

ABSTRACT
T-cell engaging biologics is a class of novel and promising immune-oncology compounds that leverage the immune system to eradicate cancer. Here, we compared and contrasted a bispecific diabody-Fc format, which displays a relatively short antigen-binding arm distance, with our bispecific IgG platform. By generating diverse panels of antigen-expressing cells where B cell maturation antigen is either tethered to the cell membrane or located to the juxtamembrane region and masked by elongated structural spacer units, we presented a systematic approach to investigate the role of antigen epitope location and molecular formats in immunological synapse formation and cytotoxicity. We demonstrated that diabody-Fc is more potent for antigen epitopes located in the membrane distal region, while bispecific IgG is more efficient for membrane-proximal epitopes. Additionally, we explored other parameters, including receptor density, antigen-binding affinity, and kinetics. Our results show that molecular format and antigen epitope location, which jointly determine the intermembrane distance between target cells and T cells, allow decoupling of cytotoxicity and cytokine release, while antigen-binding affinities appear to be positively correlated with both readouts. Our work offers new insight that could potentially lead to a wider therapeutic window for T-cell engaging biologics in general.
KEYWORDS: Bispecific engineering, cd3, t-cell engager, therapeutic window
Introduction
T cell receptors (TCRs) on lymphocytes and the major histocompatibility complex (MHC) on antigen-presenting cells (APCs) determine the specificity of T cell response to foreign antigens. TCR, composed of two highly variable heterodimeric protein chains that render antigen specificity, has a short cytoplasmic domain that lacks any known signaling motif. Therefore, upon TCR engagement of peptide-loaded MHC (pMHC), transduction of intracellular signals depends on non-covalent association of the complex with the common cluster of differentiation 3 (CD3) chains, composed of four distinct polypeptides, namely CD3ε, γ, δ, and ζ chains, paired as three dimers (εγ, εδ, and ζζ).1 The cytoplasmic tails of the CD3 chains have a total number of 10 immunoreceptor tyrosine activation motifs (ITAMs),2 with three contributed by one CD3ζ chain and one contributed by each of the other CD3 chains. Together with other secondary signals, this complexity in ITAMs allows the so-called “scalable signaling” to orchestrate diverse T cell responses, including activation, proliferation, effector function, or apoptosis.3
The clinical success and approval of blinatumomab (BLINCYTO®; anti-CD19/CD3 tandem single-chain variable fragment (scFv)) in treating relapsed or refractory acute lymphoblastic leukemia has opened new avenues for retargeting T cells for cancer treatment.4 Termed as Bispecific T-cell Engager (BiTE), this type of molecule is designed to bridge T cells and tumor cells through interactions with the invariant CD3 chain on T cells and a tumor-associated antigen (TAA) on cancer cells. On the molecular basis, through bypassing MHC/TCR recognition, this approach is capable of inducing T cell activation with no clonal restriction, as well as overcoming one of the tumor escape mechanisms of down-regulating MHC expression.5 Encouraged by the ground-breaking efficacy of blinatumomab, researchers have expanded upon the tandem scFv format to target different TAAs6–8 and have further investigated other bispecific formats,9–12 many of which contain an Fc domain for extended systemic half-life. It is worth noting that, although the interaction is very different from the TCR-pMHC ligation under physiological conditions, subsequent studies revealed that the downstream signaling cascades induced by both events resemble each other.13
Previously, through building on our charge-steering Fc heterodimerization technology,14 we engineered a bispecific IgG2 platform for T-cell engagement and demonstrated its superior pharmacological activity and minimal nonspecific activation. In this study, we sought to take a deeper dive into how epitope, binding affinity, receptor density, and kinetics drive the activity of T-cell-engaging biologics. In light of the finding that geometric configurations may play critical roles in forming immunological synapses,9 we created a new bispecific diabody-Fc (DbFc) format, which adopts a much more compact configuration between the two paratopes. More specifically, the distance between the two antigen-recognition arms was estimated to be 3–6 nm15,16 and 9–15 nm17–19 (Figure 1) for our bispecific DbFc and previously described IgG-based molecules, respectively. This difference in distance between binding sites makes them ideal tools to study the effectiveness of synapse formation as a function of distance. By using B-cell maturation antigen (BCMA) as the TAA in our model system, we first generated a series of artificial antigen-expressing cell lines whereby BCMA, a relatively small glycoprotein, is tethered to the cell surface via increasing numbers of protein domains to gradually extend its distance from the membrane, or in another scenario, is anchored to the juxtamembrane region and masked by growing numbers of structural spacer units.
Figure 1.
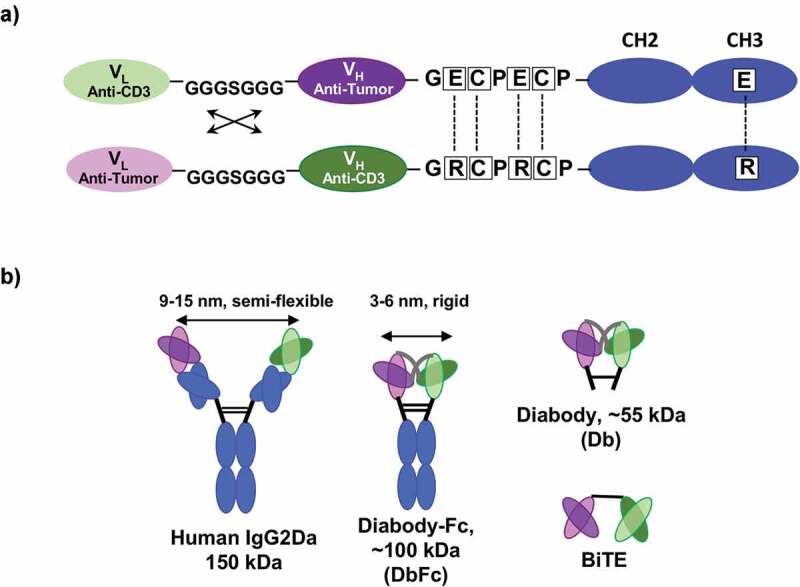
Generation and optimization of diabody-Fc (DbFc) fusion for T-cell redirection. (a) Schematic diagram of DbFc construct after optimization of the hinge and linker sequences between the variable domains; (b) Relative geometric configurations of bispecific IgG2, DbFc, diabody (Db), and BiTE
We discovered that the DbFc format, due to its short arm distance, is more effective in killing cell lines with BCMA with a long tether, when compared to IgG2. However, the trend is completely the opposite when BCMA is located at the membrane-proximal region and masked by additional protein domains. In this case, the long arm length of the bispecific IgG2 is generally superior in overcoming steric hinderance, and therefore effectively bridges both masked BCMA on antigen-expressing cells and CD3 on T cells. We then explored the potencies of both formats on a non-engineered known TAA, FMS-like tyrosine kinase 3 (FLT3), whose extracellular domain is composed of five immunoglobulin-like (Ig-like) domains. By generating antibodies against different domains of FLT3, we validated the aforementioned observations on BCMA cell lines. Lastly, we took a combinatorial approach to explore the interplay of binding affinity of either arm, receptor density, kinetics, and molecular formats in affecting cytotoxicity and cytokine release in the multi-dimensional space. Our results demonstrated successful decoupling of cytotoxicity and cytokine release. Furthermore, we integrated our comprehensive data set into a unified mechanistic mathematical model to guide affinity selection for optimal pharmacological effects. Our research highlights the importance of understanding the complexity in developing T-cell recruiting protein therapeutics. As a result, careful navigation through the optimization process is required to achieve the desired potency and safety profile.
Results
Bispecific DbFc is more potent than full-length bispecific IgG when antigen epitope is located in the membrane-distal region
In order to compare with full-length IgG, we generated and optimized a DbFc fusion molecule by separating cognate variable heavy and light domains on two polypeptide chains, which during folding are associated through preferential non-covalent VH/VL pairing and further stabilized through the hinge region and CH3 domains. Like other diabody (Db) formats reported in prior studies,20 the linker between the variable domains of the same chain was designed to be short enough to force pairing with variable domains on the other chain. Sequential optimization of hinge and linker sequences yielded a bispecific DbFc fusion molecule that displays good expression titer, product integrity and thermostability, and unaltered binding to both CD3 and TAA (Supplementary Tables 1 and Tables 2, Hinge 3, and L2). The final configuration of our DbFc format is shown in Figure 1a. Figure 1b illustrates the relative geometry of the bispecific IgG2 (our preferred full-length IgG platform), DbFc, Db, and BiTE. The most remarkable difference between IgG and DbFc is that the distance between the two antigen-binding arms of IgG molecules is usually 9–15 nm17–19 while that of Db or DbFc is ~3–6 nm,15,16 allowing investigation of the distance effect in redirected T cell cytotoxicity. Additionally, the difference in structural flexibility of IgG and Db molecules may also play a role in antigen recognition.
Epidermal growth factor (EGF)-like domains, originally identified in EGF and characterized by three intradomain disulfide bonds, are protein subunits present in a wide variety of extracellular proteins such as Notch, fibrillin, and various blood factors.21 One of their important functions is to serve as relatively rigid structural spacers,22as observed in crystal structures of tandem EGF-like domains from Notch 1 receptor (PDB ID: 5MWB)23 and Jagged-1 protein (PDB ID: 2VJ2).24 To investigate the effect of antigen epitope location in T-cell redirected lysis in a systematic fashion, we engineered cell lines that express BCMA antigen with different EGF-like domains as tethers to increase the distance to the target cell membrane. Based on the numbers of EGF-like domains used, these cell lines were named T0, T1, T2, …,T7, with T abbreviated from “tether.” Schematic views of these receptors based on structural modeling are shown in Figure 2a. Fluorescence-activated cell sorting (FACS) analysis revealed similar BCMA expression levels (~104 receptors per cell) across all eight cell lines (Supplementary Figure 1), making a direct comparison of these cell lines feasible.
Figure 2.
