

Supplemental Digital Content is available in the text.
Keywords: coronary artery disease, hypercholesterolemia, lipids, lipoproteins, metabolism
Background:
Familial hypercholesterolemia (FH) is a common autosomal codominant genetic disorder, which causes elevated levels of low-density lipoprotein cholesterol (LDL-C) and increased risk of premature atherosclerotic cardiovascular disease (ASCVD). Even among individuals with monogenic FH, there is substantial interindividual variability in LDL-C levels and risk of ASCVD. We assessed the influence of an LDL-C polygenic score on levels of LDL-C and risk of ASCVD for individuals with monogenic FH.
Methods:
We constructed a weighted LDL-C polygenic score, composed of 28 single-nucleotide variants, for individuals with monogenic FH from the British Columbia FH (n=262); Nutrition, Metabolism and Atherosclerosis Clinic (n=552); and UK Biobank cohorts (n=306). We assessed the association between LDL-C polygenic score with LDL-C levels and ASCVD risk using linear regression and Cox-proportional hazard models, respectively. ASCVD was defined as myocardial infarction, coronary or carotid revascularization, transient ischemic attack, or stroke. The results from individual cohorts were combined in fixed-effect meta-analyses.
Results:
Levels of LDL-C were significantly associated with LDL-C polygenic score in the Nutrition, Metabolism and Atherosclerosis Clinic cohort, UK Biobank cohort, and in the meta-analysis (β [95% CI]=0.13 [0.072–0.19] per a 20% increase in LDL-C polygenic score percentile, P<0.0001). Additionally, an elevated LDL-C polygenic score (≥80th percentile) was associated with a trend towards increased ASCVD risk in all 3 cohorts individually. This association was statistically significant in the meta-analysis (hazard ratio [95% CI]=1.48 [1.02–2.14], P=0.04).
Conclusions:
Polygenic contributions to LDL-C explain some of the heterogeneity in clinical presentation and ASCVD risk for individuals with FH.
Familial hypercholesterolemia (FH) is the most common life-threatening autosomal dominant genetic disorder (≈1 out of 225–250 people) and is typically caused by pathogenic genetic variants in the LDLR, APOB, or PCSK9 genes.1,2 These pathogenic variants impair the clearance of LDL-C (low-density lipoprotein cholesterol) from the blood and significantly increase the risk of atherosclerotic cardiovascular disease (ASCVD).3 Despite the availability of effective treatment strategies, it is estimated that >85% of individuals with FH remain undiagnosed and undertreated.4
A major challenge to the diagnosis and treatment of FH is the heterogeneity in the clinical presentation of the condition.5–9 The variability in presentation of FH can be attributed to age and sex,10 the type of pathogenic variant,11–13 other genetic influences,14–18 and environmental factors.6,19–21 Notably, the contribution of common polygenic variants to LDL-C levels has been well characterized14,16,22 and may help explain why family members that carry the same FH-associated variant and share similar environments can display varying severity of FH.9
Our previous study,12 and the work of others,15,23–25 suggest that polygenic determinants of LDL-C levels can modulate both the clinical phenotype (ie, LDL-C levels) and, potentially, the cardiovascular risk observed in FH. However, most work to date has been underpowered to assess whether polygenic factors contribute additional risk for adverse clinical outcomes among those with monogenic FH.11,12,21,25 Here, we used 3 independent cohorts to evaluate how polygenic contributions to LDL-C levels influence the risk of ASCVD events among individuals with monogenic FH.
Methods
A full-length description of the methods can be found in the Data Supplement. The data that support the findings of this study are available from the corresponding author upon reasonable request.
The BCFH study (British Columbia Familial Hypercholesterolemia) was approved by the Clinical Research Ethics Board of the University of British Columbia.12,26,27 The CNMA study (Nutrition, Metabolism and Atherosclerosis Clinic) was approved by the Institut de recherches cliniques de Montréal Institutional Review Board and ethical committee.28 The UK Biobank study was approved by the UK Biobank and by the Clinical Research Ethics Board of the University of British Columbia.29,30 For each study, all individuals or substitute decision-makers provided written informed consent.
Results
Cohort Characteristics
The enrollment characteristics of individuals from the BCFH, CNMA, and UK Biobank cohorts with monogenic FH are shown in Table 1, and the FH-associated variants identified in each cohort are displayed in Tables I through III in the Data Supplement. Notably, individuals from the UK Biobank cohort were older and displayed less severe hypercholesterolemia relative to the BCFH and CNMA cohorts.
