

Abstract
Background:
Disrupted placental functioning due to stress can have lifelong implications. Cumulative stress and trauma are likely to have lasting impacts on maternal physiological functioning and offspring development resulting in increased risk for later life complex disorders for which racial disparities exist.
Methods:
This study examines the association between maternal lifetime stress and placental mitochondrial DNA (mtDNA) mutational load in an urban multi-ethnic cohort. Maternal lifetime exposure to stressful events was assessed using the validated Life Stressor Checklist-Revised. Whole mtDNA sequencing was performed and mutations were determined for 365 placenta samples with complete exposure and covariate data. Multivariable regression was used to model maternal lifetime stress in relation to placental mtDNA mutational load. Racial/ethnic differences were examined by cross-product terms and contrast statements. Gene-wise analyses were conducted.
Results:
We identified 13,189 heteroplasmies (phred score >10000, MAF < 0.5, # of mutant reads > 1). Women experiencing increased psychosocial stress over their lifetime exhibited a higher number of total placental mitochondrial mutations (β=0.23, 95% CI 0.03–0.42) and heteroplasmic (β=0.18, 95% CI 0.05–0.31) but not homoplasmic (β= −0.008, 95% CI −0.03 – 0.01) mutations; the strongest associations were observed among Black women and genes coding for NADH dehydrogenase and cytochrome c oxidase subunits.
Conclusions:
Cumulative maternal lifetime stress is associated with a greater mitochondrial mutational load, particularly among Black women. The impact of racial/ethnic differences in mutational load on placental function directly impacting offspring development and/or leading to chronic disease disparities warrants further investigation.
Keywords: prenatal, heteroplasmy, mitochondrial function, disparities, trauma, psychosocial
INTRODUCTION
Research in the field of developmental origins of health and disease has underscored effects of prenatal stress on maternal and offspring physiology and function (1). Compared to prenatal stress, cumulative stress and trauma exposure are more likely to have a lasting impact on maternal physiological functioning carried into pregnancy with greater implications for fetal and offspring development and later life complex disorders (2–5). Preconception stress, even when occurring during the mother’s childhood, can have effects on maternal stress response during pregnancy (5), infant birthweight (6,7), child cognition (8), and asthma (9). Moreover, lifetime exposure to psychosocial stressors including traumatic events not only varies by race (10), but how the body physiologically and psychologically responds differs by race (11,12). Ethnic minorities experiencing more severe (i.e., meeting DSM-V PTSD Criterion A) and chronic stressors (e.g., trauma, violence, discrimination) that continue to negatively impact their life, may be more likely to demonstrate disrupted stress hormone activity (5,13,14) that, in turn, can exert lasting effects on physiological processes influencing fetal development (15).
The placenta plays a central role in optimal fetal maturation (16) and perturbations in the maternal environment can be transmitted across the placenta impacting fetal development. A hallmark of the placenta is its capacity to adapt in response to variations in the maternal-fetal environment making it an excellent target for the identification of biomarkers/mechanisms of adverse development. Although it is unclear if racial/ethnic differences in placental response to stress exist, altered placental function has been linked to racially disparate pregnancy complications and birth outcomes (17,18).
Oxidative stress (OS) is a key pathway linking stress and health including cognitive, emotional and physiological health (19). Mitochondria are major intracellular sources and targets of reactive oxygen species (ROS), making mitochondria susceptible to increases in systemic ROS and subsequent damage (20) by sustaining systemic oxidative stress (21). One key feature of mitochondria is their maternally inherited genome; mitochondrial DNA (mtDNA) contains 37 genes and, compared with nuclear DNA, has diminished repair capacity making it highly mutable and susceptible to accumulating oxidative damage. The most commonly studied mtDNA biomarker of environmental toxicants and OS in epidemiological studies is mtDNA copy number (22). Race-specific effects of maternal stress on mtDNA copy number in placenta and peripheral blood have been observed (23,24).
Within each mitochondrion multiple copies of mtDNA exist and mutations can affect all (homoplasmy) or a portion (heteroplasmy) of the molecules. Heteroplasmy has been identified as a marker of oxidative damage (25) and linked to numerous human diseases ranging in severity, prevalence, and complexity (26) and serves as a marker of inefficient cellular respiration (27). Research reporting identical mtDNA variants in placenta and cord blood support the use of placenta suggesting both are adequate sources to obtain fetal mitochondrial biomarkers (28). Interestingly, mitochondrial haplogroups (a marker of genetic ancestry) have been shown to modify the effect of environmental exposures with similar OS mechanisms of action (i.e., air pollution) on cognitive outcomes (29). Thus, we hypothesize that maternal lifetime exposure to psychosocial stress will be associated with increased placental mtDNA mutations/heteroplasmy particularly among racial/ethnic minorities.
METHODS AND MATERIALS
Study Population
Participants were from the Programming of Intergenerational Stress Mechanisms (PRISM) study, an urban pregnancy cohort designed to investigate associations among maternal psychosocial stress and other environmental actors (i.e., air pollution, smoking, diet) and pre- and postnatal child development. Pregnant English- and Spanish-speaking women aged 18 years and older carrying a singleton fetus were recruited from prenatal clinics at the Beth Israel Deaconess Medical Center and the East Boston Neighborhood Health Center in Boston, Massachusetts between the years of 2011 and 2013 and from the prenatal clinic at Mount Sinai Hospital in New York City from 2013 to 2018. Exclusion criteria included maternal intake of ≥7 alcoholic drinks per week before and/or any alcohol after pregnancy recognition as usage above these thresholds has been associated with increased risk for numerous health problems impacting child development (30–32). Birth details were ascertained by postnatal questionnaires and medical chart review. Study procedures were approved by the relevant institutional review boards. Written informed consent was obtained in the participant’s preferred language.
Maternal Lifetime Stress Exposure
Maternal lifetime exposure to traumatic and non-traumatic stressful events was assessed using the validated 30-item Life Stressor Checklist-Revised (LSC-R) during the second trimester (25.4 ± 7.0 weeks of gestation) (33) in person and by phone. The LSC-R has established test-retest reliability and validity in diverse populations(33) and assesses exposure to 30 stressful life experiences including potentially traumatic events particularly relevant to women (e.g., sexual assault, interpersonal violence). For each endorsed event, women were asked to rate the severity of the negative impact (ranging from 1 [not at all] to 5 [extremely]) of each endorsed event during the past 12 months. A maternal lifetime stress score was derived by summing the rate of severity across endorsed events [e.g., a score of 150 would mean every event was 1) endorsed and 2) severely impact their life (rated a ‘5’ on the Likert scale)] with scores ranging from 0 to 96 in our sample (LSCRwt); outliers were winzorized to reduce spurious associations. Higher scores indicate greater exposure to and impact of exposure to stressful and traumatic events.
Placental Mitochondrial DNA Sequencing
Placental Processing
Placenta samples (~1–2 cm3) were taken on the fetal side ~1 – 1.5 cm below the fetal membrane to avoid membrane contamination and approximately 4 cm from the cord insertion site, taking care to avoid large vessels (details available in online supplement). Fetal placental tissue origin was confirmed by the previously reported near-perfect agreement of placenta and cord blood samples in 64 genotyping probes used for identity verification (34).
Whole Mitochondrial DNA Sequencing
Placenta DNA extraction was conducted using the Promega Wizard Genomic DNA Purification Kit (Promega – Madison, WI, USA). Whole mtDNA sequencing was performed by the Genomics, Epigenomics and Sequencing Core at the University of Cincinnati. To amplify whole mtDNA genome (complete genome sequence ID AY495156.2, 16569 bp), Illumina (San Diego, CA) protocol (Document # 15037958 v01) was followed with modifications. For each sample two pairs of primers that cover the entire mtDNA sequence were used in two individual amplifications: MTL-F1/MTL-R1 and MTL-F2/MTL-R2. TaKaRa LA Taq (Takara Bio USA, Mountain View, CA) was used in long range PCR with 2 ng human gDNA as input in each 50 µl PCR reaction. The volumes of the two PCRs were then adjusted and pooled to make roughly equal amount of the two PCR products in the pool. After DNA clean up using Wizard SV Gel and PCR Clean-Up kit (Promega), about 100 ng DNA was used as input for library preparation using NEBNext Ultra II FS DNA Library Prep kit (NEB, Ipswich, MA). After Bioanalyzer QC analysis (Agilent, Santa Clara, CA) of the library and pPCR quantification using NEBNext Library Quant Kit (NEB), individually indexed and compatible libraries were proportionally pooled and sequenced using HiSeq 1000 sequencer (Illumina). Under the sequencing setting of paired-end 2×101 bp, about 8 million pass filter reads per sample were generated. The demultiplexed fastq files were used downstream for bioinformatic analysis. The online supplement contains information on primer sequences, amplicon sizes, and detailed methods on checking amplification specificity/yield.
