

ABSTRACT
The biosimilar concept is now well established. Clinical data accumulated pre- and post-approval have supported biosimilar uptake, in turn stimulating competition in the biologics market and increasing patient access to biologics. Following technological advances, other innovative biologics, such as “biobetters” or “value-added medicines,” are now reaching the market. These innovative biologics differ from the reference product by offering additional clinical or non-clinical benefits. We discuss these innovative biologics with reference to CT-P13, initially available as an intravenous (IV) biosimilar of reference infliximab. A subcutaneous (SC) formulation, CT-P13 SC, has now been developed. Relative to CT-P13 IV, CT-P13 SC offers clinical benefits in terms of pharmacokinetics, with comparable efficacy, safety, and immunogenicity, as well as increased convenience for patients and reduced demands on healthcare system resources. As was once the case for biosimilars, nomenclature and regulatory pathways for innovative biologics require clarification to support their uptake and ultimately benefit patients.
KEYWORDS: Biobetter, biologic, biosimilar, CT-P13, infliximab, innovative biologics, value-added medicine
Introduction
Advancing technologies have enabled further innovations in biologic development, resulting in medicines that offer benefits beyond those offered by reference biologics, or their biosimilars. However, a lack of consensus on nomenclature and regulatory approaches toward these innovative biologics threatens their successful uptake, now that increasing numbers of these medicines are reaching the market. In addition, requirements for clinical and pharmacoeconomic evaluation must also be carefully considered and met by developers, to ensure physician and patient confidence in these innovative medicines. We examine some of these issues, by drawing on experiences with biosimilars and focusing on innovative biologics such biobetters and value-added medicines (VAMs), with particular reference to the subcutaneous (SC) formulation of the infliximab biosimilar CT-P13 (CT-P13 SC).
Many biologics are available for the treatment of patients with immune-mediated inflammatory diseases (IMIDs), with SC treatment options being developed in addition to intravenous (IV) medications (Supplementary Figure 1). For the first tumor necrosis factor (TNF) inhibitor, infliximab, initial approval was gained in 1998 from the United States (US) Food and Drug Administration (FDA), for the treatment of Crohn’s disease (CD).1 Infliximab indications approved by the FDA and European Medicines Agency (EMA) now encompass rheumatoid arthritis (RA), ankylosing spondylitis (AS), psoriatic arthritis (PsA), psoriasis, CD, and ulcerative colitis (UC) in adults, as well as CD and UC in pediatric patients.2,3
Several biosimilars of TNF inhibitors are now available (Supplementary Table 1). CT-P13 was the first infliximab biosimilar to receive regulatory approval for all indications of the reference product (RP).4–6 This followed the demonstration of equivalence to infliximab RP in terms of efficacy in the Phase III PLANETRA study,7 and in terms of pharmacokinetics (PK) in the Phase I PLANETAS study.8 Randomized controlled trials of CT-P13 in over 1,200 patients have provided evidence of biosimilarity and switchability consistently, while a risk management plan encompassing more than 5,000 patients has been implemented to monitor the efficacy and safety of CT-P13 data globally (Supplementary Table 2). The advent of biosimilars has facilitated substantial cost savings for health-care systems, with market competition stimulated by the reduced costs of biosimilars relative to RPs, enabling greater patient access to biologic therapy,9 although uptake has varied globally.
While biosimilars are biologics that have no clinically meaningful differences from their RP,10 advances in biologic development have led to innovative medicines that offer improvements for patients versus the RP. “VAMs” and “biobetters” (or “biosuperiors”) are terms that have been used to describe such products, although these terms are not consistently defined or used, and definitions may overlap. The first biobetter to receive FDA approval, in 1996, was a fast-acting insulin analogue, insulin lispro, produced by altering the amino acid sequence at two positions.11,12 Subsequently, multiple strategies, often involving molecular engineering, have been used in biobetter development, while varied approaches, including drug repositioning and reformulation, have been taken to develop VAMs. Innovation has recently been achieved for infliximab with the development of CT-P13 SC (Table 113–23), with changes in both the formulation and administration route relative to the IV-administered CT-P13 (CT-P13 IV). CT-P13 SC has been approved by the EMA for the treatment of RA, AS, CD, UC, PsA, and psoriasis, and is the first SC-administered infliximab to receive regulatory approval in any territory.4,24,25
Table 1.
Overview of clinical trials of subcutaneous CT-P13
| Study | Phase I CT-P13 SC 1.5 study |
Phase I CT-P13 SC 1.6 study |
Phase I/III CT-P13 SC 3.5 study |
Phase I CT-P13 SC 1.9 study |
||
|---|---|---|---|---|---|---|
| Pilot | Part 1 (dose-finding) |
Part 2 (NI test) |
Part 1 (dose-finding) |
Part 2 (NI test) |
PK | |
| Population | Healthy subjects | CD | UC, CD | RA | Healthy subjects | |
| Primary endpoint | Safety | Dose-finding (up to W30) |
PK (W22) | Dose-finding (up to W30) | Efficacy (W22) | PK (up to W12) |
| Test, reference | CT-P13 SC (PFS), CT-P13 IV | CT-P13 SC (PFS), CT-P13 IV |
CT-P13 SC (PFS), CT-P13 IV |
CT-P13 SC (PFS), CT-P13 IV |
CT-P13 SC (PFS), CT-P13 IV |
CT-P13 SC (PFS), CT-P13 SC (AI) |
| Study duration (total including follow-up) |
8 weeks (total 12 weeks) |
54 weeks (total 62 weeks) |
54 weeks (total 62 weeks) |
54 weeks (total 62 weeks) |
54 weeks (total 62 weeks) |
8 weeks (total 12 weeks) |
| Presentations or publications | UEGW 201713 | DDW 2018 (Part 1 W30);14 ECCO 2019 (Part 1 W54)15 |
UEGW 2019 (Part 2 W30);16 ECCO 2020 (Part 2 W54)17 |
EULAR 2018 (Part 1 W30);18 EULAR 2019 (Part 1 W54)19 |
EULAR 2019 (Part 2 W30);20 ACR 2019 (Part 2 W54);21 Westhovens et al.22 |
ECCO 201923 |
ACR, American College of Rheumatology; AI, autoinjector; CD, Crohn’s disease; DDW, Digestive Disease Week®; ECCO, European Crohn’s and Colitis Organisation; EULAR, European League Against Rheumatism; IV, intravenous; NI, non-inferiority; PFS, prefilled syringe; PK, pharmacokinetics; RA, rheumatoid arthritis; SC, subcutaneous; UC, ulcerative colitis; UEGW, United European Gastroenterology Week; W, Week.
Defining innovative biologics
Biosimilars are biologic medicines that have an identical primary molecular structure and comparable quality characteristics and non-clinical profile to their RP, a biologic licensed by the relevant authority.10,26 Biosimilars have no clinically meaningful differences in safety, purity, and potency from the RP, although minor differences are likely due to the complexity of biologics and differences in manufacturing processes.10,26 Regulatory pathways for biosimilars are now well established, with 59 biosimilars currently authorized via the EMA centralized procedure (as of November 24, 2020)27 and 28 biosimilars licensed by the US FDA (as of July 7, 2020).28
Due to technology advances, the development of improved versions of approved biologics, rather than biosimilars, has become possible. A biobetter is a recombinant protein drug that is in the same class as an existing biopharmaceutical and targets the same epitope but has been modified to gain specific attributes. Relevant modifications may include: a new drug delivery method or modifications to provide clinically relevant improvement(s) versus the existing biological product (such as improved efficacy, safety or tolerability, or reduced immunogenicity).29,30 For example, the type II, glycoengineered anti-CD20 monoclonal antibody obinutuzumab is a rituximab biobetter, with a different mechanism of action compared with its type I RP.31–33 Biobetters can also be easier to administer, or may have an improved dosing regimen (e.g., due to a longer serum half-life).34,35
VAMs have been defined by Medicines for Europe as “medicines based on known molecules that address healthcare needs and deliver relevant improvement for patients, healthcare professionals (HCPs) and/or payers.”36 Relevant improvements might be related to the efficacy or safety profile, administration method or ease of use, or could result from new indications or patient populations being targeted.36 Strategies of repositioning, reformulation, or combination (with other drugs, devices, or services), collectively referred to as drug repurposing, may enable a medicine to deliver added value.36 However, definitions and/or classifications of repositioning and reformulation vary, and not all changes will provide added value.37 Repositioning may involve identifying a new indication for a drug that is anatomically and/or therapeutically distinct from the original indication, or a different pharmacological target in a similar indication.37 However, “repositioning” has also been used to describe new uses in similar indications, or drugs newly launched in major (rather than emerging) pharmaceutical markets.37 Reformulation might include changes to the release profile, pharmaceutical form, or route of administration of a drug, or changes to excipients with no impact on PK parameters.37 This definition excludes changes to the chemical structure of the drug itself, or development of a new product without altering the formulation (such as a change in dose), although these scenarios have also been described as “reformulation.”37 Drug repurposing generally involves off-patent molecules38,39 but can occur for discontinued drugs or molecules that have never been commercialized.37 VAMs are beginning to emerge in the biologic arena as medicines come off patent.39
Regulatory approaches to innovative biologics
The biosimilar regulatory landscape
Biosimilar regulatory pathways are now well established, supported by guidance documents issued by regulatory authorities such as the EMA (Supplementary Figure 2). Biosimilar development programs aim to establish “biosimilarity” based on a stepwise “totality of evidence” approach (Supplementary Figure 3).10,26 Extensive, varied testing methods are used to demonstrate that physicochemical and biological attributes of the candidate biosimilar and RP are proven identical or comparable.10,40 For example, non-clinical data supporting FDA approval of CT-P13 demonstrated that the biosimilar was highly similar to its RP.41 In addition, non-clinical studies of CT-P13 demonstrated similarity in human tissue binding, off-target toxicity, and PK/toxicokinetic profiles to the European Union (EU)-sourced RP.42 Clinical trials must then establish similarity of the candidate biosimilar to the RP in terms of PK and efficacy; pharmacodynamic, immunogenicity, and safety profiles must also be comparable.10,43 FDA approval of CT-P13 followed the demonstration that CT-P13 and the EU-sourced RP were highly similar in terms of PK, immunogenicity, safety, and efficacy across studies in patients with RA and AS,7,8 as well as the three-way comparability of PK and safety between CT-P13, EU-sourced reference infliximab, and US-sourced reference infliximab.44 The requirements of biosimilar clinical studies contrast with those for new drug development, for which the main goal is to demonstrate clinical efficacy in each indication (Supplementary Figure 3). As such, data requirements for the approval of biosimilars and new biologics differ (Supplementary Figure 4). Following regulatory approval, a risk management plan and pharmacovigilance system (for the EMA) or a risk evaluation and mitigation strategy (for the FDA) may need to be established.40,45 Real-world data have been accumulating for biosimilars,46–50 supporting increased confidence in these innovative medicines and their ongoing clinical use.
