 0.5% (v/v) Triton X-100 in PBS, and stained with 200 μL of the mouse monoclonal anti-phosphohistone H2A.X primary antibody (1:500 dilution) and an anti-mouse IgG secondary antibody conjugated to Alexa Fluor 488. Nuclei were stained with DAPI and images were captured using a Nikon A1R confocal microscope. Cells with 10 or more positive γH2A.X foci in the nucleus were considered to have significant double strand break (DSB) damage.
2.5. In vivo tumor model:
Animal experiments were carried out according to IACUC-approved procedures at the University of California San Diego. Female NCr Foxn1nu mice at 4–6 weeks of age (Charlies River, Wilmington, MA, USA) were subcutaneously injected in the right hind leg with 0.1 mL of MDA-MB-231 cells at a concentration of 1×106 per mL in a 1:1 mixture of RPMI-1640 medium and Matrigel (Corning, New York, NY, USA). Tumor-bearing mice were allocated to one of four treatment groups (PBS, PhMV-PEG, free doxorubicin, DOX-PhMV-PEG) when the tumor volume reached ~150 mm3 (n = 15 per group). The mice were intravenously injected twice per week with free doxorubicin or DOX-PhMV-PEG at a dosage of 1.0 mg doxorubicin per kg body weight. Mice in the PhMV-PEG group were injected with an equivalent number of particles. Tumor size and body weight were measured before each injection and total tumor volume was calculated using the formula ν = l × w2/2. Mice were euthanized if the tumor size reached 1000 mm3 according to IACUC guidelines.
3. Results and Discussion
3.1. Preparation and characterization of the DOX-PhMV-PEG
PhMV-based VLPs were prepared according to our established protocol by expressing the PhMV coat protein in Escherichia coli.[11a, 14] Each VLP comprises 180 coat protein subunits. The crystal structure indicates that four accessible lysine residues (K62, K143, K153, and K166) are exposed on the external surface of each coat protein, offering 720 addressable amino groups per VLP, and a single cysteine residue (C75) is exposed on the internal surface, offering 180 addressable thiol groups per VLP.[10] The absence of the viral genome allows small molecules to diffuse in and out of the VLP, making it possible to attach different components to the internal and external surfaces.[11a] We were, therefore, able to conjugate DOX-EMCH to the internal surface using a thiol-maleimide reaction, as shown in Figure 1A. DOX-EMCH (also known as INNO-206 or aldoxorubicin) is a prodrug of doxorubicin that has been tested in phase III clinical trials (https://clinicaltrials.gov/ct2/show/NCT02049905). The acid-sensitive hydrazine linker allows the release of doxorubicin in the slightly acidic tumor microenvironment or the acidic endosomal or lysosomal compartments following cellular uptake of the DDS.[15] The external surface of the DOX-PhMV particles was coated with PEG by conjugating methoxy-PEG N-hydroxysuccinimide (mPEG-NHS) to exposed amine groups by NHS esterification. The PEGylation of the DOX-PhMV particles enhances their stability and solubility, improves their biocompatibility and retention in vivo, and prevents nonspecific uptake by non-target cells.
Figure 1.
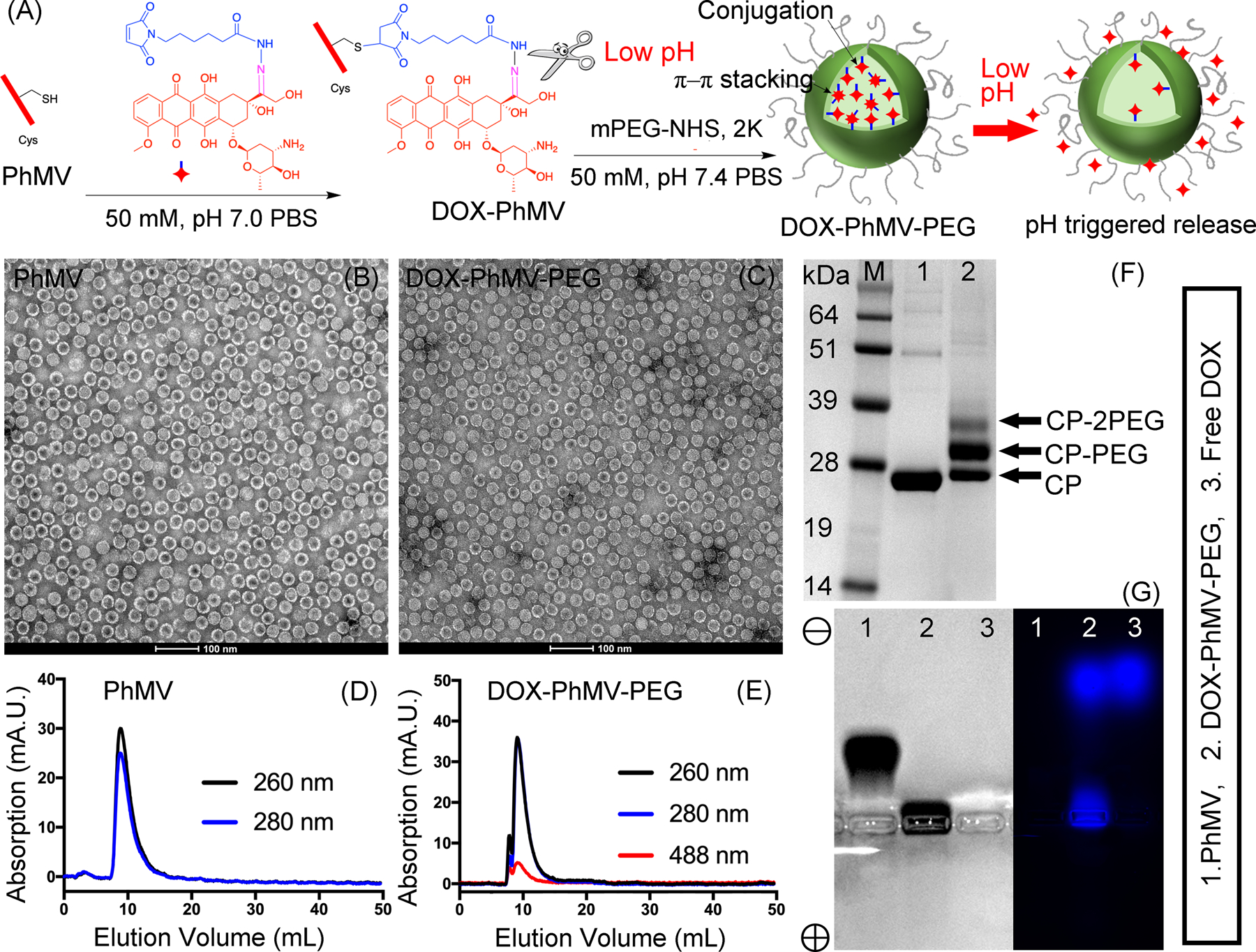
Synthesis and physicochemical properties of PhMV-DOX-PEG particles. (A) the synthesis of doxorubicin-loaded PhMV-based VLPs and the pH-induced drug release mechanism. (B, C) Transmission electron micrographs of negatively-stained (B) native PhMV-derived VLPs and (C) DOX-PhMV-PEG. (D, E) Size-exclusion chromatograms of (D) native PhMV-derived VLPs and (E) DOX-PhMV-PEG. (F) SDS-PAGE analysis of the VLPs followed by staining with Coomassie Brilliant Blue. (G) Analysis of the VLPs by agarose gel electrophoresis followed by staining with Coomassie Blue (left) and fluorescence imaging (right).