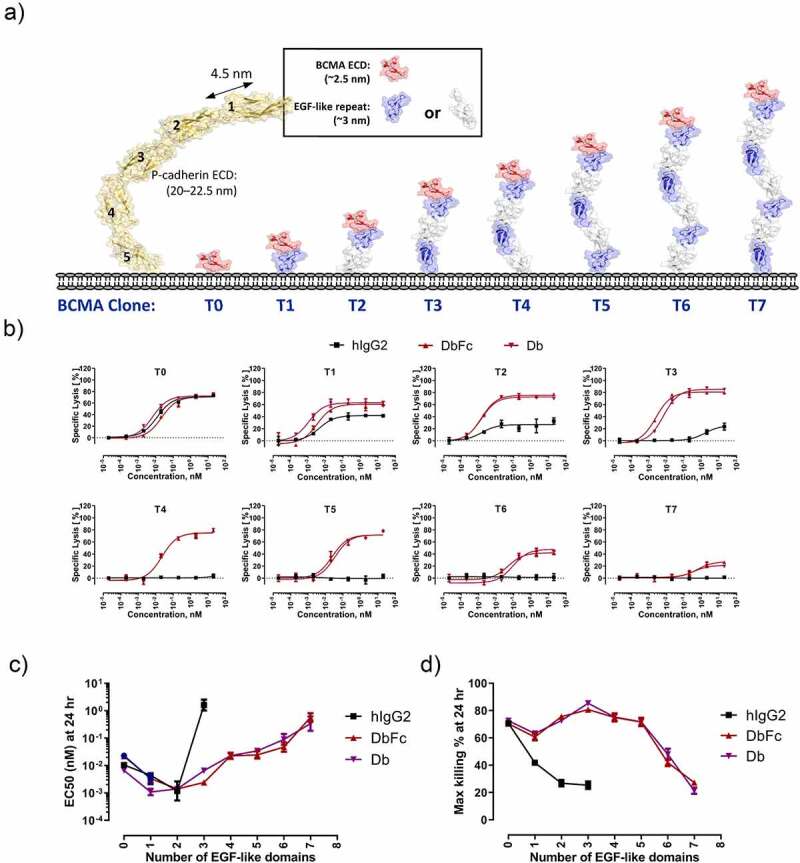
Differential cytotoxicity profiles of bispecific anti-BCMA/anti-CD3 IgG2, DbFc, and Db on engineered cell lines where BCMA is tethered to the cell surface with increasing distance from the membrane. (a) Schematic diagram of T0-T7 cell lines, with P-cadherin ectodomain structure depicted alongside to show relative dimensions; (b) Cytotoxicity of aforementioned four bispecific molecules on the eight cell lines at 24-hour timepoint; (c) EC50 values as a function of the number of EGF-like domains; (d) Maximal killing (Emax) values as a function of the number of EGF-like domains
We then evaluated the in vitro potencies of anti-CD3/anti-BCMA bispecific IgG2, DbFc (with optimized hinge and linker), and Db in lysing these cell lines in the presence of purified T cells. At the effector-to-target ratio (E:T ratio) of 5:1 at a 24-h time point, all three molecules displayed nearly equivalent activity in inducing lysis of the T0 cell line, which presents BCMA on the cell surface without any tether. As the distance to the cell surface increases with the number of EGF-like domains, the ability of bispecific IgG2 in inducing T cell redirected lysis was quickly dampened, as indicated by EC50 as well as maximal killing Emax in cell lines from T0 to T3, shown in Figure 2b. In contrast, Db and DbFc displayed comparable and robust cytotoxicity in cell lines T0 through T5, with noticeable reduction in potency in T6 and very weak activity in T7. Figure 2c and Figure 2d illustrates the overall correlation of EC50 and Emax values as a function of the number of EGF repeats for all three molecules. The monotonic decline in cytotoxicity, as the distance between the antigen epitope to target cell membrane increases due to the tether length, indicated that the efficiency of immunological synapse formation could be critically affected by the extent bispecific molecules can bring T cells and target cells into proximity. The disparity of full-length IgG and Db-derived molecules could be explained by the arm distance of the two antigen-binding sites, as the more rigid and compact structure of Db and DbFc gave rise to a relatively shorter distance between T cells and antigen-expressing cells. The rapid potency reduction between one cell line to the next may indicate a distance threshold for productive engagement and subsequent T cell activation, which we measured indirectly as cytotoxicity. In the case of the T0 cell line, full-length IgG and Db displayed similar potency, suggesting that once the distance threshold is met, making the intermembrane distance shorter does not seem to provide an additional enhancement in cytotoxicity.
In order to rule out the possibility that our observation somehow pertained to properties of the EGF-like domains rather than the simple distance effect, we engineered another series of cell lines by substituting the EGF-like domain with a different structural unit, Ig-like domain, as the tether (referring as t1ʹ, t2ʹ, t3ʹ …). Unlike the EGF-like domain, which is roughly 3 nm per unit in length, the Ig-like domain is estimated to add 5 nm per unit based on crystal structures,25 although the domains may not be oriented in a completely linear fashion with respect to each other. As depicted in Supplementary Figure 2, the dose-dependent cytotoxicity of one of the IgG2-based bispecific molecules on T2 and t1ʹ cell lines was nearly identical, with their estimated tether distances being 6 nm and 5 nm, respectively. Similarly, the activities on T3 and t2ʹ cell lines were also comparable, with their estimated tether distances being 9 nm and 10 nm, respectively. This suggests that the differences in cytotoxicity were likely entirely due to antigen epitope location and the resulting synaptic distance between T cells and target cells, regardless of which protein domains were used as tethers. Based on these data, we propose that the intermembrane breadth between target and T cells, which can be contributed by the distance of antigen epitope relative to target cell membrane as well as the format of the bispecific molecule, plays an important role in forming productive immunological synapses and eventually redirected T-cell killing.
Full-length bispecific IgG is more potent than DbFc when antigen epitope is masked at the membrane-proximal region
We then examined the opposite case, where the antigen epitope is located at the membrane-proximal region and masked by different numbers of structural units on top of it. To achieve this, we engineered another set of BCMA-expressing cell lines, with 0, 2, 4, or 7 EGF-like domains stacked above the BCMA antigen, respectively (Figure 3a). For simplicity, these cell lines will be referred as M0v, M2v, M4v, and M7v, with receptor density on the order of 105 per cell (Supplementary Figure 3). IgG2- and DbFc-based bispecific molecules were evaluated using these cell lines (Figure 3b and Figure 3c). For the cell line M0v without any masking, both molecules displayed nearly indistinguishable EC50 and Emax, at both 15 h and 24 h time points, with 24 h data clearly showing higher maximal killing and significantly left-shifted EC50. For M2v and M4v at the 15 h time point, the DbFc bispecific was considerably inferior to the IgG2-based molecule, with Emax reaching less than 20% for the former, while nearly complete lysis was observed for the latter at high concentration (10 nM). However, the dose–response cytotoxicity curves of the two molecules converge at the 24 h timepoint, indicating that the result we observed at 15 h was likely due to kinetic differences in T cell activation. Judging from the fact that two tandem EGF-like domains, presumably arranged in a linear fashion, would already exceed the arm distance of DbFc, we hypothesize that there might be some structural flexibility of the EGF-like domains that allows exposure of membrane-proximal BCMA antigen, given a sufficient amount of time. For M7v cell line, even at 24 h, Emax of DbFc plateaued at ~50%, lower than the IgG2-based molecule, indicating that in this case even flexibility may not overcome steric hinderance to compensate for the low probability of DbFc in bridging of CD3 and BCMA.
Figure 3.
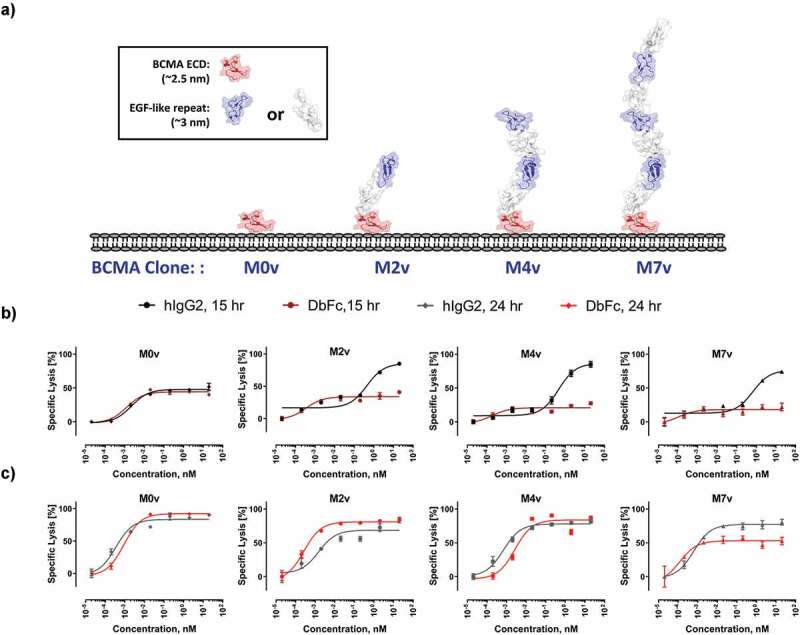
Differential cytotoxicity profiles of bispecific anti-BCMA/anti-CD3 IgG2 and DbFc engineered cell lines where BCMA is located at the membrane proximal region and masked by increasing number of EGF-like domains. (a) Schematic diagram of the M0, M2, M4, and M7 cell lines depicting how BCMA and the masking EGF-like domains may potentially orient on the cell surface; (b) Dose-dependent cytotoxicity of the IgG2 and DbFc on the four cell lines at 15-hour timepoint; (c) Dose-dependent cytotoxicity of the IgG2 and DbFc on the four cell lines at 24-hour timepoint
In vitro potencies of anti-CD3/anti-FTL3 bispecific IgG and DbFc are correlated with the location of epitope
To gain insight on how epitope location could potentially influence cytotoxicity in a less artificial setting, we engineered and investigated various anti-FLT3/anti-CD3 bispecific molecules on the EoL1 acute myeloid leukemia (AML) cell line that over-expresses FLT3. FLT3 is a receptor tyrosine kinase with an extracellular domain comprising five Ig-like domains. Crystal structures of FLT3 in complex with its ligand revealed the relative domain orientations,26 as shown in Figure 4a, although we cannot rule out the possibility that this conformation may be partially induced by ligand binding. Antibodies against different domains of FLT3 were raised and selected to pair with the same anti-CD3 clone to yield IgG2- and DbFc-based bispecific molecules. As depicted in Figure 4b, binding of IgG2 and DbFc molecules to FLT3-positive EoL1 cells are comparable, as analyzed by FACS. Consistent with our findings using an engineered BCMA-expressing series of cell lines, bispecific molecules against different domains of FLT3 displayed drastically different cytotoxicity that was strongly influenced by the molecular format (Figure 4c–g). DbFc targeting the membrane-distal domains D1 and D3 mediated considerably more potent cytotoxicity than its IgG2 counterpart (Figure 4c, e), likely due to its ability to bring the two cell membranes close together. On the other hand, IgG2 bispecific molecules against the membrane-proximal domains D4 and D5 induced specific target cell lysis, while no significant cytotoxicity was observed at concentrations up to 10 nM for the same anti-FLT3 antibody clones in DbFc format (figure 4f, g). Figure 4h shows a schematic view of a potential explanation of a lack of cytotoxicity for D4 and D5-targeted DbFc. The same trend holds true for another AML cell line MV411, which has relatively lower FLT3 expression compared to EoL1. Owing to the lower receptor density, EC50 values could not be determined for most cases, but a similar pattern of dose-response was observed (Supplementary Table 3 and Supplementary Figure 4). Collectively, these data support the notion that antigen epitope location, and hence the intermembrane spacing generated by bispecific molecules of different architectures, may regulate the efficiency of redirected T cell killing. It is worth noting that in the case of MV411, even at a 48-h timepoint, DbFc fusion was not able to overcome the masking effects, as we previously observed for the engineered M series of cells, and we hypothesize that it could be due to the discrepancy in receptor density.
Figure 4.