During clinical development of biosimilar monoclonal antibodies, the potential impacts of anti-drug antibodies (ADAs) on PK, efficacy, and safety51,52 mean that immunogenicity assessment is crucial.10,53 However, the performance of immunogenicity assays poses a challenge for inter-study comparability,54,55 with reported proportions of infliximab ADA-positive patients varying widely (0–65.3%).56 FDA guidance notes that biosimilar efficacy studies may not be required if the results of PK/pharmacodynamic and clinical immunogenicity studies provide adequate evidence for biosimilarity.10
Local biosimilar regulatory guidance applies for each jurisdiction globally, although there may be similarities between regulators due to shared scientific perspectives and collaboration.57 Regulatory approaches can vary, such as regarding requirements for submission of in vivo data,58 or the use of local versions of the RP for biosimilarity assessment.57 For example, the EMA allows RPs for clinical trials to be produced outside EU, while the FDA requires that at least one clinical PK study is conducted using the US-licensed version of the RP.10,26 Studies evaluating a biosimilar in patients of a particular ethnicity are also required by some regulatory agencies, such as in Japan:59 to support approval of CT-P13, bridging and extension studies were required in Japanese patients (Supplementary Table 2). Robust biosimilar development plans should include strategies to ensure alignment with the approaches of different regulatory agencies to minimize any inefficiencies or delays, including taking advantage of opportunities for discussion with the agencies.60–62 However, proposals to further optimize regulatory pathways have been made, involving greater alignment between agencies and re-evaluation of biosimilar regulatory approval requirements.63 One suggestion is that perhaps Phase III comparative efficacy studies should no longer be required: the decisive factor in biosimilar regulatory decisions, to date, appears to have been high similarity in terms of analytical characterization and human PK studies, rather than the outcome of efficacy trials.63 In addition, large pre-approval clinical studies may not be needed in the future as advancing analytical technologies allow more discriminatory evidence to be collected alongside comparative PK and postmarketing monitoring.64
Design of biosimilar efficacy studies: establishing equivalence margins
Biosimilar efficacy trials must be carefully designed. Studies should be conducted in populations that allow the greatest sensitivity for the detection of potential differences between the candidate biosimilar and its RP.10,43 Equivalence, rather than non-inferiority, study designs allow demonstration that the candidate biosimilar is neither inferior nor superior to its RP.10,43 Prespecified equivalence margins must be determined, reflecting acceptable variations in efficacy with reference to existing data for the RP: this is used to determine target study population size.65,66 Typically, equivalence margins are symmetrical and two-sided; the 95% confidence interval (CI) for the difference in the relevant endpoint between biosimilar and RP should be fully contained within the equivalence margin to support the conclusion of equivalence.
There is currently no standard method to define the prespecified equivalence margin. To support CT-P13 approval, EMA guidelines were consulted67,68 and a systematic literature review (unpublished) was conducted to inform determination of the equivalence margin for the pivotal PLANETRA study of CT-P13 in patients with RA.7 Treatment differences in 20% improvement in American College of Rheumatology criteria (ACR20) response rates for patients with RA (receiving concomitant methotrexate) treated with reference infliximab versus placebo ranged from 17.6% to 37.8% (Supplementary Table 3). Among other considerations, these response rates informed the 15% equivalence margin used in the PLANETRA study.7 Subsequently, equivalence of efficacy for CT-P13 and reference infliximab was concluded: the 95% CI for the difference in ACR20 response rates at Week 30 was −6% to 10%, contained within the prespecified −15% to 15% equivalence margin.7
The regulatory landscape for VAMs and biobetters
No standardized guidance is available from major regulatory agencies regarding the approval pathways for innovative biologic drugs. Varied regulatory pathways have been taken to gain approval for repurposed drugs.69 For example, obinutuzumab received a positive opinion from the EMA through the centralized procedure with an orphan medicinal product designation for untreated chronic lymphocytic leukemia (CLL),32 while FDA approval was received via a Biologics License Application (BLA) with Breakthrough Therapy designation.70 In addition, the SC formulation of rituximab received EU marketing authorization following extension applications for the new route of administration,71 while FDA approval was via BLA.72
The EMA hybrid medicines pathway evaluates applications for “a generic medicine that is based on a reference medicine but has a different strength, a different route of administration or a slightly different indication from the reference medicine” that rely on data for both the reference and new products.73 In the US, applications under the 505(b)(2) pathway sometimes concern changes to approved drugs (for example, changes to dosage, strength, route of administration, or formulation) where the applicant relies on information from published literature or previous FDA findings.74 Until March 23, 2020, the 505(b)(2) pathway was also open to follow-on versions of biologics approved viaa New Drug Application – the regulatory process for biologic approval before the Biologics Price Competition and Innovation Act was adopted.75–77 After this date, biologics differing from approved products are unlikely to be considered under the biosimilar approval pathway by the FDA and will likely require a full BLA.75 While several recombinant protein therapeutics have been approved under the 505(b)(2) pathway in the past, some have been licensed as biosimilars in the EU,77 which could suggest that they do not offer clinically meaningful benefits for patients compared with their respective RPs.
Future regulatory pathways might vary, depending on the regulatory agency’s perspective on the biobetter and VAM concepts, and might depend on the nature of differentiation from the RP (e.g., clinical outcomes vs. patient convenience). Importantly, the clinically relevant differences between biobetters and RPs mean that indication extrapolation, as permitted for biosimilars,10,43 does not apply.30 However, we suggest that indication extrapolation might be appropriate for VAMs, depending on the nature of the benefit they provide. Regardless of the availability of regulatory guidance documents, discussions with leading regulatory agencies and strategic alignment with the requirements of each agency can facilitate appropriate development decisions and help to avoid delays.
Despite uncertainty and the lack of dedicated, expedited regulatory pathways, opportunities may remain for developers to streamline development of biobetters and VAMs, particularly as placebo-controlled trials are not warranted. Existing knowledge of the drug target can reduce research and development costs, while prior development of related drugs can assist biomarker selection, boosting efficiency.78–80 In addition, safety monitoring is likely to focus on adverse events already known to be related to the established target pathway.78 However, it is essential that sufficient, novel, head-to-head clinical data are available to support comparison between the innovative biologic and RP for determination of added value.
Innovative biologic development: CT-P13 SC case study
Non-clinical and clinical development of CT-P13 SC
Both the formulation and administration route of CT-P13 SC are altered relative to CT-P13 IV. The EMA Summary of Product Characteristics indicates that several excipients differ between CT-P13 IV and CT-P13 SC, and states that CT-P13 SC was well tolerated in rabbits at the actual concentration to be used in humans in preclinical safety evaluations.4 Reflecting the need for extensive non-clinical characterization of innovative biologics, more than 20 physicochemical and 40 functional and biological tests (unpublished) were employed during the development of CT-P13 SC.41
The clinical development program for CT-P13 SC (Table 1) used CT-P13 IV as the RP. The CT-P13 SC 1.5 dose escalation study was conducted in healthy volunteers to establish the safety of CT-P13 SC; cohorts received one of the three doses, beginning with the lowest dose, with subsequent cohorts initiated based on the absence of dose-limiting toxicities.13 Across all CT-P13 SC and CT-P13 IV cohorts, bioavailability was 60.56% (95% CI: 51.93–70.63).81 The dose-linearity in PK profiles following a single CT-P13 SC injection13 paved the way for clinical studies in patient populations. The Phase I CT-P13 SC 1.6 study was conducted in patients with inflammatory bowel disease (IBD): Part 1 was a dose-finding study, conducted in patients with CD,15 and Part 2 evaluated non-inferiority in terms of PK in patients with CD or UC.16 The Phase I/III CT-P13 SC 3.5 study, conducted in patients with RA, comprised two Parts: Part 1 was dose-finding study,19 while Part 2 established non-inferiority of CT-P13 SC versus CT-P13 IV in terms of efficacy and evaluated a single switch from CT-P13 IV to CT-P13 SC (Supplementary Figure 5 shows the study design).22 While the aforementioned studies administered CT-P13 SC via prefilled syringe, administration of CT-P13 SC via autoinjector was assessed as an alternative in the Phase I CT-P13 SC 1.9 study in healthy volunteers.23
In Part 1 of the CT-P13 SC 3.5 study, efficacy profiles were similar for CT-P13 SC and CT-P13 IV arms up to Week 54.19 In Part 2, non-inferiority between treatments was concluded for the primary efficacy endpoint, change from baseline to Week 22 in Disease Activity Score in 28 joints (DAS28)-(C-reactive protein [CRP]), since the lower limit of the 95% CI for the estimate of treatment difference (0.27 [95% CI: 0.02–0.52]) exceeded the prespecified non-inferiority margin of −0.6.22 In addition, ACR response rates were similar between treatment arms up to Week 22.22 Efficacy up to Week 54 was similar between treatment arms in terms of mean DAS28 (CRP) scores, even after the CT-P13 IV group switched to CT-P13 SC at Week 30.22 In terms of PK, mean serum concentrations consistently exceeded the threshold target–therapeutic concentration in Part 119 and Part 2.22 Drug exposure was well maintained, with trough serum concentrations higher and more constant with CT-P13 SC versus CT-P13 IV:19,22 this high consistency in drug exposure could enhance treatment options for patients.19 In addition, overall safety profiles were comparable between arms for both study parts.19,22 In Part 2, the majority of localized injection site reactions, infusion-related reactions (recorded for CT-P13 IV only), and systemic injection reactions were grade 1–2 in intensity.22
Detecting immunogenicity provides valuable information about treatment efficacy, since development of ADAs can contribute to suboptimal drug concentrations and lack or loss of response,50,56,82 although the impact of low levels of ADAs on drug retention needs to be confirmed.83 It is also important to consider that different types of ADAs can have different clinical consequences: while neutralizing ADAs prevent target binding, non-neutralizing ADAs forming immune complexes may also lead to changes in clearance, as well as immune complex–mediated hypersensitivity reactions.84 A range of methods can be used to detect ADAs, with varying sensitivities and free drug tolerances, including electrochemiluminescent (ECL) immunoassay, enzyme-linked immunosorbent assay (ELISA), radioimmunoassay, and homogeneous mobility shift assay.85 Clinical studies for CT-P13 used a high-sensitivity ECL-based affinity capture elution assay to detect ADAs, offering improved sensitivity and drug tolerance compared with the ELISA method used in clinical studies for the RP.85
Immunogenicity rates for IV- and SC-administered biologics vary on a case-by-case basis: the immunogenicity rate for the IV route was higher than that for the SC route for abatacept,86,87 while contradictory findings were observed for golimumab,88–90 rituximab,91–94 tocilizumab,95,96 trastuzumab,97,98 and vedolizumab.99–101 The potential for increased immunogenicity with SC- versus IV-administered biologics has been suggested, due to factors including proximity to antigen-presenting Langerhans cells,102,103 with particular concerns for infliximab ascribed to its chimeric nature.104 For the recently approved SC infliximab, its immunogenicity appeared lower compared with its IV formulation at Week 22 in RA patients (CT-P13 SC: 50.3%; CT-P13 IV: 61.5%).22 However, features other than administration route and biologic structure influence immunogenicity, including formulation, protein concentration, dose and administration device, patient population, genotype (as recently demonstrated for infliximab105,106), immunosuppressive effects of the drug, and concomitant medications.102,107 Administration intervals, which impact drug serum concentrations, can also influence immunogenicity.107 As well as differences between IV and SC administration, a Phase I study comparing SC and intramuscular (IM) administration of an experimental infliximab formulation identified lower immunogenicity and greater efficacy with SC versus IM administration108 – a finding considered in the development of CT-P13 SC.
The proportion of ADA-positive patients was similar between the CT-P13 SC and CT-P13 IV arms in Part 2 of the CT-P13 SC 3.5 study (51.9% and 62.1%, respectively, at Week 30, prior to patients in the CT-P13 IV arm switching to CT-P13 SC).22 As discussed, factors including the change in formulation and administration intervals might contribute to any such differences in immunogenicity. Alternatively, the higher pre-dose drug concentrations maintained with CT-P13 SC versus CT-P13 IV19,22 could lead to differences in immunogenicity. Antibody responses are impacted by the antigen dose, with both very low and high doses able to induce immune tolerance (Supplementary Figure 6). In high-zone tolerance, high concentrations of a soluble protein induce long-lasting, specific immune tolerance,109,110 while lower concentrations stimulate antibody responses, exemplified by the association between low serum infliximab levels early in treatment and subsequent detection of ADAs.50,111,112 In the case of CT-P13, one hypothesis is that the higher trough concentrations achieved following SC administration (Figure 1113) remain within the high-zone tolerance range, and thereby offset the potentially more immunogenic nature of the SC route. Another possibility is that high concentrations of unbound CT-P13, achieved through frequent SC administration, might restrict the development of ADAs otherwise elicited by target–therapeutic antibody immune complexes.109
Figure 1.
