

Abstract
The novel respiratory virus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), emerged during late 2019 and spread rapidly across the world. It is now recognised that the nervous system can be affected in COVID-19, with several studies reporting long-term cognitive problems in patients. The metabolic pathway of tryptophan degradation, known as the kynurenine pathway (KP), is significantly activated in patients with COVID-19. KP metabolites have roles in regulating both inflammatory/immune responses and neurological functions. In this review, we speculate on the effects of KP activation in patients with COVID-19, and how modulation of this pathway might impact inflammation and reduce neurological symptoms.
Teaser
The kynurenine pathway of tryptophan degradation is activated in patients with Coronavirus 2019 (COVID-19). The possible effects of kynurenine metabolites on inflammation and neurocognition in COVID-19 and the potential use of kynurenine 3-monooxygenase inhibitors for treatment are discussed.
The kynurenine pathway
The KP is responsible for the degradation of ∼95% of the essential amino acid tryptophan. The initial step of this metabolic pathway involves the conversion of tryptophan to N-formyl-l-kynurenine by the enzymes idoleamine 2,3-dioxygenase 1 and 2 (IDO1 and IDO2) and tryptophan 2,3-dioxygenase (TDO) [1], followed by the enzymatic conversion of N-formyl-l-kynurenine to l-kynurenine (l-KYN). l-KYN can be further metabolised through two main branches of the KP, either to kynurenic acid (KYNA) by kynurenine aminotransferases (KAT), or to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO). 3-HK is then metabolised to quinolinic acid (QUIN) and finally to the cofactor NAD+, which has a key role in cellular metabolism [2]. It is well established that several KP metabolites are neuroactive and likely contribute to the pathogenesis of neurodegenerative and neuropsychiatric conditions [3]. For example, KYNA is neuroprotective because of its ability to block NMDA receptors (NMDARs) [4] and α7 nicotinic acetylcholine receptors (α7nAChRs) [5] and prevents neuronal cell death by inhibiting excessive glutamate signalling [6]. KYNA also protects cells from oxidative stress by acting as a free radical scavenger [7]. However, high concentrations of KYNA can be detrimental because they can cause excessive inhibition of NMDARs, resulting in cognitive impairment 8, 9. By contrast, QUIN is a NMDAR agonist that, at high concentrations, induces excitotoxicity [10] and can cause oxidative cell death by promoting the generation of free radicals [11]. In addition, high concentrations of 3-HK are neurotoxic and cause cell death through the formation of free radicals and subsequent oxidative stress [12].
Disruption of the KP in the periphery and central nervous system during inflammation
Activation of the KP by inflammation
Many studies have demonstrated that the KP is activated following immune activation and has crucial roles in the inflammatory response. Proinflammatory cytokines released by immune cells during inflammation induce the upregulation of IDO1, resulting in tryptophan depletion because tryptophan is converted to l-KYN [13]. This directly inhibits T cell proliferation given that these cells are deprived of the essential metabolite tryptophan [13], as well as indirectly inhibiting T cell proliferation by preventing the maturation of dendritic cells [14]. In addition, the KP metabolites l-KYN, 3-HK, and QUIN inhibit T cell proliferation or induce apoptosis in these cells 15, 16, 17, resulting in reduced numbers of these immune cells. By contrast, IDO expression in monocytes increases the proliferation of immunosuppressant regulatory T helper cells (Tregs) [18] and promotes the differentiation of naïve CD4+ T cells into Tregs through a mechanism that involves the binding of l-KYN to aryl hydrocarbon receptors (AHR) [19]. KYNA has also been shown to have anti-inflammatory effects. For example, it reduces the release of proinflammatory cytokines from lipopolysaccharide (LPS)-activated monocytes 20, 21. Administration of KYNA to LPS-treated mice also reduced levels of the cytokine tumour necrosis factor alpha (TNFα) and increased survival in a NMDAR-independent mechanism [22]. These anti-inflammatory effects of KYNA might be mediated through other KYNA targets in the periphery, such as the G-protein receptor 35 (GPR35), which is expressed in peripheral leukocytes [21], and the transcription factor AHR [23]. A recently identified KYNA target is the hydroxycarboxylic acid receptor 3 (HCAR3) [24], which is expressed on immune cells and, together with KYNA, might mediate immune suppression [25]. Therefore, the actions of these KP metabolites on immune cells can modulate the immune response and reduce inflammation.
The KP can be activated in both the central nervous system (CNS) and the periphery in response to inflammation. Although TDO mediates basal tryptophan degradation, IDO1 is upregulated by proinflammatory cytokines under inflammatory conditions [26]. In the periphery, the activation of the KP occurs in monocytes, which upregulate IDO1 expression and activity in response to proinflammatory cytokines, such as interferon gamma (IFNγ) [27], resulting in increased degradation of tryptophan to l-KYN. Peripheral l-KYN can then cross the blood–brain barrier (BBB) into the CNS, where l-KYN is further metabolised. In cells of the CNS, such as microglia and infiltrating macrophages, proinflammatory cytokines induce KMO activity [28], promoting the formation of neurotoxic QUIN by macrophages [28]. IDO1 expression is also upregulated in response to IFNγ in infiltrating macrophages and, to a lesser extent, in microglia [29], which can further elevate levels of l-KYN in the brain.
