

Bismuth double perovskite Cs2AgBiBr6 strong electron–acoustic phonon interactions derail its photovoltaic aspirations.
Abstract
Bismuth-based double perovskite Cs2AgBiBr6 is regarded as a potential candidate for low-toxicity, high-stability perovskite solar cells. However, its performance is far from satisfactory. Albeit being an indirect bandgap semiconductor, we observe bright emission with large bimolecular recombination coefficient (reaching 4.5 ± 0.1 × 10−11 cm3 s−1) and low charge carrier mobility (around 0.05 cm2 s−1 V−1). Besides intermediate Fröhlich couplings present in both Pb-based perovskites and Cs2AgBiBr6, we uncover evidence of strong deformation potential by acoustic phonons in the latter through transient reflection, time-resolved terahertz measurements, and density functional theory calculations. The Fröhlich and deformation potentials synergistically lead to ultrafast self-trapping of free carriers forming polarons highly localized on a few units of the lattice within a few picoseconds, which also breaks down the electronic band picture, leading to efficient radiative recombination. The strong self-trapping in Cs2AgBiBr6 could impose intrinsic limitations for its application in photovoltaics.
INTRODUCTION
The advent of solution-processed halide perovskites promises low-cost and high-efficiency optoelectronics applications. Leading the pack is lead halide perovskite solar cells with efficiencies exceeding 25% (1, 2). However, there are concerns over toxicity and stability issues of lead halide perovskites. Research focus has recently shifted to bismuth-based perovskites where both problems in lead perovskites can be simultaneously addressed (3–6). Among them, halide double perovskites A2B′B″X6 (e.g., Cs2AgBiBr6) have received the most attention because of their three-dimensional structure rather than the typical AaB″bXc (A, monovalent cation; B′, monovalent metal ion; B″, trivalent metal ion; X, halide anion; a, b, and c, integrals following the relation a + 3b = c) structure with reduced dimensionality that limits exciton dissociation and charge carrier transport (4, 5, 7). Although Cs2AgBiBr6 has shown favorable optoelectronic properties such as long carrier lifetime (4, 5, 8–10) and has successfully been used in some optoelectronic applications such as x-ray photodetection (11–14), the performance of Cs2AgBiBr6 solar cells remain disappointing, with power conversion efficiencies (PCEs) of merely 1 to 2.5% reported to date (15–19). The cause of such poor performance has been under intense debate. For example, strong electron-phonon coupling was experimentally observed (20, 21), but the strength of the Fröhlich coupling (typical for ionic/polar semiconductors) is only comparable to that of lead perovskites (20). Savory et al. (22) found that the angular momentum mismatch of the frontier atomic orbitals may lead to flat bands and large carrier effective mass, while, in other reports, the effective carrier mass is believed to be small enough for favorable charge transport (5, 8, 23). Deep defect levels such as BiAg antisites and bromine vacancies may become strong nonradiative loss channels (9, 24); however, they can be carefully tuned to be as low as 109 cm−3 under ideal growth conditions (11, 24). To further optimize their performance and evaluate their potential for solar cell applications, it is imperative to resolve the dispute and establish whether the low performance is the consequence of processing conditions or due to intrinsic limitations.
Here, we attempt to rationalize the underlying mechanisms that limit the photovoltaic performance through a detailed spectroscopic study of the photoexcitation, recombination, transport, and scattering processes in high-quality Cs2AgBiBr6 single crystals (SCs) and polycrystalline (PC) films, supported by density functional theory (DFT) calculations. Despite Cs2AgBiBr6 being an indirect bandgap semiconductor, we observe strong band-to-band emission with a maximum quantum yield >2% and bimolecular recombination coefficient of around 4.5 ± 0.1 × 10−11 cm3 s−1 at room temperature, which is only an order lower than typical direct bandgap semiconductors. In contrast to lead halide perovskites whose photoluminescence (PL) linewidths are proportional to the phonon occupation number, the temperature dependence of the PL linewidth for the double perovskite scales with , which is consistent with the prediction of electron-lattice interaction in the strong coupling regime. Concomitantly, the intrinsic carrier mobility for SCs is merely 0.05 cm2 s−1, suggesting a localization effect by the strong electron-phonon coupling. Both strong coherent optical and acoustic phonon oscillations are fast damped and are coupled to the electronic dynamics, signifying an ultrafast formation of polarons and reduction of the carrier mobility within a few picoseconds, as revealed by transient reflection spectroscopy (TRS) and time-resolved terahertz spectroscopy (TRTS). With the aid of DFT calculations, we found that the theoretical Fröhlich coupling strength is in the intermediate regime, comparable to lead halide perovskites. However, the deformation potentials by acoustic phonons for both electrons and holes in Cs2AgBiBr6 are larger than 10 eV, much stronger compared to their lead counterparts, which may assist the ultrafast and strong self-trapping of the charge carriers. From the above experimental and theoretical results, we propose that after the coupling of the charge carriers with the optical phonons, a range of acoustic phonon modes may further interact strongly with the charge carriers, which localizes the charge carriers within a few lattice units. The strong self-trapping causes the charge carriers to lose coherence and move by hopping from site to site. The electronic band picture breaks down because of the localization effect and is replaced by local polaronic picture, which circumvents the strict transition rules for bare electrons. Our findings suggest that the double perovskite has its intrinsic limitations in photovoltaic performance due to the strong self-trapping effects, which may also be applicable to other Bi-based and Sb-based semiconductors.
RESULTS
Basic properties and device performance
Cs2AgBiBr6 has a cubic double perovskite superstructure, with Ag and Bi located at alternate perovskite octahedra units (Fig. 1A) (5). We prepared high-quality Cs2AgBiBr6 SCs following our previous reports (4, 19, 25). Figure 1B is the x-ray diffraction (XRD) pattern of the powdered SC, which is in agreement with theoretical calculations and indicates a high phase purity. The PC films were fabricated by spin-coating SC solutions onto quartz substrates. The scanning electron microscopy (SEM) image shows that the PC film is pinhole-free and smooth with a large grain size of around 240 nm (Fig. 1C). Hall measurements show a background electron density of the order of 1013 to 1014 cm−3 for the PC films, suggesting a weak n-type semiconductor in contrast to a previous theoretical prediction. This is further supported by ultraviolet photoelectron spectroscopy characterization of the energy levels (figs. S1 and S2) (9). The absorption spectrum of the PC film shows a strong excitonic feature at the red side of a continuum band with absorption coefficients of around 104 to 105 cm−1 (Fig. 1D). This is attributed to the direct electronic and associated excitonic transitions above the indirect bandgap, e.g., at X point (26). SC exhibits a slight blue shift of the excitonic and continuum absorption, which is possibly due to different microstrains or lattice disorder–induced band energy shift (27). The absorption from indirect transitions is not obvious in the PC film because of their weak transition strength. With diffuse reflectance spectroscopy, we can clearly observe an absorption band at the red side of the strong direct absorption bands due to much longer light penetration depths in the SCs (Fig. 1D). We propose that it mainly arises from the indirect transition, as the extracted indirect bandgap (Egi) of around 2.18 eV through Tauc plot is consistent with previously reported theoretical and experimental results (fig. S3) (4, 5). However, band tail and defect absorption cannot be fully excluded.
Fig. 1. Basic properties of Cs2AgBiBr6 and solar cell performance.
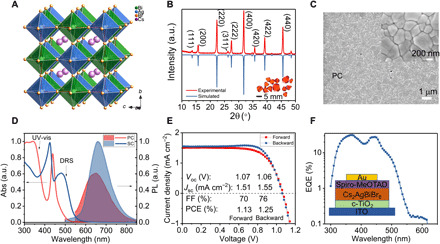
(A) Crystal structure of Cs2AgBiBr6. (B) XRD pattern of powdered Cs2AgBiBr6 SC. Inset: An image of Cs2AgBiBr6 SCs of several millimeters. a.u., arbitrary units. (C) SEM image of Cs2AgBiBr6 PC film. Inset: Zoom-in SEM image. (D) Absorption (Abs) and emission spectra of Cs2AgBiBr6 PC film and SC. UV-vis, ultraviolet-visible; DR, diffuse reflectance. (E) J-V and (F) IPCE curves of a typical Cs2AgBiBr6 solar cell with ITO/compact TiO2/Cs2AgBiBr6/Spiro-MeOTAD/Au planar structure. EQE, external quantum efficiency.