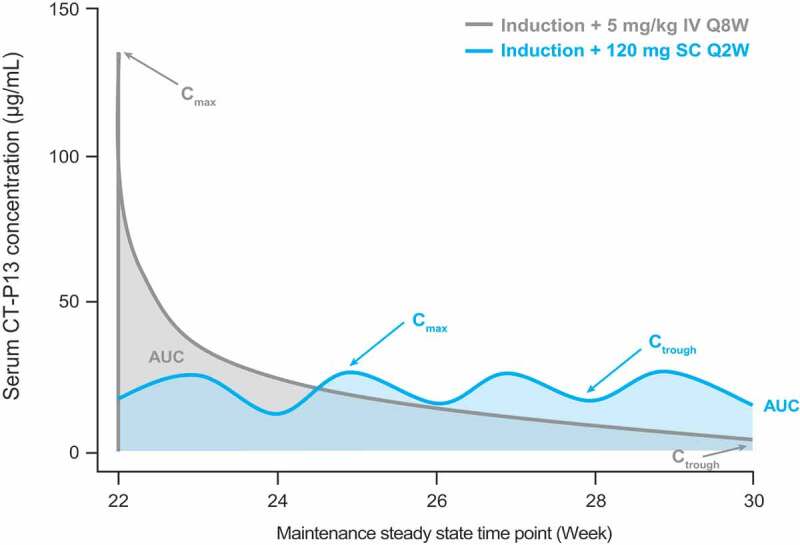
Generalized simulated serum concentration–time profile for CT-P13 following administration via IV or SC routes. Adapted with permission from Schreiber et al.113
AUC, area under the concentration–time curve; Cmax, maximum serum concentration; Ctrough, trough serum concentration; IV, intravenous; Q2W, every 2 weeks; Q8W, every 8 weeks; SC, subcutaneous.
Regulatory approval of CT-P13 SC and future plans
CT-P13 SC recently received regulatory approval in the EU, initially for the treatment of patients with RA, sought through an expanded authorization for the new formulation as a line extension.24,114 Subsequently, regulatory approval for CT-P13 SC has been extended to the adult indications approved for CT-P13 IV, encompassing IBD, AS, PsA, and psoriasis,4 following marketing authorization granted by the European Commission in July 2020.25 For regulatory approval of CT-P13 SC via an extension application,115 additional dose-finding, dose-limiting toxicity, and device studies were conducted (Table 1), following on from the assessments supporting biosimilar approval of CT-P13 IV. When FDA approval is sought for CT-P13 SC, it is likely that new drug evaluation pathways will be followed.24 In line with the conclusions of a review of regulatory perspectives toward biobetters,30 discussions with the FDA indicated that a pivotal study in each requested indication would be required for CT-P13 SC approval, rather than the biosimilar regulatory pathway being followed.24
Pharmacoeconomic considerations for innovative biologics
Adoption of biosimilars in place of RPs has enabled healthcare systems to achieve substantial cost savings, with biosimilar price discounts relative to RPs stimulating market competition.9 Prior to the introduction of CT-P13, budget impact analyses suggested that its uptake in six European countries could deliver net savings of €15.3 million or €20.8 million (depending on whether switching from the RP was permitted) over 3 years; these savings could be reinvested to finance the treatment of an additional 1,200–1,800 patients.116 In practice, the United Kingdom has saved £99,400,000 by switching to infliximab biosimilars for the treatment of patients with RA and IBD during the 2017/18 financial year (Supplementary Table 4). In addition, decreases in average European list prices have improved patient access to biologics.9 For example, following the introduction of infliximab biosimilars in Norway in 2013, the costs of infliximab therapy decreased by almost half, while the number of patients receiving infliximab biosimilars increased substantially (Supplementary Figure 7). However, the biosimilar landscape varies between countries, linked to differences in biosimilar policies and reimbursement systems.117 Differences in biosimilar discount rate relative to RPs may also influence biosimilar uptake: for example, analysis of IMS data to the second quarter of 2017 showed that discount rates exceeding 50% for CT-P13 versus the infliximab RP were linked with CT-P13 market shares approaching 100% (across all indications) in Bulgaria (from 2014) and Norway (from 2015), contrasting with discount rates and market shares both below 50% in Spain and France.
Beyond the benefits of cost savings from biosimilar uptake enabling additional patients to be treated, improved cost-effectiveness may allow biologic treatment to be extended to other patient groups or earlier in the course of disease.117 This implies that cost-effectiveness analyses conducted for RPs should be revisited following biosimilar availability.117 Analyses should use net prices to accurately assess value;118 however, limited access to pricing information, as well as variations between countries and treatment settings, can lead to biased cost-effectiveness estimates. In addition, national variations challenge extrapolation of analyses between countries;117 methodologies should be appropriate for the setting, considering the resources of the healthcare system.119 Cost savings from biosimilar uptake could also support patient access to other treatments or be reinvested in healthcare systems (such as through employing additional HCPs).49,117 Furthermore, competition between biosimilars and RPs may encourage further innovation by developers,117 leading to the development of biobetters or VAMs.
VAMs and biobetters may provide several benefits for healthcare systems (Supplementary Table 5). Irrational use of medicines – including poor adherence, off-label use, prescribing inefficiency, and medicine wastage – places a significant cost burden on healthcare systems.120,121 Medicines offering increased convenience for patients might improve adherence, while drug repositioning/reformulation and packaging optimization could limit off-label use and medicine wastage, respectively.120 Ready-to-use formulations could also reduce the medicine handling burden for HCPs.120 Alongside benefits for patients, changes in drug administration route, such as from IV to SC, might generate cost savings for healthcare systems as in-hospital treatment time and resources are reduced.122–125 The increased availability of treatment options offering benefits over RPs could impact price setting for other innovative medicines being launched for that indication, reducing their budget impact.120 Furthermore, while therapeutic advantages mean that biobetters may attract a price premium, this might be offset by other factors such as reduced dosing frequency.30
Potential budget savings could be used to provide personalized patient care, which may lead to further cost savings. Therapeutic drug monitoring, encompassing measurement of ADAs and drug PK parameters, could be used to optimize therapy for individual patients with IMIDs.126,127 Dashboards guiding personalized dosing could help this approach to deliver clinical benefits,128 as recently demonstrated in patients with IBD.129 Biomarker monitoring, such as for fecal calprotectin in patients with IBD, can also provide useful insights into disease and therapeutic status.130 The objective information gained from these techniques could be used to inform treatment decisions, ultimately improving patient care.
Delivering the benefits of innovative biologics to patients
Potential benefits of VAMs and biobetters for patients
Innovative approaches to medicine development can deliver a multitude of benefits for patients, by addressing unmet needs and offering alternatives to existing treatments. For example, the biobetter obinutuzumab demonstrated superior efficacy compared with rituximab in the treatment of CLL,131,132 with efficacy also identified in rituximab-refractory patients.133 The development of innovative medicines may also encourage investigation of their use in novel indications in which existing products are not approved. In addition, combination products could offer improved convenience and ease of medicine delivery,38 while further benefits may be achieved by replacing RPs with biobetters or VAMs in existing combination therapies.
Reformulated medicines could allow dosing to be tailored to individual patients, alongside optimizing PK and improvements in efficacy, safety, and patient convenience and experience.38,120 For CT-P13 SC, immunogenicity and PK have previously been discussed. In addition, the altered administration route for CT-P13 SC could offer patients increased flexibility and convenience: two factors contributing to patient preferences for SC- versus IV-administered anti-TNFs, alongside reduced hospital attendance and increased ease of use (Supplementary Table 5). The choice of autoinjector and prefilled syringe administration devices for CT-P13 SC could also increase treatment options for patients. In patients with UC receiving golimumab and patients with RA receiving methotrexate, patient satisfaction was high with autoinjector administration and ease of use was greater with autoinjector versus prefilled syringe administration,134,135 indicating the attractiveness of this approach. SC administration also offers the benefit of being less invasive than IV administration, although pain-free administration of larger fluid volumes and minimizing injection site reactions represent challenges of the SC route (Supplementary Table 5).
In the future, availability of both CT-P13 IV and CT-P13 SC could facilitate personalization of therapy. To date, CT-P13 SC maintenance treatment has been evaluated in the context of CT-P13 IV induction treatment. This treatment strategy is reflected in the current EU prescribing information, as CT-P13 SC treatment should be initiated as maintenance therapy following induction with CT-P13 IV.4 For ustekinumab, a pilot study recently evaluated induction therapy with the SC (rather than IV) formulation in patients with CD, establishing comparability of PK and acceptability of the approach.136
Supporting the uptake of innovative biologics through patient empowerment: dissemination of evidence and education
Dissemination of evidence and education is important to support the uptake of therapeutics developed through innovative approaches, with accumulating evidence positively influencing patient, HCP, and payer perceptions (Supplementary Figure 8). For biosimilars, patient understanding of the rationale for their development has been crucial to support successful biologic treatment initiation with, or switching to, biosimilars. Concerns surrounding the safety and efficacy of biosimilars had to be addressed,137 with real-world data helping to improve physician confidence toward biosimilar use in indications approved by extrapolation, such as IBD.138–140 In addition, nocebo effects resulting from negative patient perceptions have been observed in studies of patients with IMIDs switching from RPs to biosimilars.141 Such nocebo effects pose a risk to biosimilar uptake, treatment adherence and potential cost savings.141 However, strategies for minimizing the risk of patients with IMIDs experiencing nocebo effects have been widely discussed, including providing tailored educational initiatives for patients and HCPs (to address knowledge gaps and misconceptions).139,142 Medical society position papers and recommendations can also deliver valuable guidance and advocacy for the use of biosimilars.138,139 Furthermore, the importance of patient–HCP relationships in avoiding nocebo effects has been noted in the context of IMIDs, with positive framing, open communication, and shared decision-making often beneficial.137,139,142 Nurses may play a particularly important role in addressing concerns for patients with IBD, for example, due to their frequent interaction with patients.137,139,140 Similar educational approaches, targeting both patients and HCPs, may be important to support the introduction of VAMs and biobetters, as the concepts become established. For biosimilars, differences in regulatory requirements between countries were highlighted as a barrier to acceptance by healthcare systems, HCPs, and patients, and regulatory alignment is recommended.143 This too might be an important consideration for biobetters and VAMs.
Benefits and challenges of innovative biologics: the developers’ perspective
Developers must be cost competitive throughout the process of research, development, manufacturing, and distribution of innovative biologics, while maintaining product quality, sustainable supply, and proper pharmacovigilance systems. Balancing this investment, pricing and market access policies that support long-term market sustainability and healthy competition should be encouraged, as discussed for biosimilars.144 In the context of multiple competitors, additional innovation could enable a product to be competitive in the biologics market: this in turn might determine the future success of a biologic developer.
Targeting unmet clinical needs is fundamental to successful development of biobetters and VAMs120,145,146 – a factor shown to support price premiums in an analysis of repurposed drugs.147 Consultation with patient groups and HCPs may help to ensure that development appropriately addresses such needs.120 Once an unmet need has been identified, the size of the potential market must be quantified to ensure the viability of product development.146
Drug repositioning or reformulation could offer cost-effective and reduced-risk routes for drug development.37 Repositioned drugs may be eligible for patent protection, if sufficient evidence is available and the novelty and inventiveness of the new use can be demonstrated,148 while composition of matter patent protection may be available for medicines incorporating new formulations, delivery mechanisms, or combinations of active pharmaceutical ingredients.149 Biobetters that offer advantages over RPs and any biosimilars may be patentable, and will benefit from data and market exclusivity rights.30 Once licensed, medicines with FDA approval under the 505(b)(2) pathway could benefit from 3- or 5-year exclusivity (depending on new clinical data or chemical entities),74 while per the EMA, an additional year of data exclusivity and/or market protection might be granted for repositioned drugs (although for market protection, only if the new indication is registered within 8 years of initial approval).148 In contrast, the EMA incorporates any changes in strength, pharmaceutical form, and route of administration under the “global marketing authorization” concept, meaning that data and marketing protections are determined solely by the initial authorization.150 Regardless of patent protection for the new medicine, patents for the RP should not delay product launches for biobetters (unlike the situation for biosimilars), while increased differentiation from the RP could reduce the risk of patent litigation.30
In terms of pricing, biosimilars have been offered at discounts of up to 90% compared with RP list prices,9 while biosimilar competition has exacerbated price erosion for RPs and biosimilars.9,151,152 However, differentiation from RPs may allow biobetters to command higher prices153 and be less susceptible to price erosion than biosimilars, subject to the assessment of added value by payers. In addition, biobetters offering significant advantages over the RP could also impact the market share for biosimilars.12 In this way, companies with biologic–biosimilar pipelines may use biobetter development as a defense strategy.78 For CT-P13 SC, the SC administration route provides differentiation from all IV-administered anti-TNFs, which could result in its market impact extending beyond reference and biosimilar infliximab.
