

Abstract
Alzheimer’s disease (AD) is the most common form of dementia worldwide, characterized by a progressive decline in a variety of cognitive and non-cognitive functions. The amyloid beta protein cascade hypothesis places the formation of amyloid beta protein aggregates on the first position in the complex pathological cascade leading to neurodegeneration, and therefore AD might be considered to be a protein-misfolding disease. The Ubiquitin Proteasome System (UPS), being the primary protein degradation mechanism with a fundamental role in the maintenance of proteostasis, has been identified as a putative therapeutic target to delay and/or to decelerate the progression of neurodegenerative disorders that are characterized by accumulated/aggregated proteins. The purpose of this study was to test if the activation of proteasome in vivo can alleviate AD pathology. Specifically by using two compounds with complementary modes of proteasome activation and documented antioxidant and redox regulating properties in the 5xFAD transgenic mice model of AD, we ameliorated a number of AD related deficits. Shortly after proteasome activation we detected significantly reduced amyloid-beta load correlated with improved motor functions, reduced anxiety and frailty level. Essentially, to our knowledge this is the first report to demonstrate a dual activation of the proteasome and its downstream effects. In conclusion, these findings open up new directions for future therapeutic potential of proteasome-mediated proteolysis enhancement.
Keywords: Alzheimer’s disease, Proteasome, Behavior, Therapeutic, Frailty
1. Introduction
Alzheimer’s disease (AD) is a complex, heterogeneous and the most prevalent form of dementia, characterized by extracellular aggregates of amyloid-β (Aβ) peptides and tau-associated pathology [1-4] The amyloid cascade hypothesis that placed the formation of amyloid plaques as a key step in AD pathogenesis [5] went through certain modifications, leading to the amyloid beta protein cascade hypothesis that takes into account not only plaques, but also several different amyloid beta protein assemblies that may contribute to AD pathogenesis [6]. The amyloid precursor protein (APP) is cleaved at three sites, leading to the release of Aβ peptides and other APP fragments of different lengths [7]. Especially Aβ42 is neurotoxic, as it has a high propensity to form oligomeric aggregates and eventually deposits as plaques [8,9]. Although amyloid plaques were considered as the main culprit for cell loss and dementia [1], new evidences revealed that plaque burden does not always correspond to disrupted cognition or neurodegeneration. It seems that oligomeric forms generated in the process of fibrillization are toxic [10]. Oligomeric, but not fibrillar Aβ could lead to significant impairment of learning and memory [11], while smaller soluble Aβ oligomers lead to cognitive deficits in the absence of plaques [12]. Based on these studies, a strong inverse correlation between the size of Aβ assemblies and their toxicity has been established; as the oligomeric assembly size increases, its toxic effects decrease.
A progressive decline in cognitive functions appears in parallel with the progression of pathology. However, AD is not just a memory disorder; in addition to cognitive deficits there is a wide range of non-cognitive symptoms associated with AD pathogenesis: motor disabilities, anxiety, depression and increased frailty [13-15]. As non-cognitive disturbances also represent a huge practical and psychological burden on family members and caregivers, more emphasis should be placed on understanding and correcting these deficits, along with the cognitive aspects of the disease.
Despite considerable efforts, there are currently very limited efficient treatment options [16]. To this end, several animal models, especially mouse transgenic models, have been extremely useful to test the outcomes of numerous pharmacological interventions. For instance, 5xFAD transgenic mice recapitulate key features of Aβ pathology and provide valuable insight into the molecular and behavioral abnormalities in AD patients. Five mutations present in 5xFAD mice lead to robust intraneuronal aggregated amyloid at 1.5 months, extracellular amyloid deposition around 2 months of age and memory deficits starting at 4 months of age [17]. Several recent studies have highlighted some so far neglected behaviors other than cognitive ones, with a great potential to assess the extent or progression of neurodegeneration and to give a valuable insight about the efficacy of potential therapeutic agents. Comparatively, motor dysfunctions in 5xFAD mice have been extensively studied and successfully used for therapeutics assessment in several pre-clinical studies [18,19].
The accumulation of Aβ and tau makes AD a protein-misfolding disease, or proteopathy, and suggests that proteostasis perturbations may be directly or indirectly involved in the pathogenesis of the disease. The Ubiquitin Proteasome System (UPS) is the primary selective protein degradation mechanism and thereby plays a fundamental role in cellular physiology and the maintenance of proteostasis [20]. Interestingly, proteasome’s function gradually declines during aging, leading to increased levels of protein misfolding and aggregation, which in turn obstruct cellular function and contribute further to the progression of aging itself and of late on-set proteostasis disorders [21-23]. Indeed, many neurodegenerative diseases, including AD, are characterized by deposits of ubiquitinated and aberrant proteins [24]. In addition, there is clear genetic and biochemical evidence linking proteasome failure with the formation of protein aggregates that are involved in AD progression [25-27], while it is well established that Aβ and tau interact with and impair proteasome function [28-32]. Concomitantly, brain-specific proteasome inhibition faithfully mirrors the clinical hallmarks of neurodegenerative disorders [33-37]. Despite the pivotal role of the proteasome in aggregation-related pathologies, there are limited and not clinically applicable studies in mammals indicating that proteasome activation may reduce Aβ load [38]. Interestingly, a reversion of this phenotype has been observed in healthy centenarians as we have shown that these individuals exhibit a functional proteasome [39]. Given this remarkable observation, we have achieved to activate the proteasome, either genetically or by the use of natural compounds, and have observed a significant increase of lifespan in vitro and in vivo [22,40]. Furthermore, proteasome activation through β5 catalytic subunit overexpression [27] or with a natural proteasome activating compound [41], in a C. elegans model of Aβ-induced toxicity that mimics Alzheimer’s disease and results to paralysis, significantly delayed the paralysis phenotype and reduced total and oligomeric Aβ levels. However, these promising studies in lower eukaryotes have to be further tested in mammalian models and ultimately to humans.
Given these intriguing findings herein we have employed a combination of two compounds in the 5xFAD mouse model as an intervention to alleviate AD pathology. We report that the compound combination enhances the function of the proteasome machinery by different mechanisms and provokes long term beneficial multilevel organismal effects. Our findings thereby define proteasome activation as an important protective mechanism that can be pharmacologically manipulated to target Aβ aggregation and relieve symptoms associated with relevant proteostasis disorders.
2. Material and methods
2.1. Animals and treatments
5xFAD mice were used in this study (B6SJL-Tg (APPSwFlLon, PSEN1*M146L *L286V) 6799Vas) from The Jackson Laboratory, stock number: 34840-JAX/5XFAD [17]. The animals (3–4 mice per cage) were housed under standard conditions (23 ± 2 °C, 60–70% relative humidity, and 12 h light/dark cycles) with a free access to food and water. Two independent experiments were performed: short term (two months of treatment) and long-term (6 months of treatment). A total number of 17 animals was used (10 males and 7 females) for a long-term treatment and 14 animals for the short-term treatment (8 males, 6 females).
For both short term and long term treatment, animals were divided into two groups: 1. DMSO treated (i.e. c ontrol) animals. Although it has been shown that stress caused by handling and injection in experimental rodents disappear usually within two weeks [42] in order to exclude any possible side effects of stress caused by experimental procedure, we used control group injected with DMSO (8% DMSO solution in NaCl, given intraperitoneally every day for short term treatment and every second day for a long term treatment) and receiving water by oral gavage (100 μl/per animal).
2.18 alpha-glycyrrhetinic acid (18α-GA, Sigma) + omega-3 fatty acids (n-3FA, Dietpharm-Atlantic Group, Croatia) treated animals. 18α-GA was given intraperitoneally in a concentration of 10 μg/g of body weight in 8% DMSO solution. This concentration of 18α-GA induced exactly the same beneficial effects as the double dose (data not shown). Still, this concentration is significantly lower than the one of a stereoisomer (18β-GA) used in another study [43], and therefore excludes any harmful side effects during our long term treatment. Omega-3 fatty acids were given orally in a dosage (100 μl/per animal of the fish oil encapsulated, commercially available; fatty acid composition was given in the Supplementary Table 1) previously established as effective in 5xFAD mice memory [44,45]. In order to avoid degradation of DHA, one capsule containing 1 mL of fish oil was used for each 3–4 animals and was administered via oral gavage in no longer than 45 s. Treatment was given every day for the short treatment and every second day for the long one.
For the short-term experiment animals were sacrificed at the age of 4 months and the brain cortex and hippocampus were isolated for molecular analysis. For the long-term experiment a detailed behavioral analysis was performed at 4 and 8 months of age. At the age of 8 months, all the animals were sacrificed for molecular analysis. All animal procedures were in compliance with the Directive (2010/63/EU) on the protection of animals used for experimental and other scientific purposes and was approved by the Ethical Committee for the Use of Laboratory Animals (resolution No. 01–06/13) of the Institute for Biological Research “Sinisa Stankovic”, University of Belgrade. Minimal numbers of animals were used and all efforts were made to minimize animal suffering.
2.2. Behavioral tests
During the light phase (12h/12h light-dark cycle), mice were subjected blindly to a series of behavioral tests. First, the general motor activity was addressed following the open field (OF) test during 3 consecutive days. Then, the following analyses were conducted in sequential order: body weight measurement, grip strength and rope test for frailty assessment, followed by hind-limb clasping test.
2.3. Open field test
Motor activity and anxiety-like behavior of mice was recorded in Opto-Varimex cages (Columbus Instruments, OH). Data were analyzed using Auto-Track software (Columbus Instruments). Mice were allowed to freely explore the test arena for 30 min during the three consecutive days. Locomotor (distance traveled, DT) and vertical activity (V1B, i.e. rearing - standing on rear limbs; both free-standing and against the walls) was observed and habituation was determined. Habituation was characterized by a decrease in activity upon repeated exposure to the stimulus (activity cage). For detection of anxiety-like behavior, we used square analysis of the Auto-Track software to distinguish number of entries and time animal spent in the center of arena (4 quadrants in the center of arena, 11.5 × 10.8 cm) vs. periphery zone (12 quadrants in the periphery of the arena). Anxiety-like behavior was determined based on the first 5 min of testing.
2.4. Body weight
The mice body weight (g) was recorded individually at the beginning and regularly during the experiment. Changes in the body weight were analyzed in order to make a judgment about the effects of treatment, but also as a parameter for frailty score calculation.
2.5. Hind-limb clasping test
The clasping test (tail suspension test) for measuring functional impairments in 5xFAD mice was described by Jawhar and co-workers [46]. In mice, tail suspension test can also be used as a measure of depressive-like behaviors, i.e. behavior despair measured as a cessation of escape-oriented behaviors in response to an unpleasant and/or stressful situation [reviewed in [47, 48]]. DMSO- and 18αGA + n-3FA-treated mice were suspended from the tail for 1 min to provoke the clasping phenotype. Clasping behavior was scored in the following manner: 0 = no clasping, 1 = forepaws clasping, 2 = forepaws and one hindpaw clasping, 3 = all paws clasping.
3. Frailty analysis
3.1. Frailty criteria
For all assessments, mice were brought to the testing room prior to testing and allowed to habituate for 15 min. In accordance with phenotype frailty evaluation described by Fried and colleagues (2001), five frailty criteria are set up as appropriate for mice frailty assessment [49-51] and are presented in Table 1: body weight, physical activity, walking speed, grip strength (for endurance) and tight-rope test (for motor coordination). For physical activity, five parameters measured in the OF were taken into account (total distance traveled, DT; total duration of movement-ambulatory time, AT; percent of total time spent in moving, T/AT; average velocity of movement, DT/AT; rearing frequency i.e. vertical activity, V1B). All the frailty criteria are extensively described in 47).
Table 1.
Frailty Criteria for detecting frailty score in 5xFAD mice.
| Human Frailty Criteria | Mouse Frailty Criteria | Approach |
|---|---|---|
| Weakness/endurance | Grip strength | Grip test |
| Slowness | Walking speed | Velocity (cm/s) |
| Low activity | Physical activity | Open field test (cm/10 min) |
| Gait balance | Gait/motor coordination | Tight-Rope test |
| Unintentional weight loss | Unintentional weight loss | Weight (g) |
3.2. Phenotypic frailty assessment - frailty score
We established 20% as a cut-off point for all parameters used except for body weight that was set at 5%. For grip and rope tests we used simple evaluation, determining whether the animals passed the test or not, and based on 3 attempts in each test (passed-Y, failed-N). Then the frailty score for each experimental group (based on age and/or gender) was calculated as followed: total number of tests failed by the experimental group (A) divided by the total number of tests performed by the same group of animals (B) and expressed as a percentage, as described previously [52].
