

Microfluidic platform reveals the effects of tumor environmental stress on natural killer cell biology.
Abstract
Solid tumors generate a suppressive environment that imposes an overwhelming burden on the immune system. Nutrient depletion, waste product accumulation, hypoxia, and pH acidification severely compromise the capacity of effector immune cells such as T and natural killer (NK) cells to destroy cancer cells. However, the specific molecular mechanisms driving immune suppression, as well as the capacity of immune cells to adapt to the suppressive environment, are not completely understood. Thus, here, we used an in vitro microfluidic tumor-on-a-chip platform to evaluate how NK cells respond to the tumor-induced suppressive environment. The results demonstrated that the suppressive environment created by the tumor gradually eroded NK cell cytotoxic capacity, leading to compromised NK cell surveillance and tumor tolerance. Further, NK cell exhaustion persisted for an extended period of time after removing NK cells from the microfluidic platform. Last, the addition of checkpoint inhibitors and immunomodulatory agents alleviated NK cell exhaustion.
INTRODUCTION
Cancer is the second leading cause of death around the world and has recently surpassed cardiovascular disease as the number one result of death in developed countries (1). In the past years, multiple breakthroughs in immunology such as chimeric antigen receptor (CAR) T/natural killer (NK) cells and immune checkpoint inhibitors (ICIs) have demonstrated great success in hematological cancers (e.g., acute myeloid leukemia and acute lymphoblastic leukemia) (2). Therefore, scientists and clinicians are working to expand the success of immunotherapy in hematological cancers to solid tumors (3).
Despite the initial promising results in hematological cancers, the use of ICIs in solid tumors has achieved moderate and heterogeneous success, partially due to the immunosuppressive environmental stress generated by solid tumors (4, 5). In this context, recent studies showed that tumor-associated environmental factors such as nutrient starvation, hypoxia, waste product accumulation, and pH acidification suppress T and NK cell antitumor activity (6–9). Thus, understanding the consequences of the environmental stress on immune cells is critical to develop more effective immunotherapies (10, 11). However, capturing the complexity of the solid tumor microenvironment (TME) with traditional two-dimensional (2D) models remains challenging (12, 13).
In this context, microfluidic technologies and organotypic models offer potential to mimic critical environmental features of the TME while allowing researchers to easily monitor the evolution of tumor and immune cells (13–17). Researchers developed numerous microdevices to impose gradients of oxygen, growth factors, or pH and evaluate cell response. Commonly, microdevices generate gradients by perfusing two parallel streams of liquid (e.g., medium with 20 and 1% O2) to create a gradient across the microdevice leveraging passive diffusion (18). Thus, here, we have evaluated the influence of the tumor environmental stress on NK cell functions using a tumor-on-a-chip model (13, 19). Our tumor-on-a-chip model includes a microchamber where breast cancer cells were embedded in a 3D matrix. A lumen located at one end of the microchamber was lined with endothelial cells and perfused with medium to nourish the cells, mimicking the vasculature present in the tumor. This design allowed us to mimic nutrient, pH, proliferation, and necrosis gradients across solid tumors. Our results demonstrated that NK cells initially exhibited promising cytotoxic capacity, destroying a significant percentage of tumor cells. However, culture in the device gradually eroded their cytotoxic capacity, leading to NK cell exhaustion. Further, we explored the plasticity of NK cells and their capacity of reversing immune exhaustion. In this context, the results demonstrated that NK cells did not revert from the exhausted phenotype, showing multiple molecular and functional alterations despite removing the environmental stress generated by the tumor.
RESULTS
Tumor-on-a-chip model for cell-based onco-immunology studies
Solid tumors generate a harsh microenvironment where environmental factors such as nutrient depletion, acidic pH, or waste product accumulation might affect the immune response (Fig. 1A). Thus, we used a tumor-on-a-chip microfluidic model to evaluate NK cell response and exhaustion. The model consists of a central microchamber where breast cancer cells (i.e., MCF7) with/without NK cells (i.e., NK-92) were embedded in a collagen hydrogel (Fig. 1B). After collagen polymerization, a polydimethylsiloxane (PDMS) rod located in the chamber flank was removed to generate a lumen through the collagen hydrogel. Then, the lumen was lined with endothelial cells [i.e., human umbilical cord endothelial cells (HUVECs)] and perfused with culture medium to generate a blood vessel surrogate that nourished the cells. The presence of the lumen in one of the microdevice flanks led to an asymmetric distribution of nutrients, and depending on the cell density used, gradients of cell viability could be generated in the tumor-on-a-chip platform. This property allowed us to mimic multiple tumor scenarios, from low-density tumors, where no necrosis was observed, to high-density tumors, where cell metabolism generated a large necrotic core inside of the microdevice (fig. S1). In this study, we decided to seed MCF7 cells at 1.5 million cells/ml, which rendered a highly viable zone next to the blood vessel surrogate and a necrotic core at the innermost area (Fig. 1C). More specifically, after 7 days in cell culture, tumor cells located in the vicinity of the lumen (i.e., proximal region) showed high cell viability (>90%) and proliferated, forming multicellular clusters. Conversely, most cells located in the farthest (i.e. 15 mm far from the lumen) region of the microchamber (i.e., distal region) did not proliferate and died (cell viability of <15%). Most tumor cells located in the center of the chamber (i.e., central region) remained viable after 7 days (>85%) but exhibited slower proliferation compared with those cells located in the proximal region (i.e., threefold change in the area occupied by live cells in proximal versus central).
Fig. 1. Tumor-on-a-chip.
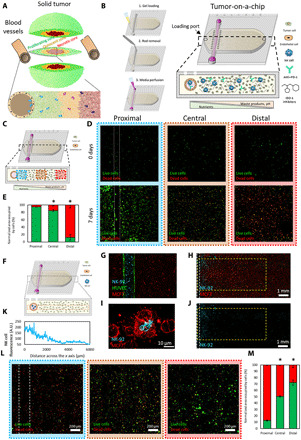
(A) Schematic representation of the different tumor phenotypes generated in a solid tumor due to nutrient starvation. (B) Scheme of the tumor-on-a-chip microdevice. The bottom panel shows the microdevice cross-section. The lumen was lined with endothelial cells (e.g., HUVECs) to generate a blood vessel surrogate, allowing the perfusion of medium, NK-92 cells, anti–PD-L1 antibodies (i.e., atezolizumab), or IDO-1 inhibitors (i.e., epacadostat). (C) Schematic representation of the microdevice, and the proximal, central, and distal regions are shown in blue, orange, and red, respectively. (D) Confocal images showing live and dead MCF7 cells in green and red, respectively, after 0 and 7 days in the device. (E) Area occupied by live cells (in green) and dead cells (in red) in the proximal, central, and distal regions. Asterisks denote P value of <0.05. (F) Scheme of the experimental setup. (G) Confocal image showing the dispersal of cells in the tumor-on-chip device. MCF7 cells (in red) are embedded in the collagen gel, while NK-92 cells (in blue) and HUVEC cells (in green) are embedded in the lumen. (H) This confocal image shows NK-92 cells (in blue) migrating across the chamber and MCF7 cells (in red). (I) Confocal image representing an NK-92 engaging with an MCF7. (J) This confocal image highlights the migration of NK cells out of the lumen and into the chamber. (K) Quantification of NK-92 migration across the x axis measured by NK cell fluorescence. A.U., arbitrary units. (L) MCF7 and NK-92 cells were cocultured for a week. The proximal region has a higher percentage of dead cells due to NK interaction close to the nutrient-rich lumen. (M) Quantification showing that distance from the lumen and the number of live cancer cells (in green) is proportional. Asterisk denotes P value of <0.05 compared with the proximal region; graphs show means ± SD.
