

SUMMARY
The epidermis regenerates continually to maintain a protective barrier at the body’s surface composed of differentiating keratinocytes. Maturation of this stratified tissue requires that keratinocytes undergo wholesale organelle degradation upon reaching the outermost tissue layers to form compacted, anucleate cells. Through live imaging of organotypic cultures of human epidermis, we find that regulated breakdown of mitochondria is critical for epidermal development. Keratinocytes in the upper layers initiate mitochondrial fragmentation, depolarization, and acidification upon upregulating the mitochondrion-tethered autophagy receptor NIX. Depleting NIX compromises epidermal maturation and impairs mitochondrial elimination, whereas ectopic NIX expression accelerates keratinocyte differentiation and induces premature mitochondrial fragmentation via the guanosine triphosphatase (GTPase) DRP1. We further demonstrate that inhibiting DRP1 blocks NIX-mediated mitochondrial breakdown and disrupts epidermal development. Our findings establish mitochondrial degradation as a key step in terminal keratinocyte differentiation and define a pathway operating via the mitophagy receptor NIX in concert with DRP1 to drive epidermal morphogenesis.
In Brief
Using live microscopy of human organotypic epidermis, Simpson et al. demonstrate how keratinocytes degrade their mitochondria in the upper tissue layers during their final stage of differentiation. By upregulating expression of the mitophagy receptor NIX, keratinocytes initiate DRP1- dependent mitochondrial fragmentation, a process critical for epidermal tissue maturation.
Graphical Abstract
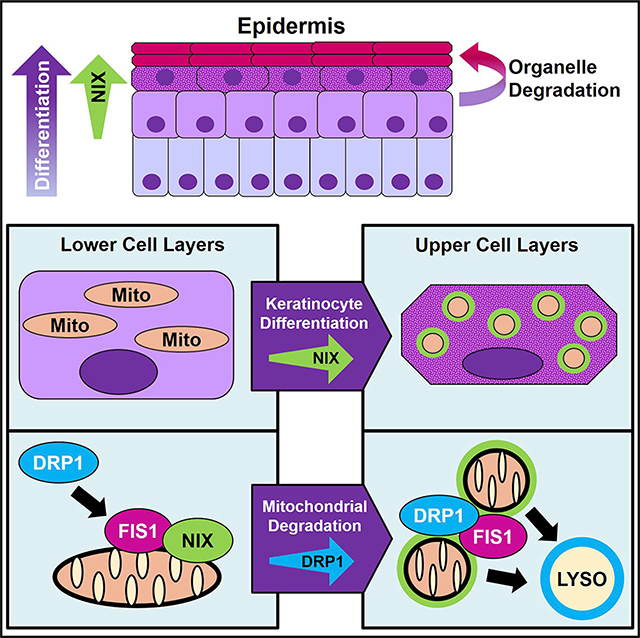
INTRODUCTION
The epidermis forms a protective barrier at the body’s surface and is primarily composed of keratinocytes, which undergo a specialized process of terminal differentiation as they journey outward in the multi-layered epithelium (Muroyama and Lechler, 2012; Simpson et al., 2011). In the outermost layers, keratinocytes flatten and initiate organelle and nuclear elimination to form compacted cornified cellular sheets (Candi et al., 2005; Matsui and Amagai, 2015), but the molecular drivers of organelle clearing in the final stages of keratinocyte differentiation remain unknown. We aimed to understand the fate of mitochondria in differentiating epidermal keratinocytes and how these dynamic organelles contribute to stratified tissue morphogenesis. Mitochondria undergo cyclic fission and fusion (Chan, 2020) and serve as versatile regulators of metabolism, oxidative stress, and cell death, including in keratinocytes (Bhaduri et al., 2015; Hamanaka and Chandel, 2013). Although epidermal mitochondria have been known to fragment late during keratinocyte differentiation (Ipponjima et al., 2020; Mellem et al., 2017), the importance of this process in epidermal morphogenesis remains untested, and the mechanisms driving mitochondrial breakdown in keratinocytes have not been elucidated.
Organelle turnover can be achieved by bulk autophagy, which has been implicated in keratinocyte differentiation (Akinduro et al., 2016; Sil et al., 2018; Yoshihara et al., 2015). Here we focused on selective autophagic degradation of mitochondria (mitophagy), in which whole organelles can be targeted for lysosomal breakdown. Mitophagy has been best characterized as a response to organelle damage in which defective mitochondria are ubiquitinated by the E3 ligase Parkin, marked by ubiquitin-binding autophagy receptors, sequestered in autophagosomes, and finally routed to lysosomes for degradation (Lazarou et al., 2015; Wong and Holzbaur, 2015; Youle and Narendra, 2011). However, distinct developmental pathways for constitutive and Parkin-independent mitophagy have also been delineated (Villa et al., 2018); these control removal of mitochondria from highly differentiated cells, such as erythrocytes (Diwan et al., 2007; Novak et al., 2010; Schweers et al., 2007). We hypothesized that keratinocytes upregulate mitophagy to trigger mitochondrial breakdown during their final stages of differentiation and that this process would be integral to constructing the mature epidermis.
To model differentiation-induced keratinocyte organelle clearing, we utilized organotypic epidermal cultures composed of primary normal human epidermal keratinocytes (NHEKs), which can be engineered for live imaging (Figure 1A). Within 1 week, organotypic cultures form a fully differentiated epidermis, including anucleate keratinized layers lacking organelles (Simpson et al., 2010), allowing mechanistic investigation of keratinocyte maturation in a native tissue architecture. Employing high-resolution confocal microscopy to image organelle dynamics in live stratified cultures, we uncovered a pathway driving mitochondrial breakdown during terminal differentiation of keratinocytes, which is critical to support morphogenesis of the epidermis.
Figure 1. Keratinocyte mitochondria undergo fragmentation, depolarization, and acidification in the upper layers of organotypic epidermis.

(A) NHEKs with labeled mitochondria (green cells) progressively differentiate as they stratify (arrow) to form organotypic epidermis, imaged by SDC microscopy.
(B) SDC images of Mito-dsRed in lower versus upper layers of epidermal cultures.
(C and D) Mitochondrial perimeter and (D) area quantification in cells relative to their z-distance from the top of the tissue (mean ± SD, n = 60 fields, 4 experiments (expts.), ***p < 0.0001).
(E) SDC images of TOM20-mCh-labeled mitochondria in the upper versus lower layers of organotypic epidermis.
(F) SDC images of GFP-labeled mitochondria and TMRE dye in transitional layers containing cells with or without fragmented mitochondria.
(G) Quantification of relative TMRE intensity in pairs of cells having fragmented versus non-fragmented mitochondria (mean ± SD, n = 24 cell pairs, 4 expts., ***p < 0.0001).
(H) The tandem fluorophores GFP-mCh are imported into mitochondria by the Cox8 targeting sequence. Under neutral pH, both fluorophores are active, whereas acidic pH quenches GFP upon routing to lysosomes (LYSO).
(I) SDC images of organotypic epidermis expressing Cox8-GFP-mCh in upper versus lower layers.
(J) TEM image of the upper layers of organotypic epidermis with spherical mitochondrial fragments within a second membrane (arrowheads), indicative of mitophagy.
White scale bars, 10 μm; black scale bar, 500 nm.
RESULTS
Keratinocyte mitochondria undergo fragmentation, depolarization, and acidification in the upper layers of organotypic epidermis
Using spinning-disk confocal (SDC) microscopy, we live-imaged stratified epidermal cultures grown from NHEKs stably expressing fluorophore-tagged mitochondrial proteins after retroviral transduction (Figure 1A). Expression of dsRed2 targeted to the mitochondrial matrix (Mito-dsRed) revealed tubular mitochondrial networks in lower-layer keratinocytes that underwent dynamic fusion and fission (Figure 1B). Mitochondria in the upper-layer cells transformed to become more spherical and remained stably fragmented, as shown by reduced mitochondrial size in more superficial layers (Figures 1C and 1D). Similar fragmentation of mitochondria was observed using mCherry (mCh) fused to TOM20 (TOM20-mCh) (Figure 1E), a mitochondrial outer membrane protein, and by SNAP tag labeling the mitochondrial matrix (Figure S1).
Mitochondrial fragmentation has been associated with aged and dysfunctional organelles (Youle and van der Bliek, 2012), so we wanted to determine whether spherical mitochondrial remnants in differentiated NHEKs remained polarized using the cell-permeable dye tetramethyl-rhodamine ethyl ester (TMRE), which concentrates in polarized mitochondria. In stratified NHEKs expressing GFP targeted to the outer mitochondrial membrane (GFP-Mito), TMRE was excluded from fragmented mitochondria in the upper layers but retained in tubular mitochondria in the lower layers; importantly, we compared TMRE intensity in neighboring cells within transitional layers that had fragmented or non-fragmented mitochondria to ensure that differences were not related to dye permeation of the tissue (Figures 1F and 1G). Our results show that late in keratinocyte maturation, mitochondria undergo programmed fragmentation and depolarize, which may drive essential metabolic changes during terminal differentiation.
Building on classic transmission electron microscopy (TEM) studies of fixed keratinized epithelia suggesting that mitochondria may be routed to lysosomes in the uppermost cell layers (Lavker et al., 1969; Lavker and Matoltsy, 1970), we coupled confocal microscopy with a live stratified tissue model to test whether fragmented mitochondria in differentiated NHEKs were routed to an acidic compartment for degradation. We used the tandem fluorophores GFP and mCh targeted to the mitochondrial matrix (Cox8-GFP-mCh; Figure 1H) as an indicator of mitophagy suited for live imaging (Rojansky et al., 2016); if the mitochondrial matrix is acidified, as occurs upon lysosomal targeting, then the GFP fluorophore is quenched, and mCh persists. Although tubular mitochondria in lower keratinocyte layers exhibited GFP and mCh signals, fragmented mitochondria in the upper layers lost GFP fluorescence, signaling acidification (Figure 1I). Moreover, in TEM images of epidermal cultures, we noted many mitochondrial fragments enclosed in double-membrane structures in the uppermost layers (Figure 1J, arrowheads), indicative of an autophagic fate.
Regulated mitochondrial depolarization and lysosomal acidification are critical for proper epidermal differentiation
We next tested whether disruption of spatiotemporally controlled mitochondrial depolarization and lysosomal acidification interfered with epidermal morphogenesis. To impair lysosomal acidification, required for effective autophagy, we chronically treated epidermal cultures with bafilomycin A1 (BafA1) (Mauvezin et al., 2015), which resulted in grossly abnormal tissue architecture (Figure 2A). Although BafA1-treated NHEKs formed a multi-layered epithelium, they exhibited cytoplasmic clearing suggestive of vacuole accumulation and failed to undergo proper differentiation. BafA1-treated cultures retained nuclei in the uppermost layers (Figure 2A, yellow arrowhead), signifying defective cornification. Moreover, BafA1 impaired formation of pre-cornified “granular” layers typified by basophilic keratohyalin (KH) granules seen in control cultures (Figure 2A, white arrowhead). KH granules contain the keratin-aggregating protein filaggrin (FLG), which was depleted in the upper layers of BafA1-treated cultures (Figure S2A). Western blotting (WB) of lysates from BafA1-treated cultures revealed accumulation of microtubule-associated protein 1A/1B light chain 3 (LC3), indicative of impaired autophagic degradation, and a reduction in early (desmoglein 1 [Dsg1], keratin 10 [K10]) and late markers (loricrin) of keratinocyte differentiation (Figure 2B).
Figure 2. Regulated mitochondrial depolarization and lysosomal acidification are critical for proper epidermal differentiation.

(A) H&E staining of DMSO- and BafA1-treated epidermal cultures after 9 days, which highlights KH granules (white arrowhead) and retained nuclei in cornified layers (yellow arrowhead).
(B) WB of LC3 and early (Dsg1, K10) and late (Loricrin) markers of epidermal differentiation in lysates from DMSO- and BafA1-treated cultures.
(C) BafA1 impairs lysosomal acidification, EACC inhibits autophagosome-LYSO fusion, and CCCP depolarizes mitochondria.
(D) H&E staining of cultures treated with DMSO or EACC, highlighting KH granules (white arrowhead) and retained nuclei (yellow arrowhead).
(E) Quantification of retained nuclei per high-powered field (hpf) (mean ± SD, n = 17 fields, **p = 0.0002) and FLG IF (mean ± SD, n = 20 fields, ***p < 0.0001) in DMSO- and EACC-treated cultures.
(F) IF of FLG in DMSO- and EACC-treated cultures.
(G) H&E staining of cultures treated with DMSO or CCCP shows KH granules (white arrowhead) and retained nuclei (yellow arrowhead).
(H) Quantification of retained nuclei (mean ± SD, n = 57 fields, ***p < 0.0001) and FLG IF (mean ± SD, n = 8 fields, *p = 0.0286) in DMSO- versus CCCP-treated cultures.
(I) IF of FLG in DMSO- and CCCP-treated cultures. Dashed lines mark the bottom of the epidermis.
White scale bars, 10 μm; black scale bars, 50 μm.