The role of the KP in viral infections and associated neurological impairments
Previous studies have reported that the KP is activated in response to viral infections, such as HIV-1 [30], hepatitis C virus (HCV) [31], and herpes simplex virus [32]. Interestingly, some of these chronic viral infections have been associated with neurological and cognitive impairments in patients, which in some cases have been at least partly attributed to the activation of the KP and the production of neuroactive KP metabolites 32, 33, 34, 35, 36. For example, several studies showed the activation of the KP in patients with HIV-1, particularly in those with HIV-1-associated dementia 34, 36. Huengsberg et al. [30] observed elevated l-KYN:tryptophan ratios in the serum of patients with HIV-1 compared with controls. Other studies detected increased levels of KP metabolites in the CNS of patients with HIV-1, such as elevated levels of neurotoxic 3-HK [35], excitotoxic QUIN [36], and KYNA [37]. Interestingly, higher levels of 3-HK and QUIN have been detected in patients with HIV-associated dementia compared with patients with HIV without dementia 35, 36. This might be the result of increased IDO activity, which has been observed in the brains of patients with HIV-associated dementia compared with patients with HIV but without dementia and healthy controls [34]. The potential detrimental effects of KP metabolites in patients with HIV-1 is supported by Kerr et al. [38], who showed in vitro that high levels of QUIN paralleling those observed in the cerebrospinal fluid (CSF) of patients with HIV-1 dementia, induced necrosis of neurons and changes in neuron morphology similar to those found in the brains of patients with HIV dementia.
HCV infections have also been associated with activation of the KP [31] and cognitive impairments [39]. Patients with HCV were shown to have increased peripheral activation of the KP compared with healthy controls, indicated by increased levels of l-KYN in the blood 31, 40, and elevated IDO1 expression in the liver [31] and peripheral monocytes [40]. Interestingly, correlations have been observed between cognitive changes and increased levels of l-KYN in the periphery of patients with HCV [33], suggesting the involvement of the KP in cognitive dysfunction in these patients. Furthermore, simulation of viral infection in pregnant rats using poly I:C was shown to disrupt the KP by activating the maternal immune system, leading to a dysfunctional KP in the periphery of preadolescent offspring, including increased levels of QUIN and reduced levels of l-KYN and KYNA [41]. A separate study also showed disruptions to the KP in the CNS of the offspring of poly I:C-treated rats in response to immune activation in the adult offspring [42]. Interestingly, increased levels of serum l-KYN and QUIN have been observed in patients with influenza-associated encephalopathy compared with patients with influenza without encephalopathy [43]. This is relevant to COVID-19 because similar brain pathology has been observed in patients with COVID-19 44, 45, 46, and indicates that KP activation might have a role in cognitive symptoms associated with COVID-19. Together, these studies indicate that activation of the KP in the periphery in response to stimulation of the immune system by viral infections might also affect the brain and contribute to cognitive dysfunction.
Neurological symptoms in patients with COVID-19
During the COVID-19 pandemic, it has been increasingly recognised that, as well as the respiratory symptoms caused by the virus, patients may have neurological and neuropsychiatric problems. An early report identified neurological symptoms in patients with COVID-19, including CNS symptoms (headaches and dizziness) and peripheral nervous system symptoms (loss of taste and smell) [47]. A separate study found that patients with severe COVID-19 had neurological symptoms, with 65% of patients exhibiting confusion and 33% of patients with disruption of executive function, including disorientation and poor attention, even after being discharged from hospital [48]. A more extensive study by Varatharaj et al. [44] examined 153 patients with COVID-19 admitted to hospitals throughout the UK who also exhibited neurological symptoms, including cerebrovascular events (stroke or intracerebral haemorrhage) and altered mental state (change in behaviour, personality, consciousness, or cognitive function). This study found that 31% of patients had an altered mental state, including encephalopathy, dementia-like cognitive impairment, and psychosis [44].
The cause of neurological symptoms in patients with COVID-19 is unknown, but might be due to the host's inflammatory response to the infection, direct effects of SARS-CoV-2, which causes COVID-19, on the CNS, or effects of the virus in the periphery affecting the brain [45]. A recent study by Kremer et al. [49] reported that some patients with COVID-19 and neurological symptoms also had markers of inflammation in the CSF, such as a high number of leukocytes, high protein levels, and increased immunoglobulin G levels. However, most studies have failed to detect SARS-CoV-2 mRNA in the CSF 48, 49, 50, indicating that peripheral inflammation might be implicated in the neurological symptoms associated with severe cases of COVID-19. Given that activation of the KP has been observed in patients with COVID-19 51, 52 it is interesting to speculate that this might lead to changes in levels of KP metabolites, resulting in modulation of the immune response and promoting cognitive dysfunction.
Activation of the KP in patients with COVID-19
Given that viral infections can result in activation of the KP, it is possible that this metabolic pathway is also activated in patients infected with SARS-CoV-2. In fact, several recent studies indicate that the KP is significantly activated in patients with SARS-CoV-2. In a study by Thomas et al. [51], targeted metabolomics revealed that patients with COVID-19 had reduced levels of tryptophan and elevated levels of l-KYN in their serum compared with controls. Overall, greater changes in metabolite levels were observed in patients with high levels of the proinflammatory cytokine interleukin-6 (IL-6) compared with patients with low IL-6 levels and with controls, indicating a general dysregulation of amino acid and fatty acid metabolic pathways in patients with COVID-19. However, in the same study, both targeted and untargeted metabolomics showed that the tryptophan pathway was the pathway most affected by SARS-CoV-2 [51]. Specifically, untargeted metabolomics revealed that tryptophan, serotonin, and indole pyruvate were significantly decreased in the serum of patients compared with controls (Figure 1 ) [51]. Serum 3-HK levels were also reduced in patients compared with controls, whereas the KP metabolites l-KYN, KYNA, picolinic acid, and nicotinic acid were significantly increased in patients with COVID-19, particularly those with high levels of IL-6 [51]. In support of these findings, another metabolomics study by Shen et al. [52] reported that levels of l-KYN were significantly increased in serum from patients with severe COVID-19 compared with healthy controls. Increased levels of KYNA and QUIN were also observed in serum from patients with severe COVID-19 compared with healthy controls, whereas levels of tryptophan were reduced in these patients [52] (Figure 1). Another study by Fraser et al. [53] showed that l-KYN was the major metabolite increased in the plasma of patients severely ill with COVID-19 compared with patients who were COVID-19 negative or with healthy controls. Interestingly, Cai et al. [54] observed gender-specific differences in correlations between serum levels of KYNA and the immune response in patients with COVID-19, with increased KYNA being associated with elevated levels of several inflammatory cytokines/chemokines and reduced T cell numbers in male patients but not in female patients. An elevated KYNA:l-KYN ratio was also associated with increased severity of the disease in male, but not female patients [54]. Kimhofer et al. [55] also observed an increased l-KYN/tryptophan ratio in the plasma of patients with COVID-19 compared with healthy controls, indicating activation of the KP in the periphery. Finally, transcriptomics analysis of peripheral blood mononuclear cells (PBMCs) isolated from patients with COVID-19 revealed increases in the expression of genes associated with tryptophan metabolism in patients compared with healthy controls [56].
