

Abstract
The vascular adventitia contains numerous cell types including fibroblasts, adipocytes, inflammatory cells, and progenitors embedded within a complex extracellular matrix (ECM) network. In response to vascular injury, adventitial progenitors and fibroblasts become activated and exhibit increased proliferative capacity and differentiate into contractile cells that remodel the ECM. These processes can lead to vascular fibrosis and disease progression. Our previous work established that the ECM protein aortic carboxypeptidase-like protein (ACLP) promotes fibrotic remodeling in the lung and is activated by vascular injury. It is currently unknown what controls vascular adventitial cell differentiation and if ACLP has a role in this process. Using purified mouse aortic adventitia Sca1+ progenitors, ACLP repressed stem cell markers (CD34, KLF4) and upregulated smooth muscle actin (SMA) and collagen I expression. ACLP enhanced myocardin-related transcription factor A (MRTFA) activity in adventitial cells by promoting MRTFA nuclear translocation. Sca1 cells from MRTFA-null mice exhibited reduced SMA and collagen expression induced by ACLP, indicating Sca1 cell differentiation is regulated in part by the ACLP-MRTFA axis. We determined that ACLP induced vessel contraction and increased adventitial collagen in an explant model. Collectively these studies identified ACLP as a mediator of adventitial cellular differentiation, which may result in pathological vessel remodeling.
Subject terms: Mechanisms of disease, Transcription factors, Cell biology, Cell signalling, Extracellular signalling molecules, Cardiovascular biology
Introduction
The adventitia, the outermost layer of muscular and elastic arteries, is bounded from the medial smooth muscle cell (SMC) layer by the external elastic lamina1–3. Originally considered a simple supportive structure, the adventitia is emerging as a key regulator of vascular diseases including atherosclerosis1,4,5 and pulmonary vascular hypertension2,6. The adventitia is an assembly of several cell types including fibroblasts, stem/progenitor cells, adipocytes, neurons, and inflammatory cells and in the normal vessel, these cells are organized within a connective tissue rich in fibrillar collagens types I and III7. In response to injury, adventitial fibroblasts and progenitor cells are activated. The activation of proliferation, migration, and differentiation programs eventually leads to adventitial fibrosis that may restrict beneficial vessel expansion8–10. Due in part to the complexity of the adventitia, the mechanisms controlling adventitial cell differentiation and remodeling have yet to be elucidated.
There are numerous similarities between fibroblast to myofibroblast differentiation and progenitor to vascular smooth muscle differentiation pathways with the notable expression of the contractile, cytoskeletal protein α-smooth muscle actin (SMA)11. In fibrotic diseases, differentiated myofibroblasts secrete collagens, including type I, and subsequently remodel it through their contractile functions. Myofibroblasts participate in vascular repair processes but also result in adventitial extracellular matrix (ECM) accumulation1,2. These same processes can also result in inward arterial remodeling, which potentiates tissue ischemia, myocardial infarction, and stroke12. Myofibroblast differentiation is regulated by numerous growth factors including transforming growth factor β (TGFβ) and ECM molecules including collagen and fibronectin13. The ECM influences myofibroblast differentiation through both biochemical and mechanical signaling11,13,14. Importantly, the transcriptional regulator, myocardin-related transcription factor A (MRTFA/Mkl1) acts as a central regulator of myofibroblast differentiation and fibrosis15,16. In response to extracellular stimuli, changes in cytoskeletal reorganization and actin polymerization can induce MRTFA nuclear translocation and transcriptional activity including the regulation of collagen17,18.
While adventitial fibroblasts are clearly important in vascular remodeling, adventitial stem cell antigen-1 (Sca1) positive progenitor cells adopt both vascular and nonvascular fates19–22. Adventitial Sca1 cells in adult mice with hypertension can give rise to collagen-producing cells and contribute to vascular fibrosis23 and Sca1 cells in the coronary adventitia are responsible for perivascular fibrosis20. In an atherosclerosis model in mice, progenitor cells in the adventitia differentiate into smooth muscle-like cells and contribute to vein graft atherosclerosis19. Importantly, adventitial Sca1 progenitors are maintained by sonic hedgehog signaling24 and when activated contribute to both vascular fibrosis and cardiovascular disease. However, the complex pathways controlling their differentiation are only partially understood.
Our previous studies identified a secreted protein, aortic carboxypeptidase-like protein (ACLP/AEBP1), which contains an evolutionarily conserved collagen binding discoidin domain and a catalytically inactive carboxypeptidase domain25–27. ACLP is highly expressed and secreted by fibroblasts and injured SMCs25–28. Several studies identified correlations of ACLP expression with cardiovascular disease processes. For example, ACLP was discovered in the ECM in a porcine model of ischemia/reperfusion cardiac remodeling29, in human abdominal aortic aneurysms30, and in the plasma following cardiac injury31. Our work has shown ACLP expression increases with SMC development26,32 and at sites of tissue injury25,26,28. In addition, mutations in the AEBP1 gene that encodes ACLP result in Ehlers Danlos Syndrome with vascular complications33,34. Taken together, there is strong evidence correlating ACLP with vascular disease processes. However, little is currently known about ACLP’s role in controlling adventitial cell biology. In this study, we show that ACLP promotes adventitial progenitor and fibroblast differentiation. We identified a novel pathway where ACLP-induced Sca1 cell differentiation is controlled in part by enhancing MRTFA nuclear localization and activity.
Results
ACLP promotes Sca1 cell differentiation into myofibroblast-like cells
It has been established that resident Sca1 positive progenitor cells in adventitia have a potential to switch their phenotype and differentiate into collagen-producing cells or smooth muscle-like cells19,23. In the adventitial niche, Sca1+ cells are heterogeneous and contribute to vascular disease and remodeling24,35,36. It is also recognized that the ECM is an essential regulator of the stem cell niche and interacts with progenitor cells to determine their differentiation capacity37,38. ACLP is a secreted ECM protein and we have previously demonstrated that ACLP promotes lung fibroblast differentiation and enhances adipose tissue stromal progenitor differentiation28,39,40. To understand the mechanisms controlling the differentiation of adventitial Sca1 progenitors with respect to contractile and ECM markers, we purified mouse vascular adventitial Sca1+ cells using microbeads. These Sca1+ cells were characterized by high expression of progenitor markers including Sca1, CD34 and transcription factor krüppel-like factor 4 (KLF4) (Fig. 1a). In culture, these cells progressively lose progenitor marker expression (Sca1, KLF4, CD34) with significant reductions by day 3 (Fig. 1a). Concomitant with the loss of stem cell markers, these cells differentiate into myofibroblast or smooth muscle-like cells characterized by significant increases in smooth muscle/myofibroblast marker gene expression including SMA and SM22α by day 7 (Fig. 1b). Importantly, as these cells differentiate, they also significantly increase their expression of ACLP and collagen I (Fig. 1c).