To evaluate the potential of the platform to study tumor immunotherapy, we used NK-92 cells, which are known for retaining cytotoxic capacity both in vitro and in vivo and have been extensively tested in clinical trials. NK-92 cells were perfused through the blood vessel surrogate to evaluate their migration through the collagen hydrogel (Fig. 1D). Although our experiments used a collagen hydrogel, the in vivo composition of the extracellular matrix is complex, including a variety of components (e.g., proteins, glycoproteins, and glycosaminoglycans). In this context, we observed that the collagen hydrogel architecture, controlled by the polymerization temperature, had an effect on NK-92 cell migration (fig. S3 and movies S1 and S2). These results highlight the potential of the tumor-on-a-chip platform to study the effect of different matrix formulations on NK cell biology. When using a collagen hydrogel, NK-92 cells penetrated through the hydrogel, and after 3 days, NK-92 cells were observed in the distal area interacting with MCF7 cells. However, NK cell density across the chamber was not homogeneous, generating a gradient of NK cell density, with most of the NK-92 cells concentrated in the proximal area and rapidly decreasing below 50% density at 2 mm (Fig. 1D). To evaluate the effect of any potential environmental gradient generated in the microdevice, we needed to guarantee that NK-92 cells were present at the distal area. Therefore, NK-92 cells were directly embedded in the hydrogel with tumor cells (0.5 million NK-92 versus 1.5 million MCF7 cells/ml) to ensure that NK-92 cells were present at the distal area and also guarantee that the initial cell density was homogeneous across the microchamber. We also tested NK-92 cell viability when NK cells were embedded in the microdevice for 1 and 7 days (fig. S2). The images demonstrated that NK-92 cells remained viable (>90%) after 24 hours in culture as well as after 7 days. In addition, the images showed that NK-92 cells proliferated significantly slower compared with MCF7 cells in the tumor-on-a-chip (figs. S1 and S2). After 7 days in coculture, the presence of NK cells led to a marked decrease in tumor cell viability in the proximal area (<15%), and no MCF7 clusters were observed. We used an apoptosis fluorescent dye in combination with multiple cell trackers to distinguish dead tumor cells from potentially dead NK cells. However, these stainings did not last for 7 days; therefore, we limited our apoptosis imaging to 48 hours after seeding the cells. The apoptosis staining revealed that most dead cells in the proximal area were MCF7 cells, whereas most NK-92 cells stained negative for the apoptosis marker, demonstrating the capacity of NK-92 cells to destroy tumor cells in this region (fig. S4). In the central area, the presence of NK-92 cells led to a moderate decrease in MCF7 cell viability (from 85% in the absence of NK cells to 50%), suggesting a decrease in NK-92 cell cytotoxic capacity in this region. Last, MCF7 cells in the distal region exhibited significant resistance to NK-92 cell cytotoxicity compared with those ones located in the distal region (cell viability of 70%) (fig. S4). These results suggested that, in the distal area of the microchamber, environmental gradients generated by cell metabolism suppressed NK cell cytotoxic capacity, allowing MCF7 cells to proliferate. MCF7 is a cell line that has been traditionally used in research laboratories. However, this cell line may present significant differences compared with patient-derived cells, which are more heterogeneous and capture more efficiently the in vivo physiology. Thus, we also evaluated the potential of the platform for precision medicine culturing patient-derived breast cancer cells. First, we evaluated the capacity of NK-92 cells to destroy patient-derived tumor cells in a traditional 2D assay (fig. S5). In these experiments, we seeded a confluent monolayer of patient-derived cancer cells in 96-well plate, and after 24 hours, we added NK-92 cells on top at different NK:tumor cell ratios. After 24 or 72 hours, we removed floating dead cells and NK-92 cells and stained viable patient-derived cancer cells. The results demonstrated that after 24 hours, NK-92 cells exhibited a moderate cytotoxicity against patient-derived cancer cells when cultured at 1 NK:1 tumor cell ratio. After 72 hours, the cytotoxicity was more evident, observing a significant decrease in the amount of viable patient-derived cancer cells at the multiple ratios analyzed. Next, we set out to evaluate the capacity of the model to generate a necrotic core using patient-derived cells. We cultured patient-derived cells in the model at different cell densities (i.e., 1, 4, and 12 million cells/ml) for 7 days (fig. S6, A to C). The viability analysis revealed that the generation of viability gradients across the hydrogel was possible, but we required a higher cell density compared with MCF7 cells. These results suggested that the patient-derived cells that we used had a lower nutrient consumption/requirement compared with MCF7 cells. Last, we cocultured NK-92 cells and patient-derived cancer cells in the tumor-on-a-chip at the same densities used in the MCF7 experiments (i.e., 0.5 × 106 NK cells/ml and 1.5 × 106 MCF7 cells/ml). In the presence of NK-92 cells, we observed a homogeneous decrease in cell viability across the proximal, central, and distal region(fig. S6, D to G). Together, these results suggested that at these cell densities, patient-derived cells did not generate enough environmental stress to affect NK-92 cell cytotoxicity (fig. S6).
NK cell exhaustion in the tumor-on-a-chip microdevice
Next, we set out to evaluate molecular changes in NK-92 cells in response to the environment generated in the tumor-on-a-chip microdevice (Fig. 2A). After 7 days in culture, we degraded the collagen hydrogel and selectively removed MCF7 cells from the cell suspension to isolate NK-92 cells (>99% purity) (Fig. 2B). The reverse transcription quantitative polymerase chain reaction (RT-qPCR) experiments and nonhierarchical clustergram analysis showed that NK-92 cells exhibited changes in gene expression after 1 day in the microdevice that led to more intense changes after 7 days (Fig. 2C; clustergram is shown at higher resolution in fig. S7). More specifically, we observed that multiple genes associated with immune exhaustion (e.g., IDO-1, PD-1, and CTLA-4) were up-regulated in the microdevice. Further, genes associated with nutrient starvation, hypoxia [e.g., hypoxia-inducible factor 1A (HIF1A)], and angiogenesis [e.g., vascular endothelial growth factor (VEGF)] were also up-regulated, suggesting that NK-92 cells became exhausted and activated several adaptative responses. On the other hand, multiple genes associated with NK cell survival, proliferation, and activation [e.g., GZMB, BCL2, FASL, and interleukin-15 (IL-15)] were significantly down-regulated, highlighting the immunosuppressive effect of the environment generated inside of the tumor-on-a-chip microdevice. After 7 days, some genes (e.g., IDO-1 and PD-1) gradually changed their expression compared with 1 day, whereas others (e.g., CTLA-4) were dysregulated only after 7 days. These observations suggested that NK-92 cell exhaustion encompasses a complex variety of molecular responses with multiple temporal dynamics. Next, we studied whether NK cells located in the different regions of the tumor-on-a-chip microdevice (i.e., proximal versus distal) exhibited different molecular adaptations. Thus, after 7 days, we isolated NK-92 cells from the proximal and distal region and compared their gene expression profile (Fig. 2E). The gene expression analysis showed that NK-92 cells from the distal region had a significantly different expression profile compared with those cells located in the proximal area (Fig. 2F). More specifically, multiple genes related to immune recruitment such as CCL4 (associated with NK cell recruitment), CCL5 (T cell recruitment), CCL18, and IRF1 (antitumor response in NK cells) were up-regulated in the distal region compared with the proximal area. In addition, the hypoxia-associated gene HIF1A was also up-regulated. On the other hand, genes associated with chemokine receptors (e.g., CCR1, CCR2, and CCR4) and NK cell survival (e.g., BLC2) were down-regulated in the distal region. Together, these results suggested that NK cells located in the distal region were less proliferative and less responsive to chemokines, whereas they up-regulated their proinflammatory response.
Fig. 2. Culture in the tumor-on-a-chip microdevice led to NK cell exhaustion.
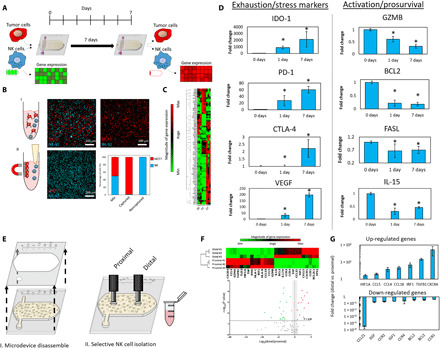
(A) Schematic representation of an experiment measuring immune exhaustion. NK cells and MCF7 cells were mixed with the collagen mixture and cocultured in a 1:3 ratio (0.5 million cells/1 ml:1.5 million cells/1 ml) for 7 days. This approach guaranteed that NK cell density was homogeneous across the hydrogel (e.g., proximal versus central versus distal area). After 7 days, the NK cells were isolated, and gene expression and other characteristics were measured. (B) Scheme of NK cell separation. MCF7 cells attach to magnetic beads, thus isolating NK cells in suspension. Confocal images represent captured MCF7 cells (in red) and isolated NK cells (in blue). The graph highlights the efficiency of NK cell isolation. (C) Cluster graph depicting gene expression after 0, 1, and 7 days. (D) Bar graphs show the up-/down-regulation of exhaustion markers and activation/prosurvival genes. (E) Scheme of spatially controlled NK cell isolation from proximal and distal regions. (F) Cluster graph and volcano plot quantifying gene expression in the proximal and distal regions. (G) Bar graphs depicting the increase of up-regulated genes and decrease of down-regulated genes in the distal versus proximal regions. Asterisk denotes P value of <0.05; graphs show means ± SD.
