

ABSTRACT
Poison frogs sequester chemical defenses from their diet of leaf litter arthropods for defense against predation. Little is known about the physiological adaptations that confer this unusual bioaccumulation ability. We conducted an alkaloid-feeding experiment with the Diablito poison frog (Oophaga sylvatica) to determine how quickly alkaloids are accumulated and how toxins modify frog physiology using quantitative proteomics. Diablito frogs rapidly accumulated the alkaloid decahydroquinoline within 4 days, and dietary alkaloid exposure altered protein abundance in the intestines, liver and skin. Many proteins that increased in abundance with decahydroquinoline accumulation are plasma glycoproteins, including the complement system and the toxin-binding protein saxiphilin. Other protein classes that change in abundance with decahydroquinoline accumulation are membrane proteins involved in small molecule transport and metabolism. Overall, this work shows that poison frogs can rapidly accumulate alkaloids, which alter carrier protein abundance, initiate an immune response, and alter small molecule transport and metabolism dynamics across tissues.
KEY WORDS: Alkaloids, Proteomics, Saxiphilin, Complement system, Cytochrome P450s
Summary: Poison frogs rapidly accumulate toxins, which changes the abundance of proteins involved in the immune system and small molecule binding and metabolism across tissues.
INTRODUCTION
Many organisms can acquire chemical defenses from external sources. These acquired chemical defenses are sometimes diet-derived and many examples can be found in invertebrates (Opitz and Müller, 2009) and less commonly in vertebrates (Savitzky et al., 2012). Poison frogs sequester lipophilic alkaloids from arthropod prey (Saporito et al., 2009), which requires specialized physiology for bioaccumulation and toxin resistance (Santos et al., 2016). The bioaccumulation of alkaloids requires coordination between several tissue systems, such as absorption in the gut, transport between tissues and storage in skin granular glands. Although sequestering defensive chemicals involves a well-studied ecological relationship between arthropod prey and frog predators (Darst et al., 2005; Saporito et al., 2011), how poison frogs sequester alkaloids is unknown.
The chemical defenses of poison frogs are mainly lipophilic alkaloids and over 800 have been characterized in poisonous amphibians (Daly et al., 2005). There is exceptional species diversity in chemical defenses, particularly in South American dendrobatid poison frogs (Saporito et al., 2011). The ability to sequester alkaloids evolved several times in dendrobatids and coincides with a dietary specialization on ants and mites (Darst et al., 2005; Santos et al., 2003). The dietary hypothesis of poison frog toxicity arose when it was noted that poison frogs reared in captivity lacked alkaloids (e.g. Daly et al., 1994a), leading to examination of their alkaloid uptake (Daly et al., 1994b; Hantak et al., 2013). Research over several decades has focused on alkaloid characterization (Saporito et al., 2011), but how poison frogs accumulate toxins remains largely unexplored.
Although alkaloids are lipophilic, these small molecules do not simply perfuse passively throughout the organism. Evidence for an uptake system in poison frogs comes from alkaloid quantification in various tissues, including the liver, muscles and oocytes (Prates et al., 2012; Stynoski et al., 2014), although alkaloids are most abundant in the skin where they are stored in granular glands (Neuwirth et al., 1979). Moreover, the vertebrate intestinal lining is well-equipped to prevent passive absorption of toxic substances using a series of membrane transporters (Zhang and Benet, 2001). Absorbed lipophilic molecules are either transported through the lymph system by chylomicrons or through the blood circulation bound to carrier proteins produced by the liver. The molecular physiology of alkaloid bioaccumulation, metabolism and transport between each of these poison frog tissue systems is unknown.
We conducted an experiment to determine how alkaloid sequestration alters frog physiology and tested the general hypothesis that alkaloid bioaccumulation alters the abundance of proteins involved in small molecule transport and metabolism. We used the Diablito frog (Oophaga sylvatica) to address this question, which complements our previous studies examining the chemical ecology of this species (Caty et al., 2019; McGugan et al., 2016; Roland et al., 2016). Frogs were fed the alkaloid decahydroquinoline (DHQ), a commercially available alkaloid they naturally sequester in the wild (Caty et al., 2019), and we examined how alkaloid sequestration influences frog physiology using quantitative proteomics. Our experiments show that poison frogs sequester alkaloids within 4 days of exposure, shifting protein abundance across tissues, including plasma glycoproteins and cytochrome P450s.
MATERIALS AND METHODS
Animals
Adult Diablito frogs [Oophaga sylvatica (Funkhouser 1956), N=12, 5 females, all of unknown age] were purchased from Indoor Ecosystems (Whitehouse, OH, USA) and housed for 3 months prior to the experiment. Frogs were housed individually on a 12 h:12 h light:dark photoperiod, immediately preceded and followed by a 10 min period of dim incandescent lights to simulate dawn and dusk. Frogs were misted with water three times daily and fed four times per week with fruit flies dusted with a vitamin supplement. The Institutional Animal Care and Use Committee of Harvard University approved all procedures (Protocols 15-02-233 and 15-07-247).
