

Abstract
Aberrant expression of human ether-a-go-go-related gene (hERG) potassium channels has been implicated in the pathophysiology of glioblastoma (GBM). Letrozole has demonstrated efficacy in pre-clinical GBM models. The objective of this research was to assess the potential for hERG inhibition by letrozole to mediate efficacy in GBM. hERG currents were assessed using patch clamp electrophysiology in an overexpression system during treatment with letrozole, exemestane, or vehicle (dimethyl sulfoxide). Relative to vehicle, peak hERG tail current density was reduced when treated with 300 nM and 1 μM letrozole but not when treated with exemestane (up to 1 μM). Cell proliferation was assessed in cultured glioblastoma cell lines (U87 and U373) treated with letrozole, exemestane, doxazosin (hERG blocker), or vehicle. Letrozole, but not exemestane, reduced cell proliferation relative to vehicle in U87 and U373 cells. The associations between expression of hERG (KCNH2), aromatase (CYP19A1), and the estrogen receptors (ESR1 and ESR2) and time to all-cause mortality were assessed in GBM patients within The Cancer Genome Atlas (TCGA) database. hERG expression was associated with reduced overall survival in the TCGA GBM cohort. Future work is warranted to investigate hERG expression as a potential biomarker to predict the therapeutic potential of hERG inhibitors in GBM.
Keywords: Carcinogenesis, Cardiac arrhythmias, Ion channels as drug targets, Human ether-a-go-go related gene, hERG, Letrozole, Glioblastoma
INTRODUCTION AND BACKGROUND
Glioblastoma (GBM) remains the most aggressive form of brain cancer with five-year survival rates under 10%.(1, 2) The standard of care for GBM management includes maximal surgical resection followed by adjuvant radiation and chemotherapy, including the alkylating agent temozolomide (TMZ).(3) However, this strategy and other treatment options are largely ineffective. Thus, the identification of molecular pathways that can be therapeutically targeted to improve outcomes in patients with GBM is an urgent clinical need.(4)
Investigations have identified a pathogenic role of the human ether-a-go-go-related gene (hERG; also called Kv.11.1; encoded by KCNH2) potassium channel in oncologic conversion and disease progression in a number of cancer types, including GBM.(5–8) hERG protein comprises the pore-forming channel subunit of the cardiac rapid delayed rectifier potassium current (IKr), which has been best characterized for its role in ventricular repolarization and the development of drug-induced arrhythmias.(9) Overexpression of hERG has been implicated in the pathophysiology of various types of cancer, wherein increased hERG expression is associated with enhanced cell proliferation, neoangiogenesis, and tissue invasiveness.(10–12) Increased hERG expression is also associated with a worse prognosis, including reduced overall survival, in GBM and other types of cancer.(5, 7)
Letrozole is a third-generation aromatase (estrogen synthase) inhibitor with durable efficacy in treating hormone receptor-positive breast cancer in postmenopausal women. More recently, pre-clinical investigations have demonstrated the efficacy of letrozole in treating cellular and animal models of GBM.(13, 14) Based on these promising results, a Phase I clinical trial (NCT03122197) is underway to assess letrozole in recurrent gliomas. However, whether letrozole’s therapeutic activity in GBM models is mediated through its inhibition of aromatase remains unknown. There are numerous reports of prolongation of the cardiac QT interval, including one case of the potentially fatal ventricular arrhythmia torsades de pointes, attributed to letrozole therapy in the U.S. Food and Drug Administration’s Adverse Event Reporting System (FAERS) database. In addition, the letrozole arm in a thorough QT study demonstrated statistically significant QT prolongation.(15) Since the predominant mechanism of drug-induced QT prolongation occurs via blockade of IKr,(16) these findings suggest that letrozole may inhibit hERG at clinically relevant concentrations.
The goal of this study was to assess whether letrozole’s beneficial mechanism in GBM may be mediated by inhibition of hERG potassium channels. The primary hypothesis was that letrozole, but not exemestane, would inhibit IhERG and demonstrate beneficial effects in GBM. Our experimental objectives were to: (1) assess the potential for letrozole and exemestane, a structurally unrelated aromatase inhibitor, to inhibit hERG current (IhERG); (2) assess the potential for letrozole and exemestane to reduce cell proliferation in cultured GBM cells lines; and (3) assess the association between expression of KCNH2, aromatase (CYP19A1), and the estrogen receptors (ESR1 and ESR2; downstream targets of aromatase inhibitors) with overall survival in GBM patients.
METHODS AND MATERIALS
The study was conducted in accordance with the Basic & Clinical Pharmacology & Toxicology policy for experimental and clinical studies.(17)
Cell Culture and Treatment
The glioblastoma cells lines, U87 and U373, were purchased from the American Type Culture Collection (ATCC®; Manassas, VA; catalog numbers: HTB-14 and HTB-17, respectively). Human embryonic kidney cells (HEK 293; ATCC® CRL-1573) that stably express hERG1a (product of KCNH2; herein referred to as hERG-HEK cells) were generously provided by a Dr. Craig January. U87 and U373 cells were cultured in modified essential medium (MEM) with 10% fetal bovine serum at 37˚C and 5% CO2. hERG-HEK cells were cultured in MEM with 10% fetal bovine serum, 1% Pen-Strep, 1% sodium pyruvate, and 1% non-essential amino acids at 37˚C and 5% CO2. Cell proliferation assays were performed following 24-hour treatment with vehicle (dimethylsulfoxide [DMSO]), letrozole (serial dilution of concentrations from 312.5 nM to 40 μM in U87 cells; 78.1 nM to 10 μM in U373 cells), exemestane (156.3 nM to 20 μM in U87 cells; 78.1 nM to 10 μM in U373 cells), or doxazosin (10 μM and 30 μM in U87 and U373 cells). Electrophysiology experiments were performed 48 hours after subculture in hERG-HEK cells treated with DMSO, letrozole (50 nM, 100 nM, 300 nM, or 1 μM), or exemestane (300 nM or 1 μM) in the pipette solution during recording. Electrophysiology experiments were performed in U87 cells with DMSO or dofetilide (100 nM) in the pipette solution during recording. Electrophysiology results were not obtainable with letrozole concentrations ≥ 2 μM due to observed cytotoxicity following attainment of the whole-cell patch clamp configuration.