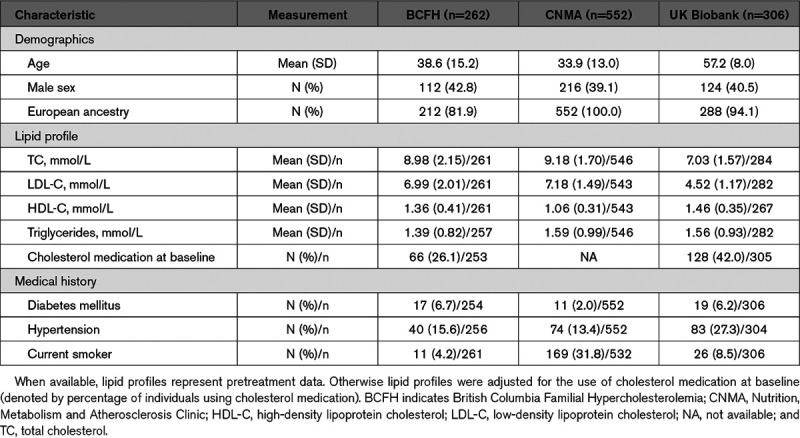
Table 1.
Demographic Characteristics of the Study Cohorts at Enrollment
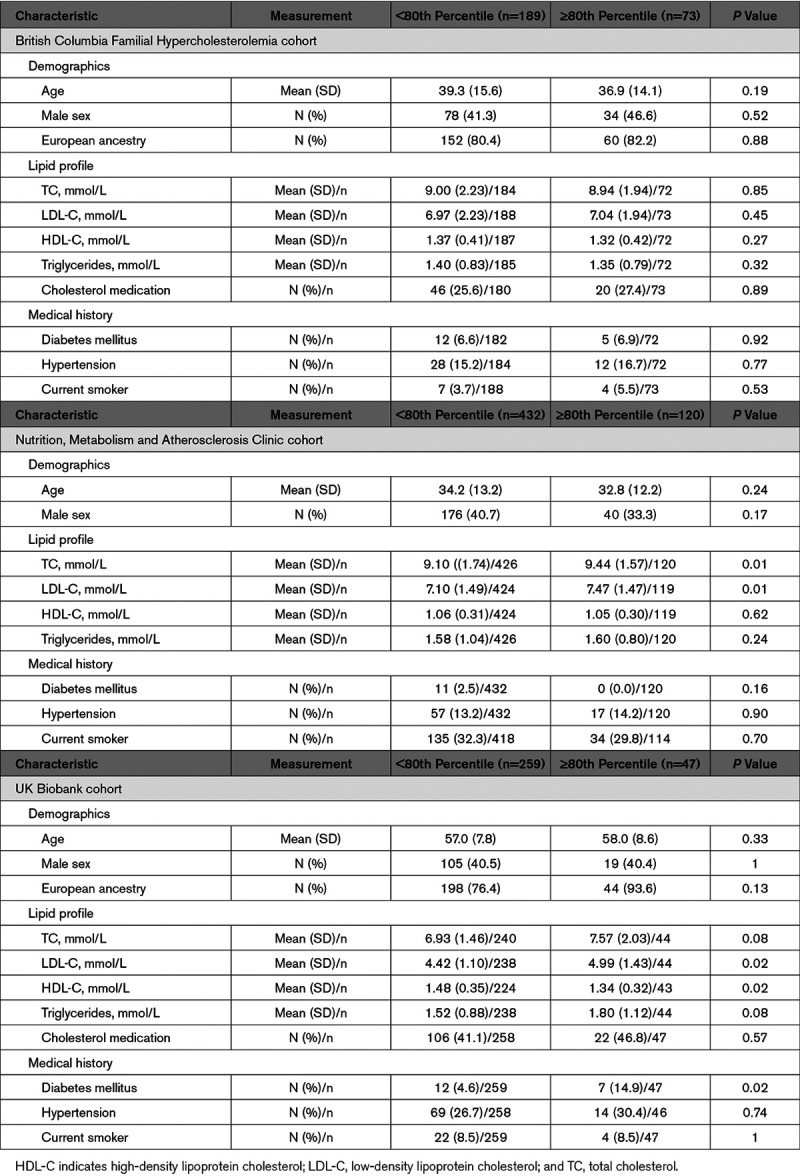
The 28 single-nucleotide variants (SNVs) used in the calculation of LDL-C polygenic score percentile (LDL-CPSP) are displayed in Table IV in the Data Supplement. The characteristics of these individuals, stratified by LDL-CPSP, are shown in Table 2. LDL-C levels were significantly higher in those with an elevated LDL-CPSP for the CNMA and UK Biobank cohorts but not the BCFH cohort. Other characteristics were similar between individuals with and without an elevated LDL-CPSP.
Table 2.
Characteristics of the Study Cohorts at Enrollment Stratified by Elevated LDL-C Polygenic Score Percentile (≥80th Percentile Using 28 Single-Nucleotide Variants)
LDL-C Polygenic Scores Associate With Measured Levels of LDL-C for Individuals With FH
A 20% increase in LDL-CPSP was associated with significantly higher levels of LDL-C in the CNMA (R2=0.084, P=0.0007) and UK Biobank cohorts (R2=0.047; P=0.004) when adjusted for age and sex (Figure 1). However, this relationship was not observed in the BCFH cohort (R2=0.0036, P=0.57; Figure 1). We used a fixed-effect meta-analysis to assess the overall association between measured levels of LDL-C and continuous LDL-CPSP. The fixed-effect meta-analysis demonstrated a significant association between LDL-CPSP and levels of LDL-C (β [95% CI]: 0.13 [0.072–0.19] per a 20% increase in LDL-CPSP, P<0.0001; Figure 1D). The interstudy heterogeneity was not statistically significant (Q=0.95 with 2 df, P=0.62).