Mitochondrial Sequencing Data Processing
A mitochondria consensus genome, based on analyses of the complete mtDNA sequence of 53 humans of diverse origins, was used for alignment (URL: https://www.ncbi.nlm.nih.gov/nuccore/AF346978.1) (35). Picard (version 2.17.8) was used to align the reads to the reference genome and remove adapters and duplicates.(36) The Genome Analysis Toolkit (GATK) was used to make variant calls.(37) Phred-based quality scores (i.e. QUAL in GATK) were computed as the sum of depth of coverage multiplied by the Phred score at the variant site. To reduce the effects of sequencing errors, we removed variant calls with a QUAL score < 10000 (i.e., probability of having an incorrect base call is less than 1 in 10,000) (38). The potential for strand bias was interrogated and ruled out (Figure S1). Repeated sequencing was carried out on 20% of our sample and concordance in mutation calls ranged from 80–95% (Online Supplement).
Insertions, deletions, and variants observed only once were ignored; variants occurring only once had lower quality scores and are likely prone to artifacts in the sequencing and/or alignments. Single nucleotide substitutions account for approximately 90% of somatic mutations in the mitochondrial genome (39) (96.3% in our data- Figure S2) and were used in this analysis to determine mutational load. Positions were considered heteroplasmic if less than 50% of the reads support alternative alleles (MAF computed as read counts of variant allele/total depth of coverage < 0.5); all other mutations were considered homoplasmic. This criteria is used to reduce the effects of potential homoplasmic mutations in the germline (40), represent risk alleles of complex disease (41), and obtain good concordances among cell types at the heteroplasmy level (42) which is important for epidemiological studies unable to analyze cell-type specific biomarkers. Total mutational load is the total number of heteroplasmic and homoplasmic mutations present in a sample. For gene-wise analyses, protein-coding genes and ribosomal RNAs were examined independently; the number of mutations observed in tRNAs was minimal (n=395) with only 5 out of 22 tRNAs accumulating more than 30 mutations. Thus, mutational loads for tRNAs were summed to create one tRNA total mutational load variable. Although we sampled the feto-placenta, it should be noted that the mutational loads observed do not necessarily correspond to mutational loads of embryonic tissue.
Covariates
Covariates included maternal race [White, Black, Hispanic, Other/multi-race], nativity status (United States vs. Other), and education status (≤ high school degree, some college or/college degree) which were ascertained at enrollment; child sex and maternal age at delivery were obtained postnatally. Mode of delivery (vaginal vs. C-section), pregnancy complications (i.e., gestational diabetes, hypertension, and/or preeclampsia), and maternal smoking have been shown to impact placental functioning/response leading to inflammation, oxidative stress, and mitochondrial dysfunction (43–47) and may be related to maternal stress response, thus these were considered confounders. Women were classified as prenatal smokers if they reported smoking at enrollment or during the third trimester. Birth weight was extracted from delivery records and gestational age was determined by reported last menstrual period and compared to the first trimester ultrasound; if there was a discrepancy of more than two weeks, obstetrical values were used. Sex-specific Fenton birthweight for gestational age z-scores were then derived (48). Genetic ancestry was determined by mitochondrial haplogroup and categorized as African (L), Native American/Asian (A, B, C, D, F, G, N, P, Y, Z, M, E, R), or European (H, I, J, K, T, U, V, W, X) (49).
Statistical Analysis
Descriptive statistics, including mutational loads, were examined for the overall sample as well as by race/ethnicity and child sex using t-tests and chi-square where appropriate. Multivariable regression was used to model maternal lifetime stress in relation to total mtDNA mutational loads; race-specific and gene-wise analyses were conducted using multivariable Poisson regression with a log link function given the count nature of the data (i.e., distribution not binomial due to fewer mutations by race or gene). Covariates/confounders were determined using augmented backward elimination (ABE) (50). Gene-specific mutational loads were used in similar regression models as outlined above. Further, given our group’s previous finding regarding the race-specific impact of maternal psychosocial stress on other mitochondrial function biomarkers (i.e., mtDNA copy number), we evaluated our models for non-additive (interaction) effects between stress and race/ethnicity (23) by including cross-product terms in the model and contrast statements to test for differences in slopes; we removed the other/multi-race category due to small sample size for these analyses. Lastly, multiple imputation using the Fully Conditional Specification method was performed and the results are similar under both conditions (i.e., using complete data vs. imputed). Thus, we report results for the complete case analysis in the manuscript. All analyses were conducted using SAS 9.4 (SAS Institute Inc., Cary, NC, USA) or RStudio v1.1.447. Multiple comparisons were addressed by controlling FDR (false discovery rate) at 0.05 (51).
RESULTS
Maternal and child characteristics for the total sample with sequencing data (N = 420) and the analytic sample with complete stress, covariate, and sequencing data (n = 365) are presented in Table 1. The average maternal age at delivery was 29.4 years; the majority of women were racial/ethnic minorities (Black, 40%; Hispanic, 19%; multi-racial, 8%) with 26% reporting less than or at most a high school degree, 9% reporting smoking during pregnancy, and 28% delivering their child via C-section; 54% of the children were male; levels of psychosocial stress did vary by race/ethnicity (p=0.0001). All three haplogroups are present with the majority (74%) of Black women belonging to the African (L) haplogroup (Table S9). There were no significant differences between the total sample and the analytic sample on maternal age, race/ethnicity, education, smoking during pregnancy, pregnancy complications, mode of delivery, nativity status, or child sex.
Table 1. Cohort characteristics for all participants with sequencing data and those included in analyses by maternal race.
| Characteristic n (%) | Total Sample (n=420) | Analytic Sample (n=365) | Characteristics by Maternal Race |
|||
|---|---|---|---|---|---|---|
| White (n=79) | Black (n=165) | Hispanic (n=104) | Other (n=17) | |||
| Maternal race a | ||||||
| White | 83 (20.6) | 79 (21.6) | -- | -- | -- | -- |
| Black | 184 (44.4) | 165 (45.2) | -- | -- | -- | -- |
| Hispanic | 124 (29.9) | 104 (28.5) | -- | -- | -- | -- |
| Other | 23 (5.6) | 17 (4.7) | -- | -- | -- | -- |
| Maternal education ≤ HS degree | 140 (35.0) | 127 (34.8) | 1 (1) | 78 (47) | 47 (45) | 1 (6) |
| Smoking during pregnancy | 36 (9.1) | 33 (9.2) | 1 (1) | 15 (9) | 16 (16) | 1 (6) |
| Pregnancy complications b | 71 (16.9) | 60 (16.4) | 11 (13.9) | 30 (18.2) | 18 (17.3) | 1 (5.8) |
| C-section delivery | 118 (28.7) | 105 (29.1) | 19 (24) | 51 (31) | 30 (29) | 5 (31) |
| Born outside of the United States | 95 (23.8) | 86 (23.6) | 15 (19) | 25 (15) | 35 (34) | 11 (65) |
| Male sex | 223 (53.1) | 193 (52.9) | 42 (53) | 90 (54) | 53 (51) | 8 (47) |
| Maternal lifetime stress score mean (SD)c | 13.4 (12.05) | 13.3 (11.96) | 7.9 (7.41) | 15.2 (13.28) | 15.6 (12.28) | 8.4 (6.67) |
| Fenton birth weight Z score mean (SD) | −0.19 (0.91) | −0.17 (0.91) | −0.006 (0.84) | −0.27 (0.98) | −0.09 (0.85) | −0.36 (0.74) |
| Maternal age at birth in years mean (SD) | 29.4 (5.79) | 29.4 (5.83) | 32.9 (4.34) | 27.9 (5.53) | 28.8 (6.07) | 31.8 (5.87) |
Six participants declined providing race.
Complications include gestational diabetes, hypertension, and/or preeclampsia.