The final DOX-PhMV-PEG particles were stable for at least 4 months in PBS pH 7.4, judging by the absence of precipitation and characterization by transmission electron microscopy (TEM) and size exclusion chromatography (SEC). TEM showed ~30 nm-sized VLPs before and after modification, confirming their structural integrity (Figure 1B, C, high magnification images in Figure S2). SEC revealed a single peak eluting at ~7.8 mL for the protein component (A280) and a corresponding peak for doxorubicin (A488) indicating that the drug was encapsulated by the DOX-PhMV-PEG particles (Figure 1D,E). Longitudinal dynamic light scattering (DLS) experiments of DOX-PhMV-PEG particles in PBS pH7.4 with 10% (v/v) serum indicated that the DOX-PhMV-PEG formulations remained stable for 4 months (Figure S3).
The composition of the particles was determined by measuring the protein concentration using a BCA protein quantitation assay kit, and the concentration of doxorubicin was determined by measuring the absorption at 488 nm (Figure S4). This revealed a relative doxorubicin concentration of ~230 µg/mg protein (equivalent to ~1570 doxorubicin molecules per particle) which is 8.9-fold higher than the theoretical maximum achieved by conjugation to 180 cysteine residues (25.9 µg/mg protein). We attributed this result to π–π stacking interactions between the conjugated doxorubicin molecules and additional free molecules in the internal cavity. In addition to π–π stacking some doxorubicin molecules may also be absorbed to the VLPs through drug-protein interactions – either via hydrophobic coupling or electrostatic binding.
SDS-PAGE analysis confirmed the successful conjugation of PEG to the surface of the PhMV coat protein, yielding a mixture of bands representing the unconjugated protein and variants with one and two PEG chains, respectively (Figure 1F). Densitometric analysis of the protein bands using an AlphaImager® gel documentation systems indicated that ~62% of the coat proteins of the DOX-PhMV-PEG particles was covered with PEG (~450 PEG chains per particle). Native agarose gel electrophoresis followed by fluorescence imaging and staining with Coomassie Brilliant Blue showed that the control VLPs migrated toward the cathode due to their strong positive charge but this was affected by the presence of doxorubicin (negatively charged) and PEG (which abolishes positively charged amines), causing a significant loss of mobility (Figure 1G). The two fluorescent bands in the lane representing DOX-PhMV-PEG confirm that the particles contain both conjugated and unconjugated doxorubicin. The lower fluorescent band with the same mobility as the matching Coomassie-stained band corresponds to the conjugated doxorubicin, whereas the upper fluorescent band with the same mobility as free doxorubicin in the adjacent lane corresponds to doxorubicin released from the particles during electrophoresis. The fluorescence intensity of the free doxorubicin is ~10-fold higher than the conjugated doxorubicin, which is consistent with the absorption data presented above and further supports that the conjugated doxorubicin interacts with additional doxorubicin molecules in the cavity by π–π stacking or protein coupling through hydrophobic or electrostatic interactions.