4. Molecular analysis
4.1. Preparation of tissues
Following 2 or 6 months of treatment, animals were anesthetized with ketamine 100 mg/kg, Ketamidor, Richter Pharma, Wels, Austria; 16 mg/kg Xylased, Bioveta, a.s.) in a dosages of 100 mg/kg. Mice were perfused with ice-cold Phosphate-buffered saline (PBS), brains harvested, and one hemi-brain drop fixed in 4% paraformaldehyde/PBS and cryo-preserved in graded sucrose solutions (10–30% w/v sucrose/PBS) for sectioning. The brains were frozen in isopentane, cooled on dry ice and stored at −80 °C until further use. Every coronal section (20 μm thick) was collected and stored at −20 °C.
4.2. Immunofluorescence
Tissue sections were blocked in 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 1h at RT, incubated overnight at 4 °C with primary anti-Aβ42 antibody (Invitrogen, cat No 700254, rabbit monoclonal, used at the 1:500 concentration in PBS), followed by secondary antibody (anti-rabbit conjugated to Alexa 568; Invitrogen) used at the 1:250 concentration in PBS for 2h at room temperature (RT). Sections incubated in parallel without primary antibody were included as negative controls for autofluorescence and background binding of the secondary antibody.
Isolated hippocampal neurons or MEFs were grown on coverslips and treated with 2 μg/ml 18α-GA and/or 3.3 μg/ml n-3 FAs or DMSO for 2h at 37oC then fixed in 2% paraformaldehyde followed by cell permeabilization with 0.2% Triton X-100 in PBS. Cells were incubated with anti-Nrf2 antibody (ab31163, abcam) at a 1:200 concentration at room temperature for 1h, followed by incubation with a rhodamine-conjugated secondary antibody (Santa Cruz Biotechnology). Slides were mounted using Anti-Fade with DAPI mounting Medium (ab188804, abcam) and analyzed using a Leica TCS SPE confocal laser scanning microscope (Leika Lasertechik). The LAS AF software was used for image acquisition.
4.3. Amyloid plaque staining
Thioflavin S (ThioS) staining was used for amyloid plaque labeling. Following protocol for Aβ42 immunostaining, sections were washed three times in PBS, and incubated in 0.01% ThioS solution in 50% ethanol for 8 min at RT. Stained sections were washed briefly in 80% and 96% ethanol, rinsed in distilled water and mounted onto glass slides using fluorescent mounting medium (Dako).
4.4. Quantitative analysis of Aβ42 clusters and ThioS labeled amyloid plaques
Images were captured on an Axio Observer Microscope Z1 using an AxioVision 4.6 software system (Carl Zeiss, Germany) at a 5 × magnification. For quantification of Aβ42 clusters, we used available software provided by the National Institute of Health (NIH, USA) - Image J, Version 1.74. We have designated a threshold of the fluorescence intensity. When setting the limit value for analysis we have opted for the automatic value (Maximum Entropy), instead of manual adjustment, in order to perform further analysis equally in all cross-sections and to reduce faults. Based on given parameters we produced values of the surface of Aβ42 clusters. Analysis of amyloid plaque number and % of area covered with ThioS staining was done using Image J with threshold processing (Max Entropy) and low surface area limit set to 50 μm2. The low surface area limit was set in order to exclude ThioS staining which corresponds to artifact. Based on given parameters we produced values of the surface of ThioS-positive amyloid plaque number and % of area. Values were further processed statistically.
4.5. Mouse embryonic cells isolation and culture
Hippocampal neurons were isolated from embryonic day 18 (E18) mouse embryos (C57B/6J, The Jackson Laboratory) using as previously described [53]. Briefly, hippocampi were removed under a dissecting microscope in CMF-HBSS (Calcium-, magnesium-free Hank’s balanced solution buffered with 10 mM HEPES, pH7.3; 14175, ThermoFischer Scientific). Whole hippocampi were incubated with 2.5% (wt/vol) trypsin (15090046, ThermoFischer Scientific) and DNase (10 mg/ml; EN0521, Sigma) for 20 min followed by inactivation of the enzymes with 10% FBS (ThermoFischer Scientific) for 5 min. Following three washing steps with CM-HBSS, hippocampi were dissociated into single neurons using a fire-polished sterile glass Pasteur pipette. Cells were plated at a density of 70,000 onto 10 mm round glass coverslips coated with 1 mg/ml polylysine solution (P9155, Sigma) or 12-well plates at a density of 500,000 cells per well, and were grown in medium containing Neurobasal (A3582901, ThermoFischer Scientific) and N21 (AR008, R&D Systems) to allow early and rapid development, and to ensure their long term survival. Neurons were used for experiments between divisions 5–7.
Primary mouse Embryonic Fibroblasts (MEFs) were isolated from C57BL/6J strain and cultured according to standard procedures [54]. Briefly, MEFs were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (v/v; Invitrogen), 2 mM glutamine and 1% non-essential amino-acids at 37oC, 5% CO2 and 95% humidity. MEFs were seeded at 100 mm plates.
Hippocampal neurons or MEFs were treated with DMSO (Sigma–Aldrich) as solvent control or 2 μg/ml 18α-GA (Sigma-Aldrich, ≥95% purity, diluted in DMSO) and/or 3.3 μg/ml n-3FA (EPA, eicosapentaenoic acid; and DHA, docosahexaenoic acid in a 2:1 ratio, Sigma-Aldrich, diluted with DMSO) for 2 or 24h, as indicated.
4.6. Real time PCR analysis
Cells were harvested after a 24h incubation period with the indicated compounds and total RNA was isolated using TRIzol (Invitrogen) and converted into cDNA with the cDNA iScript synthesis kit (Bio-Rad). The Real time PCR primer sequences are summarized in Supplementary Table 2. Quantitative RT-PCR was performed using the iQ SYBR Green Supermix (Biorad) and relative quantities of transcripts were determined using the relative standard curve method normalized to GAPDH with the iCycler iQ software Gene Expression MacroTMversion1.1 (Bio-Rad).
4.7. Immunoblot analysis
Twenty μg of protein from the cell or tissue extract were mixed with non-reducing Laemmli buffer and separated by SDS-PAGE (10% Mini-PROTEAN TGX Stain-Free precast Gels, Biorad) according to standard procedures [55]. Following gel electrophoresis, the total protein load was quantified using the StainfreeTM technology and the proteins were transferred to nitrocellulose membrane to be probed for β5 (X, MB1, Enzo Life Sciences; PW8895; 22.9 kDa). The secondary antibody (ab97051; abcam) was conjugated with horseradish peroxidase and detected the bound primary antibody by enhanced chemiluminescence using the Chemidoc XRS + imaging system. The levels of oxidized proteins were detected with the OxyBlot™ Protein Oxidation Detection Kit (S7150, Sigma-Aldrich) according to manufacturer’s specifications.
4.8. Proteasome peptidase assays
Harvested cells after a 2h incubation period with the indicated compounds or tissues were lysed in 25 mM Tris/HCl lysis buffer, pH 7,6 containing 5 mM ATP, 10% glycerol, 20 mM KCL, 1 mM EDTA, 1 mM DTT, 0.2% Nonidet P-40, 10 mM phenylmethylsulfonyl-fluoride and 10 μg/ml aprotinin [56]. CT-L, activity of the proteasome was determined after incubating 10 μg of total protein for 30 min at 37 °C with the fluorogenic peptide LLVY-AMC (Enzo Life Sciences), as previously described [57]. To test structural proteasome activation, 0.5 μg of pure 20S proteasome (Enzo) was incubated with 0.01% SDS (positive control) or 5μM MG132 (negative control) or increasing n-3 FA concentrations for 30 min at 37 °C with LLVY-AMC. Fluorescence was measured using a Tecan Safire II plate fluorescence spectrophotometer. In cells or tissue samples, proteasomal activity was determined as the difference between total activity and activity in the presence of 20 μM of the proteasome inhibitor MG132. Protein concentration was determined by Bradford Protein Assay (Biorad) using bovine serum albumin as standard.
4.9. Statistical analysis
Analysis of frailty score was performed by Pearson’s chi-squared test. Statistical analysis of habituation of locomotor and vertical activity was performed using RM One-way ANOVA, followed by Dunnett’s multiple-comparison post hoc tests. Analysis of locomotor, vertical activity and anxiety-like behavior was obtained using Mann-Whitney nonparametric test as well as analysis of body weight and clasping score. All data were expressed as mean ± SEM. Statistical analysis of RT-PCR and peptidase activity data was performed using GraphPad Software (San Diego, CA).
Significant differences for immunohistochemical data between the experimental groups were determined using STATISTICA software, Ver. 6.0, StatSoft. Further statistical analysis was made using relative values. Considering that data did not meet the criteria of a normal distribution, nonparametric Man-Whitney U test was used for comparisons among two experimental groups. Statistical significance was set at P < 0.05.
Densitometry analysis for the quantification of immunoblots was performed with Bio-Rad’s Image Lab software 6.0.1.
5. Results
Aiming to identify novel clinically translatable interventions to mitigate AD pathology, in a preliminary analysis we tested an array of compounds that have been associated with enhanced degradation of protein aggregates (data not shown). Based on this screening analysis and extensive follow-up studies in both AD-mice model and mammalian cell lines, we chose an optimal combination of two compounds, namely 18α-glycyrrhetinic acid (18α-GA) and omega-3 fatty acids (also called n-3 fatty acids, n-3FA) that, as our results demonstrate, activate protein clearance mechanisms in a complementary manner. In brief, in previous studies we have shown that 18α-GA is a potent proteasome activator that, moreover, confers lower paralysis rates in various AD nematode models, accompanied by decreased Aβ deposits [41,58]. Likewise, we have also demonstrated that short term fish-oil supplementation attenuates the AD pathology in 5xFAD mice brains when applied early [44,45]. As such, we treated 5xFAD mice with the compound combination (18α-GA + n-3FA) and performed an array of behavioral and molecular analyses during disease’s progression.
5.1. Behavioral analysis
The 5xFAD mice develop numerous non-cognitive deficits over time including slower speed, deficits in motor activity and motor coordination, abnormal reflexes like hind limb clasping, deficits in grip strength and significant weight loss. These impairments can be evaluated precisely by employing various behavioral tasks and have been used in several pre-clinical studies of potential AD therapeutics [18,19].
18α-GA + n-3FA-treated mice are characterized by higher general activity, reduced anxiety and preserved long-term memory.
The open field test is a very valuable and broadly used tool for evaluating locomotive impairment and efficacy of therapeutic drugs in animal models of various diseases. Therefore we used open field activity monitoring as a comprehensive assessment of the 18αGA+ 3-nFA effects on general motor activity, long term memory and anxiety level in the 5xFAD mice.
5.2. 18α-GA + n-3FA treatment increases locomotor and vertical activity in 5xFAD mice and preserves normal habituation
As shown in Fig. 1, the comparison of locomotor (DT) and vertical activity (V1B) during the 30 min testing period revealed that 18αGA+3-nFA treatment significantly affected locomotor and vertical performances of 5xFAD mice. Specifically, in the 4-month-old 18αGA + n-3FA-treated mice a significant increase in the locomotor activity, in comparison to the DMSO-treated counterparts was noticed (Fig. 1A) during all 3 days of testing (p = 0.0002, p = 0.0006 and p = 0.0294 respectively); in the older animals increased DT was detected only during the first testing day (p = 0.0401, Fig. 1B). Similar results were noticed in exploratory activity (Fig. 1C and D). Namely, significantly increased V1B values were detected in 4-month-old animals treated with 18αGA + n-3FA during all three days of testing, in comparison to their matching controls (1st day: p = 0.0002; 2nd day: p = 0.0001; 3rd day: 0.0003), while in the older group of mice statistically significant increase in the vertical behavior was detected on the first testing day (p = 0.05, Fig. 1D). These results point out that 18αGA + n-3FA treatment has a beneficial effect and elicits significantly higher motor and exploratory activity in 5xFAD mice, in comparison to the control groups.
Fig. 1.