The gene expression analysis showed that NK cells in the tumor-on-a-chip microdevice exhibited a progressive immune exhaustion. Next, we set out to determine whether NK cell exhaustion was caused by environmental stress generated in the microdevice or merely by chronic activation due to the presence of tumor cells for 7 days. Therefore, we cocultured MCF7 and NK-92 cells in a 3D collagen hydrogel in traditional 96-well plates using the same experimental conditions used in the tumor-on-a-chip experiments (e.g., cell-to-medium ratio, MCF7:NK-92 cell ratio, collagen density, and medium changes), and NK-92 cells were isolated after 7 days (Fig. 3A). The results demonstrated that NK cells isolated from the well plate had a different gene expression profile compared with those NK cells extracted from the tumor-on-a-chip device (Fig. 3B). Further, the NK cell phenotype in the traditional well plate was more similar to the profile obtained in naïve NK cells (i.e., NK-92 cells never exposed to the tumor cells) (Fig. 3C). In this context, multiple genes associated with immune exhaustion that were up-regulated in the tumor-on-a-chip microdevice showed no changes in the well plate experiments (e.g., IDO-1, MICA, and VEGF). In addition, genes related to NK cell activation and survival that were down-regulated in the tumor-on-a-chip showed a more moderate down-regulation (e.g., GZMB and BCL2) or, in some cases, up-regulation (e.g., FASL) (Fig. 3D). The gene set enrichment analysis (GSEA) showed that NK-92 cells cultured in the tumor-on-a-chip microdevice, compared with the well plate experiments, up-regulated pathways associated with immune migration, immune recruitment, cytokine secretion, inflammatory response, and immune exhaustion, highlighting the differences between these two sets of NK-92 cells (Fig. 3E). Combined, these results suggested that chronic activation of NK cells (i.e., coculture with MCF7 cells for 7 days) plays a minor role in regulating the changes observed in the tumor-on-a-chip, highlighting the impact of the environmental stress generated in the tumor-on-a-chip platform on NK cell exhaustion.
Fig. 3. Comparison of NK cell exhaustion in the tumor-on-a-chip microdevice with traditional 3D culture.
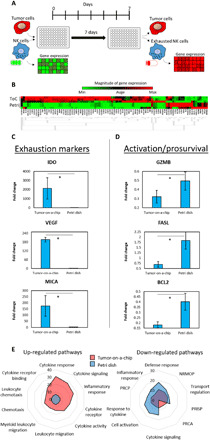
(A) Schematic representation of an experiment measuring immune exhaustion. NK cells and MCF7 cells were cocultured in a 1:3 ratio (0.5 million cells/1 ml:1.5 million cells/1 ml) for 7 days in a well plate. After 7 days, the NK cells were isolated, and gene expression and other characteristics were measured. This experiment shows similar exhaustion patterns in a well plate rather than a tumor on a chip device. (B) Clustergram showing changes in gene expression in the tumor-on-a-chip (ToC) compared with the petri dish experiments (Petri). (C and D) Bar graphs depicting the change in gene expression of exhaustion markers and activation/prosurvival genes in a tumor-on-a-chip device versus a petri dish. (E) Spider plot showing the 10 gene pathways more differentially affected in the tumor-on-a-chip microdevice compared with experiments performed in traditional well plates. NRMOP, negative regulation of multicellular organismal process; PRISP, positive regulation of immune system process; PRCA, positive regulation of cell activation; PRCP, positive regulation of cytokine production. These pathways were identified as the most affected using the GSEA software. Asterisk denotes P value of <0.05; graphs show means ± SD.
The gene expression analysis demonstrated that the tumor-on-a-chip platform led to more severe immune exhaustion, showing up-regulation of exhaustion markers such as PD-1 and IDO-1 that were not observed in traditional well plates. Therefore, we set out to evaluate the potential of the platform to test immunotherapies targeting immune exhaustion. First, we tested the effect of atezolizumab (i.e., anti–PD-L1 antibody) and epacadostat (i.e., IDO-1 inhibitor) in a traditional 2D assay, where NK-92 cells were seeded on top of a MCF7 cell monolayer in the presence/absence of these drugs. The 2D assay guarantees that tumor and immune cells are directly and homogeneously exposed to the drug, preventing any potential undesired bias caused by the presence of 3D hydrogels (e.g., drug penetration or absorption). Atezolizumab recently received U.S. Food and Drug Administration approval for breast cancer to treat tumors exhibiting immune exhaustion (i.e., PD-1/L1 expression). However, when tested in a 96-well plate assay, where no immune exhaustion was observed, atezolizumab did not affect NK-92 cell cytotoxic capacity (Fig. 4A). We also tested an IDO-1 inhibitor (i.e., epacadostat) that showed promising potential in in vitro and in vivo studies where IDO-1 was overexpressed. Similarly, in the well plate assay, where no up-regulation of IDO-1 was observed, the IDO-1 inhibitor did not improve NK-92 cell cytotoxic capacity. Further, both epacadostat and tryptophan deprivation experiments in traditional 96-well plates showed no effect on NK-92 cell growth (fig. S8). On the other hand, when tested in the tumor-on-a-chip microdevice, both atezolizumab and epacadostat showed a different response (Fig. 4B). In the presence of the IDO-1 inhibitor, we observed an increase in the amount of dead MCF7 cells, showing a displacement of the necrotic front toward the inner areas of the microchamber. Similarly, the atezolizumab increased MCF7 cell death, leading to a larger increase of the necrotic area. However, neither drug led to complete destruction of the MCF7 cells, as viable cells were still present in the most distal areas of the tumor-on-a-chip. Together, these experiments demonstrated that immunomodulatory agents such as checkpoint and metabolic inhibitors partially alleviated immune exhaustion. However, complete eradication of the tumor cells located in the innermost layers of the tumor may require new approaches targeting the effects of the tumor microenvironment.
Fig. 4. Effect of ICIs and IDO-1 inhibitors in traditional assays and the tumor-on-a-chip.
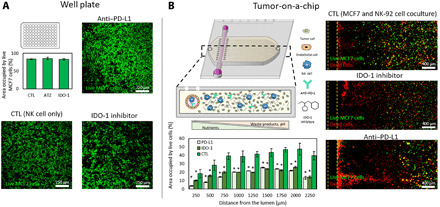
(A) The potential effect of atezolizumab (anti–PD-L1 antibody) and epacadostat (IDO-1 inhibitor) was evaluated in traditional well plates. A confluent monolayer of MCF7 was seeded on 96-well plates, and NK-92 cells were added 24 hours later (9 MCF7:1 NK ratio) with/without the IDO-1 inhibitor or the PD-L1 antibody. Confocal images showed no significant improvement on NK-92 cell cytotoxic potential. (B) Similar experiment evaluating epacadostat and atezolizumab in the tumor-on-a-chip microdevice. Confocal images demonstrated that both the PD-L1 antibody and IDO-1 inhibitor increased MCF7 cell necrosis. Dead cells (shown in red) concentrated in the vicinities of the lumen, whereas live MCF7 cells (shown in green) remained present in the farthest (distal) region. Asterisk denotes P value of <0.05; graphs show means ± SD.
Recovery of exhausted NK cells
New therapies that reverse or prevent immune exhaustion have great potential, but the mechanisms driving this process are still poorly understood. In our platform, NK-92 cells rapidly exhibited signs of progressive immune exhaustion. Thus, we set out to study whether removing these exhausted immune cells from the suppressive environment could return them to their original phenotype. To explore this hypothesis, after coculturing NK-92 cells in the tumor-on-a-chip for 7 days, NK cells were isolated from the microdevice and cultured again in a traditional flask for an additional week before analyzing their expression profile (Fig. 5A). The results demonstrated that exposed NK cells (i.e., NK cells isolated form the microdevice and subcultured in a flask) had a different molecular profile compared with naïve NK cells (i.e., NK cells never exposed to MCF7) (Fig. 5B). Exposed NK cells exhibited up-/down-regulations in a large percentage of the genes analyzed. We observed that several genes associated with immune exhaustion and stress response (e.g., IDO-1, CCl28, VEGF, and MICA), which were progressively up-regulated in cells cultured in the tumor-on-a-chip, returned to their original values after 1 week in traditional flasks (Fig. 5C). In addition, genes traditionally associated with viral and bacterial infection [e.g., Toll-like receptor 4 (TLR4) and TLR5] were up-regulated, suggesting that exposed NK cells may retain some memory state after the coculture with tumor cells. However, multiple prosurvival genes that were down-regulated (GZMB, IL-15, or BCL2) in the tumor-on-a-chip microdevice exhibited partial or no recovery after 1 week (Fig. 5D). Similarly, other exhaustion-associated genes that were gradually up-regulated in the microdevice showed partial or no recovery. Together, these results point out that NK-92 cells had a limited capacity to recover from the exposure to the environmental stress exerted by the tumor. In this context, whereas some changes were completely reversible, the majority remained present after removing NK cells from the suppressive environment, suggesting that exposure to the tumor microenvironment might exert long-lasting effects on NK cell biology. In this study, we limited our analysis to 7 days (i.e., 7 days in the microdevice followed by 7 days of recovery in a flask); thus, additional studies involving long-term culture (i.e., weeks and months) are needed to determine whether these stress-induced alterations are permanent.
Fig. 5. NK cell recovery from the tumor-on-a-chip platform.