Figure 1.

ACLP promotes adventitial Sca1 cell differentiation into myofibroblast-like cells. Sca1 cells were isolated from mouse aorta adventitia and cultured on tissue culture plastic and RNA was isolated at the given times and analyzed by qRT-PCR (a) progenitor marker genes (b) smooth muscle/contractile marker genes; (c) ECM marker genes. Results are presented as the mean ± SEM for 3 independent experiments performed in triplicate. Statistical significance was calculated using ANOVA, followed by Tukey’s multiple comparison test. *p < 0.001, #p < 0.005 and §p < 0.05. Sca1 cells were isolated from the aortic adventitia of SMA-mCherry and col3.6GFPtpz transgenic male mice and cultured for 7 days. (d) Progenitor marker genes were examined by RT-PCR. Results are presented as the mean ± SEM for 3 independent experiments performed in triplicate. Data were analyzed by ANOVA, followed by Tukey’s multiple comparison test. *p < 0.001, #p < 0.005. (e) Images of SMA-mCherry and col3.6GFPtpz reporter expression were taken by fluorescence microscopy. Representative images are shown from three independent experiments. Fluorescence intensity of reporter expression was analyzed by ImageJ and normalized by control. Data were analyzed by the Student’s t test. §p < 0.05. (f) Protein expression was examined by Western blot (left) and quantification of results from 3 independent experiments (right). Statistical significance was calculated using the Student’s t test (two tailed). §p < 0.05.
Because ACLP was upregulated during progenitor differentiation and we also know that it regulates both SMA and collagen expression in fibrotic cells, we next examined whether exogenous ACLP could enhance adventitial progenitor differentiation. To do this we studied adventitial Sca1 cells isolated from transgenic mice harboring the SMA-mCherry and collagen1-GFP-Tpz transgenes to monitor changes in gene expression. After treatment with ACLP for 7 days, we isolated RNA and expression was analyzed by qRT-PCR. We found that compared with controls, ACLP further decreased Sca1, CD34, and KLF4 gene expression (Fig. 1d). Consistent with mRNA level changes, ACLP treatment increased SMA and collagen I reporter expression using live cell imaging and quantified using ImageJ (Fig. 1e). Protein isolates from these cells revealed an increase in both collagen I and SMA by Western blot analysis (Fig. 1f). Taken together these data indicate that ACLP down regulates progenitor marker expression and promotes differentiation into myofibroblasts.
ACLP promoting Sca1 cell differentiation is mediated by MRTFA pathway
Our previous work and that of others supports the concept that MRTFA regulates type I collagen and myofibroblast gene expression through transcriptional complexes with both serum response factor (SRF) and Sp117,18. The MRTFA pathway is also required for TGFβ-induced fibroblast differentiation15,41,42. To explore whether adventitial progenitor differentiation required MRTFA activity and more specifically if ACLP stimulated this pathway, we isolated adventitial Sca1 progenitors from MRTFA+/+ and MRTFA−/− mouse aorta and cultured in the presence or absence of ACLP. We found that ACLP promoted Sca1 differentiation into myofibroblasts by increasing myofibroblast-associated protein SMA and collagen I expression. Importantly, using live cell imaging of the SMA and collagen I reporters, both were blunted in cells differentiated from MRTFA−/− Sca1 progenitors compared with controls (Fig. 2a). Consistent with the collagen and SMA reporter fluorescence, Western blotting showed that compared with MRTFA+/+ fibroblasts, MRTFA deficient fibroblasts exhibited reduced ACLP stimulated SMA, collagen I, and ACLP expression (Fig. 2b,c). Notably, ACLP stimulated its own expression in these cells. Taken together these findings indicate that ACLP-induced Sca1 cell differentiation is mediated in part through the MRTFA pathway.
Figure 2.

ACLP promotes Sca1 cell differentiation via the MRTFA pathway by promoting MRTFA nuclear translocation. Sca1 cells were isolated from MRTFA+/+ and MRTFA−/− mouse aorta adventitia and cultured in the presence or absence of ACLP (30 nM) for 7 days. (a) SMA-mCherry and col3.6GFPtpz reporter activity was monitored by fluorescence microscopy. Representative images are shown from three independent experiments. Fluorescence intensity of reporter expression was analyzed by ImageJ and normalized by control. Data were analyzed by ANOVA, followed by Tukey’s multiple comparison test. *p < 0.001, #p < 0.005. (b) Adventitial fibroblast protein expression was examined by Western blot. (c) Quantification of Western results from three independent experiments. Statistical significance was calculated using ANOVA, followed by Tukey’s multiple comparison test. *p < 0.001, #p < 0.005 and §p < 0.05. Adventitial fibroblasts were serum-starved overnight and treated with ACLP (30 nM) for 2 h, MRTFA nuclear translocation was examined by immunofluorescence staining (d) and quantified as shown in (e). Data shown represent the mean ± SEM from 3 independent experiments. Statistical significance was calculated using the Student’s t test (two tailed). §p < 0.05, #p < 0.005.
MRTFA activity is regulated through its interaction with G-actin in the cytoplasm via its RPEL domain43. In response to several stimuli including actin polymerization, MRTFA is released and rapidly translocates to the nucleus to enhance expression of its target genes15. We next asked whether ACLP regulates MRTFA activity by measuring nuclear translocation of MRTFA. Adventitial fibroblasts were treated with ACLP for 2 h and MRTFA nuclear translocation was examined by immunofluorescence staining. In the untreated control fibroblasts, MRTFA exhibited predominantly cytoplasmic location (Fig. 2d). Compared with controls, ACLP treatment increased MRTFA nuclear accumulation and more ACLP treated cells exhibited MRTFA expression in both the nucleus and cytoplasm (Fig. 2e).
