

Abstract
Abnormal protein aggregation within neurons is a key pathologic feature of Parkinson’s disease (PD). The spread of brain protein aggregates is associated with clinical disease progression, but how this occurs remains unclear. Mutations in glucosidase, beta acid 1 (GBA), which encodes glucocerebrosidase (GCase), are the most penetrant common genetic risk factor for PD and dementia with Lewy bodies and associate with faster disease progression. To explore how GBA mutations influence pathogenesis, we previously created a Drosophila model of GBA deficiency (Gba1b) that manifests neurodegeneration and accelerated protein aggregation. Proteomic analysis of Gba1b mutants revealed dysregulation of proteins involved in extracellular vesicle (EV) biology, and we found altered protein composition of EVs from Gba1b mutants. Accordingly, we hypothesized that GBA may influence pathogenic protein aggregate spread via EVs. We found that accumulation of ubiquitinated proteins and Ref(2)P, Drosophila homologue of mammalian p62, were reduced in muscle and brain tissue of Gba1b flies by ectopic expression of wildtype GCase in muscle. Neuronal GCase expression also rescued protein aggregation both cell-autonomously in brain and non-cell-autonomously in muscle. Muscle-specific GBA expression reduced the elevated levels of EV-intrinsic proteins and Ref(2)P found in EVs from Gba1b flies. Perturbing EV biogenesis through neutral sphingomyelinase (nSMase), an enzyme important for EV release and ceramide metabolism, enhanced protein aggregation when knocked down in muscle, but did not modify Gba1b mutant protein aggregation when knocked down in neurons. Lipidomic analysis of nSMase knockdown on ceramide and glucosylceramide levels suggested that Gba1b mutant protein aggregation may depend on relative depletion of specific ceramide species often enriched in EVs. Finally, we identified ectopically expressed GCase within isolated EVs. Together, our findings suggest that GCase deficiency promotes accelerated protein aggregate spread between cells and tissues via dysregulated EVs, and EV-mediated trafficking of GCase may partially account for the reduction in aggregate spread.
Author summary
Parkinson’s disease (PD) is a common neurodegenerative disease characterized by abnormal clumps of proteins (aggregates) within the brain and other tissues which can lead to cellular dysfunction and death. Mutations in the gene GBA, which encodes glucocerebrosidase (GCase), are the strongest genetic risk factor for PD, and are associated with faster disease progression. GCase-deficient mutant flies display features suggestive of PD including increased protein aggregation in brain and muscle. We found that restoring GCase protein in the muscle of mutant flies reduced protein aggregation in muscle and the brain, suggesting a mechanism involving interaction between tissues. Previous work indicated that GBA influences extracellular vesicles (EVs)–small membrane-bound structures released by cells to communicate and/or transport cargo from cell to cell. Here, we found increased aggregated proteins within EVs of mutant flies, which was reduced by restoring GCase in muscle. In addition, we found GCase within the EVs, possibly explaining how GCase in one tissue such as muscle could reduce protein aggregation in a distant tissue like the brain. Our findings suggest that GCase influences proteins within EVs, affecting the spread of protein aggregation. This may be important to understanding PD progression and could uncover new targets to slow neurodegeneration.
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, affecting 1–2% of people over 65 years of age [1]. PD is characterized by cardinal motor and non-motor symptoms, including rigidity, slowness of voluntary movements, and cognitive decline [2–4]. Intraneuronal Lewy bodies containing ubiquitinated proteins and α-synuclein are a hallmark pathologic finding in PD. The stereotypic spread of Lewy bodies in PD from the rostral brain stem to the midbrain and eventually throughout the neocortex suggests a prion-like mechanism mediating propagation of protein aggregates from neuron to neuron [5]. This temporo-spatial spread of Lewy bodies correlates with clinical progression of PD and has been replicated in several animal models [6–8]. Although much work has focused on identifying genes involved in PD, and how perturbation of these genes lead to PD pathogenesis, the mechanisms underlying PD are not yet completely understood.
Mutations in the gene glucosidase, beta acid 1 (GBA), encoding the lysosomal ceramide metabolism enzyme glucocerebrosidase (GCase), are the strongest genetic risk factor for PD and dementia with Lewy bodies, increasing risk of developing PD by approximately 5-fold in GBA mutation carriers compared to non-carriers [9–11]. GBA carriers with PD are otherwise clinically similar to idiopathic PD patients, with indistinguishable response to dopaminergic medications, slightly younger age of onset by about 4 years, and higher incidence of cognitive decline [12,13]. While some studies suggest that GBA carriers with PD may have a heavier burden of Lewy bodies than in non-carriers with PD, the neuropathologic features are similar [14]. Importantly, mutations in the gene GBA are common, having been found in 4–5% of all idiopathic PD patients [13,15]. Recent longitudinal clinical studies have revealed that in addition to increased risk, GBA carriers with PD have faster progression of both motor and cognitive symptoms compared to idiopathic PD patients [16–18].
Several models have been developed and characterized using Drosophila melanogaster to examine how GBA influences PD pathogenesis [19–22]. Our previously characterized Drosophila model of GBA deficiency (Gba1b) manifests several phenotypes reminiscent of key features of PD, including neurodegeneration, locomotor deficits, cognitive deficits, and accelerated protein aggregation in multiple tissues including the nervous system and muscle [19]. Proteomic analysis of our Gba1b mutant flies revealed that proteins involved in extracellular vesicle (EV) biology were dysregulated, and EVs isolated from Gba1b Drosophila hemolymph revealed increased levels of aggregate-prone and EV-intrinsic proteins, indicating that GBA deficiency alters the protein composition of EVs [23]. EVs are a heterogeneous group of membrane-bound vesicles secreted by cells that can have multiple functions, including intercellular communication through protein, lipid, and nucleic acid cargo and discard of cell components outside of the cell [24,25]. EVs can originate from the endosomal system as a result of fusion of the late endosome to the plasma membrane, releasing intraluminal vesicles as EVs into the extracellular matrix (exosomes), or from direct outward budding of the plasma membrane (microvesicles) [24,26,27]. EVs have been implicated in the propagation of misfolded proteins between cells in multiple neurodegenerative diseases, including PD [28–33]. α-synuclein has been found within EVs isolated from tissues of PD and dementia with Lewy bodies patients [28,34], and in vitro studies have suggested that α-synuclein within EVs may be more pathogenic than free extracellular α-synuclein [35]. Accordingly, we hypothesized that tissue-specific expression of wildtype (WT) GCase might reduce the protein aggregates that accumulate in Gba1b mutants.
Using our GBA-deficient Drosophila model [18,23], we examined whether GCase could be mediating propagation of protein aggregates from cell-to-cell via EVs. In this study, we found that tissue-specific expression of WT GCase in Gba1b mutants corrected the alterations in protein composition of GBA-deficient EVs, as well as protein aggregation in local and distant tissues. Perturbing an EV biogenesis pathway in Gba1b mutants by knocking down neutral sphingomyelinase (nSMase), encoding an enzyme important for EV release as well as ceramide metabolism, resulted in tissue-specific cell-autonomous effects on protein aggregation and changes in EV protein composition but did not rescue protein aggregation in distant tissues. Lipidomic analysis of the effect of nSMase knockdown on ceramide (Cer) and glucosylceramide (GlcCer) levels suggested that Gba1b mutant protein aggregation may be dependent on relative depletion of specific Cer species that are known to be enriched in EVs. Finally, we observed ectopically expressed GCase in EVs, suggesting that trafficking of GCase within EVs may contribute to the observed non-cell-autonomous rescue. Our findings suggest that mutations in GBA result in alterations in the lipid composition of cells and the protein composition of EVs that promote enhanced cell-to-cell transmission of pathogenic protein aggregates. Moreover, our findings indicate that GCase can be packaged into EVs and trafficked between cells to reduce protein aggregation throughout an organism. These findings suggest a possible mechanism underlying the clinical finding that GBA mutation carriers not only have increased risk of developing PD, but also faster progression of disease.
Results
Protein aggregation in Gba1b mutants can be rescued cell-autonomously and non-cell-autonomously by tissue-specific dGba1b expression
Our prior work revealed accelerated aggregate accumulation and dysregulation of EV-related proteins in Gba1b mutant flies suggesting that GCase deficiency may influence cell-to-cell spread of protein aggregates. To explore the hypothesis that mutations in GBA promote the spread of protein aggregates from peripheral tissues to the brain, we expressed WT Drosophila Gba1b (dGba1b) in non-neural tissues of Gba1b mutants and examined accumulation of ubiquitinated protein and Ref(2)P, the Drosophila homologue of mammalian p62, in the brain. Using the DMef-GAL4 driver to drive expression of WT dGba1b in muscles throughout the fly, insoluble ubiquitinated protein aggregate accumulation was reduced in both the thoraces and heads of Gba1b mutant flies (Fig 1A and 1B). This non-cell-autonomous rescue of ubiquitinated protein aggregates was dramatically apparent in the whole brains of Gba1b mutants (Fig 1C). However, because DMef-GAL4 is also expressed in muscles located in the fly head that control the proboscis and may not exclusively drive expression in only muscle cells, we repeated this experiment using the Act88F-GAL4 driver. Act88F expression is specifically in the thoracic indirect flight muscles and is not found in the head [36]. Expression of WT dGba1b using the Act88F-GAL4 driver in Gba1b mutants significantly reduced accumulation of insoluble ubiquitinated proteins in the thoraces and heads (Fig 1E and 1F) and rescued their shortened lifespan (Fig 1D and S1 Table).
Fig 1. Muscle expression of dGba1b rescues protein aggregation and lifespan in Gba1b mutants.