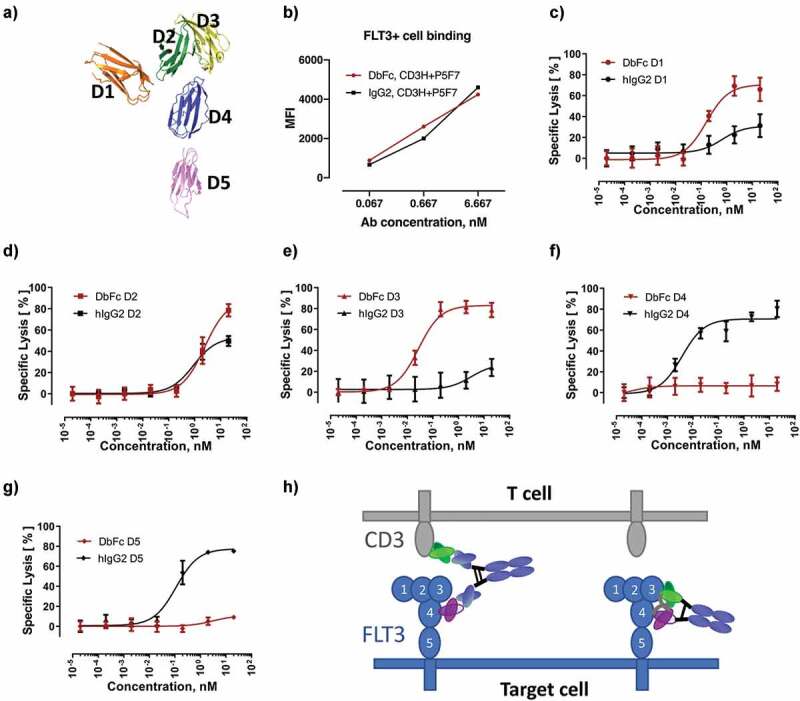
Case study using the multi-domain FTL3 as antigen demonstrating that DbFc more potently targets membrane-distal epitopes while IgG2 bispecific is more potent for membrane-proximal epitopes. (a) Crystal structure of the FLT3 extracellular region (PDB ID: 3QS9); (b) Binding of DbFc and bispecific IgG (derived from the same pairs of anti-FLT3 and anti-CD3 variable domains) to cells expressing FLT3 is equivalent over the concentration ranges tested here; (c-g) T-cell redirected lysis of the FLT3-overexpressing AML cell line EoL-1 in the presence of increasing concentrations of DbFc and IgG2-based anti-FLT3/anti-CD3 targeting domains 1, 2, 3, 4, 5 of the FLT3, respectively; (h) Schematic diagram illustrating why IgG format gives rise to more effective bridging between the epitopes located at domains 4 or 5 of FLT3 on target cell and CD3 on T cell
The interplay of receptor density, epitope location, molecular format, kinetics and binding affinities in cytotoxicity and cytokine release
Some data above may already suggest that, in addition to TAA epitope location and molecular format, there are other key parameters such as receptor density, kinetics, and affinities of both arms that could significantly affect cytotoxicity readout. These parameters can be classified into two essential categories: intrinsic factors, such as receptor density and time course, that cannot be changed; and extrinsic factors, including affinities, molecular format, antigen epitope location, that can be potentially modulated through engineering designs. Therefore, to inform rational design of bispecific T-cell engaging proteins, we took a systematic approach to interrogate how these parameters influence cytotoxicity.
First, we engineered high, intermediate, and low affinity variants for both the CD3 and BCMA arms (affinity mutants originated from the same antibody clone to ensure binding to the same epitope), namely CD3-H, CD3-M, CD3-L, BCMA-H, BCMA-M, and BCMA-L. Their binding affinities are 24.4 nM, 110 nM, 394 nM, 214 pM, 34.1 nM, and 934 nM, respectively (as measured in bispecific IgG format, similar results were obtained for bispecific DbFc, see Supplementary Table 4). All nine permutations were generated for both IgG2 and DbFc format, totaling 18 bispecific molecules. To investigate how receptor density influences cytotoxicity, a low BCMA-expressing cell line, L0, was created in addition to the T0 and M0v cell lines, which were used in previous sections. The receptor density levels are on the orders of 103, 104, and 105, respectively, for these three cell lines. Additionally, two timepoints, 24 h and 48 h, were sampled to shed light on the kinetics. This comprehensive analysis of dose-dependent cytotoxicity involves 108 assay conditions, as shown in Supplementary Figure 5. In order to account for both EC50 and Emax values, especially for dose–response curves that did not plateau or plateaued at very different levels, we chose the Area Under the Curve (AUC, also known as activity area) approach for comparing differential activities of various molecules and assay conditions. This data analysis framework was previously reported to correlate better than EC50 alone between different studies.27 We first examined how IgG and DbFc molecules compare on low, medium, and high BCMA-expressing cell lines, when the same antigen variable domains were used in both molecular formats. As shown in Figure 5a (24 h) and 5B (48 h), at medium and high receptor density, IgG and DbFc are nearly equally potent, with data points scattered along the diagonal line, irrespective of the timepoints. However, for the cell line with low receptor expression L0, DbFc is far superior to the IgG format, especially at 24 h, as data points are clustered along the Y-axis. To evaluate how binding affinity influences cytotoxicity, we conducted pair-wise comparisons by matching all other parameters (molecular format, cell line, affinity of the other arm) and plotting AUC of cytotoxicity for high, intermediate, and low affinity variants against that of the intermediate affinity molecule as the benchmark (X-axis) (Figure 5c–f). It is evident that when using the intermediate affinity CD3-M or BCMA-M as references, higher affinity, either in the CD3 arm or BCMA arm, always resulted in more potent killing (above the diagonal line) while lower affinity induced less cell lysis (below the diagonal line). Globally, data points are more clustered to the origin at 24 h compared to the 48 h timepoint, suggesting that T cell activation and subsequent lysis of target cells were not saturated within the first day.
Figure 5.
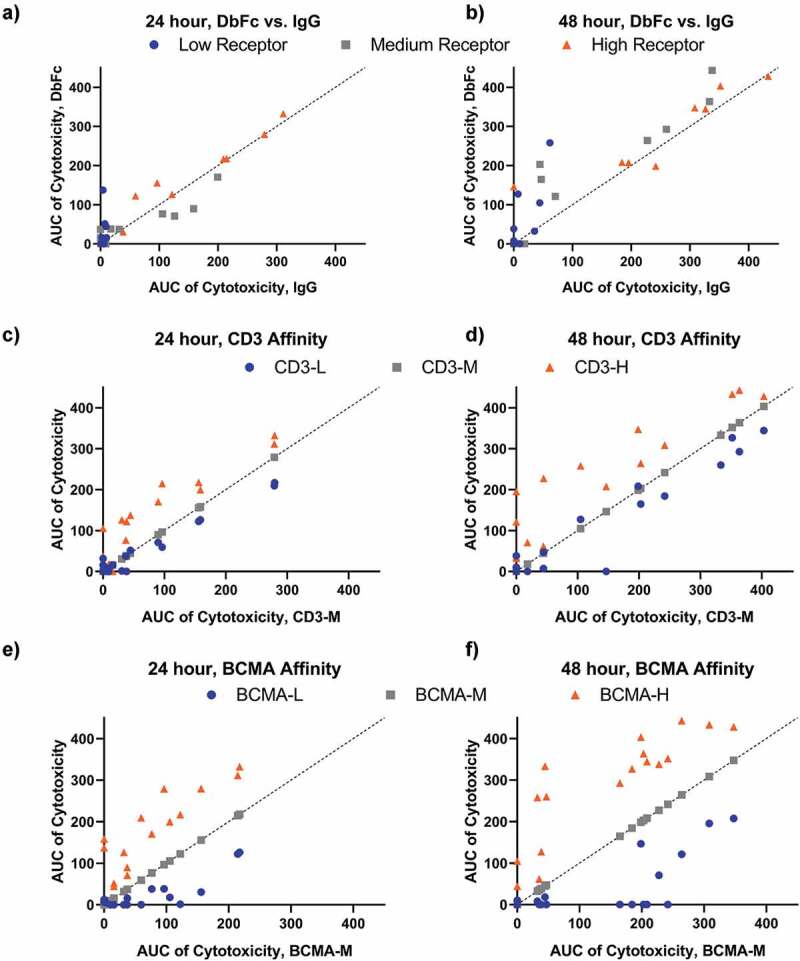
The influence of receptor density, antigen-binding affinities, molecular format, and cytotoxicity kinetics, as represented by AUC (raw data in Supplementary Figure 5). Dashed lines represent y = x.(a-b) Pair-wise comparison of cytotoxicity of DbFc versus IgG (with the same antigen-binding arms) on cell lines with low, medium, and high receptor expression at 24 and 48 h timepoints; (c–d) Pair-wise comparison of cytotoxicity of CD3 affinity variants at 24 and 48 h timepoints, with all other parameters (cell line, affinity to BCMA, molecular format, receptor density) being the same; (e–f) Pair-wise comparison of cytotoxicity of BCMA affinity variants at 24 and 48 h timepoints, with all other parameters (cell line, affinity to CD3, molecular format, receptor density) being the same
We then investigated if it is possible to decouple cytotoxicity and cytokine release through any combination of parameters that would yield strong T-cell mediated killing and minimal induction of cytokine secretion. In this experiment, CD3 binding arms with different affinities (CD3-L, CD3-M, and CD3-H) were paired with the same high-affinity BCMA arm (BCMA-H) in both IgG2 and DbFc format, and their dose-dependent cytotoxicity and cytokine release (including tumor necrosis factor (TNF), interleukin-2 (IL-2), and interferon (IFN)γ) were measured for cell lines T0, T2, T4, and T7 (Supplementary Figure 6). AUCs of cytotoxicity and cytokine release were obtained based on fitted dose–response curves and their correlation were plotted. By grouping the data set by molecular format, it is evident that the correlations of TNF, IL-2, and IFNγ with respect to cytotoxicity were very similar (Figure 6a–c). To reduce the complexity of data analysis, we decided to focus on the AUC ratio of cytotoxicity and TNF and generated tables containing subsets of data to compare molecular format (Table 1), antigen epitope location (as represented by cell lines, Table 2), and CD3 affinity (Table 3) while fixing all other variables. The larger value of the AUC ratio of cytotoxicity and TNF secretion indicates less cytokine release. As shown in Table 1, for T0 cell line, IgG2-based bispecific molecules produced much lower levels of TNF, in the cases of CD3-H and CD3-M, with an AUC ratio of cytotoxicity and TNF secretion valued at ~2, in contrast to 0.5 for equivalent molecules in DbFc format. However, as listed in Table 2, in the T2 cell line, DbFc format achieved similar AUC ratio for both CD3-H and CD3-M binders. We observed, given the same CD3 binder, the effect of increasing tether length by 2 EGF domains can be fully compensated for by decreasing the arm distance of the bispecific molecule (IgG2 to DbFc), highlighting the fact that intermembrane distance could be contributed by both molecular architecture and antigen epitope location. Moreover, in Figure 6a, there are pairs of data points that clustered together on these correlation plots (circled in dashed green lines), meaning that they conferred similar cytotoxic activity and generated similar amounts of cytokine. Closer examination of these pairs revealed that they were the following combinations: (T4, CD3-H, DbFc) and (T2, CD3-H, IgG2); (T2, CD3-M, DbFc) and (T0, CD3-M, IgG2); and (T2, CD3-H, DbFc) and (T0, CD3-H, IgG2), respectively, further supporting our hypothesis that molecular format and antigen epitope location are the key determinants in modulating cytotoxicity and cytokine secretion (Supplementary Table 5, Figure 7).