Figure 1.
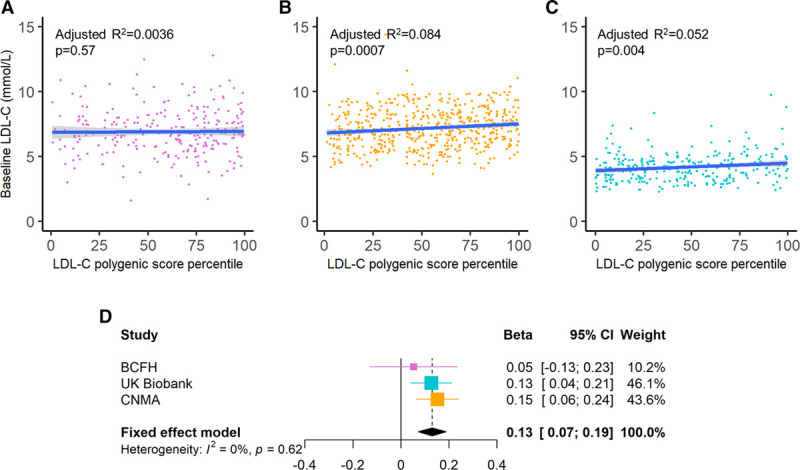
Association between LDL-C (low-density lipoprotein cholesterol) levels and LDL-C polygenic score percentile among individuals with familial hypercholesterolemia. The association between LDL-C levels and LDL-C polygenic score percentile is displayed for the (A) British Columbia Familial Hypercholesterolemia (BCFH), (B) Nutrition, Metabolism and Atherosclerosis Clinic (CNMA), and (C) UK Biobank cohorts. The blue lines and associated gray shading represent the linear regression line of best fit and 95% CI, respectively. D, A fixed-effect meta-analysis of the β coefficients and standard errors obtained from the linear regression models of LDL-C levels (mmol/L) vs continuous LDL-C polygenic score (per 20% increase) are shown. Linear regression models were adjusted for age and sex.
We also assessed the influence of the LDL-CPSP on LDL-C levels among individuals from the CNMA cohort carrying the same FH-associated variant. Similar to the analyses of overall FH-associated variants in the CNMA cohort, there was a significant correlation between LDL-C levels and LDL-CPSP for carriers of either the LDLR 15 kb promoter and exon 1 deletion (n=369, R2=0.088, P=0.02; Figure 2A) or the LDLR exon 3 missense variant p.Trp87Arg in LDLR (n=103, R2=0.15, P=0.05; Figure 2B).
Figure 2.
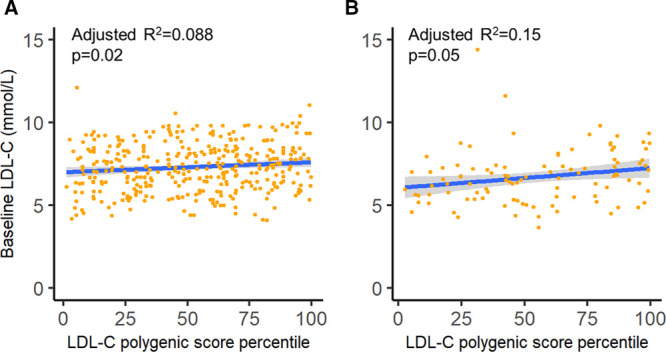
Association between LDL-C (low-density lipoprotein cholesterol) levels and LDL-C polygenic score percentile among individuals with the same familial hypercholesterolemia (FH)-associated variant. The association between LDL-C levels and LDL-C polygenic score percentile is displayed for the individuals from the Nutrition, Metabolism and Atherosclerosis Clinic (CNMA) that were carriers for (A) the LDLR 15 kb promoter and exon 1 deletion and (B) the LDLR exon 3 missense variant p.Trp87Arg. The blue lines and associated grey shading represent the linear regression line of best fit and 95% CI, respectively.
We used the UK Biobank imputed genotyping array data to compare the association between the 28 SNV polygenic score and more comprehensive LDL-C polygenic scores composed of 223 SNVs and 1.92 million SNVs among individuals of British White ancestry (n=389 127). There was a significant correlation between LDL-C levels, in mmol/L, and SD units of LDL-C polygenic score for the 28 SNV score (β [SE]: 0.820 [0.006], P<0.0001, R2=0.074), 223 SNV score (β [SE]=0.840 [0.005], P<0.0001, R2=0.102), and 1.92 million SNV score (fraction of causal variants =0.1: β [SE]: 0.866 [0.005], P<0.0001, R2=0.113) when adjusted for age and sex. These results suggest that while the 28 SNV polygenic score provides a highly significant association with LDL-C levels, more comprehensive polygenic scores will likely provide improvements in predicting LDL-C levels (Figure I in the Data Supplement).