(A-C) Using the muscle-specific driver, DMef-GAL4, wildtype (WT) Drosophila Gba1b (dGba1b) was expressed in Gba1b mutants and WT revertant controls. (A,B) Homogenates were prepared from fly thoraces and heads using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin. Representative images and quantification of ubiquitin (Ub) in (A) thoraces (one-way ANOVA: F(3,7) = 6.972, p = 0.016) and (B) heads (F(3,7) = 8.822, p = 0.009) are shown. Results are normalized to Actin loading control and control flies. (C) Representative anti-Ub immunofluorescent staining of whole brains from control, Gba1b mutant, and control and Gba1b mutants expressing dGba1b using the DMef-GAL4 driver. (D-H) Using the indirect flight muscle specific driver, Act88F-GAL4, WT dGba1b was expressed in Gba1b mutants and wildtype revertant controls. (D) Kaplan-Meier survival curves of control, Gba1b mutants, and Gba1b mutants and controls expressing WT dGba1b using the Act88F-GAL4 driver. (E-H) Homogenates were prepared from fly thoraces and heads using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin, and on the soluble fractions using antibodies to Ref(2)P and Actin. Representative images and quantification of ubiquitin in (E) thoraces (F(3,8) = 4.657, p = 0.036) and (F) heads (F(3,8) = 9.538, p = 0.005) and Ref(2)P in (G) thoraces (F(3,8) = 88.77, p < 0.001) and (H) heads (F(3,8) = 5.977, p = 0.019) of controls and Gba1b mutants with and without muscle expression of WT dGba1b are shown. Results are normalized to Actin in controls. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test.
Ref(2)P, the Drosophila ortholog of mammalian SQSTM1/p62, is important for selectively targeting ubiquitinated proteins for lysosomal degradation. While Ref(2)P is often used as a marker of autophagic flux, we have previously found it to accumulate in Gba1b mutants with no evidence of significantly impaired autophagy [23]. However, Ref(2)P is also required for aggregate formation under normal physiological conditions [37], and it has been shown to be released concomitantly with α-synuclein in EVs [34]; therefore we used its accumulation as a second readout of protein aggregation. We anticipated that Ref(2)P accumulation in Gba1b mutants could also be non-cell-autonomously rescued. We indeed found that Ref(2)P accumulation was significantly decreased in both the thoraces and heads of Gba1b mutants expressing WT dGba1b in flight muscle (Fig 1G and 1H).
To determine whether there might be a directionality or tissue-specificity to the non-cell-autonomous rescue of protein aggregation, we expressed WT dGba1b in the nervous system and assessed for protein aggregates in the body. Neuronal expression of WT dGba1b using the elav-GAL4 driver decreased both Ref(2)P accumulation and insoluble ubiquitinated proteins in both the heads and bodies of Gba1b mutants (Fig 2A–2D). In addition, neuronal expression of WT dGba1b partially rescued the shortened lifespan of Gba1b mutant flies (Fig 2E and S2 Table). Our findings indicate that GBA has both cell-autonomous and non-cell-autonomous roles in preventing protein aggregation. The non-cell-autonomous role of GBA raises the possibility that GCase may normally function to reduce the spread of protein aggregates from one cell and tissue to another.
Fig 2. Neuronal expression of dGba1b rescues protein aggregation in Gba1b mutants.

(A-D) Using the neuronal driver elav-GAL4, wildtype dGba1b was expressed in Gba1b mutants and wildtype revertant controls. Homogenates were prepared from fly heads and bodies using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 soluble fractions using antibodies to Ref(2)P and Actin and on the insoluble proteins using antibodies to ubiquitin (Ub) and Actin. Representative images and quantification of Ref(2)P in (A) bodies (one-way ANOVA: F(3,7) = 153.581, p < 0.001) and (B) heads (F(3,6) = 542.043, p < 0.001) and Ub in (C) bodies (F(3,8) = 6.099, p = 0.018) and (D) heads (F(3,8) = 16.878, p < 0.001) of control and Gba1b mutants with and without neuronal expression of dGba1b are shown. Results are normalized to Actin and control. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test. (E) Kaplan-Meier survival curves of control, Gba1b mutants, and Gba1b mutants and controls expressing WT dGba1b using the elav-GAL4 driver.
Non-cell-autonomous rescue of protein aggregation in Gba1b mutants is mediated by extracellular vesicles
There are multiple mechanisms that could mediate non-cell-autonomous interactions, including exocytosis of cytoplasmic components into the extracellular matrix, direct cell-to-cell contacts, and release of cytoplasmic components via EVs. Given that our prior proteomic analysis of Gba1b mutants found evidence of dysregulation of EVs [23], we hypothesized that GCase influences the trafficking of aggregate-prone proteins in EVs. We isolated EVs from hemolymph and confirmed that Ref(2)P was increased in EVs from Gba1b mutants compared to controls (Fig 3A), as we had seen previously [23]. We also found that EV-intrinsic proteins Rab11 and Rab7 were elevated, although the increase in Rab11 did not reach statistical significance (Fig 3B and 3C). We tested the possibility that muscle-specific expression of WT dGba1b would reduce the increased levels of Ref(2)P and EV-intrinsic proteins from Gba1b mutants. Indeed, we observed a reduction in Ref(2)P and Rab7 levels in EVs isolated from Gba1b mutants expressing WT dGba1b in flight muscle using the Act88F-Gal4 driver (Fig 3A and 3C). However, while ubiquitinated proteins were increased in Gba1b mutant whole flies, they were not significantly altered in EVs isolated from Gba1b mutants (Fig 3D). These results indicate that expression of GCase in the muscles of Gba1b mutants restores normal EV content and EVs may promote the spread of protein aggregates between tissues.
Fig 3. Muscle expression of dGba1b rescues alterations in EV-intrinsic proteins and Ref(2)P observed in EVs.

(A-D) Fly carcass homogenates and isolated extracellular vesicles (EVs) from Gba1b mutants and wildtype revertant control flies with and without wildtype dGba1b expressed in indirect flight muscle using the Act88F-GAL4 driver. Western blot analysis was performed using antibodies to (A) Ref(2)P (One-way ANOVA: F(3,8) = 7.610, p = 0.010), (B) Rab11 (F(3,8) = 3.584, p = 0.066), (C) Rab7 (F(3,8) = 8.133, p = 0.008), and (D) ubiquitin (Ub) (F(3,8) = 4.718, p = 0.035). Representative images and quantification normalized to controls are shown. Ponceau S is shown to verify equal loading. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test.
Tissue-specific Cer depletion is necessary for the spread of protein aggregation in Gba1b mutants
To examine whether EVs may be mediating the observed non-cell-autonomous rescue, we examined factors influencing the metabolism of EVs. We hypothesized that disruption of EV biogenesis might reduce protein aggregation in distant tissues in Gba1b mutants by decreasing production of dysregulated EVs that promote propagation of protein aggregates. To test this hypothesis, we examined knockdown of neutral sphingomyelinase (nSMase), encoding a lipid-modifying enzyme important for the formation and release of EVs [38,39]. nSMase hydrolyzes sphingomyelin, producing phosphocholine and ceramide. This enzyme has been implicated in multiple cellular functions, including inflammatory responses, reaction to lung and cardiac pathology, synaptic regulation, and release of EVs independent of ESCRT machinery [40]. We hypothesized that tissue-specific RNAi knockdown of nSMase in Gba1b mutants could reduce protein aggregation in distant tissues by reducing the production of dysregulated EVs promoting protein aggregation. However, if Gba1b mutant phenotypes are due to relative Cer depletion rather than accumulation of GlcCer, then nSMase knockdown might enhance the cell-autonomous protein aggregation in Gba1b mutants as Cer is known to trigger EV release [38]. We observed the latter, with increased accumulation of ubiquitinated proteins and Ref(2)P in thoraces (Fig 4A and 4C), suggesting that Cer depletion promotes cell-autonomous protein aggregation. However, we also observed a trend towards an increase in ubiquitinated proteins and Ref(2)P in the heads of Gba1b mutants with knockdown of nSMase expression in flight muscle (Fig 4B and 4D), suggesting that nSMase knockdown in muscle is not sufficient to reduce non-cell-autonomous spread of protein aggregates to other tissues, and may be enhancing non-cell-autonomous spread of protein aggregates. In contrast, knock down of nSMase in neuronal tissue did not modify Ref(2)P or ubiquitinated protein levels in the heads of Gba1b mutants (Fig 4E and 4F). These results suggest that nSMase-mediated release of EVs and/or influence on Cer metabolism may be important for the cell-autonomous regulation of protein aggregation in Gba1b mutant muscles but not neurons.
Fig 4. Tissue-specific knockdown of nSMase in Gba1b mutants differentially alters ubiquitinated proteins and Ref(2)P.

(A-F) Neutral sphingomyelinase (nSMase)-RNAi was expressed using tissue-specific drivers in Gba1b mutants. Homogenates were prepared from fly heads and thoraces using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin, and on the soluble fractions using antibodies to Ref(2)P and Actin. (A-D) Representative images and quantification of ubiquitin in (A) thoraces and (B) heads and Ref(2)P in (C) thoraces and (D) heads of flies with and without nSMase knockdown in flight muscle using the Act88F-GAL4 driver. (E,F) Representative images and quantification of (E) ubiquitin and (F) Ref(2)P in the heads of flies with and without nSMase knockdown in neurons using the elav-GAL4 driver. Results are normalized to Gba1b mutants without RNAi expression. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test.
We next examined whether EV cargo is altered in Gba1b mutants with RNAi knockdown of nSMase in muscle. We found an even further increase in Ref(2)P in EVs isolated from Gba1b mutants with nSMase knocked down in muscle (Fig 5A). Rab11 which was elevated in Gba1b mutants was significantly increased compared to controls after knockdown of nSMase in the muscle of mutant flies (Fig 5C). There was no significant change in the levels of ubiquitinated proteins or Rab7, which were already increased in EVs from Gba1b mutants (Fig 5B and 5D). This suggests that nSMase may regulate the protein cargo and abundance of specific types of EVs.