18αGA + n-3FA treatment increases locomotor (A,B) and vertical (C,D) activities and preserves habituation in 5xFAD mice (E, F). Results are expressed as mean ± SEM for 30 min registration period during 3 consecutive days for locomotor and vertical activity (*p < 0.05; ***p < 0.0001) and as mean ± SEM for 5 min registration period during 3 consecutive days for habituation (*p < 0.05; **p < 0.001).
When placed in a novel environment of the open field, mice tend to explore it for a certain period of time and to reduce the level of exploration afterwards. This reduction in both locomotor and vertical activity is known as habituation; it can occur within a single session or across sessions (intrasession and intersession habituation respectively) and represents the simplest form of learning [reviewed in 59]. Therefore to examine the effects of the compounds on cognitive status of the animals, we analyzed intersession habituation in DT and V1B activity during 3 consecutive experimental days. Detailed analysis of the open field activity showed that only 4-month-old animals treated with 18αGA + n-3FA preserved normal capacity to habituate in both locomotor and vertical activity over time (Fig. 1E and F), while the older group of mice was incapable to habituate normally, regardless of treatments they received (data not shown). Using repeated measures ANOVA (RM-ANOVA), the significant effect of time on distance traveled by 4-month-old 18αGA + n-3FA-treated animals was noticed during the first 5 min of testing (p = 0.0398, F(1.568; 14.11) = 4.419). Post-hoc Dunett’s test pointed to statistically significant difference in the locomotor activity between 1st and 3rd day of testing (p = 0.0092, Fig. 1E, grey bars). The effect of time on vertical activity was also observed in this group (p = 0.0397, F(1.831; 18.31) = 3.3982), while post-hoc Dunett’s test indicates that on a 3rd day of testing 18αGA + n-3FA-treated animals showed significantly decreased V1B activity in comparison to the 1st day (p = 0.0148); i.e. animals decreased their V1B activity from day one to day 3 (Fig. 1F, grey bars). However, this protective effect has not been detected in the older group of mice (8-month-old) after 6 months of 18αGA + n-3FA treatment, as this group failed to habituate. This result indicates that treatment cannot preserve memory when substantial deterioration of cognitive capabilities occurs i.e. when significant memory impairments are already present (see Discussion). Similarly, there were no major variations in DT and V1B activity in DMSO treated mice over time and regardless of age (Fig. 1 E, F, black bars). As intersession habituation measures long-term memory of previous exposure [59], observed results indicate that 18α-GA + n-3FA substantially improved the long-term memory in 5xFAD mice, but up to certain age.
5.3. Decreased anxiety level of 5xFAD mice under the 18α-GA + n-3FA treatment
To further evaluate the potential favorable effects of 18αGA + n-3FA treatment in 5xFAD transgenic model of Alzheimer’s disease, we assessed anxiety-like behavior. When rodents are introduced into an open-field, they show a characteristic behavior called thigmotaxis (i.e. natural tendency to stay close to the walls and explore mainly the peripheral zone of the open field); this can be used as an index of anxiety in mice, especially in examining models of AD [60]. Analysis of characteristic animals’ pattern of movement (Fig. 2A) pointed to a significant difference in several parameters that could serve as indicators of the anxiety level, namely: the number of entries into central arena (inner zone; Fig. 2B), the time spent in the central arena (Fig. 2C) and the number of entries into periphery (outer zone) of the OF apparatus (Fig. 2D). No statistically significant difference was observed in the number of entries into the inner zone of the apparatus. However, the analysis showed that all the animals treated with 18α-GA + n-3FA spent significantly more time in exploring this zone (60% and 35%, p = 0.001 and p = 0.0367, for 4 and 8 months old animals respectively; Fig. 2C). Additionally, the older group of 18α-GA + n-3FA-treated mice entered the periphery significantly less than DMSO control mice, meaning that although they did not enter more often the central arena, they spent significantly more time there (p = 0.0289, for more than 50%, Fig. 2D). Although there are conflicting reports whether 5xFAD mice are hyperactive or have reduced anxiety level in parallel to pathology [61,62]. In summary, all the parameters analyzed herein indicated that the reduced anxiety level in animals treated with 18α-GA + n-3FA represents the beneficial outcome of this treatment.
Fig. 2. 18αGA + n-3FA treatment decreases anxiety of 4- and 8-month-old 5xFAD mice.

Graphical illustration of representative animal movements (A). Number of entries (B) and time spent in the inner (C) and outer zone (D) of the open field arena. Results are expressed as mean ± SEM. *p < 0.05; ***p < 0.0001.
5.4. 18α-GA + n-3FA treatment increases body weight and decreases clasping behavior in 5xFAD mice
In addition to impaired motor activity described previously, there are other deficiencies characteristic for AD that are often mirrored in transgenic animal models. Weight loss is common in Alzheimer’s disease patients and could furthermore serve as a marker of disease progression [63]. Similarly, the reduced body weight of 5xFAD in comparison with wild-type mice that has been reported previously [46,64], could be considered as a marker of the general health status and thus may increase prognostic accuracy. We detected that 5xFAD mice treated with 18α-GA + n-3FA weighed significantly more than DMSO treated counterparts at 4 months of age (p = 0.0253). That difference was even more profound at the later time point analyzed (p = 0.0033; Fig. 3A), indicating that 18α-GA + n-3FA possess a long-lasting beneficial effect on body weight.
Fig. 3.
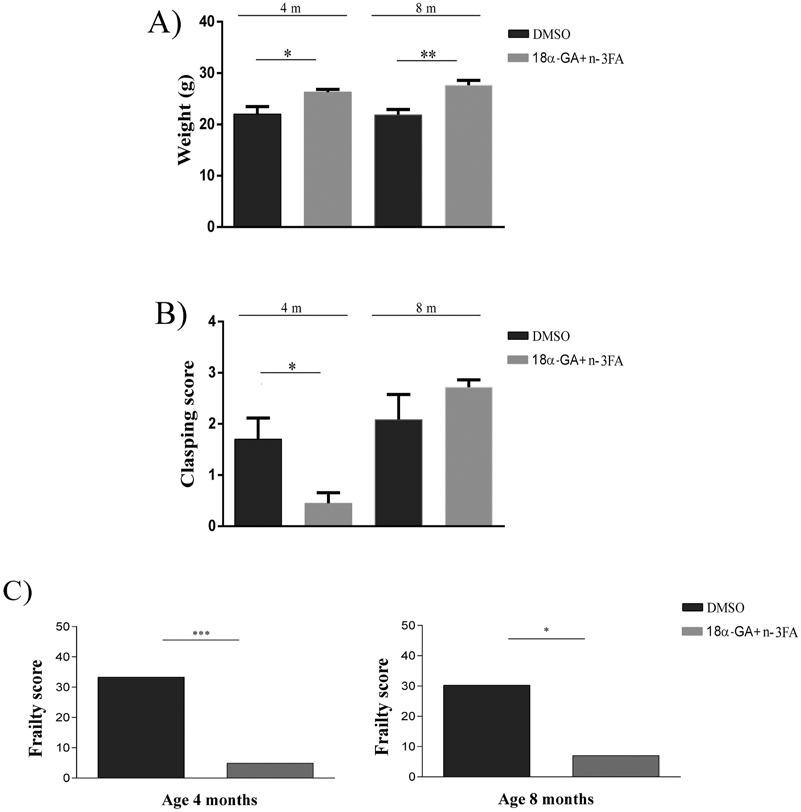
18α-GA + n-3FA treatment increases body weight (A) and decreases clasping behavior (B) and frailty level (C) in 5xFAD mice. Body weight of 4- and 8-month-old 5xFAD mice treated either with 18α-GA + n-3FA or DMSO (A). Clasping score of 5xFAD mice at 4 and 8 months of age and treated either with 18α-GA + n-3FA or DMSO (B). Graphical representation of frailty score (FS) in 4- and 8-month-old (C) 5xFAD mice treated with DMSO or 18αGA + n-3FA. Results are expressed as mean ± SEM. *p < 0.05; **p < 0.001; ***p < 0.0001.
Furthermore, 5xFAD mice develop abnormal reflexes during aging. In a tail-suspension test, higher frequencies and durations of hind-limb and fore-limb clasping can be noticed for 5xFAD mice as early as 5 months of age [64]. To measure the development of sensorimotor dysfunctions we performed hind-limb clasping tests at 4 and 8 months of age. Results are presented in Fig. 3B. When suspended by the tail, at 4-month-old 5xFAD mice treated with DMSO showed the hind-limb clasping response significantly more often than the mice treated with 18α-GA + n-3FA (Mann-Whitney, p = 0.0386). However, there were no significant differences in clasping scores between the two groups at end of the study (8 months of age).
5.5. Frailty level
Frailty level is significantly decreased under the 18α-GA + n-3FA treatment in the transgenic model of AD.
As analysis of animals’ behavior in the open field showed significantly beneficial effects of 18α-GA + n-3FA treatment on motor and cognitive abilities as well as on anxiety level, we investigated further the 18α-GA + n-3FA effects on general welfare. To this end an 8-item frailty score (FS) was calculated in order to assess the impact of 18α-GA + n-3FA treatment on general health and wellbeing of 5xFAD mice during aging (Fig. 3C). Table 2 shows the number of 5xFAD mice of two different ages exposed either to DMSO or 18α-GA + n-3FA treatment that ranked in the lowest 20% for each frailty criteria, the number of tests performed and the number tests failed. Values for 5% for body weight parameter and for 20% cut-off for other frailty parameters used to calculate frailty score are presented in Table 3. This analysis revealed that the total number of mice treated with 18α-GA + n-3FA and failed the tests was significant lower than DMSO- treated mice, regardless of the age. This resulted in the significantly lower number of frail mice in the 18α-GA + n-3FA treated groups (Table 2). These data suggest that both short and long term 18α-GA + n-3FA treatment reduce frailty level for more than 80% (p = 0.0003 and p = 0.0165 in 4- and 8-month-old animals respectively). Taken together, these results demonstrate a positive effect of 18α-GA + n-3FA treatment on frailty level in the 5xFAD mouse model.
Table 2.
Number of animals in the lowest 20% for each parameter and 5% for body weight, used to calculate frailty score. The frailty score for each experimental group was calculated as follows: total number of tests failed by the animals at each experimental group divided by the total number of tests performed by those animals, expressed in percentage.
| Age |
4-month-old |
8-month-old |
|||
|---|---|---|---|---|---|
| DMSO | 18αGA + n-3FA |
DMSO | 18αGA + n3FA |
||
| Number of animals in the lowest 20% | Distance traveled (cm) | 4 | 0 | 2 | 0 |
| Vertical activity (beams) | 3 | 1 | 2 | 1 | |
| Velocity (cm/sec) | 2 | 2 | 1 | 2 | |
| Ambulatory time (sec) | 4 | 0 | 3 | 0 | |
| Total time spent in moving (%) | 4 | 0 | 3 | 0 | |
| Grip test | 1 | 1 | 0 | 2 | |
| Rope test | 1 | 0 | 0 | 2 | |
| Weight in the lowest 5% (g) | 1 | 0 | 2 | 0 | |
| Total number of animals that failed the tests (A) | 20 | 4 | 13 | 4 | |
| Total number of tests performed (B) | 60 | 82 | 43 | 57 | |
| (A/B) | 0.333 | 0.0488 | 0.302 | 0.071 | |
| Frailty score (A/Bx100) | 33.333 | 4.878 | 30.230 | 7.010 | |
Table 3.
Values for 20% cut-off for each parameter and 5% for body weight for 4- and 8-month-old 5xFAD mice treated with DMSO and 18αGA + n-3FA.
| |
20% cut-off and 5% for body weight |
|
|---|---|---|
| Parameter |
|
|
| 4-month-old | 8-month-old | |
| Distance traveled (cm) | 1694 | 651 |
| Vertical activity (beams) | 14 | 1 |
| Velocity (cm/sec) | 9.882 | 9.017 |
| Ambulatory time (sec) | 157 | 60 |
| Total time spent moving (%) | 0.26 | 0.1 |
| Weight (g) | 16 | 19 |
6. Molecular analyses
6.1. 18α-GA + n-3FA combination decreases Aβ42 coverage in the parietal cortex and hippocampus
Given the previous findings as well as the causative links between the accumulation of amyloid beta and the progressive decline in cognitive and non-cognitive functions, next we assessed whether 18α-GA + n-3FA treatment provokes beneficial changes in the number of parameters analyzed above, by changing the amount of amyloid β (Aβ42), as well as the number of amyloid β plaques. Parietal cortex and hippocampus have been analyzed as the structures most important in AD [65]. Mann-Whitney U test of data obtained by immunohistochemical analysis revealed that short term treatment (2 months) has no effects on both the number of plaques and the surface of cortex and hippocampus covered with amyloid β (data not shown). However, immunohistochemical analysis of the effects of the long-term 18α-GA + n-3FA treatment showed a significant decrease of the total percentage of Aβ42 coverage in both parietal cortex (37.7%) and hippocampus (39.2%) of the 8 months old mice (Fig. 4). The treatment, however, did not affect the number of amyloid plaques in any structure analyzed (data not shown). These results suggest that 18α-GA + n-3FA treatment has a strong impact on Aβ42 content in both parietal cortex and hippocampus of 5xFAD mice, while it does not affect plaques’ number.