LDL-C Polygenic Scores Associate With Increased Cardiovascular Risk for Individuals With FH
We used a fixed-effect meta-analysis to assess the association between elevated LDL-CPSP and risk of ASCVD among 3 cohorts of individuals with monogenic FH (overall n=1120). ASCVD events were defined as a composite end point of myocardial infarction, coronary or carotid revascularization, transient ischemic attack, or stroke (Methods and Table V in the Data Supplement). A LDL-CPSP ≥80th percentile was associated with a trend towards increased risk of ASCVD in all 3 cohorts in analyses that were adjusted for sex and for which age was used as the timescale. This effect was statistically significant in the meta-analysis (hazard ratio [95% CI], 1.48 [1.02–2.14], P=0.04; Figure 3A), and there was no significant interstudy heterogeneity (Q=0.48 on 2 df, P=0.79).
Figure 3.
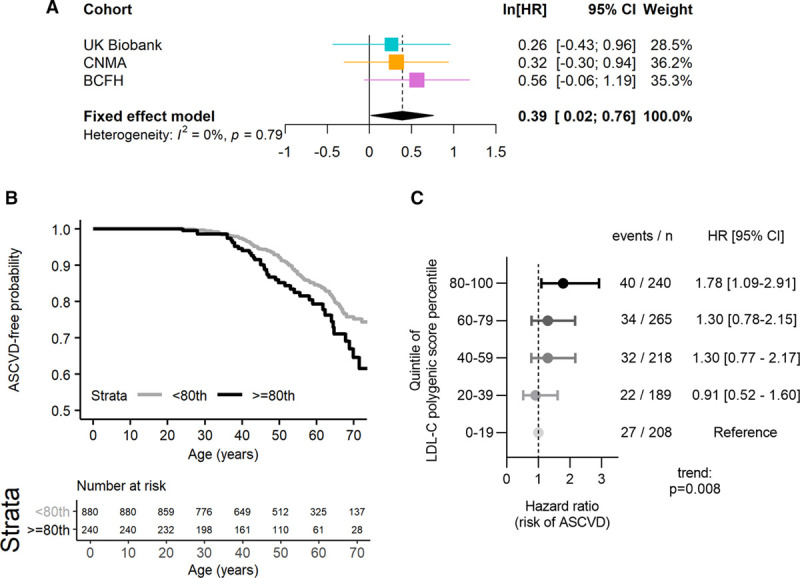
Association between risk of events and LDL-C (low-density lipoprotein cholesterol) polygenic score percentile among individuals with familial hypercholesterolemia. Atherosclerotic cardiovascular events included myocardial infarction, cardiovascular or carotid revascularization, and stroke. A, A fixed-effect meta-analyses of the β coefficients (ln[hazard ratio (HR)]) and SE obtained from Cox-proportional hazard models for risk of atherosclerotic cardiovascular events is shown for an LDL-C polygenic score ≥80th percentile score relative to and LDL-C polygenic score <80th percentile score. These Cox-proportional hazard models were adjusted for sex and used age as the time-to-event time scale. The studies included in the meta-analysis were the British Columbia Familial Hypercholesterolemia (BCFH), Nutrition, Metabolism and Atherosclerosis Clinic (CNMA), and UK Biobank cohorts. B, The time-to-atherosclerotic cardiovascular event curves are shown for the combined cohorts stratified by LDL-C polygenic score percentile above and below the 80th percentile. C, Sex-adjusted Cox-proportional hazard models are shown for the combined cohorts stratified by quintiles of LDL-C polygenic score percentile. ASCVD indicates atherosclerotic cardiovascular disease.
Additionally, when all 3 cohorts were merged, individuals with monogenic FH and LDL-CPSP ≥80th percentile displayed significantly greater risk of ASCVD compared with individuals with monogenic FH and LDL-CPSP <80th percentile (Log-rank P=0.01; Figure 3B). This finding was also observed in Cox-proportional hazard models that were unadjusted (hazard ratio [95% CI]: 1.60 [1.12–2.29], P=0.01) and adjusted for sex (hazard ratio [95% CI]: 1.58 [1.10–2.27], P=0.01). Similarly, continuous LDL-CPSP and quintiles of LDL-CPSP were significantly associated with increasing risk of ASCVD among individuals with monogenic FH in analyses adjusted for sex (hazard ratio [95% CI]: 1.15 [1.03–1.29] per 20% increase in LDL-CPSP, P=0.01; Figure 3C).
LDL-C Polygenic Score Influences the Penetrance of Monogenic FH
In the UK Biobank cohort, cases of monogenic FH were identified from the general population based on genotype, whereas individuals from the BCFH and CNMA cohorts were identified from lipid clinics based on a clinical diagnosis of FH. We observed that many individuals from the UK Biobank with monogenic FH-associated variants did not manifest a typical phenotype of severe hypercholesterolemia.31 Indeed, a majority of carriers of FH-associated variants had LDL-C levels <5 mmol/L (205 of 282 individuals with LDL-C measurements =72.7%), a commonly used cutoff for FH in many diagnostic algorithms.32 We examined whether the LDL-CPSP contributed to the incomplete penetrance of FH-associated variants in the UK Biobank cohort.