Electrophysiology
Whole-cell, patch-clamp electrophysiology experiments in the voltage-clamp mode were performed to functionally assess the effects of letrozole, exemestane, and vehicle on elicited hERG currents (IhERG) in hERG-HEK cells and a dofetilide-sensitive current in U87 cells. All recordings were performed at room temperature (~22°C) and data were acquired using an EPC-9 amplifier and Patchmaster software (HEKA Instruments Inc; Holliston, MA, USA). For experiments in hERG-HEK cells, culture media was replaced by a bath solution, containing (in mM): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose prior to recording. For experiments in U87 cells, culture media was replaced by a bath solution containing (in mM): 135 NaCl, 5 KCl, 1.8 MgCl2, 10 HEPES, and 10 glucose prior to recording. Borosilicate glass micropipettes were pulled using a P-2000 Puller (Sutter Instrument; Novato, CA, USA) to a have a resistance in solution of 2–5 MΩ. For experiments in hERG-HEK cells, micropipettes were filled with pipette solution containing (in mM): 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, and 10 HEPES. For experiments in U87 cells, micropipettes were filled with solution containing (in mM): 135 KCl, 8 MgCl2, 10 glucose, and 10 HEPES. Bath solution was adjusted to a pH of 7.4 using NaOH, and pipette solution was adjusted to a pH of 7.2 with KOH in all experiments.
The following voltage protocols were employed to functionally assess hERG current, as previously described: (1) elicited tail currents were measured to assess peak current density from a protocol with a holding potential of −80 mV that clamped at +60 mV for 1 second before a 5-second step protocol that clamped at 10 mV increments between −100 mV and + 40 mV; (2) tail currents were measured to assess the voltage dependence of activation from a protocol with a holding potential of −80 mV that stepped in 10 mV increments from −60 mV to +50 mV for 4 seconds followed before clamping at −40 mV for 4 seconds. Current recordings were acquired ~1 minute after attainment of the whole-cell patch conformation in all experiments, with fast and slow capacitance compensation being performed during this time. Recordings in hERG-HEK cells were performed in a minimum of 3 plates (3–5 cells/plate) for each drug treatment, and all treatment groups (letrozole, exemestane, vehicle) were performed on the same days of experimentation. Series resistance values were not different among groups (ANOVA p-values were 0.44 and 0.15 for letrozole and exemestane experiments, respectively), and series resistance compensation was not performed.
Cytotoxicity Assay
For the cytotoxicity assay, U87 and U373 cells were plated in a 96-multiwell plate at a density of 7,500 cells per well, in Hyclone DMEM media with 10% charcoal-stripped FBS, 20 U/mL penicillin, and 20 μg/mL streptomycin, at 37°C in a humidified atmosphere containing 5% CO2. After 24-hour incubation, the cells were treated with letrozole at concentrations ranging from 0 to 100 μmol/L and control (DMSO) for a period of 48 hours. Cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell growth assay kit (Merck KGaA, Darmstadt, Germany) per the manufacturer’s instructions. The absorbance at 570 nm was measured using a microplate reader (Bio-Tek; 800 TS; Winooski, VT, USA).
Bioinformatic Resources for KCNH2, CYP19A1, and ESR1 Expression Analyses
The Xena Functional Genomics Explorer (developed and hosted by the University of California- Santa Cruz; http://xena.ucsc.edu/) (18) and OncoLnc (http://www.oncolnc.org/),(19) an interactive self-service tool for exploring genetic-cancer survival correlations, were used for analysis of the The Cancer Genome Atlas (TCGA) data repository GBM cohort survival based on upper and lower quartiles of tumour KCNH2, CYP19A1, ESR1, and ESR2 expression (n=152 patients with expression and survival data).
Data Analysis
All electrophysiology data were analysed using FitMaster v2×73.1 (HEKA Instruments) and GraphPad Prism v7.4. Boltzmann distributions were used to fit normalized activation current vs. voltage (I-V) curves for each drug treatment with the following equations:
| (Eq. 1: Activation) |
In the equation above, I/Imax is the normalized current, V is membrane voltage, V1/2 is the voltage of half-maximal activation, and k is the slope factor. Peak current density, V1/2, and absorbance at 570 nM were compared via one-way ANOVA with Dunnett’s test post-hoc test (control group: DMSO vehicle) or independent sample t-test with Welch correction, as appropriate, using JMP Pro 13 software (SAS Institute; Cary, NC, USA). The associations between KCNH2, CYP19A1, and ESR1/2 expression and days to all-cause mortality were analysed via the log-rank test in GraphPad Prism v7.4 (GraphPad Software; San Diego, CA, USA). All data are expressed as mean ± standard error of the mean (SEM) and, alpha was set to 0.05 for all experiments.