Figure 1.

Overview of the activation of the kynurenine pathway in the periphery of patients with Coronavirus 2019 (COVID-19). Several metabolomics studies revealed that patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have reduced levels of tryptophan, serotonin, and indole pyruvate in the periphery. This indicates activation of the kynurenine pathway because tryptophan is converted to N-formyl-l-kynurenine by indoleamine 2,3-dioxygenase 1 or 2 (IDO1/2), and tryptophan 2,3-dioxygenase (TDO) and then further metabolised to l-kynurenine (l-KYN). These studies also showed that levels of l-KYN and kynurenic acid (KYNA) are increased in the periphery of patients with COVID-19 compared with healthy controls. l-KYN is converted to KYNA by the kynurenine aminotransferase enzymes (KAT) I, II, III or IV. In the other main branch of the kynurenine pathway, l-KYN is converted to 3-hydroxykynurenine (3-HK) by kynurenine 3-monooxygenase (KMO). Reduced levels of 3-HK were detected in the periphery of patients with COVID-19, compared with increased levels of downstream metabolites quinolinic acid (QUIN) and nicotinic acid.
These studies indicate that the KP is activated in the periphery of patients with severe COVID-19, resulting in increased peripheral levels of l-KYN and depletion of tryptophan. It is well established that IDO1 expression is upregulated by proinflammatory cytokines, such as IFNγ [27], and that this drives increased synthesis of l-KYN [26]. Although the expression of IDO1 has not been examined in patients with COVID-19, it is possible that increased l-KYN/tryptophan ratios result from enhanced IDO1 expression or activity, resulting in increased metabolism of tryptophan to l-KYN. It is interesting that KYNA levels are elevated in the periphery of patients with COVID-19. Given that KYNA cannot cross the BBB [57], this is unlikely to be the result of KYNA produced in the CNS, although disruption of the BBB has been detected in some patients with COVID-19 [50]. However, KYNA can also be generated in the periphery by KAT enzymes [58], as well as by the conversion of l-KYN to KYNA in the presence of reactive oxygen species [59]. Therefore, KYNA production in the periphery might result from the highly inflammatory state associated with SARS-CoV-2 infections. Furthermore, if KYNA is elevated in the CNS of patients with COVID-19 because of increased transport of peripheral l-KYN across the BBB and increased KYNA synthesis in the CNS, this might contribute to the cognitive impairments reported in some patients with SARS-CoV-2, because high concentrations of KYNA can block glutamate-mediated neurotransmission [6]. In fact, Cai et al. [54] reported that patients with COVID-19 and deteriorated health had reduced glutamate levels and increased KYNA:l-KYN ratios in their serum compared with stabilised patients. However, to date, the levels of KP metabolites in the CNS of patients with COVID-19 have not been examined. Increased levels of QUIN [52] and nicotinic acid [51] were also detected in the periphery of patients with COVID-19, indicating increased flux through the KMO branch of the KP, although 3-HK was reduced in patients with COVID-19 compared with healthy controls [51]. QUIN can cause oxidative stress and neuronal cell death 10, 11, which could contribute to the cognitive symptoms and encephalopathy reported in some patients with COVID-19 44, 45, 46.
Another important point is that levels of serotonin were shown to be reduced in patients with COVID-19 51, 52. This might have an impact on other neurological symptoms in COVID-19, such as depression. Depression has been reported in some patients with COVID-19 [60], but it remains unclear as to whether this is a direct or indirect effect of the virus and associated inflammatory response on the brain, or whether it is because of the psychological impact of having a severe illness [60]. Therefore, it remains to be elucidated whether the activation of the KP in COVID-19 resulting in the increased conversion of tryptophan to l-KYN and the depletion of serotonin increases the susceptibility of patients with COVID-19 to depression.
Targeting KMO in infections and inflammation
Inhibition of KMO has been recognised as a potential approach to reduce the production of neurotoxic KP metabolites, as well as moderating the inflammatory response during infection and inflammation. As a result of this, several KMO inhibitors have been developed (Table 1 ) 61, 62, 63, 64, 65, 66, 67, 68. Various studies have shown that KMO inhibition or the knockdown of KMO expression reduces levels of neurotoxic KP metabolites and decreases inflammation in a range of infectious and non-infectious inflammatory conditions. For example, Swainson et al. [69] demonstrated that infection of rhesus macaques with simian immunodeficiency virus (SIV) resulted in increased plasma levels of QUIN, and that this could be prevented by treatment with the KMO inhibitor CHDI-340246. Plasma levels of cytotoxic 3-HK were also reduced following KMO inhibition, whereas increased levels of l-KYN and neuroprotective KYNA were observed [69]. Interestingly, KMO inhibition in SIV-infected animals also resulted in increased numbers of naïve CD4+ and CD8+ T cells compared with controls without KMO inhibition, possibly as a result of reductions in cytotoxic KP metabolite levels [69]. In addition to viral infections, inhibition of peripheral KMO using Ro 61-8048 was shown to reduce neuroinflammation in mice with late-stage trypanosome infection in the CNS [70], and to increase survival and levels of neuroprotective KYNA in the CNS of mice with cerebral malaria [71], possibly as a result of shifting the KP away from the 3-HK branch of the pathway toward the production of KYNA.
Table 1.