ACLP stimulates fibroblast-to-myofibroblast differentiation
In addition to specific progenitors, adventitial fibroblasts also contribute to vascular remodeling1,2,44. Furthermore, the differentiated Sca1 cells take on properties similar to activated adventitial fibroblast24. We next studied these adventitial fibroblasts in culture and found that they express and secrete high levels of ACLP (Fig. 3a). The upper band in these blots is the glycosylated, secreted form of ACLP28. Similar to the experiments with Sca1 cells, mouse adventitial fibroblasts treated with ACLP or TGFβ (a positive control and known mediator of fibroblast-to-myofibroblast differentiation) significantly increased SMA and SM22 mRNA expression (Fig. 3b). Consistent with these results, ACLP also enhanced SMA protein expression (Fig. 3c). Using a siRNA loss of function approach, we found that knocking down ACLP significantly decreased SMA and collagen I protein expression (Fig. 3d). These data indicate that adventitial fibroblasts synthesize ACLP and ACLP promotes fibroblast-to-myofibroblast transition in these cells by enhancing SMA and collagen I expression.
Figure 3.

ACLP regulates myofibroblast marker gene expression. Fibroblasts were isolated from mouse adventitia and cultured in 0.5% FBS/DMEM. Two days later, cells and medium were collected and ACLP expression examined by Western blot (a). Mouse adventitial fibroblasts were treated with ACLP (30 nM) or TGFβ (1 nM) for 24 h. SMA (Acta2) and SM22 mRNA were examined by RT-PCR. Results are presented as the mean ± SEM for 3 independent experiments performed in triplicate (b). SMA protein was examined by Western Blot (c) and data are presented as the mean ± SEM for 3 independent experiments and analyzed by one way ANOVA with post hoc Tukey’s test. Mouse adventitial fibroblasts were transfected with different doses of siACLP or siControl. (d) Protein was extracted 48 h later and subjected to Western blot. Protein expression was quantified by densitometry normalized to GAPDH expression and relatively compared to untreated control cells (3 independent experiments). Statistical significance was calculated using ANOVA, followed by Tukey’s multiple comparison test *p < 0.001, #p < 0.005, §p < 0.05.
ACLP potentiates collagen gel contraction by fibroblasts
In addition to changes in cellular composition, in vascular disease adventitial fibrosis is characterized by the accumulation of collagen, which is then remodeled through cellular contractile activity1–3. We hypothesized that ACLP in the collagen-rich ECM surrounding adventitial fibroblasts promotes ECM contraction induced by fibroblasts. Mouse adventitial fibroblasts were plated into collagen gels in the presence or absence of ACLP, and then incubated overnight. The area of contracted collagen lattices was calculated as a percentage of the initial size. ACLP induced gel contraction by reducing gel size from 33 to 24% of their original size compared to controls, indicating that ACLP promotes fibroblast-induced gel contraction (Fig. 4a,b). We recently generated mice with a floxed ACLP allele (ACLPflx/flx) and crossed these mice with a tamoxifen controlled Cre mice45. After ACLP deletion by tamoxifen treatment in vitro, cells were plated into collagen gels in the presence or absence of TGFβ to stimulate contraction. As expected, TGFβ induced collagen gel contraction in ACLP+/+ fibroblasts. This effect was blunted in the ACLPΔ/Δ adventitial fibroblasts (Fig. 4c,d). Western blotting revealed that TGFβ increased ACLP levels in ACLP+/+ cells and as expected ACLPΔ/Δ fibroblasts lacked ACLP expression (Fig. 4e). Taken together, these data indicate that ACLP potentiates collagen gel contraction by fibroblasts and ACLP-containing adventitial matrix participates in regulating ECM contraction. To assess the requirement of MRTFA in ACLP mediated collagen gel contraction, adventitial fibroblasts were isolated from MRTFA+/+ and MRTFA−/− mice and were plated in collagen gels in the presence or absence of ACLP. Similar to previous results (Fig. 4a) we observed a significant enhancement of gel contraction in the MRTFA+/+ cells (Fig. 4f,g). However, both untreated and ACLP-induced gel contraction was blunted in the MRTFA−/− cells compared to MRTFA+/+ cells (Fig. 4f,g). In addition, contraction was not significantly different between control and ACLP treated MRTFA−/− cells (Fig. 4f,g) further supporting the concept MRTFA is an important mediator of collagen contractility.
Figure 4.
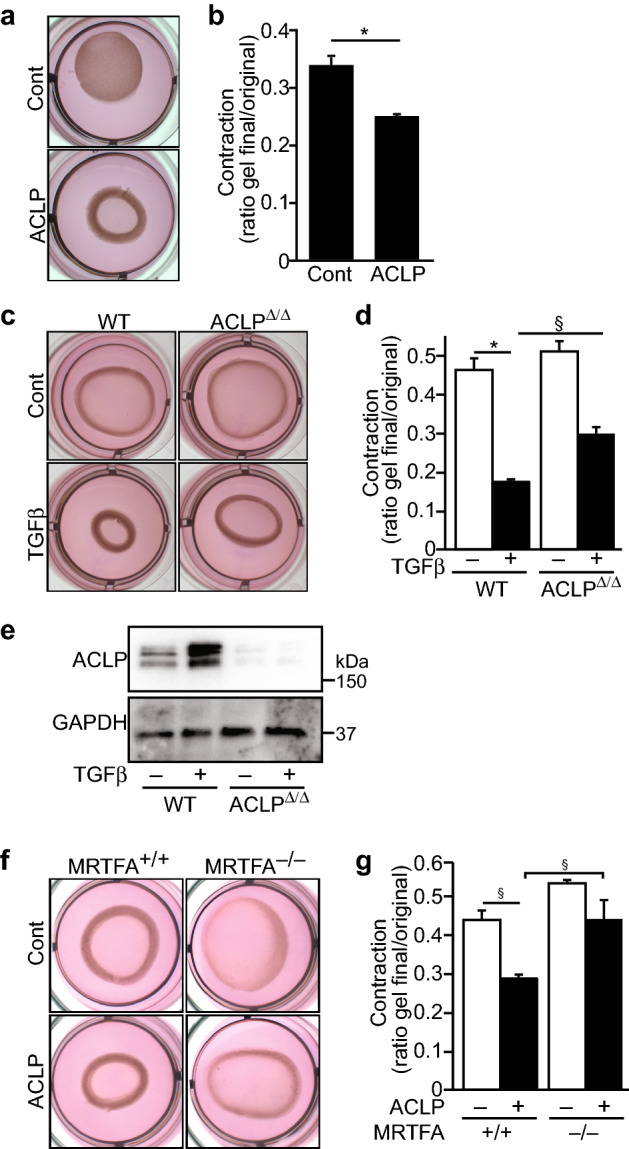
ACLP-induced gel contraction in fibroblasts is blunted by loss of MRTFA. (a) Mouse adventitial fibroblasts were plated into collagen gels in the presence or absence of ACLP (30 nM), followed by overnight incubation. The area of contracted collagen lattices was calculated as a percentage of the surface of the dish (b). Statistical significance was calculated using the Student’s t test (two tailed) (n = 3). Mouse adventitial fibroblasts were isolated from ACLPflx/flx·Cre+ mice. After ACLP deletion by tamoxifen treatment, cells were plated into a collagen gel in the presence or absence of TGFβ (1 nM) for collagen gel contraction assays (c). The area of contracted collagen lattices was calculated as a percentage of the surface of the dish (d). ACLP protein expression was analyzed by Western Blot (e). Results shown represent the mean ± SEM (n = 3). Statistical significance was calculated using ANOVA, followed by Tukey’s multiple comparison test *p < 0.001, §p < 0.05. (f) Adventitial fibroblasts from MRTFA+/+ and MRTFA−/− male mice were plated into collagen gels in the presence or absence of ACLP (30 nM), followed by overnight incubation. The area of contracted collagen lattices was calculated as a percentage of the surface of the dish (g). Data shown represent the mean ± SEM (n = 3). Statistical significance was calculated using one way ANOVA with post hoc Tukey’s test. §p < 0.05.