Fig 5. Muscle-specific knockdown of nSMase alters EV protein cargo.

(A-D) Neutral sphingomyelinase (nSMase)-RNAi was expressed using the flight muscle driver Act88F-GAL4 in Gba1b mutants and wildtype revertant controls. Isolated EVs from these flies were prepared in RIPA buffer. Quantification of western blot analysis of (A) Ref(2)P (one-way ANOVA: F(3,8) = 20.397, p < 0.001), (B) ubiquitin (F(3,8) = 55.25, p < 0.001), (C) Rab11 (F(3,8) = 6.713, p = 0.014), and (D) Rab7 (F(3,8) = 5.159, p = 0.028) in the EV fraction are shown. Results are normalized to control. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test. (Representative images are found in S1 Fig).
To investigate whether the observed effects of nSMase-RNAi on cell-autonomous protein aggregation and EV cargo are due to alterations in Cer metabolism, we performed targeted lipidomic analysis to determine Cer and GlcCer levels in Gba1b mutants and controls with and without nSMase knocked down in muscle. We detected 39 Cer and 55 GlcCer species by liquid chromatography-mass spectroscopy (LC-MS) from whole bodies of 10 day old flies. Principal component analysis (PCA) of GlcCer species in Gba1b mutants and controls with and without nSMase knocked down in muscle revealed 4 distinct groups which could be distinguished by 2 main principal components (PC) (Fig 6A). Most of the variance (94.7%) was explained by PC1, separating control and Gba1b mutants. The top 15 significant GlcCer species for the loadings for PC1 and PC2 are listed in S1 Data. We found the levels of all GlcCer species in Gba1b mutants to be significantly elevated, as highlighted by the heat map displaying clustering analysis for GlcCer species in Gba1b vs control (Fig 7). All the GlcCer species were increased in Gba1b mutants by a fold-change of ≥1.5 compared to controls (Fig 6B), and 31 (56%) GlcCer species were increased over 20-fold for a False Discovery Rate (FDR) ≤ 0.05 (Fig 6B and Table 1, and S3A Fig). Consistent with PCA results, the effect of nSMase knock down in muscle on GlcCer levels had a modest effect on both control and Gba1b mutants but tended to decrease GlcCer levels (Fig 6C and 6E). Only one GlcCer species decreased by over 2-fold in Gba1b mutants expressing nSMase-RNAi in muscle compared to Gba1b mutants, and 14 GlcCer species decreased by over 1.5-fold with FDR ≤ 0.10 (Fig 6C and Table 2 and S3B Fig). Partial Least Squares–Discriminant Analysis (PLS-DA), a supervised multivariate regression analysis, was also performed to identify key GlcCer species differing between Gba1b mutants and controls, and Gba1b mutants with nSMase-RNAi in muscle vs Gba1b mutants. PCA and PLS-DA analyses resulted in very similar group separation of Gba1b mutants versus controls and Gba1b mutants expressing nSMase-RNAi in muscle versus Gba1b mutants (S2 Fig). Only 1 GlcCer species (GlcCer d16:2/24:1) in the top 15 significant GlcCer species identified by PLS-DA in Gba1b mutants expressing nSMase-RNAi in muscle versus Gba1b mutants (R2 = .99, Q2 = 0.98) was also in the top 15 GlcCer species differing significantly between Gba1b mutants versus controls (R2 = 0.99, Q2 = 0.79) (Fig 6D and 6E).
Fig 6. Muscle-specific knockdown of nSMase minimally decreases glucosylceramide levels, which are globally elevated in Gba1b mutants.

Targeted lipidomic analysis of glucosylceramide (GlcCer) species in 10 day old whole flies of the specified genotypes. (A) Principal component analysis (PCA) scores plot of all detected GlcCer species in control, Gba1b, Gba1b; Act88F-GAL4>nSMase-RNAi, and control; Act88F-GAL4>nSMase-RNAi flies. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2, which account for 97.4% of the variance. Ovals indicate 95% confidence region. (B) GlcCer species plotted by fold change between Gba1b and controls. Pink circles represent GlcCer species above a threshold fold change of 1.5 (dotted line), FDR ≤0.05. Values are plotted on log scale so that both up-regulated and down-regulated GlcCer species can be plotted in a symmetrical way. (C) GlcCer species plotted by fold change in Gba1b versus Gba1b; Act88F-GAL4>nSMase-RNAi. Pink circles represent GlcCer species above a threshold of 1.5-fold change (dotted line), FDR ≤0.10. Values are plotted on log scale. (D) Top 15 significant GlcCer species identified by Partial Least Squares–Discriminant Analysis (PLS-DA) in Gba1b versus controls (R2 = 0.99, Q2 = 0.98, 2 components), ranked by VIP scores. Refer to S2B Fig for PLS-DA scores plot of GlcCer species in Gba1b versus control. Variable Importance in Projection (VIP) is a weighted sum of squares of the PLS loadings (S2 Data) taking into account the amount of explained Y-variation in each dimension. The colored boxes on the right indicate the relative concentrations of the corresponding GlcCer species in each genotype. The first column (1) is control; the second column (2) is Gba1b. (E) Top 15 significant GlcCer species identified by PLS-DA in Gba1b versus Gba1b; Act88F-GAL4>nSMase-RNAi as ranked by VIP score (R2 = 0.99, Q2 = 0.79, 2 components, refer to S2D Fig for scores plot). The colored boxes on the right indicate the relative concentrations of the corresponding GlcCer species in each genotype. The first column (1) is Gba1b; the second column (2) is Gba1b; Act88F-GAL4>nSMase-RNAi. The highlighted species GlcCer d16:2/24:1 is also found in the top 15 significant GlcCer species identified by PLS-DA for Gba1b versus controls (D).
Fig 7. Glucosylceramide levels are globally elevated in Gba1b mutants compared to controls.

Targeted lipidomic analysis of glucosylceramide (GlcCer) species in 10 day old whole flies. Clustering analysis represented by a heat map of all detected GlcCer species in control (columns 1–3) versus Gba1b (columns 4–6).
Table 1. Top 15 significant GlcCer species by Volcano plot in Gba1b mutants vs controls.
| Compound | Fold Change | Adjusted p value |
|---|---|---|
| GlcCer d14:1/16:0 | 66.81 | 9.68E-05 |
| GlcCer d14:0/18:1 | 48.15 | 9.68E-05 |
| GlcCer d14:1/14:0 | 36.95 | 9.68E-05 |
| GlcCer d14:1/24:0 | 26.04 | 9.68E-05 |
| GlcCer d14:0/16:0 | 22.73 | 9.68E-05 |
| GlcCer d16:2/24:1 | 7.29 | 9.68E-05 |
| GlcCer d14:2/16:0 | 32.38 | 1.22E-04 |
| GlcCer d14:0/22:1 | 27.46 | 1.26E-04 |
| GlcCer d14:1/24:1 | 26.45 | 1.38E-04 |
| GlcCer d16:1/14:0 | 60.18 | 1.42E-04 |
| GlcCer d14:1/18:0 | 51.59 | 1.49E-04 |
| GlcCer d14:0/20:0 | 23.13 | 1.63E-04 |
| GlcCer d14:1/20:0 | 21.91 | 1.63E-04 |
| GlcCer d14:0/18:0 | 33.00 | 1.85E-04 |
| GlcCer d16:1/24:0 | 39.11 | 2.00E-04 |
* adjusted for FDR ≤ 0.05
Table 2. Significant GlcCer species identified by Volcano plot in Gba1b mutants expressing nSMase-RNAi in muscle vs. Gba1b mutants.
| Compound | Fold Change | Adjusted p value |
|---|---|---|
| GlcCer d14:1/20:1 | 0.52 | 0.013 |
| GlcCer d16:1/20:1 | 0.58 | 0.013 |
| GlcCer d14:2/22:1 | 0.60 | 0.025 |
| GlcCer d14:1/18:1 | 0.59 | 0.026 |
| GlcCer d16:2/20:1 | 0.54 | 0.029 |
| GlcCer d16:1/18:0 | 0.58 | 0.034 |
| GlcCer d16:2/22:1 | 0.49 | 0.041 |
| GlcCer d16:2/18:1 | 0.56 | 0.042 |
| GlcCer d14:0/20:1 | 0.60 | 0.047 |
| GlcCer d14:1/20:0 | 0.66 | 0.058 |
| GlcCer d16:1/18:1 | 0.57 | 0.095 |
| GlcCer d16:1/16:0 | 0.65 | 0.095 |
* adjusted for FDR ≤ 0.10
PCA of Cer species in Gba1b mutants and controls with and without nSMase knocked down in muscle revealed a distinct separation between Gba1b mutants and controls, accounting for 46.5% variance, and distinct separation between Gba1b mutants expressing nSMase-RNAi in muscle versus Gba1b mutants, accounting for 20.4% variance (Fig 8A, significant Cer species for PCA loadings in S1 Data). Heat map derived from clustering analysis of Cer species in Gba1b mutants versus controls indicates that the majority of Cer species are increased in Gba1b mutants compared to controls (Fig 9), although there is a significant subset of Cer species that are decreased. Fold-change analysis revealed that the magnitude of alterations in Cer levels between Gba1b mutants and controls was much smaller in comparison to alterations of GlcCer species in Gba1b mutants versus controls (Fig 8B). Volcano plot with adjusted p-values for FDR ≤ 0.10 revealed 36% of Cer species had greater than 1.5-fold change in Gba1b mutants versus controls, with 4 Cer species decreased and 9 Cer species increased in Gba1b mutants versus controls (Table 3 and S5A Fig). PCA and PLS-DA analyses again resulted in similar discrimination between groups when comparing Gba1b mutants versus controls, and Gba1b mutants expressing nSMase RNAi in muscle versus Gba1b mutants, (S4 Fig and S1 and S2 Data). PLS-DA of Cer species in Gba1b mutants versus controls identified two Cer species (Cer d16:2/22:1, and Cer d14:2/22:1) that were significantly reduced in Gba1b mutants versus controls (R2 = 0.99, Q2 = 0.80) (Fig 8D and S6 Fig). These two Cer species were also within the top 15 loadings for PC1 in PCA of Gba1b mutants versus controls (S1 Data) and reduced by greater than 1.5 fold with adjusted p-value for FDR ≤ 0.10 in Volcano plot for Gba1b mutants versus controls (Table 3 and S5A Fig). Cer d16:2/22:1 and Cer d14:2/22:1 were found to be further significantly decreased by PLS-DA of Gba1b mutants expressing nSMase-RNAi in muscle versus Gba1b mutants (R2 = 0.99, Q2 = 0.70) (Fig 8E and Table 4), and again found to be significant contributors to PC1 in PCA of Gba1b mutants expressing nSMase-RNAi in muscle versus Gba1b mutants (S1 Data). These results confirm that the knockdown of nSMase expression by RNAi affects Cer metabolism, as the majority of Cer species in Gba1b mutants with nSMase knocked down in muscle decreased compared to Gba1b mutants, consistent with the predicted enzymatic effect of nSMase knockdown on Cer metabolism.
