

Abstract
Accumulating evidence has shown that nutrient metabolism is closely associated with the differentiation and functions of various immune cells. Cellular metabolism, including aerobic glycolysis, fatty acid oxidation, and oxidative phosphorylation, plays a key role in germinal center (GC) reaction, B-cell trafficking, and T-cell-fate decision. Furthermore, a quiescent metabolic status consolidates T-cell-dependent immunological memory. Therefore, dietary interventions such as calorie restriction, time-restricted feeding, and fasting potentially manipulate immune cell functions. For instance, intermittent fasting prevents the development of experimental autoimmune encephalomyelitis. Meanwhile, the fasting response diminishes the lymphocyte pool in gut-associated lymphoid tissue to minimize energy expenditure, leading to the attenuation of Immunoglobulin A (IgA) response. The nutritional status also influences the dynamics of several immune cell subsets. Here, we describe the current understanding of the significance of immunometabolism in the differentiation and functionality of lymphocytes and macrophages. The underlying molecular mechanisms also are discussed. These experimental observations could offer new therapeutic strategies for immunological disorders like autoimmunity.
Keywords: dietary intervention, calorie restriction, fasting, mTOR, AMPK, GCN2, metabolic rewiring
Introduction
Abnormal nutritional conditions, such as malnutrition and diet-induced obesity, considerably affect the immunological status of the body. For instance, undernourished children are highly susceptible to infectious diseases and frequently show insufficient vaccine efficacy. In addition, multiple cohort studies have indicated that overnutrition and the prevalence of a westernized diet are associated with an increased incidence of inflammatory disorders, namely, metabolic syndrome, type 2 diabetes, allergy, and autoimmune disorders. Thus, diet and nutritional status significantly influence immune response. Furthermore, accumulating evidence has shown the importance of cellular metabolism in many aspects of immune cell biology. Inflammation-related M1 macrophages rely mainly on glycolysis, while immunosuppressive M2 macrophages utilize fatty acid oxidization (FAO) (1, 2). Naïve B and T lymphocytes are characterized by quiescent cellular metabolism, mainly depending on mitochondrial respiration (3, 4). Conversely, upon antigen recognition, naïve lymphocytes undergo metabolic rewiring to aerobic glycolysis to acquire effector functions. The amount of ATP production by glycolysis is much lower than that produced by mitochondrial respiration. Nevertheless, activated lymphocytes and macrophages exploit the prompt generation of ATP by glycolysis to fulfill the metabolic requirements for their proliferation and its provision of intermediate metabolites essential for the biosynthesis of nucleic acids, amino acids, and fatty acids.
Nutrient sensing is essential for the survival of cells and whole organisms, and thus the machinery of nutrient sensing and the downstream responses are highly preserved among eukaryotes (5). Cellular metabolic status is thus markedly affected by the extracellular milieu. For example, colonocytes at the luminal surface and upper crypts utilize short-chain fatty acids (e.g., butyrate), which is abundant in the colonic lumen, as a major energy source (6). In addition, white adipose tissue (WAT)-resident memory T cells and group 2 innate lymphoid cells (ILC2s) enhance FAO when activated (7, 8). Dietary intervention such as fasting prominently changes energy metabolism systemically, which in turn affects the immune cell biology, including cellular metabolism, cell dynamics, and survival. These findings raise the possibility that dietary intervention could offer novel approaches to ameliorate inflammatory disorders by modulating the immune response through metabolic rewiring. To date, a variety of dietary intervention protocols including calorie restriction, fasting, and nutrient supplementation have been proposed to regulate body weight, aging, the intestinal barrier, specific immune functions, and cognitive ability. In this review, we discuss the immunological significance of dietary intervention as well as the underlying mechanisms at both molecular and cellular levels. We also explore immunometabolism from a clinical perspective.
The Impact of Dietary Intervention on Immune Responses
In the modern age, the food supply is generally stable thanks to agriculture and animal husbandry. Therefore, people residing in developed countries rarely face famine. Overnutrition has become a global health concern as a factor conferring a predisposition to metabolic syndrome, cancer, allergic diseases, and autoimmune disorders. Adjustment of dietary intake, frequency of meals, and dietary composition has emerged as a potential option to protect against the development of these diseases. However, recent studies have also demonstrated that certain intervention protocols occasionally lead to adverse events, such as the development of metabolic disorders (9, 10). Therefore, it is important to clarify the immunological consequences and underlying molecular mechanisms of various types of dietary intervention to maximize beneficial effects. In this section, we focus on four major types of dietary intervention in the context of immune–metabolic interaction.