Decahydroquinoline feeding experiment
Frogs were randomly assigned to one of four groups to total N=3 per group. Within-group variation was unknown during experimental design and an N=3 is standard in the quantitative proteomics field. The three experimental groups received fruit flies dusted with a 1% decahydroquinoline (DHQ; Sigma-Aldrich, St Louis, MO, USA) in a vitamin mix (Dendrocare vitamin/mineral powder, Black Jungle, Turners Falls, MA, USA) for 1, 4 or 18 days (Fig. 1A). DHQ-like alkaloids are sequestered by O. sylvatica in the wild (Caty et al., 2019) and represent an ecologically relevant treatment. Control frogs (N=3) received flies dusted with vitamin mix without DHQ. For the 1-day group, frogs received toxin-dusted flies in the morning, and then were killed the following day after receiving another dose of flies that morning (a total of 2 feedings in 36 h). Frogs were anesthetized by applying 20% benzocaine gel to the ventral side and then euthanized by decapitation. For the 4-day group, frogs were fed toxic flies on Monday, Tuesday, Thursday and Friday mornings and were killed on Friday afternoon. For the 18-day group, frogs were fed toxic flies 4 days per week for 3 weeks for a total of 12 feedings. Approximately 15–25 flies (mean±s.d.=18±4.8) flies were given to the frogs at each feeding session and the number of flies remaining the next morning were recorded. On the afternoon of the last day, the frogs were euthanized. The mean±s.d. total number of alkaloid-dusted flies eaten by each group during the duration of the experiment was 14.3±2.5 for 1-day, 52.3±4.9 for 4-day and 149.7±8.2 for 18-day frogs. As DHQ was administered by dusting prey items, and since individual frogs ate a variable number of prey, we were unable to calculate the exact DHQ dose for each individual. Dorsal skin, liver and intestines were dissected, flash frozen in liquid nitrogen and stored at −80°C for later processing.
Fig. 1.

Rapid uptake of decahydroquinoline (DHQ) in the Diablito poison frog (Oophaga sylvatica). (A) Frogs were fed DHQ-dusted flies to determine the rate of toxin bioaccumulation (N=3 per group). The timeline shows the number of times that frogs were fed with toxic flies and the endpoints. This experiment was performed once in a teaching laboratory by undergraduate students. (B) Tissues were dissected and both protein and DHQ were isolated using methanol–chloroform precipitation. (C) DHQ abundance (y-axis, log scale) in the intestine (light green), liver (cream) and skin (light orange) increases over time, with the first detectable amounts within 4 days. Data are represented in boxplots that show rectangles as the lower and upper quartiles (with the median as the line) and whiskers that indicate the maximum and minimum values. A generalized linear mixed model showed a significant effect of group (P=5.163×10−5) and tissue (P=0.001). N=9 samples per group; n=3 samples per tissue.
To isolate proteins and alkaloids from the same tissue samples, flash frozen tissues were removed from −80°C and immediately crushed to a powder in liquid nitrogen using a mortar and pestle. Tissue powder was then resuspended in a lysis buffer from the Mammalian Cell Lysis kit containing a protease inhibitor cocktail (Sigma-Aldrich). Samples were incubated in lysis buffer at 4°C for 15 min rotating and then centrifuged at 20,000 g for 10 min at 4°C to pellet cell debris. The supernatant was transferred to a chilled microcentrifuge tube and protein concentration was determined using 2 μl of lysate in a bicinchoninic acid (BCA) assay according to the manufacturer's instructions (Thermo Scientific, Waltham, MA, USA). To standardize the amount of input material in downstream assays, lysate containing 300 μg protein from each tissue type was transferred to a chilled microfuge tube for protein and alkaloid (DHQ) extraction. A chloroform–methanol extraction was used to separate the DHQ and proteins from the same tissue lysate sample. Briefly, 400 μl of 100% methanol was added to each sample and then each sample was vortexed. Then 100 μl chloroform was added to each sample, which was immediately vortexed. To precipitate protein, 200 μl nuclease-free water was added to each sample. After vortexing, samples were centrifuged at 21,000 g for 3 min at room temperature, resulting in a protein disk at the interface of the aqueous methanol top layer and the organic chloroform bottom layer. A previous pilot experiment showed roughly 90% of DHQ spiked into a protein lysate sample separated into the chloroform layer. Therefore, the methanol layer was removed and the chloroform layer was isolated into a glass vial and stored at −20°C for later quantification of DHQ. The remaining protein pellet was rinsed twice with 1 ml methanol and then stored at −80°C for further processing.
Quantification of DHQ
Liquid chromatography/mass spectrometry (LC/MS) quantification of DHQ was performed on a Bruker maXis Impact Q-TOF system (Billerica, MA) with an Agilent 1290 LC (Palo Alto, CA). A reversed-phase LC gradient method was used with a Dikma C8, 3 μm particle size, 2.1×100 mm column (Dikma, Lake Forest, CA). Mobile phase A was composed of water with 0.1% formic acid and mobile phase B was composed of acetonitrile with 0.1% formic acid. The flow rate was 0.3 ml min−1. The gradient began with 0% B for 2 min then increased linearly to 100% B at 8 min and was held until 10 min. The solvent composition was then returned to starting aqueous conditions to re-equilibrate the column for the next injection. The mass spectrometer was tuned for standard mass range analysis and data were continually acquired in the range m/z 50–3000; each run was recalibrated for the m/z scale using a post-run injection of a sodium formate solution. Electrospray positive mode ionization was used with a source drying gas of 10 liters min−1 at 200°C, nebulizer at 30 p.s.i., and capillary set at 4000 V with an endplate offset of 500 V. For the Agilent Q-TOF, the Ion Funnel electrospray positive mode source used drying gas of 14 liters min−1 at 200°C with nebulizer at 35 psi, a sheath gas flow of 11 liters min−1 at 350°C, capillary set at 3500 V, nozzle voltage of 1000 V, and Fragmentor set at 175 V. Collision energies were set at 15 and 30 eV and data were continually acquired in the range m/z 50−1700 using a reference lock mass. A DHQ standard was run along with the experimental samples to identify the correct peak and the structure was confirmed by its fragmentation pattern using tandem mass spectrometry (Fig. S1).