Figure 6.
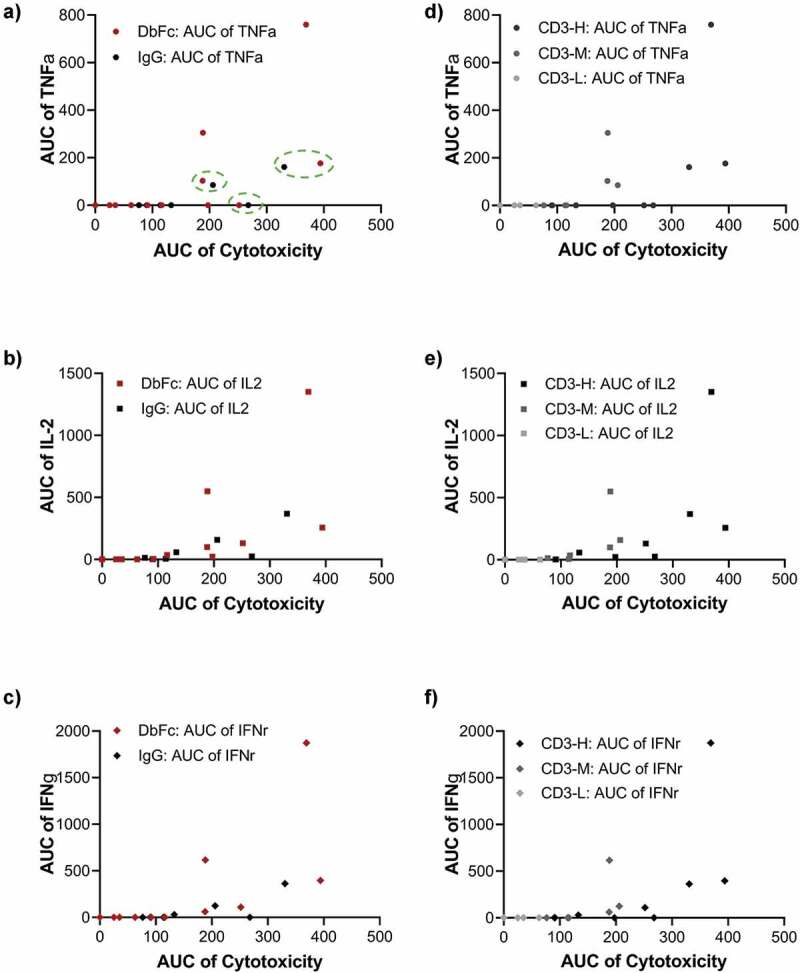
Deconvolution of cytotoxicity and cytokine release is achievable through a combination of molecular format and antigen epitope location. (a-c) Correlation of cytotoxicity versus the release of cytokines TNFα, IL-2, and IFNγ shown as AUC scatter plots, with data sets grouped based on molecular format; (d-f) Correlation of cytotoxicity versus the release of cytokines TNFα, IL-2, and IFNγ shown as AUC scatter plots, with data sets grouped based on CD3 binding affinity
Table 1.
AUC Ratios of cytotoxicity versus TNFα when comparing molecular format
| Cell line | CD3 Binder | Format | AUC |
||
|---|---|---|---|---|---|
| Cytotoxicity | TNFα | Ratio | |||
| T0 | CD3-H | DbFc | 369.1 | 759.8 | 0.5 |
| T0 | CD3-H | IgG | 330.6 | 161.0 | 2.1 |
| T0 | CD3-M | DbFc | 188.6 | 304.9 | 0.6 |
| T0 | CD3-M | IgG | 206.0 | 84.8 | 2.4 |
| T2 | CD3-H | DbFc | 394.1 | 176.4 | 2.2 |
| T2 | CD3-H | IgG | 268.0 | 0 | - |
Table 2.
AUC Ratios of cytotoxicity versus TNFα when comparing epitope location
| Cell line | CD3 Binder | Format | AUC |
||
|---|---|---|---|---|---|
| Cytotoxicity | TNFα | Ratio | |||
| T0 | CD3-H | DbFc | 369.1 | 759.8 | 0.5 |
| T2 | CD3-H | DbFc | 394.1 | 176.4 | 2.2 |
| T4 | CD3-H | DbFc | 251.7 | 0.0 | |
| T7 | CD3-H | DbFc | 197.4 | 0.0 | |
| T0 | CD3-M | DbFc | 188.6 | 304.9 | 0.6 |
| T2 | CD3-M | DbFc | 188.0 | 103.3 | 1.8 |
| T4 | CD3-M | DbFc | 116.2 | 0.0 | |
| T7 | CD3-M | DbFc | 91.7 | 0.0 | |
| T0 | CD3-H | IgG | 330.6 | 161.0 | 2.1 |
| T2 | CD3-H | IgG | 268.0 | 0.0 | |
| T4 | CD3-H | IgG | 132.9 | 0.0 | |
| T7 | CD3-H | IgG | 90.5 | 0.0 | |
| T0 | CD3-M | IgG | 206.0 | 84.8 | 2.4 |
| T2 | CD3-M | IgG | 114.0 | 0.0 | |
| T4 | CD3-M | IgG | 76.5 | 0.0 | |
| T7 | CD3-M | IgG | 0.0 | 0.0 | |
Table 3.
AUC Ratios of cytotoxicity versus TNFα when comparing CD3 affinity
| Cell line | CD3 Binder | Format | AUC |
||
|---|---|---|---|---|---|
| Cytotoxicity | TNFα | Ratio | |||
| T0 | CD3-H | DbFc | 369.1 | 759.8 | 0.5 |
| T0 | CD3-M | DbFc | 188.6 | 304.9 | 0.6 |
| T0 | CD3-L | DbFc | 35.1 | 0.0 | - |
| T2 | CD3-H | DbFc | 394.1 | 176.4 | 2.2 |
| T2 | CD3-M | DbFc | 188.0 | 103.3 | 1.8 |
| T2 | CD3-L | DbFc | 62.9 | 0.0 | |
| T0 | CD3-H | IgG | 330.6 | 161.0 | 2.1 |
| T0 | CD3-M | IgG | 206.0 | 84.8 | 2.4 |
| T2 | CD3-H | IgG | 268.0 | 0.0 | |
| T2 | CD3-M | IgG | 114.0 | 0.0 | |
Figure 7.
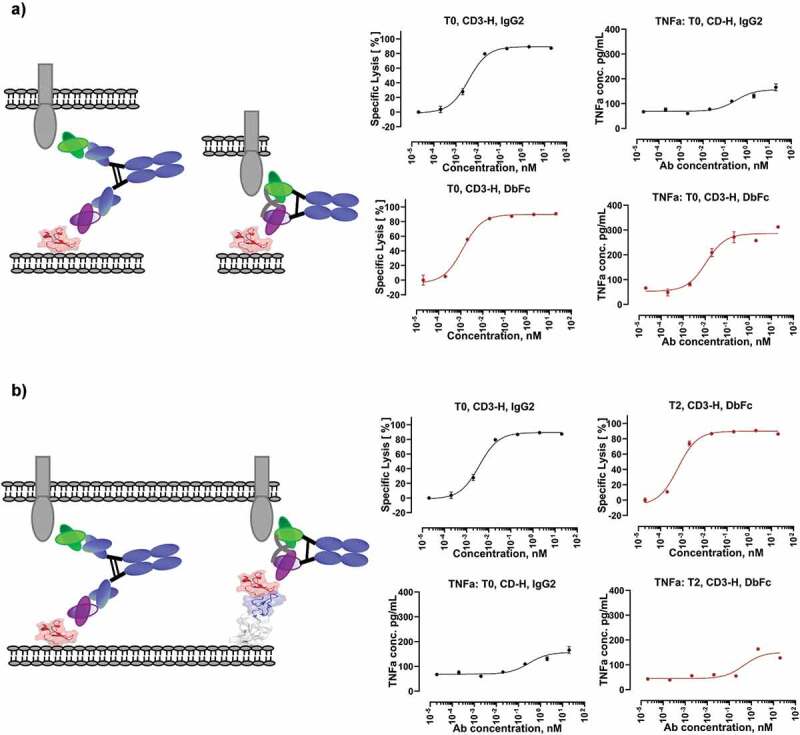
Exemplary cytotoxicity and cytokine release profiles for different scenarios. (a) Same cell line (T0) and same antibody clone with the only difference in therapeutic modality suggest that it is possible to decouple cytotoxicity and cytokine secretion by modulating therapeutic modality; (b) T0, IgG2 and T2, DbFc demonstrated similar potency and cytokine secretion, indicating that the optimal intermembrane distance can be achieved by a combination of modality and antigen epitope location
Alternatively, if we group the data set by CD3 arm affinity (Table 3 and Figure 6d–f), both cytotoxicity and cytokine release follow a similar trend with higher affinity molecules being more potent, accompanied with more cytokine secretion, suggesting that affinity to CD3 may not be a differentiating parameter to disentangle the two readouts, at least in the affinity ranges we chose. For pair-wise comparison of CD3-H and CD3-M, refer to Supplementary Figure 7.
Integration of bispecific IgG and DbFc cytotoxicity data into a mechanistic mathematical model that can guide optimal affinity selection
As the results above show, the potency of CD3 bispecifics depends on multiple target/epitope-specific and drug-specific factors. To further understand this multi-factorial dependence, we developed a unified mechanistic mathematical modeling framework that can explain the observed exposure–response relationship (in vitro cytotoxicity results) across the different CD3 bispecifics and conditions tested above.
To develop this unified framework, we constructed a mechanistic mathematical model to relate CD3 bispecific concentration to in vitro cytotoxicity similar to previous published models.28 Specifically, the model describes the formation of TAA-Bispecific-CD3 Trimers, whose number drives tumor cell killing by T-cells in the cytotoxicity assay (Figure 8a). The formation of these Trimers in our model depends on the target receptor density and CD3/target binding affinities. Further, our model assumes that the tumor killing rate is a saturable Hill-type function of the Trimer #/tumor-cell thus formed (demonstrated in Figure 8b).
Figure 8.