Calorie Restriction
Calorie restriction (CR) without malnutrition involves a chronic reduction of energy intake by 15% to 40% compared with ad libitum conditions while maintaining an adequate intake of micronutrients such as vitamins and minerals. The link between CR and immunological functions was revealed nearly half a century ago (11). Since then, a number of studies have proven that CR suppresses the development of multiple diseases, such as cardiovascular disease, diabetes, cancer, and autoimmune disorders in human disease models (12–15). The activation of effector T (Teff) cells and M1 macrophages is highly dependent on the phosphatidylinositol-3 kinase (PI3K)-Akt-mechanistic target of rapamycin (mTOR) signaling, and persistent activation of this pathway by overnutrition drives M1-skewed inflammation ( Figure 1 ). Conversely, the low-energy status conferred by CR suppresses the PI3K/Akt/mTOR axis with reciprocal activation of adenosine monophosphate-activated protein kinase (AMPK) and sirtuin family proteins (16–18) ( Figure 1 ).
Figure 1.

Overview of the nutritional signals regulating immune responses. Calorie restriction (CR) and fasting lowers plasma IGF-1 levels and downregulates PI3K/Akt/mTOR signaling pathways. At a low-energy status, two major energy sensors: adenosine monophosphate-activated protein kinase (AMPK) and sirtuin 1 (SIRT) family proteins, are activated by AMP and NAD+, respectively. GCN2 acts as a sensor of amino acid deficiency to regulate the differentiation and polarization of T cells and macrophages. β-HB also contributes to the anti-inflammatory effects by suppressing NLRP3 inflammasome activation. The white and orange boxes represent signal messengers and enzymes/transcription factors, respectively. The pathways depicted by black arrows and red bars represent the activation and inhibition by dietary restriction, respectively.
AMPK inhibits the activity of acetyl-coenzyme A carboxylase 1 (ACC1), which leads to a reduction of fatty acid synthesis (FAS) (19) ( Figures 1 and 2 ). The alteration of lipid metabolism is associated with the T-cell fate decision. For example, a 30% reduction of food intake for 4 weeks limited differentiation into Th17 cells and enhanced the development of regulatory T (Treg) cells by the inactivation of ACC1 in naïve T cells. Consequently, this treatment improved ischemic brain injury in a transient middle cerebral artery occlusion-induced ischemia model (20). Likewise, pharmacological inhibition of ACC1 by Soraphen A was reported to shape the Th17/Treg balance to improve the clinical score in an experimental autoimmune encephalomyelitis (EAE) model (21). The development of Th17 cells, but not Treg cells, requires ACC1-mediated de novo FAS. In addition, Th17 cells mainly utilize the glycolytic-lipogenic pathway to produce phospholipids for cellular membranes, whereas Treg cells actively take up exogenous fatty acids (21, 22). Furthermore, the other Teff cell subsets, such as Th1 and Th2 cells, rely on de novo FAS for their differentiation, and thus the inhibition of ACC1 can suppress their differentiation (21, 23). Conversely, ACC1 is dispensable for the activation of dendric cells and macrophages, even though de novo FAS is augmented upon mycobacterial infection (23). Therefore, ACC1 has emerged as a molecular target for drug development to regulate Teff cell-dependent inflammation.
Figure 2.
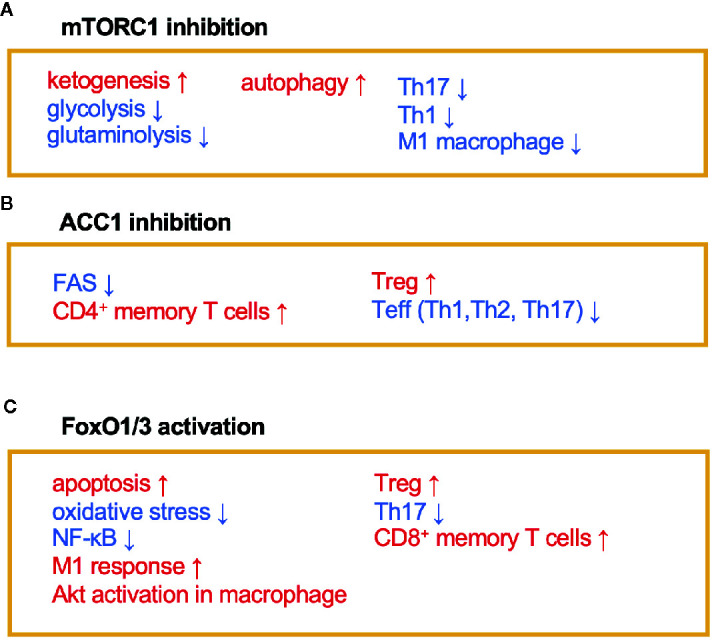
The immunomodulating effects of mTORC1, ACC1, and FoxO1/3. Fasting or calorie restriction (CR) suppresses mTORC1 and ACC1 activation and activates FoxO1/3 pathways. (A) mTORC1 inhibition enhances ketogenesis and reduces glycolysis and glutaminolysis. mTORC1 inhibition also induces autophagy in macrophage and suppresses Th1, Th17, and M1 macrophage differentiation. (B) ACC1 inhibition reduces FAS, which facilitates development of CD4+ memory T cells and Treg cells and conversely suppresses Teff (Th1, Th2, and Th17) responses. (C) Activation of FoxO1 and/or FoxO3 induces apoptosis and inactivates NF-κB. FoxO1 or FoxO3 also regulates phenotypes of macrophages, suppresses Th17 response, and induces development of CD8+ memory T cells and Treg cells.