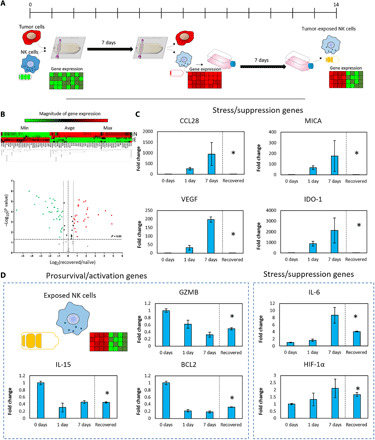
(A) Schematic representation of the experiments evaluating the recovery capacity of NK-92 cells. NK-92 and MCF7 cells were cocultured (0.5 million NK:1.5 million MCF7 cells/ml) for 7 days. Then, NK-92 cells were isolated from the tumor-on-a-chip and subcultured in traditional flasks for an additional week before analyzing their gene expression. (B) Clustergram and volcano plot comparing gene expression in exposed NK cells with naïve NK cells. (C) Bar graphs showing multiple genes that were up-/down-regulated in the tumor-on-a-chip microdevice but returned to the basal expression after 1 week. (D) Bar graphs showing genes that showed partial or no recovery to the basal expression compared with naïve NK cells. Asterisk denotes P value of <0.05; graphs show means ± SD.
Functional effects of NK cell exhaustion
The RT-qPCR analysis demonstrated that exposed NK cells had a different molecular phenotype compared with naïve NK cells, exhibiting a profile dominated by exhaustion markers (Figs. 5 and 6A). Thus, we evaluated the functional consequences of these molecular alterations in exposed NK cells. We analyzed changes in NK cell metabolism using optical metabolic imaging. More specifically, we studied changes in NK cell redox ratio and NAD(P)H (reduced form of nicotinamide adenine dinucleotide phosphate) and FAD (flavin adenine dinucleotide) fluorescence lifetimes (Fig. 6B). The analysis revealed that exposed NK cells had decreased redox ratio, decreased NAD(P)H mean lifetime (τm), and increased FAD τm compared with naïve NK cells (Fig. 6C and fig. S9). Gaussian distribution analysis of the redox ratio indicates a single population of cells for both naïve and exposed NK cells (Fig. 6C). Cells need to maintain their redox ratio to effectively perform their functions, and a decreased redox ratio has been associated with the accumulation of reactive oxygen species and functional impairment (20–22). In addition, a decreased NAD(P)H τm and an increased FAD τm indicate reduced binding activities for NAD(P)H and FAD (23). Thus, exposed NK cells exhibited rewiring of their metabolic phenotype that persisted after a week out of the tumor-on-a-chip microdevice. We used metabolic perturbation experiments to identify alterations in specific metabolic pathways that could explain the decrease in redox ratio and NAD(P)H τm and increase in FAD τm observed in the tumor-on-a-chip (Fig. 6D). In these experiments, naïve NK-92 cells were cultured in traditional well plates in the presence of metabolic inhibitors blocking glycolysis [i.e., 2-deoxyglucose (2DG)], fatty acid oxidation (i.e., etomoxir), or the electron transport chain complex V (i.e., oligomycin). In this context, inhibition of the electron transport chain complex V with oligomycin rendered the most similar trend (i.e., reduction of redox ratio and increase in FAD τm) compared with exposed NK cells. In addition, deprivation of IL-2, a known activator of NK cells, led to some of the changes observed in exposed NK cells (i.e., decreased redox ratio and increased FAD τm), suggesting that exposed NK cells might be in a more anergic state than naïve NK cells. Real-time microscopy also revealed that exposed NK cells exhibited different migratory properties (e.g., slower speed and decreased migrated distance) in 3D collagen hydrogels compared with naïve NK cells (Fig. 6, E and F, and fig. S10). However, identification of the specific metabolic pathways affected requires more in-depth studies. In this context, future studies could leverage techniques such as 1H nuclear magnetic resonance or mass spectrometry to pinpoint the metabolic alterations exhibited by these recovered NK cells. Last, we evaluated the cytotoxic capacity of exposed NK cells compared with naïve NK cells (Fig. 6G). We seeded a confluent monolayer of MCF7 cells in traditional well plates; 24 hours later, we added naïve or exposed NK-92 cells at different NK:MCF7 cell ratios; and another 24 hours later, we evaluated cell viability. The confocal images demonstrated that exposed NK cells still retained their capacity to kill tumor cells, but their cytotoxic capacity was significantly lower compared with naïve NK cells at all the ratios analyzed. Together, these results highlighted that the environmental stress generated in the tumor-on-a-chip significantly influence NK cell molecular and functional response. In addition, multiple changes persisted after removing the environmental stress, suggesting that these adaptations might last for long periods of time or even indefinitely. Although ICIs and metabolic inhibitors alleviated the exhaustion generated by the tumor microenvironment, they did not lead to a complete eradication of the tumor, indicating that new or alternative treatment combinations should be considered.
Fig. 6. Functional changes in exposed NK cells.
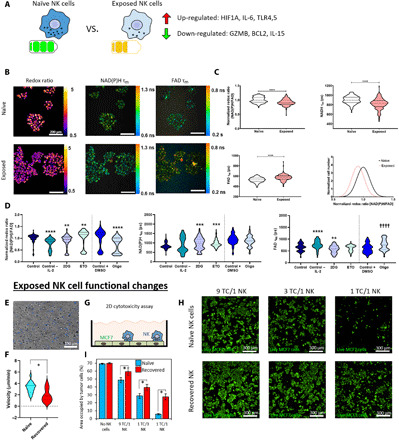
(A) Exposed NK cells isolated from the tumor-on-a-chip microdevice still exhibited multiple gene expression alterations after a week in traditional flasks. (B) Optical metabolic imaging was used to visualize intracellular NAD(P)H and FAD fluorescence intensities [redox ratio = NAD(P)H intensity divided by FAD intensity] and mean fluorescence lifetimes (τm). (C) Violin plots showing the analysis of NK cell redox ratio based on NAD(P)H and FAD intensity. Exposed NK cells had decreased redox ratio compared with naïve NK cells, indicating metabolic rewiring. In addition, exposed NK cells showed decreased NAD(P)H τm and increased FAD τm. (D) Violin plots depicted changes in redox ratio, NAD(P)H τm, and FAD τm caused by different metabolic inhibitors and chemokines: 2DG, glycolysis inhibitor; etomoxir (ETO), fatty acid oxidation inhibitor; oligomycin (Oligo), electron transport chain complex V inhibitor; DMSO, dimethyl sulfoxide. (E and F) Time-lapse microscopy analyzing exposed NK cell migration in 3D collagen hydrogels. Individual NK cell trajectories are depicted in different colors. Violin plot showing slower migration speed in exposed NK cells. (G and H) Cytotoxicity analysis of exposed and naïve NK cells. A monolayer of MCF7 was seeded in traditional well plates, and naïve or exposed NK cells were added on top. Confocal images showed the killing capacity of both exposed and naïve NK cells. (I) Bar graph analyzing the destruction of MCF7 cells shown in (H) for different ratios of tumor cells (TC) to NK cells (NK). Exposed NK cells exhibited decreased cytotoxic capacity compared with naïve NK cells. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001; graphs show means ± SD.