RESULTS
Acute Treatment with Letrozole Inhibits IhERG Function and Alters the Voltage Dependence of Activation in a Concentration-Dependent Manner
Functional assessment of IhERG during treatment with letrozole and exemestane was performed via patch clamp electrophysiology using standard tail and activation voltage protocols (shown along with representative current traces in Figure 1A). As shown in the full I-V plot in Figure 1B and in the bar graphs at the voltages of maximal inward (−100 mV) and outward (−20 mV) elicited current in Figure 1C, peak tail current density was reduced in a concentration-dependent manner during acute treatment with letrozole in the pipette solution. Peak inward and outward IhERG current density was inhibited during treatment with 300 nM (inward: −38.0 ± 5.0 pA/pF [mean ± SEM], outward: 30.5 ± 4.3 pA/pF; p<0.01 for both; n=15) and 1 μM letrozole (inward: −35.3 ± 7.8, outward: 22.6 ± 3.9; p<0.01 for both; n=9) relative to DMSO vehicle control (inward: −64.2 ± 11.7, outward: 49.3 ± 5.7; n=16 Figure 1D). Treatment with 1 μM letrozole was also associated with a hyperpolarizing shift in the voltage dependence of activation relative to vehicle (V1/2: −13.0 ± 4.9 mV [mean ± SEM] with 1 μM letrozole, n=7; V1/2: −2.7 ± 2.6 with vehicle, n=13; p<0.01; Figure 1E). Based on observed outward IhERG inhibition at −20 mV with letrozole concentrations from 50 nM to 1 μM, letrozole inhibited IhERG with an IC50 of 373 nM (Figure 1F).
Figure 1. Letrozole inhibits hERG current density and alters the voltage dependence of activation.
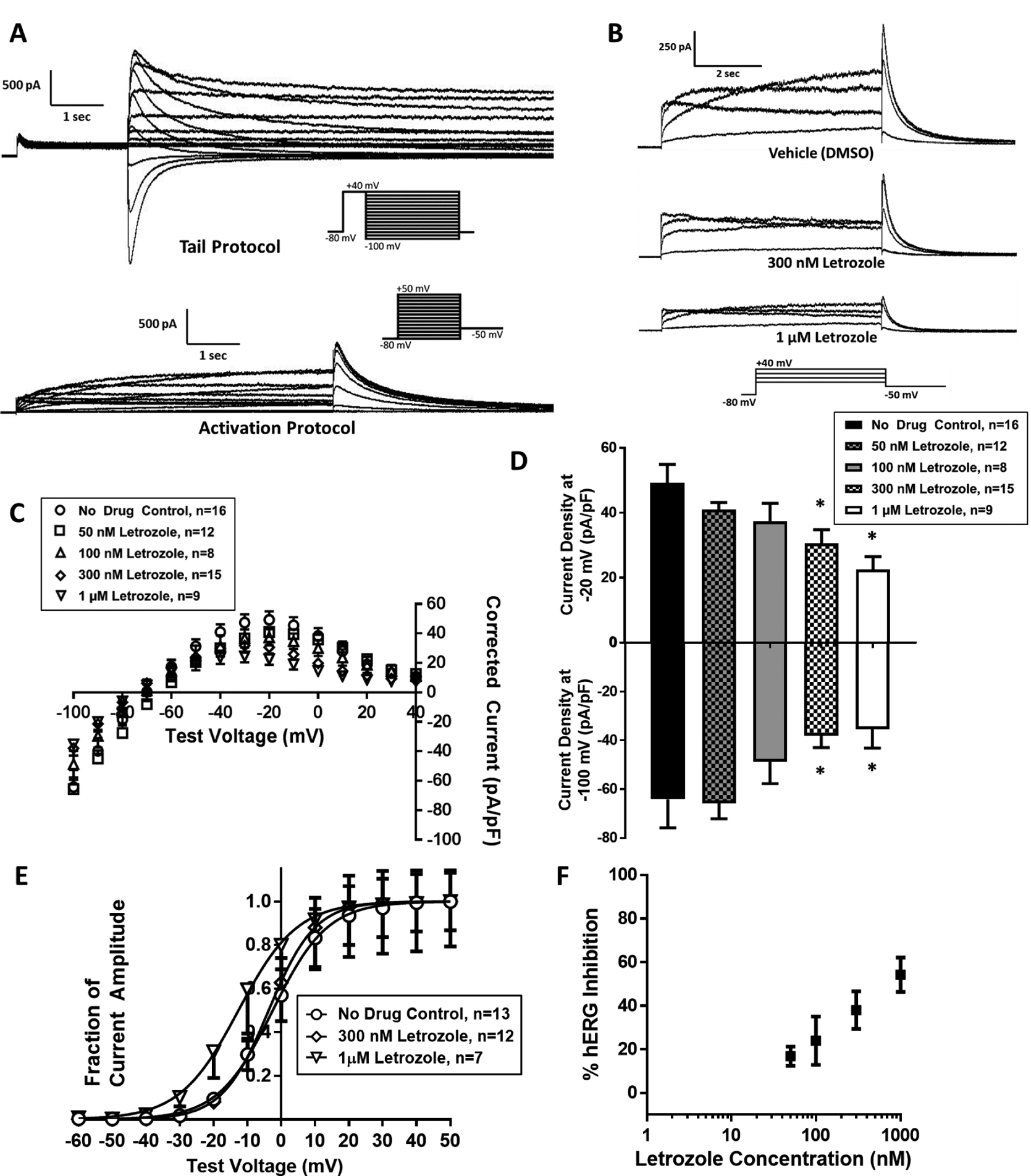
(A) Representative traces showing hERG tail (left) and activation (right) currents elicited from tail and activating voltage protocols, respectively (current protocols inset). (B) Representative activation currents elicited in the presence of vehicle (top), 300 nM letrozole (middle), and 1 μM letrozole (bottom). (C) Current vs. voltage (I-V) plot of hERG tail current density elicited from tail voltage protocol during treatment with vehicle, 50 nM, 100 nM, 300 nM, and 1 μM letrozole. (D) Bar graphs of peak hERG tail current density at maximum outward (−20 mV) and inward (−100 mV) voltages during treatment with letrozole or vehicle. (E) I-V plot of normalized hERG tail currents during an activating voltage protocol with vehicle, 300 nM, and 1 μM letrozole. (F) Concentration-effect graph presenting observed hERG inhibition with assessed letrozole concentrations. * P<0.05 vs. vehicle
Acute Treatment with Exemestane Does not Affect IhERG Function or Alter the Voltage Dependence of Activation at Concentrations Up to 1 μM
In contrast to the effect seen with letrozole, application of exemestane in the pipette solution did not inhibit peak IhERG tail current density at concentrations of 300 nM or 1μM (I-V plot shown in Figure 2A). Peak inward and outward IhERG current densities were not different among cells treated with 300 nM exemestane (inward: −101.2 ± 24.1, outward: 43.0 ± 6.5 pA/pF; inward p=0.91, outward p=0.52; n=12)or 1 μM exemestane (inward: −92.2 ± 16.0, out: 45.3 ± 5.0; inward p=0.66, outward p=0.67; n=12) relative to vehicle control (inward: −104.9 ± 24.0, outward: 49.1 ± 4.3; n=13; Figure 2B). Acute treatment with exemestane did not affect the voltage dependence of activation at concentrations up to 1μM (V1/2: 0.9 ± 0.6 mV with 1 μM letrozole, n=8; V1/2: −0.9 ± 0.7 with vehicle, n=12; p=0.09; Figure 2C).