Quantitative proteomics
Protein disks were thawed and resuspended in 10 µl acetonitrile with gentle agitation to break up the disk, followed by addition of 90 µl of 8 mol l−1 urea in 100 mmol l−1 EPPS pH 8.5 with another round of gentle vortexing. Samples were centrifuged at 13,000 g for 30 s, and sonicated in a water bath for 20 min at room temperature to complete resuspension. Proteins disulfide bonds were reduced with 5 mmol l−1 tris-(2-carboxyethyl)-phosphine (TCEP), at room temperature for 25 min, and alkylated with 10 mmol l−1 iodoacetamide at room temperature for 30 min in the dark. Excess iodoacetamide was quenched with 15 mmol l−1 dithiothreitol at room temperature for 15 min in the dark. Then a methanol–chloroform precipitation was performed again as described above. Protein disks were resuspended in 10 μl of 8 mol l−1 urea, 200 mmol l−1 EPPS pH 8.5 with gentle vortexing followed by the addition of 90 μl of 200 mmol l−1 EPPS pH 8.5 to reach 100 µl and dilute to 0.8 mol l−1 urea. Proteins were then digested at room temperature with gentle agitation for 12 h with LysC protease at a 100:1 protein to protease ratio. Then, trypsin was added at a 100:1 protein to protease ratio and the reaction temperature raised to 37°C for 6 h. Digests were centrifuged at 10,000 g for 10 min and the supernatant transferred to a new tube. Peptide concentrations were measured using the Quantitative Colorimetric Peptide assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Following quantification, 50 µg peptides from each sample were labelled with 5 µl Tandem Mass Tag 10-plex (TMT, Thermo Fisher Scientific) reagent (0.02 mg µl−1) in 200 mmol l−1 EPPS pH 8.5 at 30% acetonitrile (v/v) for 1 h before quenching the reaction with 10 µl of 5% hydroxylamine. A pool of each control sample was labelled collectively to create a bridge in an additional channel.
The TMT-labeled samples from each tissue from a given collection were then pooled in equimolar ratios, as verified by an MS2-only ratio check of an aliquot. The pooled sample was vacuum centrifuged to near dryness before resuspension in bicarbonate buffer, and separation by basic pH RP HPLC on an Agilent 1100 pump equipped with a degasser and a photodiode array (PDA) detector measuring at 220 and 280 nm wavelengths (Thermo Fisher Scientific). Peptides were subjected to a 50 min linear gradient from 8% to 80% acetonitrile in 10 mmol l−1 ammonium bicarbonate pH 8 at a flow rate of 0.6 ml min−1 over an Agilent 300Extend C18 column for 75 min (3.5 μm particles, 4.6 mm ID and 250 mm in length) to produce 96 equal volume fractions. The fractions were re-pooled down to 24 fractions, and vacuum centrifuged to reduce volume. The pooled fractions were acidified and then desalted via StageTip, vacuum centrifuged to near dryness, and reconstituted in 5% acetonitrile, 5% formic acid for injection into an Orbitrap Fusion Lumos. Twelve non-adjacent pooled fractions were analyzed via mass spectrometry.
Each fraction was separated along a 4–30% acetonitrile linear gradient over 3 h, and ionized via electrospray for analysis by mass spectrometry using a TOP10 method, where each FTMS1 scan was used to select up to 10 MS2 precursors for CID-MS2 followed by measurement in the ion trap. Each MS2 was used to select precursors (SPS ions) for the MS3 scan which measured reporter ion abundance for the 10 samples simultaneously (McAlister et al., 2014). Instrument parameter settings included an FTMS1 resolution of 120,000, ITMS2 isolation window of 0.4 m/z, 120 ms ITMS2 max ion time, ITMS2 AGC set to 2E4, ITMS2 CID energy of 35%, SPS ion count allowing up to 10 precursors, FTMS3 isolation window of 1.2 m/z, 150 ms FTMS3 max ion time, FTMS3 AGC set to 1.5E5, and FTMS3 resolution of 50,000.
Samples were searched with the Sequest algorithm (v.28) against a custom O. sylvatica proteome database. There are no high-quality genomes available for poison frogs and thus we are limited to transcriptome-based references. We confirmed this transcriptome-based method was the best approach by comparing the number of unique peptides that matched the O. sylvatica transcriptome-derived proteome compared with Xenopus and human reference proteomes (Fig. S2). All subsequent analyses were performed with a database generated from an O. sylvatica transcriptome (Caty et al., 2019) using the PHROG workflow (proteomic reference with heterogeneous RNA omitting the genome; Wühr et al., 2014), which was concatenated with their reversed sequences as decoys for FDR determination. Common contaminant sequences were similarly included. For searches, we restricted the precursor ion tolerance to 50 p.p.m., and set the product ion tolerance window to 0.9 m/z, allowed up to two missed cleavages, included static medication of lysine residues and peptide N-termini with TMT tags (+229.163 Da), static carbamidomethylation of cysteine residues (+57.021 Da), and variable oxidation of methionine residues (+15.995 Da). The search results were combined and filtered to a 1% FDR at the peptide and protein levels using linear discriminant analysis and the target-decoy strategy (Elias and Gygi, 2010; Huttlin et al., 2010). MS3 spectra were processed as signal-to-noise ratios for each reporter ion based on noise levels within a 25 Thomson window. Protein quantitation was the result of summing reporter ion intensities across all matching PSMs. PSMs with isolation specificity below 0.7, MS3 spectra with more than 8 TMT reporter ion channels missing, or MS3 spectra with a TMT reporter ion summed signal to noise ratio that is less than 200 were removed before analysis. Overall, several thousand proteins were detected in each tissue type (5328 in the intestine, 5837 in the liver and 5987 in the skin).
