

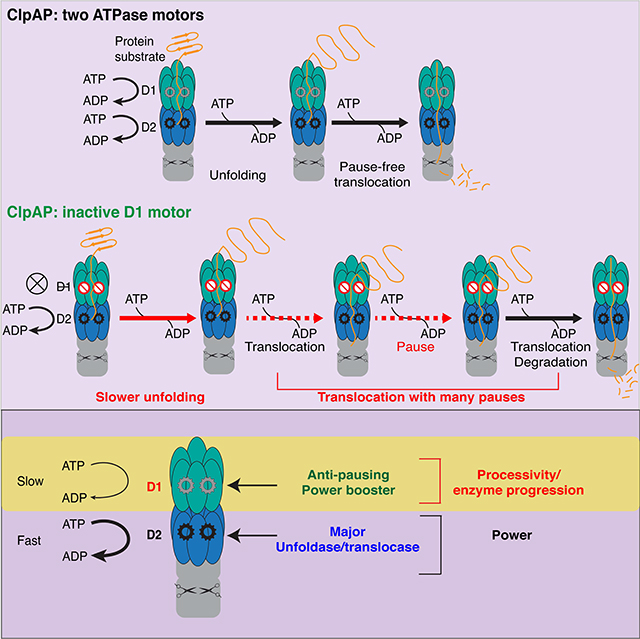
Summary
ATP-powered unfoldases containing D1 and D2 AAA+ rings play important roles in protein homeostasis, but uncertainty about the function of each ring remains. Here we use single-molecule optical-tweezers to assay mechanical unfolding and translocation by a variant of the ClpAP protease containing an ATPase-inactive D1 ring. This variant displays substantial mechanical defects both in unfolding and translocation of protein substrates. Notably, when D1 is hydrolytically inactive, ClpAP often stalls for times as long as minutes, and the substrate can “back-slip” through the enzyme when ATP concentrations are low. The inactive D1 variant also has more difficulty traveling in the N-to-C direction on a polypeptide track than moving C-to-N. These results indicate that D1 normally functions as an auxiliary/regulatory motor to promote uninterrupted enzyme advancement that is fueled largely by the D2 ring.
Keywords: AAA+ proteases, ATP motors, Single-molecule force spectroscopy, Protein unfolding, Protein translocation, Translocation pauses, Hsp104/ClpB, ClpAP
e-TOC summary
Kotamarthi et al employ optical tweezers to examine the individual rings in a double-ring AAA+ unfoldase. ClpAP inactivated in the D1 ring is partially defective in unfolding, and pauses frequently during translocation. Thus, D1 functions as a “booster motor” thereby assisting the more powerful unfoldase motor of the D2 ring.
Graphical Abstract
Introduction
AAA+ enzymes (ATPases associated with various cellular activities) perform mechanical processes in cells by operating as tiny motors that convert the chemical energy of ATP hydrolysis into work that typically involves macromolecular remodeling (Neuwald et al., 1999; Ogura and Wilkinson, 2001). In all domains of life, a subfamily of AAA+ enzymes promote protein homeostasis by degrading damaged or misfolded proteins (Olivares et al., 2016; Sauer and Baker, 2011). AAA+ proteases contain a barrel-shaped peptidase and a hexameric AAA+ unfoldase with an axial pore that can bind specific peptide tags (degrons) in target proteins. Following binding, cycles of ATP hydrolysis in the AAA+ hexamer power substrate unfolding and subsequent translocation into the peptidase chamber for degradation (Olivares et al., 2016). For example, the Escherichia coli ClpAP protease, which degrades ssrA-tagged proteins, those with N-end rule degrons, and other substrates, consists of the AAA+ ClpA hexamer and the ClpP peptidase (Gottesman et al., 1998; Katayama et al., 1988; Tobias et al., 1991).
ClpA hexamers contain 12 active sites for ATP hydrolysis, six in each of two distinct AAA+ rings; a similar double-ring architecture is present in the hexamers of related enzymes (ClpB, Hsp104, Cdc48/p97, and NSF) that function without proteolytic partners in protein remodeling. ClpA in the absence of ClpP also has remodeling activity (Wickner et al., 1994). Why ClpA and other double-ring enzymes need two rings is unclear. Some mutagenesis experiments suggest that one ring performs most of the ATP hydrolysis and functions as the dominant enzyme motor. It has been proposed that the second ring assists in the nucleotide-stabilized assembly of the hexamer (Doyle et al., 2007; Hattendorf and Lindquist, 2002; Mogk et al., 2003), but single-ring AAA+ enzymes also assemble into stable hexamers, and there would appear to be many simpler ways to stabilize the double-ring enzymes.
The top ClpA AAA+ ring, where substrates first enter the axial pore, is called D1, whereas the bottom ring, which is proximal to ClpP, is called D2 (Figure 1A) (Effantin et al., 2010; Guo et al., 2002). ATP hydrolysis in one ring or the other can be eliminated by mutating either of the conserved glutamate in the D1 Walker-B motif (E286) or in the D2 Walker-B motif (E565). Studies of these enzyme variants show: (i) D2 appears to catalyze most ATP hydrolysis, although elimination of hydrolysis in D1 can reduce ATPase activity by as much as 2-fold in the presence of some protein substrates; (ii) inactivating D1 generally has modest effects on substrate degradation, whereas inactivating D2 impairs degradation severely; and (iii) ATP hydrolysis in both rings is important for degradation of substrates with very stable local structures (Kress et al., 2009). Because the D2 ring appears to be most important for ATP-fueled degradation as well as being closely related to the well-studied ClpX AAA+ ring, we focused on determining how ATP hydrolysis in the less-understood D1 ring contributes to mechanical substrate unfolding and polypeptide translocation, critical steps in the overall degradation reaction.
Figure 1: ClpAP protease, multidomain substrates, and ATP hydrolysis.

(A) Cartoon of ClpAP and ClpAP. Each ClpA subunit contains two AAA+ ATPase modules that form the D1 (teal) and the D2 (blue) rings in the assembled hexamer. ClpP (gray) is a barrel-shaped serine peptidase; scissors schematically represent peptidase active sites. A model protein substrate engaged for unfolding in the ClpA pore is shown. ClpA (right) contains a ⦰ symbol in the D1 ring to indicate an ATP-hydrolysis defect. (B) Schematic of ssrA-tagged multidomain substrates. The ssrA tag (orange) is present at the N-terminus of ssrA-(V15P)4-Halo (red, top) or the C-terminus of Halo-(V13P)4-ssrA (blue). The sites of the V15P and V13P mutations in the titin I27 domain are represented as spheres (note the titin domains are in opposite orientations). The Halo domain, used for covalent linkage to DNA for optical trapping, is dark gray. (C) Steady-state rates of ATP hydrolysis. Rates for ClpA or ClpA (0.1 μM hexamer) were measured in the absence of ClpP and substrate (basal), in the presence of ClpP (0.2 μM tetradecamer), or in presence of ClpP (0.2 μM tetradecamer) and substrate (5 μM). Values are averages ± 1 SD of three independent measurements.
Here, we use single-molecule optical-tweezer assays to investigate how ClpAP unfolding and translocation are affected when the D1 ring cannot hydrolyze ATP as a consequence of an E286Q mutation. We find that this D1-defective ClpAP (ClpAP) displays a marked increase in the frequency of pausing/stalling during translocation as well as substantial defects in unfolding. Hence, ATP hydrolysis in the D1 ring increases enzyme productivity by stimulating both of these key mechanical components of degradation. Surprisingly, the ClpAP defects were magnified when degradation proceeded in an N-to-C direction compared to the C-to-N direction. Our results also support a role for D1 in maintaining substrate grip and preventing slipping at limiting ATP concentrations.
