

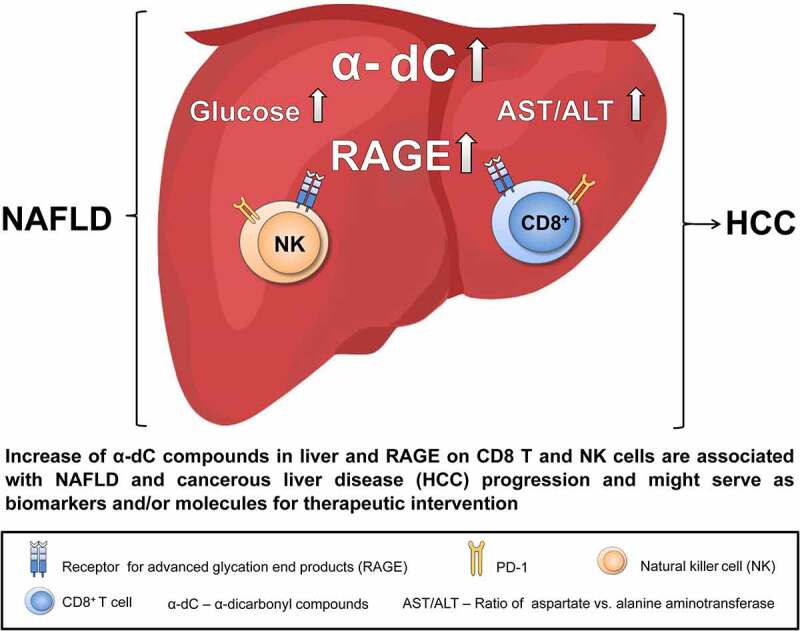
ABSTRACT
Hepatocellular carcinoma (HCC) is the most common primary malignancy of the liver with a very poor prognosis and constantly growing incidence. Among other primary risks of HCC, metabolic disorders and obesity have been extensively investigated over recent decades. The latter can promote nonalcoholic fatty liver disease (NAFLD) leading to the inflammatory form of nonalcoholic steatohepatitis (NASH), that, in turn, promotes HCC. Molecular determinants of this pathogenic progression, however, remain largely undefined. In this study, we have focussed on the investigation of α-dicarbonyl compounds (α-dC), highly reactive and tightly associated with overweight-induced metabolic disorders, and studied their potential role in NAFLD and progression toward HCC using murine models. NAFLD was induced using high-fat diet (HFD). Autochthonous HCC was induced using transposon-based stable intrahepatic overexpression of oncogenic NRASG12V in mice lacking p19Arf tumor suppressor. Our study demonstrates that the HFD regimen and HCC resulted in strong upregulation of α-dC in the liver, heart, and muscles. In addition, an increase in α-dC was confirmed in sera of NAFLD and NASH patients. Furthermore, higher expression of the receptor for advanced glycation products (RAGE) was detected exclusively on immune cells and not on stroma cells in livers of mice with liver cancer progression. Our work confirms astable interplay of liver inflammation, carbonyl stress mediated by α-dC, and upregulated RAGE expression on CD8+ Tand natural killer (NK) cells in situ in NAFLD and HCC, as key factors/determinants in liver disease progression. The obtained findings underline the role of α-dC and RAGE+CD8+ Tand RAGE+ NK cells as biomarkers and candidates for a local therapeutic intervention in NAFLD and malignant liver disease.
KEYWORDS: α-dicarbonyl compounds, nonalcoholic fatty liver disease, receptor for advanced glycation end products, precancerous liver disease, hepatocellular carcinoma
Graphical abstract
Introduction
Hepatocellular carcinoma (HCC) is the most prevalent form of liver cancer with a growing incidence and takes the fourth leading place among cancer-related deaths worldwide.1,2
The major causes of HCC development remain chronic liver diseases, cirrhosis due to infections with hepatitis B virus (HBV) and/or hepatitis C virus (HCV), consumption of aflatoxin-contaminated food, alcohol, nonalcoholic fatty liver disease (NAFLD) and steatohepatitis (NASH), diabetes, and obesity.2 In recent years, especially NAFLD became one of the leading etiologies for HCC. The initial inflammatory stage in NAFLD is defined as NASH.3 NASH that is characterized by inflammation, hepatocellular damage, and fibrosis was shown to increase the risk of HCC with high rates of mortality.4
In general, HCC belongs to inflammation-linked cancer, with more than 90% of the HCC cases arising in the context of hepatic injury and inflammation.5 Chronic inflammation, characterized by infiltration of (immature) myeloid cells, monocytes, macrophages, dysregulated production of cyto-/chemokines, are considered as one of the main triggers of HCC carcinogenesis and tumor progression.5–7 The inflammatory cells along with premalignant hepatocytes produce a vast array of cyto-/chemokines, prostaglandins, growth factors, proangiogenic factors further promoting the premalignant environment toward hepatocytes malignant transformation.5,6 In addition, the above-mentioned factors further support the survival of malignant liver cells through activation of anti-apoptotic pathways and suppression of immune surveillance.5,6
It is well documented that in premalignant livers, chronic activation of inflammatory signaling pathways is tightly associated with oxidative/nitrosative stress and generation of reactive oxygen and nitrogen species (ROS and RNS, respectively), 8,9 while carbonyl stress, induced by reactive carbonyl species (RCS), and in particular, α-dicarbonyls (α-dC) has received less attention. Endogenous RCS including acyclic dicarbonyls, methylglyoxal (MGO), and glyoxal are mostly derived from carbohydrate10 (Supplementary Fig. 1A) and lipid (Supplementary Figure 1B) oxidation11 as well as side products of glycolysis (Supplementary Figure 1C). The glyoxalase system (Glo1 and Glo2) plays a key role in the metabolism of α-dC compounds (Supplementary Fig. 1D), keeping α-dC metabolites at a nontoxic baseline. However, when adaptive limits of the α-dC detoxification system are exceeded, a variety of advanced glycation end products (AGEs) are generated.12 AGEs are poorly degraded, and therefore, an increase of their steady-state level in the body may result in activation of pro-inflammatory and pro-fibrotic pathways.12 RCS- and AGE-mediated pro-inflammatory events are resolved via activation of the specific receptor for AGEs, so called, RAGE.11,13–15 A direct link and correlation between α-dC – AGE/AGE-proteins and the activation of RAGE are supported by several literature overviews, 16,17 as introduced in the oval section of Supplementary Fig. 1. RAGE-dependent signaling cascades are considered to contribute to the progression of many chronic diseases.14,18 Upon ligand binding, RAGE activation stimulates downstream signaling pathways leading to the activation of transcription factors such as NF-κB, AP-1, CREB, STAT3, or NFAT, thereby controlling cellular processes, influencing cell viability by regulating autophagy and apoptosis (Supplementary Fig. 1E).18 Activation of RAGE has been reported to induce the expression of different NF-κB-regulated genes which encode pro-inflammatory cyto-/chemokines like TNF-α, and adhesion molecules.19–21 Moreover, activated RAGE was found in a wide range of cells like cardiomyocytes, hepatocytes, vascular endothelial cells, renal cells, etc.,22,23 and its expression has been reported in numerous immune cells including neutrophils, monocytes/macrophages, lymphocytes, and dendritic cells (DC).11,19–21,23–26 Taguchi et al. has revealed that the interaction of RAGE with its ligands and the resultant signaling plays a causative role in tumor (glioma) progression and is connected with increased metastases.27 Thus, RAGE expression may be expected to play a significant role in the development of HCC.28,29 The detailed link, however, between RAGE-mediated signaling in precancerous and cancerous livers as well as the main experimental evidence demonstrating the relevant cell types overexpressing RAGE are still missing.
Therefore, in the present study, we joined forces of biochemists, hepatologists, pathologists, and tumor immunologists and aimed to characterize the role of RCS/AGEs/RAGE signaling in precancerous and cancerous liver disease (including NAFLD, oncogene-induced precancerous disease, and HCC), as established in our previous studies.30–32 We investigated biochemical parameters of liver inflammation and α-dC levels in plasma and liver tissue. Furthermore, we explored overexpression of RAGE on liver-infiltrating innate and adaptive immune cells as well as on liver stroma cells. Our data provide evidence for a correlation between the increase of liver α-dC compounds and RAGE overexpression on several liver-infiltrating immune cells and the progression of NAFLD and HCC.
Materials and Methods
Work with Human Material
The prospective and retrospective studies using human material (human sera, and liver tissues obtained after surgeries) were conducted in accordance with the Helsinki Declaration and was approved by the Ethics Committee of the Hannover Medical School (ethical codes: 8742_BO_K_2019 and 3205). Appropriate informed consent was received from all the patients.
Animals and Experimental Conditions
Three to six weeks-old C57BL/6J female mice were purchased from Charles River Laboratories (Europe). Four weeks-old p19Arf-/- males and females were obtained in a C57BL/6J background as described previously30,32 and bred at the animal facility of the Hannover Medical School, Hannover, Germany.
For inducing NAFLD, three-weeks-old C57BL/6J female mice were exposed to a 60% (high) fat diet (HFD, Envigo Teklad TD06414) for 14 weeks long, as established.33–35 The HFD contained 18.3% protein, 21.4% carbohydrate, and 60.3% calories from lard (36% saturated, 41% monounsaturated, 23%-polyunsaturated fats), i.e., 5.1 kcal/g. The control group received a normal chow diet (NCD) composed of 10% fat.
The model of transposon-mediated stable intrahepatic transfer of oncogenic NRASG12V was used to induce oncogene-induced (OIS) precancerous liver disease (PLD) and HCC development, as established.30,32 Briefly, DNA vectors (transposon and transposase) were prepared using the QIAGEN EndoFree Maxi Kit (QIAGEN, Hilden, Germany). Transposon and transposase vectors were mixed in a 5:1 molar ratio and hydrodynamic tail vein injection (HDI) was performed as described previously.30,32 All experiments and procedures were performed in compliance with ethical regulations and the approval of the Lower Saxonian State Office for Consumer Protection and Food Safety (LAVES, Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit; AZ 18/2808, 15/1766, 13/1342).