Data analysis
All statistics and figures were generated in R Studio (v.1.1.442) running R (v.3.5.2; https://www.r-project.org/). For DHQ abundance, the integrated area under the peak from the ion chromatograph was used for sample quantification compared to the standard and values are presented in arbitrary units. We used the glmmTMB R package (https://cran.r-project.org/web/packages/glmmTMB) to run a generalized linear mixed model with a zero-inflated negative binomial distribution to test for significant differences in DHQ quantity between groups and tissue type as main effects. Frog identity was included as a random effect to account for the sampling of three tissues (intestine, liver and skin) for each frog. We then followed the model with the Anova.glmmTMB function for reported statistical values. Boxplots were generated with the ggplot function in the ggplot2 package v.3.0 in R (https://cran.r-project.org/web/packages/ggplot2).
For the proteomics data, protein contigs with fewer than two peptides were removed from the analysis, leaving 3715 protein contigs in the intestines (69.7%), 3789 in the liver (64.9%) and 3755 in the skin (62.7%). We combined the day 4 and day 18 animals with DHQ in our proteomics analysis, totalling N=6 in the ‘toxic’ group and N=3 in the ‘non-toxic’ control group. Day 1 was not included as we did not detect any DHQ in those samples and tandem mass tags for labeling was, at the time, limited to 10 tags. We used the eBayes() function in the R limma package (Ritchie et al., 2015) (v.3.36.5; https://bioconductor.org/packages/limma/) to determine significant differences between groups in each tissue (D'Angelo et al., 2017). Tissues were analysed separately as they were TMT-labeled and run separately on the mass spectrometer. We considered protein contigs significantly different if they had a moderated P-value (adjusted for multiple testing) of P<0.05 and a log fold change greater than 1. Volcano plots illustrating these cut-offs were generated with the EnhancedVolcano package v.1.4.0 (https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html). We used a principal component analysis (PCA) to characterize variance across all proteins using the prcomp function in the R base package. We tested for group differences in principal components using the aov function in the R base package and generated PCA plots using the s.class function in the ade4 package v.1.7-15 (https://cran.r-project.org/web/packages/ade4). Heatmaps for each tissue were generated with the heatmap.2 function of the gplots package v.3.0.3 (https://cran.r-project.org/web/packages/gplots/). Gene ontology enrichment analyses were performed with the R package topGO v.2.34.0 (https://bioconductor.org/packages/release/bioc/html/topGO.html) with significance set at P<0.05. Boxplots were generated with the ggplot function in the ggplot2 package in R.
RESULTS AND DISCUSSION
Poison frogs rapidly sequester toxins
We fed decahydroquinoline (DHQ) or vehicle control to non-toxic Diablito (Oophaga sylvatica) poison frogs for 1, 4 or 18 days (Fig. 1A) and quantified DHQ abundance across multiple tissues (Fig. 1B). Although DHQ was not detectable 1 day after exposure, DHQ levels increased after 4 and 18 days in all tissues (Fig. 1C). The abundance of DHQ varied across frog groups sampled at different times after the onset of toxin feeding (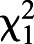 =16.387, P=5.163×10−5). DHQ abundance was also different across tissues (
=16.387, P=5.163×10−5). DHQ abundance was also different across tissues (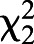 =10.331, P=0.001), with the highest amounts in the skin, where alkaloids are stored in granular glands. Uptake within 4 days is faster than is reported in previous studies that documented skin alkaloid uptake after 3 months in bufonids (Hantak et al., 2013) and in 2–6 weeks in other dendrobatid species (Daly et al., 2003). We did not detect DHQ after 1 day, suggesting either DHQ did not reach the intestine by that time or abundance was below the limit of detection. Rapid uptake of dietary alkaloid toxins has important ecological implications, as acute shifts in arthropod diet could quickly contribute to the poison frog chemical repertoire and alter palatability to predators.
=10.331, P=0.001), with the highest amounts in the skin, where alkaloids are stored in granular glands. Uptake within 4 days is faster than is reported in previous studies that documented skin alkaloid uptake after 3 months in bufonids (Hantak et al., 2013) and in 2–6 weeks in other dendrobatid species (Daly et al., 2003). We did not detect DHQ after 1 day, suggesting either DHQ did not reach the intestine by that time or abundance was below the limit of detection. Rapid uptake of dietary alkaloid toxins has important ecological implications, as acute shifts in arthropod diet could quickly contribute to the poison frog chemical repertoire and alter palatability to predators.
Decahydroquinoline exposure induces proteome shifts across tissues
As poison frogs rapidly uptake toxins, we then asked how frog physiology changes with DHQ uptake by examining protein abundance differences between control frogs and chemically defended frogs using quantitative proteomics (Fig. 2, Table S1, Figs S3–S5). We used principal component analysis to visualize overall protein variation between groups in each tissue (Figs S3–S5). In the intestines and skin, principal component (PC) 3 separated toxic and control frogs (intestines: F1=31.21, P=0.0008; skin: F1=23.36, P=0.002) and accounted for 13% of the variance in both tissues. In the liver, we did not observe significant separation of toxic versus control frogs in the first 3 PCs. Some protein contigs varied significantly with toxicity, including 276 in the intestines, 185 in the liver and 278 in the skin. Across all tissues, there was significant gene ontology enrichment in four biological processes: negative regulation of endopeptidase activity, complement activation, inflammatory response and oxidation–reduction processes.
Fig. 2.