Results
Single-molecule degradation by ClpAP
Here we used two multidomain substrates previously employed to analyze single-molecule degradation by wild-type ClpAP (Olivares et al., 2017; Olivares et al., 2014). Degradation was initiated either from the N-terminus or the C-terminus of these substrates by changing the position of the ssrA tag degron (Olivares et al., 2017; Olivares et al., 2014), which targets the protein to ClpA. For N-to-C degradation, the substrate contained an N-terminal ssrA tag, four human titin I27 domains with the mildly destabilizing V15P mutation, and a Halo domain (Figure 1B, top). For C-to-N degradation, we initially tested a substrate with four V15P domains, Halo, and a C-terminal ssrA tag, but ClpAP degraded this substrate poorly. As a consequence, we used a substrate with four titin I27 domains containing the more destabilizing V13P mutation, Halo, and the ssrA tag at the C-terminus (Figure 1B, bottom).
When protein substrate was absent, ClpA hydrolyzed ATP at essentially the same rate as ClpA, and ClpP stimulated hydrolysis to similar extents for both enzymes (Figure 1C). In the presence of either protein substrate, however, ClpAP had only 50–60% of the hydrolysis activity of WT ClpA (Figure 1C). Hence, when engaged with these substrates, the D1 ring of ClpA appears to contribute substantially, either directly or indirectly, to overall ATP-hydrolysis by the intact enzyme. Similar results were reported previously, although only certain substrates reduced ATPase activity for ClpAP in comparison with ClpAP (Kress et al., 2009).
To probe the contribution of the D1 ring to unfolding and translocation by ClpA, we performed single-molecule optical-tweezer assays (Figure 2A) (Aubin-Tam et al., 2011; Cordova et al., 2014). Biotinylated ClpAP was immobilized on a 1.25 μm streptavidin-coated polystyrene bead and the substrate Halo domain was attached, via a biotinylated DNA linker, to a 1.09 μm bead (Figure 2A) (Aubin-Tam et al., 2011; Cordova et al., 2017). The beads were trapped by two laser beams and experiments were performed in a passive force-clamp mode at forces ranging from 8 to18 pN (Greenleaf et al., 2005). After the two beads were tethered together via ClpAP on one bead binding to the substrate on the other, we monitored inter-bead distance as a function of time. Each sharp increase in bead-to-bead distance reflects unfolding of a single domain, the following gradual decreases in the distance are caused by translocation of the now unfolded polypeptide, and periods of no movement just prior to unfolding an adjacent domain are the pre-unfolding dwell times. We also collected a handful of new traces of single-molecule degradation by ClpAP, but largely relied on our previously published analyses for comparisons as there were no significant differences between the newly collected and the previously measured WT ClpAP traces (Olivares et al., 2017; Olivares et al., 2014).
Figure 2: Single-molecule degradation by ClpAP and ClpAP.

(A) Optical-trap setup with ClpA attached to one bead via ClpPplatform and a multi-domain substrate attached to the smaller bead via linkage of the Halo domain to DNA. (B) Representative single-molecule degradation traces for ClpAP and ClpAP in the N-to-C direction (left) or C-to-N direction (right). The Halo domains are shown in gray. Rectangular and elliptical boxes mark pauses and pre-unfolding dwell times, respectively. Traces were recorded at forces between 8 and18 pN and were decimated to 300 Hz. ClpAP traces are from Olivares et al (Olivares et al., 2017; Olivares et al., 2014) and are shown for comparison.
Representative single-molecule traces revealed that ClpAP takes longer than ClpAP to process one substrate domain in either the N-to-C or the C-to-N direction (Figure 2B). Processing time is defined as the interval between completion of one translocation event and completion of the next translocation event and thus includes the pre-unfolding dwell, the unfolding event, and the subsequent translocation event. The average processing time for a V15P domain during degradation initiated from the N-terminus was 36 ± 4 s for ClpAP (n = 44) versus 14 ± 1 s for ClpAP (n = 50; average ± 1 SEM). For degradation initiated at the C-terminus, these values for the V13P domain were 20 ± 3 s for ClpAP (n = 33) and 12 ± 2 s for ClpAP (n = 17).
D1 plays a major role in unfolding
Pre-unfolding dwell times, the time between the end of translocation of one domain and the start of the unfolding of the next domain, provide information on the relative difficulty of enzymatic unfolding. In experiments in which degradation and thus unfolding of V15P domains was initiated from the N-terminus, the distribution of ClpAP pre-unfolding dwell times fit well to a sum of two exponentials (R2 >0.99) with time constants of 20 s (72% amplitude) and 1.4 s (28% amplitude) as compared to a single-exponential time constant of 0.8 s for ClpAP (Figure 3A) (Olivares et al., 2017). Thus, the majority of V15P domains are unfolded at least 20-fold more slowly when the D1 domain is hydrolytically inactive. Moreover, unfolding accounted for more than 50% of the processing time for ClpAP, but less than 10% for ClpAP. Neither ClpAP nor ClpAP unfolded the Halo domain in the N-to-C direction (Supplementary Figure S1), as expected from prior studies (Olivares et al., 2017).
Figure 3: Unfolding by ClpAP and ClpAP.

(A) Cumulative-frequency distribution of pre-unfolding dwell times (n = 58 events) for ClpAP unfolding of V15P domains in the N-to-C direction. The solid red line is a double-exponential fit (R2 > 0.99), with time constants and amplitudes shown in the red rectangle. The red dashed line shows a single-exponential fit. The black line is a single-exponential fit of data for ClpAP unfolding of the same substrate taken from Olivares et al (Olivares et al., 2017; Olivares et al., 2014), with the time constant in the black rectangle. (B) ClpAP and ClpAP unfolding of V13P domains in the C-to-N direction. (For ClpAP:V13P, n = 38 events). Colors and fits are the same as panel (A), except the ClpAP data is a double-exponential fit and ClpAP data are shown in blue. (C, D) Traces, with short-lived intermediates, during the unfolding of titin domains by ClpAP in the N-to-C (C) or C-to-N (D) directions. Raw traces were decimated to 1500 Hz. (E, F) Kinetic and structural models for intermediates formed during enzymatic unfolding of titin domains by ClpAP and ClpAP. β-strands in the titin domain are labeled, and approximate structural elements in the intermediate during unfolding from each terminus are shown. Bootstrap analyses were performed on pre-unfolding dwells reported in A and B and the 95% CI of the distribution of the time constants are reported in Table 1.
During C-to-N degradation, the distribution of ClpAP pre-unfolding dwell times for V13P also fit well to a sum of two exponentials (R2 > 0.99) with unfolding time constants of 13 s (81% amplitude) and 0.9 s (19% amplitude) as compared to 4.3 s (57% amplitude) and 0.3 s (43% amplitude) for ClpAP (Figure 3B) (Olivares et al., 2014). Thus, unfolding in the C-to-N direction is ~5-fold slower when D1 is unable to hydrolyze ATP. ClpAP occasionally unfolded the Halo domain in this direction (Figure 2B), but we obtained too few traces to compile meaningful statistics. To determine the distribution of pre-unfolding dwell times, we performed bootstrap analyses and reported the values of the 95% confidence intervals (Table 1).
Table 1:
Unfolding and Translocation parameters with the substrate N-to-C V15P and C-to-N V13P
| N-to-C V15P | |||||
| Pre-unfolding dwell time (s) | Intermediate lifetime (ms) | Average translocation velocity (nm/s) | Pause-free translocation velocity (nm/s) | Step dwell time (s) | |
| ClpAP | 18.6–19.3 (68–70%)a 0.9–1.1 (30–32%) n = 58 | 24.8–26.3a
n = 76 |
0.97 ± 0.06b
n = 86 |
2.8 ± 0.1b
n = 86 |
0.83 ± 0.002b (80%), 7.4 ± 0.03b (20%) n = 798 |
| ClpAP | 0.77–0.85a | 10.56–10.62a n = 109 | 2.4 ± 0.1b,c | 2.9 ± 0.2b n = 109 | 0.55 b,c |
| C-to-N V13P | |||||
| ClpAP | 14.7–17.1 (73–78%)a 1.1–1.4 (24–26%) n = 39 | 18.4–19.2a
n = 50 |
2.1 ± 0.1b
n = 58 |
2.9 ± 0.2b
n = 58 |
0.6 ± 0.01b (92%), 7.4 ± 4.4b (8%) n = 442 |
| ClpAP | 3.0–4.2 (59–63%)a
0.24–0.28 (39–41%) |
17.3–17.6a n = 36 | 3.0 ± 0.1b | 3.5 ± 0.2b n = 35 | 0.4b, d (90%), 2 b,d (10%) |
The range indicates 95% CI of the normal distribution of fitting parameters from a bootstrap analysis of the experimental data
Errors are ± 1 SEM
From Olivares et al (Olivares et al., 2017)
From Olivares et al (Olivares et al., 2014)
As expected, in accordance to our previous data with both ClpAP and ClpXP, ClpAP unfolding of V15P from the N-terminus was faster than the unfolding of V13P from the C-terminus (Cordova et al., 2014; Olivares et al., 2017). Rapid enzymatic unfolding specifically in the N-to-C direction is logical, as the structural units near the N terminus of titinI27 are not in a very stably folded region of the domain, whereas the β-sheet structure near the C terminus is within the most stably folded region (Improta et al., 1996). Interestingly, the trend of observing faster unfolding in the “easy” N-to-C direction was not observed for ClpAP, which actually unfolded the I27 variants faster, moving C-to-N than N-to-C (Figures 2 and 3A, B). Thus, ATP hydrolysis by the D1 domain must be important to aspects of protein unfolding other than contributing to the probability of successfully unfolding the domains of high local stability.