Fig. 4.

Long-term 18αGA + n-3FA treatment significantly reduces Aβ42 in the cortex and hippocampus of 5xFAD mice. Representative images of Aβ42 immunostaining in cortex and hippocampus of DMSO (left panel) and 18αGA + n-3FA treated (right panel) 5xFAD mice (A). Quantification of Aβ42 coverage of cortex (Ctx) and hippocampus (Hpp) of 18αGA + n-3FA - vs. DMSO-treated 5xFAD mice (B). Results are expressed as mean ± SEM.*p < 0.05; **p < 0.001.
6.2. 18α-GA + n-3FA induce proteasome activity in brain regions of treated mice
The data reported so far suggest that 18α-GA + n-3FA treatment has beneficial effects on various physiological and behavioral parameters that associate with AD. Next, we focused our efforts to understand the biochemical function of these two compounds. As enhanced proteasome activity associates positively with longevity and mainly the elimination of protein aggregates that accumulate in various neurodegenerative diseases including AD [26,27,66,67], first we examined the compounds’ effects on the proteasome. In pilot experiments we have found that both compounds activate the proteasome separately although we observed a much stronger effect when they were administered in combination (data not shown). As shown in Fig. 5, 18α-GA + n-3FA activated the proteasome in different brain regions of treated animals. Specifically, the treatment significantly enhanced the chymotrypsin-like (CT-L) proteasome activity in 4-month-old animals in both cortex and hippocampus. Notably, long term exposure to the compounds partially rescued the age-related decline of proteasome function in the aged brain cortex and hippocampus of 8-month-old animals. In addition, as shown in Supplementary Fig. 1, the compound combination attenuated the age-related elevation of oxidized protein levels in the cortex of 8-month-old treated mice. In summary, these data demonstrate that use of the two compounds enhances proteasome activity in brain regions of 5xFAD mice.
Fig. 5.

18α-GA and n-3 fatty acid combinational treatment enhances proteasome activity in cortex and hippocampus of 5xFAD mice. Percentage (%) of CT-L proteasome activity in A) cortex (ctx) and B) hippocampus (hpp) of young (4 months of age) and old (8 months of age) 5xFAD animals treated with 18α-GA + n-3FA or DMSO (control). The mean value of each activity in young DMSO-treated animals was arbitrarily set to 100%. *p < 0.05.
6.3. 18α-GA and n-3FA induce the proteasome via distinct mechanisms to confer protection against AD pathology
To gain insight into the mechanism of action of the tested compounds we further studied their effects on proteasome regulation. For these studies primary hippocampal neuron cultures were employed. As shown in Fig. 6, the compound combination was more effective in increasing the CT-L activity of the proteasome, than each compound alone. Specifically, 18α-GA (2 μg/ml) or n-3FA (3.3 μg/ml) induced CT-L activity by 25.5% and 41.6% respectively in treated cells, while the combination of the two compounds increased proteasome activity by 78.5%, indicating a synergistic effect (Fig. 6A, i). As proteasome can be activated by different modes of function [68], next we examined the compounds’ effects on the transcriptional activation of its subunits. Treatment with 18α-GA and 18α-GA + n-3FA but not with n-3FA alone enhanced the protein levels of the catalytic β5 proteasome subunit to similar rates (Fig. 6A, ii). As the expression of β5 is directly regulated by Nrf2 [69], we examined the impact of the compounds on the activity of this transcription factor. We found that upon treatment with 18α-GA, Nrf2 rapidly translocates to the nucleus, while no amount of nuclear Nrf2 was detected in n-3FA or DMSO treated cells. High levels of Nrf2 were also detected in the nucleus of neurons exposed to 18αGA + n-3FA (Fig. 6B). To further confirm these findings, the expression of 2 well-known Nrf2 target genes SOD1 and NQO-1 [70], was examined following treatment with 18α-GA. As it is shown in Fig. 6C, SOD1 and NQO-1 were significantly induced in 18α-GA treated cultures, in comparison to their control counterparts. The cross tissue consistency of these results was further verified in MEFs (Supplementary Fig. 2). Thus, treatment with 18α-GA activates Nrf2, which in turn induces the expression of proteasome subunits and of other antioxidant target genes.
Fig. 6.

18α-GA and n-3 fatty acids activate the proteasome via distinct mechanisms. A) i) % CT-L proteasome activity, ii) immunoblot analysis of the catalytic β5 proteasome subunit and B) immunofluorescence imaging of Nrf2 intracellular localization, in primary hippocampal neurons treated with the solvent (DMSO), 2 μg/ml of 18α-GA or/and 3.3 μg/ml of n-3 FAs for A) 16 h or B) 2 h. Total protein load was determined using the Stainfree™ technology. 4′,6-Diamidino-2-phenylindole (DAPI) was used as a nuclear counterstain. C) Real-time PCR analysis of SOD1 and NQO-1 mRNA expression in primary hippocampal neurons treated with 2 μg/ml 18α-GA or DMSO for 16 h. GAPDH levels were used as loading control. D) Manifold of CT-L activity of pure 20S proteasome exposed to the indicated concentrations of n-3 FAs. 0.01% SDS was used as a positive control, while 5 μM MG132 was used as a negative control. Proteasome activity or mRNA and protein levels in DMSO-treated cells were arbitrary set to 100%. *p < 0.05; **p < 0.01, and ***p < 0.001, ****p < 0.0001 compared to solvent control.
Besides transcriptional activation, some compounds, like SDS, are known to enhance proteasome function by allowing the gate opening of 20S particles [71]. Therefore we sought to determine if n-3FA can structurally activate proteasome complexes. To this end, we exposed pure 20S proteasome particles to increasing concentrations of n-3FA and measured their CT-L activities. As shown in Fig. 6D, we have observed a concentration dependent increase of CT-L proteasome activity reaching to a maximum increase of 300% for 0.2 μg/ml treatment. To further verify these intriguing data, the proteasome inhibitor MG132 was able to completely abolish the enhancement of CT-L activity by n-3FA (see Fig. 6D, second bar). Collectively, these findings demonstrate that the combination of n-3FA and 18α-GA deliver a dual activation of the proteasome, by inducing conformational alterations and by enhancing the transcription of proteasome subunits, respectively.
7. Discussion
Alzheimer’s disease is a devastating form of dementia accounting up to 60–70% of all dementia cases (source: WHO). AD, as a gain-of-toxic-function disease, is characterized by an aggregation-mediated proteotoxicity that exceeds the capacity of the cellular clearance machineries. Thus, the activation of protein degradation pathways could function as a preventive or therapeutic intervention against the disease. In the present study we have shown that 18α-GA + n-3FA treatment has significant beneficial effects on body weight, motor function, cognitive abilities, anxiety, and frailty level in the 5xFAD Alzheimer mouse model. Moreover, the compound combination decreases Aβ42 coverage in both the parietal cortex and hippocampus.
The treatment was performed during the time that corresponds to the long preclinical phase of AD that has been considered as the critical time window for potential intervention. Two compounds were selected after comprehensive assessment of an array of compounds previously investigated in our laboratories and proven to be associated with enhanced degradation of protein aggregates and attenuated AD pathology. After the initial screening (data not shown), we detected a possible synergistic effect of 18α-GA and n-3FA on proteasome activation and therefore we performed a follow-up study in the 5xFAD AD-mice model. To demonstrate beneficial effects of the two selected compounds we implemented an extensive evaluation predominantly of non-cognitive AD symptoms present in 5xFAD mice as these deficits could predict future cognitive deterioration [72]. Deficits have been selected to mimic clinical expression of motor impairments in humans [60,73,74].
Shorter distance traveled, decreased rearing frequency, slower movement speed and lower percent of time spent in moving are evidenced in 5xFAD mice [19,62,75]. Anxiety is another common symptom present in AD patients; it is associated with amyloid beta burden and could be a valuable indicator for both disease progression and the efficacy of therapeutics in early AD stages [76,77]. An increased locomotor activity documented by increased DT, higher speed and an increased vertical activity, i.e. exploration of a novel environment, support the improved motor phenotype observed in animals treated with 18αGA + n-3FA. In addition, 18α-GA + n-3FA treatment significantly reduced anxiety-like behavior.
A reduced body weight of 5xFAD, compared with wild-type mice, has been reported already [46,64] resembling the weight loss observed in AD patients [78], while abnormal reflexes, especially clasping phenotype in a tail-suspension test [46] increases in parallel with a progression of AD-related pathology [48, reviewed in 62]. Increased body weight and improved motor coordination demonstrated herein, could be considered as markers of improved phenotype in 5xFAD mice treated with 18α-GA + n-3FA. The increased weight of 5xFAD mice treated with 18αGA + n-3FA could be explained by an array of pleiotropic beneficial effects of n-3 fatty acids, including their positive role in maintaining metabolic homeostasis of the body. Namely, an improved metabolic profile has been previously shown to be associated with the intake of a diet enriched in n-3 PUFA fatty acids [79,80]. In addition, it has been reported that n-3FA supplementation is capable to revert altered insulin, leptin and triglyceride levels to normal [81]. On the other hand, while there are no data about metabolic effects of 18α-GA, there are studies showing that 18β-GA, a stereoisomeric form of 18α-GA, may exert beneficial metabolic effects, via mechanisms other than proteasome activation [82]. Thus, it is quite probable that in this case and in addition to their role in proteasome activation, n-3FA and 18α-GA reverted the neurodegenerative process by modulating cellular metabolic pathways and changing metabolic homeostasis.
It is worth noting that inconsistent changes in the open field activity have been reported for some transgenic Alzheimer’s mouse models [reviewed in [48, 62, 83 [48, 62, 83]], including few reports of increased exploratory behavior and reduced anxiety as signs of an aggravation of AD-related symptoms in 5xFAD model [46,84,85]. However, in these reports abnormal locomotor activity and anxiety have been detected at later time points than 8 months analyzed herein. In addition, in the previous reports reduced anxiety was documented by the increase of the number of entries into the open arms of elevated plus maze without an increase in the time spent in exploring them, indicating behavioral disinhibition. In contrast, the 18α-GA + n-3FA -treated mice did not enter central arena of the OF more frequently than control animals, but once there, they explored it extensively over time. It should be also noticed that in several AD models with abnormal levels of anxiety, increased activity level persists even after allowing the mice to habituate to an environment [48]. Additional analysis of the time spent in center of the OF through the 3 consecutive days of testing (data not shown) documented that there was a decrease in the percent of time spent in the central OF zone for mice treated with 18α-GA + n-3FA, i.e. mice habituated over time to this zone. This result is in accordance with the habituation of locomotor and vertical activity, observed also in 18α-GA + n-3FA-treated mice. Taken these data together, the effects of treatment on anxiety level and locomotor activity cannot be taken as a deviation, but as an improvement of AD motor symptoms.
Probably the most profound finding of this work is the decreased frailty level detected in mice treated with 18α-GA + n-3FA. Physical frailty, a multidimensional syndrome often referred as a state of high vulnerability, is associated with both the level of cognition and dementia [86,87] and a very important clinical implications [88]. A more rapid rate of increasing frailty is related to a more rapid loss of cognitive functions and low response to therapy [89,90]. Frailty level correlates with AD biomarkers [91] and the higher probability for mild cognitive impairment (MCI) to progress to AD [92,93]. We have recently suggested that frailty status could be a useful marker in preclinical assessment of novel AD therapeutics [52]. Herein we have shown a significant attenuating effect of our compounds on frailty level during aging. As previous studies have shown that transgenic animals reach a state of high vulnerability and high frailty earlier than non-transgenic counterparts [52,94], it is not surprising that both 4- and 8-month-old animals were characterized by the same level of frailty. The effect of our treatment was more pronounced in younger animals with a less advantage stage of pathology, indicating that this treatment could give the best results at early stages of diseases.