Maternal lifetime stress score was derived from the number of endorsed events accounting for the reported negative impact of each event. Abbreviations: Standard deviation (SD), high school (HS).
The average depth of coverage (i.e., number of unique reads) was 1494. A list of all identified variants (n=13,335), frequencies, and Mitomap (http://www.mitomap.org) associations are available (Table S1–S3) (52). After we applied our criterion thresholds (QUAL >10000, MAF < 0.5, # of mutant reads > 1) to define heteroplasmy, we identified 13,189 heteroplasmies and 146 homoplasmies; all individuals had at least one heteroplasmy. Total mutational load did vary by race/ethnicity (p=0.001) but not child sex (p=0.24); mutational loads did not vary by child sex within race (p-values ranging 0.15–0.82). The majority of mutations were transitions (93.2%), 2,742 were nonsynonymous, 1404 have been reported in Mitomap and 801 associated with disease (43 confirmed pathogenic) (Table S3) (52). Heteroplasmies were present at low frequencies (Figure 1A) and found in all mitochondrial genes (Figure 1B).
Figure 1.
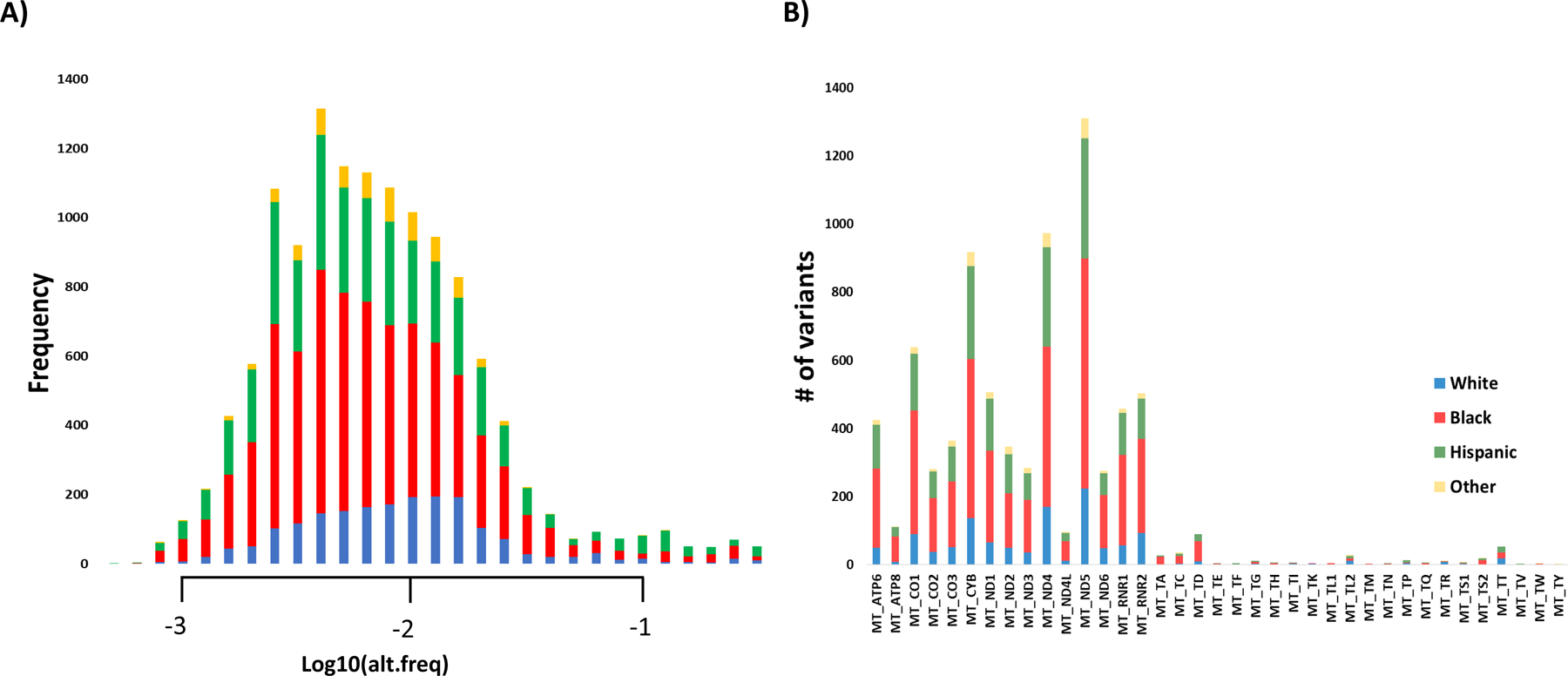
Histograms of A) mutant allele frequencies by race/ethnicity and B) number of mutant alleles by mitochondrial gene by race/ethnicity.
Maternal lifetime exposure to psychosocial stress and mutational load
Figure 2 shows the regional distribution of mutations by high and low lifetime psychosocial stress level using the median cut point for descriptive purposes only (LSCRwt score > 10 = high exposure). All regions of the genome exhibit some degree of mutational variability and the mutational load across the genome varies by lifetime psychosocial stress level and race/ethnicity (Figure 2).
Figure 2. Circos plot of mutations on the mitochondria genome by level of maternal lifetime stress exposure by race/ethnicity.
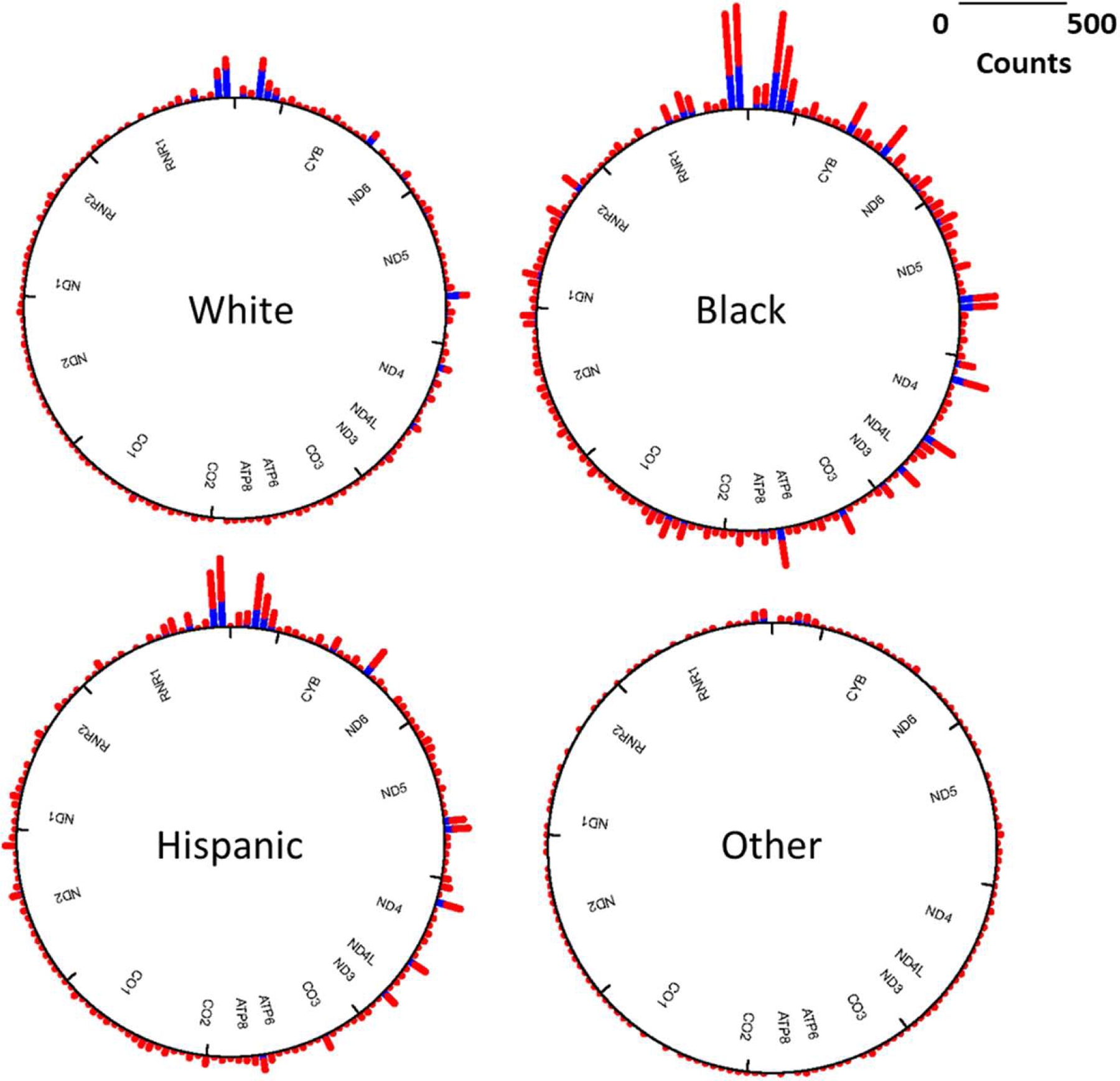
Mutational load (# of mtDNA mutations) was stratified according to the level of maternal lifetime exposure to psychosocial stress [LSCRwt score > 10 (median) is colored red, ≤ 10 is blue].