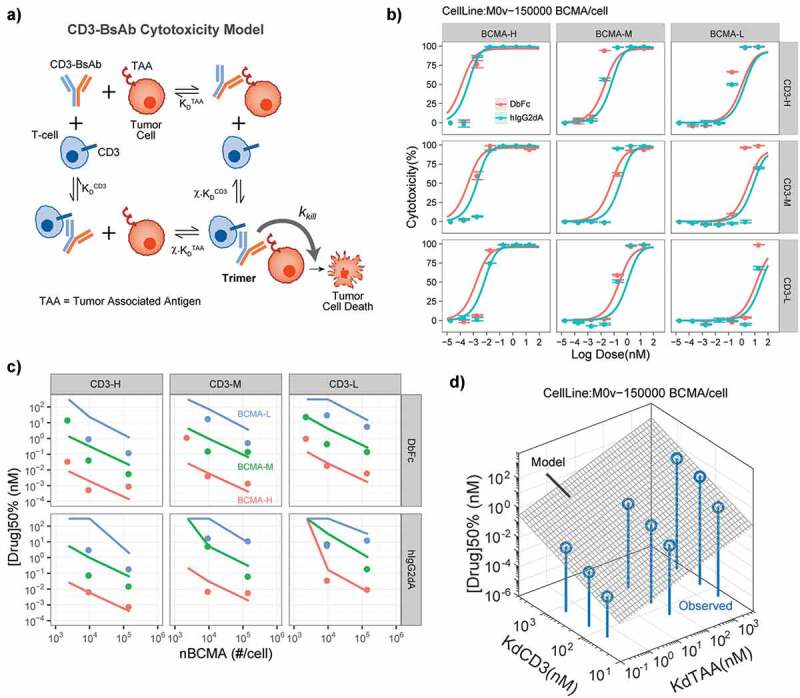
Mathematical model of Trimer-based CD3-bispecific cytotoxicity explains dependence of potency on receptor density and Tumor antigen/CD3 affinities. (a) Structure of mathematical model for TAA-Drug-CD3 Trimer formation and tumor cell in vitro cytotoxicity. (b) Model fits for BCMA-CD3 bispecifics with different combinations of CD3 and BCMA affinities. Lines and symbols show model fits and observations respectively. Colors indicate DbFc vs IgG2 formats. (c) Model explains observed potency [Drug]50% (= concentration to reach 50% cytotoxicity) dependence on BCMA density and Tumor antigen/CD3 affinities. Lines and symbols represent model predictions and observations respectively. (d) Model-predicted scaling relationship of (gray surface) explains the observed sensitivity of potency to these parameters
To parameterize the killing rate Trimer dependence in our model, we simultaneously fit the data for in vitro cytotoxicity of the BCMA-CD3 bispecifics described in the previous section and estimated the maximum kill rate (kkill) and the Trimer level for threshold for half-maximal killing (Kc50Trimer). Our results showed that the model can describe the concentration-cytotoxicity relationship for all chosen receptor density and CD3/BCMA binding affinity combinations simultaneously with a single set of Trimer-based killing parameters (Figure 8b and Supplementary Figure 8). Crucially, the concentration-cytotoxicity relationship for different modalities (DbFc vs IgG2) required different Trimer-based killing parameters (Figure 8b and Table 4). While the maximum kill-rate for both modalities was estimated to be similar, the Trimer threshold for DbFc bispecifics was ~20-fold lower than that for the IgG2 bispecifics at comparable CD3/BCMA binding affinities. These results suggest two key insights: 1) the exposure–response relationship of CD3 bispecifics across variations of receptor density and CD3/target binding affinities can indeed be predicted by a single relationship that depends on the formation of Trimers; and 2) the modality of CD3 bispecifics affects their potency in ways different than changes of binding affinities by affecting the potency of the Trimers themselves.
Table 4.
Estimated parameters of the in vitro Cytotoxicity model for CD3 Bispecifics
| Parameter | DbFc Estimate (CV%) |
hIgG2dA Estimate (CV%) |
Description |
|---|---|---|---|
| kmax(1/hr) | 0.075 (20) | 0.11 (10) | Max. Tumor cell kill rate |
| Kc50Trimer*χ (#/nM) | 3.4e+05 (58) | 6.75e+06 (<1) | Half-maximal Trimer threshold for killing rate |
| nk | 0.7 (11) | 0.74 (6) | Hill-coefficient (steepness of killing curve) |
CV% = Coefficient of Variation for parameter estimates.
Next, we wanted to determine if a general relationship exists to describe the dependence of the potency of the bispecific and its key drivers, i.e., receptor density and CD3/target binding affinities. For this, we defined [Drug]50% as the bispecific concentration that achieves 50% cytotoxicity and used it as the key measure of potency and applied sensitivity analysis to the parameterized model to predict this potency. Comparison of our model predicted [Drug]50% showed that it was able to capture its observed dependence on receptor density and CD3/target binding affinities for both DbFc and IgG2 formats (Figure 8c). Further analysis of our model equations also suggested a simple relationship that could capture these dependencies:
Where KDTAA, KDCD3, and nTAA represent the TAA, CD3 binding affinities, and TAA (here BCMA) receptor density, respectively. This model-predicted relationship implies an equivalent sensitivity of bispecific potency to TAA and CD3 binding affinities, suggesting that any loss of potency via reduction in CD3 affinity, for example, can be compensated by a proportional increase in TAA affinity. We directly validated this relationship by fitting the observed [Drug]50% at different receptor density and CD3/target affinities and found that these potencies can indeed be captured in a similar scaling relationship (Table 5) with near equivalent sensitivity to KDTAA and KDCD3.
Table 5.
Estimated parameters of the model-predicted in vitro Cytotoxicity potency scaling relationship:
| Parameter | DbFc Estimate (CV%) |
hIgG2dA Estimate (CV%) |
Description |
|---|---|---|---|
| 1 (26) | 1 (25) | KdCD3 scaling exponent | |
| 0.7 (12) | 0.8 (13) | KdTAA scaling exponent | |
| γn | 1 (17) | 1 (32) | nTAA scaling exponent |
These results indicated no specific preference for selecting either high CD3 or high target affinities for engineering a potent CD3 bispecific in vitro. However, high CD3 affinities have been previously shown to affect the in vivo pharmacokinetics (PK) of CD3 bispecifics.29 To determine if these observations can be accounted for by changes in CD3 affinity alone and what impact they have on selection of appropriate target and CD3 affinities for efficacy and toxicity, we developed a simple PK model that captures the target (CD3)-mediated disposition of CD3 bispecifics (Figure 9a). Comparison of this model to reported PK data for a CLL1-CD3 bispecific in humanized mice29 showed that the inclusion of known T-cell number, CD3 density per T-cell and CD3 internalization rates can indeed capture the KDCD3 dependent increase in apparent in vivo clearance and decrease in half-life of CD3 bispecifics in vivo (Figure 9).
Figure 9.
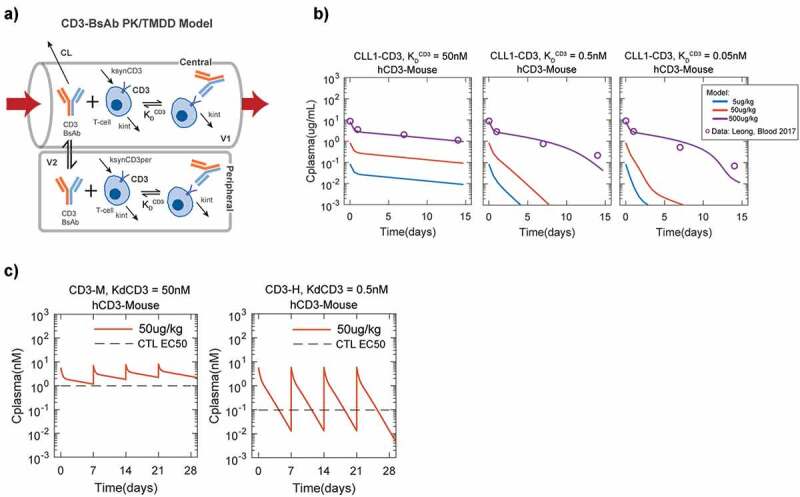
Model of CD3-bispecific pharmacokinetics (PK) shows the benefit of low CD3-affinity on potential efficacy/toxicity tradeoff. (a) Structure of in vivo PK model to explain CD3-affinity dependent PK of bispecifics. In this model, typical values of linear two-compartment parameters (V1, V2, CL and Q) are used together with physiological/measured values of T-cell number, CD3/T-cell, and CD3 internalization rate are used to predict the effect of different KDCD3 on bispecific PK. (b) Validation of model of CD3-bispecific pharmacokinetics (PK) using PK data for CLL1-CD3 bispecifics in humanized CD3 mice (hCD3-Mouse) from Leong et al., Blood 2012. Time-concentration profiles predicted by the model are consistent with the observations of CD3 bispecific PK in mice reported by Leong. Lines and circles show model predictions and reported data. (c) CD3 internalization driven elimination adversely affects the in vivo therapeutic dose of high CD3 affinity bispecifics. Despite the better in vitro CTL assay potency for CD3-H (reported CLL1-CD3 EC50 for CD3-H = 0.1 nM, EC50 for CD3-M = 1 nM), the PK model predicts that due to CD3-M’s better in vivo half-life at the same 50ug/kg IV-weekly dose, this lower affinity bispecific is better able to maintain serum concentrations high enough (i.e. >EC50) to ensure efficient killing
We next tried to contextualize the impact of high CD3 affinity by comparing the trough-level concentrations (Ctrough, i.e., drug concentration at the end of a dosing interval in vivo) achieved at a particular dose to the relevant in vitro reported cytotoxicity EC50s. As expected, the high CD3 affinity bispecific (CD3-H) was more potent (lower EC50) than a moderate CD3 affinity bispecific (CD3-M). But the shorter half-life of this CD3-H bispecific led to a comparable dose as the CD3-M needed to maintain trough-level concentrations above the cytotoxicity EC50 (Figure 9c). As a result, the Cmax/Ctrough concentration ratio at the efficacious dose is much higher for the CD3-H bispecific than for the CD3-M bispecific. This result is critical since high Cmax/Ctrough concentration ratio could represent a major risk for exaggerated cytokine release and consequent toxicity, suggesting that CD3-H bispecifics may have a significantly reduced therapeutic index and arguing for a selecting against high CD3 affinity in the design of these bispecifics.
Discussion
Here we describe a thorough investigation of the relationship between antigen epitope location and in vitro potency of T cell-engaging bispecific molecules using two in-house developed bispecific formats, IgG2-based and DbFc, with distinctive antigen-binding arm distances, in combination with several panels of artificial antigen-expressing cells we created. We found that the DbFc format is more potent when the antigen epitope is located in the membrane-distal region, while the IgG-based bispecific is more active for membrane-proximal epitopes masked with other domains. Moreover, in a more biologically relevant setting, bispecific molecules targeting different domains of FTL3 antigen corroborated these findings.