Cs2AgBiBr6 perovskite solar cells were fabricated using high-quality PC films in a planar structure with the indium tin oxide (ITO)/compact TiO2/Cs2AgBiBr6/Spiro-MeOTAD/Au configuration following our previous report (19). The best-performing solar cells have PCEs of around 1.13 and 1.24% with forward scan and backward scan, respectively (Fig. 1E). The VOC, JSC, and fill factor (FF) are 1.06(1.05) V, 1.46(1.54) mA cm−2, and 0.73 (0.77) with forward (backward) scans, respectively. The incident photon-to-electron conversion efficiency (IPCE) curve is shown in Fig. 1F with a maximum efficiency of only 30 to 35% at direct absorption region and <1% at indirect absorption region. These values are generally comparable with other reports (15–19). With carrier diffusion lengths of around 100 nm in our PC films measured using the time-resolved PL (TRPL) quenching method (19), we speculate, to some extent, that the performance may be hampered because of interfacial problems, which can be alleviated by using mesoscopic structure or other charge-transporting layers. However, the carrier diffusion lengths do not permit enough indirect absorption, and therefore, there is strong limitation of the solar cell performance. Next, we mainly focus on spectroscopic characterizations of Cs2AgBiBr6 SCs, which have much fewer defects and can therefore divulge the intrinsic photophysics of the double perovskite system.
Bright band-to-band emission and low carrier mobility
Radiative transitions at indirect band edges are forbidden, which require absorbing or emitting phonons to compensate the k-space momentum mismatch (Fig. 2A). Therefore, PL quantum yields (PLQYs) for indirect bandgap materials are typically several orders lower than their direct counterparts, e.g., 10−6 for Si and 10−5 to 10−6 for bulk MoS2 (28, 29). This limits their potential for light-emitting applications. However, we observed PL in both Cs2AgBiBr6 SCs and PC films, which is “bright” compared to typical indirect semiconductors. The broad PL spectra are centered at around 650 to 660 nm (~1.90 eV), with a Stokes shift of around 280 meV and full width at half maximum (FWHM) of around 420 and 320 meV for the PC films and SCs, respectively (Fig. 1D). The origin of the PL in Cs2AgBiBr6 has been under debate with defect/extrinsic color center, band-to-band indirect transition, and self-trapped exciton (STE) emission being proposed (4, 10, 19, 21). We will show evidence below that the PL originates from free carrier emission at the band edge at low excitation density but is dressed by the lattice deformation, i.e., the polaronic band-to-band transition, which circumvents the forbidden rule for bare electronic transitions with Δk = 0 by strong local lattice deformation. Before providing evidence on the polaron formation, below, we still refer to free carrier instead of polaron. Figure 2B presents the fluence-dependent initial PL intensity (PL|t = 0) upon above indirect bandgap excitation (530 nm). The initial PL intensity (PL|t = 0) just after photoexcitation provides an indication of the emission species within the crystal (30). The fluence has been converted to average carrier density within the effective excitation length (~ 4 μm). When the excitation density N < 1016 cm−3, the initial PL intensity scales quadratically (power law factor λ = 1.94) with carrier density, implying free electron-hole recombination. The free carrier emission dominates from 1016 cm−3 to as low as 1014 cm−3, which is in the low detection limit of our PL setup. We did not observe any emission by the doped carriers, which is monomolecular and below the free carrier emission threshold. This corresponds to a doped carrier density much lower than 1014 cm−3, consistent with the Hall measurement results (<1013 cm−3). At high carrier density regime after N > 1016 cm−3, the power law factor drops to λ = 1.26, showing the coexistence of both free carrier and exciton emission, as we observed previously for PC films (19). This is a consequence of the thermal equilibrium between the photoexcited bound excitons and unbound electron-hole plasma (free carriers) after the excitations cool to the indirect band edges according to Saha’s equation: , where ne,h and nEX are the densities of electron-hole plasma and excitons, respectively, and μEX and Eb are the effective reduced mass and the binding energy of the excitons (30). Note that we did not observe any change of the PL spectral profile with varying excitation densities. This is consistent with the fact that both bound excitons and the plasma of unbound electrons and holes emit at the exciton resonance according to a full many-body treatment in a strongly interacting many-body system (31). By fitting the fluence-dependent initial PL intensity curve, taking into account only the thermal equilibrium between the free carriers and excitons, we extract an exciton binding energy of around 94 meV, which is close to the theoretical value of 104 meV (8). Hence, we expect free carriers to be the dominant excited species (fraction larger than 70%) for an excitation density below 1016 cm−3 (fig. S4).
Fig. 2. The PL characteristics of Cs2AgBiBr6 SC and PC films.
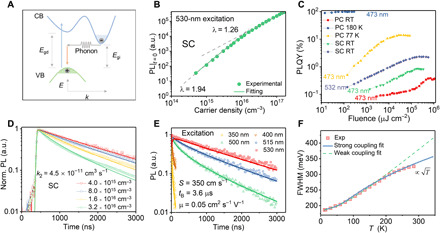
(A) Schematic of the phonon-assisted transitions at the indirect band edge for a typical indirect semiconductor. VB, valence band; CB, conduction band; Egd, direct band gap; Egi, indirect band gap. (B) Carrier density–dependent initial PL intensity of Cs2AgBiBr6 SC with 530-nm excitation. (C) The PLQYs of Cs2AgBiBr6 SC and PC film. The SC was excited using 532-nm (blue) and 473-nm (green) lasers at room temperature (RT). The PC film was excited using a 473-nm laser. (D) Fluence-dependent PL decays of Cs2AgBiBr6 SC upon 530-nm laser excitation. (E) Excitation energy–dependent PL kinetics of Cs2AgBiBr6 SC. Lines: Fitted curves with the model in the main text. (F) Temperature-dependent PL linewidth of Cs2AgBiBr6 SC. Dashed line, fitting with the conventional model for weak coupling; solid line, fitting with the model proposed by Toyozawa (39) for strong coupling.
Figure 2C presents the PLQYs of typical Cs2AgBiBr6 SC and PC film. At room temperature, both the SC and PC film undergo an initial monotonous PLQY increase with increasing photoexcitation density until reaching a plateau. This is in congruence with the dominance of emission from free carriers at low excitation density to excitons at high excitation density. By further increasing the photoexcitation density, the PLQYs start to drop because of multiparticle nonradiative recombination such as Auger recombination and exciton-exciton annihilation. With 473-nm excitation, the maximum PLQY for SC at room temperature is around 0.8%, which is further elevated to around 2% with a 532-nm laser that excites more of the bulk. Note that the PLQY of SC is probably underestimated because of the unavoidable reabsorption effect. The PC film with negligible reabsorption shows a maximum PLQY of around 0.4% at room temperature, a value lower than SC owing to faster nonradiative recombination at grain boundaries or inside the grains, which also excludes the possible origin of defect emission. As we decrease the temperature, the PLQY gradually increases and approaches unity at 77 K, which may be attributed to defect deactivation and exciton formation at low temperature.
The high PLQYs imply that the radiative recombination at room temperature for electrons and holes is in competition with the trap-assisted nonradiative recombination. Figure 2D presents the PL decay dynamics for Cs2AgBiBr6 SC that is highly dependent on the photoexcitation density, which is another important signature of band-to-band emission, as the defect or exciton emission rate is typically independent of the excitation density in the low regime. Here, we used a weak absorbing 530-nm laser as the excitation source to mainly excite the bulk part and to minimize carrier diffusion effect. In this scenario, the carrier dynamics follows the expression in a thin film: dn/dt = −k1n − k2n2 and IPL(t) ~ k2n2, where k1 and k2 are the first-order recombination rate via deep defect levels and the second-order e-h (bimolecular) recombination coefficient, respectively. Recombination via doped carriers is neglected in the presence of much lower doped carrier density (<1013 cm−3) compared to that of the photoexcited carriers (>1014 cm−3). We obtained k1 of around 2.8 ± 0.1 × 10−4 ns−1, corresponding to a bulk reccombination lifetime of 3.6 ± 0.1 μs via Shockley-Read-Hall recombination processes. The bulk carrier lifetime is the highest reported for Cs2AgBiBr6 so far, which again confirms the high quality of our SCs (4, 10). The electron-hole recombination coefficient (k2) reaches 4.5 ± 0.1 × 10−11 cm3 s−1 and does not vary much when fitted with only the experimental curves at low excitation density. The value is around an order lower than conventional direct bandgap materials (e.g., InP, 1 × 10−9 and GaAs, 7 × 10−10) but three to four orders higher than most indirect semiconductors (e.g., Si, 2 × 10−15 and GaP, 5 × 10−14) (32). The relatively large k2 for an indirect gap material can well explain the bright band-to-band emission with high PLQY in Cs2AgBiBr6.