DISCUSSION
The development of ICIs that target immune exhaustion (e.g., PD-1 and CTLA-4 antibodies) can significantly improve antitumor immune responses and patient outcomes. However, as clinicians continue to explore these therapies in clinical trials, the results point out the capacity of solid tumors to escape immunosurveillance and suppress the immune response by a variety of mechanisms (24–26). Therefore, the development of new tools that allow researchers to decipher the complex mechanisms driving immune exhaustion could accelerate the development of next-generation immunotherapies (12). In this context, our tumor-on-a-chip platform provides a simple and versatile tool to evaluate the effect of environmental stress such as nutrient deprivation, acidic pH, or waste product accumulation (13). Immune penetration into solid tumors affects tumor evolution and remains an active research area in the immunotherapy community. In this context, the presence of effector cells such as cytotoxic T cells and NK cells has been associated with good prognosis (27, 28). However, multiple studies have shown the capacity of the tumor to exclude immune cells by several mechanisms (e.g., generation of interstitial pressure), which limits the efficacy of immune cells to destroy the tumor (29). Because multiple immunotherapy treatments rely on injecting immune cells through the patient’s bloodstream, deciphering the mechanism driving immune infiltration might significantly improve immunotherapy efficiency (30). Our platform includes a biomimetic blood vessel that enables the perfusion of immune cells to study immune penetration. Our proof-of-concept experiment demonstrated the capacity of NK-92 cells to migrate several hundreds of micrometers, although most of them did not reach the distal area (Fig. 1). Future studies could include a higher tumor cell density or generate interstitial pressure using the other ports to study their effect on immune cell migration. In this context, multiple microfluidic devices have been designed to study immune/tumor interactions with blood vessels (31–33). In subsequent experiments, we directly embedded NK-92 cells in the hydrogel to ensure a homogeneous distribution across the matrix, allowing us to study immune exhaustion in the proximal, central, and distal region. The results observed in the tumor-on-a-chip platform demonstrated a correlation between the generation of a necrotic core and the presence of pH gradients. In addition, in a previous study, we observed that oxygen played a minor role in the generation of the necrotic core, leading to a moderate increase in the number of dead cells observed in the distal region (13). Our platform is permeable to oxygen, demonstrating that MCF7 cells did not generate hypoxia within the microdevice at 30 million cells/ml (19). Together, these observations suggested that waste product accumulation and nutrient consumption played a critical role in the necrosis observed in the platform. However, in this study, we did not identify the specific environmental factor, or combination of factors, driving NK cell exhaustion. Thus, the process would require a noninvasive technique capable of monitoring metabolite evolution in real time. Mass spectrometry imaging (MSI) could provide valuable information about the spatial distribution of metabolites in the platform, but MSI sample preparation imposes some challenges when working with live samples (e.g., flash-freezing) (34). We believe that recent advances in Raman and Fourier transform infrared spectroscopy/microscopy offer an exciting nondestructive alternative to monitor metabolite evolution in real time (35). Another limitation of the present study is the fact that the effect of hypoxia on NK-92 cells was not considered. Future studies could add this layer of complexity by placing the microdevice in a hypoxia incubator (i.e., 1% O2) and perfusing oxygenated medium through the lumen (1% O2). Our study demonstrated that the environmental stress generated by the tumor cells significantly impaired NK cell gene expression and cytotoxicity, especially in the innermost regions of the model. In this context, previous reports have shown that chronic activation can also impair NK cells (36, 37), and our study suggested that environmental stress might even play a major role in NK-92 cell exhaustion (38, 39). A limitation of our study was the inability to distinguish between dead MCF7 cells and dead NK-92 cells with the viability staining. Thus, the contribution of dead NK-92 cells to the total amount of dead cells remains unknown. On the other hand, the NK-92 monocultures showed that NK-92 cells proliferalted slowly in the platform (fig. S2), suggesting that the majority of dead cells observed were dead MCF7 cells (Fig. 1 and fig. S1), which was also in agreement with the apoptosis staining (fig. S3). Previous reports have studied NK cell exhaustion and dysfunction in vivo, identifying changes in multiple cell receptors, chemokines, and molecular pathways (37, 40). In vivo, exhausted NK cells frequently exhibit up-regulation of PD-1, IDO-1, TIGIT, LAG-3, TIM-3, MICA, and CCL18 and down-regulation of CD16, GZMB, IL-15, and tumor necrosis factor–α (TNF-α), among other markers (Fig. 7A). In this study, we analyzed gene expression changes in multiple pathways associated with NK cell exhaustion and dysfunction. Molecular analysis demonstrated that multiple canonical T cell exhaustion pathways such as IDO-1, CTLA-4, and PD-1 were also up-regulated in NK-92 cells. Although the PD-1 pathway has been significantly explored in NK cells, our knowledge about the CTLA-4 pathway is scarce, including conflicting reports discussing the expression of CTLA-4 in human NK cells (41, 42). In this context, our model revealed that environmental stress generated by MCF7 cells led to overexpression of CTLA-4 in NK-92 cells, suggesting new therapeutic combinations using NK-92 cells and CTLA-4 inhibitors such as ipilimumab. We also observed changes in GZMB, TNF-α, IL-15, MICA, IDO-1, and CCL-18, among other factors, in agreement with in vivo observations (Fig. 7B). However, in this study, we used RT-qPCR to analyze ≈90 genes related to NK cell response, which only represents a fraction of the total amount of alterations observed in vivo. Future studies could leverage RNA-sequencing technologies to analyze thousands of genes to provide a more in-depth molecular analysis of the exhaustion process.
Fig. 7. NK cell dysfunction marker comparison.
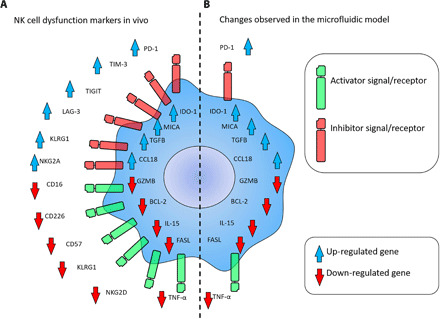
(A) Scheme shows molecular changes observed in vivo in dysfunctional (i.e., exhausted and anergic) NK cells. (B) Scheme depicts changes observed in NK-92 cells (seeding cell density: 0.5 million cells/ml) in the model after 7 days in coculture with MCF7 cells (seeding cell density: 1.5 million cells/ml).
On the other hand, our study also highlighted the importance of analyzing immunomodulatory agents (e.g., PD-1 and IDO-1 inhibitors) in a physiologically relevant context. Specifically, atezolizumab and epacadostat showed little efficacy in traditional well plates, where NK cells had no environmental stress, and showed no up-regulation of the relevant pathways. Our results also demonstrated that tumor cells located in the distal areas of the model were highly resistant to immunomodulatory treatment. Last, our study suggested that environmental stress might exert a long-term modification of NK cell biology and metabolism, leading to dysfunctional NK cells (i.e., exposed NK cells) with compromised cytotoxic capacity. Therefore, our platform could provide an exciting tool to evaluate epigenetic modifications and other molecular alterations driving these long-lasting immunosuppressive effects. Although in this study we did not identify the specific environmental factor/s (e.g., glucose, tryptophan, deprivation, and acidic pH) driving the changes observed in NK-92 cells, this platform could provide a valuable tool to identify metabolic opportunities in NK and T cell immunotherapy. NK-92 cells have been used in clinical trials against numerous malignancies, with multiple trials still ongoing (43, 44). Therefore, deciphering their response to the immunosuppressive tumor microenvironment could lead to the development of new NK-92 formulations that thrive in this harsh environment. In this work, we cultured 1.5 million MCF7 cells/ml because that density generated a necrotic core in the distal region, whereas cells next to the lumen remained viable. However, this technology could also be used to test the effect of different tumor cell densities on NK cell exhaustion. In this context, we observed that 4 million cells/ml led to a decrease in cell viability in those cells located in the proximal area, suggesting a more severe environmental stress. Several studies have demonstrated that other cell types present inside solid tumors affect the immune response [e.g., myeloid-derived suppressor cells (MDSCs), macrophages, fibroblasts, and regulatory T cells (Tregs)]. Macrophages seem to play a dual role, exerting a tumor-promoting or tumor-suppressing effect depending on the specific conditions in the tumor milieu (45). In this context, tumor-associated macrophages (TAMs) are traditionally classified into M1 (tumor-suppressing, proinflammatory role) or M2 macrophages (tumor-promoting, anti-inflammatory role) (46). Chemokines secreted by tumor and stromal cells control the balance between M1 and M2 macrophages. In this context, chemokines such as TNF-α and IL-17 promote an M1 phenotype, whereas IL-14 and IL-4 lead to a tumor-promoting M2 phenotype. MDSCs are a group of immune cells that are expanded during inflammatory processes, including cancer, and lead to immunosuppression (47). MDSCs suppress the immune system by cell-cell contact with T cells or secretion of short-lived mediators (e.g., nitric oxide, urea, and l-ornithine). Cancer-associated fibroblasts (CAFs) promote immune evasion by multiple mechanisms including modulation of adaptive and innate immune cell recruitment, cytotoxicity, and expansion (48). In addition, CAFs also contribute to MDSC and TAM expansion, exacerbating immune suppression. Similarly to MDSCs, Tregs also generate an immunosuppressive environment (49). Tregs rely on multiple cell-cell–dependent and cell-cell–independent mechanisms to regulate the immune response such as modulation of checkpoint inhibitors (e.g., PD-1, CTLA-4, and IDO-1), secretion of suppressive cytokines (e.g., IL-10 and IL-35), cytokine consumption (e.g., IL-2), or metabolic disruption. Future studies using the tumor-on-a-chip platform could include the presence of these stromal cells, providing a more comprehensive model of the tumor microenvironment. On the other hand, exposing immune cells to the tumor microenvironment could help us to decipher the adaptive and survival responses that immune cells have at their disposal. The ability of isolating these immune cells from the model enables us to design evolution-driven experiments where the same pool of immune cells is repeatedly exposed to the tumor environment, which would select the most resilient NK cells. In the long term, studying adaptations such as DNA methylation or metabolic rewiring in immune cells in the tumor microenvironment could help us to genetically engineer more efficient immune cells (e.g., pH-induced secretion of IL-2 and hypoxia-activated granzyme synthesis). These studies might also lead to the identification of new immunomodulatory agents (e.g., checkpoint inhibitors) that would allow these tumor hunters to thrive amidst the suppressive environment of solid tumors, significantly improving the reach of immunotherapy. The current healthcare pipeline requires new therapies to be validated in extensive animal studies and through clinical trials. However, differences between human and mouse immunology commonly hinder this process (50), leading to high attrition rates. Hence, we believe that tumor-on-a-chip technologies might accelerate the development of these new immunotherapies by providing a versatile tool that bridges the gap between in vitro platforms and animal models.