ACLP induces adventitial fibrosis and aorta contraction
To examine ACLP function in an ex vivo model, mouse aortas were isolated and cultured in the presence or absence of ACLP 30 nM for 7 days. We found that aorta lengths were reduced by 45% compared to that in control after ACLP treatment (Fig. 5a,b). Picrosirius staining and quantified using ImageJ revealed that collagen expression was markedly increased in the aortic adventitia treated with ACLP compared to controls (Fig. 5c). Additional sets of aortas were extracted for total protein and we observed an increase in SMA expression (Fig. 5d). We did not detect collagen in these Western blots potentially because it was primarily insoluble. However, analysis of RNA from these cultured vessel fragments resulted in a statistically significant increase in both SMA and Col1a1 mRNA expression (Fig. 5e). These data support the concept that ACLP is a critical mediator of adventitial cell differentiation and remodeling by increasing fibrosis and vessel contraction.
Figure 5.

ACLP induces aorta contraction and adventitial fibrosis. Mouse aortas were isolated and cultured in the presence or absence of ACLP 30 nM for 7 days (a). After 7 days ACLP treatment, aorta images were taken and quantified by ImageJ. Statistical significance was calculated using the Student’s t test (two tailed) (n = 9) (b). Aortas were collected and collagen expression was examined by picrosirius red staining and polarized light (c). Quantification of staining results from independent experiments (n = 5) and analyzed by ImageJ. Statistical significance was calculated using the Student’s t test. §p < 0.001. RNA and protein were extracted from aortas after ACLP treatment and SMA and Col1a1 expression were examined by Western blot (d). RNA were examined by RT-PCR (e). Results are presented as the mean ± SEM for 3 independent experiments performed in triplicate. The statistical difference between control and ACLP treatment groups was determined using two-tailed Student’s t test. *p < 0.001, #p < 0.005.
Discussion
In the present study, we demonstrate that ACLP enhances the differentiation of specific adventitial Sca1 progenitors and fibroblasts into myofibroblasts or smooth muscle-like cells. Importantly, ACLP mediates differentiation of adventitial progenitors via stimulating the nuclear translocation of MRTFA and subsequent activity. These studies uncovered a new ACLP regulated pathway that increases adventitial collagen matrix expression and contraction (Fig. 6).
Figure 6.
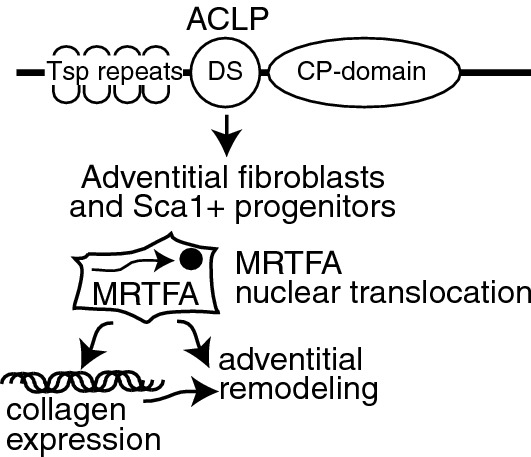
Model of aortic carboxypeptidase-like protein regulation of progenitor and fibroblast differentiation through MRTFA. ACLP structure indicated by predicated thrombospondin (Tsp) repeats, collagen-binding discoidin (DS) domain, and catalytically inactive carboxypeptidase (CP) domain. ACLP stimulates adventitial fibroblasts and Sca1+ progenitors in part through regulating the nuclear translocation of MRTFA. ACLP stimulation results in increases in collagen expression and adventitial remodeling.
Several screening studies have identified changes in ACLP expression correlating with cardiovascular disease processes. For example, Didangelos et al. found that ACLP degradation is increased in aortic aneurysms compared to controls30. ACLP was also identified in the ECM in a porcine model of cardiac ischemia/reperfusion remodeling29. In addition, increased ACLP expression parallels with severity of fibrosis in nonalcoholic steatohepatitis and idiopathic pulmonary fibrosis39,46. Our previous work determined that ACLP was highly induced in mouse vascular injury25 and increasing evidence indicates that progenitor cells exist in adventitia and differentiate into multiple types. ACLP is prominently expressed in the adventitia of mouse femoral arteries following vascular injury (Fig. S1). It has been demonstrated that adventitial Sca1 cells differentiate into collagen-producing cells and contribute to vascular fibrosis and perivascular cardiac fibrosis20,23. Several studies indicate that adventital Sca1+ cells are a heterogenous cell populations and contribute to vascular disease progression24,35,36. Our initial analysis of ACLP in the vascular adventitia by single cell RNA sequencing revealed that ACLP is co-expressed with Sca1 and also with the fibroblast makers decorin and col1a1 (Fig. S2). Adventitial Sca1 cell differentiation is regulated by ECM, growth factors and a number of cellular receptors37. The ECM is an intricate network of proteins that surrounds cells and acts as a key component of stem cell niches and participates in stem cell maintenance, proliferation, self-renewal and differentiation37,38. Our work showed that elevated ACLP alters the differentiation of these progenitors by promoting a more smooth muscle and ECM-producing myofibroblast phenotype as observed in vascular disease settings. Our studies are also consistent with a role for ACLP in maintaining vascular homeostatasis as loss of ACLP functions in humans results in a novel variant of Ehlers Danlos Syndrome with aortic dilatation33.