We further measured the diffusion and recombination properties of the double perovskite SC using the excitation energy–dependent TRPL. The PL dynamics of the SC at room temperature shows strong dependence on the excitation energies (Fig. 2E). Upon 530-nm excitation, the PL decays slowly in a monoexponential fashion. With the increase of the excitation energies (515, 500, 400, and 350 nm), the PL decays become faster and show biexponential decay behavior. Such notable change of the PL kinetics upon different excitation energies is related to the excitation light penetration depth, which leads to different volume ratios of the surface and bulk parts being excited. The set of PL decay kinetics is globally analyzed using a well-proven one-dimensional diffusion model for extracting surface recombination velocity (SRV) and carrier diffusion properties (33)
| (1) |
where x is the distance to the front surface along the incident direction, n is the carrier concentration, n0 is the carrier concentration at x = 0 and t = 0, k1 is the first-order recombination rate as discussed above, D is the carrier diffusion coefficient that is averaged over those of the electrons and holes, S is the SRV, α is the excitation light absorption coefficient, and W(x) = exp(x2)erfc(x). As we kept to a low excitation density on the order of 1014 to 1015 cm−3 to ensure k2n < <k1, the band edge electron-hole recombination term can be omitted in the fitting. The PL dynamics is then obtained using . We used a big excitation spot of ~1 mm during measurement, which is three orders larger than the carrier diffusion length in the excitation plane, and therefore, the carrier kinetics along the incident direction can be well described using the one-dimensional model. In addition, we also considered a depth of 100 μm beneath the surface for modeling to ensure that almost all the photogenerated carriers can be monitored. We obtained an SRV of 350 ± 20 cm s−1, which is comparable to the values reported for semiconductors with “good” surface, e.g., InP (~102 cm s−1) (34) and halide perovskites (MAPbBr3, MAPbI3, and CsSnI3; ~103 cm s−1) (33, 35). However, do note that the low SRV still causes a substantial drop in the carrier lifetime at the surface due to the slow Cs2AgBiBr6 SC bulk recombination rate that is easily increased by orders with recombination via surface imperfections such as dangling bonds, disorder, and impurities. This also explains why the carrier lifetime in PC films markedly decreases to tens of nanoseconds (fig. S1), which is a potential limitation for PC thin-film photovoltaic performance. Although the SC shows a very long bulk carrier lifetime promising for photovoltaic applications, we obtained a very low average carrier diffusion coefficient D = 1.2 ± 0.1 × 10−3 cm2 s−1, rendering an effective carrier mobility of around 0.05 cm2 s−1 V−1 at room temperature. This value is orders lower than conventional inorganic photovoltaic materials and is only comparable to those organic photovoltaic materials (e.g., Si, 500 to 1450 cm2 s−1 V−1; GaAs, 400 to 8000 cm2 s−1 V−1; MAPbI3, 24 to 105 cm2 s−1 V−1; and P3HT, ~10−3 cm2 s−1 V−1) (36, 37). The value is even a few times lower than that of PC film with crystal size around 240 nm using the Hall measurement (~0.14 cm2 s−1 V−1; fig. S1) or TRPL quenching method (~0.37 cm2 s−1 V−1) previously reported (19). Furthermore, the PC films with increased grain size also exhibit a decrease in Hall mobility despite the increase of the PL lifetime due to fewer grain boundaries (fig. S5). This implies that the carrier mobility is not limited by impurities and defects; otherwise, the mobility should decrease with reduced crystal size and carrier lifetime. Instead, it should be strongly limited by the intrinsic electron-lattice properties. According to , where e is the elementary charge, m is the carrier effective mass, and τ is the carrier scattering time, such low intrinsic carrier mobility either means a very large charge carrier effective mass and/or strong phonon scattering in Cs2AgBiBr6 SC. From DFT calculations, the bare effective masses for electrons and holes are 0.43 m0 and 0.38 m0, respectively, agreeing well with other reports (8, 9). These small values of the bare effective masses imply that there must be strong electron-phonon coupling in the system to increase the charge carrier effective mass and/or the scattering rate. Strong lattice deformation coupled to the photoexcitation may lead to the formation of polarons at low excitation density (<1016 cm−3) and STE at high excitation density (>1017 cm−3), which strongly localizes the charge carriers. As mentioned above, the charge carriers dressed by the local heavy phonon clouds break down the electronic band structure. The localized opposite charges can recombine with each other once they are within the Coulombic capturing radius, which significantly increase the electron-hole recombination coefficient by a few orders compared to typical inorganic semiconductors.
The electron-phonon coupling mechanisms can be established by investigating the temperature-dependent PL bandwidth broadening. In the temperature range studied (10 to 300 K), the PL spectra exhibit Gaussian line shapes rather than Lorentz line shape (fig. S6). In Fig. 2F, we find that the PL bandwidth at high temperature cannot be well fitted by the typical expression: Γ(T) = Γ0 + γacT + γLOn(T), where Γ0 is the inhomogeneous broadening arising from disorder and lattice imperfections. γac and γLO are the coupling constants of the carriers with acoustic and longitudinal optical (LO) phonons, respectively, and n(T) is the occupation number of the LO phonon involved in the coupling. We note that the above expression is only valid for electron-lattice interaction in the weak coupling regime, when the product of the phonon field fluctuation amplitude Δ and the fluctuation correlation time τc is smaller than ℏ (38). Toyozawa (39) theoretically demonstrated that in the strong-coupling regime, the linewidth reflects the statistical distribution of the fluctuation potentials of the phonon field and, therefore, the emission spectrum will show a Gaussian profile with the linewidth proportional to the square of the temperature . The temperature-dependent PL linewidth can thus be well described by (38)
| (2) |
where ΓA and ΓO arise from the scattering by acoustic and optical phonons, respectively. We have added a Γ0 term accounting for the large band energy variation by static lattice disorder in Cs2AgBiBr6. SA and SO are the coupling strengths for the acoustic and optical phonons, respectively. EA is the average acoustic phonon energy, and EO is the optical phonon energy, which we set at a fixed value EO = 22 meV corresponding to the strongest Raman peak and is attributed to the A1g stretching mode of (BiBr6)3− octahedron (fig. S7) (21). The fitting is greatly improved compared to the weak coupling model. Although the fitted SA and EA bear large uncertainties due to limited data points and too many variables in the low-temperature regime, the Gaussian emission line and the increase of the linewidth provide solid evidence of the strong coupling between the electrons and the lattice. The broad PL width at room temperature (320 meV) indicates the presence of a strong phonon field fluctuation that dresses the charge carriers.