Our method for the preparation of doxorubicin-loaded VLPs was simple, scalable, and reproducible, which should facilitate the commercial translation of this process.
3.2. Controlled release of doxorubicin
Although controlled released can be triggered by external stimuli, the delivery of drugs to tumors is more effective if the release mechanism is specific to the tumor microenvironment because this is an intrinsic process. The use of pH-sensitive linkers is effective because the tumor microenvironment is slightly acidic (pH = 6.2—6.9) and the subcellular compartments involved in particle uptake (endosome and lysosome) have a typical pH of ~5.3. The pH-sensitive hydrazone bond in the DOX-EMCH molecule allows the release of doxorubicin in the tumor microenvironment or following uptake into the endosome/lysosome.
The pH-triggered release of doxorubicin by the DOX-PhMV-PEG particles was tested in vitro by incubating the particles at a concentration of 3 mg/mL in PBS at pH 6.4 to mimic the tumor microenvironment, in citrate buffer (pH 5.2) to mimic the lysosomal compartment, or in PBS (pH 7.4) as a control representing the physiological pH of the blood (Figure 2). More than 34% of the doxorubicin was released at pH 6.4 and more than 67% at pH 5.2 after incubation for 48 h at 37 °C, with the release rate slowing thereafter for the remainder of the 192-h time course. In contrast, less than 10% of the doxorubicin was released at pH 7.4 and the release profile reached a plateau after 48 h. These results clearly showed that the DOX-PhMV-PEG particles released doxorubicin rapidly and efficiently under conditions mimicking the tumor microenvironment or endosome/lysosome but retained the cargo under conditions mimicking the normal circulation.
Figure 2.
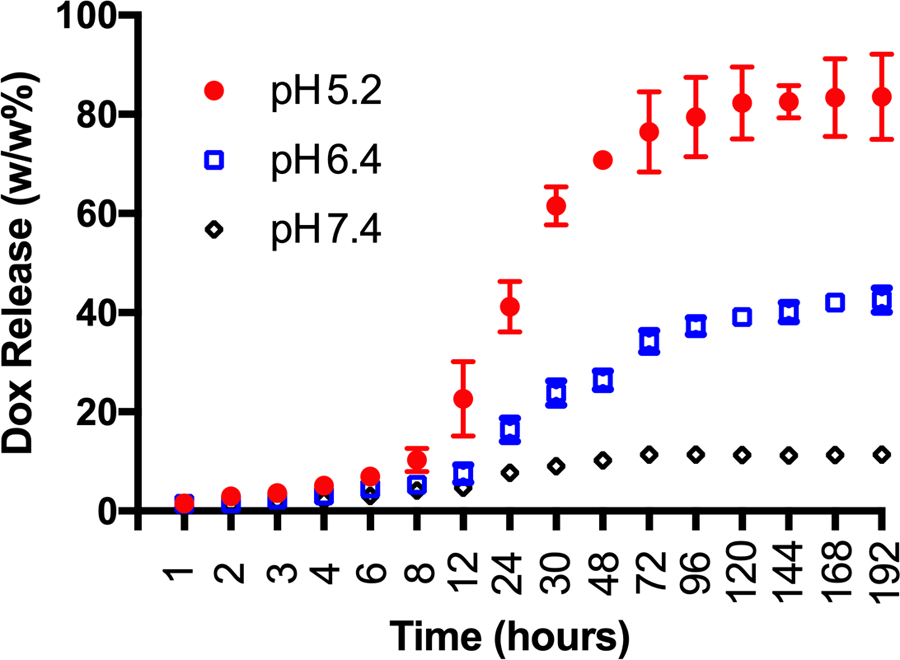
The pH-dependent controlled release of doxorubicin from the DOX-PhMV-PEG particles in three different buffers (pH = 7.4, 6.4 or 5.2) at 37 °C.
3.3. Cellular uptake, cytotoxicity and DNA damage
The uptake of DOX-PhMV-PEG particles was investigated using four cancer cell lines: MDA-MB-231 (breast cancer), PC-3 (prostate cancer), SKOV-3 (ovarian cancer) and A2780 (ovarian cancer) – all cell lines were subjected to flow cytometry analysis and cytotoxicity assays, and the MDA-MB-231 cell line was also imaged to track doxorubicin uptake and DNA damage.