Given the profound tissue defects resulting from inhibition of lysosomal acidification, we aimed to more specifically restrict autophagosome fusion with lysosomes during NHEK differentiation using a recently characterized inhibitor of syntaxin 17 (Stx17) called ethyl(2-(5-nitrothiophene-2-carboxamido)thiophene-3-carbonyl)carbamate (EACC) (Vats and Manjithaya, 2019; Figure 2C]. This treatment produced stratified cultures with large peri-nuclear vacuoles and impaired cornification (Figure 2D). Similar to BafA1, EACC treatment resulted in defective epidermal morphogenesis with loss of KH granules (Figure 2D, white arrowhead), retention of nuclei in cornified layers (Figures 2D, yellow arrowhead; quantified in 2E), and accelerated flattening of suprabasal K10-positive layers (Figure S2B). Immunofluorescence (IF) of EACC-treated cultures revealed loss of FLG in the uppermost layers (Figure 2F). Impairment of granular layer formation and failure to degrade nuclei signify dysregulated keratinocyte maturation. These data indicate that the autophagosomal/lysosomal degradation pathway is essential for epidermal differentiation.
Because mitochondrial depolarization was restricted to the uppermost tissue layers (Figure 1F), we next tested whether premature depolarization of mitochondria would affect epidermal morphogenesis. Organotypic cultures treated throughout their development with carbonyl cyanide m-chlorophenyl hydrazine (CCCP), which depolarizes mitochondria to prevent oxidative phosphorylation, were viable and stratified into multiple layers (Figure 2G). In contrast, treatment with 2-deoxyglucose (2-DG), an inhibitor of glycolysis, completely disrupted epidermal morphogenesis, producing rudimentary two- to three-layer cultures (Figure S2C), suggesting that differentiating NHEKs are highly dependent on glycolysis. However, CCCP-treated cultures exhibited aberrant morphology, especially in the uppermost layers, which lacked KH granules (Figure 2G, white arrowhead), retained nuclei in the cornified layers [Figure 2G, yellow arrowhead; quantified in Figure 2H], and lost FLG expression (Figure 2I).
Because these findings underscored the importance of regulated mitochondrial depolarization and lysosomal/autophagic degradation in epidermal morphogenesis, we next aimed to delineate the specific molecular regulators of mitochondrial breakdown during keratinocyte differentiation.
The mitochondrion-localized autophagy receptor NIX is upregulated in the upper epidermal layers
Selective autophagy targets intracellular cargo for degradation through recruitment of receptor proteins that mark organelles and bind Atg8-related proteins like LC3 to enter the autophagic pathway (Kirkin and Rogov, 2019; Mancias and Kimmelman, 2016). Using a public single-cell RNA sequence database of murine epidermis (Joost et al., 2016), we queried expression levels of known mitophagy receptors and Parkin (Figure S3A; see graphs generated from http://linnarssonlab.org/epidermis/). Expression of Parkin was minimal, and the levels of NBR1, NDP52, optineurin, and p62 did not parallel keratinocyte differentiation, but in the upper epidermal layers, we noted dramatic upregulation of NIX (also known as BNIP3L), a mitochondrial outer membrane-tethered autophagy receptor that contains an LC3-interacting region (LIR) and a BH3-like domain (Chen et al., 1997; Mazure and Pouysségur, 2009; Zhang et al., 2012). A role of NIX in the epidermis has not been demonstrated previously, but NIX has been studied in highly differentiated cells like erythrocytes, in which it supports clearance of mitochondria (Diwan et al., 2007; Novak et al., 2010; Schweers et al., 2007). We hypothesized that NIX is upregulated in keratinocytes to achieve a very different cellular fate in the epidermis, where mitochondrial elimination could drive cornification (Figure 3A).
Figure 3. The mitochondrion-localized autophagy receptor NIX is upregulated in the upper epidermal layers.

(A) Diagram of the epidermis, showing increasing NIX expression in the upper layers as keratinocytes differentiate.
(B) IF of NIX in normal human skin and organotypic cultures.
(C) IF of endogenous NIX and TOM20 in organotypic epidermis.
(D) WB of NIX in lysates from undifferentiated NHEKs versus epidermal cultures grown for 6 or 9 days (below, relative band intensity normalized to TOM20).
(E) Diagram depicting regulation of NIX, a transcriptional target of HIF-1a. Inhibition of prolyl hydroxylases (PHs) by DMOG or chelation of iron using DFP relieves repression of HIF-1a, which leads to increased NIX.
(F) WB and quantification of NIX in lysates from NHEKs treated with DMSO or DMOG (mean ± SD, n = 4 expts., *p = 0.039).
(G) IF of NIX and TOM20 in undifferentiated NHEKs treated with DFP.
(H) IF of NIX and TOM20 in NHEKs treated with DMSO, CoCl2, or EUK134.
(I) Quantification of NIX IF (relative to TOM20) in NHEKs treated with DMSO, CoCl2, or EUK134 (mean ± SD, n = 33 fields, p < 0.0001).
(J) Stimulated emission-depletion (STED) microscopy images of undifferentiated NHEKs transduced with GFP-NIX and immunostained for TOM20.
Dashed lines mark the bottom of the epidermis. White scale bars, 10 μm.
Intriguingly, IF of NIX in human skin biopsies (from 6 non-related individuals) and in organotypic epidermal cultures (from NHEKs isolated from 6 non-related individuals) revealed that its protein levels were increased greatly in the uppermost layers (Figure 3B), which are poised to initiate mitochondrial clearing, and co-localized with TOM20 (Figure 3C). This expression pattern was consistent with WB of lysates (Figure 3D). Although undifferentiated NHEKs lacked NIX, its expression was initiated after 6 days in organotypic cultures, when cornification has just begun, and was augmented greatly after 9 days, when epidermal cultures are mature.
The mechanisms regulating NIX levels in the epidermis have not been determined, but NIX expression in other tissues is governed by hypoxia-regulated signaling (Bellot et al., 2009; Mazure and Pouysségur, 2009; Figure 3E). In the epidermis, hypoxia-inducible factor 1-alpha (HIF-1a) is activated (Boutin et al., 2008) and plays an important role in the upper layers of murine epidermis, notably in regulating FLG expression (Wong et al., 2015). To determine whether hypoxia-regulated signals control NIX expression in human keratinocytes, we treated undifferentiated NHEKs with the prolyl-hydroxylase inhibitor dimethyloxalylglycine (DMOG) to stabilize HIF-1a and found that NIX protein levels were increased (Figure 3F).
Other methods of simulating hypoxia also increased NIX expression. Treatment with the iron chelator deferiprone (DFP) is known to upregulate HIF-1a expression (Baek et al., 2011; Dalle et al., 2000; Yamashita and Kanki, 2018) and induce Parkin-independent mitophagy (Allen et al., 2013). In undifferentiated NHEKs, DFP treatment increased NIX expression on TOM20-positive mitochondria (Figure 3G). Similar to mitochondrial fragments in the upper layers of epidermal cultures (Figure 1E), iron chelation produced spherical mitochondria, but these tended to cluster near the nucleus, an effect reported previously in response to hypoxia (Al-Mehdi et al., 2012). Like DFP, cobalt (CoCl2), which also simulates hypoxia, induced NIX expression in undifferentiated NHEKs with peri-nuclear clustering of NIX-positive spherical mitochondria (Figure 3H). Treatment with a chemical mimetic of superoxide dismutase (EUK134) led to an even greater increase in NIX levels (Figure 3I). These findings confirm that NIX expression in the epidermis is dependent on signaling pathways regulated by hypoxia and reactive oxygen species (ROS).
To force NIX expression in undifferentiated keratinocytes, we transduced NHEKs with NIX having its N terminus tagged with GFP (GFP-NIX). Importantly, GFP-NIX localized to the outer mitochondrial membrane and co-localized with TOM20, as seen by super-resolution microscopy (Figure 3J). Similar to the spherical mitochondrial morphology seen in the NIX-positive upper epidermal layers, GFP-NIX expression in undifferentiated NHEKs induced fragmentation of mitochondria into spherical organelles, which clustered in a peri-nuclear distribution (Figure 3J, inset). As expected for an autophagy receptor, we also found that the exogenous NIX protein level was increased by lysosomal but not proteasomal inhibition (Figure S3B). These data suggest that NIX may be actively targeted to lysosomes to prohibit its accumulation in less differentiated keratinocytes. These results reveal that NIX protein levels are highly regulated in NHEKs, including by hypoxia- and ROS-dependent signaling and lysosomal degradation, likely to prevent premature mitochondrial fragmentation in the epidermis.
Loss of NIX results in impaired keratinocyte mitophagy and defective epidermal differentiation
Our findings suggest that NIX plays a crucial role in inducing mitochondrial fragmentation within the upper epidermal layers, where its expression is enhanced prior to cornification. To test whether NIX was required for mitochondrial degradation in a stratified tissue context, we developed a human model of NIX-deficient epidermis. We used CRISPR-Cas9 gene editing in human telomerase reverse transcriptase (hTERT)-immortalized human epidermal keratinocytes (THEKs) to generate stable NIX-deficient cell lines (NIX knockout (KO)-1 and NIX KO-2). THEKs are able to undergo terminal differentiation in the organotypic epidermal model but to a lesser extent than NHEKs (Dickson et al., 2000).
Targeting of the first exon of the NIX gene by CRISPR-Cas9 was confirmed by Sanger sequencing of genomic DNA from two independent THEK lines. NIX-deficient THEKs were grown in the organotypic model alongside control CRISPR-Cas9 THEKs. Although NIX-depleted THEKs were able to generate a stratified epithelium, these tissues demonstrated clear maturation defects. Unlike control cultures, NIX-deficient keratinocytes failed to undergo typical cell flattening in the upper layers (Figures 4A and 4B), which exhibited atypical cytoplasmic vacuolization and dyskeratosis (intense eosinophilic cytoplasm and nuclear condensation), signs of abnormal cornification (Figures 2A and 2B, yellow arrowheads]. The number of dyskeratotic cells was much higher in NIX-deficient cultures (Figure 4C). We confirmed loss of NIX expression in the organotypic cultures by WB of lysates from NIX-targeted THEK lines (Figure 4D).
Figure 4. Loss of NIX results in impaired keratinocyte mitophagy and defective epidermal differentiation.

(A and B) H&E staining of organotypic cultures of control versus NIX-depleted THEKs (NIX KO-1 and KO-2), which exhibited swelling, vacuolization, and dyskeratotic cells (yellow arrowheads) in the upper layers.
(C) Quantification of dyskeratotic cells in control versus KO cultures (mean ± SD, n = 53 fields, 3 expts., ***p < 0.0001).
(D) WB of NIX in lysates from control and KO cultures.
(E) IF of K10 and Dsg1, demonstrating altered keratin expression and cell morphology in NIX KO cultures; asterisks mark example suprabasal cells lacking cytoplasmic K10 staining.
(F) Quantification of K10-negative cells in control versus KO cultures (mean ± SD, n = 20 fields, ***p < 0.0001).
(G) Quantification of mean suprabasal cell roundness in control versus KO cultures (line, mean; box, 25th–75th percentile; whiskers, 5th–95th percentile; n = 376 cells from 20 fields, **p = 0.0003).
(H) TEM images of control cultures, showing mitophagic events (green arrowheads) and clearance of membranous organelles in the uppermost layers; NIX-depleted epidermis showed retention of mitochondria (yellow arrowheads).
(I) Quantification of relative electron density of upper layers in control versus KO cultures (mean ± SD, n = 63 fields, 2 KO lines, **p = 0.0015).
Dashed lines mark the bottom of the epidermis. White scale bars, 10 μm; H&E black scale bars, 50 μm; TEM black scale bars, 100 nm.
IF of NIX-deficient cultures revealed architectural abnormalities of the suprabasal layers, including non-uniform expression of the cytokeratin K10 and Dsg1, a desmosomal cadherin that highlighted irregular cell morphology (Figure 4E). The number of K10-deficient epidermal cells was higher in cultures lacking NIX (Figure 4F). The roundness of suprabasal cells was also higher in NIX-deficient cultures (Figure 4G), reflecting a reduced ability of keratinocytes lacking NIX to undergo cellular flattening in the upper layers. In TEM of tissue sections from control THEK cultures, we found that the uppermost layers became more electron lucent, having sparse organelles and frequent examples of mitochondria within autophagosomal membranes (Figure 4H, green arrowheads]. In contrast, NIX-depleted cultures retained more electron-dense organelles, including mitochondria, within the uppermost layers, suggesting impairment of their degradation (Figure 4H, yellow arrowheads). Accordingly, the electron density of the upper epidermal layers was increased in NIX-deficient cultures (Figure 4I). These data indicate that NIX plays a critical role in differentiation of human epidermal keratinocytes.
Premature NIX expression induces mitochondrial fragmentation and accelerates epidermal differentiation
Consistent with NIX expression being limited to the uppermost epidermal layers in human skin (Figure 3B), WB of lysates of organotypic cultures showed that NIX was initially upregulated between 3 and 6 days of growth and became highly expressed upon full maturation after 9 days (Figure 5A). To determine whether restricted expression of NIX in the epidermis was essential for tissue morphogenesis, we transduced NHEKs to drive constitutive expression of GFP-NIX in less mature organotypic cultures, which exhibited transgene expression throughout the lower cell layers (Figures S4A and S4B). Epidermal cultures ectopically expressing GFP-NIX demonstrated enhanced flattening of suprabasal cells, resulting in thinning of the tissue and an increase in dyskeratotic cells, which underwent premature cornification (Figure 5B). Moreover, forced expression of GFP-NIX in NHEKs grown as submerged cell cultures led to reduced levels of the basal cell keratin K14 (Figures S4C and S4D]. These data indicate that premature onset of NIX expression is sufficient to accelerate the keratinocyte differentiation program.
Figure 5. Premature NIX expression induces mitochondrial fragmentation and accelerates epidermal differentiation.