Figure 2. Exemestane does not affect hERG current density or alter the voltage dependence of activation.
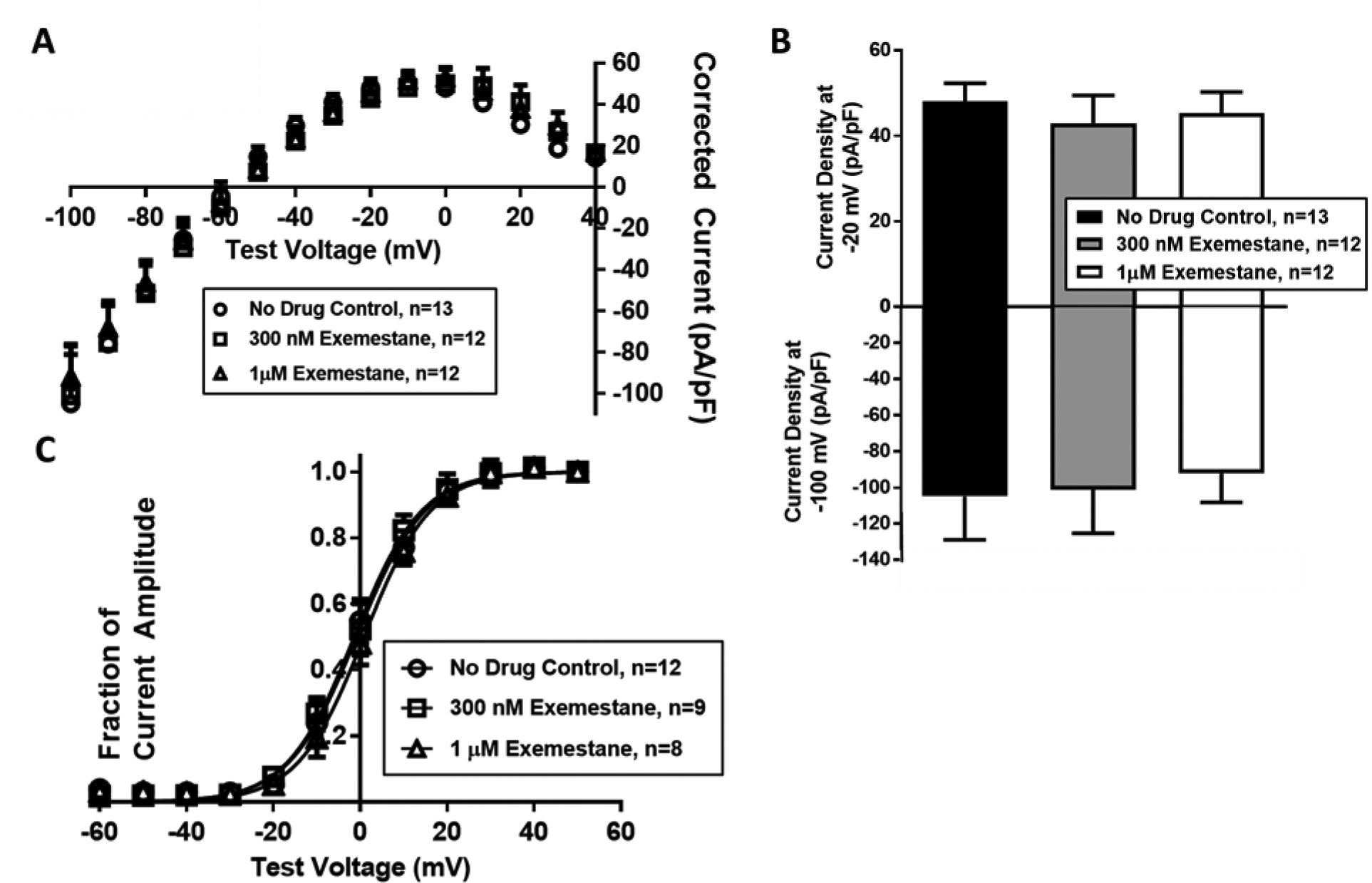
(A) I-V plot of hERG tail current density during tail voltage protocol with vehicle, 300 nM, and 1 μM exemestane. (B) Bar graphs of peak hERG tail current density at the −20 mV and −100 mV during treatment with exemestane or vehicle. (C) I-V plot of normalized hERG tail current density during an activating voltage protocol with vehicle, 300 nM, and 1 μM exemestane.