The MDA-MB-231 cells were incubated with the VLPs for 3 h at 37 °C or with 10 µg/mL of free doxorubicin as a control. The dose of doxorubicin in the VLPs was normalized to the control. The cells were characterized by laser confocal scanning microscopy (LCSM) and flow cytometry-based on the intrinsic fluorescence of doxorubicin. The representative confocal images of MDA-MB-231 cells (Figure 3A) confirmed the uptake of free and VLP-delivered doxorubicin. The strong fluorescence signal in the nuclei of cells exposed to the VLPs confirmed that the drug was released inside the cancer cells and was trafficked to the nuclei.
Figure 3.
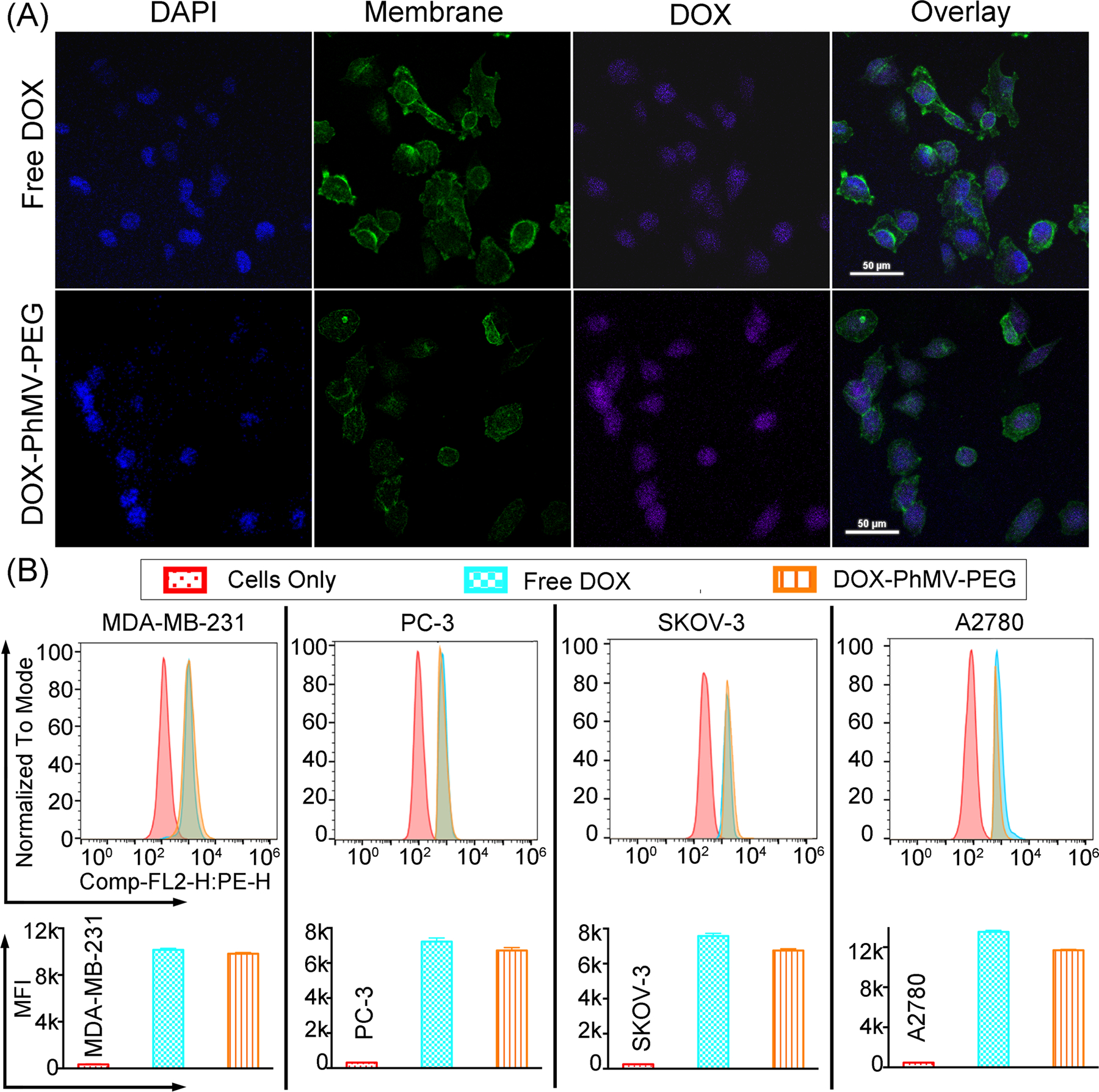
Analysis of doxorubicin uptake and intracellular trafficking in MDA-MB-231 cells. (A) Tracking of doxorubicin after 3 h incubation in the presence of the free drug (10 µg/mL) or an equivalent quantity of VLPs by confocal laser scanning microscopy. Nuclei were stained with DAPI (blue) and membranes were stained with AF647-labeled wheat germ agglutinin (green), whereas the doxorubicin is shown in purple (scale bar = 50 μm). (B) Flow cytometry showing the mean fluorescence intensities (MFIs) of cells in each sample (n = 3 ± standard deviations, p*** < 0.001). Data were analyzed using FlowJo v10 software.