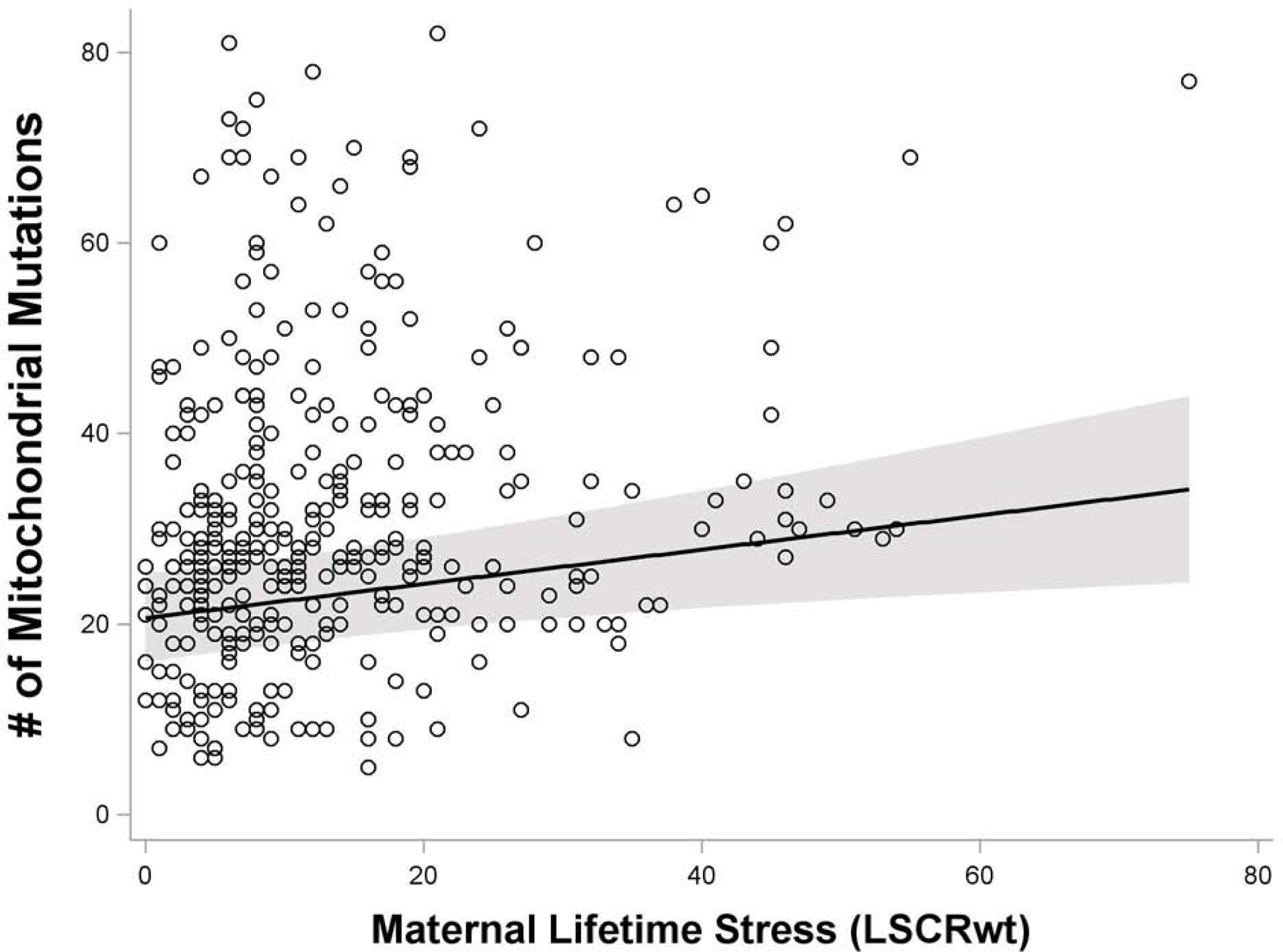
Women experiencing increased psychosocial stress over their lifetime exhibited a higher number of mitochondrial mutations in the placenta. Specifically, in regression models treating the maternal lifetime psychosocial stress score as a continuous variable, we observed increased stress to be associated with a higher mutational load across the entire genome (β=0.23, 95% CI 0.03–0.42, p=0.02) after controlling for maternal race (p < 0.0001), education (p = 0.92), and age (p= 0.11) (Figure 3, Table S5); although the effect is weaker, increased stress was also significantly associated with more nonsynonymous mutations (β=0.04, 95% CI 0.002–0.08, p=0.04) (Table S6). This association remained significant in models considering only heteroplasmic mutations (β=0.18, 95% CI 0.05–0.31, p=0.01) (Table S7) but not homoplasmic (β= −0.008, 95% CI −0.03 – 0.01, p=0.40) (Table S8) and given the fact that very few homoplasmic mutations were identified (1.1%), remaining analyses focused on total and nonsynonymous mutations.
Figure 3. Association between maternal lifetime stress exposure and number of placental mitochondrial DNA mutations (homoplasmic + heteroplasmic = mutational load).
Model is adjusted for maternal age at birth (χ2= 2.61, p = 0.11), education (χ2= 0.01, p=0.92), and race/ethnicity (χ2= 27.78, p < 0.0001) based on augmented backward elimination algorithm. Results are similar for analyses using only heteroplasmic mutations (Table S7).