Proteomic changes with toxin bioaccumulation in the Diablito poison frog. (A) Several proteins change in abundance with toxicity across all tissues, including plasma proteins alpha-1-antitrypsin, saxiphilin, complement C3 and a venom factor, as well as the small-molecule detoxification proteins nicotinamide N-methyltransferase (NNMT) and glutathione S-transferase kappa 1 (GSTK1). y-axis shows the normalized sum of the signal to noise ratio. There are also many tissue-specific membrane protein abundance changes in the (B) intestine, (C) liver and (D) skin that include small-molecule transport and metabolism. This experiment was performed once. See Table 1 for detailed statistics (*P<0.05, **P≤0.005, ***P≤0.0005) comparing non-toxic (N=3) and DHQ-containing (N=6) frogs. Data are represented in boxplots that show rectangles as the lower and upper quartiles (with the median as the line) and whiskers that indicate the maximum and minimum values. For statistics of data and full protein names of symbols in B–D, see Table 1.
Many serum proteins were differentially abundant across tissues including transferrins, globulins, complement factors and protease inhibitors (Table S1). Plasma protease inhibitors, which negatively regulate endopeptidase activity, were increased in abundance across all tissues, including alpha-1-antitrypsin (Fig. 2A, Table 1) and alpha-2-macroglobulin. Although these proteins have been associated with rattlesnake venom resistance in squirrels (Gibbs et al., 2020), whether they bind alkaloids is currently unknown. The amphibian protein saxiphilin, which binds the lethal marine alkaloid saxitoxin (Mahar et al., 1991; Yen et al., 2019), increased in abundance in the intestines and skin of toxic frogs (Fig. 2A, Table 1). We have previously shown that saxiphilin is more abundant in the plasma of non-toxic frogs and that saxiphilin may bind DHQ using thermal shift assays (Caty et al., 2019). These data support the hypothesis that saxiphilin may carry alkaloid toxins in poison frogs. Although plasma proteins may transport alkaloids through blood, binding studies are needed to determine which of these proteins bind the lipophilic alkaloids found in poison frogs.
Table 1.
Statistics for protein contigs displayed in Fig. 2

A strong pattern observed in this study was that the complement system, which plays a role in adaptive and innate immunity (Sahu and Lambris, 2001), is more active in toxic frogs (Fig. 2A). Complement C3 (CO3) and a related protein, A.superbus venom factor, were increased in toxic frogs in all tissues (Table 1). Complement C3 has been linked to poison frog toxicity previously through RNA sequencing studies (Caty et al., 2019; Sanchez et al., 2019), but this is the first proteomic evidence across tissues. The significant upregulation of a complement C3-like venom factor was particularly interesting. Venom complement factors were first discovered in cobras and later identified in other venomous animals such as spiders (Tambourgi and van den Berg, 2014). Cobra venom factor (CVF)-like proteins are co-injected with toxins from the venom glands, but are not toxic in themselves. Rather, CVF-like proteins activate complement C3 and deplete the complement system in the victim, which increases vascular permeability at the envenomation site, spreading the toxins throughout the prey faster (Vogel and Fritzinger, 2010). Although the function of CVF-like proteins in amphibians is unknown, they could have alkaloid absorption enhancing capabilities, as has been suggested for other amphibian skin peptides (Raaymakers et al., 2017), but further testing is necessary to establish this as a complement-dependent phenomenon.
Across all tissues, a number of membrane proteins involved in drug metabolism were differentially abundant between toxic and control frogs. One of the strongest protein responses to toxicity was a decrease of nicotinamide N-methyltransferase abundance in the intestines and skin of toxic frogs (Fig. 2A). This protein detoxifies xenobiotics by methylating the nitrogen in pyridine rings (Alston and Abeles, 1988). Similarly, many cytochrome P450s, well known for their small molecule metabolism (Danielson, 2002), decreased in abundance with toxicity (Fig. 2B–D, Fig. S2). These patterns were surprising, as we initially hypothesized that small-molecule metabolism capacity would increase with toxin accumulation, and indeed there are some cytochrome P450s that increase with DHQ accumulation in the skin (Fig. S5). However, this decrease in abundance of proteins that metabolize small molecules may instead reflect physiological reduction in xenobiotic metabolism so that toxins can arrive intact to the skin. On the other hand, glutathione S-transferase kappa 1, which is involved in cellular detoxification by conjugating glutathione to hydrophobic substances for export, increased in toxic frogs in the intestines and skin. Acquisition of toxins likely involves a delicate balance between storing the compound with intact potency and avoiding autotoxicity. Determining which of these enzymes modifies poison frog alkaloids would be a useful step towards understanding this physiological balance.
Abundance of many membrane transporter proteins changed with DHQ accumulation, including ion channels, ABC (ATP-binding cassette) transporters (Schinkel and Jonker, 2003) and solute carrier proteins (Lin et al., 2015). The transient receptor potential cation channel subfamily V member 2 (TRPV2) is involved in the detection of noxious chemicals (Perálvarez-Marín et al., 2013) and decreased in abundance in the intestines and skin of toxic frogs. We also note that Na+/K+-ATPase is downregulated with toxin accumulation in the skin. Mutations in this protein are associated with toxin resistance mechanisms in many animals (Ujvari et al., 2015) and changing the abundance of this protein may be an additional coping mechanism. Additionally, ABC and solute carrier proteins bind to xenobiotics in mammals, including in the context of cancer resistance to alkaloid-based anticancer therapies (Chen et al., 2016; Liu, 2009). These proteins may be important in uptake and transfer of alkaloids across tissues, as we found changes in abundance of several proteins in this family. For example, the increase in abundance of the neutral and basic amino acid transport protein (SLC31) in the intestines contributed to the separation of toxic and control frogs in the PCA. More in-depth in vitro studies with these membrane transporter proteins would be required to fully understand these trends and the extent to which these proteins can interact with poison frog alkaloids.
