

Abstract
Efficacious pharmacotherapies for the treatment of substance use disorders need to be expanded and improved. Non-neuronal cells, particularly astrocytes and microglia, have emerged as therapeutic targets for the development of pharmacotherapies to treat dependence and relapse that accompanies chronic drug use. Cytokines and chemokines are neuroimmune factors expressed in neurons, astrocytes, and microglia that demonstrate promising clinical utility as therapeutic targets for substance use disorders. In this review, we describe a role for cytokines and chemokines in the rewarding and reinforcing effects of alcohol, opioids, and psychostimulants. We also discuss emerging cytokine- and chemokine-based therapeutic strategies that differ from conventional strategies directed toward transporters and receptors within the dopamine, glutamate, GABA, serotonin, and GABA systems.
Keywords: Cytokines, chemokines, alcohol, opioids, cocaine, methamphetamine
1. Introduction
Substance use disorders are a widespread global health problem. In 2018, an estimated 269 million individuals worldwide abused drugs, representing 5.3 percent of the global population (World Drug Report 2020). In the United States, approximately 20.3 million individuals aged 12 or older in 2018 had a substance use disorder attributed to alcohol or illicit drugs (Lipari and Park-Lee, 2019).
The development of a substance use disorder involves dysregulation of neuronal circuits responsible for motivated behavior (Koob and Volkow, 2010). The role of dopamine and glutamate systems in mediating the effects of drugs of abuse is well established, but pharmacotherapies directly targeting these and other classical neurotransmitter systems has been largely unsuccessful in the treatment of substance use disorders, especially for opioids and psychostimulants. Thus, to develop effective pharmacotherapies, it is important to identify new endogenous targets, such as chemokine and cytokine systems, that underlie substance use disorders (Rogers, 2020; Paulus and Stewart, 2020; Bachtell et al., 2017; Cui et al., 2014; Crews et al., 2011).
Cytokines and chemokines are important components of the CNS neuroimmune system and contribute to neuroinflammation, neuronal activity, neuron-glia communication, neuroendocrine interactions, neurogenesis, and CNS development (Bachtell et al., 2017; Bachtell et al., 2015; Boulanger, 2009). Chemokines are chemoattractant cytokines that comprise a family of low molecular weight proteins and play a role in both the immune system and the central nervous system (CNS) (Parsadaniantz et al., 2015; Rostène et al., 2007). They are categorized into four subfamilies based on the position of the cysteine residues within the amino-terminal region: C-, CC- CXC-, and CX3C-chemokines, and mediate their effects through seven-transmembrane G protein-coupled receptors (GPCRs) designated CR1, CCR1–11, CXCR1–5, or CX3CR1, respectively (Murphy, 2002; Murphy, 2000). Several chemokines, including CCL2, CCL3, CCL5, CXCL1, CXCL8, CXCL12, and CX3CL1 are secreted in the brain (note that the “L” designation denotes ligand) (Bajetto et al., 2001; Tashiro et al., 1993). The chemokine receptors CCR1, CCR4, CCR5, CCR9, CXCR2, CXCR4, and CX3CR1 are also expressed in different brain regions, including the hippocampus, cerebral cortex, amygdala, thalamus, and basal ganglia (Gabuzda et al., 1998; Meucci et al., 1998; Bajetto et al., 1999). These GPCRs can signal through Gαi/o proteins through which they inhibit adenylate cyclase and decrease protein kinase A activity (Zheng et al., 1999), as well as through Gq proteins through which they can increase intracellular Ca2+ levels and protein kinase C via the phospholipase C pathway (Cali and Bezzi, 2010; Khan et al., 2004; Meucci et al., 1998).
The neuromodulatory properties of cytokines and chemokines have been associated with the development of substance use disorders and provide a different framework for understanding functional and behavioral changes contributing to drug addiction (Cui et al., 2014). This review describes recent advances in the role of cytokines and chemokines in alcohol, opioid and psychostimulant use disorders, including a discussion of potential therapeutic strategies targeting these systems.
2. Alcohol
2.1. Background
In 2018, 139.8 million individuals aged 12 or older in the United States were alcohol users, 67.1 million were binge drinkers, and 16.6 million were heavy drinkers the previous month (Lipari and Park-Lee, 2019). Furthermore, an estimated 401,000 adolescents, 3.4 million young adults, and 11.0 million adults had an alcohol use disorder (AUD) (Lipari and Park-Lee, 2019).
2.2. Pathophysiology of Alcohol Liver Disease
Individuals with alcohol use disorder (AUD), particularly those with alcohol liver disease (ALD), frequently experience systemic endotoxemia attributable to increased gut permeability, reactive oxygen species (ROS) generation from ethanol metabolism, and impaired hepatic clearance function (McClain et al., 1997). ROS activate toll-like receptors (TLRs), specifically TLR4, and the associated plasma membrane protein CD14 (Qin et al., 2008). This signal transduction cascade activates various transcription factors, including nuclear Factor kappa-light-chain-enhancer (NF-κB) in liver Kupffer cells, which play a critical role in cytokine gene activation (McClain et al., 1997). Notably, tumor necrosis factor alpha (TNFα) is produced, which enhances the immune response by inducing fever, activating neutrophils and macrophages, and stimulating macrophages to produce cytokines (McClain et al., 1997). These functions can then further increase intestinal permeability, endotoxemia, and ROS generation (McClain et al., 1999).
Additional cytokines that modulate systemic responses to ALD include the: (1) proinflammatory cytokine interleukin-1 (IL-1), which induces fever and stimulates growth and differentiation of immune cells; (2) anti-inflammatory cytokine interleukin 10 (IL-10), which reduces production of proinflammatory cytokines such as TNFα; (3) chemokine interleukin-8 (IL-8), which promotes neutrophil infiltration; (4) hepatic acute-phase cytokine interleukin-6 (IL-6), which stimulates inflammatory mediators and promotes maturation of antibody-secreting cells; and (5) monocyte chemoattractant protein-1 (MCP-1/CCL2), which promotes monocyte infiltration (McClain et al., 1997).
2.3. Cytokine Signaling Dysregulation in ALD
Several studies using animal models and postmortem human alcoholic brains indicate a role for innate immunity in alcohol dependence and addiction. Many differentially expressed genes found in animals chronically treated with alcohol or post-mortem brains from alcoholics contribute to the TLR4 signaling pathway (Blednov et al., 2011). Ethanol-induced microglial activation, production of inflammatory mediators, and apoptosis are prevented in microglia lacking TLR4 in vitro and microglia of TLR4-deficient mice (Fernandez-Lizarbe et al., 2009; Alfonso-Loeches et al., 2010). These studies demonstrate an important role for the TLR4 response in ethanol-mediated activation of NF-κB and subsequent transcription of proinflammatory immune genes.
Brain gene expression analyses in mice further identified NF-κB, in addition to many innate immune genes, to be differentially expressed in high and low alcohol consuming rodent lines (Mulligan et al., 2006). Increased levels of innate immune gene mRNA related to NF-κB were also shown in a study of postmortem human alcoholic brain samples (Liu et al., 2006). Studies on human alcoholics found associations between NF-κB and TNFα polymorphisms and alcohol abuse (Edenberg et al., 2008; Gonzalez et al., 2004; Kebir et al., 2011). Additional polymorphisms of genes encoding proinflammatory cytokines interleukin-1 beta (IL-1β) and IL-1, as well as anti-inflammatory cytokine IL-10, have been associated with a risk for developing alcohol use disorder (Marcos et al., 2008; Saiz et al., 2009; Pastor et al., 2005). These studies suggest that variations in NF-κB gene and innate immune gene expression influence alcoholism susceptibility.
Other studies found that ethanol treatment increases NF-κB-DNA binding in rat brain in vivo and brain slice cultures in vitro (Crews et al., 2006; Zou and Crews, 2006). Zou and Crews (2010) reported that chronic ethanol administration increases NF-κB-DNA binding and subsequent proinflammatory gene expression in rat brain slice cultures, including TNFα, IL-1β, and MCP-1. Furthermore, in the same study, NF-κB blockade and TNFα-neutralizing antibody reduced ethanol induction of proinflammatory genes. Thus, ethanol is thought to increase NF-κB-DNA binding and subsequent proinflammatory gene expression, resulting in neurodegeneration and altered neurotransmission (Zou and Crews, 2010).
2.4. Ethanol-induced Neuroinflammation and Cytokine Signaling in the Absence of ALD
Ethanol exposure has been shown to increase liver, serum, and brain cytokine production (TNFα, MCP-1, and IL-1β) and to substantially increase LPS-induced cytokine responses in mice (Qin et al., 2008). Postmortem human alcoholic brains also revealed increased expression of proinflammatory cytokines MCP-1 (He and Crews, 2008) and IL-1β (Crews, 2012). The coordination of these responses may be explained by liver Kupffer and blood monocyte cell cytokine secretion, resulting in cytokine transport across the blood-brain barrier and induction of brain proinflammatory cytokine production (Qin et al., 2008).
Brain proinflammatory cytokines remain elevated after liver and serum cytokines return to baseline levels. This finding is also accompanied by a prolonged reduction in brain anti-inflammatory cytokine IL-10 (Qin et al., 2008). Long-lasting brain induction of proinflammatory cytokines and reduction of IL-10 may contribute to central nervous system pathologies including alcoholic dementia (Qin et al., 2008). This study also found that neurogenesis was decreased in ethanol-LPS treated animals as evident from reduced proliferation of hippocampal neuroprogenitors and reduced differentiation of newly born neurons. Ethanol-induced proinflammatory cytokine cascades may therefore inhibit neurogenesis and influence the development of neurodegenerative diseases (Qin et al., 2008).