For biobetters, therapeutic advantages could potentially allow increased cost-effectiveness versus RPs or biosimilars, if these are not outweighed by increased prices. When determining pricing strategies, developers should consider the price range of existing products with the same mechanism of action, while considering the clinical profile and additional value of the innovative product, as should payers (discussed below). However, undisclosed discounts for competitor products, associated with tendering, might present a barrier to such evaluation. Meta-analysis, meta-regression, and budget impact analyses could be used to help set the price range for innovative products. Importantly, developers should consider how manufacturing and formulation choices affect drug administration costs, as this impacts cost-effectiveness.154
Developers must also be mindful of the requirements and challenges presented by health technology assessment (HTA) procedures. Pricing and reimbursement issues have been identified as key hurdles to the development of VAMs120 and explored in a white paper.155 A significant challenge is ensuring that the benefits of VAMs are recognized in HTAs, and that the level of evidence required is aligned with the level of reward from HTA bodies and payers.120 This issue is particularly relevant for VAMs benefiting patient-reported outcomes or patient preferences, in part due to HTA reliance on data from randomized controlled trials (vs. real-world data) and impact on quality-adjusted life-years, which often poorly capture these effects.120,155 Differences in HTA approach between countries adds complexity, and some perspectives used can miss benefits for society or healthcare systems as a whole.120,155 Pricing policies presenting challenges for developers also include positioning based on active substance alone, or a lack of flexibility in pricing over time (as additional evidence emerges) or between indications.38,120 For drug repositioning, a lack of pricing incentives for innovation surrounding established products means that developers are substantially more likely to pursue drug repositioning early or at a mid-stage in the product life cycle, rather than after RP patent expiration.69,148
The poorly defined regulatory landscape for biobetters and biologic VAMs, discussed previously, also poses challenges and risks for developers. Compared with abbreviated biosimilar approval pathways, development can be more time-consuming and costly. However, drug repositioning could substantially reduce clinical development time and the risk of drug failure due to safety issues.148 Another important consideration is that biobetters may not show clinical improvements versus the RP in all indications; linked to this is the lack of indication extrapolation possible for biobetters, while this is a possibility for biosimilars.30 This uncertainty should be considered during development of any biobetter candidate, as differences in benefit between indications could impact the potential market for the biobetter.
Developers have a key role in supporting stakeholders to promote confidence in the uptake of innovative biologics. Proper management of supply chains to avoid product shortages will be crucial to build trust in these medicines, with the importance of reliable supply chains previously discussed for biosimilars.156 Pharmaceutical companies must construct clinical databases to capture outcomes and long-term drug performance beyond the information required for regulatory approval, as discussed for biosimilars,157 and in line with pharmacovigilance requirements that regulatory authorities may have.40,45 In the biosimilar arena, evidence has been accumulating from real-world studies,49,158 clinical study extension phases,158 and recently, a Phase III non-inferiority study in patients with CD.159 The extension phases of the PLANETRA and PLANETAS studies reported efficacy analyses in different patient subgroups and analysis populations, and implemented alternative statistical techniques for handling missing data.160,161 Taken together, this evidence may have helped to alleviate concerns about indication extrapolation and switching from the RP in the case of CT-P13 treatment of IBD.158,162 Indeed, members of the European Crohn’s and Colitis Organisation had significantly greater confidence in biosimilars in 2015 than 2013, 2 years after CT-P13 became available in the EU.162 Shifts in the positions of medical societies have also reflected the changing evidence base.140,163 To facilitate effective communication with relevant stakeholders, developers should consider strategies such as translation into local languages or providing lay summaries of data.139
Conclusions
The introduction of biologic therapies led to a paradigm shift in the treatment of many IMIDs, with the subsequent availability of biosimilars facilitating reductions in treatment costs without compromising efficacy or safety. Ensuing cost savings provide opportunities to treat additional patients, to expand eligibility criteria (such as to patients earlier in the disease course), or to reinvest in the healthcare system (which could provide additional services for patients or increase capacity). Regulators formulated dedicated biosimilar review pathways, expediting their assessment, approval and availability for patients. In light of advancing technologies and innovative approaches, biologics with differences from the RP – offering improvements for patients, including in terms of convenience and acceptability – are being produced, which could allow developers to be competitive in a crowded biosimilars market. However, these innovative biologics face obstacles, particularly related to nomenclature, regulatory pathways, and pharmacoeconomic assessment. Terms such as “VAM” and “biobetter” have been used to describe such products, although a lack of consensus on their definitions might cause confusion regarding the differences and potential benefits that an innovative biologic may offer versus the original biologic. A lack of clear guidance and alignment between regulatory agencies in terms of requirements for the approval of VAMs or biobetters poses further challenges for developers and could hinder HCP and patient confidence in these medicines. As for biosimilars, pharmacoeconomic evaluations may need to be revisited and approaches tailored to ensure that the benefits of VAMs or biobetters for patients are recognized. Timely regulatory and healthcare policy decisions would expedite achieving the benefits of innovative medicines, including cost savings for healthcare systems, to their full potential. Thus, there is a pressing need to streamline and align regulatory decision-making processes across different regions. Ultimately, collaboration between healthcare systems, HCPs, and manufacturers is key to improving patient access to innovative medicines (Supplementary Figure 9). Long-term efficacy, safety, and immunogenicity data, including evaluation of drug retention rates, will be required to support the uptake of VAMs and biobetters, as for biosimilars.
In the case of CT-P13, an SC formulation has been developed, offering a change in administration route versus CT-P13 IV and the infliximab RP. Clinical studies have demonstrated that CT-P13 SC offers benefits in terms of PK profile compared with CT-P13 IV, with higher and more stable trough serum concentrations maintained, alongside comparable efficacy and overall safety profiles. Despite the perceived increased immunogenicity of the SC administration route, the dosing schedule evaluated to date for CT-P13 SC has elicited similar immunogenicity as CT-P13 IV treatment; high-zone tolerance provides one potential explanation for these observations. Experience with CT-P13 SC evaluation by the EMA and FDA has highlighted differences in their approaches to assessing this type of innovative biologic. The FDA has perceived CT-P13 SC as a new drug, requiring pivotal clinical trials in each indication, while the EMA required a “hybrid” dataset including clinical studies additional to those needed for the biosimilar approval pathway.
The novel formulation and change in administration route of CT-P13 SC may provide potential benefits for patients in terms of PK profile and convenience versus CT-P13 IV. Given the lack of consensus surrounding the nomenclature for innovative biologics, these features mean that it could be appropriate to describe CT-P13 SC as either a biologic VAM or a biobetter. Ultimately, regardless of the terminology used, innovative biologics like CT-P13 SC should be able to deliver additional benefits for patients compared with RPs, within the resource constraints of healthcare systems.
Supplementary Material
Acknowledgments
Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) was provided by Beatrice Tyrrell, DPhil at Aspire Scientific Limited (Bollington, UK) and funded by Celltrion Healthcare Co., Ltd (Incheon, Republic of Korea). Sang-Wook Yoon, PhD and Dong-Hyeon Kim, PhD from the Medical Communications team at Celltrion Healthcare Co., Ltd, provided suggestions for the initial concept of the article, and reviewed the article for scientific and medical accuracy. The authors had full editorial control of the article and provided final approval of all content.
Funding Statement
Medical writing support was funded by Celltrion Healthcare Co., Ltd (Incheon, Republic of Korea).
Disclosure of interest
HoUng Kim is employed by and holds stock options in Celltrion Healthcare Co., Ltd. Rieke Alten reports speaker fees and honoraria from Celltrion. Fraser Cummings reports honoraria for advisory boards/lecturing or has received research support from AbbVie, Amgen, AstraZeneca, Celltrion, Gilead, GSK, Hospira/Pfizer, Janssen, MSD, and Takeda. Silvio Danese reports consultancy fees from AbbVie, Allergan, Amgen, AstraZeneca, Athos Therapeutics, Biogen, Boehringer Ingelheim, Celgene, Celltrion, Eli Lilly, Enthera, Ferring Pharmaceuticals Inc., Gilead, Hospira, Inotrem, Janssen, Johnson & Johnson, MSD, Mundipharma, Mylan, Pfizer, Roche, Sandoz, Sublimity Therapeutics, Takeda, TiGenix, UCB Inc., and Vifor, and lecture fees from AbbVie, Amgen, Ferring Pharmaceuticals Inc., Gilead, Janssen, Mylan, Pfizer, and Takeda. Geert D’Haens has served as advisor for AbbVie, Ablynx, Active Biotech AB, Agomab Therapeutics, Allergan, Alphabiomics, Amakem, Amgen, AM Pharma, Applied Molecular Therapeutics, Arena Pharmaceuticals, AstraZeneca, Avaxia, Biogen, Bristol Myers Squibb/Celgene, Boehringer Ingelheim, Celltrion, Cosmo, DSM Pharma, Echo Pharmaceuticals, Eli Lilly, Engene, Exeliom Biosciences, Ferring, DrFALK Pharma, Galapagos, Genentech/Roche, Gilead, GlaxoSmithKline, Gossamerbio, Pfizer, Immunic, Johnson & Johnson, Kintai Therapeutics, Lycera, Medimetrics, Takeda, Medtronic, Mitsubishi Pharma, Merck Sharp & Dohme, Mundipharma, Nextbiotics, Novo Nordisk, Otsuka, Photopill, ProciseDx, Prodigest, Prometheus Laboratories/Nestlé, Progenity, Protagonist, RedHill, Robarts Clinical Trials, Salix, Samsung Bioepis, Sandoz, Seres/Nestec/Nestlé, Setpoint, Shire, Teva, Tigenix, Tillotts, Topivert, Versant, and Vifor; received speaker fees from AbbVie, Biogen, Ferring, Galapagos/Gilead, Johnson & Johnson, Merck Sharp & Dohme, Mundipharma, Norgine, Pfizer, Samsung Bioepis, Shire, Millennium/Takeda, Tillotts, and Vifor. Paul Emery has provided expert advice to AbbVie, Amgen, Bristol Myers Squibb, Celltrion, Gilead, Lilly, MSD, Novartis, Pfizer, Roche, Samsung, and Sanofi, and received grants from AbbVie and Bristol Myers Squibb. Subrata Ghosh has received consultancies from AbbVie, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Celltrion, Galapagos, Gilead, Janssen, Pfizer, Roche, and Takeda, speaker honoraria from AbbVie, Celltrion, Ferring Pharmaceuticals, Janssen, Pfizer, and Takeda, and research grants from