Ultrafast self-trapping and strong coherent phonons
We used TRS and TRTS measurements to probe the electron-phonon interaction dynamics in Cs2AgBiBr6 PCs and SCs. Figure 3A shows a pseudo color map of the transient-reflected profile over the probe wavelengths and time delay within the first few picoseconds upon 500-nm laser pumping at a fluence of around 0.4 mJ cm−2. A representative TR spectrum is shown in Fig. 3B (top), at a time delay of 1 ps after photoexcitation. The TR spectrum was further converted to transient absorption (TA) spectrum using the Kramers-Kronig transformation (Fig. 3B, bottom). The TA spectrum shows a negative peak (ΔA < 0) centered at the direct exciton (DE) resonance (around 2.85 eV) with a positive peak (ΔA > 0) located at either side. Similar features have been observed for PC films (fig. S8). These features can be well reproduced by considering the broadening of the DE optical transition introduced by carrier-exciton scattering (fig. S8 and table S1). In addition to the broadening effect, typical many-body interactions of carriers and excitons also involve the screening of the Coulomb potential of an exciton in the presence of other excitons and carriers, which results in a blue-shifted exciton transition (40). In Fig. 3C, the DE resonance undergoes a fast red shift within 10 ps, which can be seen as a recovery from the initial blue shift due to the screening effect of the photoexcited carriers. The fast recovery of the blue shift implies that the screening effect is rapidly reduced within 10 ps. Since DE is not an occupied state, the transient dynamics at DE resonance corresponds to the dynamics of the charge carriers interacting with the DE optical transition. Figure 3D presents the transient dynamics near the DE resonance monitored at 440 nm (2.82 eV) for SC, which undergoes a rapid decay within 10 ps similar to the observed recovery of the blue shift. The ultrafast TR dynamics within tens of picoseconds in semiconductors is usually attributed to fast electronic scattering and trapping processes. The early dynamics can be best fitted using a triexponential decay model with the time constants t1 = 0.25 ± 0.04 ps, t2 = 4.7 ± 0.3 ps, and t3 = 49 ± 2 ps. Similar time constants can be observed for the photoinduced absorption (PIA) at the visible and near-infrared (NIR) spectra region, which have been attributed to the cooled and hot carriers in MAPbI3, respectively (fig. S9) (41). This further consolidates our conclusion that the transient dynamics of the DE resonance reflects the carriers interacting with the DE transition. t3 becomes slower in the samples with lower defect density (annealed at 250°C) than the nonannealed ones with higher defect density and is therefore attributed to the trapping of carriers by permanent defects (fig. S10). However, the dynamics within 10 ps is almost invariant to the defect density and excitation concentration, which implies that t1 and t2 are intrinsic parameters for Cs2AgBiBr6 (fig. S10). We attribute t1 to the hot carrier relaxation via LO phonons and formation of Fröhlich polaron, as it correlates well with a period of typical LO phonon vibrations and is consistent with the rise time of the PIA in the visible region (fig. S9). The hot carriers interact with the excitons by providing a strong local electric field, which is screened by the movement of the lattice ions after they relax via carrier-phonon scattering, and therefore, the DE kinetics show a decay when they cool down (41). The intrinsic time constant t2 is more than one order larger than a period of the typical LO phonons in the double perovskite. Given that t1 approximately corresponds to a period of the dominant LO phonon, t2 is translated to a phonon energy of ~1 meV, which falls into the acoustic branch. The estimated phonon energy is also consistent with the oscillation frequency at low temperature as discussed later. Therefore, we attribute it to the electron-acoustic phonon coupling and the ultrafast self-trapping of the carriers by these acoustic phonons. Such electron-phonon interactions with a fast (subpicosecond) and a slow (a few picoseconds) component seem to be a ubiquitous feature in the electronic dynamics of bismuth-based semiconductors [i.e., as reported for Bi2Te3, Bi2Se3, BiI3, etc. (42–44)]. The self-trapping of the carriers by phonons screens the Coulomb interaction and protects the carriers from scattering (45). Intervalley scattering involving acoustic phonons could be another possible reason for t2; however, we will show later that it is not congruent with the large drop of the photoconductivity.
Fig. 3. Strong coherent phonons and ultrafast polaron formation.
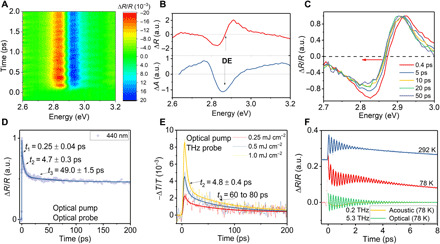
(A) Pseudo color map of the TR profile of Cs2AgBiBr6 SC. (B) Top: The TR spectrum of Cs2AgBiBr6 SC monitored at 1-ps delay upon 500-nm excitation. Bottom: The transformed TA spectrum of Cs2AgBiBr6 SC using the Kramers-Kronig relations. (C) The recovery from the blue shift of the TR spectra at early delay time. (D) The TR kinetics of Cs2AgBiBr6 SC probed near the DE resonance at 440 nm in TRS measurement. (E) The terahertz transmission kinetics of Cs2AgBiBr6 PC in TRTS measurement. (F) Strong optical and acoustic modes coupled to the electronic dynamics probed near the isochromatic point (~2.83 eV). At room temperature (292 K), only the optical mode is obvious. At low temperature (78 K), the acoustic mode becomes apparent. Blue and red lines, fitted curves using Eq. 3; green line, extracted optical mode oscillations at 78 K; yellow line, extracted acoustic mode oscillations at 78 K.
Further evidence of the ultrafast carrier self-trapping is demonstrated with TRTS measurements on Cs2AgBiBr6 PC films deposited on z-cut quartz substrates (Fig. 3E). The terahertz transmission upon optical pumping is related to the photoconductivity of a material and therefore insensitive to charge-neutral excitons. While the t1 cannot be resolved because of the limited time resolution (~ 1 ps), t2 (4.8 ± 0.4 ps) and t3 (60 to 80 ps) match well with those observed in TRS measurements. We confirm that t2 is not due to the intervalley scattering. Otherwise, it should correspond to the rise rather than the decay of the photoconductivity in TRTS measurements due to a much flatter direct band edge (X point) than the indirect edge at L and, therefore, an increase of the carrier mobility after the intervalley scattering (5, 22). Note that the similar time constants shown in both TRS and TRTS measurements implies that we are mainly monitoring the carrier (polaron) dynamics in both measurements, although excitons (STEs) are also present at high excitation density. This is a reasonable speculation, as the carriers are expected to provide a much stronger polarization than the charge-neutral excitons and therefore predominate in the TRS response. The coupling of the charge carriers with acoustic phonons causes a fast drop of 78% of the initial photoconductivity upon excitation at 1.0 mJ cm−2, highlighting the significance of self-trapping in charge carrier mobility. The photoconductivity merely reaches ~6% of its initial amplitude within 200 ps, which is in notable contrast to a large fraction of long-lived component (~50%) in TRS measurement shown in Fig. 3D. We show that the long-lived component has a lifetime that corresponds well with the PL (fig. S11). This means that the long-lived species responsible for the photovoltaic performance is highly localized after the trapping processes, which explains the low carrier mobility of 0.05 to 0.14 cm2 s−1 V−1 measured above using TRPL or Hall methods. The extracted carrier mobility in TRTS measurement taking account of the Saha’s equilibrium is merely 0.4 cm2 s−1 V−1 at 200-ps delay, dropping from the initial 6.8 cm2 s−1 V−1 upon excitation at 1.0 mJ cm−2 (corresponding to carrier density of ~1017 cm−3), which further confirms the low mobility in the system by the localization effect.
Coherent phonons generated via displacive excitation of coherent phonon and impulsive stimulated Raman scattering mechanisms upon photoexcitation provide an alternative window to observe the kinetics of the charge carrier self-trapping. In TRS measurements, the electronic kinetics is accompanied by a strong terahertz oscillation with the frequency pump and probe energy independent, and the amplitude is most pronounced near the isochromatic point ~2.83 eV at room temperature (Fig. 3F and fig. S12). Fourier transform analysis shows a dominant oscillation frequency centered at around 5.3 THz, corresponding to the A1g stretching mode of (BiBr6)3−. Hence, the terahertz oscillation can be attributed to coherent optical phonons (COPs). The same attribution of the oscillation has been given in a previous report (46). We further fitted the TR data at 2.87 eV with the following expression (47)
| (3) |
where the first term at the right side describes the fast electronic decay and the second term represents the damped oscillation. A1 and τ are the amplitude and time constant of the decay. A2, f, φ, and τp are the amplitude, frequency, phase, and damping time constant of the oscillation. The COP dephases with a time constant τp of around 0.8 ps, which lies between t1 and t2 and is therefore probably a result of the self-trapping by both optical and acoustic phonons. When we cooled the SC down to 78 K, an additional damped oscillation–like feature can be observed (Fig. 3F). The kinetics at early times can be fitted by adding a fast damped acoustic component (f ~ 0.2 THz) to the dominant optical component. The period of the oscillation is congruent with the time constant t2 above, corresponding to the main acoustic phonon modes coupled to electrons in the self-trapping process. However, the coherence of the oscillation is easily disrupted by thermal fluctuations, which explains why the oscillations become negligible at room temperature. Similar temperature-dependent coherent phonons have previously been observed for a chain metal halide material [Pt(en)2][Pt(en)2Br2]·(PF6)4 displaying strong acoustic phonon self-trapping (48). Therefore, from these results, we confirm that both optical phonons (f ~ 5.3 THz) and high-energy acoustic phonons (f ~ 0.2 THz) are involved in the self-trapping process.