MATERIALS AND METHODS
Microdevice fabrication
The tumor-on-a-chip microdevice was fabricated using a protocol more specifically detailed in (16). In summary, illustrator was used to design the template, which was then fabricated using SU-8–based lithography. The SU-8 template was used to generate the final microdevices by pouring PDMS on top and polymerizing it during 4 hours at 80°C. Following PDMS polymerization, the microdevices were removed from the SU-8 wafer, and the bottom and top layers of the microdevice were assembled together. This approach yielded a nonpermanent bonding between the two layers of PDMS, allowing the disassemble of the microdevice after the experiments to retrieve the cells. To generate the lumen structure, a 340-μm-diameter PDMS rod was inserted in the chamber. Next, the microdevices were plasma-bonded to a 60-mm glass bottom petri dish. Because top and bottom layers were already together, the plasma treatment did not affect the interface between both layers, keeping the bonding reversible. Microdevices were sterilized through 15 min of ultraviolet exposure before using them in cell culture.
Cell culture in the tumor-on-a-chip model
The NK-92 cell line was acquired from the American Type Culture Collection and cultured in X-VIVO 10 (Lonza, 04-380Q) supplemented with 20% fetal bovine serum (FBS; Lonza) and 0.02 mM folic acid (Sigma-Aldrich, F8758) dissolved in 1 N NaOH, 0.2 mM myo-inositol (Sigma-Aldrich, I7508), and IL-2 (100 U/ml; PeproTech, 200-02). NK-92 cell cytotoxicity has been shown in multiple studies and clinical trials (51–54). Breast cancer MCF7 cells were cultured in RPMI 1640 (Thermo Fisher Scientific, 21870076) supplemented with 10% FBS (Thermo Fisher Scientific, 26140079). Patient-derived invasive breast carcinoma cells (IDC; 171881-019-R-J1-PDC, passages 7 to 12) were obtained from the National Cancer Institute Patient-Derived Models Repository (NCI PDMR). Patient-derived cells required specialized medium consisting of 1× Advanced Dulbecco’s modified Eagle’s medium/F12 (Gibco, 12634010) supplemented with 5% FBS, 1% penicillin:streptomycin, 0.178 mM adenine (Sigma-Aldrich, A8626-1G), 1.61 nM EGF Recombinant Human Protein (Invitrogen, PHG0313), 1.1 μM hydrocortisone (Sigma-Aldrich, H4001-1G), 2 mM l-glutamine (Invitrogen), and 0.01 mM Y-27632 dihydrochloride (Tocris). To prepare the tumor-on-a-chip model, cells were detached using a trypsin/EDTA solution and resuspended at the desired density in NK cell medium. Next, a collagen hydrogel (4.0 mg/ml) containing MCF7 cells alone or in combination with NK-92 cells was prepared as follows: 10 μl of 10× phosphate-buffered saline (PBS), 1.2 μl of 1 M NaOH, 47.5 μl of collagen type I (8.43 mg/ml), 31.3 μl of distilled water, and 100 μl of cell suspension. The collagen hydrogel was injected into the microchamber of the microfluidic device and polymerized at room temperature for 20 min. The PDMS rod was removed using sterilized tweezers after collagen polymerization to generate a tunnel in the hydrogel through which medium was perfused to nourish the cells. NK cell medium (2.5 ml) was then added to the petri dish, and the devices were placed in an incubator at 37°C with 5% CO2.
Cell staining
HUVECs, MCF7, and NK-92 cells were stained with fluorescent markers to monitor cell migration. More specifically, we labeled HUVECs, MCF7 cells, and NK-92 cells with vibrant DiO (green; Thermo Fisher Scientific, V22886), Dil (red; Thermo Fisher Scientific, V22885), and Did (infra-red; Thermo Fisher Scientific, V22887), respectively. Briefly, cells were counted and suspended in PBS at 1 million cells/ml. Vybrant labeling agent (5 μl) was added per milliliter of cell suspension. The cell suspension was placed in the incubator for 5 min and then centrifuged (400g, 5 min) and washed twice with 15 ml of PBS to remove the staining excess. Staining in PBS was critical for NK-92 cells, because the presence of their culture medium [X-VIVO 10, 20% FBS, IL-2 (200 U/ml)] prevented the staining. DiO, Dil, and Did allowed us to distinguish the three cell populations during the first 72 hours. However, these stainings slowly leak to other surrounding cells, and at 7 days, most cells showed areas labeled in green, red, and infra-red. Apoptosis was detected with CellEvent Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific, C10423), which labels apoptotic cells in green. In these experiments, CellEvent was added to the hydrogel mixture and the culture medium at 1:1000, and HUVECs were not labeled with DiO (green) to avoid fluorescence overlapping.
Cell viability
To measure cell viability, stock solutions of calcein acetomethyl ester (CAM; 5 mg/ml) (Thermo Fisher Scientific,C3100MP) and prodidium iodide (PI; 2 mg/ml) (Thermo Fisher Scientific, P1304MP) were prepared following the supplier instructions. Working solutions were prepared dissolving at 1:1000 and 1:500, respectively, the stock solutions in PBS. The upper half of the microdevice was detached to expose the collagen hydrogel to guarantee a homogeneous staining, and the CAM/PI solution was added on top for 15 min. Afterward, cell viability was evaluated by fluorescent/confocal imaging using a Leica SP8 3× STED super-resolution confocal microscope. In 2D assays, cells were washed twice with PBS to remove floating dead cells and NK-92 cells. Next, viable tumor cells were stained with the CAM solution (no PI was used).
Optical metabolic imaging
A custom-built inverted multiphoton microscope (Bruker Fluorescence Microscopy, Middleton, WI) was used to acquire fluorescence intensity and lifetime images. The equipment consists of an ultrafast laser (Spectra Physics, Insight DSDual), an inverted microscope (Nikon, Eclipse Ti), and a 40× water immersion (1.15NA, Nikon) objective. Next, NAD(P)H and FAD images were obtained for the same field of view. FAD fluorescence was isolated using an emission bandpass filter of 550/100 nm and excitation wavelength of 890 nm. NAD(P)H fluorescence was isolated using an emission bandpass filter of 440/80 nm and an excitation wavelength of 750 nm. Subsequently, fluorescence lifetime images were collected using time-correlated single-photon counting electronics (SPC-150, Becker and Hickl) and a GaAsP photomultiplier tube (H7422P-40, Hamamatsu). Images (512 × 512 pixels) were obtained using a pixel dwell time of 4.8 μs over 60-s total integration time. To guarantee adequate photon observations for lifetime decay fits and no photobleaching, the photon count rates were maintained at 1 × 105 to 2 × 105 photons/s. The instrument response function was calculated from second harmonic generation of urea crystals excited at 900 nm, and the full width at half maximum was measured to be 244 ps. A Fluoresbrite YG microsphere (Polysciences Inc.) was imaged as a daily standard for fluorescence lifetime. The lifetime decay curves were fit to a single exponential decay, and the fluorescence lifetime was measured to be 2.1 ns (n = 7), which is consistent with published values.
Drug treatments
Metabolic inhibitors were used on NK cells to study their effect on optical metabolic imaging measurements. 2DG (10 mM) (Sigma-Aldrich, D6134), etomoxir (10 μM) (Sigma-Aldrich, E1905), and oligomycin (1 μM) (Sigma-Aldrich, 75351) were used to perturb their respective pathways. To measure how immune checkpoint and IDO-1 inhibition affected NK cytotoxicity in the microdevice, the hydrogels were treated with 40 nM atezolizumab (Selleckchem, A2004) and 50 nM epacadostat (Selleckchem, S7910) for 7 days. After these treatments, cell viability was assessed as described above.
Cell isolation from the microdevice
To selectively retrieve the NK-92 or MCF7 cells from the microdevice, the upper half of the microdevice was removed to expose the collagen hydrogel. The hydrogel was then transferred to an Eppendorf tube containing 300 μl of type I collagenase (6 mg/ml) (55). The sample was incubated on ice for 2 min to degrade the hydrogel and release the cells. Two microliters of biotinylated anti-EpCAM (D20655, Thermo Fisher Scientific) was added, and the sample was incubated at 4°C for 15 min. Ten microliters of SeraMAGS beads coupled to streptavidin was added, and the sample was incubated for another 10 min at 4°C. The SeraMAGS beads, with the MCF7 cells (EpCAM-positive), were isolated using a magnet, leaving the NK-92 cells in suspension. NK-92 cells were transferred to NK-92 cell medium for subculture experiments or lysed for PCR analysis. To isolate NK-92 cells from specific hydrogel locations (i.e., 0 and 10 mm from the lumen), 1-mm-diameter biopsy punch (33-31AA-P/25, Fisher Scientific) was used. The hydrogel punches were transferred to an Eppendorf tube and processed following the same protocol described above.