(A) WB of NIX and differentiation markers (Dsg1 and K10) in lysates from epidermal cultures grown for 3, 6, or 9 days and (bottom) quantification of relative NIX expression (mean ± SD; n = 3 expts., **p = 0.0071).
(B) H&E staining of cultures ectopically expressing GFP versus GFP-NIX and (bottom) quantification of tissue thickness (mean ± SD, n = 201 fields, 6 expts., ***p < 0.0001) and dyskeratotic cells (mean ± SD, n = 150 fields, 6 expts., ***p < 0.0001).
(C) TEM images of lower layers in control cultures, showing tubular mitochondria (green arrowheads), and in GFP-NIX-expressing cultures, showing mitochondrial constriction (blue arrowhead), partially engulfed mitochondria (magenta arrowheads), and mitophagic events (yellow arrowheads).
(D) H&E staining of cultures treated with DMSO versus EUK134 and (bottom) IF of NIX and TOM20.
(E) Quantification of tissue thickness in DMSO- and EUK134-treated cultures (mean ± SD, n = 22 fields, ***p < 0.0001).
(F) WB of lysates from cultures treated with DMSO versus EUK134 and (bottom) quantification of relative NIX protein levels (mean ± SD, n = 5 expts., **p = 0.0006).
(G) SDC images of undifferentiated NHEKs transduced with tubulin-mCh and GFP-Mito versus GFP-NIX and (right) quantification of mitochondrial length, perimeter, and AR of GFP-Mito- versus GFP-NIX-positive mitochondria (mean ± SD, n = 78 cells, 3 expts., ***p < 0.0001).
(H) Diagram showing GFP-labeled NIX constructs.
(I) SDC images of NHEKs transduced with GFP-labeled NIX constructs and incubated with TMRE.
(J) Quantification of mitochondrial length in NHEKs expressing NIX constructs (mean ± SD; n = 163 cells, 4 expts., ***p < 0.0001).
Dashed lines mark the bottom of the epidermis. White scale bars, 10 μm; H&E black scale bars, 50 μm; TEM black scale bars, 400 nm.
Based on our live epidermal imaging, mitochondrial fragments were acidified and routed to autophagosomes only in the uppermost keratinocyte layers (Figures 1I and 1J). We used TEM of cross-sections of organotypic cultures transduced with GFP-NIX to determine its effects on mitochondria in the lower layers. In control cultures, we noted tubular and elongated mitochondria (Figure 5C, green arrowheads). In contrast, epidermal cultures expressing GFP-NIX exhibited mitochondria that were more circular or had central constrictions, as seen during fission (Figure 5C, blue arrowhead); we also noted frequent mitophagic events, visualized as mitochondria partially surrounded by a second membrane [Figure 5C, magenta arrowheads] or as fully engulfed mitochondrial remnants in various stages of degradation (Figure 5C, yellow arrowheads].
Because of variability of retroviral transgene expression, we aimed to more uniformly augment NIX expression in stratified cultures using a pharmacologic approach. Given that EUK134 robustly induced NIX in NHEKs (Figures 3H and 3I), we used this compound to chronically treat organotypic cultures. EUK134-treated cultures underwent accelerated cornification and were much thinner than controls (Figures 5D and 5E). WB of lysates from EUK134-treated organotypic cultures displayed enhanced expression of NIX along with Dsg1 (Figure 5F), indicating accelerated differentiation. IF of EUK134-treated cultures revealed premature expression of NIX within the first suprabasal layer compared with control cultures, which possessed multiple NIX-negative intermediate cell layers (Figure 5D, bottom panels).
These findings demonstrate that premature initiation of NIX expression in the epidermis alters the normal timing of keratinocyte differentiation and results in aberrant tissue morphogenesis because of accelerated cornification.
NIX drives mitochondrial fragmentation via the fission GTPase DRP1
We next sought to elucidate the mechanism by which NIX contributed to mitochondrial breakdown in keratinocytes. First, to control for non-specific effects of an exogenous fluorophore-labeled outer mitochondrial membrane protein, we transduced NHEKs with a construct that linked GFP alone to the outer mitochondrial membrane utilizing the transmembrane (TM) sequence from NIX (GFP-Mito) along with mCh-labeled beta-tubulin (tubulin-mCh) to highlight microtubules. GFP-Mito-positive mitochondria were tubular, branched, and dispersed throughout the cytoplasm, whereas NHEKs expressing GFP-NIX exhibited marked fragmentation of mitochondria, which clustered near nuclei (Figure 5G). GFP-NIX-positive mitochondria were smaller in size and aspect ratio (AR), reflecting a more spherical morphology (Figure 5G, right) similar to fragmented mitochondria in the upper epidermal layers (Figures 1B–1E).
To confirm the effects of exogenous NIX in unfixed cells, we expressed GFP-tagged wild-type and mutant NIX constructs in live NHEKs to determine how NIX induced mitochondrial fragmentation (Figure 5H). We similarly found that live NHEKs expressing GFP-NIX exhibited marked fragmentation and peri-nuclear clustering of mitochondria (Figure 5I). Mutation of the critical tryptophan residue within its LIR sequence (W36A, GFP-NIX-WA) did not abrogate NIX-induced mitochondrial fragmentation, establishing this function of NIX as independent of its ability to bind Atg8 family proteins (Rogov et al., 2017). However, mutating two glycine residues in the TM domain of NIX (G204A/G208A, GFP-NIX-2GA), which are necessary for NIX dimerization (Marinković et al., 2020), impaired the protein’s ability to induce mitochondrial fragmentation. Quantifying the effect of each NIX construct confirmed that, although wild-type GFP-NIX and GFP-NIX-WA reduced mitochondrial length, organelles expressing GFP-NIX-2GA were slightly elongated (Figure 5J).
Interestingly, ectopic expression of NIX in differentiated organotypic cultures was sufficient to route mitochondrial fragments into autophagosomes (Figure 5C); however, in undifferentiated NHEKs, we noted that peri-nuclear GFP-NIX-positive mitochondrial fragments were not degraded rapidly. This suggests that additional differentiation-dependent regulators in the stratified tissue may be required to ultimately route the fragmented mitochondria for lysosomal degradation during cornification. Consistent with this, GFP-NIX-positive mitochondrial fragments in undifferentiated NHEKs remained polarized, as shown by TMRE retention (Figure 5I).
Given the fragmented and spherical appearance of NIX-positive mitochondria, we hypothesized that NIX might engage the mitochondrial fission machinery by recruiting the membrane fission GTPase DRP1 (Bleazard et al., 1999). We found enhanced localization of endogenous DRP1 to GFP-NIX-positive mitochondria, which clustered near the nucleus (Figure 6A). To quantify this effect, we calculated a peripheral bias factor for DRP1, which measured the average distance of all DRP1 particles from the nuclear rim, normalized to the average distance from the nuclear rim to the cell edge (Figure 6B). In NHEKs expressing GFP-NIX compared with non-transduced neighboring cells, DRP1 had a significantly lower peripheral bias, reflecting a tendency to cluster near the nucleus on GFP-NIX-positive mitochondria (Figure 6C).
Figure 6. NIX drives mitochondrial fragmentation via the fission GTPase DRP1.

(A) SDC images of DRP1 IF in NHEKs transduced with GFP-NIX
(B) Diagram showing a measure of peripheral bias as the ratio between the distance (marked A) from the nucleus to each particle and the distance (marked B) from the nucleus to the cell edge.
(C) Quantification of the peripheral bias of DRP1 particles in cells expressing GFP-NIX compared with neighboring non-transduced cells (mean ± SD, n = 19 cell pairs, 4 expts., *p = 0.0148).
(D) SDC images of FIS1 IF in NHEKs transduced with GFP-NIX.
(E) Model depicting a GFP-NIX-positive mitochondrion (green rim) with FIS1 stabilized on its surface, which recruits DRP1, leading to mitochondrial fragmentation.
(F) SDC images of DRP1 and Hsp60 IF in NHEKs transduced with Halo-FIS1.
(G) Quantification of mitochondrial AR and perimeter in NHEKs expressing Halo-FIS1 compared with non-transduced neighbors (mean ± SD, n = 14 cell pairs, 3 expts., ***p < 0.0001).
(H) IF of FIS1 and NIX in human skin.
(I) SDC images of NHEKs transduced with GFP-NIX alone (top panels) versus neighboring cells co-transduced with DRP1-K38A (bottom panels).
(J) Quantification of mitochondrial AR and perimeter in NHEKs expressing GFP-NIX alone or with DRP1-K38A (mean ± SD, n = 54 cells, 4 expts., ***p < 0.0001).
White scale bars, 10 μm.
To understand how NIX enhanced DRP1 recruitment to mitochondria, we tested whether GFP-NIX affected the mitochondrial localization of known protein receptors for DRP1 (Losón et al., 2013). We found that GFP-NIX co-localized with FIS1 (Figure 6D), a mitochondrial outer membrane protein that can recruit DRP1 to the surface of mitochondria to promote organelle fission (Yoon et al., 2003; Figure 6E). As seen in other cell types (Stojanovski et al., 2004), exogenous expression of FIS1 (Halo-FIS1) in NHEKs induced fragmentation and peri-nuclear clustering of mitochondria (Figure 6F). Similar to GFP-NIX, Halo-FIS1 reduced the size and AR of mitochondria (Figure 6G). To assess the in vivo relevance of these findings, we immunostained endogenous NIX and FIS1 in human skin and found co-localization of these proteins in the upper epidermal layers (Figure 6H), suggesting a cooperative role in mitochondrial fragmentation during in vivo keratinocyte differentiation.
Next we employed a dominant-negative mutant of SNAP-tagged DRP1 (Pitts et al., 1999) (DRP1-K38A) to directly test whether NIX requires DRP1 activity to drive mitochondrial fragmentation. Although GFP-NIX induced mitochondrial fragmentation and peri-nuclear clustering in NHEKs lacking the DRP1 mutant (Figure 6I, top panels), co-expression of DRP1-K38A impaired this function of GFP-NIX, resulting in highly elongated mitochondria (Figure 6I, bottom panels). Quantification confirmed that the mean AR and size of GFP-NIX-positive mitochondria were increased significantly by co-expression of DRP1-K38A (Figure 6J). These data confirm that the GTPase function of DRP1 is required for NIX-induced mitochondrial fragmentation.
Blockade of mitochondrial fission via DRP1 impairs epidermal differentiation
To extend our findings to the tissue context, we assessed the requirement for DRP1 activity in organotypic epidermis. IF of stratified cultures confirmed that DRP1 is expressed throughout the epidermal layers in the keratinocyte cytoplasm and on TOM20-positive mitochondria, including in the uppermost layers (Figure 7A). Although the specificity of the chemical Mdivi-1 has been debated (Bordt et al., 2017; Manczak et al., 2019), we found that treatment of organotypic epidermis with this DRP1 inhibitor resulted in major defects in epidermal morphogenesis with clearing of keratinocyte cytoplasm and accumulation of cornified layers with retained nuclei (Figure 7B). Interestingly, Mdivi-1-treated cultures showed some overlapping features with BafA1-treated cultures (Figure 2A), consistent with DRP1-mediated fission being required for subsequent lysosomal degradation of mitochondria. Of note, in organotypic epidermis, Mdivi-1 did not reduce endogenous DRP1 (Figure 7C), as has been reported to occur in a different cell type (Manczak et al., 2019).
Figure 7. Blockade of mitochondrial fission via DRP1 impairs epidermal differentiation.

(A) IF of DRP1 and TOM20 in mature epidermal cultures.
(B) H&E staining of organotypic epidermis treated with DMSO or Mdivi-1, showing retained nuclei in cornified layers (yellow arrowhead) and (right) quantification of retained nuclei in DMSO- versus Mdivi-1-treated cultures (mean ± SD, n = 70 fields, 4 expts., ***p < 0.0001).
(C) WB of DRP1 in lysates from organotypic cultures treated with Mdivi-1 or transduced with DRP1-K38A.
(D) Quantification of mitochondrial size in upper-layer cells transduced with TOM20-mCh along with DRP1-K38A or a viral vector (mean ± SD, n = 61 fields, 4 expts., ***p < 0.0001).
(E) SDC images of the upper layers of organotypic epidermis expressing TOM20-mCh alone (left) or with DRP1-K38A (right).
(F) H&E staining of cultures expressing a viral vector or DRP1-K38A, highlighting KH granules (white arrowhead) and retained nuclei (yellow arrowhead) and (bottom) quantification of tissue thickness (mean ± SD, n = 103 fields, 4 expts., ***p < 0.0001) and retained nuclei (mean ± SD, n = 101 fields, 4 expts., *p = 0.0115) in vector- versus DRP1-K38A-expressing cultures.
(G) IF of K10 and FLG in organotypic cultures expressing vector or DRP1-K38A and (right) quantification of K10-positive cell size (mean ± SD, n = 30 fields, ***p < 0.0001) and FLG staining (mean SD, n = 30 fields, ***p < 0.0001). Dashed lines mark the bottom of the epidermis. Black scale bars, 50 μm; white scale bars, 10 μm.
(H) Model depicting the onset of NIX expression in differentiating keratinocyte layers driven by hypoxia-regulated signaling; NIX-positive mitochondria (green rim) exhibit enhanced localization of FIS1, which recruits DRP1 to produce mitochondrion fragments that are targeted for lysosomal degradation in the uppermost epidermal layers.