AbbVie. Cyrielle Gilletta de Saint Joseph reports personal fees from AbbVie, Janssen, Pfizer, and Takeda. James O. Lindsay has served as consultant and an advisory board participant for AbbVie, Allergan (Warner Chilcott), Atlantic Healthcare, Bristol Myers Squibb, Celgene, Celltrion, Ferring Pharmaceuticals, Gilead, GSK, Janssen, Lilly, MSD, Napp, Pfizer, Shire, Takeda, and Vifor Pharma, has received speaker fees and sponsorship for academic meetings from AbbVie, Allergan (Warner Chilcott), Ferring Pharmaceuticals, Janssen, MSD, Napp, Norgine, Pfizer, Shire, Tillotts Pharma, and Takeda, and has received investigator-led research grants from AbbVie, Gilead, Pfizer, Shire, and Takeda. Elena Nikiphorou has received speaker honoraria and/or participated in advisory boards for AbbVie, Celltrion, Gilead, Lilly, Pfizer, and Sanofi. Ben Parker reports fees and honoraria from AbbVie, Bristol Myers Squibb, Celltrion, Fresenius Kabi, GSK, Lilly, Roche-Chugai, and UCB, and grants from GSK. Stefan Schreiber reports personal fees (consulting in advisory boards) from AbbVie, Arena, BMS, Biogen, Celltrion, Celgene, Falk, Fresenius, Gilead, IMAB, Janssen, MSD, Mylan, Pfizer, Protagonist, Provention Bio, Takeda, and Theravance. Steven Simoens is one of the founders of the KU Leuven Fund on Market Analysis of Biologics and Biosimilars following Loss of Exclusivity (MABEL), was involved in a stakeholder roundtable on biologics and biosimilars sponsored by Amgen, MSD, and Pfizer, has participated in advisory board meetings for Amgen and Pfizer, has contributed to studies on biologics and biosimilars for Celltrion, Hospira, Mundipharma, and Pfizer, had speaking engagements for Amgen, Celltrion and Sandoz, and is a member of the leadership team of the ISPOR Special Interest Group on Biosimilars. Rene Westhovens has participated in advisory boards and speakers bureau for Celltrion and Galapagos/Gilead. Laurent Peyrin-Biroulet reports personal fees from AbbVie, Allergan, Alma, Amgen, Applied Molecular Transport, Arena, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Celltrion, Enterome, Enthera, Ferring Pharmaceuticals, Fresenius Kabi, Genentech, Gilead, Hikma, Index Pharmaceuticals, Janssen, Lilly, MSD, Mylan, Nestlé, Norgine, Oppilan Pharma, OSE Immunotherapeutics, Pfizer, Pharmacosmos, Roche, Samsung Bioepis, Sandoz, Sterna, Sublimity Therapeutics, Takeda, Theravance, Tillotts, and Vifor, and grants from AbbVie, MSD, and Takeda. JongHyuk Lee and Ji Hoon Jeong report no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Abbreviations
- ACR
American College of Rheumatology
- ACR20
20% improvement in American College of Rheumatology criteria
- ADA
anti-drug antibody
- AI
autoinjector
- AS
ankylosing spondylitis
- AUC
area under the concentration–time curve
- BLA
Biologics License Application
- CD
Crohn’s disease
- CI
confidence interval
- CLL
chronic lymphocytic leukemia
- Cmax
maximum serum concentration
- CRP
C-reactive protein
- Ctrough
trough serum concentration
- DAS28
Disease Activity Score in 28 joints
- DDW
Digestive Disease Week®
- ECCO
European Crohn’s and Colitis Organisation
- ECL
electrochemiluminescent
- ELISA
enzyme-linked immunosorbent assay
- EMA
European Medicines Agency
- EU
European Union
- EULAR
European League Against Rheumatism
- FDA
Food and Drug Administration
- HCP
healthcare professional
- HTA
health technology assessment
- IBD
inflammatory bowel disease
- IM
intramuscular
- IMID
immune-mediated inflammatory disease
- IV
intravenous
- NI
non-inferiority
- PFS
prefilled syringe
- PK
pharmacokinetic(s)
- Q2W
every 2 weeks
- Q8W
every 8 weeks
- PsA
psoriatic arthritis
- RA
rheumatoid arthritis
- RP
reference product
- SC
subcutaneous
- TNF
tumor necrosis factor
- UC
ulcerative colitis
- UEGW
United European Gastroenterology Week
- US
United States
- VAM
value-added medicine
- W
Week
References
- 1.Melsheimer R, Geldhof A, Apaolaza I, Schaible T.. Remicade® (infliximab): 20 years of contributions to science and medicine. Biologics. 2019;13:139–16. doi: 10.2147/BTT.S207246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.European Medicines Agency . Remicade summary of product characteristics. 2019. [accessed 2019 October31]. https://www.ema.europa.eu/en/documents/product-information/remicade-epar-product-information_en.pdf
- 3.US Food and Drug Administration . Remicade prescribing information. 2018. [accessed 2019 October31]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/103772s5385lbl.pdf.
- 4.European Medicines Agency . Remsima summary of product characteristics. 2020. [accessed 2020 November26]. https://www.ema.europa.eu/en/documents/product-information/remsima-epar-product-information_en.pdf.
- 5.European Medicines Agency . Inflectra summary of product characteristics. 2019. [accessed 2019 October31]. https://www.ema.europa.eu/en/documents/product-information/inflectra-epar-product-information_en.pdf.
- 6.US Food and Drug Administration . Inflectra prescribing information. 2019. [accessed 2019 October31]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125544s009lbl.pdf.
- 7.Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, Kovalenko V, Prodanovic N, Abello-Banfi M, Gutierrez-Urena S, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park W, Hrycaj P, Jeka S, Kovalenko V, Lysenko G, Miranda P, Mikazane H, Gutierrez-Urena S, Lim M, Lee YA, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–12. doi: 10.1136/annrheumdis-2012-203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.IQVIA . The impact of biosimilar competition in Europe. 2018. [accessed 2019 May20]. https://ec.europa.eu/docsroom/documents/31642.
- 10.US Food and Drug Administration . Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. [accessed 2019 October28]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product.
- 11.US Food and Drug Administration . Humalog prescribing information. 2019. [accessed 2019 September24]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/020563s195,205747s021lbl.pdf.
- 12.Kesik‐Brodacka M. Progress in biopharmaceutical development. Biotechnology and Applied Biochemistry. 2018;65(3):306–22. doi: 10.1002/bab.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westhovens R, Yoo DH, Schreiber S, Reinisch W, Ben-Horin S, Ye BD, Kim JW, Lee SJ, Jung YJ, Suh JH, et al. Subcutaneous administration of a novel formulation of CT-P13 (infliximab biosimilar) is safe and achieves projected therapeutic drug levels: a Phase I study in healthy subjects. United European Gastroenterol J. 2017;5:A297. [Google Scholar]
- 14.Schreiber S, Jang BI, Borzan V, Lahat A, Pukitis A, Osipenko M, Mostovoy Y, Ben-Horin S, Ye BD, Lee SJ, et al. Tu2018 - Novel formulation of CT-P13 (infliximab biosimilar) for subcutaneous administration: initial results from a phase I open-label randomized controlled trial in patients with active Crohn’s disease. Gastroenterology. 2018;154(6):S–1371. doi: 10.1016/S0016-5085(18)34477-9. [DOI] [Google Scholar]
- 15.Reinisch W, Jang BI, Borzan V, Lahat A, Pukitis A, Osipenko M, Mostovoy Y, Schreiber S, Ben-Horin S, Lee SJ, et al. DOP62 A novel formulation of CT-P13 (infliximab biosimilar) for subcutaneous administration: 1-year result from a phase I open-label randomised controlled trial in patients with active Crohn’s disease. J Crohns Colitis. 2019;13(Supplement_1):S066–S067. doi: 10.1093/ecco-jcc/jjy222.096. [DOI] [Google Scholar]
- 16.Schreiber S, Leszczyszyn J, Dudkowiak R, Lahat A, Gawdis-Wojnarska B, Pukitis A, Horynski M, Farkas K, Kierkus J, Kowalski M, et al. Noninferiority of novel subcutaneous infliximab (CT-P13) to intravenous infliximab (CT-P13) in patients with active Crohn’s disease and ulcerative colitis: week 30 results from a multicentre, randomised controlled pivotal trial. United European Gastroenterol J. 2019;7(Suppl):1412 [LB02]. [Google Scholar]
- 17.Ben-Horin S, Leszczyszyn J, Dudkowiak R, Lahat A, Gawdis-Wojnarska B, Pukitis A, Horynski M, Farkas K, Kierkus J, Kowalski M, et al. OP24 A novel subcutaneous infliximab (CT-P13): 1-year results including switching results from intravenous infliximab (CT-P13) in patients with active Crohn’s disease and ulcerative colitis. J Crohns Colitis. 2020;14(Supplement_1):S021–S022. doi: 10.1093/ecco-jcc/jjz203.023. [DOI] [Google Scholar]
- 18.Westhovens R, Yoo DH, Jaworski J, Matyska-Piekarska E, Smiyan S, Ivanova D, Zielinska A, Raussi E-K, Batalov A, Lee SJ, et al. THU0191 Novel formulation of CT-P13 for subcutaneous administration in patients with rheumatoid arthritis: initial results from a phase I/III randomised controlled trial. Ann Rheum Dis. 2018;77:315.28476879 [Google Scholar]
- 19.Yoo D, Jaworski J, Matyska-Piekarska E, Smiyan S, Ivanova D, Zielinska A, Raussi E-K, Batalov A, Lee S, Suh J, et al. FRI0128 A novel formulation of CT-P13 (infliximab biosimilar) for subcutaneous administration: 1-year results from a part 1 of phase I/III randomized controlled trial in patients with active rheumatoid arthritis. Ann Rheum Dis. 2019;78:733. [Google Scholar]
- 20.Westhovens R, Wiland P, Zawadzki M, Ivanova D, Berrocal A, Chalouhi E, Balázs É, Shevchuk S, Eliseeva L, Stanislavchuk M, et al. SAT0170 A novel formulation of CT-P13 for subcutaneous administration: 30 week results from a part 2 of phase I/III randomized controlled trial in patients with rheumatoid arthritis. Ann Rheum Dis. 2019;78:1158–59. [Google Scholar]
- 21.Westhovens R, Wiland P, Zawadzki M, Ivanova D, Berrocal A, Chalouhi E, Balázs É, Shevchuk S, Eliseeva L, Stanislavchuk M, et al. Efficacy and safety of a novel subcutaneous formulation of CT P13 over the 1 year treatment period and after switching from intravenous CT P13 in patients with active rheumatoid arthritis: results from a part 2 of phase I/III randomized controlled trial. Arthritis Rheum. 2019;71(Suppl 10):932 [Abstract 548]. [Google Scholar]
- 22.Westhovens R, Wiland P, Zawadzki M, Ivanova D, Kasay AB, El-Khouri EC, Balázs É, Shevchuk S, Eliseeva L, Stanislavchuk M, et al. Efficacy, pharmacokinetics and safety of subcutaneous versus intravenous CT-P13 in rheumatoid arthritis: a randomized phase I/III trial. Rheumatology. 2020. November 23 [Epub ahead of print]. doi: 10.1093/rheumatology/keaa580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber S, Ben-Horin S, Ye BD, Westhovens R, Yoo DH, Lee SJ, Suh JH, Byeon JH, Reinisch W. P679 Development of a novel auto-injector of subcutaneous CT-P13 infliximab: phase I randomised, open-label, single-dose trial to compare the pharmacokinetics and safety to pre-filled syringe in healthy subjects. J Crohns Colitis. 2019;13(Supplement_1):S458–S459. doi: 10.1093/ecco-jcc/jjy222.803. [DOI] [Google Scholar]
- 24.Davio K. Subcutaneous formulation of celltrion’s biosimilar infliximab authorized by European Commission. 2019. [accessed 2020 November25]. https://www.centerforbiosimilars.com/news/subcutaneous-formulation-of-celltrions-biosimilar-infliximab-authorized-by-european-commission.
- 25.Biosimilar Development . European Commission grants marketing authorisation for world’s first subcutaneous formulation of infliximab, Remsima SC, for an additional five indications including for use in inflammatory bowel disease and ankylosing spondylitis. 2020. [accessed 2020 November27]. https://www.biosimilardevelopment.com/doc/european-commission-grants-marketing-authorisation-for-world-s-first-subcutaneous-0001.
- 26.European Medicines Agency . Guideline on similar biological medicinal products. 2014. [accessed 2020 May22]. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf.
- 27.European Medicines Agency . Medicines: biosimilar. 2020. [accessed 2020 November24]. https://www.ema.europa.eu/en/medicines/search_api_aggregation_ema_medicine_types/field_ema_med_biosimilar.
- 28.US Food and Drug Administration . Biosimilar product information. 2020. [accessed 2020 November24]. https://www.fda.gov/drugs/biosimilars/biosimilar-product-information.