Interestingly, alcohol consumption is limited when innate immune gene expression is interrupted (Blednov et al., 2005, 2012). Blednov et al. (2005) demonstrated that deletion of CCR2, MCP-1/CCL2 (females) or CCL3 genes in mice reduced alcohol consumption and preference. These data demonstrate that disruption of cytokine cascades at the ligand or receptor level disrupts the motivational effects of alcohol, which may be explained by a dysregulated chemokine network that facilitates an aversion to alcohol. Moreover, increased serum cytokine levels and inflammatory endotoxins are positively correlated with alcohol craving and may increase alcohol consumption (Leclercq et al., 2012; Leclercq et al., 2014).
2.5. The Role of Inflammatory Responses in the Liver
Given that the liver is essential in modulating acute inflammation, stress, or trauma and a major producer and scavenger of cytokines, it is not surprising that cytokine dysregulation largely contributes to the systemic manifestations of ALD as well as the liver injury and repair process (McClain et al. 1999; McClain et al., 1997). ALD patients can present systemically with fever, neutrophilia, and anorexia with muscle wasting (McClain et al., 1999). Clinical studies have shown high levels of IL-1, IL-6, IL-8, TNF, and MCP-1 in patients with ALD (McClain et al., 1997). IL-8 may play a role in neutrophilia and neutrophil infiltration of liver tissue in ALD, and MCP-1 may induce monocyte infiltration to ensure a sufficient phagocyte supply to fight infection (Sheron et al., 1993; McClain et al., 1997). Increased TNF may be attributed to the decreased production of the anti-inflammatory cytokine IL-10 by monocytes (McClain et al., 1997). Cytokines induce the liver to produce acute-phase proteins, which is accompanied by a marked decrease in albumin production; disrupted protein metabolism in muscle tissue presents as muscle wasting as component amino acids are required for acute-phase protein synthesis. These metabolic processes reflect a shift to host innate defenses (McClain et al., 1997). Interestingly, these complications are remarkably similar to the biologic actions of TNF, which is suggested to play an etiologic role in liver injury (McClain and Cohen, 1989).
2.6. Cytokine-based Therapeutic Approaches for ALD
Despite extensive and life-threatening complications associated with ALD, there are no FDA-approved therapies. Recommendations include smoking cessation and weight loss for obese patients as these risk factors are associated with oxidative stress and may contribute to cytokine production and ALD progression (McClain et al., 2004). Nutritional support is advised for patients suffering from malnutrition, which may decrease gut permeability and bacterial translocation and improve hepatic function (McClain et al., 2004).
Several therapeutics approaches modulating cytokine signaling pathways have been studied (McClain et al., 2004). Specific strategies include: (1) cytokine neutralization with antibodies or soluble receptors; (2) cell-bound receptor neutralization with antibodies; (3) inhibition of cytokine synthesis; (4) administration of anti-inflammatory cytokines (e.g. IL-10); and (5) removal or neutralization of proinflammatory cytokine-producing cells (McClain et al., 2004). In fact, the majority of interventions and therapeutics designed to treat ALD target cytokine metabolism (McClain et al., 1999).
Corticosteroids that inhibit proinflammatory cytokine production represent first-line drug therapy for ALD patients (McClain et al., 2004). Animal model data suggest that targeting Kupffer cell function, glut flora, or TNFα all improve outcomes of alcohol-related liver injury (McClain et al., 1997). Pilot studies in ALD patients suggested that a TNFα-soluble receptor and an anti-TNFα antibody were safe and tolerable, but human studies using infliximab (anti-TNFα antibody) and prednisone, as well as etanercept (TNFα-neutralizing molecule), have not demonstrated therapeutic efficacy and were discontinued due to increased infections (Naveau et al., 2004; Boetticher et al., 2008). The greatest limitations to existing therapeutics are infection and impaired liver regeneration as basal TNFα levels may be essential in the latter process (McClain et al., 2004). Cytokine synthesis inhibitors such as pentoxifylline, a broad phosphodiesterase inhibitor, generated promising results in patients with severe alcoholic hepatitis (AH), particularly in decreasing the risk of developing hepatorenal syndrome (Akriviadis et al., 2000; Arteel et al., 2003; De et al., 2009).
Future directions for anti-cytokine therapy will require effective strategies to reduce cytokine production while preserving therapeutic efficacy and improving safety. As previously mentioned, TNF and IL-6 are critical for liver regeneration, thus complete inhibition of these cytokines in a patient with ALD could inhibit recovery (McClain et al., 1997). While attenuation of Kupffer cell NF-κB activation with proteasome inhibitors may be a therapeutic strategy for liver injury, complete hepatocyte NF-κB inhibition may sensitize the cell to TNFα cytotoxicity (McClain et al., 1999). Furthermore, anti-cytokine therapeutics may benefit one organ system while harm another (McClain et al., 1999).
In summary, there is compelling evidence for the role of dysregulated cytokine metabolism in the systemic manifestations of ALD in both experimental animal models and patients. While anti-cytokine therapy in ALD appears promising, future clinical trials should consider numerous factors, including cytokine toxicity, drug dosing, and targeted drug delivery.
3. Opioids
3.1. Background
Opiates and opioids, which encompass synthetic opioid analgesics, play an essential role in modulating pain and antinociception (Scott, 1969; Al-Hasani and Bruchas, 2011). Approximately 10.3 million individuals aged 12 or older in the United States misused opioids in 2018 (9.9 million prescription pain reliever misusers and 808,000 heroin users), which corresponds to 3.7 percent of the population (Lipari and Park-Lee, 2019). Moreover, an estimated 2.0 million individuals aged 12 or older had an opioid use disorder, representing 0.7 percent of the population (Lipari and Park-Lee, 2019).
3.2. Opioids and Opioid Receptors
Opioid receptors are seven-transmembrane G-protein coupled receptors (GPCRs) that exist in μ-, κ-, and δ-opioid receptor (MOP, KOP, DOP) isoforms (Rogers, 2020). Opioids prescribed for pain management most often act on the MOP and exert an inhibitory effect on neuronal activity to modulate pain behavior and antinociception (Al-Hasani and Bruchas, 2011). Opioids induce analgesia by inhibiting neurons in ascending pain pathways while activating inhibitory neurons in descending pathways (Mansour et al., 1995). Receptors for opioids are expressed on multiple CNS cell types including neurons, astrocytes, and oligodendrocytes. In the context of the immune system, endogenous opioids are produced at sites of inflammation by inflammatory cells and commonly exhibit immunosuppressive activity (Rogers, 2020).
3.3. Cellular Pharmacology
Opioid receptors are primarily coupled to Gi/o signaling, and activation of opioid receptors elicits subsequent dissociation of the Gα and Gβγ subunits that lead to inhibition of adenylate cyclase and reduction of cyclic adenosine monophosphate (cAMP) levels (Pathan and Williams, 2009). These G-protein complexes further activate potassium channels and inhibit calcium channels, thereby hyperpolarizing cells and reducing neuronal excitability (Pathan and Williams, 2012). Additional downstream signals include induction of the mitogen-activated protein kinases and NF-κB signaling pathway via up-stream signaling of protein kinase A, phosphoinositide 3-kinase, or phospholipase C (Al-Hasani and Bruchas, 2011; Rogers, 2020). Following activation of downstream signaling pathways is homologous desensitization, in which the opioid receptor is phosphorylated by G protein-coupled receptor kinases (GRKs), β-arrestin is recruited to promote receptor internalization, and the desensitized receptor is degraded or re-sensitized (Rogers, 2020).
3.4. Heterologous Desensitization
GPCR desensitization may also take place by heterologous desensitization, whereby an activated GPCR desensitizes an unrelated, non-ligated receptor (Rogers, 2020). As opioid and chemokine receptors are both GPCRs, evidence demonstrating their cross-desensitization has been shown in the immune, central, and peripheral nervous systems and shown to be cell- and receptor subtype-specific (Szabo et al., 2002; Zhang et al., 2003; Chen et al., 2004; Zhang et al., 2004; Zhang et al., 2005; Chen et al. 2007; Benamar et al., 2008; Heinisch et al., 2011). Opioid and chemokine receptors have been detected on the same neurons, substantiating the potential for their interactions in the nervous system (Heinisch et al., 2011; Chen et al., 2004).
3.4.1. Opioid-induced cross-desensitization of chemokine receptors
Grimm et al. (1998) first reported opioid-induced heterologous desensitization of chemokine receptors by demonstrating that met-enkephalin, an endogenous opioid derived from proenkephalin that preferentially binds DOPs, and morphine administration inhibited the activity of CXCR1, CXCR2, CCR1, CCR5, or CCR2. Studies have shown that MOP activation induces heterologous desensitization of CCR5, but not CXCR4 (Finley et al., 2008; Szabo et al, 2003; Szabo et al., 2002). Interestingly, KOP and NOP are the only known opioid receptors to mediate cross-desensitization of CXCR4 (Rogers, 2020; Finley et al., 2008). In contrast, CXCR4 receptor activation causes desensitization of MOP and DOP receptor signaling (Chen et al., 2007; Heinisch et al., 2011). These findings identify chemokine-opioid receptor cross-talk and suggest that opioid-induced heterologous desensitization of chemokine receptors contribute to the immunosuppressive effects of opioids (Szabo et al., 2002).