Fig 8. Muscle-specific knockdown of nSMase decreases a subset of ceramide species that are decreased in Gba1b mutants.

Targeted lipidomic analysis of ceramide (Cer) species in 10 day old whole flies of the specified genotypes. (A) Principal component analysis of Cer species in control, Gba1b, Gba1b; Act88F-GAL4>nSMase-RNAi, and control; Act88F-GAL4>nSMase-RNAi flies. Ovals indicate 95% confidence region. (B) Cer species plotted by fold change in Gba1b versus controls. Pink circles represent Cer species above a 1.5-fold change threshold, FDR ≤0.10. Values are plotted on log scale so that both up-regulated and down-regulated Cer species can be plotted in a symmetrical way. (C) Cer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b, plotted by fold change. Pink circles represent Cer species above a 1.5-fold change threshold, FDR ≤0.10. Values are plotted on log scale. (D) Top 15 significant Cer species identified by Partial Least Squares–Discriminant Analysis (PLS-DA) in Gba1b versus controls (R2 = 0.99, Q2 = 0.80, 3 components), ranked by VIP score. The colored boxes on the right indicate the relative concentrations of the corresponding Cer species in each genotype. The first column (1) is control, the second column (2) is Gba1b. Refer to S4B Fig for PLS-DA scores plot of Cer species in Gba1b versus control. (E) Top 15 significant Cer species identified by PLS-DA in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b (R2 = 0.99, Q2 = 0.70, 2 components). The colored boxes on the right indicate the relative concentrations of the corresponding Cer species in each genotype. The first column (1) is Gba1b; the second column (2) is Gba1b; Act88F-GAL4>nSMase-RNAi. The highlighted species are also in the top 15 species identified by PLS-DA for Gba1b mutants versus controls (D).
Fig 9. Most ceramide species are increased in Gba1b mutants compared to controls.

Targeted lipidomic analysis of ceramide (Cer) species in 10 day old whole flies. Clustering analysis represented by a heat map of all detected Cer species in control (columns 1–3) versus Gba1b (columns 4–6).
Table 3. Significant Cer species identified by Volcano plot in Gba1b mutants vs controls.
| Compound | Fold Change | Adjusted p value* |
|---|---|---|
| Cer d14:0/20:0 | 2.506 | 0.004 |
| Cer d14:2/24:1 | 0.478 | 0.004 |
| Cer d14:0/18:0 | 1.984 | 0.004 |
| Cer d16:2/22:1 | 0.525 | 0.004 |
| Cer d14:0/22:0 | 2.620 | 0.005 |
| Cer d14:2/22:1 | 0.337 | 0.017 |
| Cer d14:1/24:0 | 1.649 | 0.017 |
| Cer d16:2/20:1 | 0.538 | 0.028 |
| Cer d14:0/24:0 | 1.600 | 0.028 |
| Cer d14:1/14:0 | 1.600 | 0.031 |
| Cer d16:1/24:1 | 1.836 | 0.043 |
| Cer d14:1/22:0 | 1.593 | 0.060 |
| Cer d16:0/20:0 | 1.641 | 0.087 |
* adjusted for FDR ≤ 0.10
Table 4. Significant Cer species identified by Volcano plot in Gba1b mutants expressing nSMase-RNAi in muscle vs Gba1b mutants.
| Compound | Fold Change | Adjusted p value* |
|---|---|---|
| Cer d14:2/20:1 | 0.541 | 0.098 |
| Cer d14:2/18:1 | 0.502 | 0.098 |
| Cer d16:2/22:0 | 2.242 | 0.098 |
* adjusted for FDR ≤ 0.10
Ectopically expressed GCase is trafficked within extracellular vesicles
Our finding that GBA expression in muscle normalizes Ref(2)P levels in EVs suggests that non-cell-autonomous rescue is mediated at least in part by preventing spread of aggregated proteins trafficked by EVs. However, EV-mediated trafficking of GCase could also be another contributing factor to the non-cell-autonomous rescue of protein aggregation in Gba1b mutants. To test this possibility, we investigated whether ectopically expressed GCase travels to distant tissues via EVs. Because there are no antisera that recognize Drosophila GCase, we examined ectopic expression of human GBA (hGBA) in Gba1b mutants. We previously found ubiquitous expression of WT hGBA (hGBAWT) to be sufficient to partially rescue the shortened lifespan of Gba1b mutants [19]. Here, we found that expression of hGBAWT in flight muscle also reduced ubiquitinated protein aggregation and Ref(2)P accumulation in Gba1b mutants in both thoraces and heads (Fig 10A–10D). EVs isolated from Gba1b mutants expressing hGBAWT in flight muscle were found to have decreased Rab11 levels compared to Gba1b mutants (Fig 11A and 11B), confirming that human GCase (hGCase) can also revert alterations in EVs due to dGba1b deficiency. We also expressed mutant hGBA bearing the common N370S mutation (hGBAN370S) in muscle, which failed to reduce the increased ubiquitin levels in the thoraces and heads of Gba1b mutant flies (Fig 10F and 10G). These results support a functional equivalence between Drosophila GCase and hGCase in reducing the formation of protein aggregates. Interestingly, we were also able to detect hGCase in both the thoraces and heads of Gba1b mutants expressing hGBAWT in flight muscle (Fig 10E), indicating that hGCase itself is able to travel to distant tissues. Furthermore, we detected hGCase in EVs isolated from hemolymph of flies expressing hGBAWT in muscle (Fig 11A). To determine whether hGCase is localized within EVs or associated on the outer surface of EVs, we added Proteinase K to the isolated EV fraction. We found hGCase to be resistant to Proteinase K digestion of the EV fraction but degraded if EVs were first treated with a detergent to disrupt vesicular membranes before Proteinase K digestion (Fig 11C). These results indicate that GCase can be incorporated into EV cargo, and that WT GCase may be trafficked by EVs to distant tissues to reduce protein aggregation.
Fig 10. Muscle expression of human WT GBA suppresses protein aggregation in Gba1b mutants.

(A-E) Using the flight muscle specific driver, Act88F-GAL4, wildtype (WT) human GBA (hGBAWT) was expressed in Gba1b mutant and WT revertant controls. Homogenates were prepared from fly heads and thoraces using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin, and on the soluble fractions using antibodies to Ref(2)P, Actin, and human glucocerebrosidase (hGCase). Representative images and quantification of ubiquitin in (A) thoraces (One-way ANOVA: F(2,6) = 19.949, p = 0.002) and (B) heads (F(2,6) = 24.854, p = 0.001) and Ref(2)P in (C) thoraces (F(2,6) = 10.609, p = 0.011) and (D) heads (F(2,6) = 23.297, p = 0.001) of Gba1b mutant flies with and without muscle expression of hGBAWT are shown. Results are normalized to Actin and control. (E) Antibody detecting hGCase in the thoraces and heads of control and Gba1b mutant flies with and without muscle expression of hGBAWT. (F,G) Using Act88F-GAL4, mutant human GBA (hGBAN370S) was expressed in Gba1b mutant and controls. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin. Quantification of ubiquitin in (F) thoraces (F(2,6) = 5.190, p = 0.049) and (G) heads (F(2,6) = 7.250, p = 0.025) of Gba1b mutant flies with and without muscle expression of hGBAN370S are shown. Results are normalized to Actin and control. (Representative images are found in S7 Fig). At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test.
Fig 11. Ectopically expressed human GCase is trafficked within EVs.