The accumulation of aggregated and oxidized proteins is a hallmark of many neurodegenerative diseases, including AD. Several lines of evidence suggest that this proteostasis disturbance may result from an age-related dysfunction of the UPS [26,27,66,67]. Indeed, proteasome activity was found to be reduced by more than 50% in both cortex and hippocampus of 8-months old 5xFAD animals in comparison to younger, 4-months old animals. As Aβ is degraded via the proteasome [25], it is tempting to speculate that the age-dependent decrease in proteasome activity may lead to its accumulation. In support, loss-of-function experiments have shown that decreased proteasomal activity promotes the neurodegenerative phenotype [95,96]. Proteasome activities [97,98] are significantly decreased in brain tissues of AD patients, further suggesting that UPS failure is related to AD pathogenesis. Taken together, these findings indicate that the age-related demise of proteasome functionality is an important determinant of AD progression.
Our data indicate that treatment with the 18α-GA + n-3FA combination significantly enhances the CT-L proteasome activity in both cortex and hippocampus of 4-months old animals and partially rescues its age-related decline in the same brain regions of 8-months old animals. This enhancement of proteasome function positively correlates with an array of beneficial effects on animals’ physiology and a reduction of amyloid beta coverage in the cortex and hippocampus. Unlike glia and other mitotic cells, neurons cannot discard damaged proteins by cell division [99]. Hence, neuronal proteasomes play a crucial role in abnormal protein clearance [100]. Distinct rodent brain regions exhibit different basal levels of proteasome activity that reflect their diverse proteome requirements [100,101]. Studies have demonstrated that the age-dependent decrease in UPS activity is more pronounced in neurons than glial cells [101]. We found an increased maintenance of proteasome activity in cortex of old 18α-GA + n-3FAs treated animals, in comparison to hippocampus, where the effect was attenuated. As cortex has a significantly higher neuron density than hippocampus [102], the preservation of proteasome activity in cortex has broad implications in the observed alleviation of the Alzheimer’s-associated pathological phenotype. Importantly, both cortex and hippocampus are the most affected brain structures in AD. There are clear hints that AD pathogenesis may also be non-cell autonomous and evidence suggests that microglial dysfunction is a primary event in sporadic AD [103]. In the 5xFAD mice, the subiculum and layer V of the cortex are the first areas where extracellular amyloid depositions appear, starting at around 2 months of age and spreading rapidly throughout the hippocampus and cortex by six months [17]. Apart from plaques, the spatiotemporal pattern of neuronal and synapse loss in this strain again points to the cortex and hippocampus as the most affected structures [104]. At the same time, previous investigations indicated that both proteasome composition and function in the brain could be affected in a specific spatial pattern by trauma [105] and ischemia [106], while UPS is either impaired or functionally insufficient in the cortex and hippocampus of the transgenic AD/GFPu mice model [107]. Furthermore, this selective impairment of the UPS was accompanied by the selective accumulation of ubiquitinated proteins in the cortex and hippocampus of AD/GFPu mice, with cortex being more vulnerable and affected than hippocampus. More importantly, it seems that UPS impairment occurs long before the formation of beta-amyloid plaques [107], suggesting that indeed this could be the first step in AD pathogenesis. However, once Aβ aggregates it further inhibits proteasome activity [25]. This might also explain why proteasome activation in 8-months old treated animals, where Aβ plaque formation is evident, is not as robust as of that displayed in younger animals.
Studies in invertebrates demonstrate that the overexpression of the β5 catalytic 20S proteasome subunit [27], or treatment with 18a-GA [41] induce the proteasome in a C. elegans AD model and confer protection against proteotoxic stress. Moreover, other groups have shown the beneficial effects of proteasome activation in the prevention of Aβ aggregation by several compounds [108-110]. However, all these preliminary studies have been performed in lower eukaryote models of AD, or cellular systems and therefore their importance remains to be further evaluated. The current study in a mammalian in vivo model system strengthens the link between the increased proteasome-mediated protein clearance and the reduced manifestation of AD pathology in several levels. It has been previously shown that proteasome stimulation reduces Aβ levels in an AD mouse model, yet the stereotaxic injection an Agouti-related peptide (AgRP) that was employed [38] is hardly applicable in AD patients. Moreover, in another AD mouse model, a proteasomal involvement in the Methylene Blue (MB)-mediated decrease in the soluble Aβ levels and the associated cognitive deficits was suggested, but the exact mechanisms of MB effects remain elusive [111]. Hence, it is noteworthy that our study not only provides a clinically applicable approach to alleviate AD pathology, but also describes in detail the mechanisms of proteasome activation by the tested compounds.
We demonstrate that the two compounds used in this study induce the proteasome activity by diverse mechanisms. 18α-GA increases the transcription of proteasome subunits via the activation of the transcription factor Nrf2, while n-3FA structurally activates the proteasome complex. To this end we isolated primary hippocampal neurons as well as MEFs from the congenic C57BL/6J strain and conducted the molecular and biochemical analyses. Specifically, 18α-GA induces the transcriptional up-regulation of proteasome subunits via Nuclear factor E2-related factor 2 (Nrf2), a well-known regulator of the Phase II antioxidant response and putative target against neurodegenerative diseases [112]. Our previous work has shown that treatment of human fibroblasts with 18α-GA up-regulates Nrf2 by reducing the levels of its inhibitor Keap-1, allowing Nrf2 to enter the nucleus and activate the expression of several proteasome subunits by binding to the AREs located on their proximal promoter regions [58]. This response to 18α-GA is also conserved on the nematode C. elegans, as treatment with the compound activates the proteasome, prolongs lifespan and reduces proteotoxicity in a SKN-1 (the Nrf2 ortholog) dependent manner [41]. In support, the present study demonstrates that upon treatment with 18α-GA, Nrf2 rapidly translocates to the nucleus to coordinate the activation of a multitude of cytoprotective genes, as confirmed by the up-regulated mRNA levels of its target genes.
The other compound used, n-3 fatty acids, has a different mode of function. Previous studies have shown that the polyunsaturated fatty acids (PUFAs) of omega-3 (n-3) and omega-6 (n-6) are essential both for brain development and brain functioning in the adult stage [reviewed in [113, 114]]. Specifically, it has been demonstrated that n-3 fatty acid supplementation is able to improve AD-associated brain pathology in several transgenic mouse models of AD, including 5xFAD mice, to reduce amyloid plaque burden and to improve learning and memory [44,45,115-117]. The majority of the beneficial properties of PUFAs have been attributed to their reported neuroprotective, anti-inflammatory, and antioxidant effects [118] and specifically to their capability to change the composition of cell membranes, replacing cholesterol and promoting the non-amyloidogenic pathway of APP processing, thus decreasing the Aβ burden. Herein, we identify a previously unknown effect of n-3FAs on cellular proteostasis, namely through the conformational activation of the proteasome. Sodium dodecyl sulfate (at concentrations ranging between 0.01 and 0.08% SDS) is the first identified structural proteasome activator that acts via partial denaturation of the latent 20S core and is characterized by inhibition at concentrations greater than 0.08% [119]. By analyzing the catalytic activity of pure 20S proteasomes under the exposure of a wide range of n-3FA concentrations, we revealed an SDS-like potential of n-3FA to structurally activate the proteasome through allosteric interactions in a concentration dependent manner. Collectively, we demonstrate here for the first time the use of two different proteasome activators that act in a complementary manner: 18α-GA enhances proteasome content and activity via the transcriptional up-regulation of Nrf2, while n-3FA promotes the conformational activation of the proteasome.
Regardless of this complementary mode of action focused on the activation of proteasome, we cannot exclude that the applied treatment exerted pleiotropic beneficial effects on the 5xFAD model, by employing additional mechanisms. Both compounds have numerous already proven favorable effects. In this context, the compound combination rescues the age-related accumulation of oxidized proteins in the cortex, as indicated in Supplementary Fig. 1. The improved metabolic balance induced by fish oil supplementation, already mentioned above, or the alteration of the phospholipid profile in 5xFAD brains, by increasing omega- 3 and decreasing omega-6 fatty acid content, as demonstrated previously [44,45], are two putative modes of action. In addition, PUFA role in maintaining brain health could be mediated by their anti-inflammatory actions [120]. Moreover, 18α-GA (and its stereoisomeric form 18β-glycyrrhetinic acid) has established anti-inflammatory and anti-tumor properties as well as it suppresses dyslipidemia and prevents oxidative stress and histological damage [43,121-123]. Therefore it is plausible that both compounds to provoke, in addition to proteasome activation, few other protective mechanisms that might be act differently over experimental time and in a complex interplay, to result the observed behavioral and molecular improvements. Besides, the proteasome is responsible for the targeted, regulated degradation of a multitude of factors, and as such a functional proteasome is required for almost all protective adaptations to stress, as it facilitates the cycling of factors that would otherwise attenuate the response [124]. Finally, apart from the possibility that more than one molecular mechanism is induced by the administration of two compounds, it is also likely that other structures, in addition to cortex and hippocampus, were affected too. While improved motor performances and preserved habituation testified in favor of improved cortical and hippocampal function, the reported behavioral results indicate that additional brain regions were benefited. For instance, basolateral amygdaloid complex which is responsible for the open-field arena-induced anxiety [125] could be tackled herein. Moreover, improved paw-clasping behavior detected at 4 months of age indicates that cerebello-cortico-reticular and cortico-striato-pallido-reticular pathways, included in this response [126] were also affected by the treatment.
In conclusion, our study identifies a combination of two natural compounds as an intervention that endows a mammalian model with an elevated proteolytic capacity to ameliorate the symptoms of AD. To our knowledge this is the first report to demonstrate a dual activation of the proteasome and its downstream effects on the symptoms of AD. Sadly, there is still no treatment to cure Alzheimer’s disease; while interventions that may help to treat symptoms are mostly ineffective in the advanced stages of the disease, when complications from severe loss of brain function could result even in death. For those reasons, it is essential to establish a preventive approach that can be applied well before the first signs of AD appear, or even in the cases that it is not clinically evident if AD will be developed at all. Therefore, an early presymptomatic phase of AD, which usually overlaps with the middle age period of human lifespan, represents a potential candidate time point to apply natural compounds that could prevent or delay the neurodegenerative process. Treatment with omega-3 fatty acids is already considered to be safe, efficient and acceptable approach in the management of neurodegenerative diseases in their early stages [116], while 18α-GA is also safe up to a daily dosage of 100 mg in humans [127]. These compounds can be readily incorporated into human dietary interventions and hence offer a unique opportunity for the prevention or alleviation of relevant neurodegenerative diseases. As human life expectancy continues to steadily increase, it is of great importance to prevent the increasingly prevalent age-related diseases, such as AD [128]. Consequently, future studies should focus on the application of such dietary interventions in clinical settings.
Supplementary Material
Acknowledgments
This work was supported by the EU COST Actions CA15214 & BM1402, Fogarty International Research Award, NIH (R03AG046216), and by Ministry of Education, Science and Technological Development of the Republic of Serbia, contract No 451-03-68/2020-14/200007.
Abbreviations:
- 18α-GA
18 alpha-glycyrrhetinic acid
- AD
Alzheimer’s disease
- AgRP
Agouti-related peptide
- APP
amyloid precursor protein
- AT
total duration of movement-ambulatory time
- Aβ
amyloid-β
- BSA
bovine serum albumin
- CT-L
chymotrypsin-like
- CTX
Cortex
- DHA
docosahexaenoic acid
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
Dimethyl sulfoxide
- DT/AT
average velocity of movement
- DT
total distance traveled
- EPA
eicosapentaenoic acid
- FA
Fatty acid
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- Hpp
Hippocampus
- MB
Methylene Blue
- MEFs
Mouse Embryonic Fibroblasts
- n-3
omega-3
- n-6
omega-6
- NIH
National Institute of Health
- NQO-1
NAD(P)H dehydrogenase [quinone]
- Nrf2
Nuclear factor E2-related factor 2
- OF
open field
- PBS
phosphate-buffered saline
- PUFAs
polyunsaturated fatty acids
- RM-ANOVA
repeated measures ANOVA
- SDS
Sodium Dodecyl Sulfate
- SOD1
superoxide dismutase 1
- T/AT
percent of total time spent in moving
- ThioS
Thioflavin S
- TRX
Thioredoxin
- UPS
Ubiquitin Proteasome System
- V1B
rearing frequency i.e. vertical activity
- WHO
World Health Organisation.