3.4.2. Chemokine-induced cross-desensitization of opioid receptors
Elevated proinflammatory cytokines at sites of chronic inflammation play a role in increased pain sensitivity (Rogers, 2020). Studies have thus explored chemokine-induced heterologous desensitization of opioid receptors. Szabo et al. (2002) found that chemokine receptors CCR2, CCR5, CCR7, and CXCR4, but not CXCR1 or CXCR2, induced cross-desensitization of MOPs and DOPs. Activation of CCR1, CCR2, and CXCR1 was later shown to desensitize MOPs in neuronal and non-neuronal cells (Zhang et al., 2004). Szabo et al. (2002) found that periaqueductal gray (PAG) pretreatment with CCR5 or CXCR4 agonists inhibited the ability of MOP to elicit an analgesic response to opioid administration as measured by the cold water tail-flick (CWTF) assay (Szabo et al., 2002). CCL5 was reported to induce hyperalgesia in the CWTF test, and its effects were blocked by an antibody to the chemokine (Benamar et al., 2008). Lee et al. (2013) reported decreased pain responses in CCR5 knockout mice to chemical or inflammatory stimuli. Furthermore, Chen et al. (2007) found that activation of CCR1, CCR5, and CXCR4 in the PAG desensitized MOP and DOP, and that CX3CR1 activation in the brain desensitized MOP, DOP, and KOP. Taken together, these studies show that activation of proinflammatory chemokine receptors downregulates opioid receptor activity and enhances the perception of pain at inflammatory sites.
3.5. Chemokine-based Therapeutic Approaches
Opioids are some of the most efficacious analgesics but pose a risk for dependence by activating dopamine reward pathways that produce euphoria (Al-Hasani and Bruchas, 2011). The U.S. opioid epidemic continues to be a major medical problem: in 2017, 47,600 drug overdose deaths involved opioids (Scholl et al., 2019). Among individuals 12 or older in 2018 who misused prescription pain relievers, 63.6% reported the main reason for their misuse was to alleviate physical pain (Lipari and Park-Lee, 2018). These concerns have driven research to pursue adjunctive and non-opioid strategies for alleviating pain in an effort to reduce the risk of opioid use disorder.
In chronic neuropathic pain, which typically involves inflammation, the analgesic efficacy of opioids is reduced (Martínez-Navarro et al., 2018). The previously mentioned chemokine-opioid studies provide a potential explanation for the reduced effectiveness of opioids against neuropathic pain: chemokines released during inflammation induce cross-desensitization of opioid receptors, resulting in downregulation of opioid signaling cascades and reducing opioid efficacy. Thus, therapeutic strategies directed toward reducing chemokine signaling may be capable of enhancing the analgesic efficacy of opioids while simultaneously reducing risks of opioid dependence and other adverse opioid effects such as constipation, respiratory depression, and tolerance.
Recent preclinical studies have indeed explored whether or not co-administration of chemokine receptor antagonists (CRAs) with morphine increases opioid analgesic potency in a preclinical model of inflammatory pain (Inan et al., 2018; Inan et al., 2019, Eisenstein, 2020). Inan et al. (2018) generated dose-response curves for morphine, a CCR5 antagonist (maraviroc), and a CXCR4 antagonist (AMD3100 [Plerixafor]), and found that co-administration of either CRA with morphine resulted in enhancement of the analgesic effect on incisional pain in rats. An additional study by Inan et al. (2019) reported that co-administration of the same two CRAs with either oxycodone or meperidine, commonly prescribed analgesics, also enhanced the analgesic potency of these opioids on incisional pain in rats. Eisenstein et al. (2020) found enhanced morphine potency when CRAs were administered with a submaximal dose of morphine in three different pain assays. Taken together, these studies demonstrate enhanced analgesia when sub-therapeutic doses of opioids are combined with CRAs. These results suggest that the same analgesic effects of opioids may be obtained in lower doses when co-administered with CRAs, with a plausible mechanisms being inhibition of chemokine-mediated cross-desensitization of opioid receptors.
Studies investigating a role for cytokine and chemokine systems in the addictive effects of opioids remain limited. However, emerging evidence suggests that the rewarding effects of morphine are influenced by inflammatory responses in the mesolimbic system. For example, inhibition of glial cell activity in the nucleus accumbens reduces morphine conditioned place preference (CPP), a learned behavior when a subject prefers an environment previously associated with a reward (Zhang et al., 2012). Regarding the involvement of specific chemokine systems, a role for the CXCL12/CXCR4 system is suggested by evidence showing that rats conditioned with morphine display elevated levels of CXCL12 mRNA and protein levels in the VTA (Liu et al., 2018). Moreover, the inhibition of CXCL12 in the VTA with a neutralizing antibody inhibits the development of morphine CPP in rats and also reduces the maintenance of CPP in rats with an established morphine preference (Liu et al., 2018). Cytokines, particularly the anti-inflammatory cytokine IL-10, may also influence the addictive effects of opioids. The delivery of plasmid DNA encoding IL-10 into the nucleus accumbens of rats reduces the self-administration of remifentanil, a potent, short-acting synthetic opioid, but does not affect sucrose self-administration, suggesting reinforcer-specific effects of IL-10 (Lacagnina et al., 2017). It will be important for future studies to further investigate the impacts of CXCL12, IL-10 and other cytokines and chemokines on the addictive effects of opioids.
In summary, sub-therapeutic doses of opioids in combination with CRAs may provide a new approach to pain in inflammatory conditions resulting from surgery or injury. Eisenstein et al. (2020) reported the most pronounced results when the drugs were provided post-surgery. The two CRAs selected for the opioid-chemokine combination studies were maraviroc and AMD3100, both of which are approved for human use by the U.S. Food and Drug Administration. Thus, the administration of opioids in lower doses post-surgery in combination with maraviroc or AMD3100 may reduce the number of individuals who subsequently develop an opioid use disorder. Use of opioids in lower doses may not only prevent tolerance and dependence, but also reduce side effects including respiratory depression and gastrointestinal transit inhibition (Eisenstein et al., 2020).
4. Cocaine
4.1. Background
Cocaine is a highly addictive psychostimulant drug derived from the Erythroxylon coca plant that is native to Central and South America (Aguayo et al., 2006). The current prevalence of cocaine use lies particularly in the western hemisphere. In 2018, an estimated 977,000 Americans had a cocaine use disorder the previous year, representing 0.4 percent of the population (Lipari and Park-Lee, 2019).
4.2. Cellular Pharmacology
Cocaine effects on reward-related brain circuits, including mesolimbic and nigrostriatal dopaminergic tracks, are well established (Trecki and Unterwald, 2009). The mesolimbic pathway is a collection of dopaminergic neurons that project form the ventral tegmental area (VTA) in the midbrain to the nucleus accumbens (NAc) in the forebrain. The nigrostriatal pathway connects the substantia nigra in the midbrain with the dorsal striatum, which includes the caudate nucleus and putamen, in the forebrain. Within these systems, cocaine acts as an indirect dopamine receptor agonist by blocking dopamine reuptake through inhibition of dopamine transporters, thereby increasing extracellular dopamine in the NAc and caudate putamen (Thomas et al., 2008; Haile et al., 2012). This enhanced dopaminergic transmission underlies motivation, attention, reward, and locomotor activity produced by cocaine (Trecki and Unterwald, 2009). Research also indicates that cocaine acts within brain reward circuits to block serotonin (5-HT) and norepinephrine reuptake and alter glutamate and GABA transmission (Hall et al., 2004; Haile et al., 2012).
4.3. Therapeutic Void
The role of dopamine, glutamate, and other classical neurotransmitter systems in the pharmacological effects of cocaine is well established, but therapeutic strategies directly targeting these systems have not yet resulted in FDA-approved medications for treating cocaine use disorder. The therapeutic gap indicates the need to explore new endogenous targets. Recent studies have explored how cocaine dysregulates the neuroimmune system (Bachtell et al., 2017; Cotto et al., 2018). Drugs that inhibit glial activation have demonstrated preclinical efficacy in reducing addictive-like behaviors, but clinical utility may be limited by undesirable immunosuppressant effects (Poland et al., 2016). Thus, studies are now investigating more specific elements within the immune system, including cytokines and chemokines, which may more selectively target behavioral effects of cocaine related to dependence and addiction (Nayak et al., 2020).
4.4. Cytokine and Chemokine Biomarkers in Predicting Cocaine Use Disorder
Cocaine regulates production of cytokines and chemokines that influence neurotransmitter systems targeted by drugs of abuse (Araos et al., 2014), suggesting that dysregulation of chemokine and cytokine signaling during cocaine exposure perpetuates cocaine use disorder (Guyon et al., 2009; Trecki and Unterwald, 2009; Trocello et al., 2011). Cocaine-induced changes in chemokines may contribute to a high prevalence of co-morbid mental disorders in cocaine users (Crews et al., 2011), as studies have suggested that deficits in immune function contribute to depression, anxiety, and neurodegenerative diseases (Stuart and Baune, 2014). In fact, the utility of chemokines as pathological biomarkers or therapeutic targets has been proposed for treating mental disorders (Stuart and Baune 2014).