Tissue Collection and Homogenization
To determine biochemical parameters, mice were anesthetized by isoflurane (Piramal Critical Care, Bethlehem, Germany), blood was collected from retro-orbital plexus, animals were euthanized using cervical dislocation, and different organs were isolated. To determine the level of α-dC compounds and the activity of Glo1, snap-frozen tissues (liver, heart, kidney, muscles) were homogenized using a T10 basic ULTRA-TURRAX homogenizer in a 1:10 ratio (mg tissue/μl of buffer) at 4°C in the medium containing 50 mM potassium phosphate buffer (PPB, pH 7.0), 0.5 mM ethylenediaminetetraacetic acid (EDTA), and 1 mM phenylmethylsulfonyl fluoride (PMSF) (final concentrations). To determine the content of lipid peroxides (LOOH), tissue samples were homogenized in 96% ice-cold ethanol at a 1:10 ratio (mg tissue/μl of ethanol). After homogenization samples were centrifuged (13,200 g, 15 min, 4°C) using the Eppendorf 5417 R centrifuge (Hamburg, Germany). Supernatants were collected and kept on ice.
Determination of metabolites in plasma
Measurement of glucose and L-lactate in plasma were performed on a cobas® 8000 modular automated clinical analyzer system with enzymatic in vitro tests using the hexokinase method (GLUC3; Roche Diagnostics, Mannheim, Germany) and the lactate oxidase/peroxidase method (LACT2; Roche Diagnostics, Mannheim, Germany), respectively.36 Plasma concentrations of total cholesterol were measured using the CHOD-PAP kit (CHOL2; Roche Diagnostics, Mannheim, Germany) as established.37
Determination of enzyme activities
The enzyme activities (U/l) of alanine aminotransferase (ALT), aspartate aminotransferase (AST), lactate dehydrogenase (LDH), and cholinesterase (ChE) were measured on a cobas® 8000 modular automatic analyzer system using standard methods (Roche Diagnostics, Mannheim, Germany): ALTPM, ASTPM, LDHI2, and ChE2 as established.38 AST, ALT, LDH activity is expressed in U/l, ChE activity is expressed in kU/l.
The activity of Glo1 in tissues was measured spectrophotometrically with a Libra S12 UV/Visible Spectrophotometer (Biochrom Ltd., Germany) by detecting the formation of S-D-lactoylglutathione at 240 nm as established.39 The S-D-lactoylglutathione extinction coefficient of 3.1 mM−1 cm−1 was used for calculations. The activity of Glo1 is expressed as U/mg protein. The BCA protein assay (Thermo Fisher Scientific, Germany) was used for the quantitation of total protein per sample.
Measurement of LOOH and α-dC compounds
The parameters were measured spectrophotometrically with a Libra S12 UV/Visible Spectrophotometer (Biochrom Ltd., Germany). The level of LOOH was measured by the ferrous-xylenol orange (FOX) method at 580 nm by using cumene hydroperoxide as a positive standard.40 The levels of LOOH are given as nanomoles of cumene hydroperoxide equivalents per gram of wet mass.
Detection of α-dC compounds was performed based on their interaction with the Girard-T reagent in 30 mM sodium tetraborate buffer (pH 9.2). The optical density of the complex formed was determined at 325 nm using the extinction coefficient of 18.8 mM−1 cm−1 for Girard’s hydrazone derivative of glyoxal as established.39,41 The results are presented as nanomoles of glyoxal equivalents per gram of wet mass.
Flow cytometry (FACS) staining and analysis
Single-cell suspensions from the liver and blood were prepared as previously described.30,32,42 Isolated single-cell suspensions were blocked with anti-CD16/32 (clone 93, Biolegend, California, San Diego, USA). Cell suspensions were further stained with the following antibodies: anti-CD68 (clone FA-11), anti-CD3 (clone 17A2), anti-PD-1 (clone 29F.1A12), anti-CD4 (clone GK1.5), anti-CD11c (clone N418), anti-CD25 (clone PC61), anti-CD44 (clone IM7), anti-NK1.1 (PK136), anti-Ly6G (clone 1A8), anti-CD146 (clone ME-9F1), and anti-IFN-ɣ (clone XMG1.2) all obtained from the Biolegend. Anti-CD8 (clone 53–6.7) and anti-CD54 (clone 3E2) were obtained from the BD Bioscience. Anti-RAGE (clone 697023) was obtained from the R&D Systems. All antibodies are listed in Supplementary Table 1A (see Supplementary Data file).
Acquisition of FACS samples was performed using an LSRII FACS analyzer (BD, Franklin Lakes, NJ, USA). The obtained data were analyzed using FlowJo software (Tree Star, Becton, Dickinson & Company, USA).
Immunofluorescent Stainings of Frozen Liver Tissue Sections
Frozen liver tissue sections were fixed in ice-cold acetone for 10 min and permeabilized (0.1% Triton™ X-100, 0.1% sodium citrate) for 2 min at 4°C. Sections were blocked with PBTB buffer (phosphate-buffered saline with 0.2% Triton-100 (PBT), 0.2% bovine serum albumin (BSA), 5% normal goat serum) for 30 min at room temperature and incubated with a mix comprising primary antibodies overnight at 4°C. Murine liver sections were stained with the mix of primary antibodies: anti-RAGE (1:100; ab228861, Abcam, UK) and anti-EOMES (1:200; 14–4875-82, eBioscience, USA). The secondary, fluorescent-conjugated antibodies: Alexa Fluor 647-conjugated goat anti-rabbit and Alexa Fluor 568-conjugated goat anti-rat (both used at 1:500 dilution, Invitrogen, USA) and a primary FITC-conjugated anti-mouse CD8a (1:100; 100705, Biolegend, USA) antibody were applied in the second mix for 2 h at room temperature. All antibodies used for the immunofluorescent staining are listed in Supplementary Table 1B (see Supplementary Data file). Thereafter, sections were counterstained with 4ʹ,6-diamidino-2-phenylindole (DAPI, 0.1 μg/ml, Sigma–Aldrich, USA) in PBS for 10 min at room temperature. Finally, samples were mounted using 70% glycerol. Fluorescent images were obtained using Zeiss LSM 700 confocal laser-scanning microscope (40x objective). Images were analyzed using ZEN 2011 and ImageJ software.
Statistical data analysis
All studied biochemical and immunological indices were tested for normality and homogeneity of variances using the Kolmogorov-Smirnov and Levine test, respectively. Not normally distributed data were subjected to Box-Cox transformation. The effects of both PLDs and HCC on the studied parameters were estimated using one-way ANOVA.
Normalized, Box-Cox transformed data were subjected to the principal component analysis (PCA) to identify the patterns of responses of the biochemical and immunological parameters studied in NAFLD. Discriminant function analysis was used to determine which variables discriminate among NAFLD and OIS groups and Mahalanobis square distances from each of the group centroids was computed to classify studied groups. Pearson’s test was used to assess correlations between the studied indices. Graphical presentation and statistical analysis of the data were performed using GraphPad Prism (Version 5.0, GraphPad software). PCA and discriminant analyses were conducted using Statistica v. 12.0 (IBM Corp., Armonk, NY, USA) software.
If not otherwise stated, the unpaired student’s t-test was used for all other statistical analyses to calculate significant differences among experimental and control groups. Experiments were repeated two-four times and data were pooled. Data are shown as mean ± standard error of the mean (SEM) with P < .05 considered statistically significant. Significance levels were denoted as *P ≤ .05, **P ≤ .01, and ***P ≤ .001.
Results
Precancerous and cancerous liver disease models
We selected three liver disease models (two precancerous ones and one cancerous) in which we studied the interrelation between α-dC and RAGE.
To induce the first type of PLD, the NAFLD, we used HFD provided to animals ad libitum 14 weeks long as described and characterized in33–35 (Figure 1a). Control mice were kept on NCD.
Figure 1.
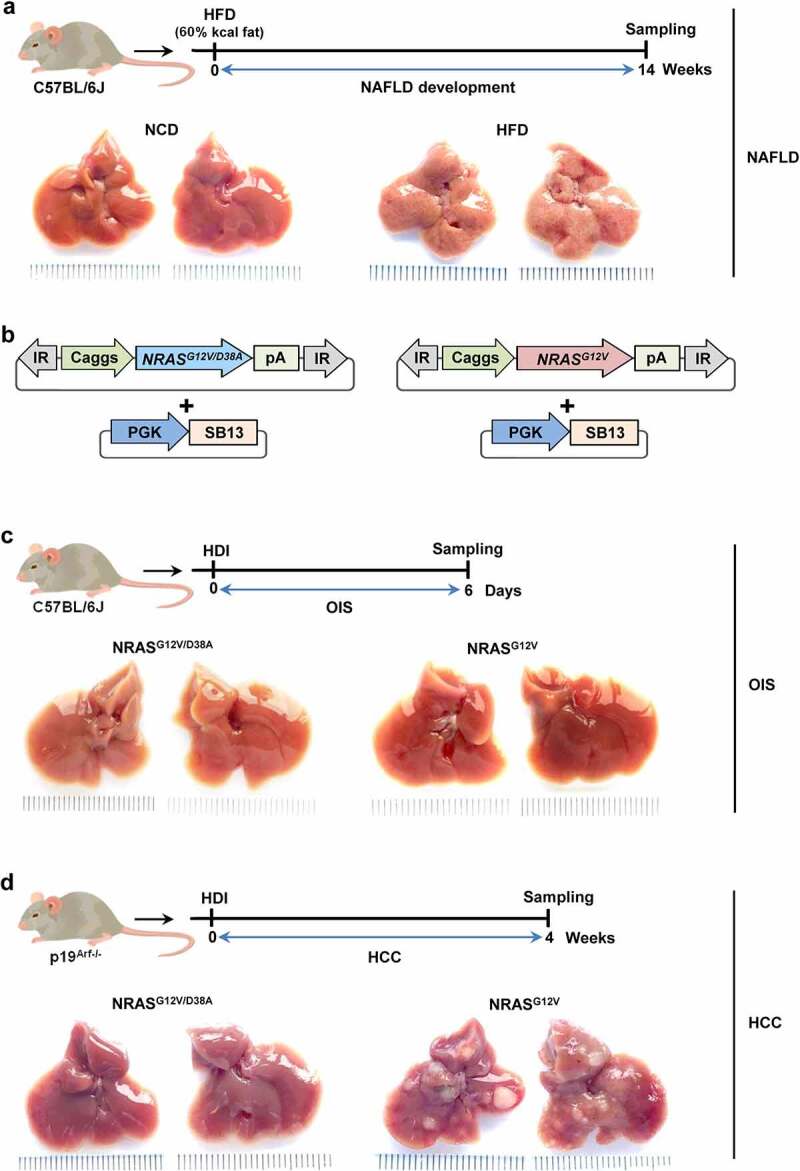
Vectors and experimental design.a, For inducing NAFLD, 3 weeks-old female C57BL/6J mice were exposed to a 60% high-fat diet (HFD) for 14 weeks. The control group was fed with a normal chow diet (NCD) composed of 10% kcal of fat. Shown are representative pictures of livers after NCD and HFD, respectively. b, NRASG12V/D38A or NRASG12V transposon constructs were delivered with SB13 into C57BL/6J or p19Arf-/- mice to induce (c) OIS or (d) HCC development, respectively. Representative pictures of livers with (a) NAFLD, (c) OIS and (d) HCC, and the corresponding controls. HDI – hydrodynamic tail vein injection, SB13 – Sleeping Beauty 13 (transposase), PGK – phosphoglycerate kinase promoter, IR – inverted repeats, pA – polyadenylation signal, Caggs – synthetic CAG promoter
The second PLD was induced via stable transposon-based overexpression of oncogenic NRASG12V directly in hepatocytes of wild type mice, leading to OIS in murine livers, as established in our previous study30 (Figure 1b-c).