Individuals with LDL-C levels ≥5 mmol/L had significantly higher LDL-C polygenic scores than individuals with LDL-C levels <5 mmol/L (Mann-Whitney P=0.04; Figure 4A). In particular, we found that 22.0 % (36 of 164) of individuals with an FH-associated variant and an LDL-CPSP below the 50th had a LDL-C ≥5 mmol/L, compared with 34.7% (41 of 118) of individuals with an FH-associated variant and a LDL-CPSP greater than the 50th percentile (χ2 P=0.02; Figure 4B). Individuals with an FH-associated variant and a LDL-CPSP greater than the 50th percentile also had a nonstatistically significant higher prevalence of parental history of heart disease relative to individuals with an FH-associated variant and a LDL-CPSP below the 50th percentile (61.9% versus 54.0%, χ2 P=0.15; Figure 4C).
Figure 4.
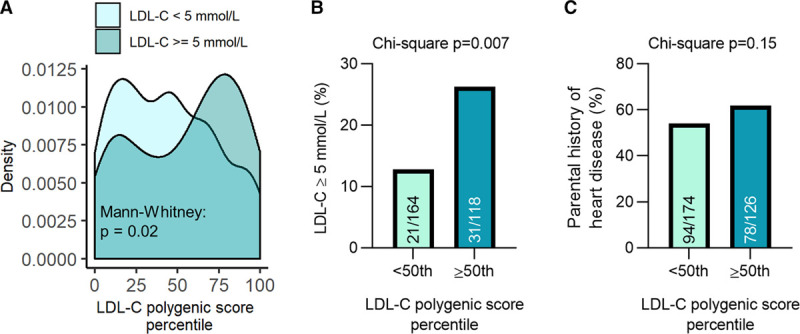
LDL-C (Low-density lipoprotein cholesterol) polygenic score associates with the penetrance of familial hypercholesterolemia (FH). A, Individuals with monogenic FH and an LDL-C ≥5 mmol/L had significantly higher LDL-C polygenic score percentiles than those with monogenic FH and an LDL-C <5 mmol/L. Individuals with monogenic FH and an LDL-C polygenic score ≥50th percentile had a greater prevalence of (B) LDL-C levels ≥5 mmol/L and (C) parental history of heart disease relative to those with monogenic FH and an LDL-C polygenic score <50th percentile.
Discussion
Here, using 3 independent cohorts, we demonstrated that LDL-C polygenic scores significantly modulate LDL-C levels and risk of ASCVD among individuals with monogenic FH. These results highlight that polygenic factors can influence the phenotype of monogenic FH, which is often viewed as a classical Mendelian disorder, and that expanded genetic testing may improve risk prediction for this condition.
Monogenic FH is a highly heterogeneous condition and individuals with the same pathogenic variant can display markedly different phenotypes.6,7,9,25 Previous studies have shown that polygenic factors can modulate the LDL-C levels observed in monogenic FH,25 but an effect on ASCVD risk has not been assessed. Individuals with FH-associated variants are known to be at greater risk of ASCVD than individuals with clinical FH without identifiable FH-associated variants,12,13,18,31 and we previously reported among individuals with monogenic FH, a superimposed elevated LDL-CPSP was associated with a trend towards increased risk of ASCVD.12 The major advance of the current study is the demonstration that polygenic modulation of LDL-C levels is significantly associated with increased risk of ASCVD in individuals with monogenic FH.
We used a limited LDL-C polygenic score, composed of 28 SNVs, to demonstrate that polygenic factors are important modulators of the penetrance of monogenic FH. Notably, this was the case even among individuals carrying the identical FH-associated variant. Broader, genome-wide polygenic scores have also been developed for LDL-C14,22 and coronary artery disease,33 and appear to enable improved risk prediction relative to more limited polygenic scores due to their ability to consolidate the associations of millions of common genetic variants. Determination of genome-wide polygenic scores for LDL-C levels22 and ASCVD risk17,33 for individuals with FH could complement standard genetic testing and further improve the risk prediction for ASCVD in this population. Specifically, Natarajan et al22 observed that a polygenic score composed of ≈2 million SNVs was associated with an ≈21.6% increase of LDL-C variance explained relative to a restricted score of 59 independent SNVs (R2 expanded=0.298 versus R2 restricted=0.245). In this study of individuals from the UK Biobank, the variance explained by both an expanded polygenic score composed of 1.92 million SNVs or restricted LDL-C polygenic score (28 or 223 independent SNVs) were notably lower. However, the relative increase in LDL-C variance explained between an expanded polygenic versus restricted score were comparable (R2=0.113 versus R2=0.074). This suggests that differences between the study population may account for the lower proportion of variance explained. One notable difference between UK Biobank and the cohorts studied by Natarajan et al22 is that individuals from the UK Biobank tended to be older and more likely to use lipid-lowering medication; both factors may impair the performance of the LDL-C polygenic score. In support of this, we note that the LDL-C polygenic score displayed the greatest performance in the CNMA cohort in which baseline LDL-C levels were measured following a washout of lipid-lowering medication.