Summary of currently available KMO inhibitors
| KMO inhibitor | IC50 (nM)a | Structures | Refs |
|---|---|---|---|
| m-NBA | 900b |  |
[61] |
| Ro 61-8048 | 37b |  |
[62] |
| UPF-648 | 40 | 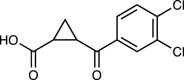 |
[63] |
| JM6 | 37b, c | 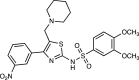 |
[64] |
| CHDI-340246 | 0.5 |  |
[65] |
| GSK180 | 6 | 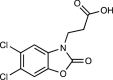 |
[66] |
| Prodrug 1b | 2600d | 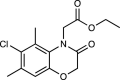 |
[67] |
IC50 values are for inhibition of human KMO unless stated otherwise.
IC50 with rat KMO.
The IC50 value for JM6 relates to the IC50 of the active compound Ro 61-8048 following release from the prodrug. Subsequent analyses of JM6 led to debate over this prodrug mechanism [68].
The IC50 value for prodrug 1b relates to the IC50 of the active compound 1 following release from the prodrug.
Recent studies also highlighted the use of KMO inhibitors to treat non-infectious inflammatory conditions. Sundaram et al. [72] showed that peripheral KMO inhibition using Ro 61-8048 in a mouse model of multiple sclerosis increased the KYNA:QUIN ratio in both the plasma and CNS, and reduced the severity of the disease, demonstrating how inhibition of KMO in the periphery can also improve symptoms in the CNS. Furthermore, knockout of KMO expression in a mouse model of colitis successfully increased levels of both l-KYN and KYNA in the colon and reduced levels of cytotoxic 3-HK compared with wild-type mice [73]. KMO-knockout mice with colitis also had significantly reduced levels of inflammation in the colon, increased levels of the anti-inflammatory cytokines IL-10 and TGF-β, and increased recruitment of regulatory T cells (Tregs) to the site of inflammation compared with wild-type mice with colitis [73]. Increased numbers of Tregs and reduced inflammation might result from the increased l-KYN levels observed in KMO-knockout mice because l-KYN can promote the differentiation of naïve CD4+ T cells into anti-inflammatory Tregs [19]. These studies demonstrate how inhibition of KMO is an important approach for modulating both inflammation and levels of cytotoxic KP metabolites in a range of inflammatory conditions.
In addition to the pulmonary effects of SARS-Cov-2 infections, patients with COVID-19 have also been reported to have damage to other organs, including the heart, kidneys, liver, gastrointestinal tract, and brain [74]. This is relevant to the activation of the KP in COVID-19 because several studies have demonstrated that KMO inhibition/knockout in animal models has positive outcomes in viral infections or pathological conditions that affect these organs. For example, Mole et al. [66] demonstrated that the KMO inhibitor GSK180 significantly reduced inflammation and damage to the lungs, kidneys, and liver in a rat model of acute pancreatitis compared with controls without KMO inhibition. Treatment of these rats with GSK180 also resulted in increased levels of l-KYN and KYNA in the plasma, whereas levels of 3-HK were reduced. The authors speculated that the observed increases in levels of KYNA might be protective in this rat model of acute pancreatitis by activating anti-inflammatory pathways, whereas the reduction in 3-HK might be responsible for reduced apoptosis seen in the lungs and kidneys of GSK180-treated rats [66]. Kubo et al. [75] demonstrated that KMO-knockout mice with viral myocarditis had increased survival, reduced macrophage infiltration, and reduced levels of chemokines compared with infected wild-type mice, possibly as a result of increased levels of l-KYN and KYNA. Finally, another study highlighted the importance of KMO inhibition in protecting against kidney damage in a mouse model of acute kidney injury [76]. Mice with knockout of KMO expression were protected from kidney damage and had lower rates of renal tubular cell apoptosis compared with wild-type mice with acute kidney injury, possibly because of reduced levels of cytotoxic 3-HK [76]. These studies are particularly relevant to COVID-19 because some patients with SARS-CoV-2 have been reported to have damage to other organs in addition to the lungs, such as the kidneys and liver, and highlight the potential benefits of KMO inhibition in protecting multiple organs during inflammation.
Potential use of KMO inhibitors to improve cognition in patients with COVID-19
As detailed earlier, COVID-19 not only affects the respiratory system, but also has detrimental effects on several other vital systems in the body, including the CNS, potentially resulting in long-term health implications for patients. In fact, some patients have reported long-lasting CNS effects of SARS-CoV-2 infection even following recovery, including fatigue, loss of concentration, memory loss, and headaches 77, 78. Therefore, interventions are required to treat these effects of the virus and improve symptoms. Several studies have shown the activation of the KP in the periphery of patients with COVID-19 51, 52, 53, 54. Although further studies are needed to examine levels of KP metabolites in the CNS of patients with COVID-19, and clinical studies are required to examine the potential link between activation of the KP and neurocognitive problems in patients with SARS-CoV-2, it is interesting to speculate how modulation of the KP could be used to reduce cognitive symptoms in patients with COVID-19.
Peripheral administration of KMO inhibitors during inflammation has been shown to elevate levels of KYNA in the CNS 71, 72, which reduces neuronal damage and improves cognition in animal models [64]. However, patients with COVID-19 already exhibit increased levels of KYNA in the periphery 51, 52, which might reflect elevated KYNA in the CNS. Therefore, although KMO inhibition might be a useful approach to modulate the immune response, it is possible that KMO inhibition could initially have detrimental effects on cognitive function because the KP is shifted toward the production of more KYNA, which, at high concentrations, can block neurotransmitter signalling crucial for memory and learning. However, reduced inflammation as a result of KMO inhibition (see later) might lead to reduced levels of proinflammatory cytokines, and feedback to normalise the KP. Alternatively, KATII inhibitors, which inhibit the conversion of l-KYN to KYNA, might be useful in reducing cognitive dysfunction in patients with COVID-19 by directly reducing levels of KYNA [79]. By contrast, IDO inhibitors could have detrimental effects by preventing the production of immunosuppressive metabolites, such as l-KYN [80], and preventing increases in neuroprotective KYNA. However, infection by SARS-CoV-2 causes the increased release of proinflammatory cytokines, which might activate the IDO1-KYN-AHR pathway 81, 82. During the initial stages of infection, positive feedback within this pathway can lead to further activation of AHR by its ligand l-KYN, resulting in a ‘cytokine storm’ 81, 82, and possibly suppression of the antiviral response [81] in some patients with COVID-19.