HCC was induced by overexpression of oncogenic NRASG12V in hepatocytes of p19Arf-/- mice, in which senescence is disabled/bypassed, 43 resulting in full-blown liver tumor development within 4 weeks, as established30–32 (Figure 1d).
HFD provoked liver injury in mice, that further increased in HCC
We first compared two PLD models (NAFLD and OIS) and HCC in terms of general inflammation and detected several biochemical parameters in the plasma of mice. In NAFLD PLD, intake of HFD provoked metabolic alterations in mice, disclosed as a higher level of glucose (by 30%), and cholesterol (by 40%) in plasma when compared to the corresponding controls (Figure 2a-b). The decreased level of glucose (by 27%) was observed in the HCC mouse model (Figure 2a). In addition, the hepatic inflammatory value in the liver, namely the AST/ALT ratio, was remarkably increased in the HFD group (by 44%) compared to the NCD-fed control and also not significantly increased in HCC (Figure 2c and Supplementary Figure 2). All other parameters, like lactate, LDH, and ChE were similar between the NCD and HFD groups (Figure 2d-f). LDH dramatically increased (by 74%) in the plasma of HCC mice (Figure 2e). In contrast, no clear difference between groups in OIS PLD (Figure 2) was reflected. Metabolic changes were also predominantly observed in NAFLD and not in OIS PLD as described in the next section (Figure 3).
Figure 2.
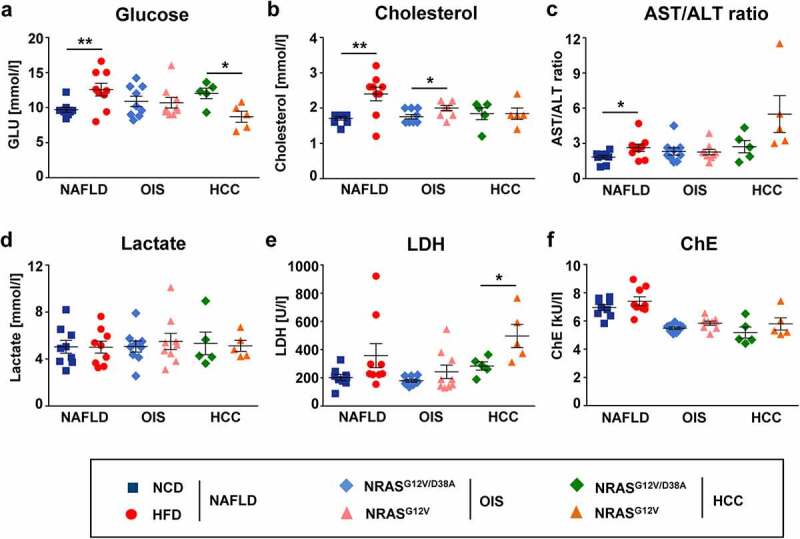
Mice with NAFLD showed a significantly increased glucose, cholesterol and AST/ALT ratio, whereas mice with HCC showed a decrease in glucose and a dramatic increase of LDH in plasma. Different biochemical parameters were measured in plasma of mice with NAFLD, OIS and HCC. Shown are concentrations of a, glucose; b, lactate; c, cholesterol; d, ratio of AST/ALT activities; e, activity of LDH; f, activity of ChE (cholinesterase). Data are presented as mean ± SEM, n = 5–9. *P < .05, **P < .01
Figure 3.
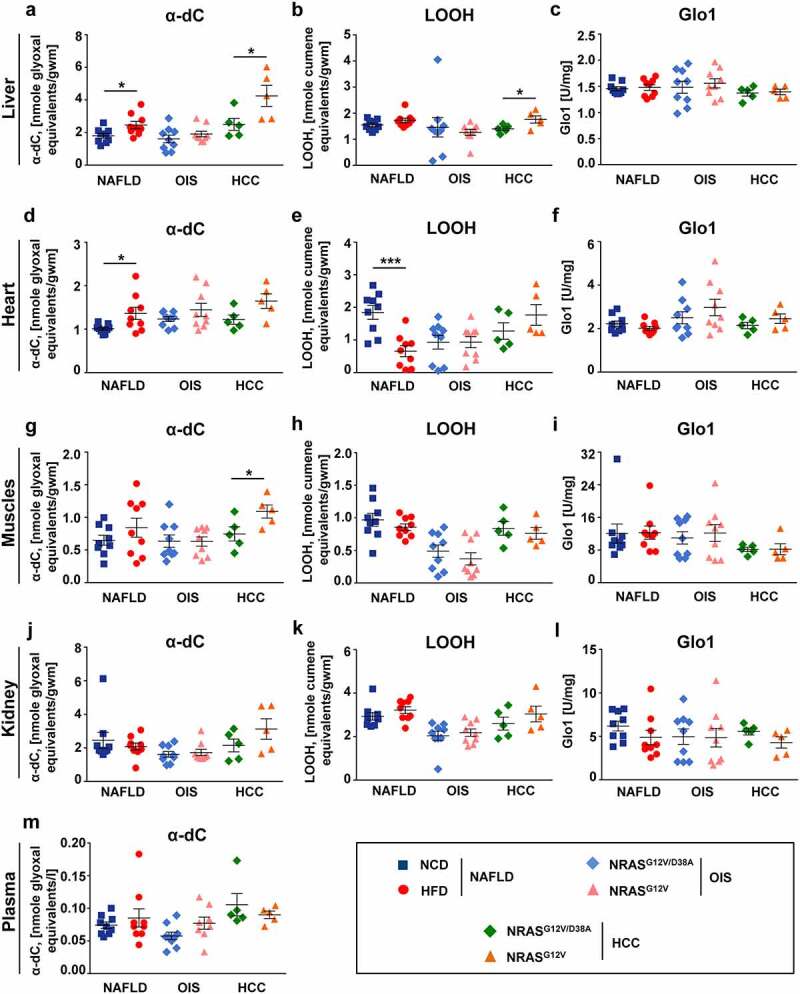
Mice with NAFLD and HCC showed increased α-dC levels in liver, heart and muscles. Different biochemical metabolic parameters were measured in different organs isolated from mice with NAFLD, OIS and HCC. a-c, Shown are parameters measured in liver: a, α-dC level; b, LOOH level; c, Glo1 activity. d-e, Shown are parameters measured in heart: d, α-dC level; e, LOOH level; f, Glo1 activity. g-i, Shown are parameters measured in muscles: g, α-dC level; h, LOOH level; i, Glo1 activity. j-l, Shown are parameters measured in kidney: j, α-dC level; k, LOOH level; l, Glo1 activity. m, Shown is α-dC level measured in plasma of mice with NAFLD, OIS and HCC. Data are presented as mean ± SEM, n = 5–9. *P < .05
Mild carbonyl stress was detected in liver, heart and muscles of mice with NAFLD and HCC
Following the lines of metabolic alterations, we further tested the parameters of oxidative and carbonyl stress in mice and measured concentrations of α-dC compounds in tissues and plasma. Importantly, in NAFLD, HFD led to the appearance of signs of oxidative and carbonyl stress in mice (Figure 3a-m). In particular, significantly elevated α-dC levels were found in the liver (by 37%) and heart (by 35%), and not significantly in muscles of mice with NAFLD (Figure 3a, d, g, and Supplementary Table 2A). In kidney or plasma of NAFLD animals α-dC kept in line with the corresponding controls (Figure 3j, m, and Supplementary Table 2A). The obtained results further clearly demonstrated the increase of α-dC not only in NAFLD but also in HCC livers (by 70%) (Figure 3a). Although not significant, α-dC were also elevated in the heart and significantly increased in muscles of mice with HCC (Figure 3d and g, respectively). Interestingly, and in consistence with NAFLD data, increased α-dC were not detected in the plasma of mice with HCC (Figure 3m). In addition, the LOOH level (parameter of oxidative stress) significantly increased in the liver of HCC mice (by 25%) (Figure 3b) and was reduced by 64% in hearts of HFD animals (Figure 3e), but kept in line with the corresponding controls in all other cases (Figure 3b, e, h, k). No other significant changes between studied indices were observed in NAFLD and HCC, including Glo1 as a component of the detoxification system (Figure 3c, f, i, l). All metabolic changes were detected exclusively in NAFLD PLD and in HCC whereas, in OIS PLD, metabolic parameters were at the levels of the respective controls (Figure 3a-m).