Finally, we transduced NHEKs with DRP1-K38A and grew them as organotypic cultures to more specifically assess the requirement for DRP1 in epidermal morphogenesis. Co-transduction of DRP1-K38A with TOM20-mCh allowed us to visualize mitochondrial morphology in live stratified cultures. Quantification of organelle morphology confirmed an increased size of mitochondria in the upper layers of organotypic cultures expressing DRP1-K38A (Figure 7D). Although, in the upper layers of control cultures, we found the expected spherical fragmented mitochondria (Figure 7E, left), in those expressing DRP1-K38A, we noted marked elongation and convolution of the tubular mitochondrial network (Figure 7E, right), similar to the effect of this dominant-negative mutant reported in other cell types (Bleazard et al., 1999; Pitts et al., 1999).
Histology of cultures expressing DRP1-K38A revealed abnormal tissue morphology: enlarged and flattened suprabasal cells, reduced KH granules (Figure 7F, white arrowhead), and retention of nuclei in the cornified layers (Figure 7F, yellow arrowhead) all signaled abnormal keratinocyte differentiation. DRP1-K38A-expressing cultures also exhibited a reduction in tissue thickness and an increase in retained nuclei. IF of the K10 cytoskeleton in DRP1 mutant cultures highlighted abnormal morphology of suprabasal keratinocytes, which were enlarged significantly compared with controls; expression of DRP1-K38A nearly abolished FLG IF (Figure 7G). These results indicate that mitochondrial fragmentation during keratinocyte differentiation requires DRP1 activity and is integral for proper morphogenesis of human epidermis.
DISCUSSION
Orderly differentiation of keratinocytes is needed to continually regenerate a functional epidermis that protects the body from fluid loss, pathogens, and toxic insults (Ramos-e-Silva and Jacques, 2012). An improved understanding of how keratinocytes form corneocytes at the end of their lifespan could uncover strategies to modulate the cutaneous barrier and restore epidermal homeostasis in skin disease. To that end, we established a live high-resolution microscopy approach to better delineate how keratinocytes degrade organelles, using an organotypic model that replicates human epidermal morphogenesis (Simpson et al., 2010). We show that keratinocytes undergo spatiotemporally controlled mitochondrial fragmentation and depolarization in the upper layers of the epidermis and that this process is critical for building a properly differentiated epithelium.
Much work on mitochondrial degradation has focused on Parkin-dependent injury-initiated mitophagy (Jin and Youle, 2012; Youle and Narendra, 2011). Although this damage-induced pathway underpins the health of neurons and is compromised in neurodegeneration, Parkin exhibits minimal expression in the epidermis (Joost et al., 2016; Figure S3A). Because keratinocytes undergo a programmed process of mitochondrial clearing during their differentiation, we postulated that the epidermis might utilize a Parkin-independent program of mitochondrial degradation to achieve a distinct cell fate via cornification. Here we identified a pathway activated in the final stages of keratinocyte maturation in which mitochondria undergo DRP1-mediated fission and are marked for degradation by NIX, a mitochondrion-tethered autophagy receptor (Figure 7H).
Versatile mitochondria regulate diverse intracellular pathways, including ATP production, ROS generation, and cell death execution (Bertero and Maack, 2018; Prudent and McBride, 2017; Youle and van der Bliek, 2012). Their cyclic fission and fusion, including in human keratinocytes (Moore et al., 2016), allow cells to adapt to metabolic needs and oxidative stress (Liesa and Shirihai, 2013; Youle and van der Bliek, 2012). In epidermis, we found that NIX stabilized the fission machinery on mitochondria, driving organelle fragmentation and degradation. Our results show that shifting the mitochondrial fission/fusion balance in NHEKs by manipulating NIX and DRP1 function has marked effects on epidermal morphology, altering tissue thickness and cornification.
Of note, premature mitochondrial depolarization, Stx17 inhibition, and DRP1 blockade all affected assembly of KH granules. These differentiation-specific sub-cellular structures have long been ascribed a role in skin barrier function and have been shown recently to drive liquid-liquid phase separation to facilitate cell flattening during keratinocyte differentiation (Quiroz et al., 2020). Moreover, loss of KH granules has been associated with skin disorders, including ichthyosis vulgaris and psoriasis, with the latter being linked to impaired autophagic flux and mitochondrial dysfunction (Akinduro et al., 2016; Brody, 1962; Douroudis et al., 2012; Therianou et al., 2019). The potential to target autophagy (Levine et al., 2015; Markaki et al., 2018) or mitochondrial dynamics (Lackner and Nunnari, 2010) for clinical benefit is under investigation and could be leveraged to treat dermatological disease (Guo et al., 2019; Yanagi et al., 2018) and even delay skin aging (Singh et al., 2018; Zhao et al., 2017).
Multiple mouse models have demonstrated an important role of mitochondrial metabolism in epidermal differentiation (Bhaduri et al., 2015; Hamanaka and Chandel, 2013; Shin et al., 2017; Weiland et al., 2018), and we find that mitochondrial polarization in human epidermal cultures is preserved until the end of the keratinocyte lifespan. Premature mitochondrial depolarization led to aberrant tissue maturation, which may be due to ATP depletion or altered levels of ROS or calcium, two key regulators of epidermal differentiation (Celli et al., 2016; Hamanaka et al., 2013). Many studies have reported important associations between mitochondria and the endoplasmic reticulum (ER) (Friedman et al., 2011; Murley and Nunnari, 2016), which can initiate mitochondrial fragmentation and intra-organellar calcium transfer (Chakrabarti et al., 2018; Prudent and McBride, 2017). Whether mitochondrial degradation in keratinocytes is dependent on the ER or related to cellular calcium flux remains to be determined.
Interesting parallels exist between erythrocytes and keratinocytes, both of which undergo differentiation culminating in organelle elimination. Although hemoglobin-rich erythrocytes are optimized for vascular transit and oxygen delivery, adherent corneocytes are compacted to form a barrier tissue. Remarkably, our results indicate that these distinct morphologic and functional cellular fates converge upon a conserved NIX-driven mitophagy pathway. Although NIX KO mice develop mild anemia with reduced mitochondrial elimination from erythrocytes (Diwan et al., 2007; Novak et al., 2010; Schweers et al., 2007), these mice do not exhibit a discernible cutaneous phenotype (data not shown), possibly because of redundancy of other mitochondrion-tethered autophagy receptors, such as BNIP3 (Moriyama et al., 2014; Ney, 2015) and FUNDC1 (Liu et al., 2012). Nevertheless, human epidermal cultures lacking NIX displayed clear maturation defects, which may reflect distinct NIX-dependent morphogenetic programming needed to generate a thicker protective epidermis in humans compared with the much thinner epidermis of fur-covered mice (Gilhar et al., 1991; Phillips et al., 2010).
Intriguingly, prior investigations revealed functional cross-talk between the epidermis and hematopoietic cells through hypoxia-induced signals and mitochondria (Boutin et al., 2008; Hamanaka et al., 2016; Rezvani et al., 2011). In fact, skin-specific KO of HIF-1a resulted in aberrant expression of FLG (Wong et al., 2015), which we noted to be decreased upon inhibition of mitochondrial polarization or fission. We also show that iron sequestration, which mimics hypoxia and induces mitophagy (Allen et al., 2013), increases NIX expression and promotes mitochondrial fragmentation. It will be interesting in future studies to determine whether the epidermis contributes to iron homeostasis via mitophagy, which could help the body reclaim mitochondrial iron prior to corneocyte shedding (Asano et al., 2017).
Finally, mitochondrial generation of ROS is a double-edged sword in the skin because it drives epidermal differentiation (Hamanaka et al., 2013, 2016) but also promotes aging (Rinnerthaler et al., 2015) and carcinogenesis in response to ultraviolet (UV) irradiation (Brand et al., 2018). Although a clinical trial for skin cancer prevention used oral nicotinamide (Chen et al., 2015), which can reduce ROS, induction of mitochondrial ROS in photodynamic therapy is used to drive apoptosis in pre-malignant skin lesions (Ji et al., 2010; Kessel and Oleinick, 2010). In fact, NIX and DRP1 have been shown to regulate mitochondrial ROS generation (Bordt et al., 2017; Ding et al., 2010), and DRP1 inhibition has been proposed as a treatment for disease (Lackner and Nunnari, 2010), including cutaneous squamous cell carcinoma (Kitamura et al., 2017; Yanagi et al., 2018). Whether mitochondrial degradation via NIX and DRP1 could be leveraged for therapeutic purposes in the epidermis remains to be tested, but future investigations of this pathway may uncover new approaches to prevent or treat UV-induced epidermal carcinogenesis and aging.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Erika L. F. Holzbaur (holzbaur@pennmedicine.upenn.edu).
Materials availability
All cell lines and plasmids generated in this study are available upon request.
Data and code availability
This study did not generate/analyze datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
All cell lines were cultured on standard 10 cm sterile tissue culture plates at 37°C and 5% CO2 in a water-jacketed cell culture incubator unless otherwise specified. Cells were passaged using trypsin upon reaching 70%–80% confluency.
J2 3T3 male murine fibroblasts and Phoenix (ΦNX) female human embryonic kidney (HEK) 293T viral packaging cells (both gifts from Dr. Kathleen Green Lab, Northwestern Univ., Chicago, IL) were cultured in DMEM supplemented with 4.5 g/L glucose (Corning #10–017-CM), 2 mM GlutaMAX (GIBCO #35050061), 100 U/mL penicillin plus 100 mg/mL streptomycin (Sigma #P0781), and 10% FBS (HyClone #SH3007103). During retroviral harvesting, ΦNX HEK293T cells were switched to grow at 32°C for 24 hr prior to medium collection. J2 3T3 cells were used only as supporting cells for organotypic epidermal keratinocyte cultures and their identity was not independently authenticated. ΦNX HEK293T cells were used only to generate retrovirus and their identity was not independently authenticated.
hTERT-immortalized male human epidermal keratinocytes (Dickson et al., 2000) (THEKs, a gift from Dr. James Rheinwald Lab, Harvard Univ., Cambridge, MA) were grown in keratinocyte serum-free medium (KSFM, GIBCO #17005042, supplemented from kit with 30 μg/ml bovine pituitary extract, 0.2 ng/ml epidermal growth factor, and 0.31 μM calcium chloride) containing 100 U/mL penicillin plus 100 mg/mL streptomycin (Sigma #P0781). THEKs were authenticated by their ability to differentiate into a stratified epidermis; no further identity authentication was performed for this study.
Primary cultures
Normal human epidermal keratinocytes (NHEKs) were isolated from deidentified, discarded foreskin of male neonates procured under a protocol approved by the University of Pennsylvania Institutional Review Board by the Penn Skin Biology and Disease Resource-based Center. Foreskin samples were incubated in 2 mL of 5 U/ml dispase II (StemCell Technologies #07913) for 18–24 hr at 4°C. Then, the epidermis was removed from the underlying dermis and mechanically dissociated using sterile forceps and was incubated in 2 mL of 0.05% trypsin-EDTA (GIBCO #25300054) at 37°C for 5 min. Trypsin was deactivated by adding 10 mL DMEM supplemented with 4.5 g/L glucose (Corning #10–017-CM) and 5% fetal bovine serum (FBS, HyClone #SH3007103). The cell suspension was then centrifuged at 1500 rpm for 5 min to collect the cell pellet. The liquid medium was aspirated and the cell pellet was re-suspended in 10 mL of medium M154 supplemented with 0.07 mM calcium chloride (GIBCO #M154CFPRF500), 5 mL human keratinocyte growth supplement (HKGS, GIBCO #S0015), and 1x Gentamicin/Amphotericin (GIBCO #R01510). NHEKs were authenticated by their ability to differentiate into a stratified epidermis; no further identity authentication was performed for this study. NHEKs were cultured on standard 10 cm sterile tissue culture plates at 37°C and 5% CO2 in a water-jacketed cell culture incubator. Cells were passaged using trypsin upon reaching 70%–80% confluency.
METHOD DETAILS
Plasmid construction
The retroviral plasmid pCLBW-Cox8-GFP-mCh (Addgene plasmid #78520) (Rojansky et al., 2016) was used to express the tandem fluorophores EGFP-mCherry linked to the C terminus of the mitochondrial targeting sequence of Cox8. All of the remaining retroviral plasmids were constructed by inserting the following tagged protein sequences into the multi-cloning site of the pLZRS retroviral vector (a gift from Dr. Kathleen Green Lab, Northwestern Univ., Chicago, IL): (1) Mito-dsRed (a gift from Dr. Thomas Schwarz Lab, Harvard Univ., Boston, MA) links the dsRed2 fluorophore to the C terminus of the Cox8 mitochondrial targeting sequence; (2) TOM20-mCh (from Addgene plasmid #55146 from Dr. Michael Davidson Lab, Florida State Univ., Tallahassee, FL) links mCherry to the C terminus of full-length human TOM20; (3) GFP-NIX was made by linking EGFP to the N terminus of the full-length human BNIP3L (NIX) AA 1–219 (from Addgene plasmid #17467) (Fei et al., 2004) with an intervening linker sequence of 5′-SDLELKL-3′; (4) GFP-NIX-WA was made by PCR-based site-directed mutagenesis of GFP-NIX to change tryptophan 36 to an alanine residue (W36A); (5) GFP-NIX-2GA was made by PCR-based site-directed mutagenesis of GFP-NIX to change glycines 204 and 208 to alanine residues (G204A/G208A); (6) GFP-Mito was made by linking EGFP to the N terminus of AA 186–219 from full-length human BNIP3L (NIX) with an intervening linker sequence of 5′-SDLELKL-3′; (7) Tubulin-mCh (a gift from Dr. Kathleen Green Lab, Northwestern Univ., Chicago, IL) links the mCherry fluorophore to the C terminus of mouse beta-tubulin; (8) SNAP-DRP1-K38A (from Addgene plasmid #45161) (Smirnova et al., 1998) links the SNAPf tag to the N terminus of human DRP1 containing a point mutation of lysine 38 to alanine (K38A); (9) Mito-SNAP was made from Mito-dsRed by replacing dsRed2 with the SNAPf tag at the C terminus of the Cox8 mitochondrial targeting sequence; and (10) Halo-FIS1 (from Addgene plasmid #111136) (Appelhans et al., 2012) links the Halo7 tag to the N terminus of human FIS1.