Intermediates in enzymatic I27 unfolding
During V15P and V13P unfolding by ClpAP, ~30% of events occurred faster than the time, resolved by the instrument, indicative of cooperative F → U unfolding, but ~70% of events displayed non-cooperative denaturation with a short-lived intermediate (I). Figures 3C and 3D show examples of non-cooperative unfolding from the N-to-C and C-to-N directions, respectively. Re-analysis of ClpAP unfolding of these domains showed similar trends. The distributions of pre-unfolding dwell times did not differ substantially for two-state and three-state unfolding (Supplementary Figure S2), indicating that the probability of either class occurring was poorly correlated with the time required for successful unfolding. The I-to-U lifetimes for C-terminal unfolding by ClpAP and ClpAP were similar (~20 ms), whereas the I-to-U lifetime for N-terminal unfolding by ClpAP (~27 ms) was longer than for ClpAP (~11 ms) (Supplementary Figure S3A; Table 1). Thus, when the D1 ring is capable of hydrolyzing ATP, the intermediate lifetime is shortened during unfolding from the N terminus. Distributions of the intermediate lifetimes were determined by bootstrap analyses and the 95% confidence intervals reported in Table 1.
Independent of the direction of ClpAP or ClpAP unfolding, the F-to-I and I-to-U distances were ~5 nm and ~9 nm, respectively (Figures 3C, 3D). These results indicate that the intermediate in N-terminal unfolding is structurally different than the intermediate during C-terminal unfolding. To determine contour lengths for each species, we fit the force dependence of extension using the worm-like-chain (WLC) model and converted these values from nm to the approximate amino acid position in I27, which allowed us to approximately map the structural folded units in each of the two intermediates (STAR methods & Supplementary Figure S3b). These results suggested that N-terminal unfolding produces an intermediate that contained the D, E, F, and G β-strands of I27 in a folded state (Figure 3E), whereas C-terminal unfolding resulted in an intermediate that contained folded the A-A′, B, C, and D β-strands (Figure 3F). Thus, we conclude that ClpAP and ClpAP, in addition to catalyzing protein unfolding, alter the unfolding pathways accessible to the I27 domains compared to non-enzymatic unfolding pathways promoted by chemical or mechanical denaturants (Best et al., 2003; Marszalek et al., 1999; Williams et al., 2003).
D1-ring activity is required for efficient translocation
We quantified translocation velocities during degradation by ClpAP and ClpAP. During N-to-C translocation, the average velocity of ClpAP (~1 nm/s) was ~60% slower than that of ClpAP (2.4 nm/s) (Figure 4A; Table 1) (Olivares et al., 2017). During C-to-N translocation, the average velocity of ClpAP (2.1 nm/s) was ~30% slower than ClpAP (3 nm/s) (Figure 4B; Table 1) (Olivares et al., 2014). Notably, during ClpAP translocation, pausing or stalling for significant periods was often observed (see boxed regions in Figure 2B). By contrast, ClpAP pausing during translocation was infrequent and short-lived, as previously observed (Olivares et al., 2017; Olivares et al., 2014).
Figure 4: N-to-C and C-to-N translocation.

(A) Average N-to-C translocation velocities and pause-free velocities for ClpAP and ClpAP. The inset shows the average number of pauses for one titin domain. Values are the mean ±1 SEM (For ClpAP, n = 86 events). (B) Average C-to-N translocation velocities and pause-free velocities. Other details are the same as in panel (A). (For ClpAP, n = 44 events) (C) Percentage of events with at least one pause during N-to-C or C-to-N translocation of one titin domain for ClpAP and ClpAP. (D) Average step dwell time(s) at each amino acid during N-to-C translocation of a titin domain by ClpAP (red) or ClpAP (black). The average pre-step dwell time at each amino acid was calculated, and a moving average over a 10-residues window is plotted. The shaded region illustrates ±1 SEM of the moving average at each amino acid. (E) Same analysis as in panel (D) except for C-to-N translocation with ClpAP data colored blue.
When ClpAP translocation progresses normally, the enzyme takes a step at least every 2.5 s, with most step dwell times being ≤ 1s (Table 1). We re-calculated average translocation velocities with pauses ≥ 2.5 s removed from each trace (a pause was defined here as no substantial enzyme movement for ≥ 2.5 s, a time ~5-fold longer than the average step dwell of ~0.5 s). These calculated “pause-free” velocities for ClpAP were ~3.0 nm/s for both N-to-C and C-to-N translocation, similar to values determined for ClpAP (Figures 4A, 4B) indicating that the slower translocation of ClpAP is almost entirely caused by more frequent and longer pauses. Additionally, we observed no force dependence for either the overall or pause-free translocation velocities during this analysis (Supplementary Figure S4a).
Directionality influences pause frequency and duration
On average, ClpAP paused more frequently during N-to-C translocation (1.7 pauses per domain) than in C-to-N translocation (0.55 pauses per domain) (insets in Figures 4A, 4B), accounting for ~75% and ~30% of the total translocation time, respectively. Furthermore, ~90% of ClpAP N-to-C translocation events had one or more pauses per domain, whereas this value was ~40% for C-to-N translocation (Figure 4C). Finally, the longest ClpAP pauses during N-to-C translocation were ~2 min, whereas this value was about 10 s during C-to-N translocation (see Figures 5E, 5F). These dramatic differences in pausing frequency and duration, dependent on the direction of movement were not observed for ClpAP translocation. Hence, directional differences in pause frequency and duration for ClpAP likely arise because ATP hydrolysis in the D1 domain alters interactions with the substrate in a manner that depends on the polarity of polypeptide translocation (see Discussion).
Figure 5:

(A) Representative stepping in ClpAP trajectories during N-to-C translocation. Raw data were decimated to 50 Hz (grey) and the chi-squared fits to the 50 Hz data are shown in red. (B) Representative stepping in ClpAP during C-to-N translocation. Raw traces decimated to 50 Hz are shown in grey and the chi-squared fits are shown in blue. (C) Distribution of step sizes during N-to-C translocation for ClpAP (dark gray) and ClpAP (red). (D) Distribution of step sizes during C-to-N translocation for ClpAP (dark gray) and ClpAP (blue). (E) Cumulative-frequency distributions of pre-step dwell times during N-to-C translocation by ClpAP. (F) Distributions of pre-step dwell times during ClpAP C-to-N translocation. In panels (E) and (F) the distributions are fit by double exponentials (gray dashed line, R2 > 0.99). Pre-step dwell times during translocation by ClpAP are shown in black for comparison (Olivares et al., 2017; Olivares et al., 2014). (For ClpAP: N-to-C, n = 798 events; ClpAP:C-to-N, n = 442 events). Boxes mark pauses, defined as step dwell times ≥ 2.5 s.