Araos et al. (2014) found plasma levels of TNFα, IL-1β, CXCL12, CCL2, and CX3CL1 to be elevated during acute cocaine exposure in mice but reduced during abstinence in human cocaine users. Furthermore, plasma levels of IL-1β, CXCL12, and CX3CL1 were positively associated with a number of criteria met for cocaine abuse and dependence in the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSMIV), suggesting that the severity of cocaine use disorder may be monitored through circulating cytokine and chemokine levels (Araos et al., 2014). Cytokines and chemokines may therefore serve as predictors of cocaine addiction, cocaine symptom severity, and psychiatric comorbidity in cocaine users (Araos et al., 2014). In summary, cocaine alters the plasma levels of inflammatory cytokines and chemokines, which may be used to predict the pathological use of cocaine and establish a better stratification of cocaine users seeking treatment (Araos et al., 2014).
4.5. Chemokine Receptor Antagonists in Reducing Behavioral Effects of Cocaine
Chemokine receptor systems are altered by cocaine exposure and contribute to the behavioral effects of cocaine (Nayak et al., 2020). A characteristic behavioral feature of cocaine resulting from increased dopamine transmission is hyperlocomotion. The administration of CXCR4 agonist CXCL12 intracerebroventricularly (icv) enhanced cocaine-induced hyperlocomotion (Trecki and Unterwald, 2009). Furthermore, synaptic dopamine levels in the dorsal striatum were enhanced following CXCL12 injection into the substantia nigra and subsequent CXCR4 receptor activation (Skrzydelski et al., 2007; Guyon, 2014). These observations are supported by data identifying CXCR4 receptor expression by dopamine neurons in the substantia nigra (Banisadr et al., 2002) and GABAergic medium spiny neurons in the nucleus accumbens (Trecki et al., 2010).
Kim et al. (2017) further demonstrated the role of the CXCL12/CXCR4 receptor system on the behavioral effects of cocaine. This preclinical study found that administration of the CXCR4 antagonist AMD3100 inhibited cocaine-induced hyperlocomotion and CPP. The researchers also reported increased CXCL12 mRNA levels in the VTA following repeated cocaine exposure, potentially identifying the VTA as the source of interaction between CXCL12 and dopamine. It is hypothesized that cocaine elevates extracellular dopamine levels in the VTA, which increases the expression of TNFα mRNA levels and in turn increases CXCL12 levels. Elevated CXCL12 then activates the CXCR4 receptor, enhancing mesolimbic dopamine activity and mediating the behavioral effects of cocaine (Kim et al., 2017). Thus, interactions between the neuroimmune and brain reward systems contribute to the abuse liability of cocaine (Kim et al., 2017). Interestingly, Oliver et al. (2018) found that AMD3100 also reduced CPP and locomotor activation following administration of the ‘bath salt’ synthetic cathinone MDPV (3,4-methlylenedioxypyrovalerone).
CCR2 and its cognate ligand, CCL2, may also contribute to behavioral and neurochemical effects of cocaine as both are constitutively expressed in dopaminergic neurons in the substantia nigra (Banisadr et al., 2002, 2005a, b, c). CCL2 administration into the rat substantia nigra increased extracellular dopamine levels and locomotor activity in the nigrostriatal pathway (Guyon et al., 2009). Trocello et al. (2011) demonstrated that locomotor activity is reduced in CCR2 knockout mice, suggesting that the CCR2 chemokine receptor system contributes to cocaine-induced sensitization. Thus, development of CCR2 antagonists may present a new therapeutic approach for individuals with cocaine use disorder (Trocello et al., 2011).
Recent evidence suggests that the CCR5 chemokine receptor system may also influence dopamine systems that contribute to cocaine dependence (Nayak et al., 2020). CCR5 knockout animals, compared to wild types, displayed fewer dopamine neurons in the substantia nigra, decreased dopamine levels in the striatum, and reduced locomotion (Choi et al., 2013). Dopamine neurons in the mesolimbic pathway have been suggested to produce and express the CCR5 agonist, CCL5 (Lanfranco et al., 2018). The CCR5 ligand CCL3 is also reportedly expressed by dopamine neurons and astrocytes in the substantia nigra (Kalkonde et al., 2007). Nayak et al. (2020) demonstrated enhanced CCR5 mRNA levels in the nucleus accumbens and VTA following repeated cocaine exposure. Furthermore, inhibition of CCR5 with the antagonist maraviroc reduced hyperlocomotion and CPP. It is hypothesized that cocaine-induced increases in dopamine transmission promotes downstream activation of the mesolimbic CCR5 system, thus increasing CCR5 gene expression and enhancing the reward and locomotor effects of cocaine. These results suggest that, similar to those observed with CXCR4 and CCR2, mesolimbic CCR5 receptors are altered by cocaine exposure and may influence behavioral effects that contribute to abuse liability (Nayak et al., 2020). As maraviroc is FDA-approved, the antagonist may serve as a potential therapeutic for individuals with cocaine use disorder.
5. Methamphetamine
5.1. Background
Methamphetamine (METH) is a psychostimulant and amphetamine derivative used in the form of a crystalline hydrochloride that causes severe cognitive impairments often persisting after discontinuation of use (Loftis et al., 2011). METH consumption increased 4-fold between 2015 and 2016, and age-adjusted drug overdose deaths involving METH increased 3-fold between 2011 and 2016 (Piper et al., 2018; Hedegaard et al., 2018). In 2018, approximately 1.1 million Americans aged 12 or older had a METH use disorder the previous year, representing 0.4 percent of the population (Lipari and Park-Lee, 2019).
5.2. Cellular Pharmacology
METH acts as an indirect agonist at dopamine, noradrenaline, and serotonin receptors, thereby increasing the release of these monoamines from nerve terminals in the central and peripheral nervous systems (Paulus and Stewart, 2020). METH dysregulates vesicular monoamine transporters (VMAT) that displaces monoamines from storage vesicles into the cytosol (Cruickshank and Dyer, 2009). This process further disrupts monoamine transporters at cell surface membranes, which results in the release of dopamine, noradrenaline, and serotonin into synapses. In addition, METH reduces the metabolism of these monoamines by inhibiting the enzyme monoamine oxidase (Cruickshank and Dyer, 2009).
5.3. Therapeutic Approaches
There are no approved medications with demonstrated efficacy for the treatment of METH use order as the neurobiology of the drug is more complex than just monoamine dysregulation, and pharmacological interventions targeting the monoaminergic pathways have largely failed (Paulus and Stewart, 2020). Preclinical and clinical research proposes a new therapeutic strategy targeting factors involved in METH-induced neuroinflammation (Beardsley et al., 2010; Birath et al., 2017; DeYoung et al., 2016; Loftis and Janowsky, 2014; Snider et al., 2013; Worley et al., 2016). Studies have demonstrated that cytokines, chemokines, and cell adhesion molecules may contribute to METH-induced neuronal injury and cognitive impairments (Loftis et al., 2011; Tocharus et al., 2010; Loftis et al., 2009; Narita et al., 2008). METH-induced neuroinflammation is mediated in part by activation of the TLR4 transmembrane protein within the VTA, which is highly expressed in microglia (Wendeln et al., 2018). This TLR4-mediated microglia activation then stimulates the secretion of cytokines (e.g. TNFα, IL-1β, and IL-6) and chemokines (e.g. CCL2) that can cause neuroinflammation, neuronal damage, and behavioral impairments (Clark et al., 2013; Loftis et al., 2011; Yamamoto et al., 2010; Yamamoto and Raudensky, 2008). In fact, Loftis et al. (2011) found increased plasma levels of CCL2 and the cellular adhesion molecule ICAM-1 following repeated METH exposure in both mice and humans. The cytokine and chemokine changes were also associated with increased cognitive impairments in human participants (Loftis et al., 2011). These studies suggest a critical role for CNS immune dysregulation in METH neurotoxicity and addiction.
Ibudilast is a nonspecific phosphodiesterase and glial activation inhibitor that has been shown to reduce METH locomotor activity, self-administration, and relapse in preclinical studies (Brensilver et al., 2013; Worley et al., 2016). Li et al. (2020) demonstrated clinically that ibudilast treatment reduced METH-induced levels of proinflammatory mediators, including adhesion molecules sICAM-1 and sVCAM-1 and the lysosomal protease cathepsin D. These findings identify glial cell activity as a critical regulator of the behavioral effects of METH and put forward ibudilast as a compound to be further studied as a putative therapeutic for METH use disorder (Li et al., 2020).
CCL2 is derived from neurons and glial cells and enhances dopamine release from neurons in the substantia nigra into the striatum (Wakida et al., 2014). It is suggested that CCL2-mediated neuroinflammation is associated with the pathogenesis of drug abuse (Crews et al., 2011). Ikegami et al. (2010) demonstrated upregulation of the CCL2 receptor CCR2 following METH administration; CCR2 may therefore be involved in maintaining sensitization to MA-induced hyperlocomotion as this behavior was significantly decreased in CCR2 knockout mice. Wakida et al. (2014) reported increased CCL2 mRNA levels following METH administration in both the prefrontal cortex and nucleus accumbens. METH-induced CPP was also suppressed by a CCR2 antagonist, RS504393 (Wakida et al., 2014). METH-induced increases in phosphorylated tyrosine hydroxylase (pTH) levels, the rate-limiting enzyme for dopamine synthesis, was also attenuated by RS504393 in the VTA. Thus, CCL2-CCR2 activation of dopamine neurons enhances physiological systems that facilitate METH addiction, and therapeutics targeting CCL2-CCR2 may be effective in treating METH dependence (Wakida et al., 2014).