Focusing on proteins that contribute to PCA separation of control and toxic frogs, we found an elevation of lipid-associated processes and a decrease in some glycolysis-related proteins, suggesting acquiring toxins shifts cellular metabolism more towards aerobic than anaerobic processes. Specifically, ATP-dependent 6-phosphofructokinase (PFK), the enzyme for the first committing step in glycolysis, is decreased in all tissues in toxic frogs, while many proteins involved in lipid transport and metabolism are increased. Chemically defended dendrobatid poison frogs have greater aerobic capacity (Santos and Cannatella, 2011) compared with non-toxic species and this study provides a potential physiological explanation for this observation. Lipid-associated proteins also point to an alternative mechanism of alkaloid transport, where lipophilic compounds can be transported via chylomicrons in the lymphatic system (Trevaskis et al., 2008). For example, levels of the fatty acid binding protein, which transports lipophilic substances, increase in the intestine of toxic frogs (Fig. 2B, Table 1). Scavenger receptor proteins involved in lipoprotein endocytosis also change in abundance in the skin of toxic frogs and provide a potential sequestration mechanism. For lipid-associated transport to be feasible, proteins that break down lipids are also needed, and indeed, lipases are also increased in the skin of toxic frogs. It is currently an open question as to whether the main route of alkaloid uptake in poison frogs is carrier mediated through blood circulation or lipid mediated via the lymphatic system.
In this study, we show that the Diablito frog can bioaccumulate dietary alkaloids within 4 days of exposure, and this rapid uptake shifts abundance of proteins that interact with xenobiotics, including plasma carrier proteins, membrane transporters, cytochrome P450s and lipoproteins. Our study also suggests that the complement system and venom factors may be involved in alkaloid delivery, although experiments involving frog predators are needed to test this idea. Our study lays a foundation for future work involving rigorous small-molecule binding, transport and metabolism experiments, which are needed to pinpoint specific physiological changes that allow poison frogs to acquire their chemical defenses.
Supplementary Material
Acknowledgements
The following 2016 LS50 Integrated Science students at Harvard University conducted the toxin feeding experiment and preliminary data analyses: Marianne T. Aguilar, Sophia M. Caldera, Jacqueline Chea, Miruna G. Cristus, Jett P. Crowdis, Bluyé DeMessie, Caroline R. DesJardins-Park, Audrey H. Effenberger, Felipe Flores, Michael Giles, Emma Y. He, Nike S. Izmaylov, ChangWon C. Lee, Nicholas A. Pagel, Krystal K. Phu, Leah U. Rosen, Danielle A. Seda, Yong Shen, Santiago Vargas and Hadley S. Weiss. We thank Aurora Alvarez-Bullya, Stephanie Caty and Nora Moskowitz for comments on early versions of this manuscript and Eva Fischer and Alexandre Roland for teaching assistance in the LS50 laboratory course. We acknowledge that our research at Stanford University occurs on the ancestral and unceded land of the Muwekma Ohlone Tribe.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: L.A.O., A.W.M.; Methodology: L.A.O., J.A.P., S.A.T., S.P.G.; Formal analysis: L.A.O.; Investigation: L.A.O., LS50, J.D.O., S.A.T.; Resources: L.A.O., J.A.P., S.A.T., S.P.G., A.W.M.; Data curation: L.A.O.; Writing - original draft: L.A.O., LS50; Writing - review & editing: J.D.O., J.A.P., S.A.T., S.P.G., A.W.M.; Visualization: L.A.O.; Supervision: L.A.O., S.P.G., A.W.M.; Project administration: L.A.O.; Funding acquisition: L.A.O., A.W.M.
Funding
This work was supported a Bauer Fellowship from Harvard University (L.A.O.), the L'Oreal For Women in Science Fellowship (L.A.O.), the National Science Foundation (IOS-1557684 to L.A.O.), a Howard Hughes Medical Institute Professor's Award (520008146 to A.W.M.) and the National Institutes of Health (R01 GM132129 to J.A.P. and GM67945 to S.P.G.). Deposited in PMC for release after 12 months.