The recent development of a brain-permeable KMO inhibitor has the advantage of directly targeting the production of toxic KP metabolites in the brain [67]. This could significantly reduce the accumulation of neurotoxic 3-HK and QUIN and increase neuroprotective KYNA in the CNS (Figure 2 ). A prodrug strategy has been used to enhance the brain penetrance of the KMO inhibitor ‘compound 1’. Pharmacokinetic profiles of the prodrug form demonstrated that compound 1 is released in both the blood and brain, with a maximal brain:blood ratio of 3.22 at 15 min, which provides KMO inhibition both in the periphery and brain [67]. Brain-permeable KMO inhibitors might also have the ability to improve cognitive function in patients with COVID-19 by reducing neuronal damage. Long-term cognitive deficits have been observed in some patients with ‘long COVID’ 77, 78. However, it is difficult to determine from current data derived from a small number of metabolomics studies in patients with COVID-19 51, 52, 53, 54 whether acute activation of the KP as a result of SARS-CoV-2 has prolonged effects on the brain, or whether chronic activation of the KP occurs during COVID-19. Therefore, it would be of interest to examine KP metabolites in these patients at different times points after the initial infection to determine whether KP activation is acute or chronic in COVID-19.
Figure 2.

Potential beneficial effects of kynurenine 3-monooxygenase (KMO) inhibition on inflammation in patients with Coronavirus 2019 (COVID-19). Administration of a peripheral KMO inhibitor might modulate the immune response in COVID-19 by further increasing levels of l-kynurenine (l-KYN) in the periphery. Increased l-KYN can reduce inflammation by suppressing the proliferation of CD4+ T cells and promoting the differentiation of naïve T cells into immunosuppressive regulatory T cells. Inhibition of kynurenine 3-monooxygenase (KMO) in the periphery could lead to increased levels of l-KYN and kynurenic acid (KYNA) in the central nervous system (CNS) because l-KYN can cross the blood–brain barrier (BBB). Brain-permeable KMO inhibitors might be useful in the treatment of COVID-19 because KMO inhibition in the CNS results in increased levels of KYNA, which is neuroprotective and anti-inflammatory. Brain-permeable KMO inhibitors might also inhibit peripheral KMO, resulting in increased levels of l-KYN and KYNA in the periphery. KMO inhibition in the CNS might reduce the production of cytotoxic kynurenine pathway (KP) metabolites, such as 3-hydroxykynurenine (3-HK), preventing neuronal cell death.
Potential use of KMO inhibitors to modulate inflammation in patients with COVID-19
Given that the activation of the KP has been demonstrated in patients with severe SARS-CoV-2 51, 52, modulation of the KP by inhibiting KMO might be a potential mechanism to reduce inflammation in severely ill patients. Peripheral administration of KMO inhibitors has previously been shown to moderate the KP in animal models of disease, resulting in reduced inflammation in both infectious and non-infectious diseases 69, 70, 71, 72. Given that the KP is activated in COVID-19, it might be possible to use KMO inhibitors to inhibit peripheral KMO and modulate the immune response by shifting the KP away from the production of cytotoxic metabolites, such as QUIN. For example, peripheral KMO inhibition has been shown to increase levels of l-KYN in the periphery 69, 73, which might be beneficial in patients with COVID-19 because of the ability of l-KYN to mediate the differentiation of naïve T cells into immunosuppressant Tregs [19], and might also help to suppress the immune response (Figure 2). Peripheral KMO inhibition might also increase levels of KYNA in the periphery, which could be beneficial in COVID-19 via the anti-inflammatory actions of this KP metabolite 20, 21, 22, 25. Inhibition of KMO in the periphery has also been demonstrated to reduce inflammation in the CNS [70], possibly because of the ability of l-KYN to cross the BBB. Inhibition of peripheral KMO also reduces levels of cytotoxic 3-HK and QUIN in the periphery 69, 73, which might be beneficial in COVID-19 by potentially reducing damage to other organs [66].
At present, the role of KP activation in patients with COVID-19 is unknown but is possibly a physiological response to SARS-CoV-2 to modulate the immune response by depleting tryptophan, increasing levels of l-KYN (thereby inhibiting CD4+ T cell proliferation), and increasing the production of neuroprotective KYNA. However, prolonged activation of KP in COVID-19 could be detrimental. It is essential that a balance is maintained between high levels of inflammation that can cause tissue damage, and suppression of the immune system, which could allow SARS-CoV-2 to replicate uncontrollably. The glucocorticoid dexamethasone is currently being used to reduce mortality in patients with severe SARS-CoV-2 [83]. Therefore, inhibition of KMO might be beneficial in severe cases of COVID-19 by further increasing levels of immunosuppressant l-KYN and KYNA, and reducing production of neurotoxic KP metabolites and, thus, inflammation.
Concluding remarks
Current studies of the activation of the KP in COVID-19 have only measured KP metabolites in the periphery (plasma/serum) of patients with COVID-19 but not the CNS 51, 52, 53, 54. However, some KP metabolites, such as KYNA, cannot cross the BBB [57], and plasma or serum levels of these metabolites might not correspond to levels in the CNS. Therefore, it will be important to examine levels of KP metabolites in the CNS of patients with COVID-19 (e.g., via CSF or postmortem brains) to confirm whether they correlate with peripheral levels. It would also be interesting to measure KP metabolite levels following administration of KMO inhibitors in a mouse model of COVID-19 [84] to examine whether KMO inhibition modulates levels of KP metabolites and reduces neuroinflammation in the CNS in vivo. Therefore, further studies are required to fully examine the role of the KP in both the inflammatory response and cognitive dysfunction in patients with COVID-19. Further clinical development of KMO inhibitors for use in patients is also needed. Given that the KP is activated in severe COVID-19 cases, it might be advantageous to use clinically approved KMO inhibitors in severe cases to reduce the inflammatory response, resulting in reduced inflammation and possibly improved cognitive function in these patients.