In addition to activation of the CCR2 chemokine receptor system, Basova et al. (2018) found enhanced CCR5 gene expression following METH administration in a THP1 human macrophage cell line. Dopamine acting on the dopamine receptor DRD1 appeared to be the main factor in suppressing the CCR5 gene promoter toward transcription, thus suggesting a role for DRD1 agonists in regulating CCR5 expression (Basova et al., 2018). Future studies should explore both the CCR2 and CCR5 chemokine receptor systems as potential therapeutic targets for METH use disorder.
6. Conclusion
Central functions of cytokines and chemokines are not restricted to neuroinflammation and chemotaxis but extend to regulation of physiological systems that underlie addiction. In the present review, we discussed how these highly versatile inflammatory proteins facilitate the intersection between the immune and central nervous systems in the context of substance use disorders. In addition to providing a critical review of cytokine and chemokine mechanisms underlying substance use disorders, we also discussed cytokine- and chemokine-based therapeutic strategies. These approaches are entirely different from more conventional strategies directed toward receptors, transporters and enzymes that facilitate the biological actions of established neurotransmitters such as dopamine, glutamate, acetylcholine, and serotonin. Although speculative, an advantage of targeting cytokine and chemokine systems is their potential capability to inhibit the rewarding and reinforcing efficacy of different classes of drugs of abuse, including alcohol, opioids and psychostimulants by reducing cytokine- and chemokine-induced increases in mesolimbic dopaminergic transmission (Fig. 1). In terms of opioids, antagonists of chemokine receptors, especially CXCR4 and CCR5, may act as bifunctional opioid modulators by 1) preventing μ-opioid receptor desensitization, thus enhancing and preserving analgesic efficacy, while 2) simultaneously reducing the abuse liability of opioids through inhibition of mesolimbic dopamine transmission. Given that antagonists of some chemokine receptors (e.g. CXCR4, CCR5) enhance opioid-induced analgesia (Inan et al., 2018, 2019), it will be important in future studies to determine how specific cytokine and chemokine receptor antagonists affect adverse effects of opioids, such as dependence, relapse, constipation, and respiratory depression. In summary, these recent advances appear promising for developing cytokine- and chemokine-based pharmacotherapies for the prevention and treatment of substance use disorders.
Fig. 1.
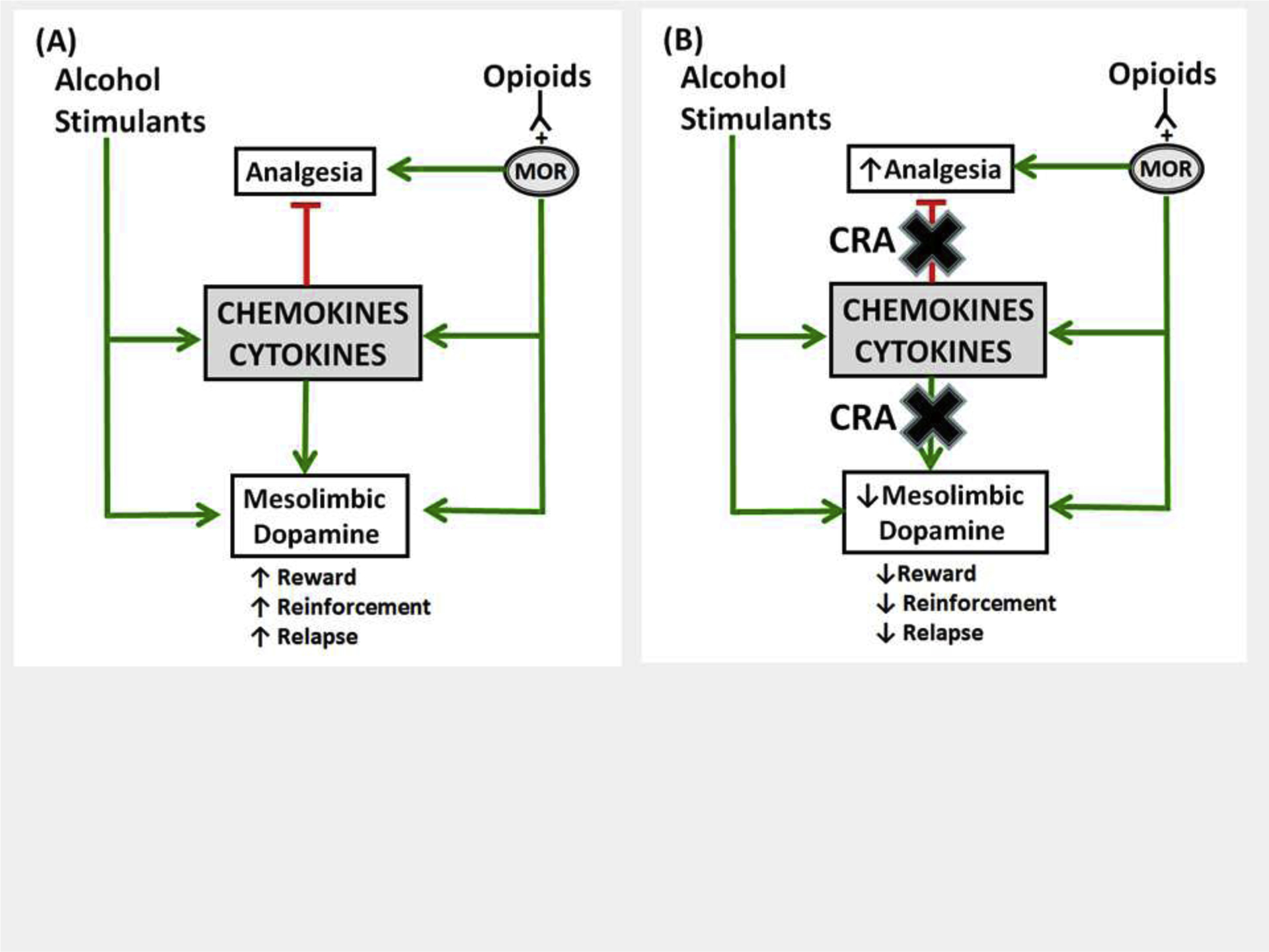
Model demonstrating potential interactions between drugs of abuse and cytokine/chemokine systems and putative therapeutic targets for substance use disorders (green lines = increase, red lines = decrease, MOR = μ-opioid receptor). (A) Exposure to different classes of drugs of abuse (psychostimulants, alcohol, opioids) causes an increase in brain levels of pro-inflammatory cytokines and chemokines (e.g. TNFα, IL-1β, CCL2, CCL5, and CXCL12). This leads to activation of specific receptors (e.g. CCR2, CCR5, and CXCR4) that enhances dopamine transmission in the limbic system, which exacerbates rewarding and reinforcing effects of drugs of abuse and relapse to drug seeking, and, in the case of opioids, reduces analgesic efficacy. (B) Cytokine and chemokine receptor antagonists (CRA) block the enhancement in dopamine transmission, thereby reducing drug reward, reinforcement and relapse, but enhance the analgesic efficacy of opioids by preventing desensitization of μ-opioid receptors (MOR).
Research Highlights.
Cytokines offer new therapeutic targets for substance use disorders.
Chemokines offer new therapeutic targets for substance use disorders.
Cytokine and chemokine dysregulation by ethanol, opioids and stimulants is reviewed.
Putative cytokine- and chemokine-based anti-addictive therapies are reviewed.
Acknowledgements
The authors would like to thank the National Institute of Health and National Institute on Drug Abuse for funding through grants R01 DA039139, R01 DA045499, R01 DA051205-01, and P30 DA013429.
Role of Funding Source
This study was supported by NIDA/NIH grants R01 DA039139, R01 DA045499, R01 DA051205-01, and P30 DA013429.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
All authors declare that they have no conflicts of interest.