24 Hour Treatment with Letrozole, but not Exemestane, Reduces Cell Proliferation in U87 and U373 Cultured Glioblastoma Cell Lines
To explore the relevance of the differential effects of letrozole and exemestane on inhibition of IhERG to human GBM, we (1) isolated voltage-sensitive tail currents in U87 cells to test for hERG activity and (2) assessed the effects of letrozole, exemestane, and doxazosin (hERG inhibitor) on reducing proliferation in GBM cell lines. Tail currents in U87 cells were elicited and tested for sensitivity to dofetilide, a specific inhibitor of hERG current. Figure 3A displays the tail current from a representative trace that was sensitive to acute treatment with 100 nM dofetilide by reducing the maximal inward voltage-sensitive tail current by an average of 78.6%. These findings suggest that U87 cells conduct voltage-stimulated tail currents that are mediated through native hERG channels. Additionally, the effects of letrozole and exemestane treatment on the proliferation of the cultured GBM cell lines U87 and U373 were assessed using the colorimetric MTT method. Relative to DMSO vehicle control, 24-hour treatment with letrozole reduced absorbance at 570 nm, an experimental marker of cellular proliferation potential, in U87 cells at concentrations from 2.5 μM (61.8% of control; n=6 in all experiments; p<0.01) to 40 μM (25.6% of control; p<0.01; Figure 3B, Left). In contrast, exemestane did not reduce absorbance at 570 nM in U87 cells at concentrations up to 10 μM (97.4% of control; p=0.60) and produced only modest reduction in 570 nM absorbance at 20 μM (80.8% of control; p=0.01; Figure 3B, Center). In U373 cells, the relative effects of letrozole versus exemestane were similar to those observed in U87 cells. Letrozole reduced absorbance at 570 nm at concentrations from 2.5 μM (62.1% of control; p<0.01) to 10 μM (24.8% of control; p<0.01; Figure 3C, Left), while exemestane did not reduce 570 nM absorbance at concentrations up to 10 μM (94.1% of control; p=0.31; Figure 3C, Center). To investigate whether the anti-proliferative effect of letrozole in GBM cells could be mediated by IhERG inhibition, the known IhERG blocker, doxazosin, was also assessed. As shown in the right panels of Figure 3B–C, 24-hour treatment with 10 μM doxazosin produced large reductions in absorbance at 570 nM relative to vehicle control (15.5% of control in U87 cells; 12.6% of control in U373 cells; p<0.01 in both). Our novel in vitro results with letrozole, in addition to our findings that corroborate the efficacy of doxazosin in reducing GBM cell proliferation, suggest that letrozole’s anti-proliferative effect may be mechanistically mediated by its ability to inhibit IhERG.
Figure 3. Letrozole, but not exemestane, reduces cell proliferation in U87 and U373 GBM cell lines at concentrations ≤10 μM.
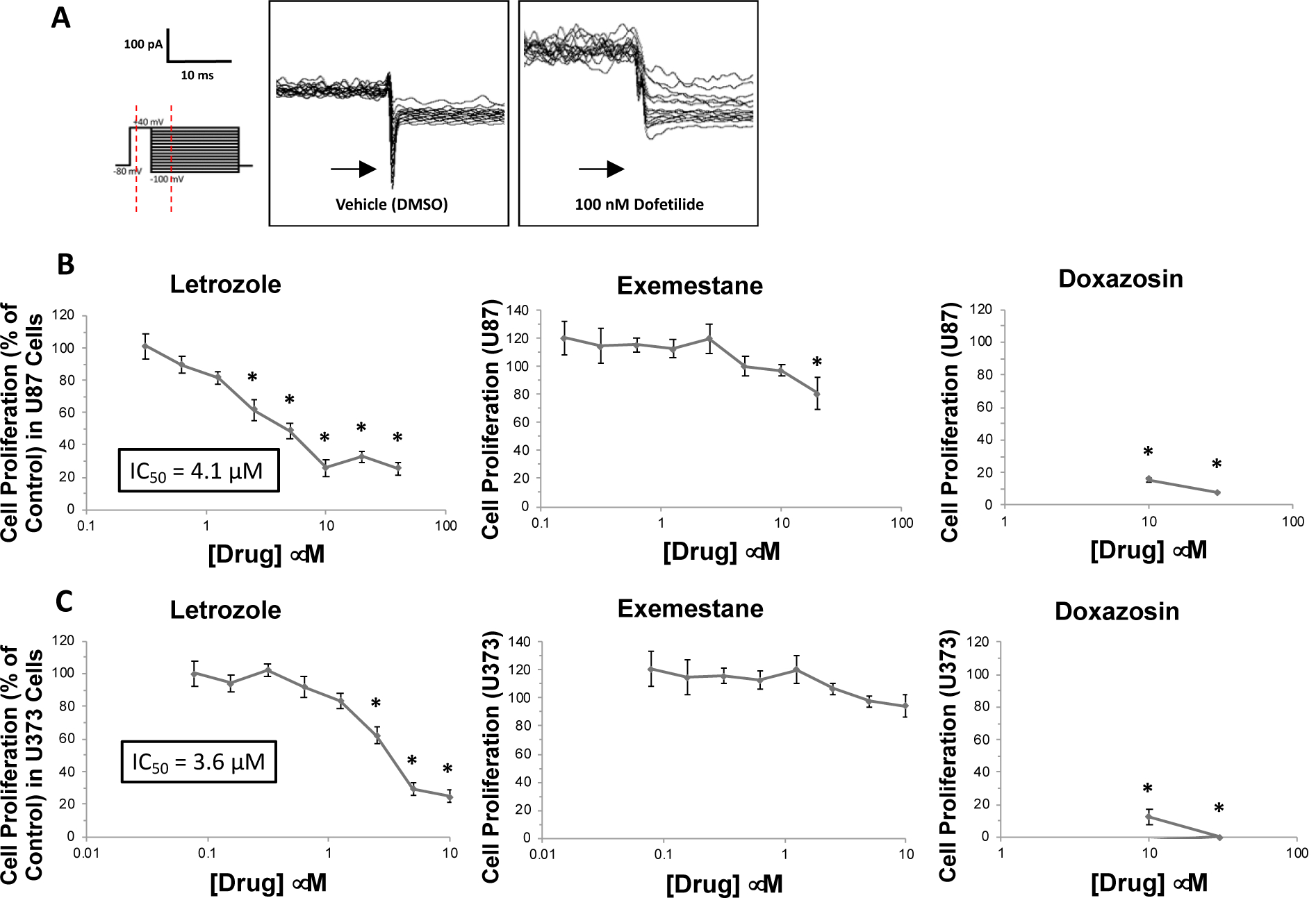
(A) Representative traces of currents elicited in U87 cells using the tail voltage protocol (protocol shown on left) during acute treatment with vehicle (DMSO) and dofetilide applied to the pipette solution. (B) Absorbance at 570 nm, the readout variable from colorimetric cell proliferation assays (MTT), in U87 cells during treatment with letrozole (concentrations up to 40 μM), exemestane (up to 20 μM), and doxazosin (up to 30 μM; positive control). All data are normalized (by percentage) to the absorbance observed in vehicle-treated cells. (C) Absorbance at 570 nm in U373 cells during treatment with letrozole (concentrations up to 10 μM), exemestane (up to 10 μM), and doxazosin (up to 30 μM; positive control). *P<0.05 vs. vehicle control.