The precise signaling mechanisms controlling adventitial progenitor differentiation are not fully elucidated. Earlier studies showed that gene expression changes in all members of the myocardin family with adventitial progenitor differentiation24 and myocardin is involved in adventitial Sca1 cell differentiation into smooth muscle cells regulated by c-Myb47. It is recognized that ACLP has direct signaling roles via the TGFβ and LRP6/Frz8-β-catenin pathways48. There are likely other ACLP-dependent pathways that are integrated with TGFβ signaling and transcriptional networks. Our previous work using inhibition of TGFβ-receptor signaling with kinase inhibitors or siRNA against Smad2/3 repressed ACLP-dependent effects in lung and adipose tissue cells39,40. There are complex interelationships with TGFβ signaling, MRTFA, and the Taz/Yap pathways in mesenchymal cells49. While previous work determined that the MRTFA signaling pathway is an important mediator of stem cell fate50,51, our work determined that the MRTFA pathway contributes to adventitial differentiation. It has been previously shown that MRTFA and its related family members MRTFB and myocardin are regulated in vascular injury and atherosclerosis models52. Although we did not examine the regulation or function of these other family members, our loss of function studies with MRTFA (Figs. 2, 4) support the concept that these proteins cannot compensate for the absence of MRTFA. Interestingly, ACLP increased its own expression that was also dependent on MRTFA (Fig. 2b,c) potentially indicating a feed-forward regulatory loop. Important insight into the role of MRTFA and its regulation in different signaling networks has come from studies examining epithelial and endothelial to mesenchymal transformation53,54. In addition to integration of TGFβ signaling pathways by MRTFA described above, MRTFA is also regulated by phosphorylation and Rho kinase pathways50,54,55. Our work found a connection between ACLP activity and enhanced MRTFA nuclear localization (Fig. 3) and in the absence of MRTFA responses to ACLP were blunted (Fig. 2). It is currently unknown whether ACLP signaling regulates Rho kinase pathways. Taken together these findings add to our understanding of pathways regulating adventitial differentiation.
Treatment of Sca1 progenitors with ACLP downregulated progenitor marker gene expression including KLF4 (Fig. 1d). KLF4 is a zinc finger protein and acts as a transcriptional repressor that blocks SRF/MRTFA-dependent transcription of multiple smooth muscle marker genes49. It has been reported that KLF4 is highly expressed in adventitial Sca1 cells and plays a critical role in the maintenance of SMC progenitors24. Once Sca1 cells differentiate, KLF4 is down-regulated and cells acquire SMC markers24,56. Consistent with previous studies, we found that KLF4 gene expression is diminished after Sca1 cell differentiation. Previous studies have demonstrated regulatory roles for KLF4, MRTFA, and TGFβ signaling in the modulation of vascular smooth musle cells in part through microRNA dependent pathways57. While there is a connection between ACLP activity and the downregulation KLF4 expression, the pathways controlling this regulatory step are currently unknown.
In addition to progenitors, fibroblasts and myofibroblasts play important roles in vascular remodeling through their collagen production and contractile abilities that are similar to SMCs11. It is widely recognized that vascular smooth muscle cells and myofibroblasts have similar gene expression patterns including the expression of SMA. The vascular SMC and myofibroblasts in their expression of both ECM genes including collagen and contractile genes including SMA and SM2215. Irrespective of their designation, the phenotype of the various cell types in the diseased vessel clearly impact vascular disease progression. Our findings support a model where ACLP contributes to adventitial fibroblast and stem cells activation and promotes the transition of fibroblasts to myofibroblasts resulting in vascular ECM contraction and remodeling. Taken together these studies have uncovered a novel pathway where ACLP regulates adventitial progenitor and fibroblast differentiation and ultimately contributes to changes in ECM remodeling. Our results connect ACLP with several critical processes that drive pathological vessel remodeling that may be amendable to inhibition through the development of specific inhibitors.
Methods
All experiments were performed in accordance with relevant guidelines, regulations, and approval by Boston University School of Medicine’s Institutional Biosafety Committee.
Mice
ACLPflx/flx mice containing loxP sites in the ACLP gene were generated at the University of California Davis mouse biology core and were crossed with the tamoxifen inducible Cre (B6.Cg-Tg) (CAG-cre/Esr1*)5Amc/j, Jackson labs). MRTFA+/+ and MRTF−/− mice (generously provided by Dr. Eric Olson)58 were crossed with mice harboring the SMA promoter driving mCherry and Col1a1 promoter driving GFP (col3.6GFPtpz), kindly provided by Dr. David Rowe59 as described17. For the MRTFA studies littermates were used for as controls and 8–12 week old male and female mice were used for Sca1 cell isolation. The Institutional Animal Care and Use Committee at Boston University School of Medicine approved all animal experimental procedures (protocol # 14769).
Histological analysis
Collagen was examined by picrosirius red staining (Electron Microscopy Sciences) following manufacturer’s instruction. Briefly, aortas were fixed and embedded in paraffin. Tissue sections were stained with picrosirius red for 1 h and sections were mounted for observation under polarized light (Olympus IX70) and results quantitated using ImageJ.
Aorta contraction assay
Thoracic aortas were isolated from 10 male mice. Each aorta was cut into 2 pieces of equal length, one piece for ACLP treatment, one for control. After culture in the presence or absence of ACLP (30 nM) for 7 days, vessel fragments were imaged using an Olympus, SZX16 microscope and analyzed using ImageJ. The lengths for ACLP treated vessel fragments were normalized to controls.
Cell culture and treatments
Mouse primary aorta adventitial fibroblasts were isolated from male mouse aorta essentially as described26. Briefly, the aortas were dissected and the adventitia was separated from the medial layer, minced, and digested in buffer containing 1 mg/ml collagenase (Worthington) and 0.125 mg/ml elastase (Sigma). Cells were maintained in DMEM containing 10% FBS (Hyclone) and antibiotics. Recombinant ACLP was expressed and purified as described39. Recombinant TGFβ was obtained from R&D Systems (240-B-002/CF). Unless otherwise indicated, cells were treated with 30 nM ACLP or 1 nM TGFβ.
Sca1 cell isolation and culture
Sca1 cells were isolated by anti-Sca1 MicroBead Kit (Miltenyi Biotec) following the manufacturer’s instructions. Briefly, male mice were euthanized and the thoracic aortic adventitia was dissected, minced, and then digested with 1× dispase (BD Falcon), 1 mg/ml type 1 collagenase (Worthington), and 4.5 μg/ml DNase at 37 °C for approximately 30 min. Cell suspensions were filtered through a 40 μm cell strainer (BD Falcon) and centrifuged at 200×g for 5 min. Cell pellets were resuspended with MACS buffer and cell numbers were counted, followed by incubation with 10 μl FITC-conjugated monoclonal anti-mouse Sca-1 antibody at 4 °C for 10 min. After washing 3 times, cells were incubated with 20 μl microbeads-conjugated monoclonal anti-FITC antibody for 15 min at 4 °C, and then passed through a magnetic cell sorting (MACS) column to isolate Sca1 cells. The yield of Sca1 cells averaged 5% in these purifications. Purified cells were maintained in DMEM supplemented with 10% FBS.