Footnotes
Declaration of competing interest
The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2020.11.038.
References
- [1].Hardy JA, Higgins GA, Alzheimer’s disease: the amyloid cascade hypothesis, Science 256 (5054) (1992) 184–185, 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- [2].Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C, The role of APP processing and trafficking pathways in the formation of amyloid beta-protein, Ann. N. Y. Acad. Sci 777 (1996) 57–64, 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- [3].Yankner BA, Lu T, Amyloid beta-protein toxicity and the pathogenesis of Alzheimer disease, J. Biol. Chem 284 (8) (2009) 4755–4759, 10.1074/jbc.R800018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ, Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model, Cell 149 (3) (2012) 708–721, 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hardy J, Allsop D, Amyloid deposition as the central event in the aetiology of Alzheimer’s disease, Trends Pharmacol. Sci 12 (10) (1991) 383–388, 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- [6].Golde TE, Dickson D, Hutton M, Filling the gaps in the abeta cascade hypothesis of Alzheimer’s disease, Curr. Alzheimer Res 3 (5) (2006) 421–430, 10.2174/156720506779025189. [DOI] [PubMed] [Google Scholar]
- [7].Small SA, Simoes-Spassov S, Mayeux R, Petsko GA, Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer’s disease, Trends Neurosci. 40 (10) (2017) 592–602, 10.1016/j.tins.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Thal DR, Walter J, Saido TC, Fändrich M, Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease, Acta Neuropathol. 129 (2) (2015) 167–182, 10.1007/s00401-014-1375-y. [DOI] [PubMed] [Google Scholar]
- [9].Wang ZX, Tan L, Liu J, Yu JT, The essential role of soluble Aβ oligomers in Alzheimer’s disease, Mol. Neurobiol 53 (3) (2016) 1905–1924, 10.1007/s12035-015-9143-0. [DOI] [PubMed] [Google Scholar]
- [10].Verma M, Vats A, Taneja V, Toxic species in amyloid disorders: oligomers or mature fibrils, Ann. Indian Acad. Neurol 18 (2) (2015) 138–145, 10.4103/0972-2327.144284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].He Y, Zheng MM, Ma Y, Han XJ, Ma XQ, Qu CQ, Du YF, Soluble oligomers and fibrillar species of amyloid β-peptide differentially affect cognitive functions and hippocampal inflammatory response, Biochem. Biophys. Res. Commun 429 (3–4) (2012) 125–130, 10.1016/j.bbrc.2012.10.129. [DOI] [PubMed] [Google Scholar]
- [12].Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, Levey AI, Krafft GA, Levy E, Cheder F, Glabe C, Bilker WB, Abel T, Schmeidler J, Ehrlich ME, Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers, Ann. Neurol 68 (2) (2010) 220–230, 10.1002/ana.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lobo A, López-Antón R, de-la-Cámara C, Quintanilla MA, Campayo A, Saz P, Non-cognitive psychopathological symptoms associated with incident mild cognitive impairment and dementia, Alzheimer’s type, Neurotox. Res 14 (2–3) (2008) 263–272, 10.1007/bf03033815. [DOI] [PubMed] [Google Scholar]
- [14].Raudino F, Non-cognitive symptoms and related conditions in the Alzheimer’s disease: a literature review, Neurol. Sci. : Off. J. Italian Neurol. Soc. Italian Soc. Clin. Neurophysiol 34 (8) (2013) 1275–1282, 10.1007/s10072-013-1424-7. [DOI] [PubMed] [Google Scholar]
- [15].Panza F, Lozupone M, Solfrizzi V, Sardone R, Dibello V, Di Lena L, D’Urso F, Stallone R, Petruzzi M, Giannelli G, Quaranta N, Bellomo A, Greco A, Daniele A, Seripa D, Logroscino G, Different cognitive frailty models and health- and cognitive-related outcomes in older age: from epidemiology to prevention, J. Alzheim. Dis. : JAD 62 (3) (2018) 993–1012, 10.3233/jad-170963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Golde TE, DeKosky ST, Galasko D, Alzheimer’s disease: the right drug, the right time, Science 362 (6420) (2018) 1250–1251, 10.1126/science.aau0437. [DOI] [PubMed] [Google Scholar]
- [17].Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R, Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation, J. Neurosci. : Off. J. Soc. Neurosci 26 (40) (2006) 10129–10140, 10.1523/jneurosci.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bilkei-Gorzo A, Genetic mouse models of brain ageing and Alzheimer’s disease, Pharmacol. Ther 142 (2) (2014) 244–257, 10.1016/j.pharmthera.2013.12.009. [DOI] [PubMed] [Google Scholar]
- [19].Sawmiller D, Li S, Mori T, Habib A, Rongo D, Delic V, Bradshaw PC, Shytle RD, Sanberg C, Bickford P, Tan J, Beneficial effects of a pyrroloquinolinequinone-containing dietary formulation on motor deficiency, cognitive decline and mitochondrial dysfunction in a mouse model of Alzheimer’s disease, Heliyon 3 (4) (2017), e00279, 10.1016/j.heliyon.2017.e00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hershko A, Ciechanover A, The ubiquitin system, Annu. Rev. Biochem 67 (1998) 425–479, 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- [21].Chondrogianni N, Sakellari M, Lefaki M, Papaevgeniou N, Gonos ES, Proteasome activation delays aging in vitro and in vivo, Free Radic. Biol. Med 71 (2014) 303–320, 10.1016/j.freeradbiomed.2014.03.031. [DOI] [PubMed] [Google Scholar]
- [22].Chondrogianni N, Voutetakis K, Kapetanou M, Delitsikou V, Papaevgeniou N, Sakellari M, Lefaki M, Filippopoulou K, Gonos ES, Proteasome activation: an innovative promising approach for delaying aging and retarding age-related diseases, Ageing Res. Rev 23 (Pt A) (2015) 37–55, 10.1016/j.arr.2014.12.003. [DOI] [PubMed] [Google Scholar]
- [23].Vanhooren V, Navarrete Santos A, Voutetakis K, Petropoulos I, Libert C, Simm A, Gonos ES, Friguet B, Protein modification and maintenance systems as biomarkers of ageing, Mech. Ageing Dev 151 (2015) 71–84, 10.1016/j.mad.2015.03.009. [DOI] [PubMed] [Google Scholar]
- [24].Selkoe DJ, Alzheimer’s disease, Cold Spring Harbor Perspect. Biol 3 (7) (2011), 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Oddo S, The ubiquitin-proteasome system in Alzheimer’s disease, J. Cell Mol. Med 12 (2) (2008) 363–373, 10.1111/j.1582-4934.2008.00276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Vilchez D, Saez I, Dillin A, The role of protein clearance mechanisms in organismal ageing and age-related diseases, Nat. Commun 5 (2014) 5659, 10.1038/ncomms6659. [DOI] [PubMed] [Google Scholar]
- [27].Chondrogianni N, Georgila K, Kourtis N, Tavernarakis N, Gonos ES, 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans, Faseb. J. : Off. Publ. Federat. Am. Soc. Exp. Biol 29 (2) (2015) 611–622, 10.1096/fj.14-252189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bence NF, Sampat RM, Kopito RR, Impairment of the ubiquitin-proteasome system by protein aggregation, Science 292 (5521) (2001) 1552–1555, 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- [29].Oh S, Hong HS, Hwang E, Sim HJ, Lee W, Shin SJ, Mook-Jung I, Amyloid peptide attenuates the proteasome activity in neuronal cells, Mech. Age. Develop 126 (12) (2005) 1292–1299, 10.1016/j.mad.2005.07.006. [DOI] [PubMed] [Google Scholar]
- [30].Cecarini V, Bonfili L, Amici M, Angeletti M, Keller JN, Eleuteri AM, Amyloid peptides in different assembly states and related effects on isolated and cellular proteasomes, Brain Res. 1209 (2008) 8–18, 10.1016/j.brainres.2008.03.003. [DOI] [PubMed] [Google Scholar]
- [31].Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM, Abeta inhibits the proteasome and enhances amyloid and tau accumulation, Neurobiol. Aging 29 (11) (2008) 1607–1618, 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Thibaudeau TA, Anderson RT, Smith DM, A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers, Nat. Commun 9 (1) (2018) 1097, 10.1038/s41467-018-03509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].McNaught KS, Belizaire R, Jenner P, Olanow CW, Isacson O, Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease, Neurosci. Lett 326 (3) (2002) 155–158, 10.1016/s0304-3940(02)00296-3. [DOI] [PubMed] [Google Scholar]
- [34].McNaught KS, Bjorklund LM, Belizaire R, Isacson O, Jenner P, Olanow CW, Proteasome inhibition causes nigral degeneration with inclusion bodies in rats, Neuroreport 13 (11) (2002) 1437–1441, 10.1097/00001756-200208070-00018. [DOI] [PubMed] [Google Scholar]
- [35].Li Z, Arnaud L, Rockwell P, Figueiredo-Pereira ME, A single amino acid substitution in a proteasome subunit triggers aggregation of ubiquitinated proteins in stressed neuronal cells, J. Neurochem 90 (1) (2004) 19–28, 10.1111/j.1471-4159.2004.02456.x. [DOI] [PubMed] [Google Scholar]
- [36].Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, Mee M, Soane T, Layfield R, Sheppard PW, Ebendal T, Usoskin D, Lowe J, Mayer RJ, Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies, J. Neurosci. : Off. J. Soc. Neurosci 28 (33) (2008) 8189–8198, 10.1523/jneurosci.2218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xie W, Li X, Li C, Zhu W, Jankovic J, Le W, Proteasome inhibition modeling nigral neuron degeneration in Parkinson’s disease, J. Neurochem 115 (1) (2010) 188–199, 10.1111/j.1471-4159.2010.06914.x. [DOI] [PubMed] [Google Scholar]
- [38].Lee NK, Park SE, Kwon SJ, Shim S, Byeon Y, Kim J-H, Na DL, Chang JW, Agouti related peptide secreted via human mesenchymal stem cells upregulates proteasome activity in an Alzheimer’s disease model, Sci. Rep 7 (1) (2017), 39340, 10.1038/srep39340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chondrogianni N, Petropoulos I, Franceschi C, Friguet B, Gonos ES, Fibroblast cultures from healthy centenarians have an active proteasome, Exp. Gerontol 35 (6–7) (2000) 721–728, 10.1016/s0531-5565(00)00137-6. [DOI] [PubMed] [Google Scholar]
- [40].Kapetanou M, Chondrogianni N, Petrakis S, Koliakos G, Gonos ES, Proteasome activation enhances stemness and lifespan of human mesenchymal stem cells, Free Radical Biol. Med 103 (2017) 226–235, 10.1016/j.freeradbiomed.2016.12.035. [DOI] [PubMed] [Google Scholar]
- [41].Papaevgeniou N, Sakellari M, Jha S, Tavernarakis N, Holmberg CI, Gonos ES, Chondrogianni N, 18alpha-Glycyrrhetinic acid proteasome activator decelerates aging and Alzheimer’s disease progression in Caenorhabditis elegans and neuronal cultures, Antioxidants Redox Signal. 25 (16) (2016) 855–869, 10.1089/ars.2015.6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ryabinin AE, Wang YM, Finn DA, Different levels of Fos immunoreactivity after repeated handling and injection stress in two inbred strains of mice, Pharmacol. Biochem. Behav 63 (1) (1999) 143–151, 10.1016/s0091-3057(98)00239-1. [DOI] [PubMed] [Google Scholar]
- [43].Kamisli S, Ciftci O, Taslidere A, Basak Turkmen N, Ozcan C, The beneficial effects of 18β-glycyrrhetinic acid on the experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mouse model, Immunopharmacol. Immunotoxicol 40 (4) (2018) 344–352, 10.1080/08923973.2018.1490318. [DOI] [PubMed] [Google Scholar]