- 29.Kumar S, Chawla S, Dutta S. Biobetters: betting on the future. J Rational Pharmacother Res. 2018;4:13–21. [Google Scholar]
- 30.Sandeep V, Parveen J, Chauhan P. Biobetters: the better biologics and their regulatory overview. Int J Drug Regul Aff. 2016;4:13–20. [Google Scholar]
- 31.Roche . Gazyva/Gazyvaro (obinutuzumab; GA101). 2020. [accessed 2019 September23]. https://www.roche.com/products/product-details.htm?productId=e37d39a7-da10-4347-991e-4d15d843a34b.
- 32.European Medicines Agency . CHMP assessment report: Gazyvaro. 2014. [accessed 2019 October29]. https://www.ema.europa.eu/en/documents/assessment-report/gazyvaro-epar-public-assessment-report_en.pdf.
- 33.Davio K. Will biobetters and biosimilars compete for market share? 2017. [accessed 2020 June18]. https://www.centerforbiosimilars.com/news/will-biobetters-and-biosimilars-compete-for-market-share.
- 34.Elgundi Z, Reslan M, Cruz E, Sifniotis V, Kayser V. The state-of-play and future of antibody therapeutics. Adv Drug Deliv Rev. 2017;122:2–19. doi: 10.1016/j.addr.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Beck A. Biosimilar, biobetter and next generation therapeutic antibodies. MAbs. 2011;3(2):107–10. doi: 10.4161/mabs.3.2.14785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medicines for Europe . What is a value added medicine? [accessed 2020January9]. https://www.medicinesforeurope.com/value-added-medicines/did-you-know/.
- 37.Murteira S, Ghezaiel Z, Karray S, Lamure M. Drug reformulations and repositioning in pharmaceutical industry and its impact on market access: reassessment of nomenclature. J Mark Access Health Policy. 2013;1:1. https://www.tandfonline.com/doi/full/10.3402/jmahp.v1i0.21131]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.IQVIA . Case studies for value added medicines. 2018. [accessed 2020 January9]. https://www.medicinesforeurope.com/wp-content/uploads/2019/04/IQVIA-MFE_Case-Studies-for-VAMs_Final-Word-Document_vUpdate2019-v3.0.pdf.
- 39.Arias A. A digital future for value added medicines. 2019. [accessed 2020 January9]. https://www.medicinesforeurope.com/wp-content/uploads/2019/09/20190924_IQVIAa-digital-future-for-value-added-medicines.pdf.
- 40.European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1). 2014. [accessed 2020 May22]. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-0.pdf.
- 41.Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Bae JS, Kim D-I, Lee SY, Chang SJ. Physicochemical characterization of remsima. MAbs. 2014;6:1163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Celltrion . CT-P13 (infliximab biosimilar): briefing document for the arthritis advisory committee. 2016. [accessed 2020 June29]. https://www.fda.gov/media/95998/download.
- 43.European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014. [accessed 2019 October28]. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf.
- 44.Park W, Lee SJ, Yun J, Yoo DH. Comparison of the pharmacokinetics and safety of three formulations of infliximab (CT-P13, EU-approved reference infliximab and the US-licensed reference infliximab) in healthy subjects: a randomized, double-blind, three-arm, parallel-group, single-dose, Phase I study. Expert Rev Clin Immunol. 2015;11(Suppl 1):S25–S31. doi: 10.1586/1744666X.2015.1090311. [DOI] [PubMed] [Google Scholar]
- 45.US Food and Drug Administration . Risk Evaluation and Mitigation Strategies (REMS). 2018. [accessed 2019 May15]. https://www.fda.gov/drugs/drug-safety-and-availability/risk-evaluation-and-mitigation-strategies-rems.
- 46.Glintborg B, Sorensen J, Hetland ML. Does a mandatory non-medical switch from originator to biosimilar infliximab lead to increased use of outpatient healthcare resources? A register-based study in patients with inflammatory arthritis. RMD Open. 2018;4(2):e000710. doi: 10.1136/rmdopen-2018-000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glintborg B, Sørensen IJ, Loft AG, Lindegaard H, Linauskas A, Hendricks O, Hansen IMJ, Jensen DV, Manilo N, Espesen J, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76(8):1426. doi: 10.1136/annrheumdis-2016-210742. [DOI] [PubMed] [Google Scholar]
- 48.Glintborg B, Loft AG, Omerovic E, Hendricks O, Linauskas A, Espesen J, Danebod K, Jensen DV, Nordin H, Dalgaard EB, et al. To switch or not to switch: results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Ann Rheum Dis. 2019;78(2):192. doi: 10.1136/annrheumdis-2018-213474. [DOI] [PubMed] [Google Scholar]
- 49.Razanskaite V, Bettey M, Downey L, Wright J, Callaghan J, Rush M, Whiteoak S, Ker S, Perry K, Underhill C, et al. Biosimilar infliximab in inflammatory bowel disease: outcomes of a managed switching programme. J Crohns Colitis. 2017;11(6):690–96. doi: 10.1093/ecco-jcc/jjw216. [DOI] [PubMed] [Google Scholar]
- 50.Kennedy NA, Heap GA, Green HD, Hamilton B, Bewshea C, Walker GJ, Thomas A, Nice R, Perry MH, Bouri S, et al. Predictors of anti-TNF treatment failure in anti-TNF-naive patients with active luminal Crohn’s disease: a prospective, multicentre, cohort study. Lancet Gastroenterol. 2019;4(5):341–53. doi: 10.1016/S2468-1253(19)30012-3. [DOI] [PubMed] [Google Scholar]
- 51.Doevendans E, Schellekens H. Immunogenicity of innovative and biosimilar monoclonal antibodies. Antibodies. 2019;8(1):21. doi: 10.3390/antib8010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jani M, Dixon WG, Chinoy H. Drug safety and immunogenicity of tumour necrosis factor inhibitors: the story so far. Rheumatology (Oxford). 2018;57:1896–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.European Medicines Agency . Guideline on similar biological medicinal products containing monoclonal antibodies – non-clinical and clinical issues. 2012. [accessed 2018 July24]. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf.
- 54.Schreitmüller T, Barton B, Zharkov A, Bakalos G. Comparative immunogenicity assessment of biosimilars. Future Oncol. 2019;15(3):319–29. doi: 10.2217/fon-2018-0553. [DOI] [PubMed] [Google Scholar]
- 55.Gorovits B, Baltrukonis DJ, Bhattacharya I, Birchler MA, Finco D, Sikkema D, Vincent MS, Lula S, Marshall L, Hickling TP. Immunoassay methods used in clinical studies for the detection of anti-drug antibodies to adalimumab and infliximab. Clin Exp Immunol. 2018;192:348–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vermeire S, Gils A, Accossato P, Lula S, Marren A. Immunogenicity of biologics in inflammatory bowel disease. Ther Adv Gastroenter. 2018;11:1756283x17750355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Webster CJ, Woollett GR. A ‘global reference’ comparator for biosimilar development. BioDrugs. 2017;31(4):279–86. doi: 10.1007/s40259-017-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chapman K, Adjei A, Baldrick P, da Silva A, De Smet K, DiCicco R, Hong SS, Jones D, Leach MW, McBlane J, et al. Waiving in vivo studies for monoclonal antibody biosimilar development: national and global challenges. MAbs. 2016;8(3):427–35. doi: 10.1080/19420862.2016.1145331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arato T. Japanese regulation of biosimilar products: past experience and current challenges. Br J Clin Pharmacol. 2016;82(1):30–40. doi: 10.1111/bcp.12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.European Medicines Agency . Tailored scientific advice to support step-by-step development of new biosimilars. 2016. [accessed 2019 July8]. https://www.ema.europa.eu/en/news/tailored-scientific-advice-support-step-step-development-new-biosimilars.
- 61.European Medicines Agency . Tailored scientific advice to support step-by-step development of new biosimilars. 2018. [accessed 2019 June18]. https://www.ema.europa.eu/en/documents/other/tailored-scientific-advice-support-step-step-development-new-biosimilars_en.pdf.
- 62.US Food and Drug Administration . Formal meetings between the FDA and sponsors or applicants of BsUFA products: guidance for industry (draft guidance). 2018. [accessed 2019 June18]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/formal-meetings-between-fda-and-sponsors-or-applicants-bsufa-products-guidance-industry.
- 63.Vulto AG. Delivering on the promise of biosimilars. BioDrugs. 2019;33(6):599–602. doi: 10.1007/s40259-019-00388-9. [DOI] [PubMed] [Google Scholar]
- 64.Schiestl M, Zabransky M, Sörgel F. Ten years of biosimilars in Europe: development and evolution of the regulatory pathways. Drug Des Devel Ther. 2017;11:1509–15. doi: 10.2147/DDDT.S130318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.US Food and Drug Administration . Guidance for industry: E9 statistical principles for clinical trials. 1998. [accessed 2019 July25]. https://www.fda.gov/media/71336/download.
- 66.European Medicines Agency . Note for guidance on statistical principles for clinical trials. 1998. [accessed 2019 July25]. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-9-statistical-principles-clinical-trials-step-5_en.pdf.
- 67.European Medicines Agency . Guideline on the choice of the non-inferiority margin. 2005. [accessed 2020 June2]. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-choice-non-inferiority-margin_en.pdf.
- 68.European Agency for the Evaluation of Medicinal Products . Points to consider on switching between superiority and non-inferiority. 2000. [accessed 2020 June2]. https://www.ema.europa.eu/en/documents/scientific-guideline/points-consider-switching-between-superiority-non-inferiority_en.pdf.
- 69.Murteira S, Millier A, Ghezaiel Z, Lamure M. Drug reformulations and repositioning in the pharmaceutical industry and their impact on market access: regulatory implications. J Mark Access Health Policy. 2014;2(1):22813. https://www.tandfonline.com/doi/full/10.3402/jmahp.v2.22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.US Food and Drug Administration . Summary review for regulatory action: Gazyva. 2013. [accessed 2019 October29]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125486Orig1s000SumR.pdf.
- 71.European Medicines Agency . Assessment report: Mabthera. 2014. [accessed 2019 October29]. https://www.ema.europa.eu/en/documents/variation-report/mabthera-h-c-165-x-83-epar-assessment-report-extension_en.pdf.
- 72.US Food and Drug Administration . BLA approval letter: Rituxan Hycela. 2017. [accessed 2019 October29]. https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2017/761064Orig1s000ltr.pdf.
- 73.European Medicines Agency . Generic and hybrid medicines. [accessed 2020. January 9]. https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/generic-hybrid-medicines.
- 74.US Food and Drug Administration . Guidance for industry: applications covered by Section 505(b)(2). 1999. [accessed 2020 January10]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/applications-covered-section-505b2.
- 75.US Food and Drug Administration . Interpretation of the “Deemed to be a License” provision of the biologics price competition and innovation act of 2009: guidance for industry. 2018. [accessed 2020 January10]. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/interpretation-deemed-be-license-provision-biologics-price-competition-and-innovation-act-2009.
- 76.Barlas S. Opening the door to follow-on proteins? Biotechnol Healthc. 2006;3:47–54. [PMC free article] [PubMed] [Google Scholar]
- 77.Mott RM. “Biosimilars” under the 505(b)(2) pathway. 2015. [accessed 2020 January10]. https://www.biologicsblog.com/biosimilars-under-505-b-2-pathway-2.