Flow cytometry and quantitative analysis of the mean fluorescence intensity (MFI) showed that the free doxorubicin was taken up slightly more efficiently than the VLPs (Figure 3B). The internalization of drugs and particles is both time-dependent and concentration-dependent.[16] Here we incubated the cells with a normalized concentration of doxorubicin (10 µg/m) equivalent to 5.4×1010 molecules of free doxorubicin or 3.4×107 DOX-PhMV-PEG particles per cell. The free drug was therefore present at a 1000-fold higher concentration in the medium, which explains the higher uptake efficiency.
Next, we tested the cytotoxicity of the VLPs compared to free doxorubicin in the same four cell lines using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Figure 4A). The 50% inhibitory concentration (IC50) was calculated following an incubation period of 48 h at 37 °C. The VLPs were most toxic towards MDA-MB-231 cells (IC50 = 0.98 µM, compared to 0.63 µM for the free drug), followed by A2780 cells (IC50 = 1.16 µM, compared to 0.35 µM for the free drug) and SKOV-3 cells (IC50 = 1.84 µM, compared to 0.33 µM for the free drug), but showed little toxicity towards PC-3 cells (IC50 = 3.85 µM, compared to 1.15 µM for the free drug). The greater toxicity of the free drug probably reflects its more efficient uptake, combined with the slow intracellular release of doxorubicin from the internalized VLPs. This is consistent with the reduced in vitro toxicity of PEGylated liposomal doxorubicin or cisplatin formulations.[17a, b]
Figure 4.
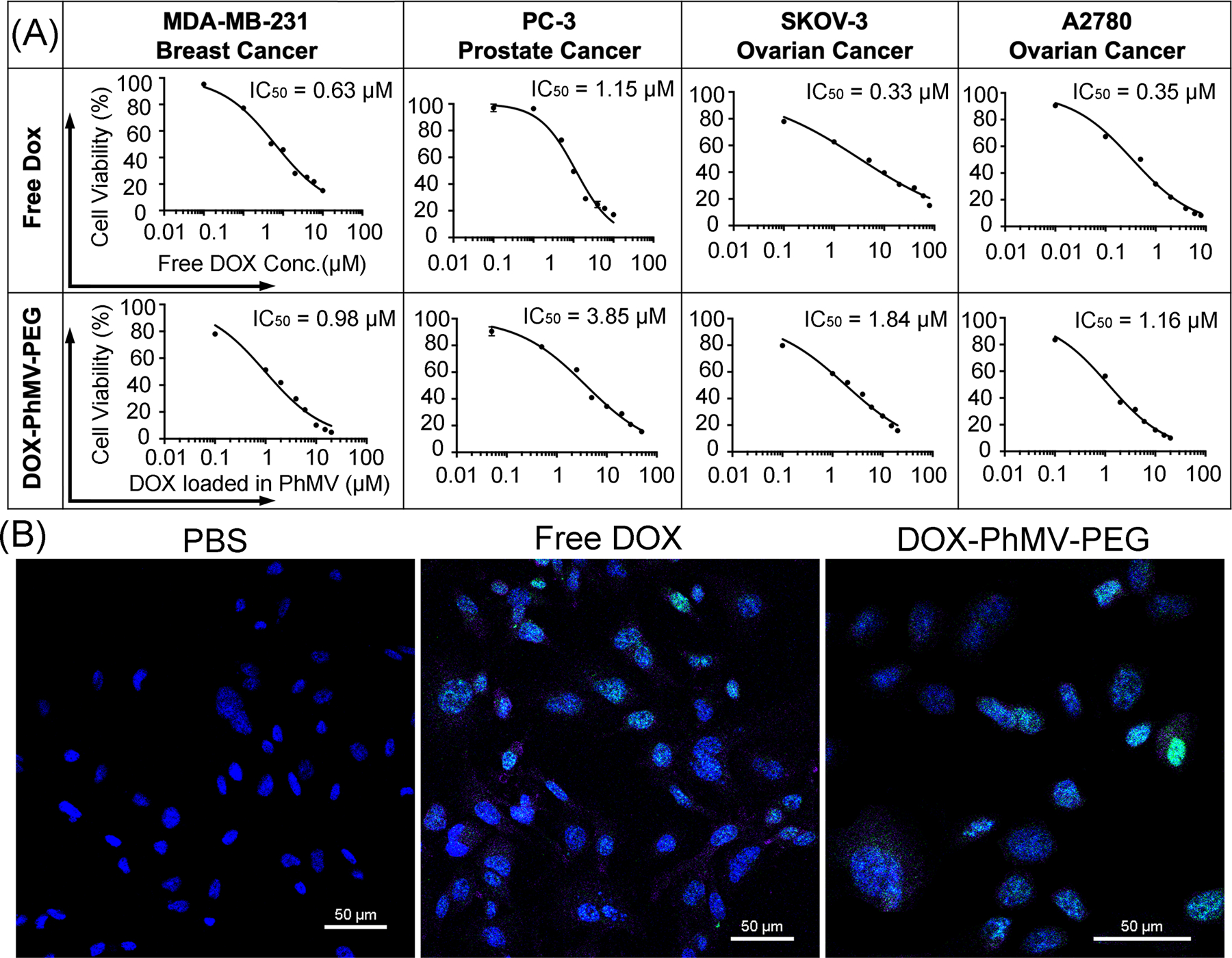
Cytotoxicity and DNA damage caused by DOX-PhMV-PEG particles. (A) The 50% inhibitory concentration (IC50) of free doxorubicin and DOX-PhMV-PEG particles against a panel of cancer cells. (B) Confocal images of MDA-MB-231 breast cancer cell reveal H2A.X phosphorylation (green) in nuclei (blue) following treatment with DOX-PhMV-PEG particles compared to free doxorubicin and PBS (scale bar = 50 µm).