KCNH2 Expression, But Not CYP19A1 or ESR1/2 Expression, is Inversely Associated with Time to All-Cause Mortality in GBM Patients
An association between expression of hERG (KCNH2), aromatase (CYP19A1), and the estrogen receptors (ESR1 and ESR2) and patient outcomes were assessed within the GBM cohort of the TCGA data repository. In patients with gene expression data and clinical follow-up within the TCGA GBM cohort (n=152), we assessed whether KCNH2, CYP19A1, and ESR1/2 expression were associated with time to all-cause mortality. As shown in Figure 4A, patients within the upper quartile of KCNH2 expression had significantly reduced time to all-cause mortality relative to those in the lowest quartile (Kaplan Meier p=0.03; n=38 in each quartile). In contrast, patients within the upper and lower quartiles of CYP19A1 expression had no difference in time to all-cause mortality (Kaplan Meier p=0.99; n=38 per quartile; Figure 4B). Expression of the estrogen receptors (ESR1 and ESR2; upper vs. lower quartile) was also not associated with time to all-cause mortality (p=0.89; n=38 per quartile; Figure 4C–D), suggesting that the action of estrogen may not mediate GBM pathophysiology.
Figure 4. KCNH2 expression, but not CYP19A1 or ESR1/2 expression, is inversely associated with time to all-cause mortality in GBM patients.
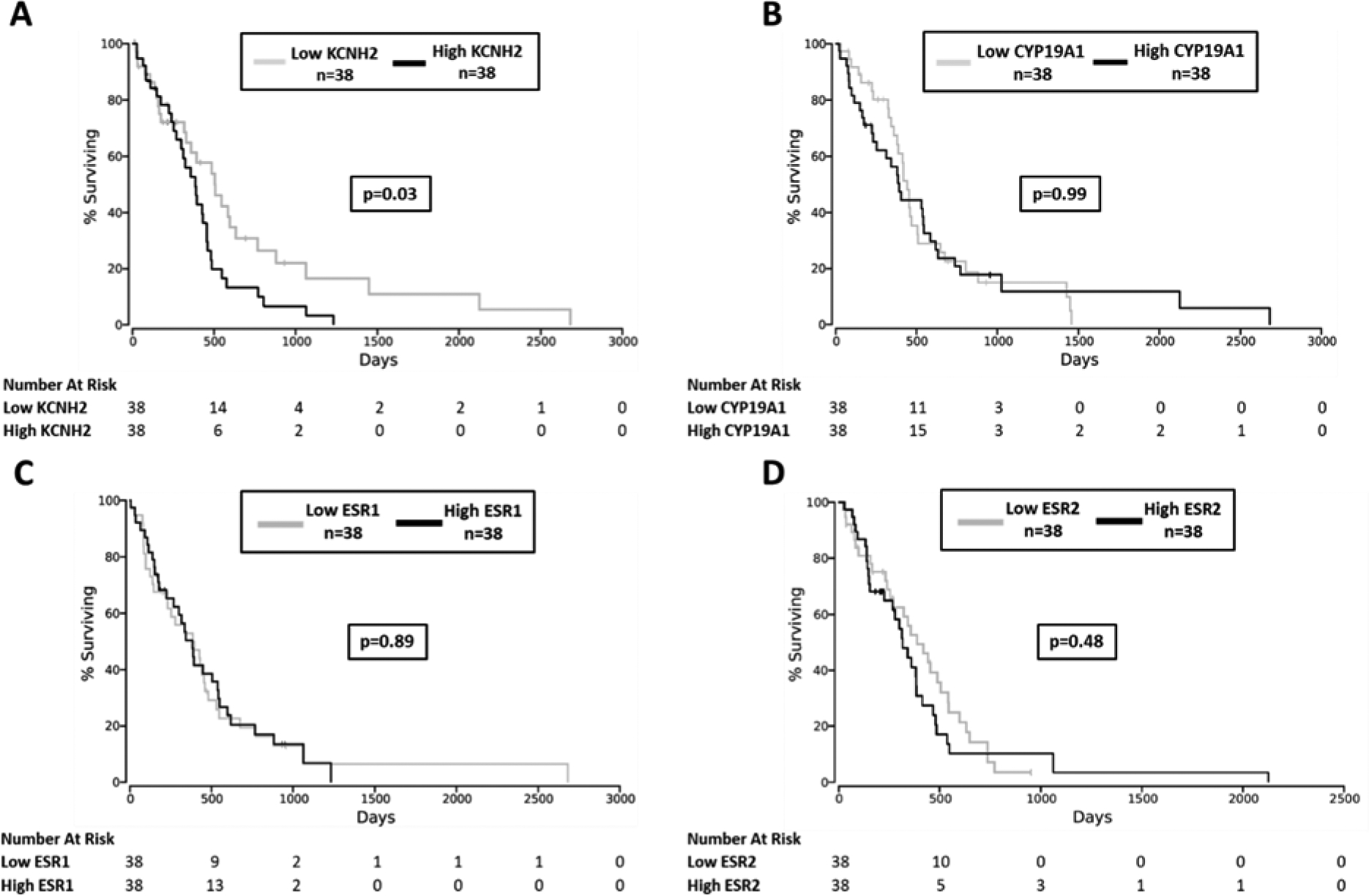
(A) Association of KCNH2 expression with time to all-cause mortality within TCGA GBM patients (n=38 patients in both the upper and lower quartiles, n=152 total). (B) Association of CYP19A1 expression (upper vs. lower quartile) with time to all-cause mortality within the TCGA GBM cohort. (C) Association of ESR1 and (D) ESR2 expression (upper vs. lower quartile) with time to all-cause mortality within the TCGA GBM cohort.