The relatively mild phenotypes of monogenic FH observed in the UK Biobank adds to evolving literature suggesting that the penetrance and expressivity of FH may be more variable than has been traditionally accepted.5–8,31 Here, we show that polygenic factors related to LDL-C may be one important modifying factor. This is supported by observations of polygenic factors related to risk of ASCVD17 and lipoprotein(a)26,34 levels being important modifiers of ASCVD risk for individuals with FH. Environmental factors such as diet, exercise, and adherence to cholesterol-lowering medication therapy are also important protective factors for ASCVD.6,20,35 Notably, the UK Biobank exhibits a healthy volunteer selection bias compared with cohorts of patients recruited from cardiology or lipid clinics, the latter of which would tend to be enriched for individuals with more severe clinical presentations.36 A better understanding of modifiers of FH phenotype among the general population may identify new and important genetic or clinical factors that are associated with risk of ASCVD which could inform the design of new therapeutic strategies for preventing ASCVD in both FH and the general population.
More comprehensive sequencing or array strategies for characterizing FH beyond targeted sequencing of candidate genes may allow improved clinical risk prediction and identify genetic diagnoses for individuals that have alternative or atypical explanation for an FH-like phenotype such as polygenic hypercholesterolemia, elevated lipoprotein(a),34 or atypical variants (eg, ABCG537). This work demonstrates that genetic testing beyond the common candidate gene analysis of LDLR, APOB, and PCSK9 may provide insights relevant to the clinical heterogeneity of FH.
This study has some notable strengths, as well as some important limitations. First, we used 3 independent cohorts of individuals with FH, which all showed similar findings in terms of the impact of the LDL-C polygenic scores on ASCVD risk. Previous studies examining LDL-C polygenic scores in FH have been underpowered to detect an effect on cardiovascular events, and we were able to overcome that limitation. Second, our study was restricted to only individuals with molecularly confirmed monogenic FH, resulting in a more well-defined population of individuals with FH, and allowing us to determine the impact of polygenic modulation of this monogenic condition. Limitations of our study include that we used a limited LDL-C polygenic score, composed of only 28 SNVs. Future studies will be needed to examine the benefit of using a genome-wide LDL-C polygenic score for risk prediction in monogenic FH. Second, the approach to determine baseline lipid profiles varied between cohorts. The CNMA cohort used a 4-week washout period before obtaining a lipid profile to reduce confounding caused by cholesterol-lowering medication. This approach is likely one of the reasons why the strongest correlation between LDL-C polygenic scores and measured LDL-C levels was observed in this cohort. In contrast, for the BCFH and UK Biobank cohorts, baseline LDL-C levels were estimated for individuals using cholesterol-lowering medication estimated by using a correction factor for 1.43.1,38 Third, while our study included individuals from both Canada and the United Kingdom, the overall study population was predominately of European ancestry. The generalizability of lipid polygenic scores to individuals with FH of other genetic ancestries requires further investigation.
In conclusion, we report that individuals with monogenic FH that have an elevated LDL-C polygenic scores tend to have higher levels of LDL-C and greater risk of ASCVD than individuals without an elevated LDL-CPSP. These results show that, even at the extremes of Mendelian disorders, polygenic factors can have an important influence on clinical phenotypes and outcomes. Our results suggest that there may be utility in broader genetic testing strategies, that go beyond candidate genes, to improve the risk stratification of individuals with monogenic FH.
Acknowledgments
We acknowledge the individuals involved in the British Columbia Familial Hypercholesterolemia; Nutrition, Metabolism and Atherosclerosis Clinic; and UK Biobank studies; the developers of software tools; and Compute Canada for establishing computational resources and providing support.
Sources of Funding
This project was supported by a Sector Improvement Program project from Genome BC. M. Trinder is supported by a Vanier Canada Graduate Scholarship. Dr Brunham is supported by a Michael Smith Foundation for Health Research Scholar Award and is a Canada Research Chair in Precision Cardiovascular Disease Prevention.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- ASCVD
- atherosclerotic cardiovascular disease
- BCFH
- British Columbia Familial Hypercholesterolemia
- CNMA
- Nutrition, Metabolism and Atherosclerosis Clinic
- FH
- familial hypercholesterolemia
- LDL-C
- low-density lipoprotein cholesterol
- LDL-CPSP
- low-density lipoprotein cholesterol polygenic score percentile
- SNVs
- single-nucleotide variants
The Data Supplement is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.120.002919.
For Sources of Funding and Disclosures, see page 522.