To map ClpAP or ClpAP pause locations, we converted translocation length to amino acid positions using the WLC model and then determined the average step dwell time at each I27 residue in traces from translocation in each direction. The average step dwell at each amino acid for N-to-C translocation was ~1.9 ± 0.7 s for ClpAP and 0.7 ± 0.3 s for ClpAP, whereas these values were 0.7 ± 0.2 s and ~0.5 ± 0.2 s, respectively, for C-to-N movement (Figures 4D, 4E). Furthermore, this analysis demonstrated that pausing by ClpAP occurs throughout the polypeptide sequence, regardless of translocation direction (Supplementary Figure S4b).
Step-size distributions and dwell times
In principle, elimination of ATP hydrolysis in the D1 ring might result in shorter translocation steps or longer dwell times between steps. To test these possibilities, we used a stepping algorithm (Kerssemakers et al., 2006) to analyze translocation-step properties (Figures 5A, B, Supplementary Figure S5). For both ClpAP and ClpAP, the step-size distributions were very similar with a peak at ~1 nm, irrespective of whether translocation was N-to-C or C-to-N (Figures 5C, D). Thus, elimination of ATP hydrolysis in the D1 ring does not alter translocation step size of the ClpAP machine in a significant fashion.
Figures 5E, 5F show the distribution of step dwell times for ClpAP and ClpAP. For both translocation directions, dwell times were slightly longer for ClpAP than for ClpAP, and this difference was greater for N-to-C translocation (Figures 5E, F and Table 1). Thus, inactivation of ATP hydrolysis in the D1 ring frequently briefly lengthens the time between translocation steps and when these longer step dwells become extended (>2.5 s), the result is enzyme pausing.
ATP dependence of ClpAP activity and substrate slipping
The studies presented above used 5 mM ATP, whereas half-maximal hydrolysis by ClpAP (in the presence of substrate) occurred at ~0.35 mM ATP (Supplementary Figure S6a). Prior studies establish that the KM of ATP for the D1 ring is tighter than that for the D2 ring (Kress et al., 2009). To determine the ATP dependence of ClpAP pausing behavior during unfolding and translocation, which should reflect D2-ring activity, we also performed single-molecule N-to-C degradation assays at 0.1 mM (n = 18), and 0.25 mM (n = 37) ATP, concentrations where the D2 ring would not be fully ATP bound. Notably, the distribution of pre-unfolding dwell times and translocation velocities were not substantially altered at these lower ATP concentrations (Figure 6A, 6B). These data strongly suggest that full saturation of the D2 ring with ATP is not required for robust mechanical function.
Figure 6:

ATP dependence of ClpAP degradation. (A) Distribution of pre-unfolding dwell times for N-to-C degradation at different ATP concentrations. The solid lines are double-exponential fits. Time constants are 20 ± 0.9 s and 1.4 ± 0.1 s for 5 mM ATP; 13 ± 1.5 s and 0.4 ± 0.1 s for 0.25 mM ATP; and 19 ± 4 s and 0.2 ± 0.1s for 0.1 mM ATP. (Values are mean ± 1 SEM). (B) Average translocation velocities at different ATP concentrations. (Values are mean ± 1 SEM). (C) Representative traces (decimated to 300 Hz) showing slipping events (orange) during N-to-C unfolding and translocation by ClpAP using 0.1 mM ATP. (D) Relative frequency of substrate slips of different lengths (number of amino acids). (E) Total substrate-slip frequencies decrease as the ATP concentration increases.
In ensemble experiments, by contrast, ClpAP degradation of the multidomain substrate in the N-to-C direction and GFP-ssrA in the C-to-N direction were both slowed at ATP concentrations below 0.75 mM (Supplementary Figures S6b, S6c). Moreover, steady-state kinetic parameters determined for ClpAP degradation of GFP-ssrA at different ATP concentrations (Supplementary Figures S6 d, e) revealed a substantial Vmax decrease at 0.1, 0.25 and 0.5 mM ATP. The differences observed between single-molecule and ensemble degradation at low ATP concentrations are likely to reflect inefficient substrate engagement by ClpAP, a step that requires ATP-bound subunits but is not monitored in the optical-trap assay.
Suboptimal ATP concentrations did, however, alter one important feature of the mechanical processes of unfolding and translocation. During N-to-C single-molecule degradation by ClpAP, we observed periodic “slips”, in which the substrate polypeptide appeared to thread backwards through the enzyme, causing a very rapid increase in bead-to-bead distance (Figure 6C). In contrast to the changes in bead-to-bead distance associated with unfolding however, slipping resulted in variable distance changes that were shorter than the I27-contour length and also often occurred before domain translocation was complete. Most slips corresponded to ~20–40 amino acid segments (Figure 6D). Slipping was more common at lower ATP concentrations with total frequencies of: 0.65 slips/min at 0.1 mM ATP; 0.37 slips/min at 0.25 mM ATP, and; 0.07 slips/min at 5 mM ATP (Figure 6E). Substrate slipping was infrequent for ClpAP, even at low ATP. In combination, these observations suggest that ClpAP has a decreased ability to grip substrates properly, and this feature is unmasked by lower ATP concentrations where the active D2 ring is unlikely to be saturated with ATP.
Discussion
Protein unfolding and remodeling enzymes that contain two AAA+ rings play important roles in proteostasis in all cells, but no clear, unifying function for the second motor ring has emerged (Gottesman et al., 1998). Prior studies show that the D2 ring of ClpAP plays the dominant role in ATP-dependent degradation (Kress et al., 2009). Our optical-tweezers studies here probe the consequences of eliminating ATP hydrolysis in the D1 ring of ClpA on single-molecule unfolding and translocation by the ClpAP protease. Similarities between ClpAP and ClpAP reveal ring functions specific to ATP hydrolysis in the D2 ring, whereas differences provide a view into mechanistic tasks that normally require ATP hydrolysis in the D1 ring.
Compared to ClpAP, ClpAP is a slower protein translocase because it pauses more frequently during polypeptide translocation, a defect that is more severe during N-to-C than C-to-N translocation. Likewise, compared to ClpAP, the average time required for ClpAP to unfold the V15P domain is ~20-times slower in the N-to-C direction, whereas unfolding of the V13P domain is ~5-times slower in the C-to-N direction. Thus, ClpAP is both a less reliable protein translocase and a slower protein unfoldase. These mechanical defects are likely to be related, as slower ClpAP unfolding and increased pausing are both intensified during N-to-C degradation; “D2 motor stalling” can reasonably explain both effects and would be expected to be more severe against the resisting force that occurs during unfolding attempts.
Prior studies of a ClpAP variant show that it degrades substrates of low and intermediate stability as efficiently as ClpAP, whereas degradation of high-stability substrates required ATP hydrolysis in both the D1 and D2 rings (Kress et al., 2009). Thermodynamically and kinetically, however, the domains studied here are metastable (V13P – ΔGu = 2.9 kcal/mol, ku = 32 min−1: V15P – ΔGu = 4.6 kcal/mol; ku = 2.3 min−1) (Kenniston et al., 2003) and yet are unfolded ~5 to ~20-times more slowly by ClpAP than by ClpAP. For comparison, the thermodynamic, kinetic and mechanical stabilities of commonly used substrates for in vitro studies on ClpAP are given in supplementary table S1. In addition, our results show that ATP hydrolysis in the D1 ring decreases the activation energy barrier for unfolding of V15P from the N terminus by 1.74 k.cal.mol−1 and for V13P from the C-terminus by 0.96 k.cal.mol−1 assuming an Arrhenius frequency factor (kA) of 109 s−1 at 291K (Bieri et al., 1999; Dietz et al., 2006). Thus, our results indicate that ATP hydrolysis in both the D1 and D2 rings can be important for enhancing unfolding of protein domains of moderate and well as high stabilities.
Although ClpAP and ClpAP catalyze similar rates of ATP hydrolysis in the absence of protein substrates as shown here and previously (Kress et al., 2009), ClpAP hydrolyzes ATP at only 50–60% of the wild-type rate in the presence of either of our protein substrates. This change in ATPase rate, at least in part, appears to be a consequence of two interrelated factors: (i) ClpAP spends more of the total time during degradation carrying out unfolding of substrate domains than does ClpAP; and (ii) ATP hydrolysis by AAA+ machines is slower during unfolding than translocation (Kenniston et al., 2003).