References
- Aguayo LG, Guzman L, Perez C, Aguayo LJ, Silva M, Becerra J, & Fuentealba J, 2006. Historical and current perspectives of neuroactive compounds derived from Latin America. Mini-Reviews in Medicinal Chemistry, 6(9), 997–1008. [DOI] [PubMed] [Google Scholar]
- Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, & Shakil O 2000. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: A double-blind, placebo-controlled trial. Gastroenterology, 119(6), 1637–1648. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, & Guerri C 2010. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. Journal of Neuroscience, 30(24), 8285–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hasani R, & Bruchas MR 2011. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology, 115(6), 1363–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araos P, Pedraz M, Serrano A, Lucena M, Barrios V, García-Marchena N, Campos-Cloute R, Ruiz JJ, Romero P, Suárez J, Baixeras E 2015. Plasma profile of pro-inflammatory cytokines and chemokines in cocaine users under outpatient treatment: Influence of cocaine symptom severity and psychiatric co-morbidity. Addiction Biology, 20(4), 756–772. [DOI] [PubMed] [Google Scholar]
- Arteel G, Marsano L, Mendez C, Bentley F, & McClain CJ 2003. Advances in alcoholic liver disease. Best Practice & Research Clinical Gastroenterology, 17(4), 625–647. [DOI] [PubMed] [Google Scholar]
- Bachtell R, Hutchinson MR, Wang X, Rice KC, Maier SF, Watkins LR 2015. Targeting the toll of drug abuse: the translational potential of Toll-like receptor 4. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders), 14(6), 692–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtell RK, Jones JD, Heinzerling KG, Beardsley PM, Comer SD 2017. Glial and neuroinflammatory targets for treating substance use disorders. Drug and alcohol dependence, 180, 156–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Florio T, Costa A, Schettini G 1999. Expression of Chemokine Receptors in the Rat Brain a. Annals of the New York Academy of Sciences, 876(1), 201–9. [DOI] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Florio T, Schettini G 2001. Chemokines and their receptors in the central nervous system. Frontiers in neuroendocrinology, 22(3), 147–84. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Fontanges P, Haour F, Kitabgi P, Rostène W, Mélik Parsadaniantz S 2002. Neuroanatomical distribution of CXCR4 in adult rat brain and its localization in cholinergic and dopaminergic neurons. European Journal of Neuroscience, 16(9), 1661–71. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin RD, Mechighel P, Rostène W, Kitabgi P, Mélik Parsadaniantz S 2005. Constitutive neuronal expression of CCR2 chemokine receptor and its colocalization with neurotransmitters in normal rat brain: functional effect of MCP-1/CCL2 on calcium mobilization in primary cultured neurons. Journal of Comparative Neurology, 492(2),178–92. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Gosselin R, Mechighel P, Kitabgi P, Rostene W, Mélik Parsadaniantz S 2005, Highly regionalized neuronal expression of MCP-1/CCL2 in rat brain. Evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol, 489, 275–92. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Rostène W, Kitabgi P, Parsadaniantz SM 2005. Chemokines and brain functions. Current Drug Targets-Inflammation & Allergy, 4(3), 387–99. [DOI] [PubMed] [Google Scholar]
- Banisadr G, Queraud-Lesaux F, Boutterin MC, Pelaprat D, Zalc B, Rostene W, Haour F, Melik Parsadaniantz S 2002. Distribution, cellular localization and functional role of CCR2 chemokine receptors in adult rat brain. Journal of neurochemistry, 81(2), 257–69. [DOI] [PubMed] [Google Scholar]
- Basova L, Najera JA, Bortell N, Wang D, Moya R, Lindsey A, Semenova S, Ellis RJ, Marcondes MC 2018. Dopamine and its receptors play a role in the modulation of CCR5 expression in innate immune cells following exposure to methamphetamine: implications to HIV infection. PloS one, 13(6), e0199861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardsley PM, Shelton KL, Hendrick E, Johnson KW 2010. The glial cell modulator and phosphodiesterase inhibitor, AV411 (ibudilast), attenuates prime-and stress-induced methamphetamine relapse. European journal of pharmacology, 637(1–3), 102–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benamar K, Geller EB, Adler MW 2008. Elevated level of the proinflammatory chemokine, RANTES/CCL5, in the periaqueductal grey causes hyperalgesia in rats. European journal of pharmacology, 592(1–3), 93–5. [DOI] [PubMed] [Google Scholar]
- Birath JB, Briones M, Amaya S, Shoptaw S, Swanson AN, Tsuang J, Furst B, Heinzerling K, Obermeit L, Maes L, McKay C 2017. Ibudilast may improve attention during early abstinence from methamphetamine. Drug and Alcohol Dependence, 178, 386–90. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA 2011. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun, 25, S92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Bergeson SE, Walker D, Ferreira VM, Kuziel WA, Harris RA 2005. Perturbation of chemokine networks by gene deletion alters the reinforcing actions of ethanol. Behavioural brain research, 165(1), 110–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Ponomarev I, Geil C, Bergeson S, Koob GF, Harris RA 2012. Neuroimmune regulation of alcohol consumption: behavioral validation of genes obtained from genomic studies. Addiction biology, 17(1), 108–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetticher NC, Peine CJ, Kwo P, Abrams GA, Patel T, Aqel B, Boardman L, Gores GJ, Harmsen WS, McClain CJ, Kamath PS 2008. A randomized, double-blinded, placebo-controlled multicenter trial of etanercept in the treatment of alcoholic hepatitis. Gastroenterology, 135(6), 1953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger LM 2009. Immune proteins in brain development and synaptic plasticity. Neuron, 64(1), 93–109. [DOI] [PubMed] [Google Scholar]
- Brensilver M, Heinzerling KG, Shoptaw S 2013. Pharmacotherapy of amphetamine-type stimulant dependence: an update. Drug and alcohol review, 32(5), 449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DY, Lee MK, Hong JT 2013. Lack of CCR5 modifies glial phenotypes and population of the nigral dopaminergic neurons, but not MPTP-induced dopaminergic neurodegeneration. Neurobiology of disease, 49, 159–68. [DOI] [PubMed] [Google Scholar]
- Calì C, Bezzi P 2010. CXCR4-mediated glutamate exocytosis from astrocytes. Journal of neuroimmunology, 224(1–2), 13–21. [DOI] [PubMed] [Google Scholar]
- Chen X, Geller EB, Rogers TJ, Adler MW 2007. Rapid heterologous desensitization of antinociceptive activity between mu or delta opioid receptors and chemokine receptors in rats. Drug and alcohol dependence, 88(1), 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Geller EB, Rogers TJ, Adler MW 2007. The chemokine CX3CL1/fractalkine interferes with the antinociceptive effect induced by opioid agonists in the periaqueductal grey of rats. Brain research, 1153, 52–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen LY 2004. Heterodimerization and cross-desensitization between the μ-opioid receptor and the chemokine CCR5 receptor. European journal of pharmacology, 483(2–3), 175–86. [DOI] [PubMed] [Google Scholar]
- Clark KH, Wiley CA, Bradberry CW 2013. Psychostimulant abuse and neuroinflammation: emerging evidence of their interconnection. Neurotoxicity research, 23(2), 174–88. [DOI] [PubMed] [Google Scholar]
- Cotto B, Li H, Tuma RF, Ward SJ, Langford D 2018. Cocaine-mediated activation of microglia and microglial MeCP2 and BDNF production. Neurobiology of disease, 117, 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews F 2012. Inflammasome-IL-1β signaling mediates ethanol inhibition of hippocampal neurogenesis. Frontiers in neuroscience, 6, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Collins MA, Dlugos C, Littleton J, Wilkins L, Neafsey EJ, Pentney R, Snell LD, Tabakoff B, Zou J, Noronha A 2004. Alcohol-induced neurodegeneration: when, where and why?. Alcoholism: Clinical and Experimental Research, 28(2), 350–64. [DOI] [PubMed] [Google Scholar]
- Crews F, Nixon K, Kim D, Joseph J, Shukitt-Hale B, Qin L, Zou J 2006. BHT blocks NF-κB activation and ethanol-induced brain damage. Alcoholism: Clinical and Experimental Research, 30(11), 1938–49. [DOI] [PubMed] [Google Scholar]
- Crews FT, Zou J, Qin L 2011. Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav Immun, 25:S4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank CC, Dyer KR 2009. A review of the clinical pharmacology of methamphetamine. Addiction, 104(7), 1085–99. [DOI] [PubMed] [Google Scholar]
- Cui C, Shurtleff D, Harris RA 2014. Neuroimmune mechanisms of alcohol and drug addiction International review of neurobiology (Vol. 118, pp. 1–12). Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De BK, Gangopadhyay S, Dutta D, Baksi SD, Pani A, Ghosh P 2009. Pentoxifylline versus prednisolone for severe alcoholic hepatitis: a randomized controlled trial. World journal of gastroenterology: WJG, 15(13), 1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYoung DZ, Heinzerling KG, Swanson AN, Tsuang J, Furst BA, Yi Y, Wu YN, Moody DE, Andrenyak DM, Shoptaw SJ 2016. Safety of intravenous methamphetamine administration during ibudilast treatment. Journal of clinical psychopharmacology, 36(4), 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Xuei X, Wetherill LF, Bierut L, Bucholz K, Dick DM, Hesselbrock V, Kuperman S, Porjesz B, Schuckit MA, Tischfield JA 2008. Association of NFKB1, which encodes a subunit of the transcription factor NF-κB, with alcohol dependence. Human molecular genetics, 17(7), 963–70. [DOI] [PubMed] [Google Scholar]
- Eisenstein TK, Chen X, Inan S, Meissler JJ, Tallarida CS, Geller EB, Rawls SM, Cowan A, Adler MW 2020. Chemokine Receptor Antagonists in Combination with Morphine as a Novel Strategy for Opioid Dose Reduction in Pain Management. Military Medicine, 185(Supplement_1), 130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Guerri C 2009. Critical role of TLR4 response in the activation of microglia induced by ethanol. The Journal of Immunology, 183(7), 4733–44. [DOI] [PubMed] [Google Scholar]