To understand the strong electron-phonon coupling mechanisms, we further carried out first principles DFT calculations on Cs2AgBiBr6. With the calculated parameters (me = 0.43 m0, mh = 0.38 m0, ε∞ = 4.96, εS = 13.83, and ELO = 13.5 meV), the Fröhlich coupling constants are found to be 2.68 and 2.52 for electrons and holes, respectively (table S2), consistent with the results calculated by Steele et al. (20). These values are comparable to those for CsPbBr3 [αe(h) = 2.64 (2.76)], which corresponds to an intermediate Fröhlich coupling (49), rather than the proposed strong coupling by Kentsch et al. (46). The theoretical polaron mobilities calculated with the Feynman-Osaka formula are 26.9 and 33.2 cm2 s−1 V−1 for electrons and holes, respectively, which are also of the same order with lead halide perovskites (table S2) (49). These results suggest that Fröhlich coupling may not play a critical role in causing the large difference in carrier mobility between the Bi-based double perovskite and lead halide perovskites. On the other hand, the deformation potential is markedly improved in Cs2AgBiBr6 compared to lead halide perovskites. We calculated the deformation potentials to be 13.7 and 14.7 eV at the valence band maximum and conduction band minimum for Cs2AgBiBr6, respectively. As a comparison, the corresponding values for CsPbBr3 are 2.2 and 6.3 eV, respectively. The deformation potentials for Cs2AgBiBr6 are one order higher than most inorganic semiconductors calculated using the same approach (50). As a result, we derived the deformation potential scattering limited carrier mobilities to be 81.4 and 96.3 cm2 s−1 V−1 for Cs2AgBiBr6, which are close to the measured value of around 6 cm2 s−1 V−1 at high carrier density (>1018 cm−3) when all the traps are filled (51). Nevertheless, these mobilities are a few orders lower compared to the deformation potential scattering limited carrier mobilities of lead halide perovskites with values in thousands of cm2 s−1 V−1. The calculated deformation potentials can be verified by performing the excitation energy–dependent coherent acoustic phonon (CAP) oscillation experiments (52). Different from the COP oscillations above, CAP oscillations (frequency on the order of gigahertz) can be generated by thermoelastic stress (σTE) and/or deformation potential–induced stress σDP in a centrosymmetric material. The former originates from the lattice thermal expansion when the photoexcited carriers have excess energy dissipating into the lattice. The latter deforms the lattice and modifies the interatomic bonding, which results in the stress. The amplitude of the CAP in TRS measurements is proportional to the sum of σTE and σDP (52)
| (4) |
where B is the bulk modulus, β is the linear expansion coefficient, Cp is the heat capacity, De(h) are the electron (hole) deformation potentials, N is the photoexcitation density, Eg is the bandgap, and E is the excitation energy. Here, we neglected the possible contribution by thermoelastic stress caused by defect trapping, which is much slower and weaker compared to the charge carrier self-trapping in Cs2AgBiBr6 as discussed above. We found that Cs2AgBiBr6 SC shows prominent CAP oscillation with the amplitude one order larger than that of MAPbBr3 (Fig. 4A and fig. S13), implying the easy deformation of the lattice upon photoexcitation. By using the electronic signal at time zero as a reference of the photoexcitation density, we found that the relative amplitude of CAP shows a dependence on the excitation energy (Fig. 4B). After fitting using B = 36.6 GPa (53), Cp = 2.89 × 105 J m−3 K−1 (53), and β = 2.78 × 10−5 K−1 (54), we extracted De + Dh = 21.2 eV, which is consistent with the large deformation potentials predicted by the calculations.
Fig. 4. Strong CAP and self-trapping by deformation potential.
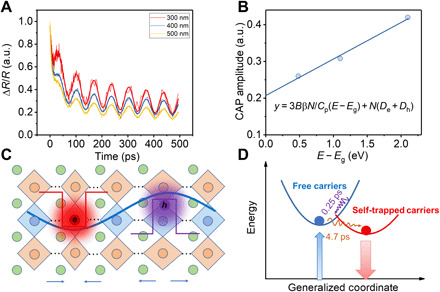
(A) Normalized TR kinetics at 740 nm for Cs2AgBiBr6 SC upon different excitations (300, 400, and 500 nm) showing different relative CAP amplitudes. (B) A linear fit of the relative CAP amplitudes versus excitation energy to extract the deformation potentials. (C) A schematic of the self-trapping of charge carriers by acoustic phonons (deformation potential). The dots between adjacent perovskite octahedra represent the omitted lattice units for simplicity. (D) A schematic of the energy diagram for carrier self-trapping under the generalized coordinate.
Toyozawa (55) introduced a factor to evaluate whether the charge carriers are self-trapped by deformation potential in a cubic lattice, which is expressed by g = Ξ2m/6ℏ2Ca, where Ξ is the deformation potential, m is the bare electron effective mass, C is the elastic constant, and a is the lattice constant. Here, we used C of around 28.6 GPa in the direction (111) calculated by the CAP frequencies shown above, we obtained g = 0.87 and 0.89 for electrons and holes using an a = 1.12 nm. With g ~ 1, it is possible that both the electrons and holes become self-trapped with a coherent length comparable to the lattice dimension [schematic and energy diagram shown in Fig. 4 (C and D)]. At high excitation density or low temperature, they may be self-trapped together, forming STEs, but this is beyond the scope of our study. The localization length can also be deduced from the acoustic phonon characteristic time. Given that the self-trapping by acoustic phonons completes within a period of the acoustic vibration (i.e., τa ~4.7 ps) and the sound velocity v = 2400 m/s, the characteristic acoustic wavelength is around 11 nm. The FWHM of a half cycle of the acoustic wavelength (~3.7 nm) is a measure of the spatial extent of the lattice deformation (48). The estimated localization length corresponds to around three repeat units of double perovskite structure, which is in close agreement with [Pt(en)2][Pt(en)2Br2]·(PF6)4, showing similar self-trapping by acoustic phonons (48).
DISCUSSION
Self-trapping of electronic excitations is initially only considered for ionic/polar semiconductors where the LO phonon modes of the lattice vibration provide a polarization for charge carriers through the long-range Coulomb interaction (Fröhlich potential). Later, the concept of self-trapping is extended to short-range interactions with acoustic phonons through deformation potential by Toyozawa (55) and in molecular crystals by Holstein (56). For charge carriers trapped by acoustic phonons, the elastic wave of the lattice vibration deforms the lattice, leading to elastic potential energy around the charge carriers: E = DΔ, where D is the deformation potential constant and Δ is the lattice volume dilation. When the potential energy is large enough, the charge carriers are trapped in the deep potential well and form bound states with correlation length comparable to the lattice constant (55). The acoustic phonons may include a range of q vectors to stabilize the self-trapping states, with the dominant phonon wavelength λ = 2π/q on the order of spatial extent of the self-trapped states (48).
In our work, the strong self-trapping in Cs2AgBiBr6 with a time constant of ~4.7 ps contrasts with the typical subpicosecond Fröhlich polaron formation time in lead halide perovskites. We attribute it to be the self-trapping by the deformation potential (acoustic phonons). The spatial extent of the lattice deformation by acoustic phonons (~3.7 nm) is comparable to the Fröhlich polaron size (diameter ~4 nm), which may not be a coincidence. It is probable that the charge carriers in Cs2AgBiBr6 are first localized by the optical phonons like what happens in lead halide perovskites forming Fröhlich polaron with a time constant of around 0.2 ps. After that, the strong distortion of the soft lattice within the Fröhlich polaron size may trigger a range of acoustic phonon modes with the average phonon wavelength λ = 2π/q on the order of the charge carrier spatial extent, which further localize the charge carriers contributing to the main loss of the photoconductivity. These processes can be seen as parts of the local lattice deformation, similar to the molecular deformation in organic semiconductors. Synergistic effects may occur between optical and acoustic modes when localizing the charge carriers. In the presence of the strong self-trapping, the band structure based on the periodic lattice breaks down and is substituted by the local polaronic picture, which is the reason why the e-h recombination can circumvent the bare electronic transition rules. Although polaron formation can also screen the recombination, the positive effect by the former predominates, leading to the efficient radiative recombination. Similar to organic semiconductors, the highly localized charge carrier transport by hopping between lattice sites with an activation energy and therefore increases with increased temperature (fig. S14). The presence of the strong self-trapping is most probably due to the Peierls-like structural instability toward lattice distortion of the semimetal Bi (57), which tends to cause strong coherent phonon oscillations in bismuth and its compounds such as Bi2Te3, BiI3, etc. (42–44, 57). In addition, the presence of silver halide soft bonds, which are frequently reported to form self-trapped carriers, may further undermine the photovoltaic performance (55). Further work may focus on doping of Cs2AgBiBr6 or incorporating it into a bulk heterojunction system, which could be a feasible strategy to reduce the strong electron-phonon coupling or the low mobility problem while maintaining the merits of the double perovskite.