RAGE strongly increased on CD8+, CD4+, NK, and NKT cells in HCC livers
We next investigated whether we can detect receptor for AGEs (RAGE) on stroma cells as well as different liver-infiltrating immune cell populations in mice with premalignant (NAFLD, OIS) and malignant (HCC) liver disease. Using a gating strategy, shown in Figure 4a, we gated on several populations of lymphocytes (CD8+, CD4+, CD3− NK1.1+, and CD3+ NK1.1+). Remarkably, a dramatic increase of RAGE was detected in the adaptive immune cell compartment, i.e., in CD8+ T cells (Figure 4b) comprising 57% in tumor-bearing versus 43% in tumor-free animals. Interestingly, in NAFLD, upregulation of RAGE was primarily demonstrated on CD8+ and not on CD4+ T cells (Figure 4b and c, respectively). In lymphocytes belonging to the innate immune cell compartment, i.e., natural killer (NK) and natural killer T (NKT) cells, we again detected a strong increase of RAGE in HCC livers (Figure 4d and e, respectively). Interestingly, in OIS values of RAGE on CD8+ T cells and NK/NKT cells remained similar to controls (Supplementary Figure 3A). Importantly, the increase in RAGE on CD8+ T cells, NK, and NKT cells was present only locally in situ in NAFLD and significantly more pronounced in HCC livers (Figure 4b-e, Supplementary Table 2B, e.g., for CD3− NK1.1+ RAGE+, P = .013944). This observation, however, was not consistent with lymphocytes in the blood (Supplementary Figure 3B-E). Only NK and NKT cells in the blood of NAFLD mice demonstrated upregulation of RAGE (Supplementary Figure 3D-E) with significant differences shown only for NK cells in the NAFLD group (Supplementary Figure 3D).
Figure 4.
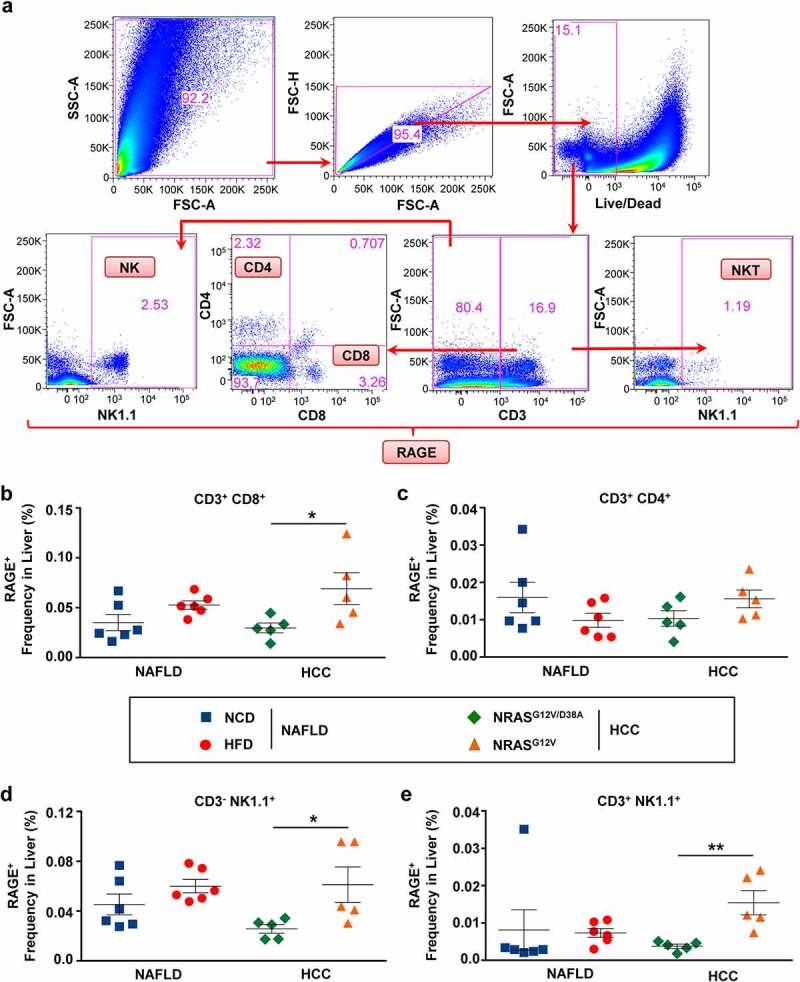
RAGE was strongly upregulated on CD8+, NK and NKT cells in HCC livers. a, Gating strategy to define RAGE expression on CD8+, CD4+, NK, and NKT cells. Single-cell suspensions were obtained from precancerous (NAFLD, OIS) and cancerous (HCC) livers. Cell suspensions were stained and analyzed via the indicated surface and intracellular markers using multicolored FACS analysis. b-e, Shown are frequencies of RAGE+ cells among (b) CD3+ CD8+ T, (c) CD3+ CD4+ T, (d) CD3− NK1.1+ NK, (e) CD3+ NK1.1+ NKT lymphocyte populations in livers of mice with NAFLD and HCC. Data were analyzed using unpaired student’s t-test. Shown are mean ± SEM, with n = 5–6 per group. *P < .05, **P < .01
We have also characterized several other cell populations, i.e., CD11c+ DC, Ly6G+ neutrophils (NΦ), CD68+ macrophages (MΦ), CD146+ CD54− liver sinusoidal endothelial cells (LSECs), and CD146− CD54+ hepatic stellate cells (HSC), in livers of mice with NAFLD, OIS, and HCC using the gating strategy shown in Supplementary Figure 4A. DC did not show any significant RAGE upregulation in both, liver, and blood in NAFLD and HCC mice (Supplementary Figure 4B-C). Moreover, none of the NΦ, MΦ, LSEC, and HSC cells investigated in situ in livers showed a significant upregulation of RAGE in NAFLD and HCC livers as determined via flow cytometry (Supplementary Figure 4D-E). As expected, in OIS we also could not detect any RAGE upregulation as shown for DC (Supplementary Figure 5).
We finally tested the presence of RAGE on liver stroma cells. We first gated on hepatocytes via FSC-A and SSC-A characteristics, thereafter we excluded all lymphoid cells (lymphocytes, NΦ, MΦ, DC, LSECs, HSCs, etc.) using the staining of pan-cell receptors (Figure 5a). Surprisingly, RAGE expression was neither detected in NAFLD nor in HCC on liver stroma cells (Figure 5b, Supplementary Table 2B).
Figure 5.
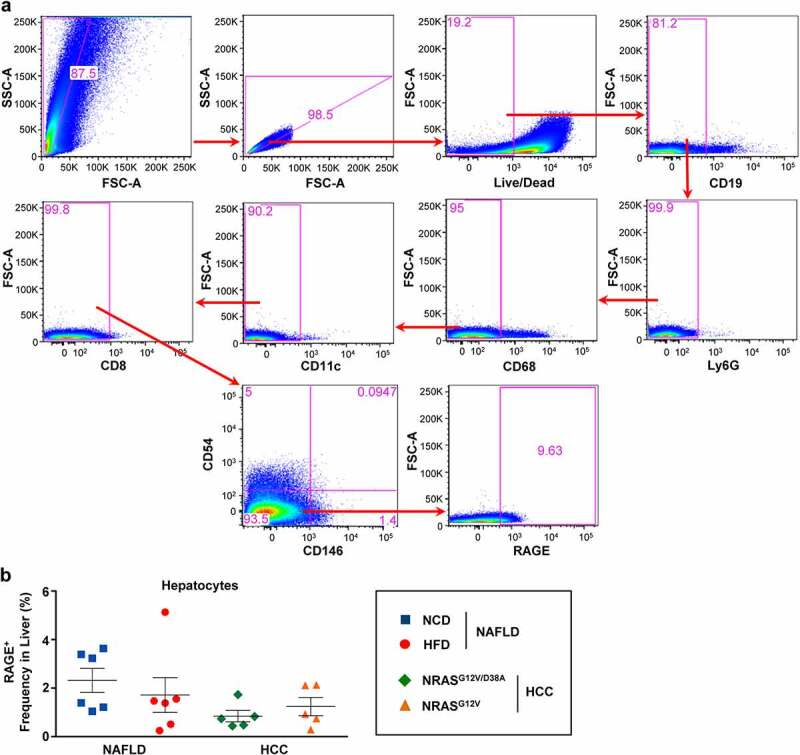
RAGE expression was not detected on liver stroma cells (hepatocytes) as confirmed using multicolored FACS analysis. a, Gating strategy to exclude all immune cells present in liver tissues and define RAGE expression on remaining stroma cells (hepatocytes). b, Shown are frequencies of RAGE++ hepatocytes in NAFLD and HCC livers. Data were analyzed using unpaired student’s t-test. Shown are mean ± SEM, with n = 5–6 per group
RAGE+ lymphocytes demonstrated significant upregulation of CD44 and PD-1 in NAFLD and HCC livers
In the next step, we aimed to characterize and detect the activation/inhibitory status of main “RAGE upregulators,” i.e., RAGE+ lymphocytes in livers of NAFLD- and HCC-harboring animals. Using the gating strategy shown in Figure 6a, we found that RAGE+ CD8+ T cells showed a significant increase in CD44 expression in NAFLD/HCC and PD-1 expression in HCC (Figure 6b). Similarly, CD4+ T cells in HCC significantly increased CD44 and PD-1 (Figure 6c). In HCC, PD-1 upregulation in RAGE+ CD8+ and in RAGE+ CD4+ T cells comprised a corresponding 3.7- and 4-fold increase, whereas in NAFLD-bearing mice, no significant differences were registered (Figure 6b and c, respectively).
Figure 6.