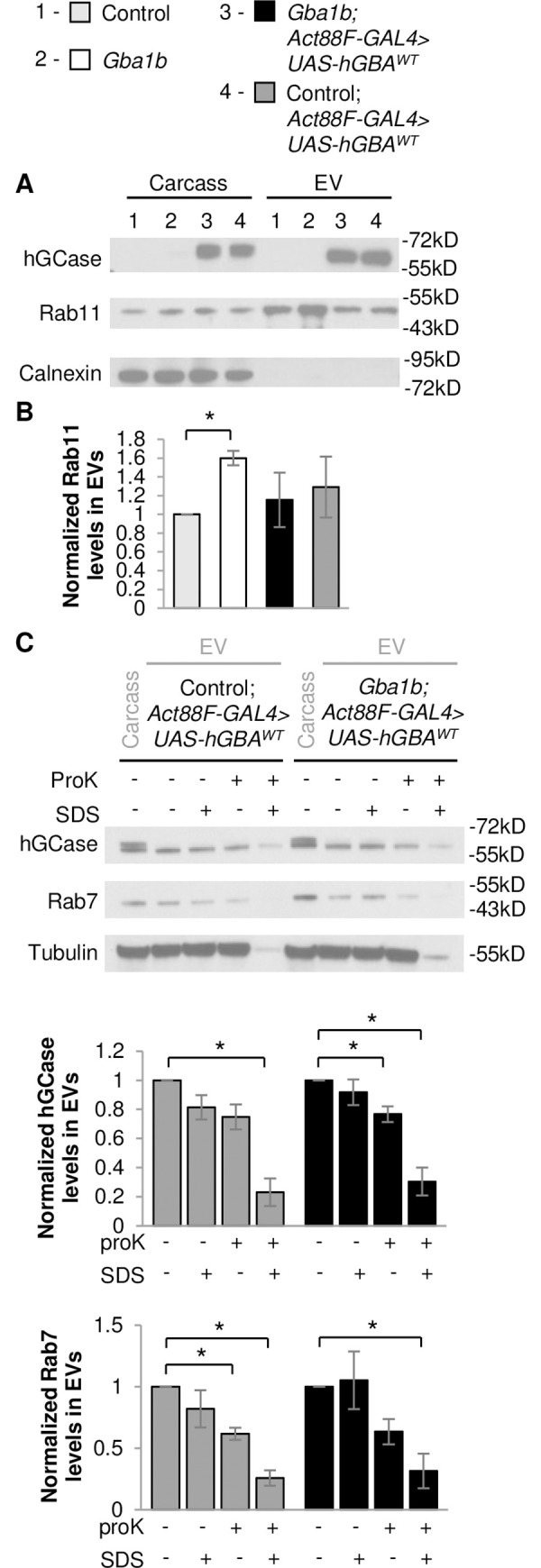
(A-B) Fly carcass homogenates and isolated extracellular vesicles (EVs) from Gba1b mutants and wildtype (WT) revertant controls with and without WT human GBA (hGBAWT) expressed in flight muscle using the Act88F-GAL4 driver were prepared using RIPA buffer. Western blot analysis was performed using antibodies to human glucocerebrosidase (hGCase), Rab11, Rab7, and Tubulin. (A) Representative images of hGCase and Rab11 in carcass and isolated EVs from control and Gba1b mutant flies with and without muscle expression of hGBAWT are shown. The blots were also probed with antibodies to Calnexin (Cnx99A) to confirm the purity of the EV samples. (B) Levels of Rab11 in the EV fraction are quantified and normalized to controls. (C) Representative images of hGCase, Rab7, and Tubulin in carcass and isolated EVs from control and Gba1b mutant flies with muscle expression of hGBAWT with and without Proteinase K (ProK) and/or SDS exposure. Levels of hGCase (One-way ANOVA, control with hGBAWT expression: (F(3,8) = 18.388, p < 0.001) and Gba1b mutants with hGBAWT expression: (F(3,8) = 19.490, p < 0.001)) and Rab7 (control with hGBAWT expression: (F(3,8) = 13.916, p = 0.002) and Gba1b mutants with hGBAWT expression: (F(3,8) = 5.550, p = 0.023)) in the EV fraction are quantified. Results are normalized to EVs without ProK or SDS exposure. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test.
Discussion
Many genetic influences of PD have now been identified, and much work has been focused on how these genes lead to protein aggregation through mechanisms such as protein misfolding and autophagy defects. However, none of these genes have been implicated in cell-to-cell spread of pathogenic protein aggregates, which closely correlates with clinical disease progression. Our proteomic analysis and non-cell-autonomous rescue of protein aggregation in Gba1b mutants has led us to hypothesize that GBA mutations may influence the rate of propagation of protein aggregates between neurons. Our work suggests a link between GBA mutations and faster spread of intracellular protein aggregates via a novel EV-mediated mechanism, possibly explaining the recent clinical finding that GBA mutations accelerate the progression of clinical disease. Using a Drosophila model of GBA deficiency that manifests accelerated protein aggregation, we found that expressing WT GCase in specific tissues of a GBA-deficient fly can not only rescue protein aggregation cell-autonomously and in distant tissues, but also rescue alterations in protein cargo observed in EVs isolated from Gba1b mutant hemolymph. Interestingly, ectopically expressed WT GCase itself was found within EVs of GBA-deficient flies, suggesting that the non-cell-autonomous rescue due to GCase expression is mediated by both reduction in aggregated proteins in EVs and trafficking of GCase via EVs to distant cells and tissues. Perturbing EV biogenesis through decreased expression of ESCRT-independent nSMase affected protein aggregation in local tissues in a tissue-dependent manner, and further decreased a subset of Cer species already reduced in Gba1b mutants. Interestingly, this subset of Cer species is known to be enriched in EV membranes [41]. Together, these findings suggest that mutations in GBA result in the accelerated spread of protein aggregates through changes in cellular lipid composition and dysregulation of proteins trafficked by EVs.
Although our model of GBA mutations promoting spread of protein aggregates via EVs is novel, the idea that proteostasis can be maintained in a non-cell-autonomous fashion is well supported in the literature. For example, in C. elegans, misfolded α- synuclein accumulating in endo-lysosomal vesicles was found to be transmitted from muscle to the hypodermis, a nearby tissue, for degradation [42]. It is possible that a non-cell-autonomous mechanism is necessary because certain tissues may be more efficient in reducing protein aggregation. This has been previously described, where overexpression of FOXO in Drosophila muscle decreased aging-related protein aggregates in muscle as well as brain and other distant tissues, but FOXO overexpression in adipose tissue was unable to prevent protein aggregation in muscle [43]. In our model, overexpressing dGba1b in Drosophila muscle or neuronal tissue prevented accumulation of protein aggregates throughout the organism, however overexpression of WT GCase in midgut and fat body did not significantly reduce protein aggregation in the brain (S8 Fig). These discrepancies could be due to tissue-specific biogenesis of EVs, which could depend on factors such as metabolic rate or endovesicular trafficking. Although dGba1b is expressed in all tissues, a second homologue of human GBA1, dGba1a, is expressed only in the midgut. Our Gba1b mutants retain ~25% expression of dGba1a [19]. Deficiency of dGba1a was found to extend lifespan [20] and does not result in significant accumulation of GlcCer [21], suggesting that there can be significant tissue-specific differences in function for GCase that could influence EV biogenesis.
Our unexpected results from perturbation of EV biogenesis suggest that the EV-mediated regulation of protein aggregation is tissue-specific and complex. Because an increase in EV-intrinsic proteins and alteration of protein cargo were observed in Gba1b mutants [23], we anticipated that genetic perturbations decreasing the biogenesis of EVs might rescue protein aggregation non-cell-autonomously by reducing the production of dysregulated EVs. However, decreased expression of ESCRT-independent nSMase in muscle did not rescue protein aggregation in heads, suggesting that a tissue-specific decrease in biogenesis of dysregulated EVs is not sufficient to reduce protein aggregation in the rest of the organism, and the cargo of EVs may need to be corrected to reduce spread of protein aggregation. In contrast, decreased expression of nSMase in the nervous system had no effect on protein aggregation in the head. This difference in outcome in perturbation of EV biogenesis in muscle and neurons could be due to cell-specific compensatory mechanisms or intrinsic metabolic demands and solicits further investigation.
A possible explanation for why decreased muscle expression of nSMase enhanced cell-autonomous protein aggregation and EV protein cargo alterations is that both GCase and nSMase enzymatically produce Cer. If GCase-deficient phenotypes are dependent on a relative reduction in Cer, decreased nSMase expression could exacerbate Gba1b mutant phenotypes. Indeed, lipidomic analysis of alterations in Cer metabolism due to nSMase knockdown revealed a further decrease in a subset of Cer species that were already significantly decreased in Gba1b mutants compared to controls. The further reduction in Cer species due to nSMase knockdown correlates with enhancement of cell-autonomous protein aggregation and EV cargo alterations, suggesting that accelerated protein aggregation in Gba1b mutants is mediated by Cer deficiency rather than GlcCer accumulation, as nSMase knockdown had a much more modest effect on the significantly increased levels of GlcCer species in Gba1b mutants compared to controls.
Cer has been implicated in the composition and biogenesis of EVs, and nSMase knockdown further altered EV cargo in Gba1b mutants, suggesting that decreased Cer levels may directly influence EV biogenesis in Gba1b mutants. However, Cer species were not globally decreased, suggesting that the regulation of Cer metabolism is complex and may be more dependent on specific Cer species. Interestingly, only 1 of the 9 Cer species significantly increased in Gba1b mutants versus controls had a monounsaturated fatty acyl group, while all 5 of the Cer species significantly decreased in Gba1b mutants versus controls had a monounsaturated fatty acyl group, suggesting GBA influences the metabolism of specific subset of Cer species that may be implicated in Gba1b mutant phenotypes. This subset of Cer species is enriched in species with long chain monounsaturated fatty acyl chains. Interestingly, lipids with monounsaturated fatty acyl groups are an abundant component in mammalian exosome membranes [40]. Investigating the alterations in lipid composition of EVs resulting from GCase deficiency and nSMase knockdown will be important in elucidating the role of Cer metabolism in Gba1b mutant phenotypes.