Chemical reagent concentrations
Bafilomycin A1 (BafA1), 100 μM (Sigma #B1793); carbonyl cyanide 3-chlorophenylhydrazone (CCCP), 10 μM (Sigma #C2759); cobalt chloride (CoCl2), 100 μM (Sigma #15862); deferiprone (DFP), 1 mM (Sigma #Y0001976); dimethyloxalylglycine (DMOG), 1 μM (Sigma #D3695); ethyl(2-(5-nitrothiophene-2-carboxamido)thiophene-3-carbonyl)carbamate (EACC), 10 μM (AOBIOUS #AOB13386); EUK134, 50 μM (Sigma #SML0743); Mdivi-1, 25 μM (Cell Signaling #CAS-338967–87-6); MG132, 10 μM (Sigma #M8699).
Vital dye concentrations
MitoTracker Deep Red, 100 nM (Invitrogen #M22426); SNAP-Cell 647-SIR, 1 μM (New England BioLabs #S9102S); JF646-Halo ligand, 100 nM (gift from Dr. L. Lavis Lab, Janelia/HHMI, Ashburn, VA); tetramethylrhodamine ethyl ester perchlorate (TMRE), 25 nM (Invitrogen #T669); Hoechst 33342, 20 μg/ml (Invitrogen #H21492).
Primary antibody dilutions
Actin, 1:200 WB (Clone C4, Santa Cruz #sc-47778); alpha-tubulin, 1:500 WB (Clone DM1A, Sigma #T9026); desmoglein 1, 1:50 WB (Clone 27B2, Abcam #Ab12077); desmoglein 1, 1:50 IHC (Clone B11, Santa Cruz #sc-137164); DRP1, 1:100 IHC (Sigma #HPA039324); DRP1, 1:50 IF (Clone 6Z-82, Santa Cruz #sc-101270); DRP1, 1:500 WB (Abcam #Ab56788); filaggrin, 1:200 IHC (Abcam #Ab3137); FIS1, 1:50 IF (Clone B-5, Santa Cruz #sc-376447); FIS1, 1:200 IHC (Sigma #HPA017430); GAPDH, 1:500 WB (Abcam #Ab9484); GFP, 1:2000 IHC (Aves #GFP-1020); Halo tag, 1:200 IF (Promega #G928A); Hsp60, 1:500 IF (Sigma #HPA001523); keratin 10, 1:5000 IHC, 1:2000 WB (Abcam #Ab76318); keratin 14, 1:25 IF (Abcam #Ab7800); LC3b, WB 1:1000 (Abcam #Ab48394); loricrin, 1:500 WB (Abcam #Ab24722); NIX/BNIP3L, 1:200 IHC, 1:200 IF (Sigma #HPA015652); NIX/BNIP3L, 1:100 WB (Clone H-8, Santa Cruz #sc-166332); NIX/BNIP3L, 1:1000 WB (Abcam #Ab109414); SNAP tag, 1:100 IF (New England BioLabs #P93105); TOM20, 1:200 WB, 1:50 IF (Clone F-10, Santa Cruz #sc-17764); TOM20, 1:500 WB (Abcam #Ab56783); TOM20, 1:250 WB, 1:100 IF, 1:500 IHC (Sigma #HPA011562).
Organotypic epidermal culture
NHEKs were grown as organotypic “raft cultures” using E-medium to induce stratification and epidermal differentiation as previously described (Simpson et al., 2010). E-medium was made from a 3:1 mixture of DMEM (Corning #10–017-CM):Ham’s F12 (Corning #10–080-CV), supplemented with 10% FBS (HyClone #SH3007103), adenine (1.8 × 10−4 M, Sigma #A2786), hydrocortisone (0.4 μg/ml, Sigma #H0888), human insulin (5 μg/ml, Sigma #91077C), cholera toxin (1 × 10−10 M, Sigma #C8052), apo-transferrin (5 μg/ml, Sigma #T1147), and 3,3′,5-tri-iodo-L-thyronine (1.36 ng/ml, Sigma #T6397) with or without supplemental human epidermal growth factor (EGF, 5 ng/ml, Sigma #E9644)
J2 3T3 murine fibroblasts were trypsinized off cell culture plates and re-suspended in DMEM+10% FBS, then counted and the required volume for a final concentration of 0.5 million cells/ml was centrifuged at 200 g for 5 min and the supernatant was aspirated. The cell pellet was re-suspended in 1/10 the final required volume (2 mL per organotypic culture) of 10X reconstitution buffer (1.1 g of NaHCO3 plus 2.39 g of HEPES in 50 mL of 0.05 N NaOH) and 1/10 the final volume of 10X DMEM (Sigma #D2554) was added on ice. High-concentration rat tail type 1 collagen (Corning #354249) was added to a final concentration of 4 mg/ml and sterile water was added to the final required volume and NaOH (0.05 N) was added to a visual pH of ~7. The collagen/fibroblast slurry was mixed via inversion, then pipetted into the top chamber of a transwell insert (Falcon #353091) placed within a deep 6-well culture plate (Falcon #355467) and allowed to solidify in the tissue culture incubator at 37°C for 60 min. The fibroblast rafts were then submerged in DMEM+10% FBS and incubated at 37°C for at least 18–24 hr.
Next, NHEKs were trypsinized off cell culture plates and re-suspended in DMEM+10% FBS, then counted and the required volume for a final concentration of 0.5 million cells/ml was centrifuged at 200 g for 5 min and the supernatant was aspirated. The cell pellet was re-suspended in E-medium with EGF 5 ng/ml to a final volume of 2 mL per organotypic culture. The DMEM was aspirated from both the upper and lower transwell chambers without disturbing the collagen-fibroblast rafts. NHEKs were seeded atop the raft (2 mL, 1 million cells) and E-medium with EGF 5 ng/ml was added to the bottom transwell chamber to submerge the insert and raft; the cultures were then incubated at 37°C for 24 hr. Next, the E-medium with EGF was removed from both chambers and 10 mL of E-medium without EGF was added only to the bottom chamber of the transwell, reaching the underside of the raft and exposing the overlying NHEK monolayer to the air.
Cultures were fed E-medium every other day until the time of imaging or harvesting for histology or protein lysis. Organotypic cultures were prepared for routine histology by removing the entire culture from the transwell using a scalpel, trimming the center of the tissue, then submerging the tissue in 10% neutral-buffered formalin (Fisher #SF100–4) for at least 24 hr prior to processing. For electron microscopy, the tissue sample was fixed in 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 at 4°C for at least 24 hr prior to processing.
Transient transfection
Cells were transfected using FuGENE6 transfection reagent (Promega #E269A) diluted in Opti-MEM (GIBCO #31985–070). Plasmid DNA (2 μg per 35 mm plate; 4 μg per 60 mm plate) was diluted in Opti-MEM (200 μl per 35 mm plate; 400 μl per 60 mm plate) and FuGENE6 (6 μl per 35 mm plate; 12 μl per 60 mm plate) was separately diluted in Opti-MEM (200 μl per 35 mm plate; 400 μl per 60 mm plate). After 5 min, the DNA solution was added to the FuGENE6 solution and incubated at room temperature for 15 min. Then, the mixed solution was pipetted drop-wise into the medium covering the adherent cells. Imaging of transfected constructs was done 24–48 hr after transfection.
Retroviral production and cell transduction
Phoenix (ΦNX) 293T viral packaging cells (originally a gift from Dr. Garry Nolan Lab, Stanford Univ., CA, USA) were used for retroviral production as previously described (Kinsella and Nolan, 1996). ΦNX 293T cells were transduced with pLZRS retroviral plasmids as above with FuGENE6. After 48 hr, cells were passaged in DMEM+10%FBS containing puromycin (1 μg/ml, Sigma #P8833) to select for cells transfected with pLZRS. After another 48 hr, the puromycin-containing medium was aspirated, plates were rinsed with PBS, and DMEM+10%FBS was added to begin virus collection. After 24 hr, the medium was collected, then centrifuged at 200 g for 5 min to pellet any dislodged cells and the supernatant was aliquoted and snap-frozen in liquid nitrogen for long-term storage.
For viral gene transduction, the cells’ native medium was aspirated and replaced with retrovirus-containing medium, which had been thawed at 37°C and supplemented with 4 μg/ml polybrene (Sigma #H9268). Cells were incubated in the virus-containing medium at 37°C for 60 min, then were rinsed in PBS, and re-fed their native medium and passaged upon reaching 70%–80% confluency. Expression of fluorophore-tagged viral constructs was visible after 24 hr.
CRISPR/Cas9 gene editing
hTERT-immortalized human epidermal keratinocytes (THEKs), an immortalized keratinocyte line (Dickson et al., 2000) (a gift from Dr. James Rheinwald Lab, Harvard Univ., Cambridge, MA) was used for generation of knock-out (KO) cell lines using non-homologous end joining (NHEJ) via the CRISPR/Cas9 system. Single guide RNA (sgRNA) target sequences for NIX/BNIP3L and TUBAP pseudogene (for control KO line) were generated using the publicly available GPP Web Portal from the Broad Institute (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). The pSpCas9 (BB)-2A-GFP (PX458) plasmid (Addgene #48138) (Ran4.2638mmet al., 2013) was used as the CRISPR/Cas9 cloning backbone. We followed the CRISPR/Cas9 protocol from our previous publication (Sarkar et al., 2018) to generate NIX/BNIP3L KO and Control CRISPR KO keratinocytes.
To generate sgRNA for NIX/BNIP3L, the following oligonucleotides were used:
BNIP3LSGRNAF1: 5′-CACCGGACATTGTCGGACAGCTGGC-3′ and
BNIP3LSGRNAR1: 5′-AAACGCCAGCTGTCCGACAATGTCC-3′.
To generate sgRNA for the control KO line, the following oligonucleotides were used:
PG5SG1F: 5′-CACCGGTATTCCGTGGGTGAACGGG-3′ and
PG5SG1R: 5′-AAACCCCGTTCACCCACGGAATACC-3′.
These complementary oligonucleotides were annealed and then ligated into PX458 plasmid. Chemically competent E. coli (Thermo-Fisher #C737303) were used to transform the ligated plasmids. After overnight transformation, colonies were randomly picked, cultured in LB medium and a mini-prep kit (QIAGEN #27106) was used to isolate plasmid DNA. Plasmids with successful sgRNA target sequence insertion was verified by Sanger sequencing and the verified plasmid was used to transfect THEKs using the TransfeX transfection kit (ATCC #ACS4005). At 24 hr post-transfection, GFP-positive single cells were sorted into 96 well plates using a MoFlo Astrios cell sorter machine. Surviving cells were expanded, DNA was extracted, and PCR amplification was performed using the following primers:
BNIP3LPCRF1: CGGACTCGGCTTGTTGTGTT and
BNIP3LPCRR1: GGGGCAGAGACTGCTCATTTT or
PG5PCRF2: CCTCGGAAAACTCTCCTTCC and
PG5PCRR2: TGGATTGCTGTCCCTCTAGC.
Amplified PCR products were purified using NucleoSpin Gel and PCR Clean-Up kit (Takara #740609) and indel mutations were analyzed by Sanger sequencing. We selected those THEK lines with homozygous and frameshift mutations.
Western blotting and quantification
To generate protein lysates from cell cultures, the medium was aspirated and cells were lysed in urea sample buffer (8 M urea, 1% SDS, 10% glycerol, 60 mM Tris, 5% β-mercaptoethanol, 0.0005% pyronin-Y, at pH 6.8) and the lysate was homogenized by vigorous pipetting using a 25-gauge needle and a 1-mL syringe. For lysing organotypic cultures, the transwell was removed from the plate and the tissue was released using a #15 scalpel; then the epidermal tissue was separated from the underlying collagen-fibroblast matrix using sterile forceps. The tissue was then transferred into urea sample buffer and homogenized by vigorous pipetting using a 25-gauge needle and a 1-mL syringe.
Lysates were subjected to SDS-PAGE on an 8% or 12% acrylamide gel, then transferred onto fluorescent PVDF membrane at 20 V for 18 hr at room temperature in transfer buffer (3 g Tris base, 14.4 g glycine, 10% methanol, diluted to 1L with deionized water). Membranes were dried for 1 hour at room temperature, then were rehydrated with methanol for 5 min, then were rinsed with deionized water. Membranes were transferred into TrueBlack WB blocking buffer (Biotium #23013) for 60 min at room temperature then were probed with primary antibodies diluted in TrueBlack WB antibody diluent (Biotium #23013) plus 0.2% Tween-20 for 18 hr at 4°C. Blots were washed in Tris-buffered saline three times, then were incubated in the dark with fluorescent secondary antibodies (Li-Cor) diluted at 1:20,000 in TrueBlack WB antibody diluent for 60 min at room temperature. Membranes were then washed in Tris-buffered saline three times and imaged on an Odyssey CLx imaging system.