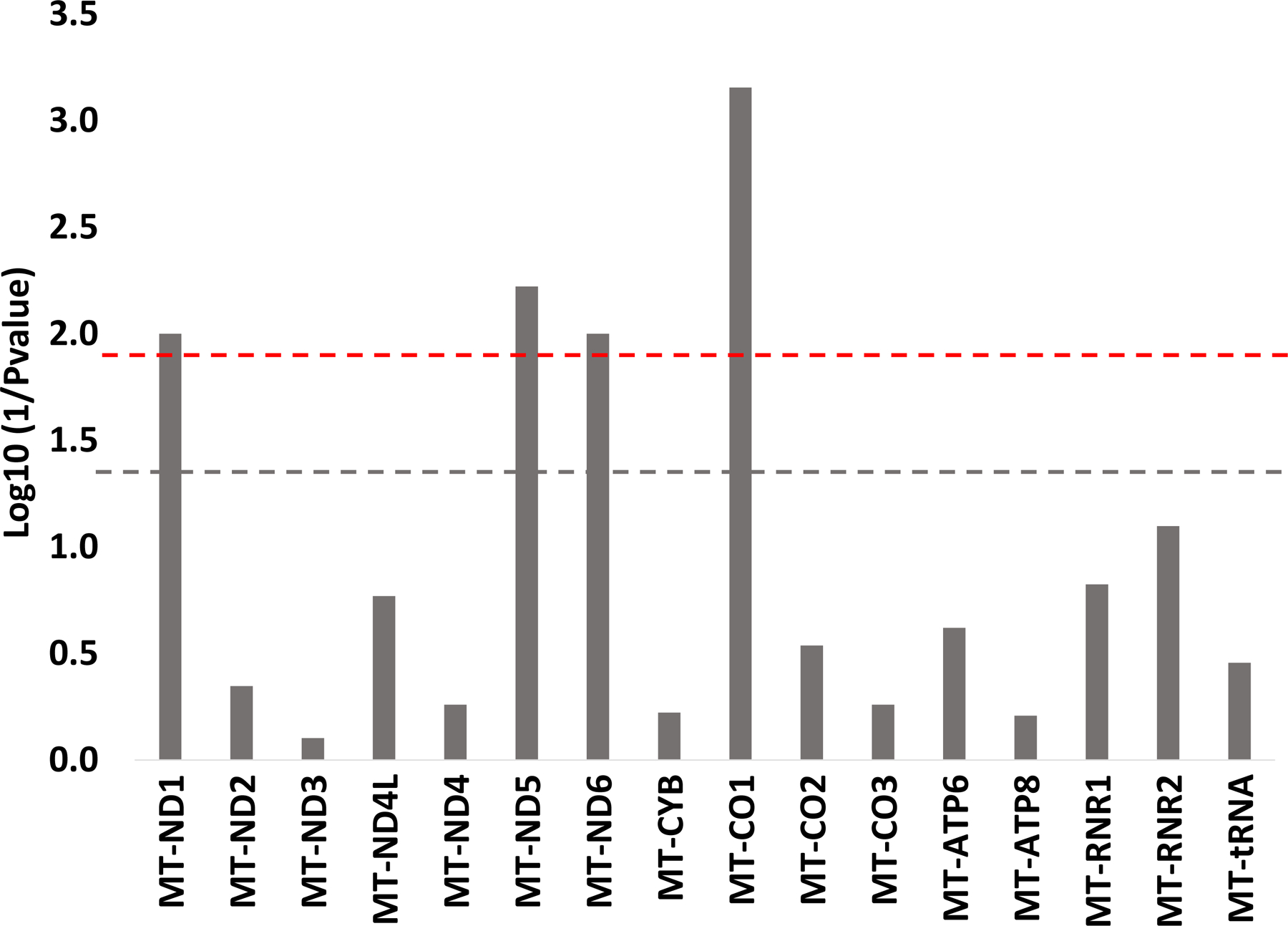
In order to determine if increased maternal lifetime psychosocial stress was more strongly related to mutations in a particular region, we conducted gene-wise regression analyses for 13 protein-coding, 2 rRNAs, and tRNA total (i.e. sum of all mutations in tRNA genes). After corrections for multiple comparisons, greater maternal lifetime stress was associated with a higher mutational load in three out of seven genes that code for various subunits of NADH dehydrogenase [Complex I of the electron transport chain (ETC)]. These include MT-ND1 (FDR=0.04), MT-ND5 (FDR=0.04), and MT-ND6 (FDR=0.04). Increased maternal lifetime stress was also significantly associated with a higher mutational load in MT-C01 (gene coding for the catalytic subunit of cytochrome c oxidase, Complex IV of the ETC) (FDR=0.01) (Figure 4). Of the 1544 variants observed in MT-ND5, 50% (n=777) were nonsynonymous. The highest percentage of disease-associated variants was observed among MT-ND1 variants (126 out of 605, 23% were nonsynonymous) and these variants were associated with metabolic, cardiac, and neurodevelopment outcomes (Table S3) (52). Table S4 provides the breakdown of the number of mutations by gene and race/ethnicity.
Figure 4. Gene-wise association between maternal lifetime stress exposure and number of placental mitochondrial DNA mutations (homoplasmic + heteroplasmic).
All models are adjusted for maternal age at birth, education, and race/ethnicity based on augmented backward elimination algorithm. Red dashed line represents FDR=0.05; gray dashed line p-value=0.05
Differences by maternal race/ethnicity or haplogroup
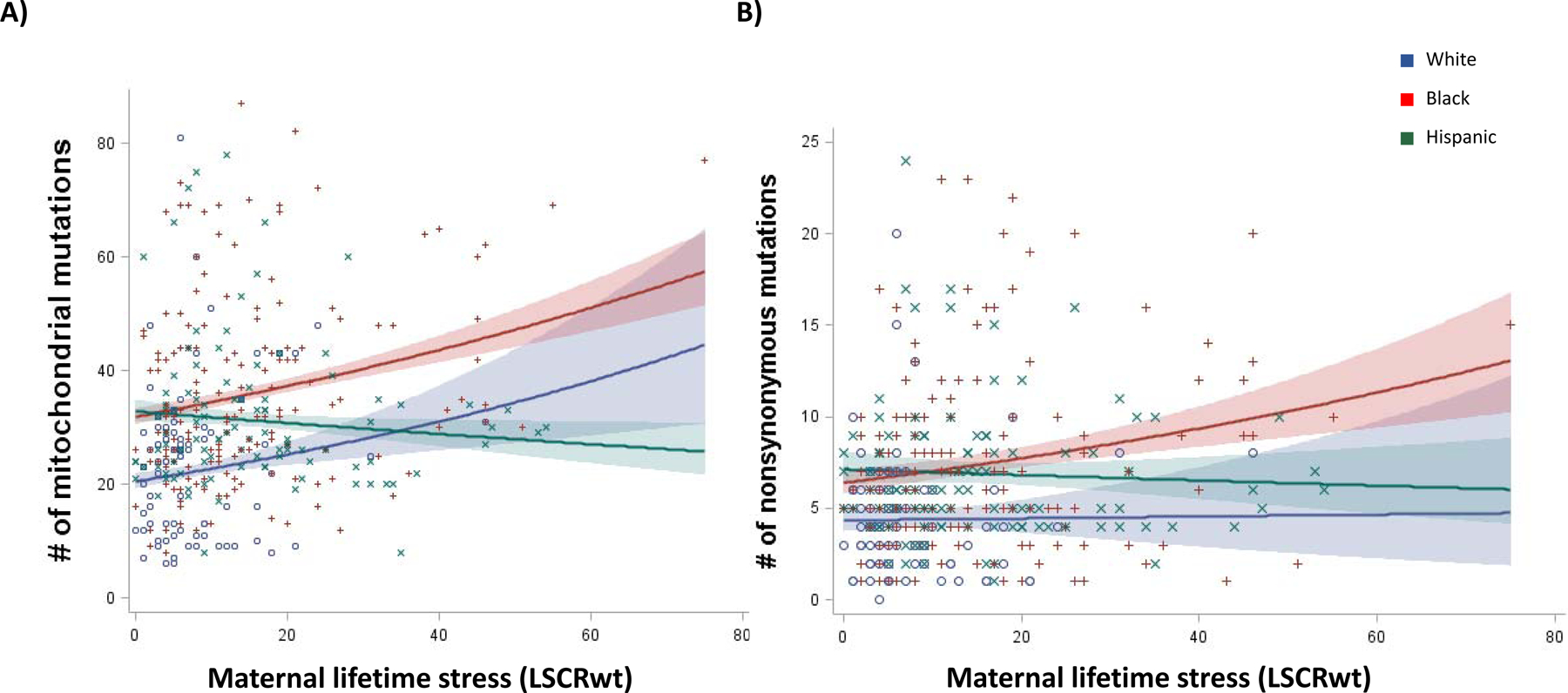
Significant racial/ethnic differences were observed in the relationship between maternal psychosocial stress and mutational load (Prace*stress < 0.0001). Compared to Hispanic participants, White (χ2= 16.91, p < 0.0001) and Black (χ2= 41.39, p < 0.0001) women reporting more lifetime psychosocial stress exhibited a greater degree of placental mtDNA mutations (Figure 5A). Compared to Hispanics, Black women were also more likely to exhibit a greater degree of nonsynonymous mutations associated with lifetime psychosocial stress (Figure 5B, χ2= 9.92, p = 0.0016) but not White women (χ2= 0.19, p =0.66). The effects observed when modeling heteroplasmic mutations only were the same (Figure S3). Haplogroup specific findings were also similar with the L (African) haplogroup being most at-risk for increased mtDNA mutations (Figure S5). Gene-wise comparisons did not reveal statistically significant racial/ethnic effects.
Figure 5. Race/ethnic differences in the association between maternal lifetime stress and placental mitochondrial A) total (homoplasmic + heteroplasmic) and B) nonsynonymous mutational load.
The likelihood ratio type 3 analysis for the three-way interaction (i.e., LSCRwt × race/ethnicity) was significant in both models (p<0.0001 and p=0.005, respectively). Contrast statements were used to test for differences in slopes across levels of race/ethnicity. Model adjusted for maternal age at birth and maternal education.
DISCUSSION
This study presents novel findings linking maternal lifetime stress to placental mtDNA mutations in a diverse urban sample. Women experiencing increased stress over their lifetime exhibited a higher number of placental mitochondrial mutations, particularly among genes coding for Complex I and IV of the ETC. Further, the effect of maternal lifetime stress was more pronounced among Black women.