Reverse transcription quantitative polymerase chain reaction
To study how NK-92 cells adapted to the microenvironment within the tumor-on-a-chip model, the expression of multiple genes related to different pathways was analyzed by RT-qPCR. Briefly, mRNA was isolated from the different punches using the Dynabeads mRNA DIRECT Purification Kit (Thermo Fisher Scientific, 61011). Isolated mRNA was quantified using a Qubit fluorometer (Thermo Fisher Scientific) and the Qubit RNA BR Assay Kit (Q10210, Thermo Fisher Scientific). mRNA was reverse-transcribed to complementary DNA (cDNA) using the RT2 PreAMP cDNA Synthesis Kit (Qiagen, 330451). cDNA was analyzed by RT-qPCR using a “Cancer Inflammation and Immunity Crosstalk” Qiagen RT2 profiler panel (Qiagen, PAHS-181Z), and data were analyzed using the Qiagen online software (https://geneglobe.qiagen.com/us/analyze/) (fig. S5).
Luminescence assay
The effect of epacadostat (i.e., IDO-1 inhibitor) and tryptophan deprivation on NK-92 cell growth was evaluated using the luminescence assay CellTiter-Glo (Promega, G7571). NK-92 cells were cultured in different concentrations of epacadostat and tryptophan, and after 72 hours, cell growth was evaluated. CellTiter-Glo reagent was dissolved in CellTiter-Glo buffer and added 1:1 in the 96-well plates containing NK-92 cells. This reagent lysed the cells, and the adenosine triphosphate (ATP) released led to the generation of light in a cell-dependent manner. After incubation for 45 min in the dark, luminescence was measured in a plate reader.
Cell migration analysis
NK-92 cell migration was evaluated in a Nikon Ti Eclipse with a top stage incubator equipped with temperature and CO2 control (set at 37°C and 5%, respectively). Naïve or exposed NK-92 cell migration was tracked for 45 min to analyze their migration capacity. NK-92 cell trajectories and kinetic parameters such as directionality, migrated distance (accumulated and Euclidean), and velocity were analyzed with Fiji (https://imagej.net/Fiji) using manual tracking plugins and the chemotaxis tool plugin (Ibidi; https://ibidi.com/chemotaxis-analysis/171-chemotaxis-and-migration-tool.html).
Image and analysis
NAD(P)H and FAD intensity and lifetime images were analyzed using SPCImage software (Becker & Hickl) as described previously in (35). The fluorescence lifetime decay curve was deconvolved with the instrument response function and fit to a two-component exponential decay model at each pixel, I(t) = α1*e(−t/τ1) + α2*e(−t/τ2) + C, where I(t) represents the fluorescence intensity at time t after the laser excitation pulse, α accounts for the fractional contribution from each component, C represents the background light, and τ is the fluorescence lifetime of each component. Because both NAD(P)H and FAD can exist in two conformational states, bound or unbound to enzymes, a two-component model was used. The short and long lifetime components reflect the bound and unbound conformations, respectively, for FAD (37). While the opposite is true for NAD(P)H, the short and long lifetime components correspond with the unbound and bound conformations, respectively. The mean lifetime (τm) was calculated using τm = α1τ1 + α2τ2 for both NAD(P)H and FAD. The optical redox ratio was determined from the NAD(P)H and FAD lifetime data by integrating the photons detected at each pixel in the image to calculate the total intensity. For each pixel, the intensity of NAD(P)H was then divided by the intensity of FAD. Using Cell Profiler, an automated cell segmentation pipeline was created. This system identified pixels belonging to nuclear regions by using a customized threshold code. Cells were recognized by propagating out from the nuclei within the image. To refine the propagation and to prevent it from continuing into background pixels, an Otsu Global threshold was used. The cell cytoplasm was defined as the cell borders minus the nucleus. Values for NADH τm, FAD τm, NAD(P)H intensity, FAD intensity, and the optical redox ratio [NAD(P)H/FAD intensity] were averaged for all pixels within each cell cytoplasm. At least 100 cells per sample were analyzed, and every experiment was repeated at least three times.
Confocal microscopy images were analyzed by Fiji. To examine molecule diffusion and cell viability, a rectangle-shape region was drawn, and the intensity profile was calculated using Fiji software.
Statistical analysis
The normal distribution assumption for statistical tests was confirmed by the Kolmogorov-Smirnov test. Statistical significance was set at P < 0.05. For nonparametric comparisons, a Kruskal-Wallis test was performed followed by the Mann-Whitney U test. For subpopulation analysis, single-cell redox ratios were binned to generate frequency histograms representing naïve and exposed NK cell redox ratio distributions. Gaussian curves were fit to the distribution data to reveal underlying populations with distinct redox ratio. The Akaike information criterion was used to assess fit quality and determine the optimal number of fitted Gaussians.
Acknowledgments
Funding: This study was supported by University of Wisconsin Carbone Cancer Center (AAB7173); Morgridge Research Institute; NIH grants R01 CA164492, R01 CA185747, and R01 CA205101; and NSF grant CBET-1642287. Author contributions: J.M.A., M.V.-M., P.H.M., S.R., P.G., and C.F. performed the experiments and analyzed the data. T.H. performed the metabolic perturbation experiments and analyzed the data. J.M.A., M.C.S., and D.J.B. designed the study and wrote the manuscript. All authors reviewed and approved the final manuscript. Competing interests: D.J.B. holds equity in Bellbrook Labs LLC, Tasso Inc., Turba LLC, Salus Discovery LLC, Stacks to the Future LLC, Lynx Biosciences Inc., and Onexio Biosystems LLC. J.M.A., M.V.-M., P.H.M., and D.J.B. are inventors on a patent application related to this work filed by Wisconsin Alumni Research Foundation (no. 17/004290, filed on 27 August 2020). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabc2331/DC1
REFERENCES AND NOTES
- 1.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2020. CA Cancer J. Clin. 70, 7–30 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Liu D., CAR-T “the living drugs”, immune checkpoint inhibitors, and precision medicine: A new era of cancer therapy. J. Hematol. Oncol. 12, 113 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schultz L., Mackall C., Driving CAR T cell translation forward. Sci. Transl. Med. 11, eaaw2127 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Grosser R., Cherkassky L., Chintala N., Adusumilli P. S., Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell 36, 471–482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mardiana S., Solomon B. J., Darcy P. K., Beavis P. A., Supercharging adoptive T cell therapy to overcome solid tumor–induced immunosuppression. Sci. Transl. Med. 11, eaaw2293 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Belladonna M. L., Puccetti P., Orabona C., Fallarino F., Vacca C., Volpi C., Gizzi S., Pallotta M. T., Fioretti M. C., Grohmann U., Immunosuppression via tryptophan catabolism: The role of kynurenine pathway enzymes. Transplantation 84, S17–S20 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Ota Y., Ishihara S., Otani K., Yasuda K., Nishikawa T., Tanaka T., Tanaka J., Kiyomatsu T., Kawai K., Hata K., Nozawa H., Kazama S., Yamaguchi H., Sunami E., Kitayama J., Watanabe T., Effect of nutrient starvation on proliferation and cytokine secretion of peripheral blood lymphocytes. Mol. Clin. Oncol. 4, 607–610 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald P. C., Chafe S. C., Dedhar S., Overcoming hypoxia-mediated tumor progression: Combinatorial approaches targeting pH regulation, angiogenesis and immune dysfunction. Front. Cell Dev. Biol. 4, 27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huber V., Camisaschi C., Berzi A., Ferro S., Lugini L., Triulzi T., Tuccitto A., Tagliabue E., Castelli C., Rivoltini L., Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 43, 74–89 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zhang J., Shi Z., Xu X., Yu Z., Mi J., The influence of microenvironment on tumor immunotherapy. FEBS J. 286, 4160–4175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.da Silva J. L., Dos Santos A. L. S., Nunes N. C. C., de Moraes Lino da Silva F., Ferreira C. G. M., de Melo A. C., Cancer immunotherapy: The art of targeting the tumor immune microenvironment. Cancer Chemother. Pharmacol. 