Reverse transcription polymerase chain reaction (RT-PCR) assays
Total RNA was extracted by GeneJet RNA purification kit (Thermo) and subjected to reverse transcription using SuperScript II kit (Invitrogen), according to the manufacturer’s instructions. qPCR was performed using Luminaris Color HiGreen High ROX qPCR Master Mix (Thermo), following the manufacturer’s instructions. Quantitative analysis was performed by real-time PCR (ABI 7300). Expression was calculated with the ΔΔCT method using 18S to normalize all samples.
Primers were used as follows (validated primer sequences designed by MGH Primer bank).
| 18S: | fwd/rev CGGCTACCACATCCAAGGAA | TTTTCGTCACTACCTCCCCG |
| Sca1 (Ly6a): | fwd/rev AGGAGGCAGCAGTTATTGTGG | CGTTGACCTTAGTACCCAGGA |
| Klf4: | fwd/rev GTGCCCCGACTAACCGTTG | GTCGTTGAACTCCTCGGTCT |
| CD34: | fwd/rev ATCCCCATCAGTTCCTACCAAT | TGGTGTGGTCTTACTGCTGTC |
| SMA (Acta2): | fwd/rev TGACGCTGAAGTATCCGATAGA | GTACGTCCAGAGGCATAGAGG |
| SM22 (Tagln): | fwd/rev CAACAAGGGTCCATCCTACGG | ATCTGGGCGGCCTACATCA |
| Col1a1: | fwd/rev CTGGCGGTTCAGGTCCAAT | TTCCAGGCAATCCACGAGC |
| ACLP/aebp1: | fwd/rev TCCCCAACTATGATGACTTGGA | GGGGCTACTACCCTCCTTTTTG |
RNA interference
Synthetic small interference RNA (siRNA) targeting mouse ACLP was purchased from Dharmacon (5′ GGCUCAAGAUCUACGCAAU 3′). A siRNA with a non-targeting sequence (scramble siRNA, Dharmacon) was used as a negative control. The siRNAs were transfected at indicated doses using RNAi Max (Invitrogen) according to manufacturer’s instructions. Primary cells were transfected with siACLP (1, 2 and 4 nM) or siControl (4 nM) and 48 h after transfection, protein was isolated, subjected to Western blot.
Immunoblot assays
Cells were lysed in extraction buffer (25 mM Tris, pH 7.4, 50 mM sodium chloride, 0.5% sodium deoxycholate, 2% NP-40, 0.2% SDS) containing protease inhibitors. Total cell lysates were separated on 4–20% SDS-PAGE (Invitrogen), transferred to nitrocellulose membranes (Millipore), immunoblotted with antibodies, and visualized using a SuperSignal West Dura Extended Duration Substrate (Thermo Scientific). The antibodies used in this study are anti-SMA (Sigma, A2547, 1:4000 dilution), anti-type I collagen (Rockland, 600-401-103, 1:1000 dilution), anti-MRTFA (Santa Cruz, sc-21558, 1:250 dilution), anti-ACLP (1:4000 dilution)27 and anti-GAPDH Sigma, G9545, 1:10,000 dilution). Western blots were quantitated using ImageJ and normalized to GAPDH. The results are expressed as fold change versus control.
Immunofluorescence staining
Sca1 cells were plated onto glass chamber slides and incubated in the presence or absence of ACLP (30 nM). After 7 days, cells were washed, fixed in 4% paraformaldehyde (PFA), permeabilized with 0.1% Triton X-100, incubated with anti-SMA antibodies (Sigma, A2547, 1:500 dilution) followed by Alexa-Fluor-568 conjugated secondary antibodies (Invitrogen, A11031, 1:500 dilution). Samples were counterstained with DAPI and mounted. Images were taken at identical exposures using an Olympus 1X70 microscope and an Optronics camera and analyzed by ImageJ. To study MRTFA translocation, adventitial fibroblasts were serum starved overnight and treated with ACLP (30 nM) for 2 h and examined by immunofluorescence as described55,60. Briefly, after treatment, cells were washed in warm PBS and fixed in 4% paraformaldehyde (PFA). Cells were then incubated with an anti-MRTF-A (1:50; Cat# sc-21558, Santa Cruz Biotechnology Inc., Santa Cruz, CA), followed by the appropriate secondary antibody conjugated to AlexaFluor 568 (1:500; Cat#A-11057, Thermo Scientific, Waltham, MA 02451). Pictures of at least four random fields were captured and analyzed by ImageJ. For each image, distribution of nuclear and cytoplasmic of MRTFA staining was determined and the ratios of the average nuclear and cytoplasmic staining distribution in total cells of each field were calculated using established methods55.
Collagen gel contraction assays
Collagen matrix contraction assay was performed essentially as described28. Mouse adventitial fibroblasts isolated from male ACLPflx/flx Cre+, MRTFA+/+ and MRTF−/− mice. ACLPflx/flx Cre+ were treated with tamoxifen in vitro for 3 days to delete ACLP. Tamoxifen was washed out for 2 additional days and fibroblasts were embedded in attached collagen matrices, followed by control, TGFβ, or ACLP treatment overnight. Upon releasing the collagen lattice from the culture dish, the embedded cells become free to contract the deformable collagen lattice and images were taken at the end of incubation. The area of contracted collagen lattices was calculated as a percentage of the surface of the dish.
Statistical analysis
The results presented are the average of at least three independent experiments. All data are plotted as the mean ± SEM and analyzed using GraphPad Prism software. Statistical significance was calculated using analysis of variance (ANOVA), followed by Tukey’s multiple comparison test or by Student’s t-test as indicated in figure legends. Significance was accepted at p < 0.05.
Supplementary Information
Acknowledgements
This work was supported in part by National Institutes of Health Grants HL078869 (MDL), DK117161 (SRF), and a grant from the American Heart Association 14GRNT18690001 (MDL). The single cell sequencing studies were supported by the Evans Center for Interdisciplinary Research Fibrosis Affinity Research Collaborative. We thank Yuriy Alexeyev, Josh Campbell, and Tianmu Hu for their contributions to the single cell experiments.