Doxorubicin forms covalent bonds with DNA via the 3′ NH2 group of the daunosamine sugar moiety, disrupting the activity of topoisomerase II and causing damage to the DNA strands.[12] We, therefore, assessed the prevalence of DNA double strand breaks (DSBs) by confocal imaging of phosphorylated histone H2A.X foci after the treatment of MDA-MB-231 cells with 2 µM free doxorubicin or VLPs with the same normalized drug concentration for 24 h. We observed few DSBs in the PBS control group, but significantly more in the cells treated with free doxorubicin or the VLPs, leading to extensive cell death (Figure 4B). These results are consistent with the data from the cellular uptake studies reported above and again confirmed that the DOX-PhMV-PEG particles were able to release the cargo of doxorubicin successfully and trigger DNA damage, even at a low dose. The intracellular on-demand release of doxorubicin could help to restore the efficacy of chemotherapy in multi-drug-resistant cancers.
3.4. In vivo anti-tumor efficacy
Although DOX-PhMV-PEG was slightly less toxic than free doxorubicin in vitro, the results were nevertheless encouraging because a small decline in toxicity is acceptable if the VLPs perform better in vivo. This might be anticipated based on our earlier characterization of the PhMV vehicle, which revealed that the particles are biocompatible and biodegradable, with a long circulation half live of ~44 h, and efficient tumor homing properties achieving delivery of up to 6% of the injection dose (ID%) to solid tumors.[11a, b] We, therefore, tested the efficacy of the VLPs against a triple-negative breast cancer (MDA-MB-231) xenograft tumor model in NCr nude mice (n = 15). Tumors were allowed to reach a volume of ~150 mm3 before the animals were randomly assigned to one of four groups, each of which received an intravenous bolus injected twice weekly with a dose equivalent to 1.0 mg doxorubicin per kg body weight. The first group received free doxorubicin and the second received DOX-PhMV-PEG particles. The third group received PhMV-PEG without a drug cargo at the same particle concentration as the second group. The fourth group was injected with PBS as a control. The disease burden and side effects were monitored twice weekly by examining the physical condition and body weight of the mice, and by assessing their behavior according to IACUC recommendations.
The average and individual tumor sizes (Figure 5A,B) increased sharply in the PBS group, exceeding 1000 mm3 in 24 days. Tumor growth was slightly delayed in the PhMV-PEG particle control group, which may indicate that the VLPs can independently elicit an innate immune response in the tumor microenvironment.[18] Tumor growth was inhibited in the mice treated with free doxorubicin but the effect was short-lived. In contrast, tumor growth was strongly inhibited in the mice treated with DOX-PhMV-PEG particles, with the tumor volume never exceeding 260 mm3 and indeed disappearing completely in most of the mice, suggesting that the particles accumulated within the tumors, released doxorubicin on-demand, and killed the tumor cells with great efficacy. In clinics, large tumors (~500 mm3) are difficult to treat and survival rates are poor.[19] Comparable results are observed in preclinical mouse models, but our experiments with the DOX-PhMV-PEG particles achieved remarkable therapeutic efficacy in terms of extended survival and eradication. Furthermore, the dose of 1.0 mg doxorubicin/kg body weight used in our experiments is substantially lower than the recommended clinical dose (60–75 mg/m²) for multiple cancers (https://reference.medscape.com/drug/doxorubicin-342120). No recurrence was observed in the cured mice during the 20-day treatment-free period following the final injection.
Figure 5.