Regardless of direction, the majority of I27-unfolding events catalyzed by ClpAP or ClpAP occur via an intermediate. The intermediate is different during N-to-C than C-to-N unfolding, and is also longer-lived during N-to-C unfolding by ClpAP. By contrast, non-enzymatic I27 unfolding is generally cooperative or two-state; and rare intermediates when observed, are clearly distinct from those characterized here (Best et al., 2003; Marszalek et al., 1999; Williams et al., 2003). These differences highlight the fact that AAA+ enzymes, like ClpAP change protein-unfolding pathways by pulling directly on one end of the target molecule, a very different process than spontaneous, non-enzymatic force and/or chemical-induced denaturation.
During N-to-C or C-to-N translocation, the step sizes taken by ClpAP and ClpAP are indistinguishable (average ~1.25 nm) and the pause-free velocities are similar (~3 nm/s). Thus, motor activity in the D2-ring alone is sufficient for normal step sizes and near normal translocation rates when pausing is ignored. In the absence of ATP hydrolysis in the D1 ring, however, pausing slowed the overall rate of translocation more than 2-fold in the N-to-C direction and ~30% in the C-to-N direction.
Compared to wild-type ClpAP translocation, the ClpAP defects are prominent in the N-to-C direction as shown by (i) higher frequencies of translocation pauses; (ii) longer pause durations; and (iii) slightly longer step dwells. Thus, the wild-type D1 of ClpA ring facilitates more efficient mechanical processing of substrate proteins, especially in the N-to-C direction. Previous instances of AAA+ proteases degrading substrates more efficiently from one terminus have been typically ascribed to the substrate structure, such that unfolding of a given protein is “easier” from one end and thus the rates of degradation from the “easy” terminus are faster than those from the “difficult” end (Kress et al., 2009; Lee et al., 2001; Olivares et al., 2017). Our results, however, show substantial D1-dependent differences in translocation (in addition to the unfolding effects) in the two directions using the same protein substrates.
We propose that interactions between the D1 ring and the substrate polypeptide are more favorable for the N-to-C polarity because of stereo-chemical differences in enzyme-substrate contacts. This idea will need testing with different D1-mutant classes, especially as the ATPase-defective Walker B mutant variants used here are generally considered to be able to grip substrates (especially in the presence of ATPγS). It will thus be interesting to explore the preferences in substrate polarity with mutant variants of ClpA in which the pore-1 loops, key parts of the enzyme needed to grip the substrate, of the D1ring and those of the D2 ring are altered. D1 pore1-impared ClpAP may show an even greater defect in N-to-C substrate processes. This result would favor the idea that the D2 ring has the opposite-polarity preference due to a differently shaped enzyme-substrate gripping surface, thus explaining why D1 motor defective enzymes have a harder time carrying out mechanical processes when traveling N-to-C on a polypeptide. It may be especially important for ClpAP to have a mechanism that specifically assists mechanical functions in the N-to C direction. Some ClpAP substrates are recognized via N-degrons and others via C-degrons. In fact, ClpAP with the adaptor ClpS appears to be the only E. coli protease responsible for degrading proteins tagged for degradation by the N-end rule pathway and degradation of N-end rule substrates has been proposed to be physiologically important (Erbse et al., 2006).
How does an active D1 ring suppress pausing? Because ClpAP (ATPase defective D2 ring) degrades some target proteins slowly (Kress et al., 2009), and the D1 ring contains AAA+ motifs required for motor activity, it is likely that the wild-type D1 ring can support translocation through the ClpA pore and into ClpP. However, ClpAP hydrolyzes ATP at least 10-fold more slowly than either ClpAP or ClpAP (Kress et al., 2009), likely accounting for its strong defect in degradation. Together, these observations establish that D2 is the principal unfoldase/translocase engine, whereas D1 serves as an auxiliary motor. Why then is the slow and inefficient D1 motor required for robust translocation and unfolding by wild-type ClpAP? One possibility is that the D1 and D2 motors function independently. Thus, when the dominant D2 motor fails, the slower D1 motor fills in and prevents pausing. Independence is supported by the fact that the contributions of the D1 and D2 rings to overall ATP hydrolysis are roughly additive under numerous conditions (Kress et al., 2009). Moreover, the greater importance of the D1 ring during N-to-C than C-to-N translocation and unfolding is also consistent with relatively independently functioning unfolding/translocation motors in the D1 and D2 rings. A second and non-exclusive possibility is that the two motor rings are allosterically coupled, as proposed for Hsp104 (Franzmann et al., 2011). For example, allosteric coupling might allow ATP hydrolysis in the D1 ring to restart the D2 motor when it stalls.
The D1 ring appears to play an additional important role in gripping protein substrates. For example, ClpAP typically unfolds substrates faster but translocates them more slowly than ClpXP, which has just one AAA+ ring (Olivares et al., 2014). Faster unfolding by ClpAP would be expected if it generates a similar unfolding force but can grip substrates more tightly using two rings compared to the single ring of ClpXP. Within the pores of AAA+ unfoldases there are highly conserved pore-1-loops (consensus sequence GYVG for the ClpAD2 and the ClpX rings, and KYR for the D1 ring of ClpA) that are especially important for substrate binding within, and translocation through the unfoldase. In support of the idea that the larger AAA+ enzymes may grip substrates better throughout their pores, cryo-EM structures of the double-ring Hsp104 enzyme reveal from 7 to 10 contacts between pore-1-loops and substrate, whereas only 4 or 5 equivalent contacts are observed in structures of single-ring enzymes (Puchades et al., 2017; Yokom et al., 2016). Our experiments here strengthen the model that D1 and D2 both contribute to substrate grip, and that the sum of the different features of each ring provide a better grasp of the target substrate by the double-ring enzymes, including ClpAP. At low concentrations of ATP, where the D2 ring is expected to be incompletely nucleotide bound (its KM for ATP hydrolysis is ~13-fold weaker than that of D1 (Kress et al., 2009)), we observe polypeptide “back slipping” through the enzyme pore of ClpAP but not ClpAP. Thus, ATP hydrolysis in the D1 ring of wild-type ClpAP appears to help the enzyme maintain a grip on the substrate and prevent back-slipping when D2 is not fully saturated with ATP. Interestingly, compared to ClpXP, ClpAP plays a more substantial role in degradation during stationary phase (Farrell et al., 2005), a condition where ATP concentrations can drop considerably (Peterson et al., 2012).
Remarkably, like ClpA, other well-studied double-ring AAA+ unfoldases/remodeling enzymes have one ring that is the dominant, faster ATPase, whereas the other ring is much slower (Wendler et al., 2012). In most cases, as with ClpA, it is the D2 ring that is dominant, although this architecture appears to be flipped for yeast Hsp104 (Schirmer et al., 1998). In two cases (Hsp104 and Cdc48), the faster ATPase is thought to be the major translocation motor, as with ClpA, and emerging evidence favors models where the auxiliary ring has an important role in substrate release from the enzyme (Bodnar and Rapoport, 2017; Schaupp et al., 2007). Interestingly, the two rings in ClpAP evolved from different subfamilies of AAA+ unfoldases (Erzberger and Berger, 2006), rather than duplication of the AAA+ module of a single-ring enzyme, further supporting the view that each ring performs unique functions in the double-motor enzyme. The D1 ring of ClpA is a member of the classic clade of AAA+ enzyme (Erzberger and Berger, 2006). Some members of this clade, including YME1, PAN, and Rpt1–6, are thought to operate by strictly sequential ATP-hydrolysis mechanisms that result in two-residue translocation steps (de la Pena et al., 2018; Kim et al., 2015; Puchades et al., 2017). The D2 of ClpA ring, is a member of the HCLR clade, together with ClpX, HslU, and Lon (Erzberger and Berger, 2006). ClpX and HslU appear to operate by probabilistic mechanisms of ATP hydrolysis, and ClpX takes basic translocation steps of ~6 residues, similar to preformed during translocation by ClpAP and ClpAP (Aubin-Tam et al., 2011; Baker and Sauer, 2012; Baytshtok et al., 2017). It is straightforward to picture situations in which having an engine of each type in a two-motor machine might be advantageous. Further dissection of individual AAA+ enzymes and their rings should reveal how differences in mechanism support the various biological functions carried out by this large and diverse family of protein unfoldases and remodeling machines.