DISCUSSION
Letrozole has shown efficacy in treating cellular and animal models of GBM and is being actively investigated in human clinical trials.(13, 14) The primary results of this study contribute to the mechanistic explanation for the efficacy of letrozole in pre-clinical GBM models. In contrast to exemestane, a steroidal aromatase inhibitor that is structurally unrelated to letrozole, letrozole significantly inhibited IhERG at therapeutically relevant concentrations. Similarly, letrozole, but not exemestane, reduced cell proliferation in a manner similar to the hERG-blocker doxazosin. Together, these findings suggest that (1) letrozole, in addition to its effect on inhibiting aromatase, also inhibits hERG potassium currents and (2) letrozole’s ability to inhibit IhERG mediates its efficacy in reducing proliferation in cultured GBM cells. These findings are supported by previous investigations within cellular and animal GBM models.(7, 20) Work by Staudacher, et al. demonstrated the potential of hERG inhibition to reduce cell proliferation and induce apoptosis in cultured GBM cells via treatment with doxazosin.(20) Additionally, an investigation by Pointer, et al. found that the chemical hERG inhibitor E-4031 reduced sphere formation, a marker of tumour proliferation, within patient-derived GBM xenografts in mice.(7)
The concentrations of letrozole demonstrated to inhibit IhERG (IC50 ~300 nM or ~85 ng/mL) in this study were comparable to the median plasma letrozole concentrations in 241 postmenopasual women with breast cancer following a standard 2.5 mg daily dose of letrozole (average concentration reported at 88.4 ng/mL).(21) The plasma letrozole concentrations observed in that clinical trial were as high as 300 ng/mL (~1.0 μM) at this standard dose.(21) In addition, letrozole readily crosses the blood brain barrier, an important criterion for its potential use in the treatment of gliomas.(22) Letrozole effectively reduced tumour volume in rats implanted with C6 gliomas with the ratio of brain to unbound plasma letrozole concentrations ranging between 0.51 to 0.98 at the various doses tested.(22)
To further explore the potential of IhERG inhibition to contribute to efficacy in GBM, the associations between KCNH2 (hERG), CYP19A1 (aromatase), and ESR1/2 (estrogen receptor) expression with time to all-cause mortality were assessed in the TCGA GBM cohort. KCNH2 expression was inversely associated with time to all-cause mortality; in contrast, there was no association with CYP19A1. The potential for aromatase inhibition, which acts through depletion of the systemic pool of circulating estrogens,(23) to mediate letrozole efficacy in GBM is also contradicted by the fact that expression of ESR1/2, the receptors responsible for cellular transduction of estrogenic signalling, were not associated with GBM mortality. These findings support our in vitro data, which indicate that letrozole’s efficacy in GBM is at least partially mediated by inhibition of hERG rather than inhibition of aromatase.
A limitation of the gene expression data in the TCGA database is the retrospective nature of the analysis. Though treatment data are relatively sparse within the TCGA GBM cohort, it is unlikely that significant treatment bias influenced our analyses since the current medical standard of care in GBM is the cytotoxic agent TMZ (60.4% of TCGA patients received TMZ), and administration of hormonal agents was only reported in 5.6% of patients in our cohort. Additionally, our findings are supported by other retrospective clinical investigations. Specifically, concomitant therapy with medications with off-target hERG blocking effects (e.g. fluoxetine, phenytoin) was associated with prolonged overall survival in GBM patients with high but not low KCNH2 expression (determined by immunohistochemistry in tumor microarrays).(7)
In summary, our results support that the efficacy of letrozole in pre-clinical GBM models may be at least partially mediated via inhibition of hERG potassium channels rather than through inhibition of aromatase. This finding warrants further investigation since it may have both clinical and research implications. Firstly, it supports a growing body of evidence regarding the therapeutic potential of hERG inhibitors, including letrozole, in GBM. Also, the potential for letrozole to inhibit hERG in addition to aromatase may be important in other cancer settings, such as hormone receptor-positive breast cancer, wherein KCNH2 expression may be a useful biomarker to guide precision oncology (i.e. preferential selection of letrozole in patients with high KCNH2 expression). Therefore, future work is warranted to better elucidate the therapeutic potential of hERG inhibitors, including letrozole, in GBM and other cancers that involve KCNH2.