Author contributions
D.W. performed all research in (Figs. 1, 2, 3, 4, 5). S-F.Y. and M.D.L. performed the studies in Fig. S1. N.R and M.D.L. performed the studies in Fig. S2. D.W., S.R.F. and M.D.L. wrote the main manuscript text. D.W. and M.D.L prepared all figures. All authors reviewed the manuscript.
Data availability
Data generated or analyzed during this study are included in this published article and in the Supplementary information.
Competing interests
M.D.L is listed as a co-inventor on a patent application submitted by Boston University for research related to ACLP entitled: “Inhibitors of Fibroproliferative Diseases and Cancer”. The other authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-82941-7.
References
- 1.Havelka GE, Kibbe MR. The vascular adventitia: Its role in the arterial injury response. Vasc. Endovasc. Surg. 2011;45:381–390. doi: 10.1177/1538574411407698. [DOI] [PubMed] [Google Scholar]
- 2.Strauss BH, Rabinovitch M. Adventitial fibroblasts: Defining a role in vessel wall remodeling. Am. J. Respir. Cell Mol. Biol. 2000;22:1–3. doi: 10.1165/ajrcmb.22.1.f172. [DOI] [PubMed] [Google Scholar]
- 3.Tinajero MG, Gotlieb AI. Recent developments in vascular adventitial pathobiology. Am. J. Pathol. 2019 doi: 10.1016/j.ajpath.2019.10.021. [DOI] [PubMed] [Google Scholar]
- 4.Corselli M, et al. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells Dev. 2012;21:1299–1308. doi: 10.1089/scd.2011.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Staab ME, et al. Arterial remodeling after experimental percutaneous injury is highly dependent on adventitial injury and histopathology. Int. J. Cardiol. 1997;58:31–40. doi: 10.1016/s0167-5273(96)02844-6. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell RN, Libby P. Vascular remodeling in transplant vasculopathy. Circ. Res. 2007;100:967–978. doi: 10.1161/01.Res.0000261982.76892.09. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt MR, Maeng M, Kristiansen SB, Andersen HR, Falk E. The natural history of collagen and α-actin expression after coronary angioplasty. Cardiovasc. Pathol. 2004;13:260–267. doi: 10.1016/j.carpath.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Arbustini E, Roberts WC. Morphological observations in the epicardial coronary arteries and their surroundings late after cardiac transplantation (allograft vascular disease) Am. J. Cardiol. 1996;78:814–820. doi: 10.1016/s0002-9149(96)00427-4. [DOI] [PubMed] [Google Scholar]
- 9.Labinaz M, Pels K, Hoffert C, Aggarwal S, O'Brien ER. Time course and importance of neoadventitial formation in arterial remodeling following balloon angioplasty of porcine coronary arteries. Cardiovasc. Res. 1999;41:255–266. doi: 10.1016/s0008-6363(98)00203-x. [DOI] [PubMed] [Google Scholar]
- 10.Majesky MW, Dong XR, Hoglund V, Mahoney WM, Jr, Daum G. The adventitia: A dynamic interface containing resident progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2011;31:1530–1539. doi: 10.1161/atvbaha.110.221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinz B, et al. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan ST, Koteliansky VE, Gotwals PJ, Lindner V. Transforming growth factor-β-dependent events in vascular remodeling following arterial injury. J. Vasc. Res. 2003;40:37–46. doi: 10.1159/000068937. [DOI] [PubMed] [Google Scholar]
- 13.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 14.Wynn TA. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Small EM. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J. Cardiovasc. Transl. Res. 2012;5:794–804. doi: 10.1007/s12265-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 16.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luchsinger LL, Patenaude CA, Smith BD, Layne MD. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J. Biol. Chem. 2011;286:44116–44125. doi: 10.1074/jbc.M111.276931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Small EM, et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010;107:294–304. doi: 10.1161/circresaha.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, et al. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J. Clin. Investig. 2004;113:1258–1265. doi: 10.1172/jci19628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ieronimakis N, et al. Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFβ1 signaling in the mdx mouse model of Duchenne muscular dystrophy. J. Mol. Cell Cardiol. 2013;63:122–134. doi: 10.1016/j.yjmcc.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Majesky MW, Dong XR, Hoglund V, Daum G, Mahoney WM., Jr The adventitia: A progenitor cell niche for the vessel wall. Cells Tissues Organs. 2012;195:73–81. doi: 10.1159/000331413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zengin E, et al. Vascular wall resident progenitor cells: A source for postnatal vasculogenesis. Development. 2006;133:1543–1551. doi: 10.1242/dev.02315. [DOI] [PubMed] [Google Scholar]
- 23.Wu J, et al. Origin of matrix-producing cells that contribute to aortic fibrosis in hypertension. Hypertension. 2016;67:461–468. doi: 10.1161/hypertensionaha.115.06123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Passman JN, et al. A sonic hedgehog signaling domain in the arterial adventitia supports resident Sca1+ smooth muscle progenitor cells. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9349–9354. doi: 10.1073/pnas.0711382105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Layne MD, et al. Characterization of the mouse aortic carboxypeptidase-like protein promoter reveals activity in differentiated and dedifferentiated vascular smooth muscle cells. Circ. Res. 2002;90:728–736. doi: 10.1161/01.res.0000013289.97650.c8. [DOI] [PubMed] [Google Scholar]
- 26.Layne MD, et al. Impaired abdominal wall development and deficient wound healing in mice lacking aortic carboxypeptidase-like protein. Mol. Cell Biol. 2001;21:5256–5261. doi: 10.1128/mcb.21.15.5256-5261.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Layne MD, et al. Aortic carboxypeptidase-like protein, a novel protein with discoidin and carboxypeptidase-like domains, is up-regulated during vascular smooth muscle cell differentiation. J. Biol. Chem. 1998;273:15654–15660. doi: 10.1074/jbc.273.25.15654. [DOI] [PubMed] [Google Scholar]
- 28.Schissel SL, et al. Aortic carboxypeptidase-like protein is expressed in fibrotic human lung and its absence protects against bleomycin-induced lung fibrosis. Am. J. Pathol. 2009;174:818–828. doi: 10.2353/ajpath.2009.080856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barallobre-Barreiro J, et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 2012;125:789–802. doi: 10.1161/circulationaha.111.056952. [DOI] [PubMed] [Google Scholar]