Conflict of interest
The authors declare no competing nonfinancial interests but the following competing financial interests: a patent [Kynurenine 3-monooxygenase (KMO) inhibitors, and uses and compositions thereof] from the University of Leicester and University of Manchester by N.S.S. and F.G. is pending, application number 18811898.8.
Acknowledgements
M.C. and F.G. are supported by funding from the National Institute of Mental Health (Silvio O. Conte Center for Translational Mental Health Research – MH-103222). S.Z. and N.S.S. are supported by the UK Engineering and Physical Sciences Research Council (awards EP/S01778X/1 and EP/S030336/1).
References
- 1.Ball H.J., et al. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396:203–213. doi: 10.1016/j.gene.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 2.Braidy N., et al. Effects of kynurenine pathway inhibition on NAD metabolism and cell viability in human primary astrocytes and neurons. Int J Tryptophan Res. 2011;4:29–37. doi: 10.4137/IJTR.S7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maddison D.C., Giorgini F. The kynurenine pathway and neurodegenerative disease. Semin Cell Dev Biol. 2015;40:134–141. doi: 10.1016/j.semcdb.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Kessler M., et al. A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem. 1989;52:1319–1328. doi: 10.1111/j.1471-4159.1989.tb01881.x. [DOI] [PubMed] [Google Scholar]
- 5.Hilmas C., et al. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21:7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pozzo Miller L.D., et al. Spontaneous pyramidal cell death in organotypic slice cultures from rat hippocampus is prevented by glutamate receptor antagonists. Neuroscience. 1994;63:471–487. doi: 10.1016/0306-4522(94)90544-4. [DOI] [PubMed] [Google Scholar]
- 7.Lugo-Huitrón R., et al. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol. 2011;33:538–547. doi: 10.1016/j.ntt.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 8.Phenis D., et al. Activation of alpha7 nicotinic and NMDA receptors is necessary for performance in a working memory task. Psychopharmacology (Berl) 2020;237:1723–1735. doi: 10.1007/s00213-020-05495-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alexander K.S., et al. Acute elevations of brain kynurenic acid impair cognitive flexibility: normalization by the alpha7 positive modulator galantamine. Psychopharmacology (Berl) 2012;220:627–637. doi: 10.1007/s00213-011-2539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwarcz R., et al. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–318. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- 11.Braidy N., et al. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res. 2009;16:77–86. doi: 10.1007/s12640-009-9051-z. [DOI] [PubMed] [Google Scholar]
- 12.Okuda S., et al. 3-Hydroxykynurenine, an endogenous oxidative stress generator, causes neuronal cell death with apoptotic features and region selectivity. J Neurochem. 1998;70:299–307. doi: 10.1046/j.1471-4159.1998.70010299.x. [DOI] [PubMed] [Google Scholar]
- 13.Munn D.H., et al. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bracho-Sanchez E., et al. Dendritic cells treated with exogenous indoleamine 2,3-dioxygenase maintain an immature phenotype and suppress antigen-specific T cell proliferation. J Immunol Regen Med. 2019;5:100015. doi: 10.1016/j.regen.2019.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fallarino F., et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 16.Frumento G., et al. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terness P., et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jürgens B., et al. Interferon-gamma-triggered indoleamine 2,3-dioxygenase competence in human monocyte-derived dendritic cells induces regulatory activity in allogeneic T cells. Blood. 2009;114:3235–3243. doi: 10.1182/blood-2008-12-195073. [DOI] [PubMed] [Google Scholar]
- 19.Mezrich J.D., et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moroni F., et al. Kynurenic acid actions in brain and periphery. Int Congress Ser. 2007;1304:305–313. [Google Scholar]
- 21.Wang J., et al. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem. 2006;281:22021–22028. doi: 10.1074/jbc.M603503200. [DOI] [PubMed] [Google Scholar]
- 22.Moroni F., et al. Kynurenic acid: a metabolite with multiple actions and multiple targets in brain and periphery. J Neural Transm (Vienna) 2012;119:133–139. doi: 10.1007/s00702-011-0763-x. [DOI] [PubMed] [Google Scholar]
- 23.DiNatale B.C., et al. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol Sci. 2010;115:89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapolka N.J., et al. DCyFIR: a high-throughput CRISPR platform for multiplexed G protein-coupled receptor profiling and ligand discovery. Proc Natl Acad Sci U S A. 2020;117:13117–13126. doi: 10.1073/pnas.2000430117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapolka N.J., Isom D.G. HCAR3: an underexplored metabolite sensor. Nat Rev Drug Discov. 2020;19:745. doi: 10.1038/d41573-020-00173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larkin P.B., et al. Tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase 1 make separate, tissue-specific contributions to basal and inflammation-induced kynurenine pathway metabolism in mice. Biochim Biophys Acta. 2016;1860:2345–2354. doi: 10.1016/j.bbagen.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edelstein M.P., et al. Synergistic effects of phorbol ester and INF-gamma on the induction of indoleamine 2,3-dioxygenase in THP-1 monocytic leukemia cells. J Immunol. 1989;143:2969–2973. [PubMed] [Google Scholar]
- 28.Alberati-Giani D., et al. Regulation of the kynurenine metabolic pathway by interferon-gamma in murine cloned macrophages and microglial cells. J Neurochemistry. 1996;66:996–1004. doi: 10.1046/j.1471-4159.1996.66030996.x. [DOI] [PubMed] [Google Scholar]
- 29.Guillemin G.J., et al. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol. 2003;527:105–112. doi: 10.1007/978-1-4615-0135-0_12. [DOI] [PubMed] [Google Scholar]
- 30.Huengsberg M., et al. Serum kynurenine-to-tryptophan ratio increases with progressive disease in HIV-infected patients. Clin Chem. 1998;44:858–862. [PubMed] [Google Scholar]