Data availability
Mass spectrometry data are available from the PRoteomics IDEntifications (PRIDE) database: PXD021216 (http://www.ebi.ac.uk/pride/archive/projects/PXD021216)
Supplementary information
Supplementary information available online at https://jeb.biologists.org/lookup/doi/10.1242/jeb.230342.supplemental
References
- Alston, T. A. and Abeles, R. H. (1988). Substrate specificity of nicotinamide methyltransferase isolated from porcine liver. Arch. Biochem. Biophys. 260, 601-608. 10.1016/0003-9861(88)90487-0 [DOI] [PubMed] [Google Scholar]
- Caty, S. N., Alvarez-Buylla, A., Byrd, G. D., Vidoudez, C., Roland, A. B., Tapia, E. E., Budnik, B., Trauger, S. A., Coloma, L. A. and O'Connell, L. A. (2019). Molecular physiology of chemical defenses in a poison frog. J. Exp. Biol. 222, 1-12. 10.1242/jeb.204149 [DOI] [PubMed] [Google Scholar]
- Chen, Z., Shi, T., Zhang, L., Zhu, P., Deng, M., Huang, C., Hu, T., Jiang, L. and Li, J. (2016). Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: a review of the past decade. Cancer Lett. 370, 153-164. 10.1016/j.canlet.2015.10.010 [DOI] [PubMed] [Google Scholar]
- Daly, J. W., Martin Garraffo, H., Spande, T. F., Jaramillo, C. and Stanley Rand, A. (1994a). Dietary source for skin alkaloids of poison frogs (Dendrobatidae)? J. Chem. Ecol. 20, 943-955. 10.1007/BF02059589 [DOI] [PubMed] [Google Scholar]
- Daly, J. W., Secunda, S. I., Garraffo, H. M., Spande, T. F., Wisnieski, A. and Cover, J. F.Jr (1994b). An uptake system for dietary alkaloids in poison frogs (Dendrobatidae). Toxicon 32, 657-663. 10.1016/0041-0101(94)90335-2 [DOI] [PubMed] [Google Scholar]
- Daly, J. W., Garraffo, H. M., Spande, T. F., Clark, V. C., Ma, J., Ziffer, H. and Cover, J. F.Jr (2003). Evidence for an enantioselective pumiliotoxin 7-hydroxylase in dendrobatid poison frogs of the genus Dendrobates. Proc. Natl. Acad. Sci. USA 100, 11092-11097. 10.1073/pnas.1834430100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly, J. W., Spande, T. F. and Garraffo, H. M. (2005). Alkaloids from amphibian skin: a tabulation of over eight-hundred compounds. J. Nat. Prod. 68, 1556-1575. 10.1021/np0580560 [DOI] [PubMed] [Google Scholar]
- D'Angelo, G., Chaerkady, R., Yu, W., Hizal, D. B., Hess, S., Zhao, W., Lekstrom, K., Guo, X., White, W. I., Roskos, L.et al. (2017). Statistical models for the analysis of isobaric tags multiplexed quantitative proteomics. J. Proteome Res. 16, 3124-3136. 10.1021/acs.jproteome.6b01050 [DOI] [PubMed] [Google Scholar]
- Danielson, P. B. (2002). The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 3, 561-597. 10.2174/1389200023337054 [DOI] [PubMed] [Google Scholar]
- Darst, C. R., Menéndez-Guerrero, P. A., Coloma, L. A. and Cannatella, D. C. (2005). Evolution of dietary specialization and chemical defense in poison frogs (Dendrobatidae): a comparative analysis. Am. Nat. 165, 56-69. 10.1086/426599 [DOI] [PubMed] [Google Scholar]
- Elias, J. E. and Gygi, S. P. (2010). Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 604, 55-71. 10.1007/978-1-60761-444-9_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs, H. L., Sanz, L., Pérez, A., Ochoa, A., Hassinger, A. T. B., Holding, M. L. and Calvete, J. J. (2020). The molecular basis of venom resistance in a rattlesnake-squirrel predator-prey system. Mol. Ecol. 177, S46 10.1016/j.toxicon.2019.12.090 [DOI] [PubMed] [Google Scholar]
- Hantak, M. M., Grant, T., Reinsch, S., Mcginnity, D., Loring, M., Toyooka, N. and Saporito, R. A. (2013). Dietary alkaloid sequestration in a poison frog: an experimental test of alkaloid uptake in Melanophryniscus stelzneri (Bufonidae). J. Chem. Ecol. 39, 1400-1406. 10.1007/s10886-013-0361-5 [DOI] [PubMed] [Google Scholar]
- Huttlin, E. L., Jedrychowski, M. P., Elias, J. E., Goswami, T., Rad, R., Beausoleil, S. A., Villén, J., Haas, W., Sowa, M. E. and Gygi, S. P. (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174-1189. 10.1016/j.cell.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, L., Yee, S. W., Kim, R. B. and Giacomini, K. M. (2015). SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 14, 543-560. 10.1038/nrd4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F.-S. (2009). Mechanisms of chemotherapeutic drug resistance in cancer therapy—a quick review. Taiwan. J. Obstet. Gynecol. 48, 239-244. 10.1016/S1028-4559(09)60296-5 [DOI] [PubMed] [Google Scholar]
- Mahar, J., Lukács, G. L., Li, L., Hall, S. and Moczydlowski, E. (1991). Pharmacological and biochemical properties of saxiphilin, a soluble saxitoxin-binding protein from the bullfrog (Rana catesbeiana). Toxicon 29, 53-71. 10.1016/0041-0101(91)90039-T [DOI] [PubMed] [Google Scholar]
- McAlister, G. C., Nusinow, D. P., Jedrychowski, M. P., Wühr, M., Huttlin, E. L., Erickson, B. K., Rad, R., Haas, W. and Gygi, S. P. (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150-7158. 10.1021/ac502040v [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGugan, J. R., Byrd, G. D., Roland, A. B., Caty, S. N., Kabir, N., Tapia, E. E., Trauger, S. A., Coloma, L. A. and O'Connell, L. A. (2016). Ant and mite diversity drives toxin variation in the little devil poison frog. J. Chem. Ecol. 42, 537-551. 10.1007/s10886-016-0715-x [DOI] [PubMed] [Google Scholar]
- Neuwirth, M., Daly, J. W., Myers, C. W. and Tice, L. W. (1979). Morphology of the granular secretory glands in skin of poison-dart frogs (Dendrobatidae). Tissue Cell 11, 755-771. 10.1016/0040-8166(79)90029-6 [DOI] [PubMed] [Google Scholar]
- Opitz, S. E. W. and Müller, C. (2009). Plant chemistry and insect sequestration. Chemoecology 19, 117-154. 10.1007/s00049-009-0018-6 [DOI] [Google Scholar]