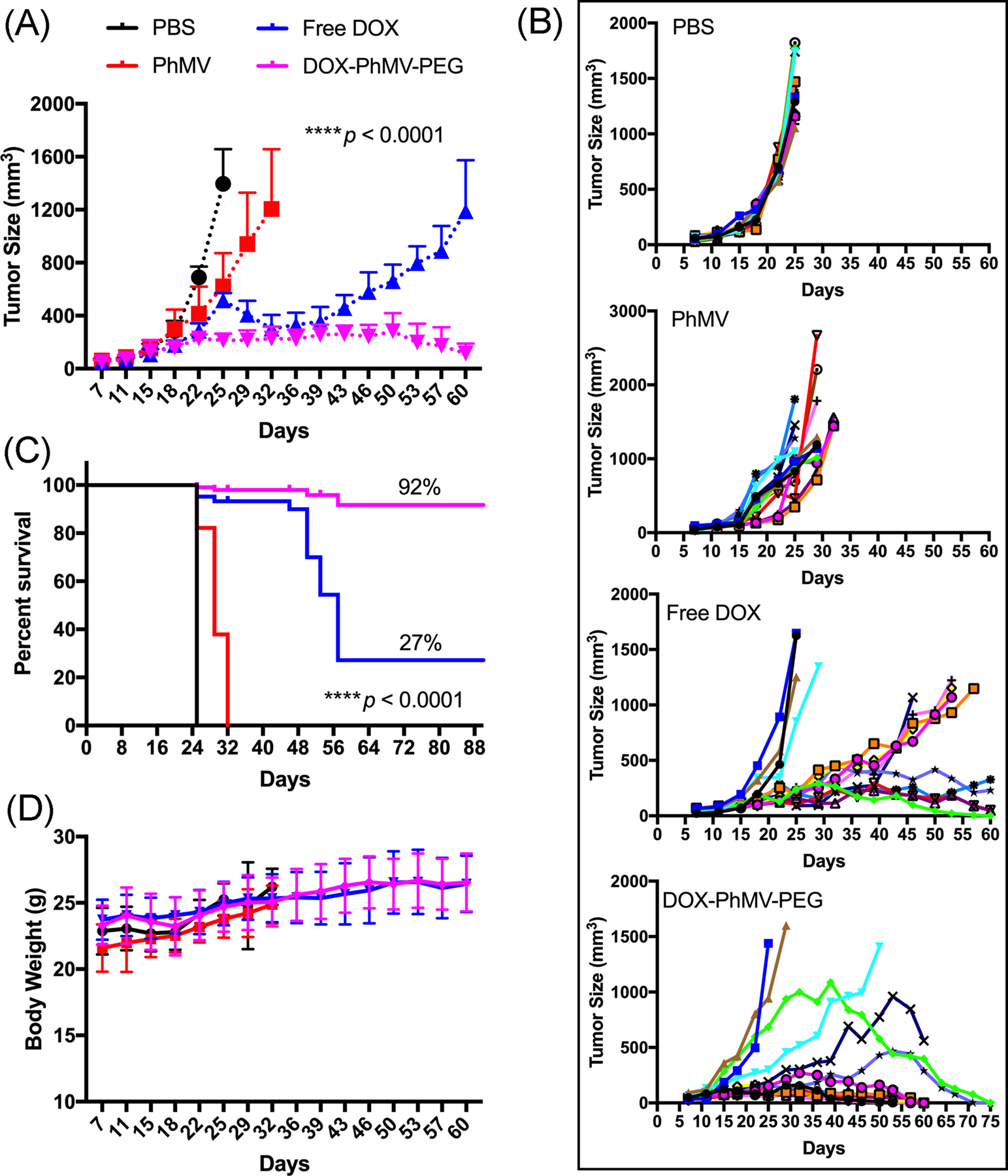
Inhibition of tumor growth in athymic mice (n = 15) with MDA-MB-231 xenografts following treatment with different drug formulations. The treatment began when the tumor volume reached ~150 mm3 and involved a twice weekly intravenous bolus of 1.0 mg free doxorubicin per kg body weight or DOX-PhMV-PEG particles normalized to the same drug dose. A particle control received the equivalent quantity of PhMV-PEG particles with no drug cargo, and an overall negative control group received PBS. Tumor volume and body weight were measured before each injection. Statistical analysis was carried out by two-way ANOVA (p****<0.0001). Mean tumor volumes and standard errors of the mean are shown. (A) Mean tumor size of all mice. (B) Individual tumor sizes in each group. (C) Statistical analysis of survival curves using the log-rank (Mantel–Cox) test (p****< 0.0001). (D) Bodyweight of treated tumor-bearing mice during the study.
Survival data were analyzed using the log-rank (Mantel–Cox) test, revealing that all mice in the DOX-PhMV-PEG treatment group remained alive when the last mouse in the PBS group had succumbed (Figure 5C). The overall survival rate among the mice in the DOX-PhMV-PEG group was 92%, which is 3.4-fold higher than the 27% observed in the free doxorubicin group under the same conditions. Among the surviving mice at the end of the experiment, 80% of the tumors were completely cured and there was no evidence of relapse. Throughout the study, we observed no weight loss or abnormal behavior in the DOX-PhMV-PEG group, indicating the biocompatibility of the DOX-PhMV-PEG particles (Figure 5D). Taken together, our results, therefore, suggest that the new DDS based on DOX-PhMV-PEG particles is a safe and highly efficacious platform for cancer therapy.
In conclusion, we developed a simple, scalable and reproducible strategy to synthesize a new DDS based on DOX-PhMV-PEG particles. The systematic analysis of these VLPs in vitro, in cancer cell lines, and in a xenograft tumor model in vivo confirmed the sensitive pH-triggered release of doxorubicin. The toxicity of the VLPs toward multiple cancer cell lines encouraged us to test the formulation in a mouse xenograft tumor model. We found that the DOX-PhMV-PEG particles achieved significantly greater antitumor efficacy than free doxorubicin in this model, leading to the complete eradication of tumors in almost all of the treated mice. Our new DDS, therefore, offers a promising new approach for efficacious cancer therapy.