- [44].Milanovic D, Petrovic S, Brkic M, Avramovic V, Perovic M, Ivkovic S, Glibetic M, Kanazir S, Short-term fish oil treatment changes the composition of phospholipids while not affecting the expression of Mfsd2a omega-3 transporter in the brain and liver of the 5xFAD mouse model of Alzheimer’s disease, Nutrients 10 (9) (2018), 10.3390/nu10091250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jović M, Lončarević-Vasiljković N, Ivković S, Dinić J, Milanović D, Zlokovic B, Kanazir S, Short-term fish oil supplementation applied in presymptomatic stage of Alzheimer’s disease enhances microglial/macrophage barrier and prevents neuritic dystrophy in parietal cortex of 5xFAD mouse model, PloS One 14 (5) (2019), e0216726, 10.1371/journal.pone.0216726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O, Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease, Neurobiol. Aging 33 (1) (2012), 10.1016/j.neurobiolaging.2010.05.027, 196.e29–40. [DOI] [PubMed] [Google Scholar]
- [47].Crowley JJ, Jones MD, O’Leary OF, Lucki I, Automated tests for measuring the effects of antidepressants in mice, Pharmacol. Biochem. Behav 78 (2) (2004) 269–274, 10.1016/j.pbb.2004.03.014. [DOI] [PubMed] [Google Scholar]
- [48].Kosel F, Torres Munoz P, Yang JR, Wong AA, Franklin TB, Age-related changes in social behaviours in the 5xFAD mouse model of Alzheimer’s disease, Behav. Brain Res 362 (2019) 160–172, 10.1016/j.bbr.2019.01.029. [DOI] [PubMed] [Google Scholar]
- [49].Miquel J, Blasco M, A simple technique for evaluation of vitality loss in aging mice, by testing their muscular coordination and vigor, Exp. Gerontol 13 (6) (1978) 389–396, 10.1016/0531-5565(78)90049-9. [DOI] [PubMed] [Google Scholar]
- [50].Liu H, Graber TG, Ferguson-Stegall L, Thompson LV, Clinically relevant frailty index for mice, J Gerontol A Biol Sci Med Sci 69 (12) (2014) 1485–1491, 10.1093/gerona/glt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Miller MG, Thangthaeng N, Shukitt-Hale B, A clinically relevant frailty index for aging rats, J Gerontol A Biol Sci Med Sci 72 (7) (2017) 892–896, 10.1093/gerona/glw338. [DOI] [PubMed] [Google Scholar]
- [52].Todorovic S, Loncarevic-Vasiljkovic N, Jovic M, Sokanovic S, Kanazir S, Mladenovic Djordjevic A, Frailty index and phenotype frailty score: sex- and age-related differences in 5XFAD transgenic mouse model of Alzheimer’s disease, Mech. Age. Develop 185 (2020), 111195, 10.1016/j.mad.2019.111195. [DOI] [PubMed] [Google Scholar]
- [53].Kaech S, Banker G, Culturing hippocampal neurons, Nat. Protoc 1 (5) (2006) 2406–2415, 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- [54].Qiu L-Q, Lai WS, Stumpo DJ, Blackshear PJ, Mouse embryonic fibroblast cell culture and stimulation, Bio Protoc 6 (13) (2016), el859, 10.21769/BioProtoc.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Palmer HM, Using antibodies: a laboratory manual, J. Antimicrob. Chemother 45 (3) (2000), 10.1093/jac/45.3.413, 413–413. [DOI] [Google Scholar]
- [56].Rivett AJ, Savory PJ, Djaballah H, [24] Multicatalytic Endopeptidase Complex: Proteasome, Methods in Enzymology, Academic Press; 1994, pp. 331–350. [DOI] [PubMed] [Google Scholar]
- [57].Georgila K, Voutetakis K, Delitsikou V, Chondrogianni N, Gonos ES, Optimization of in vitro measurement of proteasome activity in mammalian cells using fluorogenic substrates, Free Radical Biol. Med 75 (Suppl 1) (2014) S31, 10.1016/j.freeradbiomed.2014.10.762. [DOI] [PubMed] [Google Scholar]
- [58].Kapeta S, Chondrogianni N, Gonos ES, Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts, J. Biol. Chem 285 (11) (2010) 8171–8184, 10.1074/jbc.M109.031575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bolivar VJ, Intrasession and intersession habituation in mice: from inbred strain variability to linkage analysis, Neurobiol. Learn. Mem 92 (2) (2009) 206–214, 10.1016/j.nlm.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Seibenhener ML, Wooten MC, Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice, JoVE : JoVE 96 (2015), e52434, 10.3791/52434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Flanigan TJ, Xue Y, Kishan Rao S, Dhanushkodi A, McDonald MP, Abnormal vibrissa-related behavior and loss of barrel field inhibitory neurons in 5xFAD transgenics, Gene Brain Behav. 13 (5) (2014) 488–500, 10.1111/gbb.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].O’Leary TP, Mantolino HM, Stover KR, Brown RE, Age-related deterioration of motor function in male and female 5xFAD mice from 3 to 16 months of age, Gene Brain Behav. 19 (3) (2020), e12538, 10.1111/gbb.12538. [DOI] [PubMed] [Google Scholar]
- [63].Cova I, Clerici F, Rossi A, Cucumo V, Ghiretti R, Maggiore L, Pomati S, Galimberti D, Scarpini E, Mariani C, Caracciolo B, Weight loss predicts progression of mild cognitive impairment to Alzheimer’s disease, PloS One 11 (3) (2016), e0151710, 10.1371/journal.pone.0151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Richard BC, Kurdakova A, Baches S, Bayer TA, Weggen S, Wirths O, Gene dosage dependent aggravation of the neurological phenotype in the 5XFAD mouse model of Alzheimer’s disease, J. Alzheim. Dis. : JAD 45 (4) (2015) 1223–1236, 10.3233/jad-143120. [DOI] [PubMed] [Google Scholar]
- [65].Apostolova LG, Thompson PM, Mapping progressive brain structural changes in early Alzheimer’s disease and mild cognitive impairment, Neuropsychologia 46 (6) (2008) 1597–1612, 10.1016/j.neuropsychologia.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bonet-Costa V, Pomatto LC-D, Davies KJA, The proteasome and oxidative stress in Alzheimer’s disease, Antioxidants Redox Signal. 25 (16) (2016) 886–901, 10.1089/ars.2016.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gonos ES, Kapetanou M, Sereikaite J, Bartosz G, Naparlo K, Grzesik M, Sadowska-Bartosz I, Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging, Aging 10 (5) (2018) 868–901, 10.18632/aging.101450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chondrogianni N, Vasilopoulou MA, Kapetanou M, Gonos ES, Proteasome modulation: a way to delay aging? in: Rattan SIS (Ed.), Encyclopedia of Biomedical Gerontology Academic Press, Oxford, 2020, pp. 92–104. [Google Scholar]
- [69].Kwak M-K, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW, Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway, Mol. Cell Biol 23 (23) (2003) 8786–8794, 10.1128/mcb.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tu W, Wang H, Li S, Liu Q, Sha H, The anti-inflammatory and anti-oxidant mechanisms of the keap1/nrf2/ARE signaling pathway in chronic diseases, Aging Dis 10 (3) (2019) 637–651, 10.14336/AD.2018.0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tanaka K, Yoshimura T, Ichihara A, Role of substrate in reversible activation of proteasomes (multi-protease complexes) by sodium dodecyl sulfate, J. Biochem 106 (3) (1989) 495–500, 10.1093/oxfordjournals.jbchem.a122880. [DOI] [PubMed] [Google Scholar]
- [72].Bottiggi K, Harrison AL, The association between change in motor function and cognition in older adults: a descriptive review, Phys. Ther. Rev 13 (2) (2008) 91–101, 10.1179/174328808X252055. [DOI] [Google Scholar]
- [73].Goldman WP, Baty JD, Buckles VD, Sahrmann S, Morris JC, Motor dysfunction in mildly demented AD individuals without extrapyramidal signs, Neurology 53 (5) (1999) 956–962, 10.1212/wnl.53.5.956. [DOI] [PubMed] [Google Scholar]
- [74].Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, Seeman T, Tracy R, Kop WJ, Burke G, McBurnie MA, Frailty in older adults: evidence for a phenotype, the journals of gerontology, Series A, Biol. Sci. Med. Sci 56 (3) (2001) M146–M156, 10.1093/gerona/56.3.m146. [DOI] [PubMed] [Google Scholar]
- [75].Wagner JM, Sichler ME, Schleicher EM, Franke TN, Irwin C, Löw MJ, Beindorff N, Bouter C, Bayer TA, Bouter Y, Analysis of motor function in the tg4-42 mouse model of Alzheimer’s disease, Front. Behav. Neurosci 13 (2019) 107, 10.3389/fnbeh.2019.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kaiser NC, Liang LJ, Melrose RJ, Wilkins SS, Sultzer DL, Mendez MF, Differences in anxiety among patients with early- versus late-onset Alzheimer’s disease, J. Neuropsychiatry Clin. Neurosci 26 (1) (2014) 73–80, 10.1176/appi.neuropsych.12100240. [DOI] [PubMed] [Google Scholar]
- [77].Donovan NJ, Locascio JJ, Marshall GA, Gatchel J, Hanseeuw BJ, Rentz DM, Johnson KA, Sperling RA, Longitudinal association of amyloid beta and anxious-depressive symptoms in cognitively normal older adults, Am. J. Psychiatr 175 (6) (2018) 530–537, 10.1176/appi.ajp.2017.17040442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bhattacharya S, Haertel C, Maelicke A, Montag D, Galantamine slows down plaque formation and behavioral decline in the 5XFAD mouse model of Alzheimer’s disease, PloS One 9 (2) (2014), e89454, 10.1371/journal.pone.0089454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Bjursell M, Xu X, Admyre T, Böttcher G, Lundin S, Nilsson R, Stone VM, Morgan NG, Lam YY, Storlien LH, Lindén D, Smith DM, Bohlooly-Y M, Oscarsson J, The beneficial effects of n-3 polyunsaturated fatty acids on diet induced obesity and impaired glucose control do not require Gpr120, PloS One 9 (12) (2014), 10.1371/journal.pone.0114942 e114942–e114942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yang Z-H, Inoue S, Taniguchi Y, Miyahara H, Iwasaki Y, Takeo J, Sakaue H, Nakaya Y, Long-term dietary supplementation with saury oil attenuates metabolic abnormalities in mice fed a high-fat diet: combined beneficial effect of omega-3 fatty acids and long-chain monounsaturated fatty acids, Lipids Health Dis. (2015) 155, 10.1186/s12944-015-0161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ramos-Romero S, Molinar-Toribio E, Pérez-Jiménez J, Taltavull N, Dasilva G, Romeu M, Medina I, Torres JL, The combined action of omega-3 polyunsaturated fatty acids and grape proanthocyanidins on a rat model of diet-induced metabolic alterations, Food Funct 7 (8) (2016) 3516–3523, 10.1039/c6fo00679e. [DOI] [PubMed] [Google Scholar]
- [82].Park M, Lee JH, Choi JK, Hong YD, Bae IH, Lim KM, Park YH, Ha H, 18β-glycyrrhetinic acid attenuates anandamide-induced adiposity and high-fat diet induced obesity, Mol. Nutr. Food Res 58 (7) (2014) 1436–1446, 10.1002/mnfr.201300763. [DOI] [PubMed] [Google Scholar]
- [83].Flanigan TJ, Xue Y, Kishan Rao S, Dhanushkodi A, McDonald MP, Abnormal vibrissa-related behavior and loss of barrel field inhibitory neurons in 5xFAD transgenics, Gene Brain Behav. 13 (5) (2014) 488–500, 10.1111/gbb.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Peters OM, Shelkovnikova T, Tarasova T, Springe S, Kukharsky MS, Smith GA, Brooks S, Kozin SA, Kotelevtsev Y, Bachurin SO, Ninkina N, Buchman VL, Chronic administration of Dimebon does not ameliorate amyloid-β pathology in 5xFAD transgenic mice, J. Alzheim. Dis. : JAD 36 (3) (2013) 589–596, 10.3233/jad-130071. [DOI] [PubMed] [Google Scholar]
- [85].Schneider F, Baldauf K, Wetzel W, Reymann KG, Behavioral and EEG changes in male 5xFAD mice, Physiol. Behav 135 (2014) 25–33, 10.1016/j.physbeh.2014.05.041. [DOI] [PubMed] [Google Scholar]