- Finley MJ, Chen X, Bardi G, Davey P, Geller EB, Zhang L, Adler MW, Rogers TJ 2008. Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the κ-opioid receptor. Journal of neuroimmunology, 197(2), 114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabuzda D, He J, Ohagen A, Vallat AV 1998. Chemokine receptors in HIV-1 infection of the central nervous system In Seminars in immunology (Vol. 10, No. 3, pp. 203–213). Academic Press. [DOI] [PubMed] [Google Scholar]
- González-Quintela A, Dominguez-Santalla MJ, Loidi L, Quinteiro C, Perez LF 2004. Relation of tumor necrosis factor (TNF) gene polymorphisms with serum concentrations and in vitro production of TNF-alpha and interleukin-8 in heavy drinkers. Alcohol, 34(2–3), 273–7. [DOI] [PubMed] [Google Scholar]
- Grimm MC, Ben-Baruch A, Taub DD, Howard OM, Resau JH, Wang JM, Ali H, Richardson R, Snyderman R, Oppenheim JJ 1998. Opiates transdeactivate chemokine receptors: δ and μ opiate receptor–mediated heterologous desensitization. The Journal of experimental medicine, 188(2),317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon A 2014. CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Frontiers in cellular neuroscience, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyon A, Skrzydelski D, De Giry I, Rovere C, Conductier G, Trocello JM, Dauge V, Kitabgi P, Rostene W, Nahon JL, Parsadaniantz SM 2009. Long term exposure to the chemokine CCL2 activates the nigrostriatal dopamine system: a novel mechanism for the control of dopamine release. Neuroscience, 162(4),1072–80. [DOI] [PubMed] [Google Scholar]
- Haile CN, Mahoney III JJ, Newton TF, De La Garza R II 2012. Pharmacotherapeutics directed at deficiencies associated with cocaine dependence: focus on dopamine, norepinephrine and glutamate. Pharmacology & therapeutics, 134(2), 260–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Sora I, Drgonova J, LI XF, Goeb M, Uhl GR 2004. Molecular mechanisms underlying the rewarding effects of cocaine. Annals of the New York Academy of Sciences, 1025(1), 47–56. [DOI] [PubMed] [Google Scholar]
- He J, Crews FT 2008. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Experimental neurology, 210(2), 349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedegaard H, Bastian BA, Trinidad JP, Spencer M, Warner M 2018. Drugs most frequently involved in drug overdose deaths: United States, 2011–2016. Natl Vital Stat Rep, 67(9), 1–14. [PubMed] [Google Scholar]
- Heinisch S, Palma J, Kirby LG 2011. Interactions between chemokine and mu-opioid receptors: anatomical findings and electrophysiological studies in the rat periaqueductal grey. Brain Behav Immun, 25(2), 360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami D, Narita M, Imai S, Miyashita K, Tamura R, Narita M, Takagi S, Yokomizo A, Takeshima H, Ando T, Igarashi K 2010. PRECLINICAL STUDY: BRIEF REPORT: Epigenetic modulation at the CCR2 gene correlates with the maintenance of behavioral sensitization to methamphetamine. Addiction biology, 15(3), 358–61. [DOI] [PubMed] [Google Scholar]
- Inan S, Watson MN, Doura M, Tallarida CS, Meissler JJ, Chen X, Geller EB, Cowan A, Rawls SM, Eisenstein TK, Adler MW 2019. Chemokine receptor antagonists enhance the antinociceptive activity of oxycodone and meperidine on incisional pain in rats. British journal of anaesthesia, 122(6), e213–5. [DOI] [PubMed] [Google Scholar]
- Kalkonde YV, Morgan WW, Sigala J, Maffi SK, Condello C, Kuziel W, Ahuja SS, Ahuja SK 2007. Chemokines in the MPTP model of Parkinson’s disease: absence of CCL2 and its receptor CCR2 does not protect against striatal neurodegeneration. Brain research, 1128, 1–1. [DOI] [PubMed] [Google Scholar]
- Kebir O, Gorsane MA, Blecha L, Krebs MO, Reynaud M, Benyamina A 2011. Association of inflammation genes with alcohol dependence/abuse: a systematic review and a meta-analysis. European addiction research, 17(3),146–53. [DOI] [PubMed] [Google Scholar]
- Khan MZ, Brandimarti R, Patel JP, Huynh N, Wang J, Huang Z, Fatatis A, Meucci O 2004. Apoptotic and antiapoptotic effects of CXCR4: is it a matter of intrinsic efficacy? Implications for HIV neuropathogenesis. AIDS Research & Human Retroviruses, 20(10), 1063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer F, Jahn H, Schick M, Wiedemann K 2002. Alcohol intake, tumour necrosis factor-α, leptin and craving: factors of a possibly vicious circle? Alcohol and Alcoholism, 37(4), 401–4. [DOI] [PubMed] [Google Scholar]
- Kim J, Connelly KL, Unterwald EM, Rawls SM 2017. Chemokines and cocaine: CXCR4 receptor antagonist AMD3100 attenuates cocaine place preference and locomotor stimulation in rats. Brain Behav Immun., 62, 30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND 2010. Neurocircuitry of addiction. Neuropsychopharmacology, 35(1), 217–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacagnina MJ, Kopec AM, Cox SS, Hanamsagar R, Wells C, Slade S, Grace PM, Watkins LR, Levin ED, Bilbo SD 2017. Opioid Self-Administration is Attenuated by Early-Life Experience and Gene Therapy for Anti-Inflammatory IL-10 in the Nucleus Accumbens of Male Rats. Neuropsychopharmacology. 42(11), 2128–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranco MF, Mocchetti I, Burns MP, Villapol S 2018. Glial-and neuronal-specific expression of CCL5 mRNA in the rat brain. Frontiers in Neuroanatomy, 11, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq S, Cani PD, Neyrinck AM, Stärkel P, Jamar F, Mikolajczak M, Delzenne NM, de Timary P 2012. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun, 26(6), 911–8. [DOI] [PubMed] [Google Scholar]
- Leclercq S, De Saeger C, Delzenne N, de Timary P, Stärkel P 2014. Role of inflammatory pathways, blood mononuclear cells, and gut-derived bacterial products in alcohol dependence. Biological psychiatry, 76(9), 725–33. [DOI] [PubMed] [Google Scholar]
- Li MJ, Briones MS, Heinzerling KG, Kalmin MM, Shoptaw SJ 2020. Ibudilast attenuates peripheral inflammatory effects of methamphetamine in patients with methamphetamine use disorder. Drug and Alcohol Dependence, 206, 107776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipari R, Park-Lee E 2019. Key Substance Use and Mental Health Indicators in the United States: Results from the 2018 National Surgery on Drug Use and Health (NSDUH). Washington, D.C. Substance Abuse and Mental Health Services Administration, DHHS; Available at https://store.samhsa.gov/product/key-substance-use-and-mental-health-indicators-in-the-united-states-results-from-the-2018-national-survey-on-Drug-Use-and-Health/PEP19-5068; accessed July 27, 2020. [Google Scholar]
- Liu H, Wei J, Liu M, Wu S, Ma C, Liu C, Huang K, Zhang X, Guo R, Zhang K, Xin W, 2018. Epigenetic upregulation of CXCL12 expression contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Exp Neurol, 306, 55–63. [DOI] [PubMed] [Google Scholar]
- Liu J, Lewohl JM, Harris RA, Iyer VR, Dodd PR, Randall PK, Mayfield RD 2006. Patterns of gene expression in the frontal cortex discriminate alcoholic from nonalcoholic individuals. Neuropsychopharmacology, 31(7), 1574–82. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL, Elnabawi A, Merchenthaler I, Sieghart W, Aurelian L 2011. Binge alcohol drinking is associated with GABAA α2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proceedings of the National Academy of Sciences, 108(11), 4465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis JM, Choi D, Hoffman W, Huckans MS 2011. Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotoxicity research, 20(1), 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis JM, Huckans MS, Williams A 79. 2009. Methamphetamine administration causes increased neuroinflammation accompanied by peripheral immunosuppression in mice. Brain Behavior and Immunity, (23):S46–7. [Google Scholar]
- Loftis JM, Janowsky A 2014. Neuroimmune basis of methamphetamine toxicity International review of neurobiology (Vol. 118, pp. 165–197). Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ 1995. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends in neurosciences, 18(1), 22–9. [DOI] [PubMed] [Google Scholar]
- Marcos M, Pastor I, González-Sarmiento R, Laso FJ 2008. Interleukin-10 gene polymorphism is associated with alcoholism but not with alcoholic liver disease. Alcohol & Alcoholism, 43(5), 523–8. [DOI] [PubMed] [Google Scholar]
- Martínez-Navarro M, Maldonado R, Baños JE 2019. Why mu-opioid agonists have less analgesic efficacy in neuropathic pain?. European Journal of Pain, 23(3), 435–54. [DOI] [PubMed] [Google Scholar]
- Mayfield J, Ferguson L, Harris RA 2013. Neuroimmune signaling: a key component of alcohol abuse. Current opinion in neurobiology, 23(4), 513–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D 1999. Cytokines in alcoholic liver disease In Seminars in liver disease (Vol. 19, No. 02, pp. 205–219). © 1999 by Thieme Medical Publishers, Inc.. [DOI] [PubMed] [Google Scholar]
- McClain CJ, Cohen DA 1989. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology, 9(3), 349–51. [DOI] [PubMed] [Google Scholar]
- McClain CJ, Shedlofsky S, Barve S, Hill DB 1997. Cytokines and alcoholic liver disease. Alcohol Health and Research World, 21(4), 317. [PMC free article] [PubMed] [Google Scholar]
- McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I 2004. Recent advances in alcoholic liver disease IV. Dysregulated cytokine metabolism in alcoholic liver disease. American Journal of Physiology-Gastrointestinal and Liver Physiology, 287(3), G497–502. [DOI] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ 1998. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proceedings of the National Academy of Sciences, 95(24), 14500–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA, Crabbe JC, Blednov YA, Grahame NJ, Phillips TJ, Finn DA 2006. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proceedings of the National Academy of Sciences, 103(16), 6368–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PM 2002. International Union of Pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacological reviews, 54(2), 227–9. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Baggiolini M, Charo IF, Hébert CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ, Power CA 2000. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacological reviews, 52(1), 145–76. [PubMed] [Google Scholar]