Mitochondria are life-sustaining organelles that sense, assimilate, and relay environmental information including that of stress exposure. Steroidogenesis of glucocorticoids, the adrenal steroid stress response hormones, is partly regulated by mitochondria (53,54) and, in turn, can regulate placental mitochondrial oxidation and expression of mitochondrial genes (55,56). Under cumulative stress conditions and altered glucocorticoid levels, mtDNA can accumulate damage (57) which may reduce the proliferation and migration of trophoblast cells, thereby affecting placental formation and development (58,59). Animal studies support the hypothesis that psychological stress induces mitochondrial dysfunction; however, the number of human studies pales in comparison (60). Most human studies focus on the impact of Adverse Childhood Experiences (ACEs) suggesting adults who experience ACEs and/or have a history of depression have significantly higher levels of mtDNAcn in whole blood compared to those who do not experience childhood adversity or depression (61,62). Maternal lifetime stress exposure has been shown to impact placental mtDNAcn (23); however, its impact on mtDNA mutations/heteroplasmy is unclear. Using a human/experimental paradigm, Cai and colleagues (2015) (63) identified increased rates of heteroplasmy in women with major depression disorder and support their finding using mice to suggest the relationship is likely due to stress. Mitochondrial mutations have been reported in blood, placenta, cord blood, and buccal cells in the context of aging (64). This is the first study to examine and find an association between maternal lifetime psychosocial stress exposure and increased placental mutational load.
Greater maternal lifetime stress was predominantly associated with a higher placental mutational load in three out of seven genes (i.e., MT-ND1, MT-ND5, MT-ND6) that code for various subunits of NADH dehydrogenase (Complex 1 of the ETC) and MT-C01, catalytic subunit of cytochrome c oxidase (complex IV of the ETC). Complex I is a large enzyme that catalyzes the first step of the ETC that oxidizes NADH. Cytochrome C oxidase is the final enzyme in the ETC and plays a key role in the regulation of aerobic energy production. Cytochrome C oxidase and NADH dehydrogenase are targets of stress response. Hypoxic placentas have been shown to suppress complexes 1 and 4 compromising energy metabolism and potentially leading to impaired fetal growth (65). Abnormal electron transport and excessive oxidative stress may be related to the pathogenesis of early onset preeclampsia (66). Mitochondrial genetic variants located in MT-ND1 and MT-C01 have also been linked to body mass index in adults (67). Mutations in MT-ND6 have been observed among psychiatric cases of bipolar disorder, schizophrenia, and major depressive disorder (68); variants in MT-ND5 may also influence Autism Spectrum Disorder phenotypes (69). In addition to obesity and neurological outcomes, mtDNA mutational load might be associated with atherosclerosis (70). Interestingly, 50% and 23% of the mutations in MT-ND5 and MT-ND1, respectively, were nonsynonymous which may have direct impact on energy production capacity, aging, and systemic metabolism that could have later-life health consequences but should be confirmed in future studies.
Higher stress-related mtDNA mutational loads were observed for Black and White women compared to Hispanics; Black mothers also exhibited more nonsynonymous mutations. Given the disproportionate exposure to stressors among racial minorities and women of lower socioeconomic status, it is possible that stress-related variations in mtDNA contribute to disparities in health as has been observed for other outcomes such as cancer (71), insulin sensitivity (72), and low resting energy expenditure (a risk factor for obesity) (73) with African American women being at increased risk. Another explanation could be the “Hispanic Paradox”- that despite exposure to more stress, sociocultural dynamics specific to Hispanics may attenuate psychophysiological response to stress leading to better outcomes (74). Further, differences in oxidative capacity and mitochondrial respiration in other tissues, such as skeletal muscle, have been documented between African American and Caucasian women(75). We did not observe racial/ethnic differences in our gene-wise analyses. Perhaps racial/ethnic differences are more related to systemic oxidative damage and cumulative damage rather than differential mechanisms at the gene level. Our haplogroup results mimic our self-reported race/ethnicity findings that participants of the African (L) haplogroup exhibit more stress-related mutations. Haplogroups have been shown to modify the effects of other oxidative stress generating exposures (i.e., air pollution) on biomarkers of systemic inflammation (76). Additional studies linking stress, genetic ancestry and culture, mitochondrial dysfunction and subsequent health disparities are needed.
To our knowledge this is the first epidemiological study to sequence the mitochondrial genome and examine the impact of maternal lifetime psychosocial stress on mtDNA mutations. The urban minority population provides the opportunity to examine this relationship among women experiencing trauma, community and domestic violence, discrimination and disrupted stress response (5,13,14). There are limitations worth noting. We were unable to adjust for the cell-type heterogeneity as placental cell reference-based estimates for mitochondrial heteroplasmy are not available. We were unable to characterize origins of mutations (i.e., inherited or acquired) and patterns of single-cell mtDNA mutations in relation to child health outcomes. While the mechanisms around the transmission of mutations from mother to fetus are unclear, research suggests that the frequency of de novo mutations in the placenta is low (28). Further work is needed to delineate not only the mechanisms involved in transmission (e.g., bottlenecking during oogenesis vs. postnatal oocyte maturation, accumulated mutational rates associated with aging) but also the critical timing of stress exposure to inform interventions. Although pregnancy complications did not impact results, the frequency, clustering, and severity of pregnancy complications should be explored more deeply in studies with extensive obstetric data. Further, although 93.2% of the mutations identified in our study are transitions and this is in line with other literature in regards to inherited polymorphisms and disease-causing somatic mutations (Tables S2–S3),-we do not have adequate power to investigate other mutations such as transversions which have been shown to have larger regulatory effects (77). However, our analysis did see an association between lifetime stress and increased nonsynonymous variants. We also observed 20% discordance among mutations with low (< 5) read counts, likely due to sequencing errors, and mutations not present in MitoMap which could be due to the study population for which sequencing was conducted (i.e. healthy pregnant women), the tissue source (i.e. placenta rather than blood), and the use of a consensus genome which accounts for the genetic diversity of our population rather than the Revised Cambridge Reference Sequence which is based on European decent. We also detected relatively low levels of heteroplasmy which likely will not lead to gross mitochondrial function and disease (78); however, they may represent a reservoir of mtDNA variants that can alter the functional capacity of mitochondria and perhaps be used as novel biomarkers reflecting stress exposures that ultimately impact child development. Further, inherited heteroplasmies can persist across many generations once they reach intermediate frequencies suggesting potential for transgenerational effects beyond the contemporaneous period (40). A deeper investigation into the role of mitochondrial function and the intergenerational (and potentially transgenerational) consequences of women’s experiences of cumulative stress and trauma on disparities of childhood and later-life outcomes among offspring is warranted.
Supplementary Material
KEY RESOURCES TABLE.