- [86].Buchman AS, Bennett DA, Loss of motor function in preclinical Alzheimer’s disease, Expert Rev. Neurother 11 (5) (2011) 665–676, 10.1586/ern.11.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Inglés M, Mas-Bargues C, Gimeno-Mallench L, Cruz-Guerrero R, García-García FJ, Gambini J, Borrás C, Rodríguez-Mañas L, Viña J, Relation between genetic factors and frailty in older adults, J. Am. Med. Dir. Assoc 20 (11) (2019) 1451–1457, 10.1016/j.jamda.2019.03.011. [DOI] [PubMed] [Google Scholar]
- [88].Bisset ES, Howlett SE, The biology of frailty in humans and animals: understanding frailty and promoting translation, Aging Med (Milton) 2 (1) (2019) 27–34, 10.1002/agm2.12058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Buchman AS, Boyle PA, Wilson RS, Tang Y, Bennett DA, Frailty is associated with incident Alzheimer’s disease and cognitive decline in the elderly, Psychosom. Med 69 (5) (2007) 483–489, 10.1097/psy.0b013e318068de1d. [DOI] [PubMed] [Google Scholar]
- [90].Kojima G, Liljas A, Iliffe S, Walters K, Prevalence of frailty in mild to moderate Alzheimer’s disease: a systematic review and meta-analysis, Curr. Alzheimer Res 14 (12) (2017) 1256–1263, 10.2174/1567205014666170417104236. [DOI] [PubMed] [Google Scholar]
- [91].Wallace L, Theou O, Rockwood K, Andrew MK, Relationship between frailty and Alzheimer’s disease biomarkers: a scoping review, Alzheimers Dement (Arnst) 10 (2018)394–401, 10.1016/j.dadm.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kelaiditi E, Canevelli M, Andrieu S, Del Campo N, Soto ME, Vellas B, Cesari M, Frailty index and cognitive decline in Alzheimer’s disease: data from the impact of cholinergic treatment USe study, J. Am. Geriatr. Soc 64 (6) (2016) 1165–1170, 10.1111/jgs.13956. [DOI] [PubMed] [Google Scholar]
- [93].Trebbastoni A, Canevelli M, D’Antonio F, Imbriano L, Podda L, Rendace L, Campanelli A, Celano V, Bruno G, de Lena C, The impact of frailty on the risk of conversion from mild cognitive impairment to Alzheimer’s disease: evidences from a 5-year observational study, Front. Med 4 (2017) 178, 10.3389/fmed.2017.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kane AE, Shin S, Wong AA, Fertan E, Faustova NS, Howlett SE, Brown RE, Sex differences in healthspan predict lifespan in the 3xTg-AD mouse model of Alzheimer’s disease, Front. Aging Neurosci 10 (2018) 172, 10.3389/fnagi.2018.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Tonoki A, Kuranaga E, Tomioka T, Hamazaki J, Murata S, Tanaka K, Miura M, Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process, Mol. Cell Biol 29 (4) (2009) 1095–1106, 10.1128/mcb.01227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Vilchez D, Morantte I, Liu Z, Douglas PM, Merkwirth C, Rodrigues AP, Manning G, Dillin A, RPN-6 determines C. elegans longevity under proteotoxic stress conditions, Nature 489 (7415) (2012) 263–268, 10.1038/nature11315. [DOI] [PubMed] [Google Scholar]
- [97].Keller JN, Hanni KB, Markesbery WR, Impaired proteasome function in Alzheimer’s disease, J. Neurochem 75 (1) (2000) 436–439, 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- [98].Lopez Salon M, Morelli L, Castano EM, Soto EF, Pasquini JM, Defective ubiquitination of cerebral proteins in Alzheimer’s disease, J. Neurosci. Res 62 (2) (2000)302–310, . [DOI] [PubMed] [Google Scholar]
- [99].Aguilaniu H, Gustafsson L, Rigoulet M, Nyström T, Asymmetric inheritance of oxidatively damaged proteins during cytokinesis, Science 299 (5613) (2003) 1751–1753, 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- [100].Tai H-C, Besche H, Goldberg AL, Schuman EM, Characterization of the brain 26S proteasome and its interacting proteins, Front. Mol. Neurosci 3 (2010) 12, 10.3389/fnmol.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Erokhov PA, Lyupina YV, Radchenko AS, Kolacheva AA, Nikishina YO, Sharova NP, Detection of active proteasome structures in brain extracts: proteasome features of August rat brain with violations in monoamine metabolism, Oncotarget 8 (41) (2017) 70941–70957, 10.18632/oncotarget.20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Keller D, Erö C, Markram H, Cell densities in the mouse brain: a systematic review, Front. Neuroanat 12 (2018), 10.3389/fnana.2018.00083, 83–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Streit WJ, Braak H, Xue QS, Bechmann I, Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease, Acta Neuropathol. 118 (4) (2009) 475–485, 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Neuman KM, Molina-Campos E, Musial TF, Price AL, Oh K-J, Wolke ML, Buss EW, Scheff SW, Mufson EJ, Nicholson DA, Evidence for Alzheimer’s disease-linked synapse loss and compensation in mouse and human hippocampal CA1 pyramidal neurons, Brain Struct. Funct 220 (6) (2015)3143–3165, 10.1007/s00429-014-0848-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yao X, Liu J, McCabe JT, Alterations of cerebral cortex and hippocampal proteasome subunit expression and function in a traumatic brain injury rat model, J. Neurochem 104 (2) (2008) 353–363, 10.1111/j.1471-4159.2007.04970.x. [DOI] [PubMed] [Google Scholar]
- [106].Asai A, Tanahashi N, Qiu JH, Saito N, Chi S, Kawahara N, Tanaka K, Kirino T, Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia, J. Cerebr. Blood Flow Metabol. : Off. J. Int. Soc. Cerebral Blood Flow Metabol 22 (6) (2002) 705–710, 10.1097/00004647-200206000-00009. [DOI] [PubMed] [Google Scholar]
- [107].Liu Y, Hettinger CL, Zhang D, Rezvani K, Wang X, Wang H, The proteasome function reporter GFPu accumulates in young brains of the APPswe/PS1dE9 Alzheimer’s disease mouse model, Cell. Mol. Neurobiol 34 (3) (2014) 315–322, 10.1007/s10571-013-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Marambaud P, Zhao H, Davies P, Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides, J. Biol. Chem 280 (45) (2005) 37377–37382, 10.1074/jbc.M508246200. [DOI] [PubMed] [Google Scholar]
- [109].Chakrabortee S, Liu Y, Zhang L, Matthews HR, Zhang H, Pan N, Cheng CR, Guan SH, Guo DA, Huang Z, Zheng Y, Tunnacliffe A, Macromolecular and small-molecule modulation of intracellular Abeta42 aggregation and associated toxicity, Biochem. J 442 (3) (2012) 507–515, 10.1042/bj20111661. [DOI] [PubMed] [Google Scholar]
- [110].Regitz C, Fitzenberger E, Mahn FL, Dußling LM, Wenzel U, Resveratrol reduces amyloid-beta (Aβ1–42)-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans, Eur. J. Nutr 55 (2) (2016) 741–747, 10.1007/s00394-015-0894-1. [DOI] [PubMed] [Google Scholar]
- [111].Medina DX, Caccamo A, Oddo S, Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity, Brain Pathol. 21 (2) (2011) 140–149, 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Cores Á, Piquero M, Villacampa M, León R, Menéndez JC, NRF2 regulation processes as a source of potential drug targets against neurodegenerative diseases, Biomolecules 10 (6) (2020), 10.3390/biom10060904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Dyall SC, Long-chain omega-3 fatty acids and the brain: a review of the independent and shared effects of EPA, DPA and DHA, Front. Aging Neurosci 7 (2015) 52, 10.3389/fnagi.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Wysoczański T, Sokoła-Wysoczańska E, Pękala J, Lochyński S, Czyż K, Bodkowski R, Herbinger G, Patkowska-Sokoła B, Librowski T, Omega-3 fatty acids and their role in central nervous system - a review, Curr. Med. Chem 23 (8) (2016) 816–831, 10.2174/0929867323666160122114439. [DOI] [PubMed] [Google Scholar]
- [115].Devassy JG, Leng S, Gabbs M, Monirujjaman M, Aukema HM, Omega-3 polyunsaturated fatty acids and oxylipins in neuroinflammation and management of alzheimer disease, Adv Nutr 7 (5) (2016) 905–916, 10.3945/an.116.012187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Avallone R, Vitale G, Bertolotti M, Omega-3 fatty acids and neurodegenerative diseases: new evidence in clinical trials, Int. J. Mol. Sci 20 (17) (2019), 10.3390/ijms20174256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Edwards III GA, Gamez N, Escobedo G Jr., Calderon O, Moreno-Gonzalez I, Modifiable risk factors for Alzheimer’s disease, Front. Aging Neurosci 11 (2019) 146, 10.3389/fnagi.2019.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Chitre NM, Moniri NH, Murnane KS, Omega-3 fatty acids as druggable therapeutics for neurodegenerative disorders, CNS Neurol. Disord. - Drug Targets 18 (10) (2019) 735–749, 10.2174/1871527318666191114093749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Tanaka K, Yoshimura T, Kumatori A, Ichihara A, Ikai A, Nishigai M, Kameyama K, Takagi T, Proteasomes (multi-protease complexes) as 20 S ring-shaped particles in a variety of eukaryotic cells, J. Biol. Chem 263 (31) (1988) 16209–16217. [PubMed] [Google Scholar]
- [120].Calder PC, Omega-3 polyunsaturated fatty acids and inflammatory processes: nutrition or pharmacology? Br. J. Clin. Pharmacol 75 (3) (2013) 645–662, 10.1111/j.1365-2125.2012.04374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Xiao Y, Xu J, Mao C, Jin M, Wu Q, Zou J, Gu Q, Zhang Y, Zhang Y, 18Beta-glycyrrhetinic acid ameliorates acute Propionibacterium acnes-induced liver injury through inhibition of macrophage inflammatory protein-1alpha, J. Biol. Chem 285 (2) (2010) 1128–1137, 10.1074/jbc.M109.037705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Shetty AV, Thirugnanam S, Dakshinamoorthy G, Samykutty A, Zheng G, Chen A, Bosland MC, Kajdacsy-Balia A, Gnanasekar M, 18α-glycyrrhetinic acid targets prostate cancer cells by down-regulating inflammation-related genes, Int. J. Oncol 39 (3) (2011) 635–640, 10.3892/ijo.2011.1061. [DOI] [PubMed] [Google Scholar]
- [123].Oztanir MN, Ciftci O, Cetin A, Durak MA, Basak N, Akyuva Y, The beneficial effects of 18β-glycyrrhetinic acid following oxidative and neuronal damage in brain tissue caused by global cerebral ischemia/reperfusion in a C57BL/J6 mouse model, Neurol. Sci.: Off. J. Italian Neurol. Soc. Italian Soc. Clin. Neurophysiol 35 (8) (2014) 1221–1228, 10.1007/s10072-014-1685-9. [DOI] [PubMed] [Google Scholar]
- [124].Hipp MS, Kasturi P, Hard FU, The proteostasis network and its decline in ageing, Nat. Rev. Mol. Cell Biol 20 (7) (2019) 421–435, 10.1038/S41580-019-0101-y. [DOI] [PubMed] [Google Scholar]
- [125].Hale MW, Hay-Schmidt A, Mikkelsen JD, Poulsen B, Shekhar A, Lowry CA, Exposure to an open-field arena increases c-Fos expression in a distributed anxiety-related system projecting to the basolateral amygdaloid complex, Neuroscience 155 (3) (2008) 659–672, 10.1016/j.neuroscience.2008.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Lalonde R, Strazielle C, Brain regions and genes affecting limb-clasping responses, Brain Res. Rev 67 (1–2) (2011) 252–259, 10.1016/j.brainresrev.2011.02.005. [DOI] [PubMed] [Google Scholar]
- [127].Størmer FC, Reistad R, Alexander J, Glycyrrhizic acid in liquorice—evaluation of health hazard, Food Chem. Toxicol 31 (4) (1993) 303–312, 10.1016/0278-6915(93)90080-I. [DOI] [PubMed] [Google Scholar]
- [128].Viña J, Sanz-Ros J, Alzheimer’s disease: only prevention makes sense, Eur. J. Clin. Invest 48 (10) (2018), e13005, 10.1111/eci.13005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.