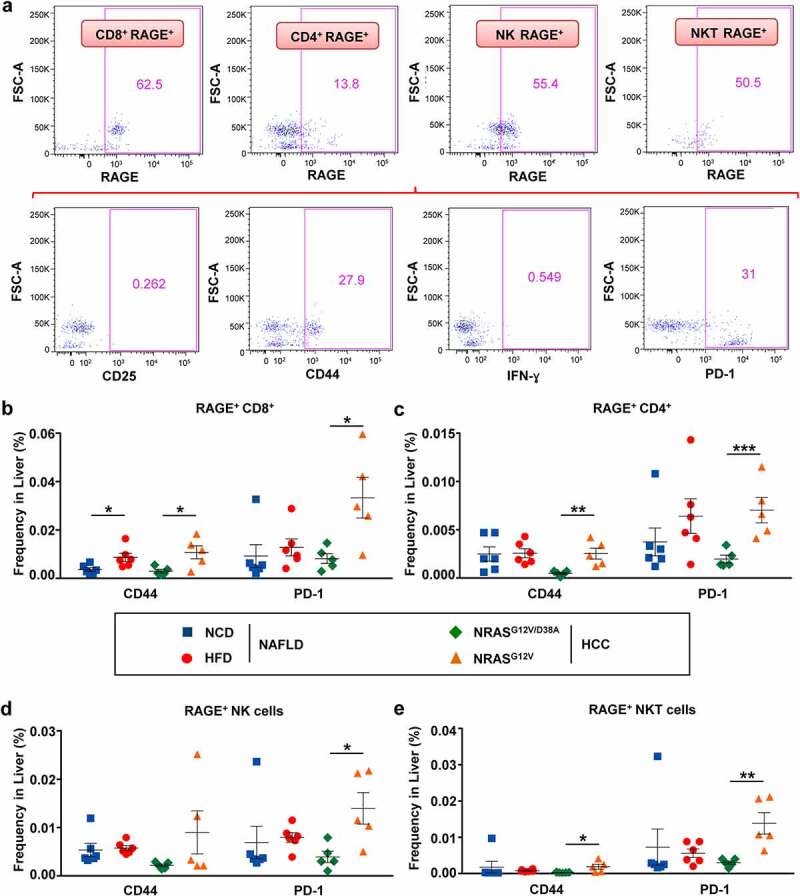
RAGE+ T, NK, and NKT cells showed a significant increase in CD44 and PD-1 in livers of mice with NAFLD and HCC. a, Gating strategy to identify and characterize the phenotype of RAGE+ cells in NAFLD and HCC livers. b-e, Overview of receptors (CD44, PD-1) on (b) RAGE+ CD3+ CD8+ T, (c) RAGE+ CD3+ CD4+ T, (d) RAGE+ CD3− NK1.1+ NK (e) RAGE+ CD3+ NK1.1+ NKT lymphocyte populations in livers of mice with NAFLD and HCC. Data were analyzed using unpaired student’s t-test. Shown are mean ± SEM, with n = 5–6 per group. *P < .05, **P < .01, ***P < .001
Remarkably, both RAGE+ NK and NKT cells significantly upregulated PD-1 in HCC livers (3.6-fold for NK, and 4.6-fold for NKT, as shown in Figure 6d and e, respectively). In HCC, NKT cells also significantly upregulated CD44 (Figure 6e).
We further checked the activation marker CD25 and intracellular IFN-ɣ in RAGE+ CD8+ and CD4+ T cells. In both NAFLD and HCC livers, CD8+ T cells demonstrated an activated phenotype with an upregulation of CD25 and IFN-ɣ. The differences to control groups, however, were not significant (Supplementary Figure 6A). Importantly, RAGE+ CD4+ T cells strongly upregulated IFN-ɣ in NAFLD livers but did not show any increase in CD25 in comparison to healthy controls (Supplementary Figure 6B). Neither IFN-ɣ nor significant upregulation of CD25 was observed in RAGE+ CD4+ T cells in HCC livers (Supplementary Figure 6B). RAGE+ NK and NKT did not demonstrate any increase in CD25 or IFN-ɣ expression in NAFLD and HCC livers (data not shown).
We aimed to further evaluate the function of CD8+ RAGE+ T cells and tested the presence of the transcription factor EOMES on these cells using an immunofluorescent staining of frozen murine liver tissue sections. Although not significant, we could detect upregulation of EOMES on CD8+ RAGE+ T cells in HCC in murine samples and not in mice with NAFLD (Supplementary Figure 7).
Multivariate statistical analysis proved a direct link between liver inflammation markers, increase in α-dC compounds, and RAGE+ overexpression on immune cells in NAFLD
We further aimed to study interrelations between biochemical and immunological parameters measured in this study in liver diseases. To achieve this aim, we performed first the discriminant analysis and compared data between two PLD models. The discriminant analysis showed that the NRASG12V/D38A/NRASG12V animals grouped closely together in the plane of the Root 2, when NAFLD groups from NCD and HFD showed significant estrangement according to Mahalanobis square distance (Figure 7a). The OIS and NAFLD groups significantly separated from each other along the Root 1 axis (Figure 7a).
Figure 7.
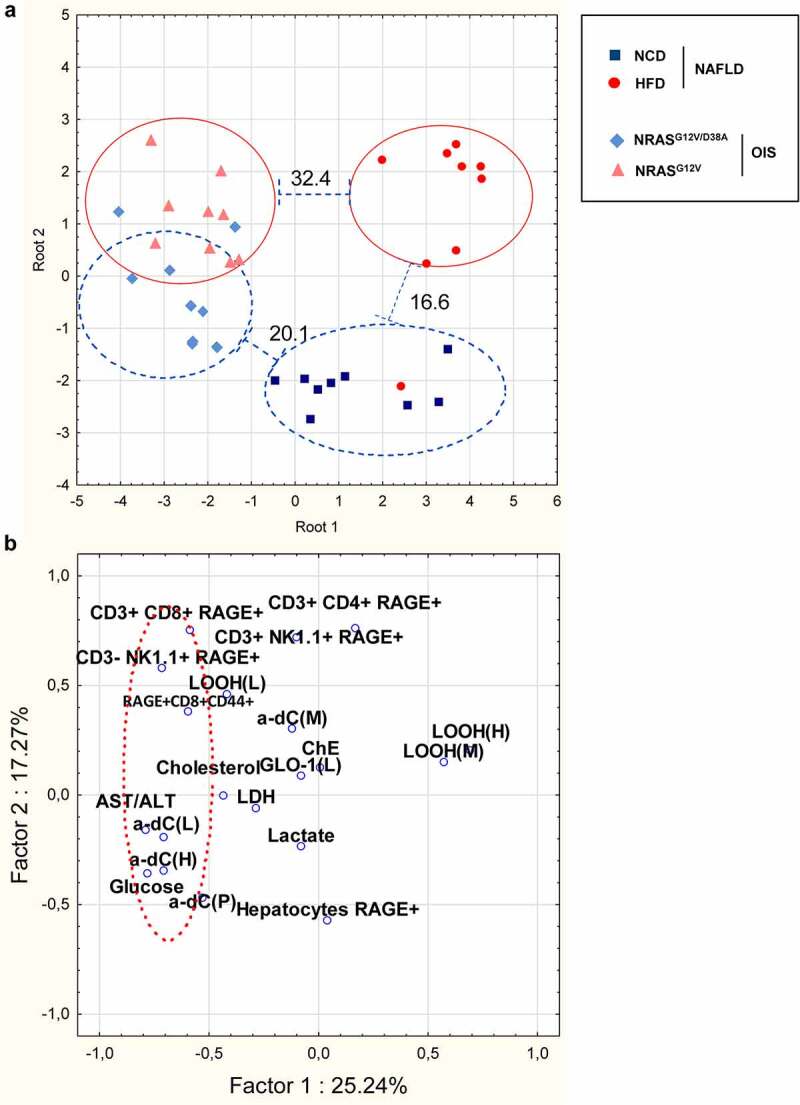
Multivariate statistical analyses of the studied biochemical and immunological indices in mice among i) NAFLD and OIS PLD groups and ii) NAFLD and HCC groups showed a direct link between liver inflammation markers,α-dC compounds, and RAGE+ CD8+ T and RAGE+ NK cells in NAFLD. a, Layout of PLD groups (NAFLD and OIS) in the plain of two main roots in the discriminant analysis module based on linear combination of signs that separates studied groups (F36,62 = 4.2078, P < .0001). Numbers above lines indicate Mahalanobis square distance between two adjacent groups. Data set contains glucose, lactate, cholesterol, AST/ALT activities ratio, α-dC compounds in plasma, liver, heart, and muscles, as well as RAGE+ immune cells (CD11c+ DC, CD8+ T cells, CD3− NK1.1+ NK cells, and CD3+ NK1.1+ NKT cells). b, Graphical representation of the PCA results based on the intercorrelation of biochemical and immunological indices in mice with NAFLD. The parameters α-dC compounds in liver and heart, glucose, AST/ALT ratio, as well as RAGE+ immune cells (CD3+ CD8+ T and CD3+ CD8+ CD44+ T cell populations and CD3− NK1.1+ NK cells) build up a common cluster
We further compared the interrelation between biochemical and immunological parameters in NAFLD using the principal component analysis (PCA). PCA is generally used to visualize relatedness between studied indices.44 PCA distinguished two main principal components (factors) with the eigenvalues >2 (Figure 7b, Supplementary Table 3). Factor 1 (25.24% of the variation) had high positive loadings (>0.6) of the oxidative stress parameter, namely LOOH in the heart. Factor 1 had high negative loading of several biochemical and immune parameters, including: α-dC level in liver and heart, AST/ALT ratio, glucose concentration in plasma, CD3− NK1.1+ RAGE+, RAGE+ CD8+ CD44+. Factor 2 (17.27% of the variation) had a high positive loading of CD3+ CD8+ RAGE+, CD3+ CD4+ RAGE+, CD3+ NK1.1+ RAGE+. CD3+ CD8+ RAGE+ and Hepatocytes RAGE+ possessed factor loading very closed to the most reliable one (>±0.57). Importantly, parameters allocation along factor 1 emphasizes close relation and a cluster between signs of carbonyl stress (α-dC), liver inflammation (AST/ALT), and RAGE overexpression on particular immune cells (RAGE+ CD8+ T cells, including a CD44+ population, and RAGE+ NK cells) in mice with NAFLD (Figure 7b, Supplementary Table 3).
Further, in order to identify interactions between biochemical and immunological parameters specific to carbonyl stress, we used the Pearson correlation test. It revealed a clear association between α-dC, particularly in liver, and RAGE+ CD4+ IFN-ɣ+, CD3+ CD8+ RAGE+, CD3− NK1.1+ RAGE+ (Supplementary Table 4, e.g., CD3− NK1.1+ RAGE+ (L) vs. α-dC (L) r = 0.04933, P = .038). AST/ALT ratio and α-dC compounds in NAFLD livers revealed the highest rate of positive/negative correlations to biochemical and immunological indices.