The first evidence that epitope distance to target cell membrane is critical in T cell-mediated cytotoxicity was reported in 2010 by a group of Micromet researchers.30 In their study, they chose melanoma chondroitin sulfate proteoglycan (MCSP, also known as NG2), which has >2,000 amino acids in its extracellular domain, as the TAA. MCSP is a large melanoma surface antigen composed of the cysteine-rich globular domain D1 (30–668), cysteine-free rod-like domain D2 (669–1537), and the cysteine-rich globular domain D3 (1538–2221). By generating a panel of BiTE molecules against different subdomains of MCSP, they discovered that BiTE molecules targeting the most membrane-proximal domain D3 are the most potent in lysing cell lines with full-length MCSP antigen. Building on these findings, they engineered Chinese hamster ovary (CHO) cell lines to express another TAA, epithelial cell adhesion molecule (EpCAM), alone and EpCAM fused to different combinations of MCSP subdomains and evaluated the ability of the anti-EpCAM/anti-CD3 BiTE molecule MT110 to lyse these CHO transfectants. More than a 1000-fold difference in potency was observed for the cell line expressing EpCAM alone and any other cell lines where EpCAM is tethered on top of any MCSP domain or domain combinations, further supporting the pivotal role of antigen epitope distance in effective T cell engagement. One of the major limitations of their system was the large sizes of MCSP subdomains – previous electron microscopy of rat MCSP revealed that D1 and D3 are ~30 nm in diameter and the rod-like D2 ranges from 30 to 110 nm.31 It remained unclear whether there is a gradual or abrupt decrease in potency when the antigen epitope distance to the target cell membrane is increased. Three more recent publications, using other TAAs, including FcRH5,13 ROR1,32 and CD33,33 further substantiated the findings that targeting membrane-proximal epitopes tends to yield molecules with greater potency. Here, using small EGF-like domain of ~3 nm as the structural spacer unit and two bispecific molecules with different arm lengths of the paratopes, we offer a more detailed picture of how increasing antigen epitope distance to target cell membrane affects cytotoxicity for both DbFc and IgG-based formats (Figure 2).
We then examined the effects of antigen receptor density, affinity of either antigen-binding arm, molecular formats, kinetics, and antigen epitope location on T-cell redirected cytotoxicity. We evaluated cell lines with 3 BCMA expression levels (low, medium, and high), affinity variants for both CD3 and BCMA arms (low, intermediate, and high affinity, a total of 9 pairs), 2 formats (DbFc and IgG), and two timepoints (24 and 48 h), which constituted a total of 108 assay conditions. To minimize assay variability and account for both EC50 and maximal killing for this complex data set, we adopted the AUC approach by integrating between the concentration ranges evaluated. In good agreement with previous results,13 the same molecule is usually less potent on cell lines with lower receptor expression, especially at the early timepoint. Notably, for the lowest antigen expression cell line (receptor copy number ~103), DbFc is more effective in triggering T cells than the IgG-based equivalent molecule, possibly due to its ability to form tight immunological synapses. For cell lines with medium and high receptor copy numbers, DbFc and IgG-based bispecifics often displayed comparable activities. Global analysis of the entire data set revealed that the affinity of either antigen-binding arm appears to be positively correlated with cytotoxicity, which is in agreement with previous reports, as reviewed recently.34 It is worth noting that the affinity range we chose here is very wide, where we investigated the previously unexplored area of very weak TAA affinities.
The remarkable clinical efficacy and severe toxicity observed for some bispecific T-cell engaging biologics or chimeric antigen receptor-modified T cells are often viewed as a double-edged sword.35 Cytokine release syndrome (CRS), documented as the major toxicity in patients treated with novel T cell-redirected therapies, can range from mild to life-threatening. Early data indicated that development of CRS might be correlated with clinical response. However, further study has not revealed any strong association between the severity of CRS and response to therapy.36 This has been validated by two independent studies using CAR T cell therapy,37,38 as well as a recent report by Genentech researchers investigating CD3-bispecific antibodies in rodent models.39 These observations lead to the question: can we somehow modulate the supraphysiologic signaling induced by T-cell engaging antibodies to dampen T cell activation and proliferation in order to maintain potency while reducing toxicity?
It was previously reported that there seems to be a dual activation threshold for cytotoxic T cells for cytotoxicity and cytokine production,40 and only the latter requires the formation of a stable immunological synapse. Here, we used a simplified in vitro system to examine cytotoxicity and cytokine release readouts while varying CD3 affinity (CD3-H, CD3-M, and CD3-L), molecular format (DbFc versus IgG), and antigen epitope location (as represented on different engineered cell lines) using purified T cells. Cytokines, including TNF, IL-2, and IFNγ, were selected as hallmarks of T cell activation status. All three cytokines demonstrated very similar trends. The most striking result that emerged from this data set is that certain combinations of molecular format and antigen epitope location may allow strong cytotoxic activity and low cytokine secretion. More specifically, taking CD3-H in combination with DbFc format as an example, cytotoxicity EC50 on the T0 cell line and T2 cell line were comparable (1.24 pM and 0.61 pM, respectively, both reaching nearly complete maximal killing), while the EC50 of TNF secretion had >40x difference (11 pM and 450 pM, respectively) and 2x difference in Emax (233 versus 107 pg/mL, respectively). Compared to T0 cell line, BCMA was tethered on T2 cell line with two extra EGF-like domains, leading to an increased intramembrane distance of the immunological synapses between T cells and target cells. Interestingly, this antigen epitope location effect can be fully compensated by using an IgG-based bispecific molecule on the T0 cell line, with EC50 of cytotoxicity 3.7 pM and EC50 of TNF secretion 300 pM, Emax 88 pg/mL, similar to the values for DbFc on the T2 cell line. This observation can be nicely explained by the differences in the distance between the two antigen-binding arms for IgG (9–15 nm) and DbFc (3–6 nm).
In contrast, manipulating CD3 binding affinity alone, at least in the ranges we chose, did not seem to achieve similar results, with higher affinity associated with greater potency and increased cytokine secretion, as shown in scatter plots in Figure 6d–f. In a higher CD3 affinity range that we did not cover here, it was reported that Emax of cytokine secretion could be 2x higher for a bispecific with 1.9 nM affinity to CD3 than that with a 7 nM affinity.41 Though we did not evaluate the impact of TAA affinity in cytokine secretion, a recent publication on HER2/CD3 bispecific demonstrated that higher HER2 affinity leads to stronger cytotoxicity as well as lower tolerability.42 One could argue that for a bispecific platform, it is possible to optimize the antigen-targeting epitope to achieve potent cytotoxic activity and relatively low cytokine release, if the antigen is large enough to offer epitopes with a range of distances to the cellular membrane. On the other hand, in the case when the antigen is small, certain bispecific platforms may be inherently suboptimal, for example, as demonstrated by the DbFc format in the T0 cell line, in which case cytotoxicity always goes hand in hand with cytokine release, regardless of the affinity of the CD3 binder chosen. Although it is still debatable whether a threshold of effector cytokine secretion exists for in vivo efficacy, the previous lack of tools to investigate this matter makes it difficult to widen the therapeutic window. As far as we know, the only other case that appeared to successfully decouple in vitro potency and cytokine release was described in a recent publication through screening a diverse repertoire of CD3 antibodies to identify unique epitopes.43 Coincidently, these authors also selected BCMA as the TAA. They observed that one of their less potent molecules (EC50 = 364 pM in BCMA+ NCI-H929 cell line), binding to a different CD3 epitope, induced less IL-2 (600 pg/mL) and IFNγ (3500 pg/mL) at saturation killing dose (10 nM), when compared to another more potent compound (EC50 = 4.8 pM) with both cytokines >6000 pg/mL at its saturation killing dose (0.1 nM). Caution needs to be taken in directly comparing the exact values of the cytokine release with those we reported here due to differences in the assay conditions, such as target cell lines, cell density, E:T ratio, donor variabilities, and timepoint.
The kinetic segregation model of T cell activation44 may explain why intermembrane distance could be a key determinant in T cell signaling. In this model, in resting T cells, TCRs are constantly phosphorylated by tyrosine kinase Lck and dephosphorylated by tyrosine phosphatase CD45 such that the net TCR signal is very low. During T cell activation, the immunological synapse formed by TCR and pMHC (or bridged by CD3 bispecifics) makes a contact zone that is too narrow for relatively large CD45 to fit into, which shifts the equilibrium to TCR phosphorylation. The discovery of the dual activation threshold for T cell cytotoxicity and cytokine secretion40,45 suggested that the number of TCR-pMHC complex in the contact region may orchestrate the activation status, and that there is a window to induce strong cytotoxicity without maximum cytokine secretion. We hypothesize that modulating the intermembrane distance, which potentially determines the degree of CD45 exclusion, can lead to decoupling of the two effects.
Lastly, we incorporated our experimental findings into one single unified mathematical model that can be used to predict the affinities of both arms needed to achieve certain potency. When it comes to the choice of affinity, it is worth noting that target-mediated clearance can play an important role. In particular, bispecific molecules with high affinity to CD3 are known to display fast clearance and shorter PK.29 Our model further validated this observation and could be used to guide selection of optimal affinities of bispecifics with desired pharmacological activity.
In summary, our data provides a general framework for optimization of T-cell engaging protein therapeutics, with focuses on antigen epitope location and molecular format. Such an approach in screening and ranking different molecular architecture was recently used by Cheung’s group, with the emphasis mostly on potency and not the safety profile of bispecific T cell engaging molecules.46 Further studies need to be carried out to investigate whether our deconvolution of cytotoxicity and cytokine release can indeed translate into wider therapeutic window in vivo, given that only pan-T cells were used in our cytokine release assay, and thus the effects of other peripheral blood mononuclear cell types were excluded. Nevertheless, minimizing supraphysiological cytokine secretion from T cells while maintaining strong cytotoxicity could have profound impacts on T-cell engaging therapies.
Materials and methods
Bispecific antibody generation
Variable heavy and light domains from in-house developed anti-BCMA and anti-CD3 antibodies were cloned into human IgG2Δa D265A, Db or DbFc format with IgG2 Fc and hinge harboring appropriate mutations to favor Fc heterodimer formation.14 During the optimization of DbFc, hinge and linker variants, as listed in Supplementary Tables 1 and Tables 2, were produced by incorporating the sequence elements using Gibson Assembly protocol (NEBuild HiFi DNA Assembly). For full-length IgG bispecifics, to ensure proper heavy and light chain pairing, monospecific IgGs were first transiently expressed in Expi293 cells (Thermo Fisher Scientific) and purified by protein A capture (MabSelect SuRe) followed by size exclusion chromatography before undergoing mild reduction and oxidation to form heterodimer. For Db and DbFc constructs, co-expression of both chains and purification via protein A was sufficient.
The optimal DbFc variant was selected based on a combination of expression titer and product integrity. Size-exclusion chromatography with in-line multi-angle light scattering was conducted on an Agilent 1260 system with a variable wavelength UV detector operated at 280 nm (Agilent Technologies, Santa Clara, USA), followed by a miniDAWN detector (Wyatt Technology, USA). Separation was performed with a Superdex 200 Increase 10/30 GL column using phosphate-buffered saline (PBS) as an aqueous phase. Each sample was injected twice, and data were collected and processed using the ASTRA® software V7.2 (Wyatt Technology, Santa Barbara, USA). Melting temperature (Tm) measurements were performed using a VP-Capillary DSC system (Microcal Inc., acquired by Malvern Instruments) at 1 mg/mL sample concentration in 1xPBS, pH 7.4, where the samples were heated from 20°C to 90°C at 1°C per min to monitor thermal unfolding. Kinetic binding of various bispecific molecules to CD3 and TAA was conducted using Biacore T200 using recombinant his-tagged CD3 εδ antigen or human BCMA at 37 °C.