Our work suggests that GCase deficiency influences EV biogenesis to promote faster propagation of pathogenic protein aggregates throughout the tissues of an organism, which may be a compensatory response to cell-autonomous lysosomal stress. In the initial characterization of our Drosophila GBA-deficient model we found accelerated insoluble ubiquitinated protein aggregates, accumulation of Ref(2)P, and oligomerization of ectopically expressed human α-synuclein in Gba1b mutants, suggesting an impairment in lysosomal degradation [19]. A similar GBA-deficient Drosophila model also found evidence of lysosomal dysfunction, including enlarged lysosomes in GBA-deficient brains [20]. However, our proteomic analysis of Gba1b mutants did not support a profound impairment in autophagy, but instead suggested dysregulation of EVs with altered protein cargo which could be suppressed locally with knockdown of genes encoding ESCRT machinery important for EV biogenesis [23]. Based on these results, we believe that our initial observations of increased insoluble ubiquitinated proteins and Ref(2)P in Gba1b mutants are due to lysosomal stress. One possible explanation for our proteomic findings is that there may be a compensatory increase in EV biogenesis and packaging of autophagy substrates within EVs for discard outside of the cell in Gba1b mutants. Such an increase may have prevented us from detecting defects in autophagy. Upregulation of EV biogenesis may be cell-autonomously neuroprotective in the setting of lysosomal stress, particularly in aggregation-prone neurodegenerative diseases such as PD [44]. It was recently demonstrated in a human neuronal cell culture model of PD that inhibiting macroautophagy protects against α-synuclein-induced cell death by promoting the release of α-synuclein-containing EVs [45]. However, it remains possible that upregulating EV biogenesis may relieve lysosomal stress within cells containing aggregate-prone proteins, while simultaneously promoting the spread of protein aggregates between cells and throughout the organism.
Our work suggests a novel mechanism for GBA in reducing the spread of pathogenic protein aggregation from cell-to-cell via regulation of EV protein cargo, but many key questions remain. To better understand the progression of neurodegenerative diseases, we must uncover the mechanisms by which GCase deficiency alters EV protein content and biogenesis, identify the specific changes in EVs facilitating propagation of pathogenic protein aggregates, and determine how these changes influence recipient cells internalizing dysregulated EVs. GCase is a critical enzyme in ceramide metabolism, hydrolyzing glucosylceramide into glucose and ceramide. Ceramides are a key component of EV membranes, and alterations in ceramide metabolism due to GCase deficiency may directly influence EV biogenesis and protein cargo trafficked via EVs. Further studies using this Drosophila model and mammalian cell culture models should better elucidate how GCase deficiency alters the protein cargo of EVs to induce propagation of pathogenic protein aggregates, as well as whether endogenous GCase is enzymatically functional when trafficked to distant tissues via EVs. Understanding this mechanism could have broad implications in understanding the pathogenesis of aggregate-prone neurodegenerative diseases and reveal new therapeutic targets to slow or halt disease progression.
Materials and methods
Drosophila strains and culture
Fly stocks were maintained on standard cornmeal-molasses food at 25°C. The Gba1b homozygous null mutant (Gba1bΔTT), isogenic control (Gba1b+), and UAS-dGba1b alleles have been previously described [19]. All other strains and alleles were obtained from the Bloomington Stock Center: elav-GAL4 (458); Act88F-GAL4 (38459); DMef-GAL4 (27390); UAS-nSMase-RNAi (36759); Gba1bMB03039(23602). In Figs 4–7, we used the following genotypes for the experiments involving the nSMase-RNAi transgene: control = Gba1b+/Gba1bMB03039; Gba1b mutant = Gba1bΔTT/Gba1bMB03039. This combination of Gba1b mutant alleles, which we used for ease of recombination with the transgene, produce the same biochemical abnormalities found in Gba1bΔTT homozygotes [23].
Lifespan analysis
Longevity assays were conducted at 25°C. Groups of 10–20 age-matched flies were collected at 0–24 hours old and transferred to fresh standard food every 2–3 days. The number of dead flies was recorded during each transfer. Transfers were continued until all flies died. Kaplan-Meier lifespan curves were generated using Stata (StataCorp, College Station, TX), and analyzed by Cox proportional hazard models for statistically significant differences in survival between tested genotypes.
Preparation of Triton-soluble and insoluble fractions
Tissues from 10-day-old flies (6 females and 6 males per sample) were homogenized in Triton lysis buffer (50 mM Tris-HCl pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA), and then spun at 15,000 x g for 20 min. The detergent-soluble supernatant was collected and mixed with an equal volume of 2x Laemmli buffer (4% SDS, 20% glycerol, 120 mM Tris-Cl pH 6.8, 0.02% bromophenol blue, 2% β-mercaptoethanol), and the same buffer was used to resuspend the Triton-insoluble pellet. All samples were boiled for 10 minutes. The Triton-insoluble protein extracts were then cleared of debris by centrifugation at 15,000 x g for 10 minutes, followed by collection of the supernatant. At least three independent experiments were performed.
Western blotting
Proteins were separated by SDS-PAGE on 4%-20% MOPS-acrylamide gels (GenScript Express Plus, M42012) and electrophoretically transferred onto Immobilon PVDF membranes (Fisher, IPVH00010). Immunodetection was performed using the iBind Flex Western Device (Thermo Fisher, SLF2000). Antibodies were used at the following concentrations: 1:10,000 mouse anti-Actin (Chemicon/Bioscience Research Reagents, MAB1501), 1:250 mouse anti-Rab11 (BD Transduction Laboratories, 610657), 1:50 mouse anti-Rab7 (DSHB Rab7), 1:200 rabbit anti-Ref(2)P (Abcam, ab178440), 1:500 mouse anti-ubiquitin (Santa Cruz, sc-8017), 1:10,000 mouse anti-tubulin (DSHB 12G10), 1:1500 rabbit anti-human GBA1 (Sigma, G4171), 1:800 mouse anti-Cnx99A (DSHB, Cnx99A 6-2-1). HRP secondary antibodies were used as follows: 1:500 to 1:1000 anti-mouse (BioRad, 170–6516) and 1:500 to 1:1000 anti-rabbit (BioRad, 172–1019). Signal was detected using Pierce ECL Western Blotting Substrate (Fisher, 32106). Densitometry measurements of the western blot images were performed using Fiji software [46]. (See uncropped western blot images in S4 Data.) For homogenates, signal was normalized either to Actin or Ponceau S [47,48]. Normalized western blot data were log-transformed when necessary to stabilize variance before means were compared using one-way ANOVA and Student t-test. Each experiment was performed at least three times.
Immunohistochemistry
10-day-old adult brains were dissected in cold Schneider’s Drosophila medium (Thermo Fisher, 21720), and fixed in 4% paraformaldehyde/PBS for 30 min. Samples were washed in 0.1% Triton X-100/PBS. Fixed brains were stained with 1:200 mouse anti-poly Ubiquitin FK2 (Enzo, BML-PW8810), then anti-mouse Alexa 488 (1:200) and were mounted using ProLong Gold anti-fade medium (Molecular Probes, P10144).
Extraction of hemolymph and preparation of EV fractions
Hemolymph was obtained from 30 flies (15 males and 15 females, 10 to 11 days old) per sample. All flies were frozen with liquid nitrogen and decapitated by vortexing. Frozen flies were placed in a 1.7-mL tube containing a volume of PBS scaled to the number of flies used (2 μL/fly) and thawed for 5 min at room temperature. The tubes were then centrifuged at 5000 x g for 5 min at 4°C, after which the extracted hemolymph (supernatant) was centrifuged for 30 min at 10,000 x g at 4°C to remove cell debris and the cell-free supernatant was collected. The supernatant was then filtered with Ultrafree 0.22 μm spin filters (Fisher, UFC30GV0S) and centrifuged at 3000 x g for 5 min at 4°C; this was the EV fraction. In order to obtain whole-fly homogenate from the same animals used for collection of hemolymph, the bodies from four flies (2 males and 2 females) were homogenized in RIPA buffer (150mM NaCl, 1% Nonidet P-40, 0.5% Sodium deoxycholate, 0.1% SDS, 50mM Tris pH 8, diluted 1:1 with ddH20), centrifuged at 10,000 x g at 4°C for 5 min, and then the supernatant was transferred to a new tube. An equal volume of 2x Laemmli buffer (4% SDS, 20% glycerol, 120 mM Tris-Cl pH 6.8, 0.02% bromophenol blue, 2% β-mercaptoethanol) was added to the EV fractions and also to the whole-fly protein homogenates, and all samples were boiled for 10 min and then stored at −80°C. The experiment was repeated at least three times.
To determine if proteins were contained within EVs, before adding the Laemmli buffer, the EV fractions were incubated with PBS for 5 min at room temperature and then 0.5 μg/μL Proteinase K was added for 30 min at 4°C to digest external proteins or with 1% sodium dodecyl sulfate (SDS) for 5 min at room temperature and then 0.5 μg/μL Proteinase K was added for 30 min at 4°C to disrupt vesicular membranes and digest proteins. After incubation, Laemmli buffer was added and samples were boiled preparing them for western blotting.
Lipidomic analysis
Fly tissue samples were prepared for LC-MS analysis by adding an equal volume of zirconium oxide beads to each sample of fly tissue (10 male and 10 female whole 10-day old flies). 200 μL water was added to each sample and homogenized using a Next Advance Bullet Blender at 4°C. 100 μL of homogenate was set aside for each sample for protein quantification. 50 μL of the isotope mix (Lipidyzer internal standard 1 μM d9-CER and 0.5 μM d9-HCER), 575 μL methyl tert-butyl ether (MTBE), and 160 μL methanol were added to the remaining 100 ul of each sample. All samples were vortexed and shaken for 30 minutes at room temperature at a 500 RPM setting. An additional 200 μL of water were added. The samples were centrifuged for 3 minutes at 12,000 x g at room temperature. The upper layers were transferred into glass culture tubes using glass pipettes. A second extraction was performed by adding 300 μL MTBE, 100 μL methanol, and 100 μL water. The samples were shaken for 5 minutes and centrifuged at the same settings as before. The upper layers were combined into the respective glass culture tubes and dried under a gentle stream of nitrogen. The samples were reconstituted in 250 μL of mobile phase B (60% acetonitrile, 40% isopropyl alcohol, 0.2% formic acid), and transferred to LC vials with inserts for LC-MS analysis.