- 30.Didangelos A, et al. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: A proteomics approach. Mol. Cell Proteomics. 2011;10:M111.008128. doi: 10.1074/mcp.M111.008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Addona TA, et al. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 2011;29:635–643. doi: 10.1038/nbt.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ith B, Wei J, Yet SF, Perrella MA, Layne MD. Aortic carboxypeptidase-like protein is expressed in collagen-rich tissues during mouse embryonic development. Gene Expr. Patterns. 2005;5:533–537. doi: 10.1016/j.modgep.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Blackburn PR, et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2018;102:696–705. doi: 10.1016/j.ajhg.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vishwanath N, et al. Mechanisms of aortic carboxypeptidase-like protein secretion and identification of an intracellularly retained variant associated with Ehlers-Danlos syndrome. J. Biol. Chem. 2020;295:9725–9735. doi: 10.1074/jbc.RA120.013902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kokkinopoulos I, et al. Adventitial SCA-1(+) progenitor cell gene sequencing reveals the mechanisms of cell migration in response to hyperlipidemia. Stem Cell Rep. 2017;9:681–696. doi: 10.1016/j.stemcr.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu W, et al. Adventitial cell atlas of wt (wild type) and ApoE (Apolipoprotein E)-deficient mice defined by single-cell RNA sequencing. Arterioscler. Thromb. Vasc. Biol. 2019;39:1055–1071. doi: 10.1161/ATVBAHA.119.312399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta. 1840;2506–2519:2014. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watt FM, Huck WT. Role of the extracellular matrix in regulating stem cell fate. Nat. Rev. Mol. Cell Biol. 2013;14:467–473. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]
- 39.Tumelty KE, Smith BD, Nugent MA, Layne MD. Aortic carboxypeptidase-like protein (ACLP) enhances lung myofibroblast differentiation through transforming growth factor β receptor-dependent and -independent pathways. J. Biol. Chem. 2014;289:2526–2536. doi: 10.1074/jbc.M113.502617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jager M, et al. Aortic carboxypeptidase-like protein enhances adipose tissue stromal progenitor differentiation into myofibroblasts and is upregulated in fibrotic white adipose tissue. PLoS ONE. 2018;13:e0197777. doi: 10.1371/journal.pone.0197777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Connor JW, Gomez EW. Cell adhesion and shape regulate TGF-β1-induced epithelial-myofibroblast transition via MRTF-A signaling. PLoS ONE. 2013;8:e83188. doi: 10.1371/journal.pone.0083188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Velasquez LS, et al. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc. Natl. Acad. Sci. U.S.A. 2013;110:16850–16855. doi: 10.1073/pnas.1316764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mouilleron S, Guettler S, Langer CA, Treisman R, McDonald NQ. Molecular basis for G-actin binding to RPEL motifs from the serum response factor coactivator MAL. EMBO J. 2008;27:3198–3208. doi: 10.1038/emboj.2008.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuwabara JT, Tallquist MD. Tracking adventitial fibroblast contribution to disease: A review of current methods to identify resident fibroblasts. Arterioscler. Thromb. Vasc. Biol. 2017;37:1598–1607. doi: 10.1161/atvbaha.117.308199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- 46.Gerhard GS, et al. AEBP1 expression increases with severity of fibrosis in NASH and is regulated by glucose, palmitate, and miR-372-3p. PLoS ONE. 2019;14:e0219764. doi: 10.1371/journal.pone.0219764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shikatani EA, et al. c-Myb regulates proliferation and differentiation of adventitial Sca1+ vascular smooth muscle cell progenitors by transactivation of myocardin. Arterioscler. Thromb. Vasc. Biol. 2016;36:1367–1376. doi: 10.1161/atvbaha.115.307116. [DOI] [PubMed] [Google Scholar]
- 48.Teratani T, et al. Aortic carboxypeptidase-like protein, a WNT ligand, exacerbates nonalcoholic steatohepatitis. J. Clin. Investig. 2018;128:1581–1596. doi: 10.1172/jci92863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miranda MZ, et al. TGF-β1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017;292:14902–14920. doi: 10.1074/jbc.M117.780502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeon ES, et al. A Rho kinase/myocardin-related transcription factor-A-dependent mechanism underlies the sphingosylphosphorylcholine-induced differentiation of mesenchymal stem cells into contractile smooth muscle cells. Circ. Res. 2008;103:635–642. doi: 10.1161/circresaha.108.180885. [DOI] [PubMed] [Google Scholar]
- 51.Wang N, et al. Myocardin-related transcription factor-A is a key regulator in retinoic acid-induced neural-like differentiation of adult bone marrow-derived mesenchymal stem cells. Gene. 2013;523:178–186. doi: 10.1016/j.gene.2013.03.043. [DOI] [PubMed] [Google Scholar]
- 52.Minami T, et al. Reciprocal expression of MRTF-A and myocardin is crucial for pathological vascular remodelling in mice. EMBO J. 2012;31:4428–4440. doi: 10.1038/emboj.2012.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan L, et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol. Biol. Cell. 2007;18:1083–1097. doi: 10.1091/mbc.e06-07-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mihira H, et al. TGF-β-induced mesenchymal transition of MS-1 endothelial cells requires Smad-dependent cooperative activation of Rho signals and MRTF-A. J. Biochem. 2012;151:145–156. doi: 10.1093/jb/mvr121. [DOI] [PubMed] [Google Scholar]
- 55.Panayiotou R, et al. Phosphorylation acts positively and negatively to regulate MRTF-A subcellular localisation and activity. Elife. 2016 doi: 10.7554/eLife.15460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Majesky MW, et al. Differentiated smooth muscle cells generate a subpopulation of resident vascular progenitor cells in the adventitia regulated by Klf4. Circ. Res. 2017;120:296–311. doi: 10.1161/circresaha.116.309322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Davis-Dusenbery BN, et al. Down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-β and bone morphogenetic protein 4. J. Biol. Chem. 2011;286:28097–28110. doi: 10.1074/jbc.M111.236950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li S, Chang S, Qi X, Richardson JA, Olson EN. Requirement of a myocardin-related transcription factor for development of mammary myoepithelial cells. Mol. Cell Biol. 2006;26:5797–5808. doi: 10.1128/mcb.00211-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kalajzic Z, et al. Use of an α-smooth muscle actin GFP reporter to identify an osteoprogenitor population. Bone. 2008;43:501–510. doi: 10.1016/j.bone.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gupta M, Korol A, West-Mays JA. Nuclear translocation of myocardin-related transcription factor-A during transforming growth factor β-induced epithelial to mesenchymal transition of lens epithelial cells. Mol. Vis. 2013;19:1017–1028. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated or analyzed during this study are included in this published article and in the Supplementary information.