Elevated α-dC levels were detected in sera of patients with NAFLD, NASH and in HCC tissues
In the next step, we aimed to analyze sera of patients with NAFLD and NASH and detected a strong and significant increase of α-dC in those patients in comparison to healthy controls (Figure 8a). In addition, an increase of transaminases has been observed in patients with NAFLD (AST 37.20 ± 6.953 U/L; ALT 56.60 ± 14.83 U/L) and NASH (AST 52.20 ± 7.618 U/L; ALT 85.40 ± 26.09 U/L).
Figure 8.
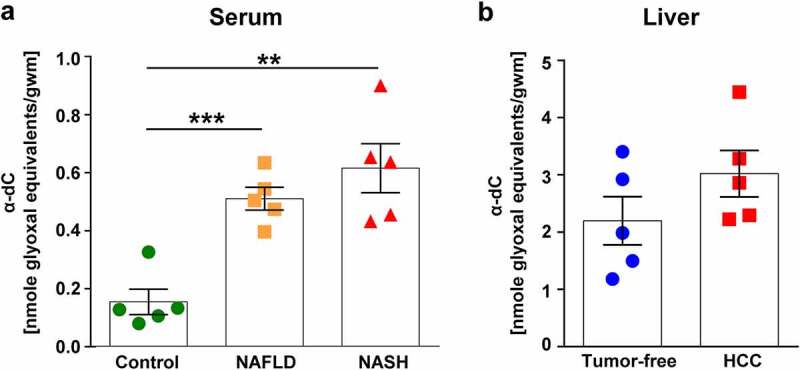
A significant increase of serum α-dCs was detected in NAFLD and NASH patients in comparison to healthy donors. Elevated α-dC were demonstrated in HCC liver tissues in comparison to non-cancerous liver tissues. Shown is the level of α-dCs measured in: a, serum of NAFLD and NASH patients in comparison to healthy donors; b, frozen tumor-free and HCC liver tissues obtained upon HCC surgeries. Data are presented as mean ± SEM, n = 5. **P < .01, ***P < .001
Further, we analyzed available liver tissue material obtained after R0 HCC resections. Although not significant, levels of α-dCs were found upregulated in HCC in comparison to tumor-free liver tissue of the same patient (Figure 8b).
Discussion
In the present study, we investigated the interplay between liver inflammation, metabolic parameters, oxidative/carbonyl stress, and NAFLD as well as liver cancer progression. Following the summarized pathways of generation and detoxification of α-dC compounds and their direct connection with a downstream activation of RAGE, we studied the expression levels of several metabolic markers in NAFLD and compared them to OIS. Our data clearly demonstrate that HFD consumption induces more severe liver injury than OIS. HCC showed the most severe liver injury (increased AST/ALT and high LDH). Further, we detected increased levels of α-dC compounds in livers, hearts, and muscles of mice with NAFLD and HCC. Our data obtained in patients with NAFLD and NASH further provide evidence for increased levels of α-dC levels detected systemically in patients sera. We also detected higher α-dC levels in resected HCC tissues, however, the latter data still has to be compared to healthy liver tissues, which remains highly limited material.
Essentially, all changes in metabolic and oxidative stress markers were observed in NAFLD only, but not in OIS, which was also consistent with our discriminant analysis showing strong differences among HFD-fed and healthy groups, whereas such differences were not supported in OIS. Importantly, almost all previous studies declared increased glucose, cholesterol, lactate, AST/ALT, and LDH levels not only in NAFLD but also in HCC using several murine models and human studies as summarized in Supplementary Table 5. These reports are consistent with our data with few exceptions, which could be related to the specificities of the respective animal models. Among carbonyl stress, the level of protein carbonyls is the most measured marker of interest and usually increased in both clinical and experimental models.45–47 The decreased level of glucose that we observed in HCC is further consistent with previous reports.48,49
We also found that the LOOH level was reduced in the hearts of HFD animals. This finding is contrary to the previous studies which have suggested that lipid peroxides consistently increase in both clinical and experimental measurements in murine and human NAFLD/NASH models.50 However, almost all data regarding an increase in lipid peroxidation in NAFLD was obtained using liver tissue samples.51 Therefore, the pattern of lipid peroxidation in the heart and its contribution to oxidative stress in NAFLD remained unclear. Moreover, in recent decades some authors among them Jaeschke and Woolbright52 emphasized on the relativism of lipid peroxidation as a unique marker of severity of oxidative stress and injury, because it might not directly reflect the profundity of reported damage. Our findings about LOOH in the heart presented here shed new light on the role of oxidative stress in the pathogenesis of NAFLD and we hope these findings will get the attention of cardiologists. It is worth pointing out that the LOOH level was increased in HCC livers. The latter findings are in line with previous reports in human samples and murine models.53
We further performed a PCA analysis and confirmed that in liver and heart, α-dC composed a joint group with AST/ALT as a marker of liver injury and significantly correlated with each other in the NAFLD model. Importantly, we also confirmed higher levels of α-dC in the NRASG12V/p19Arf-/- HCC model. Obviously, the close relation between α-dC and inflammation discloses systemic metabolic disorders under NAFLD. However, only a few studies have focussed on the enhancement of dicarbonyls and AGEs at NAFLD.54 In particular, plasma and liver levels of MGO were increased in RAGE−/− mice fed with a Western-type diet.55 Also, it has been shown that HFD was associated with increased AGE levels in the liver, heart, and kidney of rats.56
The role of AGEs, RAGE, and particularly their interaction has been in focus last decades due to the potential regulation of the NF-κB inflammatory pathway by AGE-RAGE. To our knowledge, this is the first study to investigate the interdependence between RAGE, α-dC (as the precursors of AGEs), and inflammatory indices on the one hand and progression of PLD (NAFLD) and HCC on the other. The liver has been recently recognized as an important metabolic center of AGEs in the body.15 To date, there are already several literature overviews supporting a direct link between α-dC – AGE/AGE-proteins and the activation of RAGE16,17 as well as a recent review on colorectal cancer describing the involvement of AGEs and their precursors (α-dC) and, especially, RAGE in cancer progression.57
RAGE, a multiligand cell surface receptor, has been reported to correlate with the poor therapeutic outcomes and malignancy of HCC as comprehensively reviewed by Takino et al.58 Moreover, in the same study, the researchers underlined that interactions between toxic AGEs and RAGE-induced oxidative stress, which may in turn lead to adverse effects in tumor cells and HSC during liver disease progression.58 In their study, Li and colleagues confirmed RAGE overexpression in HCC and its important role in cancerous disease progression, associated with proliferation and resistance to sorafenib.59 Although it has been reported that RAGE is expressed in HSC, hepatocytes, and hepatoma cells, 60,61 our data for HCC and NAFLD do not demonstrate RAGE on hepatocytes. Importantly, in full-blown malignant liver disease (HCC), our results confirmed the RAGE overexpression primarily on immune cells (CD8+ T cells, NK, and NKT), but not on stroma cells. These data are in line with a report showing RAGE overexpression in CD45+ inflammatory cells (liver-infiltrating leukocytes) and not in hepatocytes.62 In the majority of NAFLD and HCC studies published so far, 63 however, RAGE was tested in whole liver tissue and a thorough characterization of particular “RAGE cellular upregulators” was not performed (see Supplementary Table 5 for an overview). As mentioned above, our study newly identifies the main players in NAFLD and HCC in terms of RAGE upregulation. Among many cell populations tested, we detected RAGE upregulation predominantly on three cell populations: CD8+ T cells, NK, and NKT in both, NAFLD and (even more pronounced) in the malignant liver disease (HCC) progression.
Our results on RAGE+ CD8+ T cells are in line with the study of Akirav et al. demonstrating RAGE overexpression on CD4+ and CD8+ T cells in another chronic metabolic disorder, i.e., in patients with type 1 diabetes.64 In our work, being strongly upregulated in HCC livers, RAGE+ CD8+ T cells seem to possess an activated phenotype, strongly overexpressing the activation/memory marker CD4465,66 and the immune checkpoint marker PD-1. PD-1 expression is normally up-regulated following T cell activation.66,67 Our data on RAGE expression on CD44+ T cells are consistent with the work of Kierdorf et al.23 While overexpressing PD-1, RAGE+ CD8+ T cells also showed (not significantly) an increase in the activation marker CD25 and in IFN-ɣ secretion in both NAFLD and HCC. Generally and confirmed by a vast amount of studies, CD8+ T cells overexpressing PD-1 in NAFLD and HCC demonstrate an exhaustion phenotype and disability in cytotoxic killing.68,69 Further, the upregulation of EOMES detected by us on CD8+ RAGE+ T cells in HCC and not in NAFLD tissue suggests that these cells possess an exhausted phenotype particularly in liver cancer. The latter hypothesis is supported by several research studies.70,71 It remains to be defined in future validation studies whether RAGE expression on PD-1+ CD8+ T cells interferes and/or impacts their cytotoxic capacity.
Similarly to RAGE+ CD8+ T cells, NK, and NKT cells which showed significant upregulation in RAGE in malignant livers in our study, also exhibited a strong upregulation in PD-1 expression on both cell types, and the activation marker CD4467 was enhanced on NKT in HCC livers. It has been reported that the activation receptor CD44 can also play a leading role in metabolic diseases.72 Such, in obese patients and also in rodent models, CD44 expression has been demonstrated to augment in liver tissue and to correlate with liver steatosis and type 2 diabetes.65,72,73 Therefore, the role of CD44 and activation of NKT and T cells still needs to be tested in follow-up studies.
Importantly, PCA analysis demonstrated a direct link between RAGE+ CD8+ (also CD44+ population), RAGE+ NK cells, liver inflammation markers, and α-dC compounds in liver and heart in NAFLD. Our results are in conflict with previous findings indicating that RAGE deficiency does not affect NASH and atherosclerosis in Western-type diet-fed Ldlr−/−/Ldlr−/− RAGE−/− mice.55 This discrepancy may be reasonably attributed to the differences in the animal models used, namely RAGE-deficiency. It is important to mention that several studies recognized the role of RAGE in liver diseases, 60–62 including a very recent report on RAGE contribution to acute liver injury.74
Although RAGE or the AGE/RAGE pathway have been already suggested as a potential target for therapeutic intervention in NASH and HCC, 58,59,62 our data extend the previous view and suggest that a targeted neutralization of α-DC and RAGE in livers may promise a stronger therapeutic potential. However, the latter still needs to be confirmed experimentally.