Lipids were analyzed using a Shimadzu Nexera X2 HPLC (Shimadzu, Japan) coupled to an AB Sciex QTRAP 5500 hybrid triple quadrupole/linear ion trap mass spectrometer (AB Sciex LLC, USA) operating in positive electrospray ionization (ESI) mode. 39 ceramides and 55 glucosylceramides were monitored by multiple-reaction-mode (MRM) in a single batch run.
Lipidomic data were exported as csv files, grouped according to Drosophila strain and culture, and placed into MetaboAnalyst 4.0 web server (https://www.metaboanalyst.ca) for data processing and statistical analysis [49]. (See all lipidomic data in S3 Data.) Data was normalized by mass per sample, log-transformed and auto-scaled. The data were evaluated by univariate analysis methods (Fold Change analysis, T-tests, and Volcano plot) and multivariate analysis methods (principal component analysis and partial least square–discriminant analysis), and hierarchical clustering analysis. The False Discovery Rate (FDR), based on the Benjamini-Hochberg procedure, was set to <0.1 and used to select important Cer and GlcCer species. The FDR-adjusted p-value of <0.10 was used to control the number of false positives for multiple comparisons.
Supporting information
(A-D) Neutral sphingomyelinase (nSMase)-RNAi was expressed using the flight muscle driver Act88F-GAL4 in Gba1b mutants and wildtype revertant controls. Isolated EVs from these flies were prepared in RIPA buffer. Representative images of (A) Ref(2)P, (B) ubiquitin, (C) Rab11, and (D) Rab7 in the EV fraction are shown.
(TIF)
(A) PCA scores plot of GlcCer species in Gba1b versus control flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2, which account for 99.2% of the variance. (B) PLS-DA scores plot for PC1 and 2 for analysis of GlcCer species in Gba1b versus control flies (R2 = 0.99, Q2 = 0.98, 2 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2. (C) PCA scores plot of GlcCer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2. (D) PLS-DA scores plot for analysis of GlcCer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies (R2 = 0.99, Q2 = 0.80, 2 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2.
(TIF)
(A) Volcano plot of GlcCer species in Gba1b mutants versus controls with fold-change threshold set at 20 (vertical dotted lines) on the x-axis and t-test threshold of 0.05 (horizontal dotted line) on the y-axis. Pink circles represent GlcCer species above both thresholds. Note both fold changes and p-values are log transformed. The further its position away from the (0,0), the more significant the GlcCer species is. (B) Volcano plot with fold change threshold 1.5 on the x-axis and t-test threshold 0.1 on the y-axis. The pink circles represent GlcCer species above the threshold. Note both fold changes and p values are log transformed.
(TIF)
(A) PCA scores plot of Cer species in Gba1b versus control flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2, which account for 99.2% of the variance. (B) PLS-DA scores plot for analysis of Cer species in Gba1b versus control flies (R2 = 0.99, Q2 = 0.80, 3 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2. (C) PCA scores plot of Cer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2. (D) PLS-DA scores plot for analysis of GlcCer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies (R2 = 0.99, Q2 = 0.70, 2 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2.
(TIF)
(A) Volcano plot of Cer species in Gba1b mutants versus controls with fold-change threshold 1.5 on the x-axis and t-tests threshold 0.1 on the y-axis. Pink circles represent Cer species above both thresholds. Note both fold changes and p values are log transformed. The further its position away from the (0,0), the more significant the Cer species is. (B) Volcano plot with fold change threshold 1.5 on the x-axis and t-test threshold 0.1 on the y-axis. Pink circles represent Cer species above both thresholds. Note both fold changes and p values are log transformed. The further its position away from the (0,0), the more significant the feature is.
(TIF)
(TIF)
Using Act88F-GAL4, mutant human GBA (hGBAN370S) was expressed in Gba1b mutant and WT revertant controls. Homogenates were prepared from fly heads and thoraces using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin. Representative images of ubiquitin in (A) thoraces and (B) heads of Gba1b mutant flies with and without muscle expression of hGBAN370S are shown.
(TIF)
(A) Using the midgut driver Npc1b-GAL4, wildtype dGba1b was expressed in Gba1b mutants and wildtype revertant controls. Homogenates were prepared from fly heads using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin. Quantification of Ub in (A) heads of control and Gba1b mutants with and without midgut expression of dGba1b are shown. (B) Quantification of Ub in the heads of flies with and without dGba1b expression in fat body using the Lsp2-GAL4 driver. Results are normalized to Actin and control. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test.
(TIF)
(PDF)
(PDF)
(XLSX)
(XLSX)
(XLSX)
(PDF)
Acknowledgments
Kim Miller (Digital Microscopy Center, Virginia Merrill Bloedel Hearing Research Center, University of Washington) for technical assistance with confocal imaging and analysis; Northwest Metabolomics Research Center for developing a method specifically detecting Drosophila ceramides and analytical assistance with lipidomic analysis; Biorender for Drosophila icon figures; Raja Estes, Emma Thuline, Nathan Johnston, and Kim Kramers for technical assistance; and Evelyn S. Vincow, Colby L. Samstag, and all members of the Pallanck and Davis labs for critical review and discussion of this work and manuscript.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was generously supported by grants to LJP from the National Institutes of Health (NIH) (R01NS094252 and R01AG060963; www.nih.gov) and by a VA Career Development Award-2 to MYD from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Service (IK2 BX003244; www.va.gov). KAJ was supported by the NIH/National Institute on Aging Genetic Approaches to Aging Training Grant (T32 AG000057; www.nia.nih.gov). Development of lipidomic methodology at Northwest Metabolomics Research Center was supported by NIH grant #1S10OD021562-01; www.nih.gov). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
References
- 1.Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. European journal of epidemiology. 2011;26 Suppl 1:S1–58. Epub 2011/06/03. 10.1007/s10654-011-9581-6 . [DOI] [PubMed] [Google Scholar]
- 2.Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet. 2004;363(9423):1783–93. 10.1016/S0140-6736(04)16305-8 . [DOI] [PubMed] [Google Scholar]
- 3.Parkinson J. An essay on the shaking palsy. 1817. The Journal of neuropsychiatry and clinical neurosciences. 2002;14(2):223–36; discussion 2. 10.1176/jnp.14.2.223 . [DOI] [PubMed] [Google Scholar]
- 4.Ziemssen T, Reichmann H. Non-motor dysfunction in Parkinson’s disease. Parkinsonism & related disorders. 2007;13(6):323–32. 10.1016/j.parkreldis.2006.12.014 . [DOI] [PubMed] [Google Scholar]
- 5.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. 10.1016/s0197-4580(02)00065-9 . [DOI] [PubMed] [Google Scholar]
- 6.Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338(6109):949–53. Epub 2012/11/20. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, et al. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med. 2016;213(9):1759–78. Epub 2016/08/10. 10.1084/jem.20160368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu Y, Muller S, Tavares A, Barret O, Alagille D, Seibyl J, et al. Intrastriatal alpha-synuclein fibrils in monkeys: spreading, imaging and neuropathological changes. Brain. 2019;142(11):3565–79. Epub 2019/10/04. 10.1093/brain/awz296 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sidransky E, Samaddar T, Tayebi N. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology. 2009;73(17):1424–5, author reply 5–6. Epub 2009/10/28. 10.1212/WNL.0b013e3181b28601 . [DOI] [PubMed] [Google Scholar]
- 10.Pankratz N, Beecham GW, DeStefano AL, Dawson TM, Doheny KF, Factor SA, et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann Neurol. 2012;71(3):370–84. Epub 2012/03/28. 10.1002/ana.22687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology. 2012;79(19):1944–50. Epub 2012/10/05. 10.1212/WNL.0b013e3182735e9a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tayebi N, Callahan M, Madike V, Stubblefield BK, Orvisky E, Krasnewich D, et al. Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Molecular genetics and metabolism. 2001;73(4):313–21. 10.1006/mgme.2001.3201 . [DOI] [PubMed] [Google Scholar]
- 13.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361(17):1651–61. 10.1056/NEJMoa0901281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark LN, Kartsaklis LA, Wolf Gilbert R, Dorado B, Ross BM, Kisselev S, et al. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch Neurol. 2009;66(5):578–83. Epub 2009/05/13. 10.1001/archneurol.2009.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology. 2008;70(24):2277–83. 10.1212/01.wnl.0000304039.11891.29 . [DOI] [PubMed] [Google Scholar]
- 16.Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, et al. GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord. 2015;30(3):407–11. Epub 2014/12/03. 10.1002/mds.26071 . [DOI] [PubMed] [Google Scholar]
- 17.Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiol Aging. 2016;37:209 e1- e7. Epub 2015/11/26. 10.1016/j.neurobiolaging.2015.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016;73(10):1217–1224. Epub 2016/08/30. 10.1001/jamaneurol.2016.2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis MY, Trinh K, Thomas RE, Yu S, Germanos AA, Whitley BN, et al. Glucocerebrosidase Deficiency in Drosophila Results in alpha-Synuclein-Independent Protein Aggregation and Neurodegeneration. PLoS Genet. 2016;12(3):e1005944 Epub 2016/03/29. 10.1371/journal.pgen.1005944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kinghorn KJ, Gronke S, Castillo-Quan JI, Woodling NS, Li L, Sirka E, et al. A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J Neurosci. 2016;36(46):11654–70. Epub 2016/11/18. 10.1523/JNEUROSCI.4527-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawasaki H, Suzuki T, Ito K, Takahara T, Goto-Inoue N, Setou M, et al. Minos-insertion mutant of the Drosophila GBA gene homologue showed abnormal phenotypes of climbing ability, sleep and life span with accumulation of hydroxy-glucocerebroside. Gene. 2017;614:49–55. Epub 2017/03/14. 10.1016/j.gene.2017.03.004 . [DOI] [PubMed] [Google Scholar]