- 31.Larrea E., et al. Upregulation of indoleamine 2,3-dioxygenase in hepatitis C virus infection. J Virol. 2007;81:3662–3666. doi: 10.1128/JVI.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atlas A., et al. Sustained elevation of kynurenic acid in the cerebrospinal fluid of patients with herpes simplex virus type 1 encephalitis. Int J Tryptophan Res. 2013;6:89–96. doi: 10.4137/IJTR.S13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinstein A.A., et al. Relationships among neurotransmitters, cytokines and cognitive performance for individuals with hepatitis C achieving sustained virologic response: a pilot study. J Neuroimmunol. 2019;335:577022. doi: 10.1016/j.jneuroim.2019.577022. [DOI] [PubMed] [Google Scholar]
- 34.Sardar A.M., Reynolds G.P. Frontal cortex indoleamine-2,3-dioxygenase activity is increased in HIV-1-associated dementia. Neurosci Lett. 1995;187:9–12. doi: 10.1016/0304-3940(95)11324-p. [DOI] [PubMed] [Google Scholar]
- 35.Sardar A.M., et al. Increased concentrations of the neurotoxin 3-hydroxykynurenine in the frontal cortex of HIV-1-positive patients. J Neurochem. 1995;64:932–935. doi: 10.1046/j.1471-4159.1995.64020932.x. [DOI] [PubMed] [Google Scholar]
- 36.Heyes M.P., et al. Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: relationship to clinical and neurological status. Ann Neurol. 1991;29:202–209. doi: 10.1002/ana.410290215. [DOI] [PubMed] [Google Scholar]
- 37.Baran H., et al. Kynurenic acid metabolism in various types of brain pathology in HIV-1 infected patients. Int J Tryptophan Res. 2012;5:49–64. doi: 10.4137/IJTR.S10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerr S.J., et al. Chronic exposure of human neurons to quinolinic acid results in neuronal changes consistent with AIDS dementia complex. AIDS. 1998;12:355–363. doi: 10.1097/00002030-199804000-00003. [DOI] [PubMed] [Google Scholar]
- 39.Fontana R.J., et al. Cognitive function in hepatitis C patients with advanced fibrosis enrolled in the HALT-C trial. J Hepatol. 2005;43:614–622. doi: 10.1016/j.jhep.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Schulz S., et al. Indolamine 2,3-dioxygenase expression by monocytes and dendritic cell populations in hepatitis C patients. Clin Exp Immunol. 2015;180:484–498. doi: 10.1111/cei.12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zavitsanou K., et al. Effect of maternal immune activation on the kynurenine pathway in preadolescent rat offspring and on MK801-induced hyperlocomotion in adulthood: amelioration by COX-2 inhibition. Brain Behav Immun. 2014;41:173–181. doi: 10.1016/j.bbi.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 42.Clark S.M., et al. Maternal immune activation in rats blunts brain cytokine and kynurenine pathway responses to a second immune challenge in early adulthood. Prog Neuropsychopharmacol Biol Psychiatry. 2019;89:286–294. doi: 10.1016/j.pnpbp.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torii Y., et al. Quantitative metabolome profiling reveals the involvement of the kynurenine pathway in influenza-associated encephalopathy. Metabolomics. 2016;12:84. [Google Scholar]
- 44.Varatharaj A., et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. Lancet Psychiatry. 2020;7:875–882. doi: 10.1016/S2215-0366(20)30287-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellul M.A., et al. Neurological associations of COVID-19. Lancet Neurol. 2020;19:767–783. doi: 10.1016/S1474-4422(20)30221-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berlit P., et al. ‘Neurological manifestations of COVID-19’ – guideline of the German society of neurology. Neurol Res Pract. 2020;2:51. doi: 10.1186/s42466-020-00097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao L., et al. Neurologic manifestations of hospitalized patients with Coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77:683–690. doi: 10.1001/jamaneurol.2020.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helms J., et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. 2020;382:2268–2270. doi: 10.1056/NEJMc2008597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kremer S., et al. Neurologic and neuroimaging findings in patients with COVID-19: a retrospective multicenter study. Neurology. 2020;95:e1868–e1882. doi: 10.1212/WNL.0000000000010112. [DOI] [PubMed] [Google Scholar]
- 50.Bellon M., et al. Cerebrospinal fluid features in SARS-CoV-2 RT-PCR positive patients. Clin Infect Dis. 2020 doi: 10.1093/cid/ciaa1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas T., et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. J Clin Investig Insight. 2020;5:e140327. doi: 10.1172/jci.insight.140327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen B., et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182:59–72. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fraser D.D., et al. Metabolomics profiling of critically ill Coronavirus disease 2019 patients: identification of diagnostic and prognostic biomarkers. Crit Care Explor. 2020;2:e0272. doi: 10.1097/CCE.0000000000000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai Y., et al. Kynurenic acid underlies sex-specific immune responses to COVID-19. medRxiv 2020. 2020 doi: 10.1126/scisignal.abf8483. 2020.09.06.20189159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kimhofer T., et al. Integrative modelling of quantitative plasma lipoprotein, metabolic and amino acid data reveals a multi-organ pathological signature of SARS-CoV-2 infection. J Proteome Res. 2020;19:4442–4454. doi: 10.1021/acs.jproteome.0c00519. [DOI] [PubMed] [Google Scholar]
- 56.Gardinassi L.G., et al. Immune and metabolic signatures of COVID-19 revealed by transcriptomics data reuse. Front Immunol. 2020;11:1636. doi: 10.3389/fimmu.2020.01636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fukui S., et al. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J Neurochem. 1991;56:2007–2017. doi: 10.1111/j.1471-4159.1991.tb03460.x. [DOI] [PubMed] [Google Scholar]