Potential applications in the future may include the systemic and local detection of α-dC compounds and RAGE (over-) expression on immune cells (CD8+ T, NK, NKT) in liver biopsies as biomarkers of disease progression (as shown in Graphical Abstract). However, a direct confirmation from the clinical specimen (in sera and liver biopsies obtained from NAFLD, NAFLD-related HCC patients, and healthy donors) will be required and will be performed in the follow-up studies.
In summary, our data provide evidence for a direct link between upregulation of α-dC compounds and overexpression of RAGE on CD8+ T and NK cells locally in the liver in NAFLD and in liver cancer progression. This finding can be considered for the future translational application using both α-dC compounds and RAGE as biomarkers of NAFLD and HCC progression and as molecules for a targeted therapeutic intervention.
Supplementary Material
Acknowledgments
We thank D. Jonigk for providing the infrastructure at the Institute of Pathology, at the Hannover Medical School. We thank Maria Holovchak, Lesia Sishchuk (Department of Biochemistry and Biotechnology, Vasyl Stefanyk Precarpathian National University, Ivano-Frankivsk, Ukraine), Oihane Mauleon, Blanca Smits Ortuño (Department of Gastroenterology, Hepatology and Endocrinology, Hannover Medical School, Hannover, Germany) for excellent technical assistance. We are grateful to Ulyana Dzhaman (Department of Biochemistry and Biotechnology, Vasyl Stefanyk Precarpathian National University, Ivano-Frankivsk, Ukraine) and DAAD, respectively, for the initiation and support of the German-Ukrainian collaboration. Special thanks to Gaby Feldmann and Prof. Timo Ulrichs (Akkon University of Human Sciences, Berlin, Germany) for the support of the junior researcher’s mobility.
Funding Statement
This work was supported by the German Academic Exchange Service (Deutscher Akademischer Austauschdienst, DAAD) in the scope of Leonhard Euler Programme under Grant (Project-ID 57376448 to V.L. and T.Y.; Project-ID 57431922 to H.S. and T.Y.; and Project-ID 57482320 to H.S. and T.Y.); by the DAAD Doctoral Program in Germany 2019/2020 under Grant (Project-ID 91736778 to N.P.). T.Y. acknowledges the support of the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) under Grant (YE 151/2-1). H.B. acknowledges the support of the DFG under Grant (BA-2092/11-1). This work was also supported in part by the Fritz Thyssen Foundation (Fritz Thyssen Stiftung) under Grant (REF.10.16.1.031MN) and Wilhelm-Sander Foundation (Wilhelm-Sander Stiftung) under Grant (2013.107.1) and Gilead Sciences International Research Scholars Program in Liver Disease (Research Award) to T.Y.
Author contributions
Conceptualization, V.L., H.S., T.Y.; Methodology, N.P., L.N, M.V., Hu.S.; I.H., R.H., C.P, A.H., A.Y.; Investigation, N.P., L.N., M.V., K.T., Hu.S., I.H., R.H., A.H., A.Y., R.L., H.F., V.L., H.B., H.S., T.Y.; Formal Analysis, N.P., L.N., M.V., A.H., H.F., H.S., T.Y.; Writing – Original Draft, N.P., M.V., H.F., H.S., T.Y.; Writing – Revised Draft, N.P., L.N, M.V., H.F., T.Y.; Funding Acquisition, R.L., V.L., H.S., T.Y.; Resources, L.N., K.T., R.L., Ha.S., H.W., M.P.M., H.B., T.Y.; Supervision, L.N., R.L., Ha.S., H.W., V.L., M.P.M., H.S., T.Y.
Disclosure statement
The authors report no conflict of interest. H.W. is a consultant by Bayer, Eisai, MSD and Roche, H.W. is a clinical trial investigator by Bayer and Roche.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M,Richardson PA, Li L, Desiderio R, Thrift AP, Asch SM, et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology. 2018;155(e2):1828–16. doi: 10.1053/j.gastro.2018.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sagnelli E, Macera M, Russo A, Coppola N, Sagnelli C.. Epidemiological and etiological variations in hepatocellular carcinoma. Infection. 2020;48:7–17. doi: 10.1007/s15010-019-01345-y. [DOI] [PubMed] [Google Scholar]
- 3.Bellentani S. The epidemiology of non-alcoholic fatty liver disease. Liver International: Official Journal of the International Association for the Study of the Liver. 2017;37(Suppl 1):81–84. doi: 10.1111/liv.13299. [DOI] [PubMed] [Google Scholar]
- 4.Kutlu O, Kaleli HN, Ozer E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis- (NASH-) Related Hepatocellular Carcinoma. Canadian Journal of Gastroenterology & Hepatology. 2018;2018:8543763. doi: 10.1155/2018/8543763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu LX, Ling Y, Wang HY. Role of nonresolving inflammation in hepatocellular carcinoma development and progression. NPJ Precision Oncology. 2018;2:6. doi: 10.1038/s41698-018-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Refolo MG, Messa C, Guerra V, Carr BI, D’Alessandro R. Inflammatory Mechanisms of HCC Development. Cancers. 2020;12:641. doi: 10.3390/cancers12030641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koo SY, Park EJ, Lee CW. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp Mol Med. 2020;52:1209–1219. doi: 10.1038/s12276-020-0480-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aishima S, Kubo Y, Tanaka Y, Oda Y. Histological features of precancerous and early cancerous lesions of biliary tract carcinoma. J Hepatobiliary Pancreat Sci. 2014;21:448–452. doi: 10.1002/jhbp.71. [DOI] [PubMed] [Google Scholar]
- 9.Cho W, Jin X, Pang J, Wang Y, Mivechi NF, Moskophidis D. The Molecular Chaperone Heat Shock Protein 70 Controls Liver Cancer Initiation and Progression by Regulating Adaptive DNA Damage and Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Signaling Pathways. Mol Cell Biol. 2019;39. doi: 10.1128/MCB.00391-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rabbani N, Xue M, Thornalley PJ. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj J. 2016;33:513–525. doi: 10.1007/s10719-016-9705-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garaschuk O, Semchyshyn HM, Lushchak VI. Healthy brain aging: interplay between reactive species, inflammation and energy supply. Ageing Res Rev. 2018;43:26–45. doi: 10.1016/j.arr.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Fernando DH, Forbes JM, Angus PW, Herath CB. Development and Progression of Non-Alcoholic Fatty Liver Disease: the Role of Advanced Glycation End Products. Int J Mol Sci. 2019;20. doi: 10.3390/ijms20205037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hollenbach M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). Int J Mol Sci. 2017;18. doi: 10.3390/ijms18112466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egana-Gorrono L, Lopez-Diez R, Yepuri G, Ramirez LS, Reverdatto S, et al. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: insights From Human Subjects and Animal Models. Frontiers in Cardiovascular Medicine. 2020;7:37. doi: 10.3389/fcvm.2020.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pang Q, Sun Z, Shao C, Cai H, Bao Z,Wang L, Li L, Jing L, Zhang L and Wang Z. CML/RAGE Signal Bridges a Common Pathogenesis Between Atherosclerosis and Non-alcoholic Fatty Liver. Frontiers in Medicine. 2020;7:583943. doi: 10.3389/fmed.2020.583943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D,et al. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: bridging Association and Causality. Cell Metab. 2018;28:337–352. doi: 10.1016/j.cmet.2018.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroter D, Hohn A. Role of Advanced Glycation End Products in Carcinogenesis and their Therapeutic Implications. Curr Pharm Des. 2018;24:5245–5251. doi: 10.2174/1381612825666190130145549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta. 2013;1833:101–109. doi: 10.1016/j.bbamcr.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 19.Chuah YK, Basir R, Talib H, Tie TH, Nordin N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int J Inflam. 2013;2013:403460. doi: 10.1155/2013/403460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohtsu A, Shibutani Y, Seno K, Iwata H, Kuwayama T, Shirasuna K. Advanced glycation end products and lipopolysaccharides stimulate interleukin-6 secretion via the RAGE/TLR4-NF-kappaB-ROS pathways and resveratrol attenuates these inflammatory responses in mouse macrophages. Exp Ther Med. 2017;14:4363–4370. doi: 10.3892/etm.2017.5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014;2:411–429. doi: 10.1016/j.redox.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riuzzi F, Sorci G, Sagheddu R, Chiappalupi S, Salvadori L, Donato R. RAGE in the pathophysiology of skeletal muscle. J Cachexia Sarcopenia Muscle. 2018;9:1213–1234. doi: 10.1002/jcsm.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol. 2013;94:55–68. doi: 10.1189/jlb.1012519. [DOI] [PubMed] [Google Scholar]
- 24.Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med. 2009;87:235–247. doi: 10.1007/s00109-009-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plotkin LI, Essex AL, Davis HM. RAGE Signaling in Skeletal Biology. Curr Osteoporos Rep. 2019;17:16–25. doi: 10.1007/s11914-019-00499-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan SF, Yan SD, Ramasamy R, Schmidt AM. Tempering the wrath of RAGE: an emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann Med. 2009;41:408–422. doi: 10.1080/07853890902806576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taguchi A, Blood DC, Del Toro G, Canet A, Lee DC, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 28.Hiwatashi K, Ueno S, Abeyama K, Kubo F, Sakoda M,et al. A novel function of the receptor for advanced glycation end-products (RAGE) in association with tumorigenesis and tumor differentiation of HCC. Ann Surg Oncol. 2008;15:923–933. doi: 10.1245/s10434-007-9698-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yaser AM, Huang Y, Zhou RR, Hu GS, Xiao MF,et al. The Role of receptor for Advanced Glycation End Products (RAGE) in the proliferation of hepatocellular carcinoma. Int J Mol Sci. 2012;13:5982–5997. doi: 10.3390/ijms13055982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T,et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 31.Eggert T, Wolter K, Ji J, Ma C, Yevsa T,et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016;30:533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dauch D, Rudalska R, Cossa G, Nault JC, Kang TW,et al. A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat Med. 2016;22:744–753. doi: 10.1038/nm.4107. [DOI] [PubMed] [Google Scholar]