- 22.Maor G, Cabasso O, Krivoruk O, Rodriguez J, Steller H, Segal D, et al. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum Mol Genet. 2016;25(13):2712–27. Epub 2016/05/11. 10.1093/hmg/ddw129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas RE, Vincow ES, Merrihew GE, MacCoss MJ, Davis MY, Pallanck LJ. Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles. PLoS Genet. 2018;14(9):e1007694 Epub 2018/09/27. 10.1371/journal.pgen.1007694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–89. Epub 2014/10/08. 10.1146/annurev-cellbio-101512-122326 . [DOI] [PubMed] [Google Scholar]
- 25.van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–28. Epub 2018/01/18. 10.1038/nrm.2017.125 . [DOI] [PubMed] [Google Scholar]
- 26.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–83. Epub 2013/02/20. 10.1083/jcb.201211138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hessvik NP, Llorente A. Current knowledge on exosome biogenesis and release. Cell Mol Life Sci. 2018;75(2):193–208. Epub 2017/07/25. 10.1007/s00018-017-2595-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, Danzer KM, et al. Induction of alpha-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain. 2016;139(Pt 2):481–94. Epub 2015/12/10. 10.1093/brain/awv346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao T, Zhang W, Jiao B, Pan CZ, Liu X, Shen L. The role of exosomes in the pathogenesis of Alzheimer’ disease. Transl Neurodegener. 2017;6:3 Epub 2017/02/12. 10.1186/s40035-017-0072-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia L, Qiu Q, Zhang H, Chu L, Du Y, Zhang J, et al. Concordance between the assessment of Abeta42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019;15(8):1071–80. Epub 2019/08/20. 10.1016/j.jalz.2019.05.002 . [DOI] [PubMed] [Google Scholar]
- 31.Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem. 2013;288(22):15699–711. Epub 2013/04/18. 10.1074/jbc.M112.425066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101(26):9683–8. Epub 2004/06/24. 10.1073/pnas.0308413101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robertson C, Booth SA, Beniac DR, Coulthart MB, Booth TF, McNicol A. Cellular prion protein is released on exosomes from activated platelets. Blood. 2006;107(10):3907–11. Epub 2006/01/26. 10.1182/blood-2005-02-0802 . [DOI] [PubMed] [Google Scholar]
- 34.Minakaki G, Menges S, Kittel A, Emmanouilidou E, Schaeffner I, Barkovits K, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14(1):98–119. Epub 2017/12/05. 10.1080/15548627.2017.1395992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42 10.1186/1750-1326-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nongthomba U, Pasalodos-Sanchez S, Clark S, Clayton JD, Sparrow JC. Expression and function of the Drosophila ACT88F actin isoform is not restricted to the indirect flight muscles. J Muscle Res Cell Motil. 2001;22(2):111–9. Epub 2001/08/25. 10.1023/a:1010308326890 . [DOI] [PubMed] [Google Scholar]
- 37.Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180(6):1065–71. Epub 2008/03/19. 10.1083/jcb.200711108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319(5867):1244–7. Epub 2008/03/01. 10.1126/science.1153124 . [DOI] [PubMed] [Google Scholar]
- 39.Guo BB, Bellingham SA, Hill AF. The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J Biol Chem. 2015;290(6):3455–67. Epub 2014/12/17. 10.1074/jbc.M114.605253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shamseddine AA, Airola MV, Hannun YA. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv Biol Regul. 2015;57:24–41. Epub 2014/12/04. 10.1016/j.jbior.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skotland T, Sandvig K, Llorente A. Lipids in exosomes: Current knowledge and the way forward. Prog Lipid Res. 2017;66:30–41. Epub 2017/03/28. 10.1016/j.plipres.2017.03.001 . [DOI] [PubMed] [Google Scholar]
- 42.Sandhof CA, Hoppe SO, Druffel-Augustin S, Gallrein C, Kirstein J, Voisine C, et al. Reducing INS-IGF1 signaling protects against non-cell autonomous vesicle rupture caused by SNCA spreading. Autophagy. 2019:1–22. Epub 2019/07/30. 10.1080/15548627.2019.1643657 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143(5):813–25. Epub 2010/11/30. 10.1016/j.cell.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mathews PM, Levy E. Exosome Production Is Key to Neuronal Endosomal Pathway Integrity in Neurodegenerative Diseases. Front Neurosci. 2019;13:1347 Epub 2020/01/09. 10.3389/fnins.2019.01347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fussi N, Hollerhage M, Chakroun T, Nykanen NP, Rosler TW, Koeglsperger T, et al. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018;9(7):757 Epub 2018/07/11. 10.1038/s41419-018-0816-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gross JC, Chaudhary V, Bartscherer K, Boutros M. Active Wnt proteins are secreted on exosomes. Nat Cell Biol. 2012;14(10):1036–45. Epub 2012/09/18. 10.1038/ncb2574 . [DOI] [PubMed] [Google Scholar]
- 47.Romero-Calvo I, Ocon B, Martinez-Moya P, Suarez MD, Zarzuelo A, Martinez-Augustin O, et al. Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal Biochem. 2010;401(2):318–20. Epub 2010/03/09. 10.1016/j.ab.2010.02.036 . [DOI] [PubMed] [Google Scholar]
- 48.Thacker JS, Yeung DH, Staines WR, Mielke JG. Total protein or high-abundance protein: Which offers the best loading control for Western blotting? Anal Biochem. 2016;496:76–8. Epub 2015/12/27. 10.1016/j.ab.2015.11.022 . [DOI] [PubMed] [Google Scholar]
- 49.Xia J, Wishart DS. Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr Protoc Bioinformatics. 2016;55:14 0 1–0 91. Epub 2016/09/08. 10.1002/cpbi.11 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A-D) Neutral sphingomyelinase (nSMase)-RNAi was expressed using the flight muscle driver Act88F-GAL4 in Gba1b mutants and wildtype revertant controls. Isolated EVs from these flies were prepared in RIPA buffer. Representative images of (A) Ref(2)P, (B) ubiquitin, (C) Rab11, and (D) Rab7 in the EV fraction are shown.
(TIF)
(A) PCA scores plot of GlcCer species in Gba1b versus control flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2, which account for 99.2% of the variance. (B) PLS-DA scores plot for PC1 and 2 for analysis of GlcCer species in Gba1b versus control flies (R2 = 0.99, Q2 = 0.98, 2 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2. (C) PCA scores plot of GlcCer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2. (D) PLS-DA scores plot for analysis of GlcCer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies (R2 = 0.99, Q2 = 0.80, 2 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2.
(TIF)
(A) Volcano plot of GlcCer species in Gba1b mutants versus controls with fold-change threshold set at 20 (vertical dotted lines) on the x-axis and t-test threshold of 0.05 (horizontal dotted line) on the y-axis. Pink circles represent GlcCer species above both thresholds. Note both fold changes and p-values are log transformed. The further its position away from the (0,0), the more significant the GlcCer species is. (B) Volcano plot with fold change threshold 1.5 on the x-axis and t-test threshold 0.1 on the y-axis. The pink circles represent GlcCer species above the threshold. Note both fold changes and p values are log transformed.
(TIF)
(A) PCA scores plot of Cer species in Gba1b versus control flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2, which account for 99.2% of the variance. (B) PLS-DA scores plot for analysis of Cer species in Gba1b versus control flies (R2 = 0.99, Q2 = 0.80, 3 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2. (C) PCA scores plot of Cer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies. Ovals indicate 95% confidence region. Refer to S1 Data for significant compounds (in bold) contributing to PC1 and PC2. (D) PLS-DA scores plot for analysis of GlcCer species in Gba1b; Act88F-GAL4>nSMase-RNAi versus Gba1b flies (R2 = 0.99, Q2 = 0.70, 2 components). Ovals indicate 95% confidence region. Refer to S2 Data for significant compounds (in bold) contributing to PC1 and PC2.
(TIF)
(A) Volcano plot of Cer species in Gba1b mutants versus controls with fold-change threshold 1.5 on the x-axis and t-tests threshold 0.1 on the y-axis. Pink circles represent Cer species above both thresholds. Note both fold changes and p values are log transformed. The further its position away from the (0,0), the more significant the Cer species is. (B) Volcano plot with fold change threshold 1.5 on the x-axis and t-test threshold 0.1 on the y-axis. Pink circles represent Cer species above both thresholds. Note both fold changes and p values are log transformed. The further its position away from the (0,0), the more significant the feature is.
(TIF)
(TIF)
Using Act88F-GAL4, mutant human GBA (hGBAN370S) was expressed in Gba1b mutant and WT revertant controls. Homogenates were prepared from fly heads and thoraces using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin. Representative images of ubiquitin in (A) thoraces and (B) heads of Gba1b mutant flies with and without muscle expression of hGBAN370S are shown.
(TIF)
(A) Using the midgut driver Npc1b-GAL4, wildtype dGba1b was expressed in Gba1b mutants and wildtype revertant controls. Homogenates were prepared from fly heads using 1% Triton X-100 lysis buffer. Western blot analysis was performed on the Triton X-100 insoluble proteins using antibodies to ubiquitin (Ub) and Actin. Quantification of Ub in (A) heads of control and Gba1b mutants with and without midgut expression of dGba1b are shown. (B) Quantification of Ub in the heads of flies with and without dGba1b expression in fat body using the Lsp2-GAL4 driver. Results are normalized to Actin and control. At least 3 independent experiments were performed. Error bars represent SEM. *p < 0.05 by Student t-test.
(TIF)
(PDF)
(PDF)
(XLSX)
(XLSX)
(XLSX)
(PDF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.