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Commercial Assay Or Kit | TaKaRa LA Taq DNA Polymerase | Takara Bio | RR002B | |
| Commercial Assay Or Kit | Wizard SV Gel and PCR Clean-Up kit | Promega | A9282 | |
| Commercial Assay Or Kit | NEBNext® Ultra™ II FS DNA Library Prep Kit | New England Biolabs | E7805L | |
| Commercial Assay Or Kit | Bioanalyzer High Sensitivity DNA Kit | Agilent | 5067–4626 | |
| Commercial Assay Or Kit | NEBNext Library Quant Kit | New England Biolabs | E7630L | |
| Commercial Assay Or Kit | TruSeq SBS KIT v3 - HS (200 CYCLES) | Illumina | FC-401-3001 | |
| Deposited Data; Public Database | ||||
| Genetic Reagent | ||||
| Organism/Strain | ||||
| Peptide, Recombinant Protein | ||||
| Recombinant DNA | ||||
| Sequence-Based Reagent | Primer for PCR | This paper | ||
| Software; Algorithm | ||||
| Transfected Construct | ||||
| Other |
Acknowledgments:
We would like to thank the participants of the PRISM cohort and our funding agencies. Specifically, this work was supported by the National Heart, Lung, and Blood Institute under grants R01HL095606 (RJ Wright) and R01HL114396 (RJ Wright); the National Institute of Environmental Health Sciences (NIEHS) under grants R00ES024116 (KJ Brunst), P30ES006096 (KJ Brunst), and P30ES023515 (RJ Wright). Biobanking infrastructure was supported by the Mount Sinai Health System Clinical Translational Science Award from the National Center for Advancing Translational Sciences under grant UL1 TR001433 (RJ Wright).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement: The authors report no biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.Graignic-Philippe R, Dayan J, Chokron S, Jacquet AY, Tordjman S. (2014): Effects of prenatal stress on fetal and child development: A critical literature review. Neurosci Biobehav Rev 43:137–162. [DOI] [PubMed] [Google Scholar]
- 2.Lane RH. (2014): Fetal programming, epigenetics, and adult onset disease. Clinics in perinatology 41(4):815–831. [DOI] [PubMed] [Google Scholar]
- 3.Enlow MB, Devick KL, Brunst KJ, Lipton LR, Coull BA, Wright RJ. (2017): Maternal lifetime trauma exposure, prenatal cortisol, and infant negative affectivity. Infancy 22(4):492–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keenan K, Hipwell AE, Class QA, Mbayiwa K. (2018): Extending the developmental origins of disease model: Impact of preconception stress exposure on offspring neurodevelopment. Dev Psychobiol 60(7):753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreier HM, Enlow MB, Ritz T, Coull BA, Gennings C, Wright RO, et al. (2016): Lifetime exposure to traumatic and other stressful life events and hair cortisol in a multi-racial/ethnic sample of pregnant women. Stress (Amsterdam, Netherlands) 19(1):45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Witt WP, Cheng ER, Wisk LE, Litzelman K, Chatterjee D, Mandell K, et al. (2014): Maternal stressful life events prior to conception and the impact on infant birth weight in the united states. Am J Public Health 104 Suppl 1:S81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flom JD, Chiu YM, Hsu HL, Devick KL, Brunst KJ, Campbell R, et al. (2018): Maternal lifetime trauma and birthweight: Effect modification by in utero cortisol and child sex. J Pediatr. [DOI] [PMC free article] [PubMed]
- 8.Bosquet Enlow M, Petty CR, Svelnys C, Gusman M, Huezo M, Malin A, et al. (2019): Differential effects of stress exposures, caregiving quality, and temperament in early life on working memory versus inhibitory control in preschool-aged children. Dev Neuropsychol 44(4):339–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Magnus MC, Wright RJ, Roysamb E, Parr CL, Karlstad O, Page CM, et al. (2018): Association of maternal psychosocial stress with increased risk of asthma development in offspring. Am J Epidemiol 187(6):1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatch SL, Dohrenwend BP. (2007): Distribution of traumatic and other stressful life events by race/ethnicity, gender, ses and age: A review of the research. Am J Community Psychol 40(3–4):313–332. [DOI] [PubMed] [Google Scholar]
- 11.Mukherjee S, Trepka MJ, Pierre-Victor D, Bahelah R, Avent T. (2016): Racial/ethnic disparities in antenatal depression in the united states: A systematic review. Matern Child Health J 20(9):1780–1797. [DOI] [PubMed] [Google Scholar]
- 12.Seng JS, Kohn-Wood LP, McPherson MD, Sperlich M. (2011): Disparity in posttraumatic stress disorder diagnosis among african american pregnant women. Arch Womens Ment Health 14(4):295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger M, Sarnyai Z. (2015): “More than skin deep”: Stress neurobiology and mental health consequences of racial discrimination. Stress (Amsterdam, Netherlands) 18(1):1–10. [DOI] [PubMed] [Google Scholar]
- 14.Suglia SF, Staudenmayer J, Cohen S, Enlow MB, Rich-Edwards JW, Wright RJ. (2010): Cumulative stress and cortisol disruption among black and hispanic pregnant women in an urban cohort. Psychol Trauma 2(4):326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Racine N, Plamondon A, Madigan S, McDonald S, Tough S. (2018): Maternal adverse childhood experiences and infant development. Pediatrics 141(4). [DOI] [PubMed] [Google Scholar]
- 16.Nugent BM, Bale TL. (2015): The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front Neuroendocrinol 39:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh G, Grewal J, Mannisto T, Mendola P, Chen Z, Xie Y, et al. (2014): Racial/ethnic differences in pregnancy-related hypertensive disease in nulliparous women. Ethn Dis 24(3):283–289. [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Kershaw T, Ettinger AS, Higgins C, Lu MC, Chao SM. (2015): Association between life event stressors and low birth weight in african american and white populations: Findings from the 2007 and 2010 los angeles mommy and baby (lamb) surveys. Matern Child Health J 19(10):2195–2205. [DOI] [PubMed] [Google Scholar]
- 19.Gingrich JA. (2005): Oxidative stress is the new stress. Nat Med 11(12):1281–1282. [DOI] [PubMed] [Google Scholar]
- 20.Sinha K, Das J, Pal PB, Sil PC. (2013): Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Archives of toxicology 87(7):1157–1180. [DOI] [PubMed] [Google Scholar]
- 21.Lee HC, Wei YH. (2000): Mitochondrial role in life and death of the cell. J Biomed Sci 7(1):2–15. [DOI] [PubMed] [Google Scholar]
- 22.Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, et al. (2014): Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect 122(12):1271–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunst KJ, Sanchez Guerra M, Gennings C, Hacker M, Jara C, Bosquet Enlow M, et al. (2017): Maternal lifetime stress and prenatal psychological functioning and decreased placental mitochondrial DNA copy number in the prism study. Am J Epidemiol 186(11):1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picard M, Prather AA, Puterman E, Cuillerier A, Coccia M, Aschbacher K, et al. (2018): A mitochondrial health index sensitive to mood and caregiving stress. Biol Psychiatry 84(1):9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sondheimer N, Glatz CE, Tirone JE, Deardorff MA, Krieger AM, Hakonarson H. (2011): Neutral mitochondrial heteroplasmy and the influence of aging. Hum Mol Genet 20(8):1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart JB, Chinnery PF. (2015): The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat Rev Genet 16(9):530–542. [DOI] [PubMed] [Google Scholar]
- 27.Kopinski PK, Janssen KA, Schaefer PM, Trefely S, Perry CE, Potluri P, et al. (2019): Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc Natl Acad Sci U S A 116(32):16028–16035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J, Purcell H, Showalter L, Aagaard KM. (2015): Mitochondrial DNA sequence variation is largely conserved at birth with rare de novo mutations in neonates. Am J Obstet Gynecol 212(4):530 e531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colicino E, Power MC, Cox DG, Weisskopf MG, Hou L, Alexeeff SE, et al. (2014): Mitochondrial haplogroups modify the effect of black carbon on age-related cognitive impairment. Environmental health : a global access science source 13(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kesmodel US, Kjaersgaard MI, Denny CH, Bertrand J, Skogerbo A, Eriksen HL, et al. (2015): The association of pre-pregnancy alcohol drinking with child neuropsychological functioning. BJOG 122(13):1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murray J, Burgess S, Zuccolo L, Hickman M, Gray R, Lewis SJ. (2016): Moderate alcohol drinking in pregnancy increases risk for children’s persistent conduct problems: Causal effects in a mendelian randomisation study. J Child Psychol Psychiatry 57(5):575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bandoli G, Coles CD, Kable JA, Wertelecki W, Yevtushok L, Zymak-Zakutnya N, et al. (2019): Patterns of prenatal alcohol use that predict infant growth and development. Pediatrics 143(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolfe J, Kimerling R. Gender issues in assessment of posttraumatic stress disorder. New York: Guilford; 1997. [Google Scholar]
- 34.Brunst KJ, Tignor N, Just A, Liu Z, Lin X, Hacker MR, et al. (2018): Cumulative lifetime maternal stress and epigenome-wide placental DNA methylation in the prism cohort. Epigenetics 13(6):665–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ingman M, Kaessmann H, Paabo S, Gyllensten U. (2000): Mitochondrial genome variation and the origin of modern humans. Nature 408(6813):708–713. [DOI] [PubMed] [Google Scholar]
- 36.Picard tools. GitHub Repository; 2018. http://broadinstitute.github.io/picard/.