- 58.Braidy N., et al. Changes in kynurenine pathway metabolism in the brain, liver and kidney of aged female Wistar rats. FEBS J. 2011;278:4425–4434. doi: 10.1111/j.1742-4658.2011.08366.x. [DOI] [PubMed] [Google Scholar]
- 59.Blanco Ayala T., et al. Alternative kynurenic acid synthesis routes studied in the rat cerebellum. Front Cell Neurosci. 2015;18:178. doi: 10.3389/fncel.2015.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazza M.G., et al. Anxiety and depression in COVID-19 survivors: role of inflammatory and clinical predictors. Brain Behav Immun. 2020;89:594–600. doi: 10.1016/j.bbi.2020.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pellicciari R., et al. Modulation of the kynurenine pathway in search for new neuroprotective agents. Synthesis and preliminary evaluation of (m-nitrobenzoyl)alanine, a potent inhibitor of kynurenine-3-hydroxylase. J Med Chem. 1994;37:647–655. doi: 10.1021/jm00031a015. [DOI] [PubMed] [Google Scholar]
- 62.Röver S., et al. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J Med Chem. 1997;40:4378–4385. doi: 10.1021/jm970467t. [DOI] [PubMed] [Google Scholar]
- 63.Pellicciari R., et al. Modulation of the kynurine pathway of tryptophan metabolism in search for neuroprotective agents. Focus on kynurenine-3-hydroxylase. Adv Exp Med Biol. 2003;527:621–628. doi: 10.1007/978-1-4615-0135-0_71. [DOI] [PubMed] [Google Scholar]
- 64.Zwilling D., et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell. 2011;145:863–874. doi: 10.1016/j.cell.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Toledo-Sherman L.M., et al. Development of a series of aryl pyrimidine kynurenine monooxygenase inhibitors as potential therapeutic agents for the treatment of Huntington's disease. J Med Chem. 2015;58:1159–1183. doi: 10.1021/jm501350y. [DOI] [PubMed] [Google Scholar]
- 66.Mole D.J., et al. Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat Med. 2016;22:202–209. doi: 10.1038/nm.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang S., et al. A brain-permeable inhibitor of the neurodegenerative disease target kynurenine 3-monooxygenase prevents accumulation of neurotoxic metabolites. Commun Biol. 2019;2:271. doi: 10.1038/s42003-019-0520-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beconi M.G., et al. Metabolism and pharmacokinetics of JM6 in mice: JM6 is not a prodrug for Ro-61-8048. Drug Metab Dispos. 2012;40:2297–2306. doi: 10.1124/dmd.112.046532. [DOI] [PubMed] [Google Scholar]
- 69.Swainson L.A., et al. Kynurenine 3-monooxygenase inhibition during acute Simian immunodeficiency virus infection lowers PD-1 expression and improves post-combination antiretroviral therapy CD4+ T cell counts and body weight. J Immunol. 2019;203:899–910. doi: 10.4049/jimmunol.1801649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodgers J., et al. Kynurenine pathway inhibition reduces central nervous system inflammation in a model of human African trypanosomiasis. Brain. 2009;132:1259–1267. doi: 10.1093/brain/awp074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clark C.J., et al. Prolonged survival of a murine model of cerebral malaria by kynurenine pathway inhibition. Infect Immun. 2005;73:5249–5251. doi: 10.1128/IAI.73.8.5249-5251.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sundaram G., et al. Kynurenine pathway modulation reverses the experimental autoimmune encephalomyelitis mouse disease progression. J Neuroinflammation. 2020;17:176. doi: 10.1186/s12974-020-01844-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tashita C., et al. Kynurenine plays an immunosuppressive role in 2,4,6-trinitrobenzene sulfate-induced colitis in mice. World J Gastroenterol. 2020;26:918–932. doi: 10.3748/wjg.v26.i9.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gupta A., et al. Extrapulmonary manifestations of COVID-19. Nat Med. 2020;26:1017–1032. doi: 10.1038/s41591-020-0968-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kubo H., et al. Absence of kynurenine 3-monooxygenase reduces mortality of acute viral myocarditis in mice. Immunol Lett. 2017;181:94–100. doi: 10.1016/j.imlet.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 76.Zheng X., et al. Kynurenine 3-monooxygenase is a critical regulator of renal ischemia-reperfusion injury. Exp Mol Med. 2019;51:1–14. doi: 10.1038/s12276-019-0210-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carfì A., et al. Persistent symptoms in patients after acute COVID-19. JAMA. 2020;324:603–605. doi: 10.1001/jama.2020.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Garrigues E., et al. Post-discharge persistent symptoms and health-related quality of life after hospitalization for COVID-19. J Infect. 2020;81:e4–e6. doi: 10.1016/j.jinf.2020.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pocivavsek A., et al. Inhibition of kynurenine aminotransferase II attenuates hippocampus-dependent memory deficit in adult rats treated prenatally with kynurenine. Hippocampus. 2019;29:73–77. doi: 10.1002/hipo.23040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fox J.M., et al. Inhibition of indoleamine 2,3-dioxygenase enhances the T-cell response to influenza virus infection. J Gen Virol. 2013;94:1451–1461. doi: 10.1099/vir.0.053124-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anderson G., et al. Aryl hydrocarbon receptor role in co-ordinating SARS-CoV-2 entry and symptomatology: linking cytotoxicity changes in COVID-19 and cancers; modulation by racial discrimination stress. Biology. 2020;9:249. doi: 10.3390/biology9090249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Turski W.A., et al. AhR and IDO1 in pathogenesis of Covid-19 and the ‘Systemic AhR Activation Syndrome:’ a translational review and therapeutic perspectives. Restor Neurol Neurosci. 2020;38:343–354. doi: 10.3233/RNN-201042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horby P., et al. Dexamethasone in hospitalized patients with Covid-19 – preliminary report. N Engl J Med. 2020 doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brumeanu T.D., et al. A human-immune-system mouse model for COVID-19 research (DRAGA mouse: HLA-A2.HLA-DR4.Rag1KO.IL-2Rγc KO.NOD) bioRxiv. 2020 doi: 10.1080/21645515.2022.2048622. 2020.2020.08.19.251249. [DOI] [PMC free article] [PubMed] [Google Scholar]