References
- 1.Benn M, Watts GF, Tybjærg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the copenhagen general population study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37:1384–1394. doi: 10.1093/eurheartj/ehw028 [DOI] [PubMed] [Google Scholar]
- 2.Akioyamen LE, Genest J, Shan SD, Reel RL, Albaum JM, Chu A, Tu JV. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open. 2017;7:e016461 doi: 10.1136/bmjopen-2017-016461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. 2017;38:2459–2472. doi: 10.1093/eurheartj/ehx144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, et al. ; European Atherosclerosis Society Consensus Panel. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European atherosclerosis society. Eur Heart J. 2013;34:3478–390a. doi: 10.1093/eurheartj/eht273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berberich AJ, Hegele RA. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol. 2019;16:9–20. doi: 10.1038/s41569-018-0052-6 [DOI] [PubMed] [Google Scholar]
- 6.Pimstone SN, Sun XM, du Souich C, Frohlich JJ, Hayden MR, Soutar AK. Phenotypic variation in heterozygous familial hypercholesterolemia: a comparison of Chinese patients with the same or similar mutations in the LDL receptor gene in China or Canada. Arterioscler Thromb Vasc Biol. 1998;18:309–315. doi: 10.1161/01.atv.18.2.309 [DOI] [PubMed] [Google Scholar]
- 7.Mszar R, Grandhi GR, Valero-Elizondo J, Virani SS, Blankstein R, Blaha M, Mata P, Miname MH, Al Rasadi K, Krumholz HM, et al. Absence of coronary artery calcification in middle-aged familial hypercholesterolemia patients without atherosclerotic cardiovascular disease. JACC Cardiovasc Imaging. 2020;13:1090–1092. doi: 10.1016/j.jcmg.2019.11.001 [DOI] [PubMed] [Google Scholar]
- 8.Raal FJ, Sjouke B, Hovingh GK, Isaac BF. Phenotype diversity among patients with homozygous familial hypercholesterolemia: a cohort study. Atherosclerosis. 2016;248:238–244. doi: 10.1016/j.atherosclerosis.2016.03.009 [DOI] [PubMed] [Google Scholar]
- 9.Magaña Torres MT, Mora-Hernández S, Vázquez Cárdenas NA, González Jaimes A. Homozygous familial hypercholesterolemia: the c.1055G>A mutation in the LDLR gene and clinical heterogeneity. J Clin Lipidol. 2014;8:525–527. doi: 10.1016/j.jacl.2014.05.002 [DOI] [PubMed] [Google Scholar]
- 10.Brunham LR, Ruel I, Aljenedil S, Rivière JB, Baass A, Tu JV, Mancini GBJ, Raggi P, Gupta M, Couture P, et al. Canadian cardiovascular society position statement on familial hypercholesterolemia: update 2018. Can J Cardiol. 2018;34:1553–1563. doi: 10.1016/j.cjca.2018.09.005 [DOI] [PubMed] [Google Scholar]
- 11.Sun YV, Damrauer SM, Hui Q, Assimes TL, Ho YL, Natarajan P, Klarin D, Huang J, Lynch J, DuVall SL, et al. Effects of genetic variants associated with familial hypercholesterolemia on low-density lipoprotein-cholesterol levels and cardiovascular outcomes in the million veteran program. Circ Genom Precis Med. 2018;11:e002192 doi: 10.1161/CIRCGEN.118.002192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trinder M, Li X, DeCastro ML, Cermakova L, Sadananda S, Jackson LM, Azizi H, Mancini GBJ, Francis GA, Frohlich J, et al. Risk of premature atherosclerotic disease in patients with monogenic versus polygenic familial hypercholesterolemia. J Am Coll Cardiol. 2019;74:512–522. doi: 10.1016/j.jacc.2019.05.043 [DOI] [PubMed] [Google Scholar]
- 13.Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. doi: 10.1016/j.jacc.2016.03.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM, et al. ; Global Lipids Genetics Consortium; Myocardial Infarction Genetics (MIGen) Consortium; Geisinger-Regeneron DiscovEHR Collaboration; VA Million Veteran Program. Genetics of blood lipids among ~300,000 multi-ethnic participants of the million veteran program. Nat Genet. 2018;50:1514–1523. doi: 10.1038/s41588-018-0222-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. ; Global Lipids Genetics Consortium. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fahed AC, Wang M, Homburger JR, Patel AP, Bick AG, Neben CL, Lai C, Brockman D, Philippakis A, Ellinor PT, et al. Polygenic background modifies penetrance of monogenic variants conferring risk for coronary artery disease, breast cancer, or colorectal cancer. Nature Commun. 2020;11:3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Dron JS, Ban MR, Robinson JF, McIntyre AD, Alazzam M, Zhao PJ, Dilliott AA, Cao H, Huff MW, et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler Thromb Vasc Biol. 2016;36:2439–2445. doi: 10.1161/ATVBAHA.116.308027 [DOI] [PubMed] [Google Scholar]
- 19.Sijbrands EJ, Westendorp RG, Defesche JC, de Meier PH, Smelt AH, Kastelein JJ. Mortality over two centuries in large pedigree with familial hypercholesterolaemia: family tree mortality study. BMJ. 2001;322:1019–1023. doi: 10.1136/bmj.322.7293.1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams RR, Hasstedt SJ, Wilson DE, Ash KO, Yanowitz FF, Reiber GE, Kuida H. Evidence that men with familial hypercholesterolemia can avoid early coronary death. An analysis of 77 gene carriers in four Utah pedigrees. JAMA. 1986;255:219–224 [PubMed] [Google Scholar]