STAR METHODS:
LEAD CONTACT AND MATERIAL AVAILABILITY:
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Tania Baker (tabaker@mit.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS:
ClpAΔC9/E286Q, ClpAΔC9, Halo-(V13P)4-ssrA, Halo-(V15P)4 are expressed in E.coli BL21(DE3) strain and ClpP, ClpPM5A are expessed in E.coli JK10 strain. The detailed growth conditions and expression protocols for individual constructs are given in method details.
METHOD DETAILS:
Protein Purification:
ClpAΔC9, a variant of E. coli ClpA – containing the ΔC9 deletion(Maglica et al., 2008), which prevents autodegradation without affecting activity, and ClpAΔC9, E286Q Walker-B mutation – was cloned, expressed and purified as described (Baytshtok et al., 2015; Olivares et al., 2017; Olivares et al., 2014). Briefly, a pET23b plasmid carrying the gene encoding ClpA was transformed into a BL21(DE3) strain with the wild-type clpA gene deleted. Cells were grown to OD600 ~1 at 30 °C, induced with 0.4 mM IPTG, grown for additional 2 h, and harvested. The cell paste was resuspended in Lysis buffer (50 mM Tris-Cl, pH 7.5, 2 mM EDTA, 10% glycerol, 2 mM DTT) and stored at −80 °C. After lysis by French press, the lysate was clarified by centrifugation at 30,000 × g for 30 min, and 40% (w/v) ammonium sulfate was added to precipitate ClpA. The pellet was resuspended in SBA buffer (25 mM HEPES, pH 7.5, 200 mM KCl, 0.1 mM EDTA, 10% glycerol, 2 mM DTT), and ClpA was purified by ion-exchange chromatography using S-Sepharose and MonoQ HR 5/5 columns (GE Healthcare). Purified ClpA was dialyzed into storage buffer (50 mM HEPES, pH 7.5, 20 mM MgCl2, 0.3 M NaCl, 300 mM arginine, 10% glycerol, 0.5 mM DTT), divided into aliquots, and stored at −80 °C. ClpAΔC9 was used as the wild-type background for ClpA here and previously(Olivares et al., 2014).
E.coli ClpP, and ClpPplatform (used for optical-trap experiments) were expressed, and purified as described (Kim et al., 2000; Olivares et al., 2014). ClpPplatform is a tetradecameric ClpP variant consisting of one heptameric ring of wildtype ClpP and one heptameric ring of ClpPM5A-bAP(biotin acceptor peptide)-His. Both ClpPs were grown at 30 °C to O.D ~ 0.5 and induced with 0.5 mM IPTG. Both were purified by Ni-NTA affinity chromatography ClpPM5A-bAP-His was exogenously biotinylated using BirA enzyme and His tag of wt.ClpP was removed by TEV protease. An excess of ClpPM5A-bAP-His was mixed with wt.ClpP and dialyzed at 4 °C against a buffer containing 150 mM ammonium sulfate allowing the exchange of heptameric rings. Tetradecameric ClpPPlatform was purified by Ni-NTA chromatography. Only ClpP/ClpPM5A-bAP-His species binds to ClpA and not ClpPM5A-bAP-His/ ClpPM5A-bAP-His due to M5A mutation.
E.coli BL21DE3 cells containing pFN18A plasmid carrying Halo-(V13P)4-ssrA substrate (C-to-N) (Cordova et al., 2014; Olivares et al., 2014) and Cys-(V15P)4-Halo substrate (N-to-C) (Olivares et al., 2017) were expressed at 37°C and induced using 1 mM IPTG. These multidomain substrates were purified by tandem Ni-NTA affinity and size exclusion chromatography. A partial ssrA peptide containing an N-terminal maleimide (maleoyl-β-Ala-NYALAA-coo−)was subsequently attached to the N-terminus of Cys-(V15P)4-Halo to generate ssrA-(V15P)4-Halo (Olivares et al., 2017).
Single-molecule optical-trapping:
For optical-trap experiments, ClpA/ClpPplatform was attached to 1.25 micron bead and a Halo-titin-ssrA substrate was attached to the 1.09 micron bead as described (Aubin-Tam et al., 2011; Cordova et al., 2017; Cordova et al., 2014; Olivares et al., 2014). Briefly, a 3500 base pairs dsDNA(M13MP18) was modified using oligonucleotides to have biotin at one end and amino group at the other end. Further the amino group reacted with HaloTag Succinimidyl Ester ligand O4 to produce biotin-DNA-Haloligand. One end of a biotin-DNA-Haloligand was attached to a 1.09 μm streptavidin-coated polystyrene bead on one end and the other end was attached to Halo-domain of the multidomain substrate. The 1.09 μm bead was tethered to a glass coverslip using a DNA-linked glass-binding peptide aptamer (using sulfo-SMCC). Biotinylated ClpPplatform was attached to a 1.25 μm streptavidin-coated polystyrene bead (Spherotech), and ClpA was added in presence of 5 mM ATP (except for the analysis of ATP-concentration effects). After complex formation, free ClpA was removed by centrifugation and washing of the beads. Experiments were performed at 17–20 °C in PD-T buffer (25 mM HEPES, pH 7.6, 100 mM KCl, 10 mM MgCl2, 10% glycerol, 0.1% Tween-20, and 1 mM Tris-2-carboxyethyl-phosphine) containing ATP-regeneration and oxygen-scavenging systems containing glucose oxidase and catalase as well as 1 mg/mL bovine serum albumin. The beads were trapped by two 1064-nm lasers in a passive-force clamp mode, and tethers between ClpAP and the substrate were formed by moving the weakly-trapped larger bead. All the data were collected on distinct substrate and enzyme molecules.
Biochemical Assays:
ATP hydrolysis assays:
Steady-state rates of ATP hydrolysis were measured at 24 °C in an NADH-coupled assay by monitoring loss of absorbance at 340 nm (Kim et al., 2001) using 100 nM ClpA6 or ClpA6, 200 nM ClpP14, and 5 μM substrate. The final reaction mixtures were in PD-T buffer supplemented with 5 mM ATP (unless noted in legends), 0.7 mM NADH, 6.25 mM phosphoenolpyruvate, 23.5 U/mL pyruvate kinase, and 20 U/mL lactate dehydrogenase.
Ensemble protein degradation assays:
The kinetics of degradation of GFP-ssrA by ClpA6 (0.6 μM) and ClpP14 (1.6 μM) were assayed by monitoring loss of GFP fluorescence at 540 nm (excitation 470 nm) at 24 °C in PD-T buffer. The ssrA-(V15P)4-Halo substrate (100 μL; 1 μM) was fluorescently labeled by incubation with HaloTag TMR ligand (0.5 μL; 0.5 mM; Promega) in 25 mM HEPES (pH 7.5), 100 mM KCl, 10 mM MgCl2, 10% glycerol, 0.1% Tween-20, and 1 mM DTT at 30 °C for 15 min. Degradation reactions at room temperature contained ClpA6 or ClpA6 (200 nM), ClpP14 (400 nM), ATP (0.1 to 5 mM), and an ATP-regeneration system. Samples were taken at different time points, and degradation was quenched by addition of SDS-sample buffer and flash freezing in liquid N2. Samples were thawed, boiled for 5 min, and a fraction of each reaction was electrophoresed on a Mini-PROTEAN TGX 4–15% (w/v) precast gel (Bio-Rad) prior to scanning the gel using a Typhoon FLA 9500 scanner (GE Healthcare).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Analysis:
Data acquisition was carried out at 3 kHz as described (Cordova et al., 2014; Olivares et al., 2014). Custom MATLAB scripts were used to calculate the inter-bead distance, unfolding distance, pre-unfolding dwell times, and to determine step sizes and step-dwell times as described (Cordova et al., 2014; Olivares et al., 2017). Average translocation velocities were determined by dividing the sum of step sizes by the sum of step dwells for translocation including pauses for individual domains. For determining steps, data were decimated to 50 Hz, and individual steps were detected using χ2 minimization method as described (Kerssemakers et al., 2006). Steps < 0.75 nm and backward steps were combined, and the dwell time preceding a combined step was added to the dwell time of the following step. Any step dwell ≥ 2.5 s was considered a pause and removed from the total step-dwell time when calculating pause-free translocation velocities.