In conclusion, through studying the photophysics of bismuth-based double perovskite Cs2AgBiBr6, we uncover that the indirect bandgap semiconductor has strong band-to-band radiative recombination with a temperature-dependent bandwidth, which is attributed to the electron-phonon coupling in the strong regime that breaks the classical electronic band picture. Ultrafast spectroscopy reveals that strong carrier self-trapping by acoustic phonons occurs with a time constant of around 4 ps, leading to a drop of more than 70% of the initial photoconductivity after the subpicosecond electron-LO phonon coupling, which is consistent with a carrier mobility that is as low as 0.05 cm2 V−1 s−1. DFT calculations further reveal that the large deformation potential >10 eV by acoustic phonons may be the cause of the large discrepancy of carrier properties between the Bi-based double perovskite and lead perovskites. These findings cast fresh doubts on the suitability of Bi-based double perovskites with soft lattice as potential photovoltaic materials.
MATERIALS AND METHODS
Sample preparation and characterization
In the SC fabrication, CsBr (213 mg, 1.00 mmol), BiBr3 (225 mg, 0.5 mmol), and AgBr (94 mg, 0.5 mmol) were dissolved in 3 ml of 47% HBr. After reacting at 120°C for 24 hours, the Cs2AgBiBr6 SCs appeared with slow cooling. For PC film fabrication, the Cs2AgBiBr6 SCs were dissolved in dimethyl sulfoxide with a temperature of 100° to 130°C. The solution was then spin-coated onto quartz substrate for spectroscopic characterizations. The as-casted films were then annealed at different temperatures (100°, 150°, 200°, and 250°C) for 5 min.
Ultraviolet-visible absorption measurements
The absorption, reflectance, and transmittance spectra were obtained using an ultraviolet (UV)–visible spectrometer (SHIMADZU UV-3600 UV-Vis-NIR Spectrophotometer) with an integrating sphere (ISR-3100).
PL, TRPL, and PLQY measurements
PL and TRPL measurements were performed using femtosecond excitation pulses (>50 fs) generated from a femtosecond laser system configured by Coherent Libra (50 fs; seed and amplifier integrated system) + OPerA Solo (optical parametric amplifier). The emitted light was collected at a backscattering angle by a spectrometer (Acton, SpectraPro-2500i) and charge-coupled device (CCD) (Princeton Instruments, PIXIS 400B) in PL measurements and by an Optronis OptoScope streak camera system, which has an ultimate temporal resolution of 6 ps in TRPL measurements. PLQY measurements were performed with a 473-nm continuous wave diode laser as excitation source. The PL was collected by an integrating sphere and sent to the PL detection system (Acton, SpectraPro-2500i).
Raman measurement
The Raman measurement was performed using a 633-nm (red) line from a He-Ne gas laser (8 mW) as the excitation source. The laser beam was focused on the sample using WITec alpha300 RAS microscope with long working distance 20× objective (spot size of ∼2 μm). The backscattered Raman signal was collected with an Acton spectrometer (1800 g/mm) and a thermoelectrically cooled Andor CCD detector.
AC Hall measurement
Hall measurements were performed on the samples, which are deposited on the glass substrate with four-point gold electrode contacts on each corner according to van der Pauw technique (58). The hall system uses a rotating parallel dipole line magnet that generates AC field with pure harmonic, unidirectional, and strong magnetic field followed by Fourier spectral analysis and lock-in detection of the Hall signal (58).
Transient reflection/transmission spectroscopy
Transient reflection/transmission measurements were performed with a Helios setup (Ultrafast Systems LLC). The pump pulses were generated from a Ti-sapphire regenerative amplifier (800 nm, 50 fs, 4 mJ, and 1 kHz; Coherent Libra) that passed through an optical parametric amplifier (Coherent OPerA Solo). The probe pulses were generated by passing a small fraction of the 800-nm pulses from the regenerative amplifier through a CaF2 crystal. In the transient reflection measurements, the reflected probe beam was collected at a small incident angle (<10°) using a complementary metal-oxide semiconductor sensor.
Time-resolved terahertz spectroscopy
The TRTS measurements were carried out using an optical pump–terahertz probe setup that is based on ZnTe nonlinear terahertz generation and detection. Femtosecond laser beam from an amplifier laser system (800 nm, 6 mJ, 35 fs, and 1 kHz) was split into two parts with one being used for pumping the ZnTe crystal for terahertz generation detection and the other being frequency doubled (400 nm) using a beta barium borate (BBO) crystal for pumping the samples. The sample used for TRTS measurement was high-quality PC films deposited on z-cut quartz substrate with a thickness of around 200 nm.
DFT calculations
For the calculations of the crystal structure optimization and electron structures of Cs2AgBiBr6, we used the all-electron-like projector augmented wave method and the Perdew-Burke-Ernzerhof revised for solids exchange correlation potential as implemented in the Vienna Ab initio Simulation Package code (59). The semicore of Bi atoms (5d orbital), Ag atoms (4p orbital), and Cs atom (5s and 5p orbitals) are treated as valence electrons, i.e., 15 valence electrons for Bi (5d106s26p3) atom, 17 valence electrons for Ag (4p64d105s) atom, and 9 valence electrons for Cs (5s25p66s) atom, respectively. The cutoff energy for the plane wave expansion of the wave functions is 800 eV. The primitive cell was completely optimized, including its lattice vectors and atomic positions. The Hellman-Feynman forces are less than of 1 meV/Å. The 4 × 4 × 4 Monkhorst-Pack grid of k-points for Brillouin zone integration of Cs2AgBiBr6 was used in the calculations of structure optimization. The optimized lattice parameter is 11.193 Å. The calculation method of deformation potential used was the Wei and Zunger’s method (50, 60). Spin-orbit coupling effect was taken into consideration because of the presence of heavy valence electrons from Bi and Ag ions. The dielectric constants and force constants were calculated on the basis of the density functional perturbation theory. We used the Phonopy code (61) to calculate the phonon spectrum within the harmonic approximation. A 2 × 2 × 2 supercell was used to ensure that the boundary atoms were not affected by the effects of the atomic displacement.