Supplementary Material
Acknowledgement
We thank Dr. Hema Masarapu, Sri Venkateswara University, Tirupati, India for providing the plasmids for PhMV production. This work was funded in part by a grant from the National Institutes of Health (R01-CA224605) to N.F.S.
Abbreviations:
- DOX-EMCH
6-maleimidocaproyl-hydrazone doxorubicin
- VLPs
virus-like particles
- PhMV
physalis mottle virus
- PEG
polyethylene glycol
- DDS
drug delivery system
- FDA
Food and Drug Administration
- TEM
transmission electron microscopy
- SEC
size-exclusion chromatography
- LCSM
laser confocal scanning microscopy
- DSB
double strand break
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- IC50
inhibitory concentration
- PBS
phosphate buffered saline
- DLS
dynamic light scattering
- BCA
bicinchoninic acid
- SDS-PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- MFI
mean fluorescence intensity
- IACUC
institutional animal care and use committee.
Footnotes
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
5 References
- [1].Pushpakom S, Iorio F, Eyers PA, Escott KJ, et al. , Nat. Rev. Drug Discov 2018, 18, 41. [DOI] [PubMed] [Google Scholar]
- [2].Han Y, Shchukin D, Yang J, Simon CR, et al. , ACS Nano 2010, 4, 2838. [DOI] [PubMed] [Google Scholar]
- [3].Wang N, Cheng X, Li N, Wang H, Chen H, Adv. Healthcare Mater 2019, 8, e1801002. [DOI] [PubMed] [Google Scholar]
- [4].Hossen S, Hossain MK, Basher MK, Mia MNH, et al. , J. Adv. Res 2019, 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Theato P, Sumerlin BS, O’Reilly RK, Epps IIITH, Chem. Soc. Rev 2013, 42, 7055; [DOI] [PubMed] [Google Scholar]; b) Doshi N, Mitragotri S, Adv. Funct. Mater 2009, 19, 3843. [Google Scholar]
- [6].a) Eiben S, Koch C, Altintoprak K, Southan A, et al. , Adv. Drug Deliv. Rev 2018, 145, 96; [DOI] [PubMed] [Google Scholar]; b) Ma Y, Nolte RJM, Cornelissen JJLM, Adv. Drug Deliv. Rev 2012, 64, 811; [DOI] [PubMed] [Google Scholar]; c) Wen AM, Steinmetz NF, Chem. Soc. Rev 2016, 45, 4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Franke CE, Czapar AE, Patel RB, Steinmetz NF, Mol. Pharmaceutics 2018, 15, 2922; [DOI] [PubMed] [Google Scholar]; b) Czapar AE, Zheng Y-R, Riddell IA, Shukla S, et al. , ACS Nano 2016, 10, 4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Lam P, Steinmetz NF, Biomater. Sci 2019, 7, 3138; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cai H, Shukla S, Steinmetz NF, Adv. Funct. Mater 2020, 30, 1908743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aljabali AAA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ, Mol. Pharmaceutics 2013, 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sri Krishna S, Sastri M, Savithri HS, Murthy MRN, J. Mol. Biol 2001, 307, 1035. [DOI] [PubMed] [Google Scholar]
- [11].a) Hu H, Masarapu H, Gu Y, Zhang Y, et al. , ACS Appl. Mater. Interfaces 2019, 11, 18213; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Masarapu H, Patel BK, Chariou PL, Hu H, et al. , Biomacromolecules 2017, 18, 4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tacar O, Sriamornsak P, Dass CR, J. Pharm. Pharmacol 2013, 65, 157. [DOI] [PubMed] [Google Scholar]
- [13].Kratz F, Curr. Bioact. Compd 2011, 7, 33. [Google Scholar]
- [14].Ranjith-Kumar CT, Gopinath K, Jacob ANK, Srividhya V, et al. , Arch Virol 1998, 143, 1489. [DOI] [PubMed] [Google Scholar]
- [15].Graeser R, Esser N, Unger H, Fichtner I, et al. , Invest. New Drug 2010, 28, 14. [DOI] [PubMed] [Google Scholar]
- [16].Mosquera J, García I, Liz-Marzán LM, Acc.Chem. Res 2018, 51, 2305. [DOI] [PubMed] [Google Scholar]
- [17].a) Rifkin RM, Gregory SA, Mohrbacher A, Hussein MA, Cancer 2006, 106, 848; [DOI] [PubMed] [Google Scholar]; b) Lu Z-R, Qiao P, Mol. Pharmaceutics 2018, 15, 3603. [DOI] [PubMed] [Google Scholar]
- [18].Lizotte PH, Wen AM, Sheen MR, Fields J, et al. , Nat. Nanotechnol 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hanigan MH, Frierson HF, Swanson PE, De Young BR, Hum. Pathol 1999, 30, 300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.