Bootstrap analysis:
To estimate the distribution and error in the pre-unfolding dwells and the intermediate dwells we carried out nonparametric bootstrap analysis (Efron, 1982; Wiita et al., 2007). These analyses are performed using a statistical analysis tool XLSTAT (Addinsoft (2019). (XLSTAT statistical and data analysis, Boston, USA; https://www.xlstat.com). From a given experimental set of pre-unfolding dwell times and intermediate dwells for ClpAP and ClpAP in N-to-C and C-to-N directions, sample data sets of equal size consisting of members randomly chosen from the experimental set were created. Each data set was then individually fit to either a single or double exponential and R2 values for the resulting fit to the data were calculated. 256 such iterations were performed. The fitting parameters from all the iterations were normally distributed and the 95% confidence intervals of these distributions were reported. Double-exponential fitting parameters were used if the mean of the R2 values for a single exponential fit was < 0.95.
Intermediate structures in titinI27 unfolding:
The extensions (x) for F→I, I→U, and F→U transitions were arranged in increasing order of experimental force (F), and a moving average of three data points was fit to the WLC model (persistence length(Lp), 0.36 nm) to determine the change in the contour length (ΔLc) associated with each transition. The WLC model is given by . ΔLc was converted into number of amino-acid residues using a value of 0.365 nm/residue and mapped onto terminal structural units of the titinI27 domain. TitinI27 contains 89 residues in the structured region and 4 addition residues have been used as linkers for the multi-domain substrate. All the distances on titinI27 structure are calculated based on PDB:1TIT structure.
For F→U transition, The contour length change (ΔLc) was given by the equation, ΔLc = Lc – df where df is the length of titinI27 folded domain. Lc is 93*0.365 nm ~34 nm and the length of folded titinI27 domain is ~ 4.2 nm from the PDB 1TIT. The expected ΔLc is ~29.8 nm. The observed ΔLc values for N-to-C and C-to-N unfolding are 31 and 29 nm respectively, well within the range of expected ΔLc values.
For F→I transition, ΔLc = miLaa + dr − df is the number of residues unfolded, dr is the distance between the residue mi (or 89-mi for C-to-N unfolding) and the terminal residue in the remaining folded state (residue 89 for N-to-C unfolding and residue 1 for C-to-N unfolding), df is the length of titinI27 folded domain. We have iterated m based on the distance values from 1TIT structure, calculating the ΔLc and selecting the mi with ΔLccalculated ~ ΔLcobserved (minimum RMSD). Residue 38 and residue 55 gave the lowest RMSD for N-to-C and C-to-N unfolding respectively, indicating the unfolding of three β-strands in both cases. These residue numbers are rough approximations based on the structure available and approximate value of 0.365 nm/aa.
DATA AND CODE AVAILABILITY
Original raw data for the figures 2–6 in the paper is available from the corresponding author on request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E.coli BL21DE3 | New England Biolabs | C2527H |
| E.coli JK10 ((clpP::cat, Δlon, slyD::kan, λDE3)) | Kenniston et al, 2003 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ssrA partial peptide for N-terminal substrate maleoyl-β-Ala-NYALAA-coo− | Synthesized using Fmoc technique and an Apex 396 solid-phase peptide synthesizer | N/A |
| Glass binding peptide CGGRSGRRRSHHHRL | Synthesized using Fmoc technique and an Apex 396 solid-phase peptide synthesizer | N/A |
| HaloTag Succinimidyl Ester ligand O4 | Promega | P6751 |
| Sulfosuccinimidyl 4-[N-maleimidomethyl] cyclohexane-1-carboxylate | Fischer Sci Co | PI22622 |
| Glucose Oxidase, Recombinant | EMD Millipore | 345386 |
| Catalase from Bovine liver | Sigma Aldrich | C30 |
| Bovine serum albumin | New England Biolabs | B9000S |
| BirA, Biotin ligase | Sigma-Aldrich | SRP0417 |
| Oligonucleotides | ||
| M13MP18 double stranded DNA | Bayou Biolabs | P-105 |
| 5BiosG/AATCCGCTTTGCTTCTGAC | IDTDNA, USA | N/A |
| 5AmMC6/TTGAAATACCGACCGTGTG | IDTDNA, USA | N/A |
| Recombinant DNA | ||
| pFN18A plasmid containing Halo- (V13P)4-ssrA | Cordova et al, 2014 | N/A |
| pFN18A plasmid containing Cys- (V15P)4-Halo | Olivares et al, 2017 | N/A |
| pET23b plasmid containing ClpAE286Q/ΔC9 | This study | N/A |
| pET23b plasmid containing ClpAΔC9 | Olivares et al, 2014 | N/A |
| pKS028 plasmid containing ClpPM5A | Olivares et al, 2014 | N/A |
| pKS028 plasmid containing ClpP | Olivares et al, 2014 | N/A |
| Software and Algorithms | ||
| Step finding algorithm | Kerssemakers et al, 2006 | N/A |
| Other | ||
| 1.25 micron streptavidin coated polystyrene beads | Spherotech | SVP-10–5 1%w/v, 1.25 |
| 1.09 micron streptavidin coated polystyrene beads | Spherotech | SVP-10–5 1%w/v, 1.09 |
Highlights:
Single-molecule studies with ClpAP protease reveal roles for its two ATPase rings
D1-ring-inactive ClpAP has mechanical unfolding and translocation defects
A dramatic effect is that D1-defective ClpAP pauses frequently during translocation
Thus, D1 functions as an anti-pausing motor to assist the more powerful D2 ring
Acknowledgments:
We thank Adrian Olivares, Kristin Zuromski, Sora Kim, and Gina Mawla for materials, advice, and helpful discussions. This work was supported by NIH Grants GM-101988 and AI-15706 and the Howard Hughes Medical Institute. H.C.K and T.A.B. are employees of the Howard Hughes Medical Institute.