- 78.Anour R. Biosimilars versus ‘biobetters’—a regulator’s perspective. GaBI J. 2014;3(4):166–67. doi: 10.5639/gabij.2014.0304.039. [DOI] [Google Scholar]
- 79.Burchiel SW, Aspbury R, Munday J. The search for biosimilars and biobetters. Drug Discov Today. 2019;24(5):1087–91. doi: 10.1016/j.drudis.2019.03.016. [DOI] [PubMed] [Google Scholar]
- 80.Zhao X, Modur V, Carayannopoulos LN, Laterza OF. Biomarkers in pharmaceutical research. Clin Chem. 2015;61(11):1343–53. doi: 10.1373/clinchem.2014.231712. [DOI] [PubMed] [Google Scholar]
- 81.Westhovens R, Yoo DH, Schreiber S, Reinisch W, Ben-Horin S, Ye BD, Kim JW, Lee SJ, Jung YJ, Suh JH, et al. Subcutaneous administration of a novel formulation of CT-P13 (infliximab biosimilar) is safe and achieves projected therapeutic drug levels: a Phase I study in healthy subjects. Poster presented at: the 25th UEG Week; 2017. Oct 28–Nov 1, Barcelona, Spain. [Google Scholar]
- 82.Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol. 2017;13(12):707. doi: 10.1038/nrrheum.2017.187. [DOI] [PubMed] [Google Scholar]
- 83.Papamichael K, Vajravelu RK, Osterman MT, Cheifetz AS. Long-term outcome of infliximab optimization for overcoming immunogenicity in patients with inflammatory bowel disease. Dig Dis Sci. 2018;63(3):761–67. doi: 10.1007/s10620-018-4917-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Krishna M, Nadler SG. Immunogenicity to biotherapeutics - the role of anti-drug immune complexes. Front Immunol. 2016;7:21. doi: 10.3389/fimmu.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim JS, Kim SH, Kwon B, Hong S. Comparison of immunogenicity test methods used in clinical studies of infliximab and its biosimilar (CT-P13). Expert Rev Clin Immunol. 2015;11(Suppl 1):33–41. doi: 10.1586/1744666X.2015.1090312. [DOI] [PubMed] [Google Scholar]
- 86.Genovese MC, Covarrubias A, Leon G, Mysler E, Keiserman M, Valente R, Nash P, Simon-Campos JA, Porawska W, Box J, et al. Subcutaneous abatacept versus intravenous abatacept: a phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum. 2011;63(10):2854–64. doi: 10.1002/art.30463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.US Food and Drug Administration . Orencia prescribing information. 2019. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125118s224lbl.pdf.
- 88.US Food and Drug Administration . Simponi Aria prescribing information. 2019. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125433s028lbl.pdf.
- 89.US Food and Drug Administration . Simponi prescribing information. 2019. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125289s146lbl.pdf.
- 90.European Medicines Agency . Simponi summary of product characteristics. 2020. [accessed 2020 June3]. https://www.ema.europa.eu/en/documents/product-information/simponi-epar-product-information_en.pdf.
- 91.US Food and Drug Administration . Rituxan Hycela prescribing information. 2020. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761064s008s010lbl.pdf.
- 92.US Food and Drug Administration . Rituxan prescribing information. 2020. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/103705Orig1s5458lbl.pdf.
- 93.Emery P, Fleischmann R, Filipowicz-Sosnowska A, Schechtman J, Szczepanski L, Kavanaugh A, Racewicz AJ, van Vollenhoven RF, Li NF, Agarwal S, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54(5):1390–400. doi: 10.1002/art.21778. [DOI] [PubMed] [Google Scholar]
- 94.Assouline S, Buccheri V, Delmer A, Gaidano G, McIntyre C, Brewster M, Catalani O, Hourcade-Potelleret F, Sayyed P, Badoux X. Pharmacokinetics and safety of subcutaneous rituximab plus fludarabine and cyclophosphamide for patients with chronic lymphocytic leukaemia. Br J Clin Pharmacol. 2015;80(5):1001–09. doi: 10.1111/bcp.12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burmester GR, Choy E, Kivitz A, Ogata A, Bao M, Nomura A, Lacey S, Pei J, Reiss W, Pethoe-Schramm A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1078–85. doi: 10.1136/annrheumdis-2016-210297. [DOI] [PubMed] [Google Scholar]
- 96.US Food and Drug Administration . Actemra prescribing information. 2020. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125276s129,125472s042lbl.pdf.
- 97.US Food and Drug Administration . Herceptin Hylecta prescribing information. 2019. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761106Orig1s000lbl.pdf.
- 98.US Food and Drug Administration . Herceptin prescribing information. 2018. [accessed 2020 June3]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/103792s5345lbl.pdf.
- 99.Sandborn WJ, Baert F, Danese S, Krznarić Ž, Kobayashi T, Yao X, Chen J, Rosario M, Bhatia S, Kisfalvi K, et al. Efficacy and safety of vedolizumab subcutaneous formulation in a randomized trial of patients with ulcerative colitis. Gastroenterology. 2020;158(3):562–572.e512. doi: 10.1053/j.gastro.2019.08.027. [DOI] [PubMed] [Google Scholar]
- 100.Australian Government Therapeutic Goods Administration . Australian public assessment report for vedolizumab (rch). 2014. [accessed 2020 June3]. https://www.tga.gov.au/sites/default/files/auspar-vedolizumab-141117-cer.pdf.
- 101.European Medicines Agency . Entyvio summary of product characteristics. 2020. [accessed 2020 June3]. https://www.ema.europa.eu/en/documents/product-information/entyvio-epar-product-information_en.pdf.
- 102.Hamuro L, Kijanka G, Kinderman F, Kropshofer H, Bu DX, Zepeda M, Jawa V. Perspectives on subcutaneous route of administration as an immunogenicity risk factor for therapeutic proteins. J Pharm Sci. 2017;106(10):2946–54. doi: 10.1016/j.xphs.2017.05.030. [DOI] [PubMed] [Google Scholar]
- 103.Viola M, Sequeira J, Seica R, Veiga F, Serra J, Santos AC, Ribeiro AJ. Subcutaneous delivery of monoclonal antibodies: how do we get there? J Control Release. 2018;286:301–14. doi: 10.1016/j.jconrel.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 104.Baert F, De Vos M, Louis E, Vermeire S. Immunogenicity of infliximab: how to handle the problem? Acta Gastroenterol Belg. 2007;70:163–70. [PubMed] [Google Scholar]
- 105.Sazonovs A, Kennedy NA, Moutsianas L, Heap GA, Rice DL, Reppell M, Bewshea CM, Chanchlani N, Walker GJ, Perry MH, et al. HLA-DQA1*05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with Crohn’s disease. Gastroenterology. 2020;158(1):189–99. doi: 10.1053/j.gastro.2019.09.041. [DOI] [PubMed] [Google Scholar]
- 106.Powell Doherty RD, Liao H, Satsangi JJ, Ternette N. Extended analysis identifies drug-specific association of 2 distinct HLA class II haplotypes for development of immunogenicity to adalimumab and infliximab. Gastroenterology. 2020;159(2):784–87. doi: 10.1053/j.gastro.2020.03.073. [DOI] [PubMed] [Google Scholar]
- 107.Strand V, Balsa A, Al-Saleh J, Barile-Fabris L, Horiuchi T, Takeuchi T, Lula S, Hawes C, Kola B, Marshall L. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. BioDrugs. 2017;31:299–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Westhovens R, Houssiau F, Joly J, Everitt DE, Zhu Y, Sisco D, Van Hartingsveldt B, Mascelli MA, Graham MA, Durez P, et al. A phase I study assessing the safety, clinical response, and pharmacokinetics of an experimental infliximab formulation for subcutaneous or intramuscular administration in patients with rheumatoid arthritis. J Rheumatol. 2006;33(5):847–53. [PubMed] [Google Scholar]
- 109.Chaigne B, Watier H. Monoclonal antibodies in excess: A simple way to avoid immunogenicity in patients? J Allergy Clin Immunol. 2015;136(3):814–16. doi: 10.1016/j.jaci.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 110.Somerfield J, Hill-Cawthorne GA, Lin A, Zandi MS, McCarthy C, Jones JL, Willcox M, Shaw D, Thompson SAJ, Compston AS, et al. A novel strategy to reduce the immunogenicity of biological therapies. J Immunol. 2010;185(1):763. doi: 10.4049/jimmunol.1000422. [DOI] [PubMed] [Google Scholar]
- 111.Bendtzen K, Geborek P, Svenson M, Larsson L, Kapetanovic MC, Saxne T. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 2006;54(12):3782–89. doi: 10.1002/art.22214. [DOI] [PubMed] [Google Scholar]
- 112.Vermeire S, Noman M, Van Assche G, Baert F, D’Haens G, Rutgeerts P. Effectiveness of concomitant immunosuppressive therapy in suppressing the formation of antibodies to infliximab in Crohn’s disease. Gut. 2007;56(9):1226–31. doi: 10.1136/gut.2006.099978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schreiber S, Jang BI, Borzan V, Lahat A, Pukitis A, Osipenko M, Mostovoy Y, Ben-Horin S, Ye BD, Lee SJ, et al. Tu2018 - Novel formulation of CT-P13 (infliximab biosimilar) for subcutaneous administration: initial results from a phase I open-label randomized controlled trial in patients with active Crohn’s disease. Poster presented at: Digestive Disease Week, 2018. June 2-5, Washington, DC. [Google Scholar]
- 114.Davio K. Looking to the future, biosimilar pioneer Celltrion is banking on biobetters. 2019. [accessed 2020 January9]. https://www.centerforbiosimilars.com/conferences/ft-conference/looking-to-the-future-biosimilar-pioneer-celltrion-is-banking-on-biobetters.
- 115.European Commission . Guideline on the categorisation of extension applications (EA) versus variations applications (V). 2019. [accessed 2020 July27]. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-2/c/v2c_ea_v__10_2003_en.pdf.