Acknowledgments
Funding: This research was supported by Nanyang Technological University under its start-up grant (M4080514), the Ministry of Education under its AcRF Tier 2 grants (MOE2016-T2-1-034), the Science and Technology Program of Guangzhou (no. 2019050001), the National Natural Science Foundation of China (NFSC) (grant no. 51802331), the National Research Foundation (NRF) Singapore under its Competitive Research Programme (NRF-CRP14-2014-03), the NRF Investigatorship (NRF-NRFI-2018-04), the Guangdong Provincial Key Laboratory of Optical Information Materials and Technology (grant no. 2017B030301007), the National Center for International Research on Green Optoelectronics, and the 111 Project. F.G. acknowledges the financial support from the Swedish Energy Agency (2018-004357) and Knut och Alice Wallenbergs Stiftelse (Dnr KAW 2019.0082). F.G. is a Wallenberg Academy Fellow. The computational work for this article was (fully/partially) performed on resources of the National Supercomputing Centre, Singapore (www.nscc.sg). R.S. and M.M. acknowledge the funding support from Ministry of Education, Singapore grant numbers AcRF Tier 1, RG191/17, and MOE2017. Author contributions: B.W. and T.C.S. conceived the idea for the manuscript and designed the experiments. B.W. developed the basic concepts and conducted the spectroscopic characterization. F.G., W.N., F.W., and S.Y. developed the synthesis procedures, performed physical characterization, and fabricated the solar cell devices. Q.X. performed the first principles DFT calculations. T.Y. and Z.X.S. measured the Raman spectra. S.L. and P.C.H. did the AC Hall measurements. Y.K.E.T., J.F., and M.F. helped with the PLQY and some of the other spectroscopic measurements. M.M. and R.S. performed the TRTS measurements. T.W.G. performed the UPS characterization. F.H. synthesized some of the crystals for comparison. T.C.S., F.G., G.Z., and B.W. guided the experiments, discussed the data, and led the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Data is available at https://doi.org/10.21979/N9/LTPBM2. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabd3160/DC1
REFERENCES AND NOTES
- 1.Green M. A., Ho-Baillie A., Snaith H. J., The emergence of perovskite solar cells. Nat. Photonics 8, 506–514 (2014). [Google Scholar]
- 2.Sahli F., Werner J., Kamino B. A., Bräuninger M., Monnard R., Paviet-Salomon B., Barraud L., Ding L., Leon J. J. D., Sacchetto D., Cattaneo G., Despeisse M., Boccard M., Nicolay S., Jeangros Q., Niesen B., Ballif C., Fully textured monolithic perovskite/silicon tandem solar cells with 25.2% power conversion efficiency. Nat. Mater. 17, 820–826 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Park B.-W., Philippe B., Zhang X., Rensmo H., Boschloo G., Johansson E. M. J., Bismuth based hybrid perovskites A3Bi2I9 (A: methylammonium or cesium) for solar cell application. Adv. Mater. 27, 6806–6813 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Slavney A. H., Hu T., Lindenberg A. M., Karunadasa H. I., A bismuth-halide double perovskite with long carrier recombination lifetime for photovoltaic applications. J. Am. Chem. Soc. 138, 2138–2141 (2016). [DOI] [PubMed] [Google Scholar]
- 5.McClure E. T., Ball M. R., Windl W., Woodward P. M., Cs2AgBiX6 (X = Br, Cl): New visible light absorbing, lead-free halide perovskite semiconductors. Chem. Mater. 28, 1348–1354 (2016). [Google Scholar]
- 6.Ning W., Gao F., Structural and functional diversity in lead-free halide perovskite materials. Adv. Mater. 31, 1900326 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Luo J., Wang X., Li S., Liu J., Guo Y., Niu G., Yao L., Fu Y., Gao L., Dong Q., Zhao C., Leng M., Ma F., Liang W., Wang L., Jin S., Han J., Zhang L., Etheridge J., Wang J., Yan Y., Sargent E. H., Tang J., Efficient and stable emission of warm-white light from lead-free halide double perovskites. Nature 563, 541–545 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Zhao X.-G., Yang J.-H., Fu Y., Yang D., Xu Q., Yu L., Wei S.-H., Zhang L., Design of lead-free inorganic halide perovskites for solar cells via cation-transmutation. J. Am. Chem. Soc. 139, 2630–2638 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Xiao Z., Meng W., Wang J., Yan Y., Thermodynamic stability and defect chemistry of bismuth-based lead-free double perovskites. ChemSusChem 9, 2628–2633 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Hoye R. L. Z., Eyre L., Wei F., Brivio F., Sadhanala A., Sun S., Li W., Zhang K. H. L., MacManus-Driscoll J. L., Bristowe P. D., Friend R. H., Cheetham A. K., Deschler F., Fundamental carrier lifetime exceeding 1 μs in Cs2AgBiBr6 double perovskite. Adv. Mater. Interfaces 5, 1800464 (2018). [Google Scholar]
- 11.Pan W., Wu H., Luo J., Deng Z., Ge C., Chen C., Jiang X., Yin W.-J., Niu G., Zhu L., Yin L., Zhou Y., Xie Q., Ke X., Sui M., Tang J., Cs2AgBiBr6 single-crystal x-ray detectors with a low detection limit. Nat. Photonics 11, 726–732 (2017). [Google Scholar]
- 12.Zhou L., Xu Y.-F., Chen B.-X., Kuang D.-B., Su C.-Y., Synthesis and photocatalytic application of stable lead-free Cs2AgBiBr6 perovskite nanocrystals. Small 14, 1703762 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Yang B., Pan W., Wu H., Niu G., Yuan J.-H., Xue K.-H., Yin L., Du X., Miao X.-S., Yang X., Xie Q., Tang J., Heteroepitaxial passivation of Cs2AgBiBr6 wafers with suppressed ionic migration for x-ray imaging. Nat. Commun. 10, 1989 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steele J. A., Pan W., Martin C., Keshavarz M., Debroye E., Yuan H., Banerjee S., Fron E., Jonckheere D., Kim C. W., Baekelant W., Niu G., Tang J., Vanacken J., Van der Auweraer M., Hofkens J., Roeffaers M. B. J., Photophysical pathways in highly sensitive Cs2AgBiBr6 double-perovskite single-crystal x-ray detectors. Adv. Mater. 30, 1804450 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Greul E., Petrus M. L., Binek A., Docampo P., Bein T., Highly stable, phase pure Cs2AgBiBr6 double perovskite thin films for optoelectronic applications. J. Mater. Chem. A 5, 19972–19981 (2017). [Google Scholar]
- 16.Gao W., Ran C., Xi J., Jiao B., Zhang W., Wu M., Hou X., Wu Z., High-quality Cs2AgBiBr6 double perovskite film for lead-free inverted planar heterojunction solar cells with 2.2 % efficiency. ChemPhysChem 19, 1696–1700 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Igbari F., Wang R., Wang Z.-K., Ma X.-J., Wang Q., Wang K.-L., Zhang Y., Liao L.-S., Yang Y., Composition stoichiometry of Cs2AgBiBr6 films for highly efficient lead-free perovskite solar cells. Nano Lett. 19, 2066–2073 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Wang M., Zeng P., Bai S., Gu J., Li F., Yang Z., Liu M., High-quality sequential-vapor-deposited Cs2AgBiBr6 thin films for lead-free perovskite solar cells. Solar RRL 2, 1800217 (2018). [Google Scholar]
- 19.Ning W., Wang F., Wu B., Lu J., Yan Z., Liu X., Tao Y., Liu J.-M., Huang W., Fahlman M., Hultman L., Sum T. C., Gao F., Long electron-hole diffusion length in high-quality lead-free double perovskite films. Adv. Mater. 30, 1706246 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Steele J. A., Puech P., Keshavarz M., Yang R., Banerjee S., Debroye E., Kim C. W., Yuan H., Heo N. H., Vanacken J., Walsh A., Hofkens J., Roeffaers M. B. J., Giant electron-phonon coupling and deep conduction band resonance in metal halide double perovskite. ACS Nano 12, 8081–8090 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Zelewski S. J., Urban J. M., Surrente A., Maude D. K., Kuc A., Schade L., Johnson R. D., Dollmann M., Nayak P. K., Snaith H. J., Radaelli P., Kudrawiec R., Nicholas R. J., Plochocka P., Baranowski M., Revealing the nature of photoluminescence emission in the metal-halide double perovskite Cs2AgBiBr6. J. Mater. Chem. C 7, 8350–8356 (2019). [Google Scholar]