Images of blots were analyzed using Fiji software. For quantification of band intensity, equivalent rectangular areas were selected from the background and from bands in each lane and the integrated intensity was collected. The net intensity of each band was calculated as the integrated density of the band minus the integrated density of the background. Band intensities were normalized to a protein loading control run on the same gel and pooled values from multiple experiments were reported as mean+/−standard deviation (SD).
Fluorescent immunocytochemistry
For fixing cells in 35 mm glass-bottom dishes, the medium was aspirated and replaced with warmed (37°C) 4% paraformaldehyde in phosphate-buffered saline (PBS), then incubated at 37°C for 10 min. The fixative was removed and cells were washed with PBS, then permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature. The detergent was aspirated and rinsed with PBS. For staining of intermediate filaments, cells were fixed in 100% methanol at −20°C for 2 min, then re-hydrated in PBS for 5 min. Cells were blocked in 0.5% bovine serum albumin (BSA, Sigma #A9647) plus 10% normal goat serum (NGS, Sigma #G9023) in PBS for 30 min at 37°C. The blocking solution was removed, then cells were rinsed with PBS. Cells were then incubated in primary antibodies diluted in 0.5% BSA in PBS for 18 hr at 4°C. The antibody solution was removed and cells were rinsed in PBS 3 times, then incubated in secondary antibodies diluted in 0.5% BSA in PBS (with or without Hoechst) for 30 min at 37°C. The antibody solution was removed and cells were rinsed in PBS 3 times. Cells were re-submerged in PBS in the glass-bottom dish and then used for microscopic imaging.
Fluorescent immunohistochemistry
Paraffin-embedded formalin-fixed tissue sections on glass slides were pre-heated at 65°C for 2 hr prior to staining. The slides were then submerged in 3 successive baths of Xylenes (Fisher #X3F) for 5 min each, then 3 successive baths of 95% ethanol for 5 min each, then 70% ethanol for 5 min, then 3 successive baths of PBS for 5 min each. For antigen retrieval, slides were transferred into 0.1 M sodium citrate buffer (pH 6.0) with 0.05% Tween-20, which was warmed to 95°C for 15 min, then allowed to cool to room temperature. Slides were then rinsed in PBS. A Pap pen was used to encircle the tissue sections, then blocking solution (0.5% BSA plus 10% NGS in PBS) was added onto the slides, which were incubated in a humidified chamber for 30 min at 37°C. Slides were then rinsed in PBS, then were removed and primary antibody (diluted in 0.5% BSA in PBS) was added onto the tissue sections and the slides were incubated in a humidified chamber for 18 hr at 4°C. Slides were then rinsed in 3 successive baths of PBS, then secondary antibody (diluted in 0.5% BSA in PBS) was added onto the tissue sections and the slides were incubated in a humidified chamber for 60 min at 37°C. Slides were then rinsed in 3 successive PBS baths. Slides were then removed from the PBS and Prolong Gold mounting medium (Invitrogen #P36930) was used to cover the tissue sections and a glass coverslip (Fisher #12–543A) was applied and images were captured as below after the slides dried for 24 hr.
Spinning-disk confocal microscopy
SDC microscopy was performed utilizing a Nikon Eclipse Ti inverted microscope equipped with an UltraVIEW VOX confocal system (Perkin Elmer) and an EM-CCD camera (Hamamatsu) housed within a sealed chamber at 37°C. Cells in their native medium were excited using laser wavelengths of 405 nm, 488 nm, 561 nm, and/or 640 nm and fluorescence was detected by either a 100X/1.49NA apo TIRF objective (Nikon) or 60X/1.4NA Plan Apo objective (Nikon) with standard emission filters. For confocal imaging of live submerged cultures, cells were transduced and/or transfected with fluorophore-tagged constructs and then seeded into glass-bottom dishes (MatTek #P35G-1.5–20-C) at least 24 hr prior to imaging in their native medium. For imaging of live stratified cultures, organotypic epidermis was transferred from the transwell into a 35 mm glass-bottom dish with the top of the tissue against the glass. Multi-color Z stack images were acquired using a piezo motor with a step size of 200 nm.
Stimulated emission depletion microscopy
The Cell and Developmental Biology Microscopy Core of the University of Pennsylvania was utilized for STED imaging of fixed cultures. Cells were transduced and/or transfected with tagged constructs and then seeded into glass-bottom dishes (MatTek #P35G-1.5–20-C) at least 24 hr prior to imaging. Cells were then fixed with paraformaldehyde, permeabilized, and immunostained as above. STED microscopy was performed using a Leica TCS SP8 STED 3X super-resolution microscope equipped with two depletion lasers (592 nm and 660 nm) and pulsed white-light laser for tunable excitation from 470 to 670 nm that allowed super-resolution imaging of both green (488 nm) and red (594 nm) Alexa fluorophores.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
The n values/definitions, definition of center, dispersion measures, and statistical tests used for experimental analyses are included in each figure legend. Statistical p values are denoted by asterisks on figure graphs and are explained in each figure legend. Statistical analyses and graphs were generated using Prism version 8.1.1 (GraphPad). Datasets were tested for normality using the D’Agostino-Pearson test. To compare the means of two normally distributed datasets, a two-tailed unpaired Student’s t test was used; in experiments in which paired values (e.g., control versus non-transduced cells) were collected from the same image, a paired Student’s t test was used for normally distributed datasets or a Wilcoxon signed-rank test was used for non-normally distributed datasets. To compare the means of more than two normally distributed datasets, one-way ordinary ANOVA was used. The means of two datasets found to be non-normally distributed were compared using the Mann-Whitney test. For comparing the means of more than two datasets that were non-normally distributed, the Kruskal-Wallis test was used. A p value of ≤ 0.05 served as the cut-off for statistical significance.
Mitochondrial morphology analysis
Quantification of mitochondrial morphology in NHEKs or organotypic epidermis was performed with Fiji software in a blinded manner using non-visibly identifiable SDC microscopy images; in organotypic epidermis, image stacks were taken from varying z-positions within the same X-Y position in the tissue. Files were converted to binary images using the “Make Binary” function followed by segmentation of clustered organelles using the “Watershed” function. The Fiji “Analyze Particles” function was used to calculate the area, aspect ratio (AR), length (major axis), width (minor axis), and/or perimeter of the segmented organelles. These parameters were averaged across each cell and these measures were pooled from multiple independent experiments.
Relative TMRE intensity calculation
Quantification of TMRE fluorescence intensity was performed in a blinded manner using non-visibly identifiable SDC microscopy images from the upper cell layers of organotypic epidermis transduced with GFP-Mito to allow visualization of mitochondrial morphology of each cell within the image. Cells were categorized as having fragmented or non-fragmented mitochondria, then a pair of cells (one fragmented and one non-fragmented) was circumscribed within the same image and the Fiji “Measure” function was used to calculate the integrated density of the TMRE signal, which was divided by the measured area to obtain the mean fluorescence intensity from which the mean background fluorescence intensity for each image was subtracted. The TMRE intensity for each cell pair was normalized to a mean value of 1 for the non-fragmented cell and the pooled values were averaged across multiple experiments.
Fluorescence immunocytochemistry intensity
Quantification of the fluorescence intensity of NIX relative to TOM20 was performed in a blinded manner using non-visibly identifiable SDC microscopy images of NHEKs treated with DMSO or experimental drug treatments. The Fiji “Measure” function was used to calculate the integrated density of NIX across the entire high-powered field (hpf), which was divided by the integrated density of TOM20 across the same hpf. The ratio of NIX/TOM20 for each treatment was averaged across multiple independent fields. K14 fluorescence intensity in individual NHEKs was quantified using the Fiji “Measure” function to calculate the integrated density for regions of interest drawn by hand to encompass entire cells found to express either GFP-Mito or GFP-NIX.
DRP1 distribution analysis
Quantification of the peripheral bias of DRP1 fluorescence signal was performed in a blinded manner using non-visibly identifiable SDC microscopy images of NHEKs transduced with GFP-NIX, which were compared to non-transduced cells within the same hpf. The radius of the nucleus was measured by hand for each analyzed cell and the average radius of the whole cell was calculated using the formula, area = πr2, in which the area was measured by circumscribing the cell in Fiji. Files were then converted to binary images using the “Make Binary” function, then the XY position of each positive pixel for DRP1 signal was obtained using the Fiji “Save XY Coordinates” function. The XY coordinates of the center of the nucleus was identified and the distance of each pixel of DRP1 signal from the nuclear center (NUC) was calculated by the formula (distance = square root of [(XDRP1-XNUC)2 + (YDRP1-YNUC)2]. The mean of this net distance (distance from nuclear center minus nuclear radius, shown as vector “A” in Figure 6B) was calculated for each DRP1-positive pixel in each cell and was divided by the cell radius minus the nuclear radius (shown as vector “B” in Figure 6B). The “peripheral bias” of all DRP1-positive pixels was calculated as the mean value for vector A divided by vector B for each cell.
Histologic microscopy
Hematoxylin and eosin (H&E) stained tissue sections on glass slides were visualized on an EVOS FL Imaging System (Invitrogen) using an EVOS LWD achromatic phase contrast 40X/0.65NA/3.1WD objective (Invitrogen #AMEP4635) and photographed using the embedded high-sensitivity interline CCD color camera.
Tissue morphologic quantification
Tissue thickness was determined using Fiji to circumscribe the entire tissue section contained within each hpf, then the “Measure” function was used to calculate the area, which was divided by the length to calculate the average tissue thickness (width). Counts of dyskeratotic cells within the epidermis and retained nuclei within the cornified layers were performed by hand in a blinded fashion using non-visibly labeled H&E histologic images to identify and count the number of dyskeratotic cells or retained nuclei per non-overlapping hpf for each condition, which were averaged across multiple experiments.
Fluorescent tissue staining quantification
Fluorescence microscopy images of immunostained tissue sections were captured by SDC microscopy as above and TIF images files were opened in Fiji for quantitative analysis.
Quantification of filaggrin (FLG) IF was performed in a blinded manner using non-visibly labeled images from epidermal tissue sections immunostained for FLG. The Fiji “Measure” function was used to calculate the integrated density of the FLG signal, which was divided by the measured area to obtain the mean fluorescence intensity, then the mean background fluorescence intensity for each image was subtracted. The FLG intensity was averaged across multiple non-overlapping hpf and all intensity values from treated cultures were normalized to an average value of 1 for control cultures.
Counts of K10-negative cells within the epidermis were performed by hand in a blinded manner using non-visibly labeled fluorescence images from epidermal tissue sections immunostained for K10 and Hoechst. The Fiji “Make Binary” function was utilized and the number of nucleated epidermal cells lacking K10 per non-overlapping hpf for each condition was recorded and averaged across control and NIX-deficient (KO) organotypic cultures.
Mean suprabasal cell roundness was calculated using fluorescence images from epidermal tissue sections immunostained for K10, Dsg1, and Hoechst. In Fiji, all nucleated cells in each tissue section were outlined by hand. The Fiji “Measure” function was used to calculate the roundness (1/aspect ratio) of all nucleated cell profiles in each field, which was averaged across multiple experiments.
Electron microscopy
Samples were prepared for transmission electron microscopy (TEM) examination by the Electron Microscopy Resource Lab of the University of Pennsylvania. Tissues were placed in fixative (2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate, pH 7.4) at 4°C for at least 24 hr prior to processing. After additional washing with fixative buffer, the samples were post-fixed in 2.0% osmium tetroxide with 1.5% K3Fe(CN)6 for 1 hour at room temperature then were rinsed in deionized water. After dehydration of samples through a graded series of ethanol, the tissues were infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences #14120). Thin tissue sections were stained with uranyl acetate and SATO lead, then examined with a JEOL 1010 electron microscope using a Hamamatsu digital camera; images were acquired on AMT Advantage NanoSprint500 software.