A low-energy status during CR and fasting raises an intracellular level of nicotinamide adenine dinucleotide (NAD+) (24, 25). NAD+ is essential for glycolysis as well as oxidative phosphorylation (OXPHOS) as an electron transmitter. NAD+ also serves as a co-substrate for poly (ADP-ribose) polymerases (PARPs) and the sirtuin family (26–28). The intracellular NAD+ level is one of the critical determinants of differentiation and functions in macrophages. In human monocyte-derived macrophages, treatment with lipopolysaccharide (LPS) suppresses de novo NAD+ synthesis by inhibiting the kynurenine pathway and promotes the utilization of NAD+ by PARPs, lowering the intracellular level of NAD+ (29). Consequently, the LPS-induced decrease in NAD+ inactivates mitochondrial SIRT3, increases mitochondrial reactive oxygen species (ROS), suppresses mitochondrial respiration, and reciprocally activates glycolysis-dependent energy metabolism. Notably, such metabolic rewiring in response to the NAD+ levels substantially influences M1/M2 macrophage polarization. The augmentation of intracellular NAD+ by either overexpression of quinoline phosphoribosyltransferase (QPRT), a rate-limiting enzyme in the kynurenine pathway, or by supplementation of an NAD+ precursor rescues mitochondrial respiration in LPS-stimulated macrophages and thereby induces polarization to immunosuppressive M2 phenotype. Although the reduction of intercellular NAD+ pool facilitates differentiation into M1 phenotype, depletion of NAD+ leads to functional defect or reduces the viability of M1 macrophage due to the arrest of glycolysis (30). Indeed, NAD+ is integral to the simultaneous phosphorylation and oxidation of glyceraldehyde-3-phosphate to 1, 3-biphosphoglycerate, an essential step in glycolysis. Therefore, M1 macrophages utilize the NAD+ salvage pathway to maintain glycolysis and their functions, such as the production of pro-inflammatory cytokines (30). Inhibition of NAD+ salvage pathway by FK866 in LPS and/or IFN-γ-treated macrophages suppressed IL-1β and IL-6 production and decreased viability in vitro. Further, administration of FK866 ameliorated disease severity in an LPS-induced sepsis model.
Sirtuin 1 (SIRT1) is one of the NAD+-dependent deacetylases and serves as a significant regulator of metabolism and immune response. SIRT1 protein level increases in multiple cells and tissues in response to food deprivation and CR (31–33) ( Figure 1 ). SIRT1 deacetylates nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB), peroxisome proliferator-activated receptor γ (PPARγ), and hypoxia-induced factor 1α (HIF-1α) to regulate both innate and adaptive immune responses. In macrophages, deficiency in SIRT1 results in the hyperactivation of NF-κB, followed by the upregulation of pro-inflammatory cytokines such as TNF-α and IL-1β (34). In a syncytial virus infection model, the upregulation of SIRT1 in the lung was found to be essential to elicit respiratory immune responses and alleviate tissue damage (35). In this model, SIRT1 in dendritic cells promoted the production of Th1-inducing cytokines (e.g., IL-12 and TNF-α) and conversely suppressed the Th2 cytokines, leading to viral clearance and the resolution of inflammation. Furthermore, SIRT1 activation by NAD+ administration was found to ameliorate EAE symptoms by downregulating key transcription factors for Th1 (T-bet) and Th17 (RORγT and STAT3) (36). SIRT1 also inactivates HIF-1α through its deacetylating activity (37). Accumulating evidence has indicated the importance of HIF-1α for the Warburg-like metabolic rewiring toward aerobic glycolysis in immune cells. Therefore, SIRT1 may suppress the proinflammatory responses by inhibiting the HIF-1α-dependent metabolic rewiring (38). These findings raise the possibility that SIRT1 plays a role in the CR/fasting-dependent mitigation of inflammatory responses.