Footnotes
Authors declare no competing interests
Data Availability:
All the data sets generated and analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- Aubin-Tam ME, Olivares AO, Sauer RT, Baker TA, and Lang MJ (2011). Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145, 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, and Sauer RT (2012). ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta 1823, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baytshtok V, Baker TA, and Sauer RT (2015). Assaying the kinetics of protein denaturation catalyzed by AAA+ unfolding machines and proteases. Proc Natl Acad Sci U S A 112, 5377–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baytshtok V, Chen J, Glynn SE, Nager AR, Grant RA, Baker TA, and Sauer RT (2017). Covalently linked HslU hexamers support a probabilistic mechanism that links ATP hydrolysis to protein unfolding and translocation. J Biol Chem 292, 5695–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Fowler SB, Herrera JL, Steward A, Paci E, and Clarke J (2003). Mechanical unfolding of a titin Ig domain: structure of transition state revealed by combining atomic force microscopy, protein engineering and molecular dynamics simulations. J Mol Biol 330, 867–877. [DOI] [PubMed] [Google Scholar]
- Bieri O, Wirz J, Hellrung B, Schutkowski M, Drewello M, and Kiefhaber T (1999). The speed limit for protein folding measured by triplet-triplet energy transfer. Proc Natl Acad Sci U S A 96, 9597–9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar NO, and Rapoport TA (2017). Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 169, 722–735 e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordova JC, Olivares AO, and Lang MJ (2017). Mechanically Watching the ClpXP Proteolytic Machinery. Methods Mol Biol 1486, 317–341. [DOI] [PubMed] [Google Scholar]
- Cordova JC, Olivares AO, Shin Y, Stinson BM, Calmat S, Schmitz KR, Aubin-Tam ME, Baker TA, Lang MJ, and Sauer RT (2014). Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell 158, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Pena AH, Goodall EA, Gates SN, Lander GC, and Martin A (2018). Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H, Berkemeier F, Bertz M, and Rief M (2006). Anisotropic deformation response of single protein molecules. Proc Natl Acad Sci U S A 103, 12724–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H, and Rief M (2004). Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc Natl Acad Sci U S A 101, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SM, Shorter J, Zolkiewski M, Hoskins JR, Lindquist S, and Wickner S (2007). Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat Struct Mol Biol 14, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effantin G, Ishikawa T, De Donatis GM, Maurizi MR, and Steven AC (2010). Local and global mobility in the ClpA AAA+ chaperone detected by cryo-electron microscopy: functional connotations. Structure 18, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron B (1982). The Jackknife, The Bootstrap, And OtherResampling Plans (Society for Industrial and Applied Mathematics, Philadelphia, PA.). [Google Scholar]
- Erbse A, Schmidt R, Bornemann T, Schneider-Mergener J, Mogk A, Zahn R, Dougan DA, and Bukau B (2006). ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 439, 753–756. [DOI] [PubMed] [Google Scholar]
- Erzberger JP, and Berger JM (2006). Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct 35, 93–114. [DOI] [PubMed] [Google Scholar]
- Farrell CM, Grossman AD, and Sauer RT (2005). Cytoplasmic degradation of ssrA-tagged proteins. Mol Microbiol 57, 1750–1761. [DOI] [PubMed] [Google Scholar]
- Franzmann TM, Czekalla A, and Walter SG (2011). Regulatory circuits of the AAA+ disaggregase Hsp104. J Biol Chem 286, 17992–18001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Word JM, Burton RE, Richardson JS, and Oas TG (1998). Folding kinetics of a fluorescent variant of monomeric lambda repressor. Biochemistry 37, 9179–9185. [DOI] [PubMed] [Google Scholar]
- Gottesman S, Roche E, Zhou Y, and Sauer RT (1998). The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev 12, 1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenleaf WJ, Woodside MT, Abbondanzieri EA, and Block SM (2005). Passive all-optical force clamp for high-resolution laser trapping. Phys Rev Lett 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Maurizi MR, Esser L, and Xia D (2002). Crystal structure of ClpA, an Hsp100 chaperone and regulator of ClpAP protease. J Biol Chem 277, 46743–46752. [DOI] [PubMed] [Google Scholar]
- Hattendorf DA, and Lindquist SL (2002). Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J 21, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Improta S, Politou AS, and Pastore A (1996). Immunoglobulin-like modules from titin I-band: extensible components of muscle elasticity. Structure 4, 323–337. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Gottesman S, Pumphrey J, Rudikoff S, Clark WP, and Maurizi MR (1988). The two-component, ATP-dependent Clp protease of Escherichia coli. Purification, cloning, and mutational analysis of the ATP-binding component. J Biol Chem 263, 15226–15236. [PubMed] [Google Scholar]
- Kato H, Vu ND, Feng H, Zhou Z, and Bai Y (2007). The folding pathway of T4 lysozyme: an on-pathway hidden folding intermediate. J Mol Biol 365, 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Fernandez JM, and Sauer RT (2003). Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell 114, 511–520. [DOI] [PubMed] [Google Scholar]
- Kerssemakers JW, Munteanu EL, Laan L, Noetzel TL, Janson ME, and Dogterom M (2006). Assembly dynamics of microtubules at molecular resolution. Nature 442, 709–712. [DOI] [PubMed] [Google Scholar]
- Kim YC, Snoberger A, Schupp J, and Smith DM (2015). ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat Commun 6, 8520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI, Burton RE, Burton BM, Sauer RT, and Baker TA (2000). Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Mol Cell 5, 639–648. [DOI] [PubMed] [Google Scholar]
- Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, and Baker TA (2001). Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol 8, 230–233. [DOI] [PubMed] [Google Scholar]
- Kress W, Mutschler H, and Weber-Ban E (2009). Both ATPase domains of ClpA are critical for processing of stable protein structures. J Biol Chem 284, 31441–31452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Schwartz MP, Prakash S, Iwakura M, and Matouschek A (2001). ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell 7, 627–637. [DOI] [PubMed] [Google Scholar]
- Lim WA, Farruggio DC, and Sauer RT (1992). Structural and energetic consequences of disruptive mutations in a protein core. Biochemistry 31, 4324–4333. [DOI] [PubMed] [Google Scholar]
- Llinas M, and Marqusee S (1998). Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci 7, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglica Z, Striebel F, and Weber-Ban E (2008). An intrinsic degradation tag on the ClpA C-terminus regulates the balance of ClpAP complexes with different substrate specificity. J Mol Biol 384, 503–511. [DOI] [PubMed] [Google Scholar]
- Marszalek PE, Lu H, Li H, Carrion-Vazquez M, Oberhauser AF, Schulten K, and Fernandez JM (1999). Mechanical unfolding intermediates in titin modules. Nature 402, 100–103. [DOI] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Strub C, Rist W, Weibezahn J, and Bukau B (2003). Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J Biol Chem 278, 17615–17624. [DOI] [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, and Koonin EV (1999). AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9, 27–43. [PubMed] [Google Scholar]
- Ogura T, and Wilkinson AJ (2001). AAA+ superfamily ATPases: common structure--diverse function. Genes Cells 6, 575–597. [DOI] [PubMed] [Google Scholar]
- Olivares AO, Baker TA, and Sauer RT (2016). Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat Rev Microbiol 14, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares AO, Kotamarthi HC, Stein BJ, Sauer RT, and Baker TA (2017). Effect of directional pulling on mechanical protein degradation by ATP-dependent proteolytic machines. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares AO, Nager AR, Iosefson O, Sauer RT, and Baker TA (2014). Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol 21, 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, and Li H (2008). Atomic force microscopy reveals parallel mechanical unfolding pathways of T4 lysozyme: evidence for a kinetic partitioning mechanism. Proc Natl Acad Sci U S A 105, 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CN, Levchenko I, Rabinowitz JD, Baker TA, and Silhavy TJ (2012). RpoS proteolysis is controlled directly by ATP levels in Escherichia coli. Genes Dev 26, 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puchades C, Rampello AJ, Shin M, Giuliano CJ, Wiseman RL, Glynn SE, and Lander GC (2017). Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, and Baker TA (2011). AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80, 587–612. [DOI] [PubMed] [Google Scholar]
- Schaupp A, Marcinowski M, Grimminger V, Bosl B, and Walter S (2007). Processing of proteins by the molecular chaperone Hsp104. J Mol Biol 370, 674–686. [DOI] [PubMed] [Google Scholar]
- Schirmer EC, Queitsch C, Kowal AS, Parsell DA, and Lindquist S (1998). The ATPase activity of Hsp104, effects of environmental conditions and mutations. J Biol Chem 273, 15546–15552. [DOI] [PubMed] [Google Scholar]
- Tobias JW, Shrader TE, Rocap G, and Varshavsky A (1991). The N-end rule in bacteria. Science 254, 1374–1377. [DOI] [PubMed] [Google Scholar]
- Wendler P, Ciniawsky S, Kock M, and Kube S (2012). Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta 1823, 2–14. [DOI] [PubMed] [Google Scholar]
- Wickner S, Gottesman S, Skowyra D, Hoskins J, McKenney K, and Maurizi MR (1994). A molecular chaperone, ClpA, functions like DnaK and DnaJ. Proc Natl Acad Sci U S A 91, 12218–12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiita AP, Perez-Jimenez R, Walther KA, Grater F, Berne BJ, Holmgren A, Sanchez-Ruiz JM, and Fernandez JM (2007). Probing the chemistry of thioredoxin catalysis with force. Nature 450, 124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PM, Fowler SB, Best RB, Toca-Herrera JL, Scott KA, Steward A, and Clarke J (2003). Hidden complexity in the mechanical properties of titin. Nature 422, 446–449. [DOI] [PubMed] [Google Scholar]
- Yokom AL, Gates SN, Jackrel ME, Mack KL, Su M, Shorter J, and Southworth DR (2016). Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat Struct Mol Biol 23, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original raw data for the figures 2–6 in the paper is available from the corresponding author on request.
All the data sets generated and analyzed during the current study are available from the corresponding author on reasonable request.