EM quantification
Quantification of TEM images was performed in Fiji in a blinded manner using non-visibly labeled images from the uppermost layers of epidermal tissue sections. The area of epidermal tissue was circumscribed, then the Fiji “Measure” function was used to calculate the integrated density of the region, which was divided by the measured area to obtain the mean electron density from which the mean background electron density for each image was subtracted. The measured tissue electron density was averaged across multiple non-overlapping hpf and all intensity values were normalized to an average value of 1 for control cultures.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Actin | Santa Cruz | Cat#sc-47778; clone C4; RRID:AB_2714189 |
| Alexa Fluor goat anti-chicken IgY 488 nm | Invitrogen | Cat#A32931; RRID:AB_2762843 |
| Alexa Fluor goat anti-mouse IgG 405 nm | Invitrogen | Cat#A31553; RRID:AB_221604 |
| Alexa Fluor goat anti-mouse IgG 488 nm | Invitrogen | Cat#A32723; RRID:AB_2633275 |
| Alexa Fluor goat anti-mouse IgG 594 nm | Invitrogen | Cat#A32742; RRID:AB_2762825 |
| Alexa Fluor goat anti-mouse IgG 633 nm | Invitrogen | Cat#A21052; RRID:AB_2535719 |
| Alexa Fluor goat anti-rabbit IgG 405 nm | Invitrogen | Cat#A31556; RRID:AB_221605 |
| Alexa Fluor goat anti-rabbit IgG 488 nm | Invitrogen | Cat#A32731; RRID:AB_2633280 |
| Alexa Fluor goat anti-rabbit IgG 594 nm | Invitrogen | Cat#A32740; RRID:AB_2762824 |
| Alexa Fluor goat anti-rabbit IgG 633 nm | Invitrogen | Cat#A21071; RRID:AB_2535732 |
| Alpha-tubulin | Sigma | Cat#T9026; clone DM1A; RRID:AB_477593 |
| Desmoglein 1 | Abcam | Cat#Ab12077; Clone 27B2; RRID:AB_2093304 |
| Desmoglein 1 | Santa Cruz | Cat#sc-137164; Clone B11; RRID:AB_2093310 |
| DRP1 | Abcam | Cat#Ab56788; RRID:AB_941306 |
| DRP1 | Santa Cruz | Cat#sc-101270; Clone 6Z-82; RRID:AB_2093545 |
| DRP1 | Sigma | Cat#HPA039324; RRID:AB_2676443 |
| Filaggrin | Abcam | Cat#Ab3137; RRID:AB_303542 |
| FIS1 | Santa Cruz | Cat#sc-376447; Clone B-5; RRID:AB_11149382 |
| FIS1 | Sigma | Cat#HPA017430; RRID:AB_1848608 |
| GAPDH | Abcam | Cat#Ab9484; RRID:AB_307274 |
| GFP | Aves | Cat#GFP-1020; RRID:AB_10000240 |
| Halo tag | Promega | Cat#G9281; RRID:AB_713650 |
| Hsp60 | Sigma | Cat#HPA001523; RRID:AB_1079028 |
| Keratin 10 | Abcam | Cat#Ab76318; RRID:AB_1523465 |
| Keratin 14 | Abcam | Cat#Ab7800; RRID:AB_306091 |
| LC3b | Abcam | Cat#Ab48394; RRID:AB_881433 |
| Loricrin | Abcam | Cat#Ab24722; RRID:AB_448258 |
| NIX/BNIP3L | Sigma | Cat#HPA015652; RRID:AB_1845406 |
| NIX/BNIP3L | Santa Cruz | Cat#sc-166332; Clone H-8; RRID:AB_2066782 |
| NIX/BNIP3L | Abcam | Cat#Ab109414; RRID:AB_10863865 |
| SNAP tag | New England BioLabs | Cat#P9310S; RRID:AB_10631145 |
| TOM20 | Santa Cruz | Cat#sc-17764; Clone F-10; RRID:AB_628381 |
| TOM20 | Abcam | Cat#Ab56783; RRID:AB_945896 |
| TOM20 | Sigma | Cat#HPA011562; RRID:AB_1080326 |
| Bacterial and virus strains | ||
| Competent E. coli | Thermo-Fisher | #C737303 |
| Biological samples | ||
| Human skin tissue | Univ. of Pennsylvania Skin Biology and Disease Resource-based Center | https://dermatology.upenn.edu/sbdrc/ |
| Normal human epidermal keratinocytes | Univ. of Pennsylvania Skin Biology and Disease Resource-based Center | https://dermatology.upenn.edu/sbdrc/ |
| Chemicals, peptides, and recombinant proteins | ||
| 3,3′,5-tri-iodo-L-thyronine | Sigma | Cat#T6397 |
| Adenine | Sigma | Cat#A2786 |
| Apo-transferrin | Sigma | Cat# T1147 |
| Bafilomycin A1 (BafA1) | Sigma | Cat#B1793 |
| Bovine serum albumin (BSA) | Sigma | Cat#A9647 |
| Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) | Sigma | Cat#C2759 |
| Cholera toxin | Sigma | Cat#C8052 |
| Cobalt chloride (CoCl2) | Sigma | Cat#15862 |
| Collagen type 1, high-concentration rat tail | Corning | Cat#354249 |
| Deferiprone (DFP) | Sigma | Cat#Y0001976 |
| Dimethyloxalylglycine (DMOG) | Sigma | Cat#D3695 |
| Dimethyl sulfoxide (DMSO) | Sigma | Cat#D8418 |
| Dispase II | StemCell Technologies | Cat#07913 |
| DMEM | Corning | Cat#10–017-CM |
| DMEM 10X | Sigma | Cat#D2554 |
| Fetal bovine serum (FBS) | HyClone | Cat#SH3007103 |
| JF646-Halo Ligand | Lavis Lab, HHMI Janelia Research Campus, Ashburn, VA, USA | https://www.janelia.org/open-science/janelia-fluor-dyes |
| EMbed-812 | Electron Microscopy Sciences | Cat#14120 |
| Ethyl(2-(5-nitrothiophene-2-carboxamido)thiophene-3-carbonyl)carbamate (EACC) | AOBIOUS | Cat#AOB13386 |
| EUK134 | Sigma | Cat#SML0743 |
| Formalin, 10%, neutral buffered | Fisher | Cat#SF100–4 |
| FuGENE6 transfection reagent | Promega | Cat#E269A |
| Gentamicin/Amphotericin | GIBCO | Cat#R01510 |
| GlutaMAX | GIBCO | Cat#35050061 |
| Ham’s F12 medium | Corning | Cat#10–080-CV |
| Hoechst 33342 | Invitrogen | Cat#H21492 |
| Human epidermal growth factor (EGF) | Sigma | Cat#E9644 |
| Human insulin | Sigma | Cat#91077C |
| Human keratinocyte growth supplement | GIBCO | Cat#S0015 |
| Hydrocortisone | Sigma | Cat#H0888 |
| Keratinocyte medium M154 | GIBCO | Cat#M154CFPRF500 |
| Keratinocyte serum-free medium kit (including bovine pituitary extract; epidermal growth factor; calcium chloride) | GIBCO | Cat#17005042 |
| Mdivi-1 | Cell Signaling | Cat#CAS-338967–87-6 |
| MG132 | Sigma | Cat#M8699) |
| MitoTracker Deep Red | Invitrogen | Cat#M22426 |
| Normal goat serum (NGS) | Sigma | Cat#G9023 |
| Opti-MEM | GIBCO | Cat#31985–070 |
| Penicillin/streptomycin | Sigma | Cat#P0781 |
| Polybrene | Sigma | Cat#H9268 |
| Prolong Gold mounting medium | Invitrogen | Cat#P36930 |
| Puromycin | Sigma | Cat# P8833 |
| SNAP-Cell 647-SIR | New England BioLabs | Cat#S9102S |
| Tetramethylrhodamine Ethyl Ester Perchlorate (TMRE) | Invitrogen | Cat#T669 |
| TrueBlack WB blocking buffer kit | Biotium | Cat#23013 |
| Trypsin-EDTA | GIBCO | Cat#25300054 |
| Xylenes | Fisher | Cat#X3F |
| Critical commercial assays | ||
| NucleoSpin Gel and PCR Clean-Up kit | Takara | Cat#740609 |
| Plasmid DNA Mini-prep Kit | QIAGEN | Cat#27106 |
| TransfeX Transfection Kit | ATCC | Cat#ACS4005 |
| Experimental models: cell lines | ||
| J2 3T3 murine fibroblasts | James Rheinwald Lab, Harvard Univ., Cambridge, MA, USA | N/A |
| hTERT-immortalized human epidermal keratinocytes (THEK) | James Rheinwald Lab, Harvard Univ., Cambridge, MA, USA | N/A |
| Phoenix HEK293T Phoenix viral packaging cells | Garry Nolan Lab, Stanford Univ., Palo Alto, CA, USA | N/A |
| THEK CRISPR Control KO line PG5 | This paper | N/A |
| THEK CRISPR NIX KO line 1 | This paper | N/A |
| THEK CRISPR NIX KO line 2 | This paper | N/A |
| Oligonucleotides | ||
| sgRNA control forward oligo: PG5SG1F | This paper | 5′-CACCGGTATTCCGTGGGTGAACGGG-3′ |
| sgRNA control reverse oligo: PG5SG1R | This paper | 5′-AAACCCCGTTCACCCACGGAATACC-3′. |
| sgRNA for NIX/BNIP3L forward oligo: BNIP3LSGRNAF1 | This paper | 5′-CACCGGACATTGTCGGACAGCTGGC-3′ |
| sgRNA for NIX/BNIP3L reverse oligo: BNIP3LSGRNAR1 | This paper | 5′-AAACGCCAGCTGTCCGACAATGTCC-3′ |
| PCR forward primer for control KO sequencing: PG5PCRF2 | This paper | 5′-CCTCGGAAAACTCTCCTTCC-3′ |
| PCR reverse primary for control KO sequencing: PG5PCRR2 | This paper | 5′-TGGATTGCTGTCCCTCTAGC-3′ |
| PCR forward primer for NIX/BNIP3L KO sequencing: BNIP3LPCRF1 | This paper | 5′- CGGACTCGGCTTGTTGTGTT-3′ |
| PCR reverse primary for NIX/BNIP3L KO sequencing: BNIP3LPCRR1 | This paper | 5′-GGGGCAGAGACTGCTCATTTT-3′ |
| Recombinant DNA | ||
| pcDNA3-SNAP-DRP1-K38A | Smirnova et al. 1998 | Addgene plasmid #45161 |
| pcDNA3.1-GFP-BNIP3L | Fei et al. 2004 | Addgene plasmid #17467 |
| pCLBW-Cox8-GFP-mCh | Rojansky et al. 2016 | Addgene plasmid #78520 |
| pdsRed2-Mito | Thomas Schwarz Lab, Harvard Univ., Boston, MA, USA | N/A |
| pLZRS | Kathleen Green Lab, Northwestern Univ., Chicago, IL, USA | N/A |
| pLZRS-beta-tubulin-mCh | Kathleen Green Lab, Northwestern Univ., Chicago, IL, USA | N/A |
| pLZRS-dsRed-Mito | This paper | N/A |
| pLZRS-GFP-Mito | This paper | N/A |
| pLZRS-GFP-NIX | This paper | N/A |
| pLZRS-GFP-NIX-2GA | This paper | N/A |
| pLZRS-GFP-NIX-WA | This paper | N/A |
| pLZRS-Halo-FIS1 | This paper | N/A |
| pLZRS-Mito-SNAP | This paper | N/A |
| pLZRS-SNAP-DRP1-K38A | This paper | N/A |
| pLZRS-TOM20-mCh | This paper | N/A |
| pmCherry-TOM20 | Michael Davidson Lab, Florida State Univ., Tallahassee, FL, USA | Addgene plasmid #55146 |
| pSEMS-Halo7Tag-hFIS1 | Appelhans et al. 2012 | Addgene plasmid #111136 |
| pSpCas9 (BB)-2A-GFP (PX458) | Ran et al. 2013 | Addgene plasmid #48138 |
| Software and algorithms | ||
| CRISPR/Cas9 Guide RNA Design | GPP Web Portal, Broad Institute, Cambridge, MA, USA | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| Fiji Image Anlysis | ImageJ | https://imagej.net/Fiji |
| Prism version 8.1.1 | GraphPad | N/A |
| Other | ||
| Deep 6-well culture plates | Falcon | Cat#355467 |
| Glass coverslips | Fisher | Cat#12–543A |
| Glass-bottom tissue culture dishes | MatTek | Cat#P35G-1.5–20-C |
| Transwell inserts | Falcon | Cat#353091 |
Highlights.
Differentiating keratinocytes upregulate NIX prior to programmed organelle clearing
The mitophagy receptor NIX triggers keratinocyte mitochondrial degradation via DRP1
Disrupting NIX expression or DRP1 activity impairs epidermal tissue morphogenesis
ACKNOWLEDGMENTS
We thank the Penn Skin Biology & Disease Resource-Based Center (NIH P30-AR069589) for supporting this work via a mini-grant, histology services (Core A, Stephen Prouty), and NHEK isolation (Core B, Christine Marshall). We thank the Penn Electron Microscopy Resource Lab and CDB Microscopy Core for imaging support. C.L.S. is supported by NIH K08-AR075846 and previously by NIH T32-AR007465, the Dermatology Foundation, the National Psoriasis Foundation, the American Skin Association, and La Roche-Posay North American Foundation. J.E.G., M.K.S., and R.U. are supported by NIH P30-AR075043 and R01-AR060802. E.L.F.H. is supported by NIH R35-GM126950 and R37-NS060698.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108689.