- 22.Savory C. N., Walsh A., Scanlon D. O., Can Pb-free halide double perovskites support high-efficiency solar cells? ACS Energy Lett. 1, 949–955 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Volonakis G., Filip M. R., Haghighirad A. A., Sakai N., Wenger B., Snaith H. J., Giustino F., Lead-free halide double perovskites via heterovalent substitution of noble metals. J. Phys. Chem. Lett. 7, 1254–1259 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Li T., Zhao X., Yang D., Du M.-H., Zhang L., Intrinsic defect properties in halide double perovskites for optoelectronic applications. Phys. Rev. Appl. 10, 041001 (2018). [Google Scholar]
- 25.Ning W., Zhao X.-G., Klarbring J., Bai S., Ji F., Wang F., Simak S. I., Tao Y., Ren X.-M., Zhang L., Huang W., Abrikosov I. A., Gao F., Thermochromic lead-free halide double perovskites. Adv. Funct. Mater. 29, 1807375 (2019). [Google Scholar]
- 26.Schade L., Wright A. D., Johnson R. D., Dollmann M., Wenger B., Nayak P. K., Prabhakaran D., Herz L. M., Nicholas R., Snaith H. J., Structural and optical properties of Cs2AgBiBr6 double perovskite. ACS Energy Lett. 4, 299–305 (2018). [Google Scholar]
- 27.Yang J., Zhang P., Wei S.-H., Band structure engineering of Cs2AgBiBr6 perovskite through order–disordered transition: A first-principle study. J. Phys. Chem. Lett. 9, 31–35 (2018). [DOI] [PubMed] [Google Scholar]
- 28.L. Pavesi, S. V. Gaponenko, L. Dal Negro, Towards the First Silicon Laser (Springer Science & Business Media, 2012). [Google Scholar]
- 29.Mak K. F., Lee C., Hone J., Shan J., Heinz T. F., Atomically thin MoS2: A new direct-gap semiconductor. Phys. Rev. Lett. 105, 136805 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Saba M., Cadelano M., Marongiu D., Chen F., Sarritzu V., Sestu N., Figus C., Aresti M., Piras R., Lehmann A. G., Cannas C., Musinu A., Quochi F., Mura A., Bongiovanni G., Correlated electron-hole plasma in organometal perovskites. Nat. Commun. 5, 5049 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Koch S. W., Kira M., Khitrova G., Gibbs H. M., Semiconductor excitons in new light. Nat. Mater. 5, 523–531 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Varshni Y. P., Band-to-band radiative recombination in groups IV, VI, and III-V semiconductors (I). Phys. Stat. Solidi B 19, 459–514 (1967). [Google Scholar]
- 33.Yang Y., Yan Y., Yang M., Choi S., Zhu K., Luther J. M., Beard M. C., Low surface recombination velocity in solution-grown CH3NH3PbBr3 perovskite single crystal. Nat. Commun. 6, 7961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenwaks Y., Shapira Y., Huppert D., Picosecond time-resolved luminescence studies of surface and bulk recombination processes in InP. Phys. Rev. B 45, 9108–9119 (1992). [DOI] [PubMed] [Google Scholar]
- 35.Wu B., Zhou Y., Xing G., Xu Q., Garces H. F., Solanki A., Goh T. W., Padture N. P., Sum T. C., Long minority-carrier diffusion length and low surface-recombination velocity in inorganic lead-free CsSnI3 perovskite crystal for solar cells. Adv. Funct. Mater. 27, 1604818 (2017). [Google Scholar]
- 36.Brenner T. M., Egger D. A., Kronik L., Hodes G., Cahen D., Hybrid organic-inorganic perovskites: Low-cost semiconductors with intriguing charge-transport properties. Nat. Rev. Mater. 1, 15007 (2016). [Google Scholar]
- 37.Proctor C. M., Kuik M., Nguyen T.-Q., Charge carrier recombination in organic solar cells. Prog. Polym. Sci. 38, 1941–1960 (2013). [Google Scholar]
- 38.Vuong T. Q. P., Cassabois G., Valvin P., Liu S., Edgar J. H., Gil B., Exciton-phonon interaction in the strong-coupling regime in hexagonal boron nitride. Phys. Rev. B 95, 201202(R) (2017). [Google Scholar]
- 39.Toyozawa Y., Theory of line-shapes of the exciton absorption bands. Prog. Theor. Phys. 20, 53–81 (1958). [Google Scholar]
- 40.Wu X., Trinh M. T., Zhu X.-Y., Excitonic many-body interactions in two-dimensional lead iodide perovskite quantum wells. J. Phys. Chem. C 119, 14714–14721 (2015). [Google Scholar]
- 41.Trinh M. T., Wu X., Niesner D., Zhu X.-Y., Many-body interactions in photo-excited lead iodide perovskite. J. Mater. Chem. A 3, 9285–9290 (2015). [Google Scholar]
- 42.Qi J., Chen X., Yu W., Cadden-Zimansky P., Smirnov D., Tolk N. H., Miotkowski I., Cao H., Chen Y. P., Wu Y., Qiao S., Jiang Z., Ultrafast carrier and phonon dynamics in Bi2Se3 crystals. Appl. Phys. Lett. 97, 182102 (2010). [Google Scholar]
- 43.Jia L., Ma W. G., Zhang X., Ultrafast carrier dynamics in polycrystalline bismuth telluride nanofilm. Appl. Phys. Lett. 104, 241911 (2014). [Google Scholar]
- 44.Scholz M., Oum K., Lenzer T., Pronounced exciton and coherent phonon dynamics in BiI3. Phys. Chem. Chem. Phys. 20, 10677–10685 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Zhu X.-Y., Podzorov V., Charge carriers in hybrid organic-inorganic lead halide perovskites might be protected as large polarons. J. Phys. Chem. Lett. 6, 4758–4761 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Kentsch R., Scholz M., Horn J., Schlettwein D., Oum K., Lenzer T., Exciton dynamics and electron–phonon coupling affect the photovoltaic performance of the Cs2AgBiBr6 double perovskite. J. Phys. Chem. C 122, 25940–25947 (2018). [Google Scholar]
- 47.Lakehal M., Paul I., Microscopic description of displacive coherent phonons. Phys. Rev. B 99, 035131 (2019). [Google Scholar]
- 48.Morrissey F. X., Dexheimer S. L., Coherent acoustic phonon generation in exciton self-trapping. Phys. Rev. B 81, 094302 (2010). [Google Scholar]
- 49.Miyata K., Meggiolaro D., Trinh M. T., Joshi P. P., Mosconi E., Jones S. C., De Angelis F., Zhu X.-Y., Large polarons in lead halide perovskites. Sci. Adv. 3, e1701217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei S.-H., Zunger A., Predicted band-gap pressure coefficients of all diamond and zinc-blende semiconductors: Chemical trends. Phys. Rev. B 60, 5404–5411 (1999). [Google Scholar]
- 51.Delor M., Slavney A. H., Wolf N. R., Filip M. R., Neaton J. B., Karunadasa H. I., Ginsberg N. S., Carrier diffusion lengths exceeding 1 μm despite trap-limited transport in halide double perovskites. ACS Energy Lett. 5, 1337–1345 (2020). [Google Scholar]
- 52.Mante P.-A., Stoumpos C. C., Kanatzidis M. G., Yartsev A., Electron-acoustic phonon coupling in single crystal CH3NH3PbI3 perovskites revealed by coherent acoustic phonons. Nat. Commun. 8, 14398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su J., Zhang Z., Hou J., Liu M., Lin Z., Hu Z., Chang J., Hao Y., Pressure-dependent mechanical and thermal properties of lead-free halide double perovskite Cs2AgB″X6(B″═In, Bi; X═Cl, Br, I). Adv. Theory Simul. 2, 1900164 (2019). [Google Scholar]
- 54.Dong L., Sun S., Deng Z., Li W., Wei F., Qi Y., Li Y., Li X., Lu P., Ramamurty U., Elastic properties and thermal expansion of lead-free halide double perovskite Cs2AgBiBr6. Comput. Mater. Sci. 141, 49–58 (2018). [Google Scholar]
- 55.Toyozawa Y., Self-trapping of an electron by the acoustical mode of lattice vibration. I. Prog. Theor. Phys. 26, 29–44 (1961). [Google Scholar]
- 56.Holstein T., Studies of polaron motion : Part II. The “small” polaron. Ann. Phys. 8, 325–343 (1959). [Google Scholar]
- 57.Papalazarou E., Faure J., Mauchain J., Marsi M., Taleb-Ibrahimi A., Reshetnyak I., van Roekeghem A., Timrov I., Vast N., Arnaud B., Perfetti L., Coherent phonon coupling to individual Bloch states in photoexcited bismuth. Phys. Rev. Lett. 108, 256808 (2012). [DOI] [PubMed] [Google Scholar]
- 58.Gunawan O., Virgus Y., Tai K. F., A parallel dipole line system. Appl. Phys. Lett. 106, 062407 (2015). [Google Scholar]
- 59.Kresse G., Furthmüller J., Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996). [DOI] [PubMed] [Google Scholar]
- 60.Franceschetti A., Wei S.-H., Zunger A., Absolute deformation potentials of Al, Si, and NaCl. Phys. Rev. B 50, 17797–17801 (1994). [DOI] [PubMed] [Google Scholar]
- 61.Togo A., Tanaka I., First principles phonon calculations in materials science. Scripta Mater. 108, 1–5 (2015). [Google Scholar]
- 62.Feynman R. P., Hellwarth R. W., Iddings C. K., Platzman P. M., Mobility of slow electrons in a polar crystal. Phys. Rev. 127, 1004–1017 (1962). [Google Scholar]
- 63.Ōsaka Y., Polaron state at a finite temperature. Prog. Theor. Phys. 22, 437–446 (1959). [Google Scholar]
- 64.Frost J. M., Calculating polaron mobility in halide perovskites. Phys. Rev. B 96, 195202 (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabd3160/DC1