Affinity variants of anti-BCMA and anti-CD3 were generated using saturation mutagenesis on critical residues within the complementarity-determining regions of the parental clones. The affinities between BCMA-CD3 bispecifics and BCMA were determined on a Biacore T200 surface plasmon resonance (SPR) instrument (Cytiva Life Sciences) equipped with a Series S CM4 sensor chip immobilized with anti-human-Fc antibody. The sensor chip surface was prepared with HBS-P+ (0.01 M Hepes pH 7.4, 0.15 M NaCl, 0.05% v/v Surfactant P20) running buffer. All channels and flow cells were activated with a 1:1 (v/v) mixture of 0.4 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and 0.1 M N-hydroxysuccinimide (NHS) for 7 min at 10 µL/min. Anti-human-Fc (Southern Biotech, CAT# 2014–01) antibody was then injected at 50 µg/mL in 10 mM Acetate pH 4.5 for 7 min at 20 µL/min to allow for amine-coupling to the surface. The surface was then blocked with 0.1 M ethylenediamine (EDA) in 0.2 M borate buffer pH 8.5 for 7 min at 10 µL/min, and conditioned with three 60-s injections of 75 mM phosphoric acid at 10 µL/min. Multicycle kinetic assays were performed at 37°C with HBS-P+ supplemented with 1 mg/mL bovine serum albumin (BSA) as the running and sample dilution buffer. Purified BCMA-CD3 bispecifics were captured at 10 µg/mL for 2 min at 10 µL/min. Then, BCMA analyte (0, 1.2, 3.7, 11.1, 33.3, 100, and 300 nM) was injected for 2 min at 30 µL/min for kinetic data. Alternatively, BCMA analyte (0, 1.2, 3.7, 11.1, 33.3, 100, 300, 2700, and 8100 nM) was injected for 1 min 30 µL/min for steady-state affinity. After the analyte injections, dissociation was monitored for 10 min for kinetic data, and 2 min for steady-state affinity. The surface was then regenerated with three 60-s injections of 75 mM phosphoric acid at 10 µL/min prior to the next analysis cycle. The affinities between BCMA-CD3 bispecifics and CD3 were determined on a Bicore T200 SPR instrument, with a Series S CM4 chip immobilized with anti-His-Tag antibody (R&D Systems, CAT#MAB050). The sensor chip surface was prepared with HBS-P + . All channels and flow cells were activated with EDC:NHS. Anti-His-Tag was injected at 100 µg/mL in 10 mM Acetate pH 5.0. The surface was blocked with EDA, and conditioned with two 30-s injections of 10 mM glycine pH 1.7. Multicycle kinetic assays were performed at 37°C with HBS-P+ supplemented with 1 mg/mL BSA as the running and sample dilution buffer. CD3εδ antigen was captured at 0.125 µg/mL for 1 min at 10 µL/min. Then, BCMA-CD3 bispecifics (0,12, 37, 111, 333, 1000, and 3000 nM) were injected for 1 min at 30 µL/min. After the analyte injections, dissociation was monitored for 3 min. The surface was then regenerated with two cycles of 30-s injections of 10 mM glycine pH 1.7 at 10 µL/min prior to the next analysis cycle. Kinetics and affinity data analyses were performed with T200 Evaluation software version 2.0.
Cell line generation and receptor density quantification
Reh human lymphoblastic leukemia cell line (non-T, non-B) was obtained from American Type Culture Collection (Rockville, MD, USA). In light of natural shedding of BCMA by γ-secretase in transmembrane domain,47 only the extracellular domain of BCMA (aa 1–54) was cloned in frame with transmembrane domain of human tumor-associated calcium signal transducer 2 in a plasmid with cytomegalovirus-driven promoter. For T or M series of cell lines, plasmids were constructed by inserting an increasing number of EGF-like domains from human LRP-1 (uniprot ID: Q07954, aa 4147–4409, EGF-like domain 16–22) into corresponding locations, either N-terminal or C-terminal to the BCMA extracellular domain. For electroporation procedure, 2 × 106 cells were centrifuged at 900xg for 10 min at room temperature (RT). The supernatant was completely removed, and then the cell pellet was resuspended carefully at RT in 100 μl Cell Line Nucleofector® Solution V (supplied by Amaxa® Cell Line Nucleofector® Kit V) with 2 μg DNA. The cell/DNA suspension was transferred into certified cuvette, and the appropriate Nucleofector® Program L-013 (L-13 for Nucleofector® I Device) was selected. The cuvette with cell/DNA suspension into the Nucleofector® cuvette holder was then inserted and the electroporation program was applied After electroporation was completed, ~500 μL of the pre-equilibrated culture medium was immediately added to the cuvette, and the sample was gently transferred into the 12-well plate and incubated in humidified 37°C/5% CO2 incubator until analysis. Gene expression is determined by FACS. After expression confirmed, single-cell clones were selected by FACS sorting.
For BCMA receptor expression quantification, a volume containing 106 cells was centrifuged, supernatant removed, and cells incubated at 4°C for 30 min with 50 μl of a pure mouse monoclonal anti-BCMA mIgG2a (clone 19F2, BioLegend). Subsequently, the samples were washed twice with PBS/1% BSA/0.1% NaN3 and incubated at 4°C for 30 min in the dark with 50 μL fluorescein isothiocyanate (FITC)-labeled goat anti-mouse IgG diluted 1:50 (QIFIKIT; Dako, Glostrup, Denmark). Afterward, cells were washed twice with PBS/1% BSA/0.1% NaN3 and resuspended in 1 ml of the same solution. Blanks were processed in parallel with the samples in the same manner, except that anti-LDL receptor antibody in PBS/1% BSA/0.1% NaN3 was used. A calibration curve linking the intensity of fluorescence and the number of antigenic sites was established using QIFIKIT. Setup and calibration beads (50 μl each) were washed as recommended by the manufacturer and incubated at 4°C for 30 min in the dark with 50 μl goat FITC-labeled anti-mouse IgG diluted 1:50. Then, they were washed and suspended in PBS before flow cytometer analysis. Measurements were performed on a flow cytometer (Beckman Coulter) and the FITC signal was recorded for each sample. The fluorescence related to specific binding to BCMA was obtained by subtracting the mean fluorescence of the blank from the mean fluorescence of the sample. The number of antigenic sites per cell was calculated from the calibration curve derived from QIFIKIT beads. The coefficient of variation evaluating the reproducibility of the assay was 5%.
In vitro cytotoxicity assay
Target cell lines were either custom made in-house as described in previous section or provided by American Type Culture Collection (ATCC). Effector cells used here were freshly prepared by first isolating PBMCs from fresh whole blood supplied by healthy donors at Stanford hospital or a Pfizer-affiliated clinic (donors gave informed consent and the donation process followed the regulations and standard operating procedures of the clinic) and then purifying with a Pan T cell isolation kit (Miltenyi Biotec, Cat. No. 130–096-535), which contains a cocktail of antibodies to deplete other non-desired cell types. Target and purified T cells were seeded in 96-well, round bottom plates in triplicates, at 5:1 effector-to-target (E:T) cell ratio, in cell culture medium containing 5% fetal bovine serum. Bispecific IgG2 or Db molecules were added to the wells as 10-fold dilution series and incubated for 24 or 48 h at 37°C, supplemented with 5% CO2, before cell viabilities were assessed based on lactate dehydrogenase (LDH) levels in the supernatant using Promega ONE-Glo™ luciferase assay system (Cat. No. E6120). Percentage of specific lysis was calculated by benchmarking the LDH level in each well against the spontaneous background lysis (without antibody added) and total maximal level of LDH release by adding Triton X-100 to break down all cells. All cytotoxicity assays were at least repeated twice.
Measurement of cytokine panel using MSD
Frozen supernatants (−80°C) from incubating T0, T2, T4, T7 cell lines with freshly isolated human T cells and bispecific agents for ~16–20 h were thawed at RT for cytokine analysis using Meso Scale Discovery (MSD) V-PLEX human pro-inflammatory panel 1 (CAT# K15049D-2), 10 spot kits. This kit can be used to measure IFN- γ, IL-1b, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p70, IL-13, and TNF. V-PLEX plates were washed 2x with 150 μL ELISA wash buffer before use. Calibrators were prepared per MSD instruction manual (kit lot number K0081189, see product information sheet for specific pg/mL concentrations per cytokine). Each vial of lyophilized calibrator powder was resuspended with 1000 μL of Diluent 2. Vials were inverted at least 6x, then incubated at RT for 30 min. This resuspended solution was used as the top concentration for the calibration series. The resuspended calibrators were vortexed and serially diluted 4x. Solutions were vortexed between each serial dilution step. This was repeated until there were 7 serially diluted solutions and an 8th diluent only tube. 50 μL/well of calibrators were plated top to bottom in duplicate in columns 1 and 2 for all V-PLEX kit plates. 25 μL of Diluent 2 was added to columns 3 through 12. 25 μL of thawed media was then transferred to the plates, so that all wells had 50 μL sample. Plates were placed on shaker for at least 30 seconds, then incubated overnight at 4°C. Detection antibody mixture was prepared by diluting the 10 provided SULFO-Tag antibodies 50x in Diluent 3. Plates were washed 3x with 150 μL ELISA wash buffer. Detection antibody mixture was added to all wells at 25 μL/well. Plates were protected from light and incubated at RT for 2 h with shaking. “2x Read Buffer T” was prepare by diluting the provided 4x Read Buffer T with equal volume deionized water. Plates were washed 3x with 150 μL ELISA wash buffer. 150 μL of “2x Read Buffer T” was added per well and read immediately on the MSD instrument. Raw data was analyzed on MSD Discovery Workbench 4.0 software, using a built-in plate layout for human pro-inflammatory panel 1 and inputting calibrator information, to obtain results in pg/mL. Results were then plotted on GraphPad Prism for Windows (GraphPad Software, San Diego, California USA). Each sample was measured in duplicates.
Supplementary Material
Acknowledgments
We acknowledge critical review of the manuscript by Michelle Wagner and Robert Hollingsworth from Pfizer Center of Therapeutic Innovation and Oncology Research Unit, respectively.
Abbreviations
| AML | acute myeloid leukemia |
| APC | antigen-presenting cell |
| AUC | area under the curve |
| BCMA | B cell maturation antigen |
| BiTE | bispecific T-cell engager |
| CD3 | cluster of differentiation 3 |
| Db | Diabody |
| DbFc | Diabody Fc |
| EC50 | half maximum effective concentration |
| Emax | maximum effect |
| EGF | epidermal growth factor |
| FACS | Fluorescence-activated cell sorting |
| FLT3 | FMS-like receptor tyrosine kinase-3 |
| IgG | immunoglobin G |
| IFN-γ | interferon γ |
| IL-2 | interleukin 2 |
| MHC | major histocompatibility complex |
| PBMC | peripheral blood mononuclear cell |
| scFv | single-chain variable fragment |
| TAA | tumor-associated antigen |
| TCR | T cell receptor |
| TNF | tumor necrosis factor |
Author Contributions
W.C. and J.C.-R. conceived the experimental design; A.D. and W.C. designed and generated the cell lines; E.P., I.N., S.D., M.L.C., A.P., T.V.B., and X.J. generated and qualified protein reagents; K.C.L. designed the biosensor affinity measurements and C. W. executed on them; W.C. and C.W. performed the in vitro cytotoxicity assays and cytokine level determination; W.C. and P.J.D. investigated the mechanism of synapse formation; J.N. built the integrated mathematical model; W.C., F.Y., J.N., M.L.C., and J.C.-R. participated in data interpretation and manuscript drafting. J.C.-R., Y.A.Y., and T.V.B supervised the project; all authors reviewed and approved the final version of the manuscript.