ACKNOWLEDGEMENTS
We wish to thank Dr. Craig January for his generous contribution of the hERG-HEK cells used in our experiments. This work was supported by NIH NHLBI HL095655.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts to disclose.
REFERENCES
- 1.Tykocki T, Eltayeb M. Ten-year survival in glioblastoma. A systematic review. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2018;54:7–13. [DOI] [PubMed] [Google Scholar]
- 2.Di Carlo DT, Cagnazzo F, Benedetto N, Morganti R, Perrini P. Multiple high-grade gliomas: epidemiology, management, and outcome. A systematic review and meta-analysis. Neurosurgical review. 2017. [DOI] [PubMed] [Google Scholar]
- 3.Paolillo M, Boselli C, Schinelli S. Glioblastoma under Siege: An Overview of Current Therapeutic Strategies. Brain sciences. 2018;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Touat M, Idbaih A, Sanson M, Ligon KL. Glioblastoma targeted therapy: updated approaches from recent biological insights. Annals of oncology : official journal of the European Society for Medical Oncology. 2017;28(7):1457–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding XW, Luo HS, Luo B, Xu DQ, Gao S. Overexpression of hERG1 in resected esophageal squamous cell carcinomas: a marker for poor prognosis. Journal of surgical oncology. 2008;97(1):57–62. [DOI] [PubMed] [Google Scholar]
- 6.Masi A, Becchetti A, Restano-Cassulini R, Polvani S, Hofmann G, Buccoliero AM, et al. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br J Cancer. 2005;93(7):781–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pointer KB, Clark PA, Eliceiri KW, Salamat MS, Robertson GA, Kuo JS. Administration of Non-Torsadogenic human Ether-a-go-go-Related Gene Inhibitors Is Associated with Better Survival for High hERG-Expressing Glioblastoma Patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(1):73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao XD, Wu KC, Guo XZ, Xie MJ, Zhang J, Fan DM. Expression and significance of HERG protein in gastric cancer. Cancer biology & therapy. 2008;7(1):45–50. [DOI] [PubMed] [Google Scholar]
- 9.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81(2):299–307. [DOI] [PubMed] [Google Scholar]
- 10.Comes N, Serrano-Albarras A, Capera J, Serrano-Novillo C, Condom E, Ramon YCS, et al. Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochimica et biophysica acta. 2015;1848(10 Pt B):2477–92. [DOI] [PubMed] [Google Scholar]
- 11.Lastraioli E, Guasti L, Crociani O, Polvani S, Hofmann G, Witchel H, et al. herg1 gene and HERG1 protein are overexpressed in colorectal cancers and regulate cell invasion of tumor cells. Cancer research. 2004;64(2):606–11. [DOI] [PubMed] [Google Scholar]
- 12.Jehle J, Schweizer PA, Katus HA, Thomas D. Novel roles for hERG K(+) channels in cell proliferation and apoptosis. Cell death & disease. 2011;2:e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tivnan A, Heilinger T, Ramsey JM, O’Connor G, Pokorny JL, Sarkaria JN, et al. Anti-GD2-ch14.18/CHO coated nanoparticles mediate glioblastoma (GBM)-specific delivery of the aromatase inhibitor, Letrozole, reducing proliferation, migration and chemoresistance in patient-derived GBM tumor cells. Oncotarget. 2017;8(10):16605–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dave N, Chow LM, Gudelsky GA, LaSance K, Qi X, Desai PB. Preclinical pharmacological evaluation of letrozole as a novel treatment for gliomas. Molecular cancer therapeutics. 2015;14(4):857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Durairaj C, Ruiz-Garcia A, Gauthier ER, Huang X, Lu DR, Hoffman JT, et al. Palbociclib has no clinically relevant effect on the QTc interval in patients with advanced breast cancer. Anti-cancer drugs. 2018;29(3):271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz PJ, Woosley RL. Predicting the Unpredictable: Drug-Induced QT Prolongation and Torsades de Pointes. Journal of the American College of Cardiology. 2016;67(13):1639–50. [DOI] [PubMed] [Google Scholar]
- 17.Tveden-Nyborg P, Bergmann TK, Lykkesfeldt J. Basic & Clinical Pharmacology & Toxicology Policy for Experimental and Clinical studies. Basic Clin Pharmacol Toxicol. 2018;123(3):233–5. [DOI] [PubMed] [Google Scholar]
- 18.Goldman M, Craft B, Hastie M, Repecka K, Kamath A, McDade F, et al. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv. 2019;326470. [Google Scholar]
- 19.Anaya J OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Computer Science. 2016;2(e67). [Google Scholar]
- 20.Staudacher I, Jehle J, Staudacher K, Pledl HW, Lemke D, Schweizer PA, et al. HERG K+ channel-dependent apoptosis and cell cycle arrest in human glioblastoma cells. PloS one. 2014;9(2):e88164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desta Z, Kreutz Y, Nguyen AT, Li L, Skaar T, Kamdem LK, et al. Plasma letrozole concentrations in postmenopausal women with breast cancer are associated with CYP2A6 genetic variants, body mass index, and age. Clinical pharmacology and therapeutics. 2011;90(5):693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dave N, Gudelsky GA, Desai PB. The pharmacokinetics of letrozole in brain and brain tumor in rats with orthotopically implanted C6 glioma, assessed using intracerebral microdialysis. Cancer Chemother Pharmacol. 2013;72(2):349–57. [DOI] [PubMed] [Google Scholar]
- 23.Geisler J, Haynes B, Anker G, Dowsett M, Lonning PE. Influence of letrozole and anastrozole on total body aromatization and plasma estrogen levels in postmenopausal breast cancer patients evaluated in a randomized, cross-over study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2002;20(3):751–7. [DOI] [PubMed] [Google Scholar]