- Narita M, Suzuki M, Kuzumaki N, Miyatake M, Suzuki T 2008. Implication of activated astrocytes in the development of drug dependence: differences between methamphetamine and morphine. Annals of the New York Academy of Sciences, 1141(1), 96–104. [DOI] [PubMed] [Google Scholar]
- Nash B, Meucci O 2014. Functions of the chemokine receptor CXCR4 in the central nervous system and its regulation by μ-opioid receptors International review of neurobiology (Vol. 118, pp. 105–128). Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, Davion T, Oberti F, Broët P, Emilie D 2004. Foie-Alcool group of the Association Française pour l’Etude du Foie (AFEF). A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology, 39(5), 1390–7. [DOI] [PubMed] [Google Scholar]
- Nayak SU, Cicalese S, Tallarida C, Oliver CF, Rawls SM 2020. Chemokine CCR5 and cocaine interactions in the brain: Cocaine enhances mesolimbic CCR5 mRNA levels and produces place preference and locomotor activation that are reduced by a CCR5 antagonist. Brain Behav Immun., 83, 288–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CF, Simmons SJ, Nayak SU, Smith GR, Reitz AB, Rawls SM 2018. Chemokines and ‘bath salts’: CXCR4 receptor antagonist reduces rewarding and locomotor-stimulant effects of the designer cathinone MDPV in rats. Drug and alcohol dependence, 186, 75–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsadaniantz SM, Rivat C, Rostène W, Réaux-Le Goazigo A 2015. Opioid and chemokine receptor crosstalk: a promising target for pain therapy?. Nature Reviews Neuroscience, 16(2), 69–78. [DOI] [PubMed] [Google Scholar]
- Pastor IJ, Laso FJ, Àvila JJ, Rodriguez RE, González-Sarrniento R 2000. Polymorphism in the Interleukin-1 receptor antagonist gene is associated with alcoholism in spanish men. Alcoholism: Clinical and Experimental Research, 24(10), 1479–82. [PubMed] [Google Scholar]
- Pastor IJ, Laso FJ, Romero A, González-Sarmiento R 2005. Interleukin-1 gene cluster polymorphisms and alcoholism in Spanish men. Alcohol and Alcoholism, 40(3), 181–6. [DOI] [PubMed] [Google Scholar]
- Pathan H, Williams J 2012. Basic opioid pharmacology: an update. British journal of pain, 6(1), 11–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus MP, Stewart JL 2020. Neurobiology, Clinical Presentation, and Treatment of Methamphetamine Use Disorder: A Review. JAMA psychiatry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper BJ, Ogden CL, Simoyan OM, Chung DY, Caggiano JF, Nichols SD, McCall KL 2018. Trends in use of prescription stimulants in the United States and Territories, 2006 to 2016. PloS one, 13(11), e0206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland RS, Hahn YK, Knapp PE, Beardsley PM, Bowers MS 2016. Ibudilast attenuates expression of behavioral sensitization to cocaine in male and female rats. Neuropharmacology, 109, 281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT 2008. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. Journal of neuroinflammation, 5(1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TJ 2020. Bidirectional Regulation of Opioid and Chemokine Function. Front. Immunol. 11:94. doi: 10.3389/fimmu.2020.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostène W, Kitabgi P, Parsadaniantz SM 2007. Chemokines: a new class of neuromodulator?. Nature Reviews Neuroscience, 8(11), 895–903. [DOI] [PubMed] [Google Scholar]
- Saiz PA, Garcia-Portilla MP, Florez G, Corcoran P, Arango C, Morales B, Leza JC, Alvarez S, Díaz EM, Alvarez V, Coto E 2009. Polymorphisms of the IL-1 gene complex are associated with alcohol dependence in spanish caucasians: Data from an association study. Alcoholism: Clinical and Experimental Research, 33(12), 2147–53. [DOI] [PubMed] [Google Scholar]
- Scholl L, Seth P, Kariisa M, Wilson N, Baldwin G 2019. Drug and opioid-involved overdose deaths—United States, 2013–2017. Morbidity and Mortality Weekly Report, 67(51–52), 1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JM 1969. The white poppy. A history of opium. Cox and Wyman Ltd., London, England. [Google Scholar]
- Sheron N, Bird G, Koskinas J, Portmann B, Ceska M, Lindley I, William R 1993. Circulating and tissue levels of the neutrophil chemotaxin interleukin-8 are elevated in severe acute alcoholic hepatitis, and tissue levels correlate with neutrophil infiltration. Hepatology, 18(1), 41–6. [PubMed] [Google Scholar]
- Skrzydelski D, Guyon A, Dauge V, Rovere C, Apartis E, Kitabgi P, Nahon JL, Rostene W, Parsadaniantz SM 2007. The chemokine stromal cell-derived factor-1/CXCL12 activates the nigrostriatal dopamine system. Journal of neurochemistry, 102(4), 1175–83. [DOI] [PubMed] [Google Scholar]
- Snider SE, Hendrick ES, Beardsley PM 2013. Glial cell modulators attenuate methamphetamine self-administration in the rat. European journal of pharmacology, 701(1–3), 124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Rahim RT, Davey PC, Bednar F, Bardi G, Zhang L, Zhang N, Oppenheim JJ, Rogers TJ 2011. Protein kinase Cζ mediates μ-opioid receptor-induced cross-desensitization of chemokine receptor CCR5. Journal of Biological Chemistry, 286(23), 20354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart MJ, Baune BT 2014. Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: a systematic review of biomarker studies. Neuroscience & Biobehavioral Reviews, 42, 93–115. [DOI] [PubMed] [Google Scholar]
- Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ 2002. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proceedings of the National Academy of Sciences, 99(16), 10276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen LY, Bednar F, Henderson EE, Howard OZ, Oppenheim JJ 2003. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. Journal of leukocyte biology, 74(6), 1074–82. [DOI] [PubMed] [Google Scholar]
- Tashiro K, Tada H, Heilker R, Shirozu M, Nakano T, Honjo T 1993. Signal sequence trap: a cloning strategy for secreted proteins and type I membrane proteins. Science, 261(5121), 600–3. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y 2008. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. British journal of pharmacology, 154(2), 327–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocharus J, Khonthun C, Chongthammakun S, Govitrapong P 2010. Melatonin attenuates methamphetamine-induced overexpression of pro-inflammatory cytokines in microglial cell lines. Journal of pineal research, 48(4), 347–52. [DOI] [PubMed] [Google Scholar]
- Trecki J, Brailoiu GC, Unterwald EM 2010. Localization of CXCR4 in the forebrain of the adult rat. Brain research, 1315, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trecki J, Unterwald EM 2009. Modulation of cocaine-induced activity by intracerebral administration of CXCL12. Neuroscience, 161(1), 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trocello JM, Rostene W, Melik-Parsadaniantz S, Godefroy D, Roze E, Kitabgi P, Kuziel WA, Chalon S, Caboche J, Apartis E 2011. Implication of CCR2 chemokine receptor in cocaine-induced sensitization. Journal of Molecular Neuroscience, 44(3), 147–51. [DOI] [PubMed] [Google Scholar]
- Wakida N, Kiguchi N, Saika F, Nishiue H, Kobayashi Y, Kishioka S 2014. CC-chemokine ligand 2 facilitates conditioned place preference to methamphetamine through the activation of dopamine systems. Journal of pharmacological sciences, 125(1), 68–73. [DOI] [PubMed] [Google Scholar]
- Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Häsler LM, Wild K, Skodras A, Blank T 2018. Innate immune memory in the brain shapes neurological disease hallmarks. Nature, 556(7701), 332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Drug Report 2020. (United Nations publication, Sales No. E.20.XI.6). Available at https://wdr.unodc.org/wdr2020/field/WDR20_BOOKLET_1.pdf; accessed August 8, 2020.
- Worley MJ, Heinzerling KG, Roche DJ, Shoptaw S 2016. Ibudilast attenuates subjective effects of methamphetamine in a placebo-controlled inpatient study. Drug and alcohol dependence, 162, 245–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Moszczynska A, Gudelsky GA 2010. Amphetamine toxicities Classical and bRemerging mechanisms. Annals of the New York Academy of Sciences, 1187, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Raudensky J 2008. The role of oxidative stress, metabolic compromise, and inflammation in neuronal injury produced by amphetamine-related drugs of abuse. Journal of Neuroimmune Pharmacology, 3(4), 203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XQ, Cui Y, Cui Y, Chen Y, Na XD, Chen FY, Wei XH, Li YY, Liu XG, Xin WJ, 2012. Activation of p38 signaling in the microglia in the nucleus accumbens contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Brain Behav Immun, 26(2), 318–25. [DOI] [PubMed] [Google Scholar]
- Zhang N, Hodge D, Rogers TJ, Oppenheim JJ 2003. Ca2+-independent protein kinase Cs mediate heterologous desensitization of leukocyte chemokine receptors by opioid receptors. Journal of Biological Chemistry, 278(15), 12729–36. [DOI] [PubMed] [Google Scholar]
- Zhang N, Rogers TJ, Caterina M, Oppenheim JJ 2004. Proinflammatory chemokines, such as CC chemokine ligand 3, desensitize μ-opioid receptors on dorsal root ganglia neurons. The Journal of Immunology, 173(1):594–9. [DOI] [PubMed] [Google Scholar]
- Zheng J, Thylin MR, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng YC, Gelbard HA, Shepard RB 1999. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. Journal of neuroimmunology, 98(2), 185–200. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews F 2006. CREB and NF-κB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cellular and molecular neurobiology, 26(4–6), 383–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Crews F 2010. Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-κB and proinflammatory cytokines. Alcoholism: Clinical and Experimental Research, 34(5), 777–89. [DOI] [PubMed] [Google Scholar]