- 21.Patel AP, Wang M, Fahed AC, Mason-Suares H, Brockman D, Pelletier R, Amr S, Machini K, Hawley M, Witkowski L, et al. Association of rare pathogenic DNA variants for familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and lynch syndrome with disease risk in adults according to family history. JAMA Netw Open. 2020;3:e203959 doi: 10.1001/jamanetworkopen.2020.3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Natarajan P, Peloso GM, Zekavat SM, Montasser M, Ganna A, Chaffin M, Khera AV, Zhou W, Bloom JM, Engreitz JM, et al. ; NHLBI TOPMed Lipids Working Group. Deep-coverage whole genome sequences and blood lipids among 16,324 individuals. Nat Commun. 2018;9:3391 doi: 10.1038/s41467-018-05747-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, Harrison SC, Li K, Drenos F, Karpe F, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet. 2013;381:1293–1301. doi: 10.1016/S0140-6736(12)62127-8 [DOI] [PubMed] [Google Scholar]
- 24.Futema M, Shah S, Cooper JA, Li K, Whittall RA, Sharifi M, Goldberg O, Drogari E, Mollaki V, Wiegman A, et al. Refinement of variant selection for the LDL cholesterol genetic risk score in the diagnosis of the polygenic form of clinical familial hypercholesterolemia and replication in samples from 6 countries. Clin Chem. 2015;61:231–238. doi: 10.1373/clinchem.2014.231365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oetjens MT, Kelly MA, Sturm AC, Martin CL, Ledbetter DH. Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat Commun. 2019;10:4897 doi: 10.1038/s41467-019-12869-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holmes DT, Schick BA, Humphries KH, Frohlich J. Lipoprotein(a) is an independent risk factor for cardiovascular disease in heterozygous familial hypercholesterolemia. Clin Chem. 2005;51:2067–2073. doi: 10.1373/clinchem.2005.055228 [DOI] [PubMed] [Google Scholar]
- 27.Brunham LR, Cermakova L, Lee T, Priecelova I, Alloul K, de Chantal M, Francis GA, Frohlich J. Contemporary trends in the management and outcomes of patients with familial hypercholesterolemia in Canada: a prospective observational study. Can J Cardiol. 2017;33:385–392. doi: 10.1016/j.cjca.2016.08.016 [DOI] [PubMed] [Google Scholar]
- 28.Paquette M, Chong M, Thériault S, Dufour R, Paré G, Baass A. Polygenic risk score predicts prevalence of cardiovascular disease in patients with familial hypercholesterolemia. J Clin Lipidol. 2017;11:725–732.e5. doi: 10.1016/j.jacl.2017.03.019 [DOI] [PubMed] [Google Scholar]
- 29.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Hout CV, Tachmazidou I, Backman JD, Hoffman JX, Ye B, Pandey AK, Gonzaga-Jauregui C, Khalid S, Liu D, Banerjee N, et al. Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. bioRxiv. 2019572347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trinder M, Francis GA, Brunham LR. Association of monogenic vs polygenic hypercholesterolemia with risk of atherosclerotic cardiovascular disease. JAMA Cardiol. 2020;5:390–399. doi: 10.1001/jamacardio.2019.5954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruel I, Brisson D, Aljenedil S, Awan Z, Baass A, Bélanger A, Bergeron J, Bewick D, Brophy JM, Brunham LR, et al. Simplified Canadian definition for familial hypercholesterolemia. Can J Cardiol. 2018;34:1210–1214. doi: 10.1016/j.cjca.2018.05.015 [DOI] [PubMed] [Google Scholar]
- 33.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langsted A, Kamstrup PR, Benn M, Tybjærg-Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4:577–587. doi: 10.1016/S2213-8587(16)30042-0 [DOI] [PubMed] [Google Scholar]
- 35.Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, Chasman DI, Baber U, Mehran R, Rader DJ, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–2358. doi: 10.1056/NEJMoa1605086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, Collins R, Allen NE. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am J Epidemiol. 2017;186:1026–1034. doi: 10.1093/aje/kwx246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nomura A, Emdin CA, Won HH, Peloso GM, Natarajan P, Ardissino D, Danesh J, Schunkert H, Correa A, Bown MJ, et al. Heterozygous ATP-binding cassette transporter G5 gene deficiency and risk of coronary artery disease. Circ Genom Precis Med. 2020. doi: 10.1161/CIRCGEN.119.002871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones PH, Davidson MH, Stein EA, Bays HE, McKenney JM, Miller E, Cain VA, Blasetto JW; STELLAR Study Group. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92:152–160. doi: 10.1016/s0002-9149(03)00530-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.