- Perálvarez-Marín, A., Doñate-Macian, P. and Gaudet, R. (2013). What do we know about the transient receptor potential vanilloid 2 (TRPV2) ion channel? FEBS J. 280, 5471-5487. 10.1111/febs.12302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prates, I., Antoniazzi, M. M., Sciani, J. M., Pimenta, D. C., Toledo, L. F., Haddad, C. F. B. and Jared, C. (2012). Skin glands, poison and mimicry in dendrobatid and leptodactylid amphibians. J. Morphol. 273, 279-290. 10.1002/jmor.11021 [DOI] [PubMed] [Google Scholar]
- Raaymakers, C., Verbrugghe, E., Hernot, S., Hellebuyck, T., Betti, C., Peleman, C., Claeys, M., Bert, W., Caveliers, V., Ballet, S.et al. (2017). Antimicrobial peptides in frog poisons constitute a molecular toxin delivery system against predators. Nat. Commun. 8, 1495 10.1038/s41467-017-01710-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W. and Smyth, G. K. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland, A. B., Santos, J. C., Carriker, B. C., Caty, S. N., Tapia, E. E., Coloma, L. A. and O'Connell, L. A. (2016). Radiation and hybridization of the Little Devil poison frog (Oophaga sylvatica) in Ecuador. Ecol. Evol. 7, 9750-9762. 10.1002/ece3.3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu, A. and Lambris, J. D. (2001). Structure and biology of complement protein C3, a connecting link between innate and acquired immunity. Immunol. Rev. 180, 35-48. 10.1034/j.1600-065X.2001.1800103.x [DOI] [PubMed] [Google Scholar]
- Sanchez, E., Rodríguez, A., Grau, J. H., Lötters, S., Künzel, S., Saporito, R. A., Ringler, E., Schulz, S., Wollenberg Valero, K. C. and Vences, M. (2019). Transcriptomic signatures of experimental alkaloid consumption in a poison frog. Genes 10, 733 10.3390/genes10100733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, J. C. and Cannatella, D. C. (2011). Phenotypic integration emerges from aposematism and scale in poison frogs. Proc. Natl. Acad. Sci. USA 108, 6175-6180. 10.1073/pnas.1010952108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, J. C., Coloma, L. A. and Cannatella, D. C. (2003). Multiple, recurring origins of aposematism and diet specialization in poison frogs. Proc. Natl. Acad. Sci. USA 100, 12792-12797. 10.1073/pnas.2133521100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, J. C., Tarvin, R. D. and O'Connell, L. A. (2016). A review of chemical defense in poison frogs (Dendrobatidae): ecology, pharmacokinetics, and autoresistance. Chem. Signal. Vertebr. 13, 305-337. 10.1007/978-3-319-22026-0_21 [DOI] [Google Scholar]
- Saporito, R. A., Spande, T. F., Martin Garraffo, H. and Donnelly, M. A. (2009). Arthropod alkaloids in poison frogs: a review of the “dietary hypothesis”. Heterocycles 79, 277-297. 10.3987/REV-08-SR(D)11 [DOI] [Google Scholar]
- Saporito, R. A., Donnelly, M. A., Spande, T. F. and Martin Garraffo, H. (2011). A review of chemical ecology in poison frogs. Chemoecology 22, 159-168. 10.1007/s00049-011-0088-0 [DOI] [Google Scholar]
- Savitzky, A. H., Mori, A., Hutchinson, D. A., Saporito, R. A., Burghardt, G. M., Lillywhite, H. B. and Meinwald, J. (2012). Sequestered defensive toxins in tetrapod vertebrates: principles, patterns, and prospects for future studies. Chemoecology 22, 141-158. 10.1007/s00049-012-0112-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel, A. H. and Jonker, J. W. (2003). Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Deliv. Rev. 55, 3-29. 10.1016/S0169-409X(02)00169-2 [DOI] [PubMed] [Google Scholar]
- Stynoski, J. L., Torres-Mendoza, Y., Sasa-Marin, M. and Saporito, R. A. (2014). Evidence of maternal provisioning of alkaloid-based chemical defenses in the strawberry poison frog Oophaga pumilio. Ecology 95, 587-593. 10.1890/13-0927.1 [DOI] [PubMed] [Google Scholar]
- Tambourgi, D. V. and van den Berg, C. W. (2014). Animal venoms/toxins and the complement system. Mol. Immunol. 61, 153-162. 10.1016/j.molimm.2014.06.020 [DOI] [PubMed] [Google Scholar]
- Trevaskis, N. L., Charman, W. N. and Porter, C. J. H. (2008). Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv. Drug Deliv. Rev. 60, 702-716. 10.1016/j.addr.2007.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujvari, B., Casewell, N. R., Sunagar, K., Arbuckle, K., Wüster, W., Lo, N., O'Meally, D., Beckmann, C., King, G. F., Deplazes, E.et al. (2015). Widespread convergence in toxin resistance by predictable molecular evolution. Proc. Natl. Acad. Sci. USA 112, 11911-11916. 10.1073/pnas.1511706112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, C.-W. and Fritzinger, D. C. (2010). Cobra venom factor: Structure, function, and humanization for therapeutic complement depletion. Toxicon 56, 1198-1222. 10.1016/j.toxicon.2010.04.007 [DOI] [PubMed] [Google Scholar]
- Wühr, M., Freeman, R. M.Jr., Presler, M., Horb, M. E., Peshkin, L., Gygi, S. P. and Kirschner, M. W. (2014). Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr. Biol. 24, 1467-1475. 10.1016/j.cub.2014.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen, T.-J., Lolicato, M., Thomas-Tran, R., Du Bois, J. and Minor, D. L.Jr (2019). Structure of the saxiphilin:saxitoxin (STX) complex reveals a convergent molecular recognition strategy for paralytic toxins. Sci. Adv. 5, eaax2650 10.1126/sciadv.aax2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. and Benet, L. Z. (2001). The Gut as a barrier to drug absorption. Clin. Pharmacokinet. 40, 159-168. 10.2165/00003088-200140030-00002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.