84, 227–240 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Boussommier-Calleja A., Li R., Chen M. B., Wong S. C., Kamm R. D., Microfluidics: A new tool for modeling cancer-immune interactions. Trends Cancer 2, 6–19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayuso J. M., Virumbrales-Munoz M., McMinn P. H., Rehman S., Gomez I., Karim M. R., Trusttchel R., Wisinski K. B., Beebe D. J., Skala M. C., Tumor-on-a-chip: A microfluidic model to study cell response to environmental gradients. Lab Chip 19, 3461–3471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pavesi A., Tan A. T., Koh S., Chia A., Colombo M., Antonecchia E., Miccolis C., Ceccarello E., Adriani G., Raimondi M. T., Kamm R. D., Bertoletti A., A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight 2, e89762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adriani G., Pavesi A., Kamm R. D., Studying TCR T cell anti-tumor activity in a microfluidic intrahepatic tumor model. Methods Cell Biol. 146, 199–214 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Ayuso J. M., Truttschel R., Gong M. M., Humayun M., Virumbrales-Munoz M., Vitek R., Felder M., Gillies S. D., Sondel P., Wisinski K. B., Patankar M., Beebe D. J., Skala M. C., Evaluating natural killer cell cytotoxicity against solid tumors using a microfluidic model. Oncoimmunology 8, 1553477 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virumbrales-Munoz M., Ayuso J. M., Gong M. M., Humayun M., Livingston M. K., Lugo-Cintrón K. M., Minn P. M., Álvarez-García Y. R., Beebe D. J., Microfluidic lumen-based systems for advancing tubular organ modeling. Chem. Soc. Rev. 49, 6402–6442 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sackmann E. K., Fulton A. L., Beebe D. J., The present and future role of microfluidics in biomedical research. Nature 507, 181–189 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Ayuso J. M., Rehman S., Farooqui M., Virumbrales-Muñoz M., Setaluri V., Skala M. C., Beebe D. J., Microfluidic tumor-on-a-chip model to study tumor metabolic vulnerability. Int. J. Mol. Sci. 21, 9075 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang S., Zhao Y., Heaster T. M., Fischer M. A., Stengel K. R., Zhou X., Ramsey H., Zhou M.-M., Savona M. R., Skala M. C., Hiebert S. W., BET inhibitors reduce cell size and induce reversible cell cycle arrest in AML. J. Cell. Biochem. 120, 7309–7322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones J. D., Ramser H. E., Woessner A. E., Quinn K. P., In vivo multiphoton microscopy detects longitudinal metabolic changes associated with delayed skin wound healing. Commun. Biol. 1, 198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Podsednik A., Jacob A., Li L. Z., Xu H. N., Relationship between optical redox status and reactive oxygen species in cancer cells. React. Oxyg. Species (Apex) 9, 95–108 (2020). [PMC free article] [PubMed] [Google Scholar]
- 23.Meleshina A. V., Dudenkova V. V., Bystrova A. S., Kuznetsova D. S., Shirmanova M. V., Zagaynova E. V., Two-photon FLIM of NAD(P)H and FAD in mesenchymal stem cells undergoing either osteogenic or chondrogenic differentiation. Stem Cell Res. Ther. 8, 15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhatia A., Kumar Y., Cellular and molecular mechanisms in cancer immune escape: A comprehensive review. Expert Rev. Clin. Immunol. 10, 41–62 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Beatty G. L., Gladney W. L., Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21, 687–692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iorgulescu J. B., Braun D., Oliveira G., Keskin D. B., Wu C. J., Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. 10, 87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gatti-Mays M. E., Balko J. M., Gameiro S. R., Bear H. D., Prabhakaran S., Fukui J., Disis M. L., Nanda R., Gulley J. L., Kalinsky K., Abdul Sater H., Sparano J. A., Cescon D., Page D. B., McArthur H., Adams S., Mittendorf E. A., If we build it they will come: Targeting the immune response to breast cancer. NPJ Breast Cancer 5, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossi J.-F., Ceballos P., Lu Z.-Y., Immune precision medicine for cancer: A novel insight based on the efficiency of immune effector cells. Cancer Commun. (Lond.) 39, 34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pai S. I., Cesano A., Marincola F. M., The paradox of cancer immune exclusion: Immune oncology next frontier. Cancer Treat. Res. 180, 173–195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hegde P. S., Chen D. S., Top 10 challenges in cancer immunotherapy. Immunity 52, 17–35 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Virumbrales-Munoz M., Ayuso J. M., Olave M., Monge R., de Miguel D., Martínez-Lostao L., Gac S. L., Doblare M., Ochoa I., Fernandez L. J., Multiwell capillarity-based microfluidic device for the study of 3D tumour tissue-2D endothelium interactions and drug screening in co-culture models. Sci. Rep. 7, 11998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon J. S., Bersini S., Gilardi M., Dubini G., Charest J. L., Moretti M., Kamm R. D., Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. U.S.A. 112, 214–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen M. B., Whisler J. A., Fröse J., Yu C., Shin Y., Kamm R. D., On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc. 12, 865–880 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchberger A. R., DeLaney K., Johnson J., Li L., Mass spectrometry imaging: A review of emerging advancements and future insights. Anal. Chem. 90, 240–265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Surmacki J. M., Woodhams B. J., Haslehurst A., Ponder B. A. J., Bohndiek S. E., Raman micro-spectroscopy for accurate identification of primary human bronchial epithelial cells. Sci. Rep. 8, 12604 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schafer J. L., Muller-Trutwin M. C., Reeves R. K., NK cell exhaustion: Bad news for chronic disease? Oncotarget 6, 21797–21798 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bi J., Tian Z., NK Cell exhaustion. Front. Immunol. 8, 760 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melaiu O., Lucarini V., Cifaldi L., Fruci D., Influence of the tumor microenvironment on NK cell function in solid tumors. Front. Immunol. 10, 3038 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terren I., Orrantia A., Vitalle J., Zenarruzabeitia O., Borrego F., NK cell metabolism and tumor microenvironment. Front. Immunol. 10, 2278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Judge S. J., Murphy W. J., Canter R. J., Characterizing the dysfunctional NK cell: Assessing the clinical relevance of exhaustion, anergy, and senescence. Front. Cell. Infect. Microbiol. 10, 49 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stojanovic A., Fiegler N., Brunner-Weinzierl M., Cerwenka A., CTLA-4 is expressed by activated mouse NK cells and inhibits NK Cell IFN-γ production in response to mature dendritic cells. J. Immunol. 192, 4184–4191 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Sanseviero E., O’Brien E. M., Karras J. R., Shabaneh T. B., Aksoy B. A., Xu W., Zheng C., Yin X., Xu X., Karakousis G. C., Amaravadi R. K., Nam B., Turk M. J., Hammerbacher J., Rubinstein M. P., Schuchter L. M., Mitchell T. C., Liu Q., Stone E. L., Anti–CTLA-4 activates intratumoral NK cells and combined with IL15/IL15Rα complexes enhances tumor control. Cancer Immunol. Res. 7, 1371–1380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang X., Yang L., Li Z., Nalin A. P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W., Chen G., Zhu X., Wu D., Yu J., First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 8, 1083–1089 (2018). [PMC free article] [PubMed] [Google Scholar]
- 44.Williams B. A., Law A. D., Routy B., denHollander N., Gupta V., Wang X. H., Chaboureau A., Viswanathan S., Keating A., A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 8, 89256–89268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galdiero M. R., Bonavita E., Barajon I., Garlanda C., Mantovani A., Jaillon S., Tumor associated macrophages and neutrophils in cancer. Immunobiology 218, 1402–1410 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Ley K., M1 means kill; M2 means heal. J. Immunol. 199, 2191–2193 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Tesi R. J., MDSC; the most important cell you have never heard of. Trends Pharmacol. Sci. 40, 4–7 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Liu T., Zhou L., Li D., Andl T., Zhang Y., Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 7, 60 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Togashi Y., Shitara K., Nishikawa H., Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 16, 356–371 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Mestas J., Hughes C. C., Of mice and not men: Differences between mouse and human immunology. J. Immunol. 172, 2731–2738 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Klingemann H. G., Miyagawa B., Purging of malignant cells from blood after short ex vivo incubation with NK-92 cells. Blood 87, 4913–4914 (1996). [PubMed] [Google Scholar]
- 52.Gong J. H., Maki G., Klingemann H. G., Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 8, 652–658 (1994). [PubMed] [Google Scholar]
- 53.Tonn T., Schwabe D., Klingemann H. G., Becker S., Esser R., Koehl U., Suttorp M., Seifried E., Ottmann O. G., Bug G., Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 15, 1563–1570 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Arai S., Meagher R., Swearingen M., Myint H., Rich E., Martinson J., Klingemann H., Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 10, 625–632 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Virumbrales-Munoz M., Ayuso J. M., Lacueva A., Randelovic T., Livingston M. K., Beebe D. J., Oliván S., Pereboom D., Doblare M., Fernández L., Ochoa I., Enabling cell recovery from 3D cell culture microfluidic devices for tumour microenvironment biomarker profiling. Sci. Rep. 9, 6199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/8/eabc2331/DC1