- 33.Ohashi T, Nakade Y, Ibusuki M, Kitano R, Yamauchi T,et al. Conophylline inhibits high fat diet-induced non-alcoholic fatty liver disease in mice. PloS One. 2019;14:e0210068. doi: 10.1371/journal.pone.0210068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffler U, Hobbie K, Wilson R, Bai R, Rahman A,et al. Diet-induced obesity is associated with hyperleptinemia, hyperinsulinemia, hepatic steatosis, and glomerulopathy in C57Bl/6J mice. Endocrine. 2009;36:311–325. doi: 10.1007/s12020-009-9224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartolome F, Antequera D, de la Cueva M, Rubio-Fernandez M, Castro N,et al. Endothelial-specific deficiency of megalin in the brain protects mice against high-fat diet challenge. J Neuroinflammation. 2020;17:22. doi: 10.1186/s12974-020-1702-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khaladj N, Teebken OE, Hagl C, Wilhelmi MH, Tschan C,et al. The role of cerebrospinal fluid S100 and lactate to predict clinically evident spinal cord ischaemia in thoraco-abdominal aortic surgery. European Journal of Vascular and Endovascular Surgery: The Official Journal of the European Society for Vascular Surgery. 2008;36:11–19. doi: 10.1016/j.ejvs.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 37.Mang B, Wolters M, Schmitt B, Kelb K, Lichtinghagen R,et al. Effects of a cinnamon extract on plasma glucose, HbA, and serum lipids in diabetes mellitus type 2. Eur J Clin Invest. 2006;36:340–344. doi: 10.1111/j.1365-2362.2006.01629.x. [DOI] [PubMed] [Google Scholar]
- 38.Lichtinghagen R, Pietsch D, Bantel H, Manns MP, Brand K, Bahr MJ. The Enhanced Liver Fibrosis (ELF) score: normal values, influence factors and proposed cut-off values. J Hepatol. 2013;59:236–242. doi: 10.1016/j.jhep.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 39.Semchyshyn HM, Miedzobrodzki J, Bayliak MM, Lozinska LM, Homza BV. Fructose compared with glucose is more a potent glycoxidation agent in vitro, but not under carbohydrate-induced stress in vivo: potential role of antioxidant and antiglycation enzymes. Carbohydr Res. 2014;384:61–69. doi: 10.1016/j.carres.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 40.Monserrat JM, Geracitano LA, Pinho GL, Vinagre TM, Faleiros M,et al. Determination of lipid peroxides in invertebrates tissues using the Fe(III) xylenol orange complex formation. Arch Environ Contam Toxicol. 2003;45:177–183. doi: 10.1007/s00244-003-0073-x. [DOI] [PubMed] [Google Scholar]
- 41.Mitchel RE, Birnboim HC. The use of Girard-T reagent in a rapid and sensitive methods for measuring glyoxal and certain other alpha-dicarbonyl compounds. Anal Biochem. 1977;81:47–56. doi: 10.1016/0003-2697(77)90597-8. [DOI] [PubMed] [Google Scholar]
- 42.Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE,et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253–257. doi: 10.1038/nature16969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–2222. [PubMed] [Google Scholar]
- 44.Zhang Z, Castello A. Principal components analysis in clinical studies. Annals of Translational Medicine. 2017;5:351. doi: 10.21037/atm.2017.07.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Videla LA, Rodrigo R, Araya J, Poniachik J. Oxidative stress and depletion of hepatic long-chain polyunsaturated fatty acids may contribute to nonalcoholic fatty liver disease. Free Radic Biol Med. 2004;37:1499–1507. doi: 10.1016/j.freeradbiomed.2004.06.033. [DOI] [PubMed] [Google Scholar]
- 46.Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol. 2002;37:56–62. doi: 10.1016/S0168-8278(02)00073-9. [DOI] [PubMed] [Google Scholar]
- 47.Ciapaite J, van den Broek NM, Te Brinke H, Nicolay K, Jeneson JA,et al. Differential effects of short- and long-term high-fat diet feeding on hepatic fatty acid metabolism in rats. Biochim Biophys Acta. 2011;1811:441–451. doi: 10.1016/j.bbalip.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Gjorgjieva M, Mithieux G, Rajas F. Hepatic stress associated with pathologies characterized by disturbed glucose production. Cell Stress. 2019;3:86–99. doi: 10.15698/cst2019.03.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai CY, Chou SC, Liu HT, Lin JD, Lin YC. Persistent hypoglycemia as an early, atypical presentation of hepatocellular carcinoma: A case report and systematic review of the literature. Oncol Lett. 2014;8:1810–1814. doi: 10.3892/ol.2014.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ore A, Akinloye OA, Stress O. Antioxidant Biomarkers in Clinical and Experimental Models of Non-Alcoholic Fatty Liver Disease. Medicina. 2019;55. doi: 10.3390/medicina55020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020;152:116–141. doi: 10.1016/j.freeradbiomed.2020.02.025. [DOI] [PubMed] [Google Scholar]
- 52.Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev. 2012;26:103–114. doi: 10.1016/j.trre.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu Y, Chung FL. Oxidative stress and hepatocarcinogenesis. Hepatoma Research. 2018;4. doi: 10.20517/2394-5079.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spanos C, Maldonado EM, Fisher CP, Leenutaphong P, Oviedo-Orta E,et al. Proteomic identification and characterization of hepatic glyoxalase 1 dysregulation in non-alcoholic fatty liver disease. Proteome Sci. 2018;16:4. doi: 10.1186/s12953-018-0131-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bijnen M, Beelen N, Wetzels S, Gaar JV, Vroomen M,et al. RAGE deficiency does not affect non-alcoholic steatohepatitis and atherosclerosis in Western type diet-fed Ldlr(-/-) mice. Sci Rep. 2018;8:15256. doi: 10.1038/s41598-018-33661-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li SY, Liu Y, Sigmon VK, McCort A, Ren J. High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetes Obes Metab. 2005;7:448–454. doi: 10.1111/j.1463-1326.2004.00387.x. [DOI] [PubMed] [Google Scholar]
- 57.Azizian-Farsani F, Abedpoor N, Hasan Sheikhha M, Gure AO, Nasr-Esfahani MH, Ghaedi K. Receptor for Advanced Glycation End Products Acts as a Fuel to Colorectal Cancer Development. Front Oncol. 2020;10:552283. doi: 10.3389/fonc.2020.552283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takino J, Nagamine K, Hori T, Sakasai-Sakai A, Takeuchi M. Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J Hepatol. 2015;7:2459–2469. doi: 10.4254/wjh.v7.i23.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li J, Wu PW, Zhou Y, Dai B, Zhang PF,et al. Rage induces hepatocellular carcinoma proliferation and sorafenib resistance by modulating autophagy. Cell Death Dis. 2018;9:225. doi: 10.1038/s41419-018-0329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hyogo H, Yamagishi S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr Pharm Des. 2008;14:969–972. doi: 10.2174/138161208784139701. [DOI] [PubMed] [Google Scholar]
- 61.Basta G, Navarra T, De Simone P, Del Turco S, Gastaldelli A, Filipponi F. What Is the Role of the Receptor for Advanced Glycation End Products-ligand Axis in Liver Injury? Liver Transplantation. Official Publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society. 2011;17:633–640. [DOI] [PubMed] [Google Scholar]
- 62.Pusterla T, Nemeth J, Stein I, Wiechert L, Knigin D,et al. Receptor for advanced glycation endproducts (RAGE) is a key regulator of oval cell activation and inflammation-associated liver carcinogenesis in mice. Hepatology. 2013;58:363–373. doi: 10.1002/hep.26395. [DOI] [PubMed] [Google Scholar]
- 63.Song F. Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, Xu X, et al. RAGE Regulates the Metabolic and Inflammatory Response to High-fat Feeding in Mice Diabetes. 2014;63:1948–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akirav EM, Preston-Hurlburt P, Garyu J, Henegariu O, Clynes R,et al. RAGE expression in human T cells: a link between environmental factors and adaptive immune responses. PloS One. 2012;7:e34698. doi: 10.1371/journal.pone.0034698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu X, Taftaf R, Kawaguchi M, Chang YF, Chen W,et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019;9:96–113. doi: 10.1158/2159-8290.CD-18-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6:e1792. doi: 10.1038/cddis.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahn E, Araki K, Hashimoto M, Li W, Riley JL,Cheung J, Sharpe AH, Freeman GJ, Irving BA, and Ahmed R. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A. 2018;115:4749–4754. doi: 10.1073/pnas.1718217115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma J, Zheng B, Goswami S, Meng L, Zhang D,Cao C, Li T, Zhu F, Ma L, Zhang Z, et al. PD1(Hi) CD8(+) T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. Journal for Immunotherapy of Cancer. 2019;7:331. doi: 10.1186/s40425-019-0814-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sachdeva M, Chawla YK, Arora SK. Immunology of hepatocellular carcinoma. World J Hepatol. 2015;7:2080–2090. doi: 10.4254/wjh.v7.i17.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA,Michaëlsson J, Lund O, Hejdeman B, Jansson M, Sönnerborg A, et al. T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog. 2014;10:e1004251. doi: 10.1371/journal.ppat.1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li J, He Y, Hao J, Ni L, Dong C. High Levels of Eomes Promote Exhaustion of Anti-tumor CD8(+) T Cells. Front Immunol. 2018;9:2981. doi: 10.3389/fimmu.2018.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bertola A, Deveaux V, Bonnafous S, Rousseau D, Anty R,Wakkach A, Dahman M, Tordjman J, Clément K, McQuaid SE, et al. Elevated expression of osteopontin may be related to adipose tissue macrophage accumulation and liver steatosis in morbid obesity. Diabetes. 2009;58:125–133. doi: 10.2337/db08-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kodama K, Horikoshi M, Toda K, Yamada S, Hara K,Irie J, Sirota M, Morgan AA, Chen R and Ohtsu H, et al. Expression-based genome-wide association study links the receptor CD44 in adipose tissue with type 2 diabetes. Proc Natl Acad Sci U S A. 2012;109:7049–7054. doi: 10.1073/pnas.1114513109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weinhage T, Wirth T, Schutz P, Becker P, Lueken A,et al. The Receptor for Advanced Glycation Endproducts (RAGE) Contributes to Severe Inflammatory Liver Injury in Mice. Front Immunol. 2020;11:1157. doi: 10.3389/fimmu.2020.01157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.