- 116.Brodszky V, Baji P, Balogh O, Pentek M. Budget impact analysis of biosimilar infliximab (CT-P13) for the treatment of rheumatoid arthritis in six central and eastern European countries. Eur J Health Econ. 2014;15(Suppl1):S65–S71. doi: 10.1007/s10198-014-0595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dutta B, Huys I, Vulto AG, Simoens S. Identifying key benefits in European off-patent biologics and biosimilar markets: it is not only about price! BioDrugs. 2020;34(2):159–70. doi: 10.1007/s40259-019-00395-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moorkens E, Broux H, Huys I, Vulto AG, Simoens S. Economic evaluation of biosimilars for reimbursement purposes - what, when, how? J Mark Access Health Policy. 2020;8(1):1739509. doi: 10.1080/20016689.2020.1739509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Inotai A, Csanádi M, Vitezic D, Francetic I, Tesar T, Bochenek T, Lorenzovici L, Dylst P, Kaló Z. Policy practices to maximise social benefit from biosimilars. J Bioequivalence Bioavailab. 2017;9:467–72. [Google Scholar]
- 120.Toumi M, Rémuzat C. Value added medicines: what value repurposed medicines might bring to society? J Mark Access Health Policy. 2017;5(1):1264717. doi: 10.1080/20016689.2017.1264717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cutler RL, Fernandez-Llimos F, Frommer M, Benrimoj C, Garcia-Cardenas V. Economic impact of medication non-adherence by disease groups: a systematic review. BMJ Open. 2018;8(1):e016982. doi: 10.1136/bmjopen-2017-016982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Anderson KC, Landgren O, Arend RC, Chou J, Jacobs IA. Humanistic and economic impact of subcutaneous versus intravenous administration of oncology biologics. Future Oncol. 2019;15(28):3267–81. doi: 10.2217/fon-2019-0368. [DOI] [PubMed] [Google Scholar]
- 123.Hedayati E, Fracheboud L, Srikant V, Greber D, Wallberg S, Linder Stragliotto C. Economic benefits of subcutaneous trastuzumab administration: A single institutional study from Karolinska University Hospital in Sweden. PLoS One. 2019;14(2):e0211783. doi: 10.1371/journal.pone.0211783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tjalma WAA, Van den Mooter T, Mertens T, Bastiaens V, Huizing MT, Papadimitriou K. Subcutaneous trastuzumab (Herceptin) versus intravenous trastuzumab for the treatment of patients with HER2-positive breast cancer: A time, motion and cost assessment study in a lean operating day care oncology unit. Eur J Obstet Gynecol Reprod Biol. 2018;221:46–51. doi: 10.1016/j.ejogrb.2017.12.006. [DOI] [PubMed] [Google Scholar]
- 125.Lopez-Vivanco G, Salvador J, Diez R, López D, De Salas-Cansado M, Navarro B, De la Haba-Rodríguez J. Cost minimization analysis of treatment with intravenous or subcutaneous trastuzumab in patients with HER2-positive breast cancer in Spain. Clin Transl Oncol. 2017;19(12):1454–61. doi: 10.1007/s12094-017-1684-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mitrev N, Vande Casteele N, Seow CH, Andrews JM, Connor SJ, Moore GT, Barclay M, Begun J, Bryant R, Chan W, et al. Review article: consensus statements on therapeutic drug monitoring of anti-tumour necrosis factor therapy in inflammatory bowel diseases. Aliment Pharmacol Ther. 2017;46(11–12):1037–53. doi: 10.1111/apt.14368. [DOI] [PubMed] [Google Scholar]
- 127.Papamichael K, Juncadella A, Wong D, Rakowsky S, Sattler LA, Campbell JP, Vaughn BP, Cheifetz AS. Proactive therapeutic drug monitoring of adalimumab is associated with better long-term outcomes compared to standard of care in patients with inflammatory bowel disease. J Crohns Colitis. 2019;13(8):976–81. doi: 10.1093/ecco-jcc/jjz018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Strik AS, Berends SE, Löwenberg M. Therapeutic drug monitoring-based dosing of TNF inhibitors in inflammatory bowel disease: the way forward? Exp Rev Clin Pharmacol. 2019;12(9):885–91. doi: 10.1080/17512433.2019.1642745. [DOI] [PubMed] [Google Scholar]
- 129.Strik A, Berends S, Mould D, Mathôt R, Ponsioen C, van den Brande J, Jansen J, Hoekman D, Brandse J, Löwenberg M, et al. DOP56 Dashboard driven vs. conventional dosing of infliximab in inflammatory bowel disease patients: the PRECISION trial. J Crohns Colitis. 2019;13(Supplement_1):S063. doi: 10.1093/ecco-jcc/jjy222.090. [DOI] [Google Scholar]
- 130.Reenaers C, Bossuyt P, Hindryckx P, Vanpoucke H, Cremer A, Baert F. Expert opinion for use of faecal calprotectin in diagnosis and monitoring of inflammatory bowel disease in daily clinical practice. United European Gastroenterol J. 2018;6(8):1117–25. doi: 10.1177/2050640618784046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, Chagorova T, de la Serna J, Dilhuydy M-S, Illmer T, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med. 2014;370(12):1101–10. doi: 10.1056/NEJMoa1313984. [DOI] [PubMed] [Google Scholar]
- 132.Goede V, Fischer K, Engelke A, Schlag R, Lepretre S, Montero LFC, Montillo M, Fegan C, Asikanius E, Humphrey K, et al. Obinutuzumab as frontline treatment of chronic lymphocytic leukemia: updated results of the CLL11 study. Leukemia. 2015;29(7):1602. doi: 10.1038/leu.2015.14. [DOI] [PubMed] [Google Scholar]
- 133.Sehn LH, Chua N, Mayer J, Dueck G, Trneny M, Bouabdallah K, Fowler N, Delwail V, Press O, Salles G, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081–93. doi: 10.1016/S1470-2045(16)30097-3. [DOI] [PubMed] [Google Scholar]
- 134.Vermeire S, D’Heygere F, Nakad A, Franchimont D, Fontaine F, Louis E, Van Hootegem P, Dewit O, Lambrecht G, Strubbe B, et al. Preference for a prefilled syringe or an auto-injection device for delivering golimumab in patients with moderate-to-severe ulcerative colitis: a randomized crossover study. Patient Prefer Adherence. 2018;12:1193–202. doi: 10.2147/PPA.S154181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Saraux A, Hudry C, Zinovieva E, Herman-Demars H. Use of auto-injector for methotrexate subcutaneous self-injections: high satisfaction level and good compliance in SELF-I study, a randomized, open-label, parallel group study. Rheumatol Ther. 2019;6(1):47–60. doi: 10.1007/s40744-018-0134-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rowan CR, Keegan D, Byrne K, Cullen G, Mulcahy HE, Sheridan J, Ryan EJ, de Vries A, D’Haens G, Doherty GA. Subcutaneous rather than intravenous ustekinumab induction is associated with comparable circulating drug levels and early clinical response: a pilot study. Aliment Pharm Ther. 2018;48(3):333–39. doi: 10.1111/apt.14834. [DOI] [PubMed] [Google Scholar]
- 137.Gecse KB, Cumming F, D’Haens G. Biosimilars for inflammatory bowel disease: how can healthcare professionals help address patients’ concerns? Expert Rev Gastroenterol Hepatol. 2019;13(2):143–55. doi: 10.1080/17474124.2019.1553617. [DOI] [PubMed] [Google Scholar]
- 138.Baumgart DC, Misery L, Naeyaert S, Taylor PC. Biological therapies in immune-mediated inflammatory diseases: can biosimilars reduce access inequities? Front Pharmacol. 2019;10:279. doi: 10.3389/fphar.2019.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Edwards CJ, Hercogová J, Albrand H, Amiot A. Switching to biosimilars: current perspectives in immune-mediated inflammatory diseases. Expert Opin Biol Ther. 2019;19(10):1001–14. doi: 10.1080/14712598.2019.1610381. [DOI] [PubMed] [Google Scholar]
- 140.Danese S, Fiorino G, Raine T, Ferrante M, Kemp K, Kierkus J, Lakatos PL, Mantzaris G, van der Woude J, Panes J, et al. ECCO position statement on the use of biosimilars for inflammatory bowel disease-an update. J Crohns Colitis. 2017;11(1):26–34. doi: 10.1093/ecco-jcc/jjw198. [DOI] [PubMed] [Google Scholar]
- 141.Kristensen LE, Alten R, Puig L, Philipp S, Kvien TK, Mangues MA, van den Hoogen F, Pavelka K, Vulto AG. Non-pharmacological effects in switching medication: the nocebo effect in switching from originator to biosimilar agent. BioDrugs. 2018;32(5):397–404. doi: 10.1007/s40259-018-0306-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pouillon L, Danese S, Hart A, Fiorino G, Argollo M, Selmi C, Carlo-Stella C, Loeuille D, Costanzo A, Lopez A, et al. Consensus report: clinical recommendations for the prevention and management of the nocebo effect in biosimilar-treated IBD patients. Aliment Pharmacol Ther. 2019;49(9):1181–87. doi: 10.1111/apt.15223. [DOI] [PubMed] [Google Scholar]
- 143.Cazap E, Jacobs I, McBride A, Popovian R, Sikora K. Global acceptance of biosimilars: importance of regulatory consistency, education, and trust. Oncologist. 2018;23(10):1188–98. doi: 10.1634/theoncologist.2017-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Simon Kucher & Partners . Payers’ price & market access policies supporting a sustainable biosimilar medicines market. 2016. [accessed 2019 June19]. https://www.medicinesforeurope.com/wp-content/uploads/2016/09/Simon-Kucher-2016-Policy-requirements-for-a-sustainable-biosimilar-market-FINAL-report_for-publication2.pdf.
- 145.Wright J. Are biobetters better? 2017. [accessed 2019 November14]. https://social.eyeforpharma.com/column/are-biobetters-better.
- 146.Financier Worldwide . Competitive strategies in life sciences: biobetters versus biosimilars. 2011. [accessed 2019 November14]. https://www.financierworldwide.com/competitive-strategies-in-life-sciences-biobetters-versus-biosimilars#.Xc1crHd2s2w.
- 147.Murteira S, Millier A, Toumi M. Drug repurposing in pharmaceutical industry and its impact on market access: market access implications. J Mark Access Health Policy. 2014:2(1):22814. doi: 10.3402/jmahp.v2.22814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nayroles G, Frybourg S, Gabriel S, Kornfeld Å, Antoñanzas-Villar F, Espín J, Jommi C, Martini N, de Pouvourville G, Tolley K, et al. Unlocking the potential of established products: toward new incentives rewarding innovation in Europe. J Mark Access Health Policy. 2017;5(1):1298190. doi: 10.1080/20016689.2017.1298190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Smith RB. Repositioned drugs: integrating intellectual property and regulatory strategies. Drug Discov Today. 2011;8:131–37. [Google Scholar]
- 150.European Medicines Agency . European Medicines Agency pre-authorisation procedural advice for users of the centralised procedure. 2018. [accessed 2020 February21]. https://www.emwa.org/media/2923/european-medicines-agency-pre-authorisation-procedural-advice-users-centralised-procedure_en-0-1.pdf.
- 151.Farfan-Portet M-I, Gerkens S, Lepage-Nefkens I, Vinck I, Hulstaert F. Are biosimilars the next tool to guarantee cost-containment for pharmaceutical expenditures? Eur J Health Econ. 2014;15(3):223–28. doi: 10.1007/s10198-013-0538-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.San-Juan-Rodriguez A, Gellad WF, Good CB, Hernandez I. Trends in list prices, net prices, and discounts for originator biologics facing biosimilar competition. JAMA Netw Open. 2019;2(12):e1917379. doi: 10.1001/jamanetworkopen.2019.17379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Barei F, Le Pen C, Simoens S. From generic to biosimilar drugs: why take an innovative pace? Farmeconomia Health Economics and Therapeutic Pathways. 2012;13(Suppl 3):21–27. doi: 10.7175/fe.v13i3S.328. [DOI] [Google Scholar]
- 154.Tetteh EK, Morris S. Evaluating the administration costs of biologic drugs: development of a cost algorithm. Health Econ Rev. 2014;4(1):26. doi: 10.1186/s13561-014-0026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Toumi M, Rémuzat C, Borissov B, Dankó D, Espin J, Antoñanzas F, Władysiuk M, Pavlovic M, Persson U, Ruggeri M.. Value added medicines: time to adjust the HTA decision frameworks. 2017. [accessed 2020 January9]. http://prescriptia.com/wp-content/uploads/2017/09/White-paper_HTAonValueaddedmedicines.pdf.
- 156.Li E, Subramanian J, Anderson S, Thomas D, McKinley J, Jacobs IA. Development of biosimilars in an era of oncologic drug shortages. Drug Des Devel Ther. 2015;9:3247–55. doi: 10.2147/DDDT.S75219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kim WS, Ogura M, Kwon H-C, Choi D. Looking to the future and learning lessons from the recent past: changing stakeholder perceptions of biosimilars in cancer. Future Oncol. 2017;13(15s):17–29. doi: 10.2217/fon-2017-0154. [DOI] [PubMed] [Google Scholar]
- 158.Braun J, Kudrin A. Switching to biosimilar infliximab (CT-P13): evidence of clinical safety, effectiveness and impact on public health. Biologicals. 2016;44(4):257–66. doi: 10.1016/j.biologicals.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 159.Ye BD, Pesegova M, Alexeeva O, Osipenko M, Lahat A, Dorofeyev A, Fishman S, Levchenko O, Cheon JH, Scribano ML, et al. Efficacy and safety of biosimilar CT-P13 compared with originator infliximab in patients with active Crohn’s disease: an international, randomised, double-blind, phase 3 non-inferiority study. Lancet. 2019;393(10182):1699–707. doi: 10.1016/S0140-6736(18)32196-2. [DOI] [PubMed] [Google Scholar]
- 160.Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Urena S, Mikazane H, Lee YA, Smiyan S, Lim MJ, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76(2):346–54. doi: 10.1136/annrheumdis-2015-208783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, Baranauskaite A, Wiland P, Abud-Mendoza C, Oparanov B, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76(2):355–63. doi: 10.1136/annrheumdis-2015-208786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Danese S, Fiorino G, Michetti P. Changes in biosimilar knowledge among European Crohn’s Colitis Organization [ECCO] members: an updated survey. J Crohns Colitis. 2016;10(11):1362–65. doi: 10.1093/ecco-jcc/jjw090. [DOI] [PubMed] [Google Scholar]
- 163.Lamb CA, Kennedy NA, Raine T, Hendy PA, Smith PJ, Limdi JK, Hayee BH, Lomer MCE, Parkes GC, Selinger C, et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut. 2019;68:s1–s106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.