Declaration of Interest Statement
All authors are current or former employees of Pfizer.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Guy CS, Vignali DA.. Organization of proximal signal initiation at the TCR:CD3 complex. Immunol Rev. 2009;232(1):7–18. doi: 10.1111/j.1600-065X.2009.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reth M. Antigen receptor tail clue. Nature. 1989;338(6214):383–84. doi: 10.1038/338383b0. [DOI] [PubMed] [Google Scholar]
- 3.Combadiere B, Freedman M, Chen L, Shores EW, Love P, Lenardo MJ. Qualitative and quantitative contributions of the T cell receptor zeta chain to mature T cell apoptosis. J Exp Med. 1996;183(5):2109–17. doi: 10.1084/jem.183.5.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–77. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 5.Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43(6):763–71. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T, Raum T, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43(8):1129–43. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 7.Oberst MD, Fuhrmann S, Mulgrew K, Amann M, Cheng L, Lutterbuese P, Richman L, Coats S, Baeuerle PA, Hammond SA, et al. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. MAbs. 2014;6(6):1571–84. doi: 10.4161/19420862.2014.975660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedrich M, et al. Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol Cancer Ther. 2012;11:2664–73. doi: 10.1158/1535-7163.MCT-12-0042. [DOI] [PubMed] [Google Scholar]
- 9.Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, Gorlatov S, Veri MC, Aggarwal S, Yang Y, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood. 2011;117(17):4542–51. doi: 10.1182/blood-2010-09-306449. [DOI] [PubMed] [Google Scholar]
- 10.Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ, et al. A novel carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22(13):3286–97. doi: 10.1158/1078-0432.CCR-15-1696. [DOI] [PubMed] [Google Scholar]
- 11.Asano R, Shimomura I, Konno S, Ito A, Masakari Y, Orimo R, Taki S, Arai K, Ogata H, Okada M, et al. Rearranging the domain order of a diabody-based IgG-like bispecific antibody enhances its antitumor activity and improves its degradation resistance and pharmacokinetics. MAbs. 2014;6(5):1243–54. doi: 10.4161/mabs.29445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith EJ, Olson K, Haber LJ, Varghese B, Duramad P, Tustian AD, Oyejide A, Kirshner JR, Canova L, Menon J, et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep. 2015;5(1):17943. doi: 10.1038/srep17943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, Clark V, Hristopoulos M, Cook R, Slaga D, et al. Membrane-proximal epitope facilitates efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31(3):383–95. doi: 10.1016/j.ccell.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strop P, Ho W-H, Boustany LM, Abdiche YN, Lindquist KC, Farias SE, Rickert M, Appah CT, Pascua E, Radcliffe T, et al. Generating bispecific human IgG1 and IgG2 antibodies from any antibody pair. J Mol Biol. 2012;420(3):204–19. doi: 10.1016/j.jmb.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 15.Perisic O, Webb PA, Holliger P, Winter G, Williams RL. Crystal structure of a diabody, a bivalent antibody fragment. Structure. 1994;2(12):1217–26. doi: 10.1016/s0969-2126(94)00123-5. [DOI] [PubMed] [Google Scholar]
- 16.Root AR, et al. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART molecule with extended half-life for the treatment of cancer. Antibodies (Basel. 2016;5. doi: 10.3390/antib5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Werner TC, Bunting JR, Cathou RE. The shape of immunoglobulin G molecules in solution. Proc Natl Acad Sci U S A. 1972;69(4):795–99. doi: 10.1073/pnas.69.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sosnick TR, Benjamin DC, Novotny J, Seeger PA, Trewhella J. Distances between the antigen-binding sites of three murine antibody subclasses measured using neutron and X-ray scattering. Biochemistry. 1992;31(6):1779–86. doi: 10.1021/bi00121a028. [DOI] [PubMed] [Google Scholar]
- 19.Klein JS, Bjorkman PJ, Rall GF. Few and far between: how HIV may be evading antibody avidity. PLoS Pathog. 2010;6(5):e1000908. doi: 10.1371/journal.ppat.1000908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A. 1993;90(14):6444–48. doi: 10.1073/pnas.90.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campbell ID, Bork P. Epidermal growth factor-like modules. Current Opinion in Structural Biology. 1993;3(3):385–92. doi: 10.1016/S0959-440X(05)80111-3. [DOI] [Google Scholar]
- 22.Wouters MA, Rigoutsos I, Chu CK, Feng LL, Sparrow DB, Dunwoodie SL. Evolution of distinct EGF domains with specific functions. Protein Sci. 2005;14(4):1091–103. doi: 10.1110/ps.041207005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suckling RJ, Korona B, Whiteman P, Chillakuri C, Holt L, Handford PA, Lea SM. Structural and functional dissection of the interplay between lipid and Notch binding by human Notch ligands. Embo J. 2017;36(15):2204–15. doi: 10.15252/embj.201796632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cordle J, Johnson S, Zi Yan Tay J, Roversi P, Wilkin MB, de Madrid BH, Shimizu H, Jensen S, Whiteman P, Jin B, et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat Struct Mol Biol. 2008;15(8):849–57. doi: 10.1038/nsmb.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulahin N, Kristensen O, Rasmussen KK, Olsen L, Rydberg P, Vestergaard B, Kastrup JS, Berezin V, Bock E, Walmod PS, et al. Structural model and trans-interaction of the entire ectodomain of the olfactory cell adhesion molecule. Structure. 2011;19(2):203–11. doi: 10.1016/j.str.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 26.Verstraete K, Vandriessche G, Januar M, Elegheert J, Shkumatov AV, Desfosses A, Van Craenenbroeck K, Svergun DI, Gutsche I, Vergauwen B, et al. Structural insights into the extracellular assembly of the hematopoietic Flt3 signaling complex. Blood. 2011;118(1):60–68. doi: 10.1182/blood-2011-01-329532. [DOI] [PubMed] [Google Scholar]
- 27.Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJWL, Quackenbush J. Inconsistency in large pharmacogenomic studies. Nature. 2013;504(7480):389–93. doi: 10.1038/nature12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Haddish-Berhane N, Moore P, Clark T, Yang Y, Li H, Xuan D, Barton HA, Betts AM, Barletta F, et al. Mechanistic projection of first-in-human dose for bispecific immunomodulatory P-Cadherin LP-DART: an Integrated PK/PD modeling approach. Clin Pharmacol Ther. 2016;100(3):232–41. doi: 10.1002/cpt.393. [DOI] [PubMed] [Google Scholar]
- 29.Leong SR, Sukumaran S, Hristopoulos M, Totpal K, Stainton S, Lu E, Wong A, Tam L, Newman R, Vuillemenot BR, et al. An anti-CD3/anti–CLL-1 bispecific antibody for the treatment of acute myeloid leukemia. Blood. 2017;129(5):609–18. doi: 10.1182/blood-2016-08-735365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bluemel C, Hausmann S, Fluhr P, Sriskandarajah M, Stallcup WB, Baeuerle PA, Kufer P. Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol Immunother. 2010;59(8):1197–209. doi: 10.1007/s00262-010-0844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tillet E, Ruggiero F, Nishiyama A, Stallcup WB. The membrane-spanning proteoglycan NG2 binds to collagens V and VI through the central nonglobular domain of its core protein. J Biol Chem. 1997;272(16):10769–76. doi: 10.1074/jbc.272.16.10769. [DOI] [PubMed] [Google Scholar]
- 32.Qi J, Li X, Peng H, Cook EM, Dadashian EL, Wiestner A, Park H, Rader C. Potent and selective antitumor activity of a T cell-engaging bispecific antibody targeting a membrane-proximal epitope of ROR1. Proc Natl Acad Sci U S A. 2018;115(24):E5467–E5476. doi: 10.1073/pnas.1719905115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nair-Gupta P, Diem M, Reeves D, Wang W, Schulingkamp R, Sproesser K, Mattson B, Heidrich B, Mendonça M, Joseph J, et al. A novel C2 domain binding CD33xCD3 bispecific antibody with potent T-cell redirection activity against acute myeloid leukemia. Blood Advances. 2020;4(5):906–19. doi: 10.1182/bloodadvances.2019001188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellerman D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. 2019;154:102–17. doi: 10.1016/j.ymeth.2018.10.026. [DOI] [PubMed] [Google Scholar]
- 35.Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20(2):119–22. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017–23. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, Guo X, Liu H, Ding N, Zhang T, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25(6):947–53. doi: 10.1038/s41591-019-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feucht J, Sun J, Eyquem J, Ho Y-J, Zhao Z, Leibold J, Dobrin A, Cabriolu A, Hamieh M, Sadelain M, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25(1):82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Piskol R, Ybarra R, Chen YJJ, Li J, Slaga D, Hristopoulos M, Clark R, Modrusan Z, Totpal K, et al. CD3 bispecific antibody–induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med. 2019;11(508):eaax8861. doi: 10.1126/scitranslmed.aax8861. [DOI] [PubMed] [Google Scholar]
- 40.Faroudi M, Utzny C, Salio M, Cerundolo V, Guiraud M, Muller S, Valitutti S. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A. 2003;100(24):14145–50. doi: 10.1073/pnas.2334336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zuch de Zafra CL, Fajardo F, Zhong W, Bernett MJ, Muchhal US, Moore GL, Stevens J, Case R, Pearson JT, Liu S, et al. Targeting multiple myeloma with AMG 424, a novel anti-CD38/CD3 Bispecific T-cell-recruiting antibody optimized for cytotoxicity and cytokine release. Clin Cancer Res. 2019;25:3921–33. doi: 10.1158/1078-0432.CCR-18-2752. [DOI] [PubMed] [Google Scholar]
- 42.Staflin K, Zuch de Zafra CL, Schutt LK, Clark V, Zhong F, Hristopoulos M, Clark R, Li J, Mathieu M, Chen X, et al. Target arm affinities determine preclinical efficacy and safety of anti-HER2/CD3 bispecific antibody. JCI Insight. 2020;5(7). doi: 10.1172/jci.insight.133757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trinklein ND, Pham D, Schellenberger U, Buelow B, Boudreau A, Choudhry P, Clarke SC, Dang K, Harris KE, Iyer S, et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs. 2019;11(4):639–52. doi: 10.1080/19420862.2019.1574521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. 2012;487(7405):64–69. doi: 10.1038/nature11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM. T cell killing does not require the formation of a stable mature immunological synapse. Nat Immunol. 2004;5(5):524–30. doi: 10.1038/ni1058. [DOI] [PubMed] [Google Scholar]
- 46.Santich BH, Park JA, Tran H, Guo H-F, Huse M, Cheung NKV. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci Transl Med. 2020;12(534):eaax1315. doi: 10.1126/scitranslmed.aax1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laurent SA, Hoffmann FS, Kuhn P-H, Cheng Q, Chu Y, Schmidt-Supprian M, Hauck SM, Schuh E, Krumbholz M, Rübsamen H, et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6(1):7333. doi: 10.1038/ncomms8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.