REFERENCES
- Akinduro O, Sully K, Patel A, Robinson DJ, Chikh A, McPhail G, Braun KM, Philpott MP, Harwood CA, Byrne C, et al. (2016). Constitutive Autophagy and Nucleophagy during Epidermal Differentiation. J. Invest. Dermatol. 136, 1460–1470. [DOI] [PubMed] [Google Scholar]
- Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, and Gillespie MN (2012). Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 5, ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GF, Toth R, James J, and Ganley IG (2013). Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 14, 1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelhans T, Richter CP, Wilkens V, Hess ST, Piehler J, and Busch KB (2012). Nanoscale organization of mitochondrial microcompartments revealed by combining tracking and localization microscopy. Nano Lett. 12, 610–616. [DOI] [PubMed] [Google Scholar]
- Asano M, Yamasaki K, Yamauchi T, Terui T, and Aiba S (2017). Epidermal iron metabolism for iron salvage. J. Dermatol. Sci. 87, 101–109. [DOI] [PubMed] [Google Scholar]
- Baek JH, Reiter CE, Manalo DJ, Buehler PW, Hider RC, and Alayash AI (2011). Induction of hypoxia inducible factor (HIF-1α) in rat kidneys by iron chelation with the hydroxypyridinone, CP94. Biochim. Biophys. Acta 1809, 262–268. [DOI] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, and Mazure NM (2009). Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29, 2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero E, and Maack C (2018). Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 122, 1460–1478. [DOI] [PubMed] [Google Scholar]
- Bhaduri A, Ungewickell A, Boxer LD, Lopez-Pajares V, Zarnegar BJ, and Khavari PA (2015). Network Analysis Identifies Mitochondrial Regulation of Epidermal Differentiation by MPZL3 and FDXR. Dev. Cell 35, 444–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, and Shaw JM (1999). The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1, 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, et al. (2017). The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 40, 583–594.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin AT, Weidemann A, Fu Z, Mesropian L, Gradin K, Jamora C, Wiesener M, Eckardt KU, Koch CJ, Ellies LG, et al. (2008). Epidermal sensing of oxygen is essential for systemic hypoxic response. Cell 133, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand RM, Wipf P, Durham A, Epperly MW, Greenberger JS, and Falo LD Jr. (2018). Targeting Mitochondrial Oxidative Stress to Mitigate UV-Induced Skin Damage. Front. Pharmacol. 9, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody I (1962). Electron microscopic demonstration of mitochondria and alpha-cytomembranes with negative contrast in the horny layer of parakeratotic psoriatic epidermis. J. Ultrastruct. Res. 7, 346–358. [DOI] [PubMed] [Google Scholar]
- Candi E, Schmidt R, and Melino G (2005). The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 6, 328–340. [DOI] [PubMed] [Google Scholar]
- Celli A, Crumrine D, Meyer JM, and Mauro TM (2016). Endoplasmic Reticulum Calcium Regulates Epidermal Barrier Response and Desmosomal Structure. J. Invest. Dermatol. 136, 1840–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, and Higgs HN (2018). INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 217, 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2020). Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 15, 235–259. [DOI] [PubMed] [Google Scholar]
- Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, Saxena S, Gietz RD, and Greenberg AH (1997). The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. 186, 1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AC, Martin AJ, Choy B, Fernández-Peñas P, Dalziell RA, McKenzie CA, Scolyer RA, Dhillon HM, Vardy JL, Kricker A, et al. (2015). A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N. Engl. J. Med. 373, 1618–1626. [DOI] [PubMed] [Google Scholar]
- Dalle B, Payen E, and Beuzard Y (2000). Modulation of transduced erythropoietin expression by iron. Exp. Hematol. 28, 760–764. [DOI] [PubMed] [Google Scholar]
- Dickson MA, Hahn WC, Ino Y, Ronfard V, Wu JY, Weinberg RA, Louis DN, Li FP, and Rheinwald JG (2000). Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol. Cell. Biol. 20, 1436–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW 2nd, and Yin XM (2010). Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 285, 27879–27890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, Daria D, Jegga AG, Geiger H, Aronow BJ, et al. (2007). Unrestrained erythroblast development in Nix−/− mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. USA 104, 6794–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douroudis K, Kingo K, Traks T, Reimann E, Raud K, Rätsep R, Mössner R, Silm H, Vasar E, and Kõks S (2012). Polymorphisms in the ATG16L1 gene are associated with psoriasis vulgaris. Acta Derm. Venereol. 92, 85–87. [DOI] [PubMed] [Google Scholar]
- Fei P, Wang W, Kim SH, Wang S, Burns TF, Sax JK, Buzzai M, Dicker DT, McKenna WG, Bernhard EJ, and El-Deiry WS (2004). Bnip3L is induced by p53 under hypoxia, and its knockdown promotes tumor growth. Cancer Cell 6, 597–609. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, and Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhar A, Pillar T, and David M (1991). Aged versus young skin before and after transplantation onto nude mice. Br. J. Dermatol. 124, 168–171. [DOI] [PubMed] [Google Scholar]
- Guo Y, Zhang X, Wu T, Hu X, Su J, and Chen X (2019). Autophagy in Skin Diseases. Dermatology 235, 380–389. [DOI] [PubMed] [Google Scholar]
- Hamanaka RB, and Chandel NS (2013). Mitochondrial metabolism as a regulator of keratinocyte differentiation. Cell. Logist. 3, e25456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Glasauer A, Hoover P, Yang S, Blatt H, Mullen AR, Getsios S, Gottardi CJ, DeBerardinis RJ, Lavker RM, and Chandel NS (2013). Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci. Signal. 6, ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka RB, Weinberg SE, Reczek CR, and Chandel NS (2016). The Mitochondrial Respiratory Chain Is Required for Organismal Adaptation to Hypoxia. Cell Rep. 15, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipponjima S, Umino Y, Nagayama M, and Denda M (2020). Live imaging of alterations in cellular morphology and organelles during cornification using an epidermal equivalent model. Sci. Rep. 10, 5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji HT, Chien LT, Lin YH, Chien HF, and Chen CT (2010). 5-ALA mediated photodynamic therapy induces autophagic cell death via AMP-activated protein kinase. Mol. Cancer 9, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SM, and Youle RJ (2012). PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joost S, Zeisel A, Jacob T, Sun X, La Manno G, Lönnerberg P, Linnarsson S, and Kasper M (2016). Single-Cell Transcriptomics Reveals that Differentiation and Spatial Signatures Shape Epidermal and Hair Follicle Heterogeneity. Cell Syst. 3, 221–237.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel D, and Oleinick NL (2010). Photodynamic therapy and cell death pathways. Methods Mol. Biol. 635, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella TM, and Nolan GP (1996). Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7, 1405–1413. [DOI] [PubMed] [Google Scholar]
- Kirkin V, and Rogov VV (2019). A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 76, 268–285. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Yanagi T, Imafuku K, Hata H, Abe R, and Shimizu H (2017). Drp1 regulates mitochondrial morphology and cell proliferation in cutaneous squamous cell carcinoma. J. Dermatol. Sci. 88, 298–307. [DOI] [PubMed] [Google Scholar]
- Lackner LL, and Nunnari J (2010). Small molecule inhibitors of mitochondrial division: tools that translate basic biological research into medicine. Chem. Biol. 17, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavker RM, and Matoltsy AG (1970). Formation of horny cells: the fate of cell organelles and differentiation products in ruminal epithelium. J. Cell Biol. 44, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavker R, Chalupa W, and Dickey JF (1969). An electron microscopic investigation of rumen mucosa. J. Ultrastruct. Res. 28, 1–15. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Packer M, and Codogno P (2015). Development of autophagy inducers in clinical medicine. J. Clin. Invest. 125, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, and Shirihai OS (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. [DOI] [PubMed] [Google Scholar]
- Losón OC, Song Z, Chen H, and Chan DC (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, and Kimmelman AC (2016). Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 428 (9 Pt A), 1659–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Kandimalla R, Yin X, and Reddy PH (2019). Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 28, 177–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinković M, Šprung M, and Novak I (2020). Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markaki M, Palikaras K, and Tavernarakis N (2018). Novel Insights Into the Anti-aging Role of Mitophagy. Int. Rev. Cell Mol. Biol. 340, 169–208. [DOI] [PubMed] [Google Scholar]
- Matsui T, and Amagai M (2015). Dissecting the formation, structure and barrier function of the stratum corneum. Int. Immunol. 27, 269–280. [DOI] [PubMed] [Google Scholar]
- Mauvezin C, Nagy P, Juhász G, and Neufeld TP (2015). Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 6, 7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure NM, and Pouysségur J (2009). Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy 5, 868–869. [DOI] [PubMed] [Google Scholar]
- Mellem D, Sattler M, Pagel-Wolff S, Jaspers S, Wenck H, Rübhausen MA, and Fischer F (2017). Fragmentation of the mitochondrial network in skin in vivo. PLoS ONE 12, e0174469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, Wong YC, Simpson CL, and Holzbaur EL (2016). Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 7, 12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M, Moriyama H, Uda J, Matsuyama A, Osawa M, and Hayakawa T (2014). BNIP3 plays crucial roles in the differentiation and maintenance of epidermal keratinocytes. J. Invest. Dermatol. 134, 1627–1635. [DOI] [PubMed] [Google Scholar]
- Murley A, and Nunnari J (2016). The Emerging Network of Mitochondria-Organelle Contacts. Mol. Cell 61, 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muroyama A, and Lechler T (2012). Polarity and stratification of the epidermis. Semin. Cell Dev. Biol. 23, 890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney PA (2015). Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 1853 (10 Pt B), 2775–2783. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips KG, Samatham R, Choudhury N, Gladish JC, Thuillier P, and Jacques SL (2010). In vivo measurement of epidermal thickness changes associated with tumor promotion in murine models. J. Biomed. Opt. 15, 041514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts KR, Yoon Y, Krueger EW, and McNiven MA (1999). The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell 10, 4403–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudent J, and McBride HM (2017). The mitochondria-endoplasmic reticulum contact sites: a signalling platform for cell death. Curr. Opin. Cell Biol. 47, 52–63. [DOI] [PubMed] [Google Scholar]
- Quiroz FG, Fiore VF, Levorse J, Polak L, Wong E, Pasolli HA, and Fuchs E (2020). Liquid-liquid phase separation drives skin barrier formation. Science 367, eaax9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-e-Silva M, and Jacques Cd. (2012). Epidermal barrier function and systemic diseases. Clin. Dermatol. 30, 277–279. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvani HR, Ali N, Nissen LJ, Harfouche G, de Verneuil H, Taïeb A, and Mazurier F (2011). HIF-1α in epidermis: oxygen sensing, cutaneous angiogenesis, cancer, and non-cancer disorders. J. Invest. Dermatol. 131, 1793–1805. [DOI] [PubMed] [Google Scholar]
- Rinnerthaler M, Bischof J, Streubel MK, Trost A, and Richter K (2015). Oxidative stress in aging human skin. Biomolecules 5, 545–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov VV, Suzuki H, Marinković M, Lang V, Kato R, Kawasaki M, Buljubašić M, Šprung M, Rogova N, Wakatsuki S, et al. (2017). Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 7, 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojansky R, Cha MY, and Chan DC (2016). Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife 5, e17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, Berthier CC, Swindell WR, Patrick MT, Shao S, et al. (2018). Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann. Rheum. Dis. 77, 1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, and Ney PA (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 104, 19500–19505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JM, Choi DK, Sohn KC, Kim JY, Im M, Lee Y, Seo YJ, Shong M, Lee JH, and Kim CD (2017). Targeted deletion of Crif1 in mouse epidermis impairs skin homeostasis and hair morphogenesis. Sci. Rep. 7, 44828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sil P, Wong SW, and Martinez J (2018). More Than Skin Deep: Autophagy Is Vital for Skin Barrier Function. Front. Immunol. 9, 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson CL, Kojima S, and Getsios S (2010). RNA interference in keratinocytes and an organotypic model of human epidermis. Methods Mol. Biol. 585, 127–146. [DOI] [PubMed] [Google Scholar]
- Simpson CL, Patel DM, and Green KJ (2011). Deconstructing the skin: cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Biol. 12, 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Schoeb TR, Bajpai P, Slominski A, and Singh KK (2018). Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 9, 735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Shurland DL, Ryazantsev SN, and van der Bliek AM (1998). A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 143, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D, Koutsopoulos OS, Okamoto K, and Ryan MT (2004). Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 117, 1201–1210. [DOI] [PubMed] [Google Scholar]
- Therianou A, Vasiadi M, Delivanis DA, Petrakopoulou T, Katsarou-Katsari A, Antoniou C, Stratigos A, Tsilioni I, Katsambas A, Rigopoulos D, and Theoharides TC (2019). Mitochondrial dysfunction in affected skin and increased mitochondrial DNA in serum from patients with psoriasis. Exp. Dermatol. 28, 72–75. [DOI] [PubMed] [Google Scholar]
- Vats S, and Manjithaya R (2019). A reversible autophagy inhibitor blocks autophagosome-lysosome fusion by preventing Stx17 loading onto autophagosomes. Mol. Biol. Cell 30, 2283–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa E, Marchetti S, and Ricci JE (2018). No Parkin Zone: Mitophagy without Parkin. Trends Cell Biol. 28, 882–895. [DOI] [PubMed] [Google Scholar]
- Weiland D, Brachvogel B, Hornig-Do HT, Neuhaus JFG, Holzer T, Tobin DJ, Niessen CM, Wiesner RJ, and Baris OR (2018). Imbalance of Mitochondrial Respiratory Chain Complexes in the Epidermis Induces Severe Skin Inflammation. J. Invest. Dermatol. 138, 132–140. [DOI] [PubMed] [Google Scholar]
- Wong YC, and Holzbaur EL (2015). Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci. 128, 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WJ, Richardson T, Seykora JT, Cotsarelis G, and Simon MC (2015). Hypoxia-inducible factors regulate filaggrin expression and epidermal barrier function. J. Invest. Dermatol. 135, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita SI, and Kanki T (2018). Detection of Hypoxia-Induced and Iron Depletion-Induced Mitophagy in Mammalian Cells. Methods Mol. Biol. 1759, 141–149. [DOI] [PubMed] [Google Scholar]
- Yanagi T, Kitamura S, and Hata H (2018). Novel Therapeutic Targets in Cutaneous Squamous Cell Carcinoma. Front. Oncol. 8, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, and McNiven MA (2003). The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23, 5409–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara N, Ueno T, Takagi A, Oliva Trejo JA, Haruna K, Suga Y, Komatsu M, Tanaka K, and Ikeda S (2015). The significant role of autophagy in the granular layer in normal skin differentiation and hair growth. Arch. Dermatol. Res. 307, 159–169. [DOI] [PubMed] [Google Scholar]
- Youle RJ, and Narendra DP (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, and van der Bliek AM (2012). Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, and Ney PA (2012). A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy 8, 1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Sui BD, Liu N, Lv YJ, Zheng CX, Lu YB, Huang WT, Zhou CH, Chen J, Pang DL, et al. (2017). Anti-aging pharmacology in cutaneous wound healing: effects of metformin, resveratrol, and rapamycin by local application. Aging Cell 16, 1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.