- 37.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. (2011): A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. (2010): The sanger fastq file format for sequences with quality scores, and the solexa/illumina fastq variants. Nucleic Acids Res 38(6):1767–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoang ML, Kinde I, Tomasetti C, McMahon KW, Rosenquist TA, Grollman AP, et al. (2016): Genome-wide quantification of rare somatic mutations in normal human tissues using massively parallel sequencing. Proc Natl Acad Sci U S A 113(35):9846–9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaidi AA, Wilton PR, Su MS, Paul IM, Arbeithuber B, Anthony K, et al. (2019): Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed]
- 41.Kido T, Sikora-Wohlfeld W, Kawashima M, Kikuchi S, Kamatani N, Patwardhan A, et al. (2018): Are minor alleles more likely to be risk alleles? BMC Med Genomics 11(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang P, Samuels DC, Wang J, Zhao S, Shyr Y, Guo Y. (2016): Mitochondria single nucleotide variation across six blood cell types. Mitochondrion 28:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Y, Huang K, Sun Y, Wang J, Xu Y, Yan S, et al. (2017): Placenta response of inflammation and oxidative stress in low-risk term childbirth: The implication of delivery mode. BMC Pregnancy Childbirth 17(1):407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huuskonen P, Amezaga MR, Bellingham M, Jones LH, Storvik M, Hakkinen M, et al. (2016): The human placental proteome is affected by maternal smoking. Reprod Toxicol 63:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segura MT, Demmelmair H, Krauss-Etschmann S, Nathan P, Dehmel S, Padilla MC, et al. (2017): Maternal bmi and gestational diabetes alter placental lipid transporters and fatty acid composition. Placenta 57:144–151. [DOI] [PubMed] [Google Scholar]
- 46.Aouache R, Biquard L, Vaiman D, Miralles F. (2018): Oxidative stress in preeclampsia and placental diseases. Int J Mol Sci 19(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holland O, Dekker Nitert M, Gallo LA, Vejzovic M, Fisher JJ, Perkins AV. (2017): Review: Placental mitochondrial function and structure in gestational disorders. Placenta 54:2–9. [DOI] [PubMed] [Google Scholar]
- 48.Fenton TR, Kim JH. (2013): A systematic review and meta-analysis to revise the fenton growth chart for preterm infants. BMC Pediatr 13:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitchell SL, Goodloe R, Brown-Gentry K, Pendergrass SA, Murdock DG, Crawford DC. (2014): Characterization of mitochondrial haplogroups in a large population-based sample from the united states. Hum Genet 133(7):861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dunkler D, Plischke M, Leffondre K, Heinze G. (2014): Augmented backward elimination: A pragmatic and purposeful way to develop statistical models. PLoS One 9(11):e113677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benjamini Y, Hochberg Y. (1995): Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal Royal Statistical Society Series B 57:289–300. [Google Scholar]
- 52.Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, et al. (2013): Mtdna variation and analysis using mitomap and mitomaster. Curr Protoc Bioinformatics 44:1 23 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Midzak A, Papadopoulos V. (2016): Adrenal mitochondria and steroidogenesis: From individual proteins to functional protein assemblies. Front Endocrinol (Lausanne) 7:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller WL. (2013): Steroid hormone synthesis in mitochondria. Mol Cell Endocrinol 379(1–2):62–73. [DOI] [PubMed] [Google Scholar]
- 55.Bartho LA, Holland OJ, Moritz KM, Perkins AV, Cuffe JSM. (2019): Maternal corticosterone in the mouse alters oxidative stress markers, antioxidant function and mitochondrial content in placentas of female fetuses. J Physiol 597(12):3053–3067. [DOI] [PubMed] [Google Scholar]
- 56.Puscheck EE, Awonuga AO, Yang Y, Jiang Z, Rappolee DA. (2015): Molecular biology of the stress response in the early embryo and its stem cells. Adv Exp Med Biol 843:77–128. [DOI] [PubMed] [Google Scholar]
- 57.Picard M, McEwen BS. (2018): Psychological stress and mitochondria: A conceptual framework. Psychosom Med 80(2):126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anton L, DeVine A, Polyak E, Olarerin-George A, Brown AG, Falk MJ, et al. (2019): Hif-1alpha stabilization increases mir-210 eliciting first trimester extravillous trophoblast mitochondrial dysfunction. Front Physiol 10:699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fisher JJ, McKeating DR, Cuffe JS, Bianco-Miotto T, Holland OJ, Perkins AV. (2019): Proteomic analysis of placental mitochondria following trophoblast differentiation. Front Physiol 10:1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Picard M, McEwen BS. (2018): Psychological stress and mitochondria: A systematic review. Psychosom Med 80(2):141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tyrka AR, Parade SH, Price LH, Kao HT, Porton B, Philip NS, et al. (2016): Alterations of mitochondrial DNA copy number and telomere length with early adversity and psychopathology. Biol Psychiatry 79(2):78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cai N, Chang S, Li Y, Li Q, Hu J, Liang J, et al. (2015): Molecular signatures of major depression. Curr Biol 25(9):1146–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cai N, Li Y, Chang S, Liang J, Lin C, Zhang X, et al. (2015): Genetic control over mtdna and its relationship to major depressive disorder. Curr Biol 25(24):3170–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rebolledo-Jaramillo B, Su MS, Stoler N, McElhoe JA, Dickins B, Blankenberg D, et al. (2014): Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A 111(43):15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Colleoni F, Padmanabhan N, Yung HW, Watson ED, Cetin I, Tissot van Patot MC, et al. (2013): Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: A role for mirna-210 and protein synthesis inhibition. PLoS One 8(1):e55194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu Z, Jin X, Cai W, Zhou M, Shao P, Yang Z, et al. (2018): Proteomics analysis reveals abnormal electron transport and excessive oxidative stress cause mitochondrial dysfunction in placental tissues of early-onset preeclampsia. Proteomics Clin Appl 12(5):e1700165. [DOI] [PubMed] [Google Scholar]
- 67.Flaquer A, Baumbach C, Kriebel J, Meitinger T, Peters A, Waldenberger M, et al. (2014): Mitochondrial genetic variants identified to be associated with bmi in adults. PLoS One 9(8):e105116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sequeira A, Rollins B, Magnan C, van Oven M, Baldi P, Myers RM, et al. (2015): Mitochondrial mutations in subjects with psychiatric disorders. PLoS One 10(5):e0127280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patowary A, Nesbitt R, Archer M, Bernier R, Brkanac Z. (2017): Next generation sequencing mitochondrial DNA analysis in autism spectrum disorder. Autism Res 10(8):1338–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piotrowska-Nowak A, Elson JL, Sobczyk-Kopciol A, Piwonska A, Puch-Walczak A, Drygas W, et al. (2018): New mtdna association model, mutpred variant load, suggests individuals with multiple mildly deleterious mtdna variants are more likely to suffer from atherosclerosis. Front Genet 9:702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choudhury AR, Singh KK. (2017): Mitochondrial determinants of cancer health disparities. Semin Cancer Biol 47:125–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DeLany JP, Dube JJ, Standley RA, Distefano G, Goodpaster BH, Stefanovic-Racic M, et al. (2014): Racial differences in peripheral insulin sensitivity and mitochondrial capacity in the absence of obesity. J Clin Endocrinol Metab 99(11):4307–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimm SY, Glynn NW, Aston CE, Damcott CM, Poehlman ET, Daniels SR, et al. (2002): Racial differences in the relation between uncoupling protein genes and resting energy expenditure. Am J Clin Nutr 75(4):714–719. [DOI] [PubMed] [Google Scholar]
- 74.Ruiz JM, Sbarra D, Steffen PR. (2018): Hispanic ethnicity, stress psychophysiology and paradoxical health outcomes: A review with conceptual considerations and a call for research. Int J Psychophysiol 131:24–29. [DOI] [PubMed] [Google Scholar]
- 75.Toledo FGS, Dube JJ, Goodpaster BH, Stefanovic-Racic M, Coen PM, DeLany JP. (2018): Mitochondrial respiration is associated with lower energy expenditure and lower aerobic capacity in african american women. Obesity (Silver Spring) 26(5):903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wittkopp S, Staimer N, Tjoa T, Gillen D, Daher N, Shafer M, et al. (2013): Mitochondrial genetic background modifies the relationship between traffic-related air pollution exposure and systemic biomarkers of inflammation. PLoS One 8(5):e64444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo C, McDowell IC, Nodzenski M, Scholtens DM, Allen AS, Lowe WL, et al. (2017): Transversions have larger regulatory effects than transitions. BMC Genomics 18(1):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wallace DC, Chalkia D. (2013): Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol 5(11):a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.