The immunoregulatory effect of CR may also be mediated by the Forkhead transcription factors O class 1 and 3 (FoxO1 and FoxO3) ( Figures 1 and 2 ). Since CR alleviates PI3K/Akt signaling, which is a negative regulator of FoxO1 and FoxO3, CR eventually upregulates these transcription factors (39). In CD4+ T cells, FoxO1 and FoxO3 upregulate the expression of Foxp3, the master regulator of regulatory T (Treg) cells (40–42). Treg cells play a critical role in suppressing excessive immune responses by expressing immunosuppressive molecules (e.g., IL-10 and CTLA-4) and a high-affinity IL-2 receptor, CD25, which provokes IL-2 deprivation. T-cell-specific deletion of FoxO1 attenuates the TGF-β-induced differentiation of Treg cells (43). FoxO3 suppresses the proliferation and activation of Teff cells by inhibiting NF-κB (44), and also induces apoptosis by upregulating Puma and Bim (45). FoxO3-deficient mice showed spontaneous lymphoproliferation, associated with inflammation of the lung, kidney, and salivary gland (44). Such inflammation was found to correlate with the presence of hyperactivated Th1 and Th2 cells. In addition, mice carrying T cells deficient in FoxO1 and FoxO3 develop severe systemic autoimmune diseases mainly because of a defect in Treg cells and the activation of Teff cells (46). FoxO3 also suppresses the production of IL-6 from dendritic cells, which also contributes to the inhibition of Teff cells (47).
FoxO1 activation in macrophages exacerbates inflammatory responses. Exogenous expression of a constitutively active form of Foxo1 in RAW264.7 cells potentiated LPS-induced TLR4 signaling pathway leading to phosphorylation of NF-κB, and vice versa, knockdown of Foxo1 suppressed the TLR4 signaling pathway (48). Additionally, monocyte/macrophage-specific FoxO1-deficient mice (LysMcre/+Foxo1fl/fl) impaired TLR2-mediated response in liver-resident macrophages and failed to induce Th1 and Th17 response during Staphylococcus aureus infection (49). Interestingly, FoxO1-induced TLR2 and TLR4 signals induced Akt phosphorylation leading to FoxO1 inactivation. This negative feedback may constrain excessive inflammatory responses in macrophages and induce resolution of inflammation. Besides their immunomodulatory functions, FoxOs prevent tissue damage by suppressing oxidative stress and accelerating wound healing (50–53). This raises the possibility that CR-dependent activation of FoxOs may also promote wound healing, although further investigations are required to prove this.
In addition to anti-inflammatory effects, CR may consolidate immunological memory in response to vaccination. This possibility has been raised by findings from two recent independent studies: 1) 50% dietary restriction reinforced the functions of CD8+ memory T cells to protect against secondary bacterial infection and tumorigenesis (54) and 2) severe CR enhanced the proliferative response and cytokine production by T cells upon secondary infection with influenza (55). These effects by CR may be attributable to the inactivation of mTOR signaling that orchestrates glycolysis, glutaminolysis, and fatty acid biosynthesis ( Figures 1 and 2 ). In support of this, low-dose treatment with an mTOR inhibitor, rapamycin, also facilitated the development and maintenance of memory T cells and conferred protection against viral infection (56). Similarly, in vitro culture of tumor-specific memory T cells under CR conditions enhanced anti-tumor functions, accompanied by the inactivation of mTOR signaling (57). Notably, ACC1 negatively regulates the transition of antigen-specific CD4+ T cells from effector to memory cell types ( Figure 2 ). Therefore, genetic ablation or pharmacological inhibition of ACC1 enforces CD4+ memory T-cell formation in response to helminth infection (58). IL-7 is a cytokine essential for the survival of naïve and CD8+ memory T cells. Importantly, FoxO1 upregulates Il7r genes in these T-cell subsets to reinforce IL-7/IL-7R signaling, by interacting with other transcription factors: GABP and Gfi-1 (59, 60). Thus, CR is considered to strengthen the development and functionality of the memory T-cell subset by regulating multiple mechanisms. In addition to these molecular mechanisms in specific cell types, CR or fasting affects systemic metabolism, hormone release (e.g., insulin, glucagon, adipokines, and glucocorticoids), and nerve systems. Consequently, the pleiotropic effects of CR could be attributable to integration of the alterations of metabolic, endocrine, and nerve systems. Interestingly, the efficacy of vaccines is attenuated by both malnutrition and obesity (61–64), illustrating that an appropriate energy balance is a prerequisite to induce immune responses to vaccination fully.
Fasting and Refeeding
Fasting refers to completely abstaining from food intake for certain periods ranging from several hours a day to a few weeks. There are many different fasting practices, including time-restricted feeding (TRF), intermittent fasting (IF), and periodic fasting (PF)/long-term fasting. Among them, TRF represents the daily restriction of food intake, usually for 12 to 20 h (65, 66). In IF, subjects or laboratory animals take little or no calories at least two days per week, but are allowed ad libitum feeding on the remaining days. IF protocols include alternative day fasting (ADF), in which 24-h fasting is repeated every other day (67, 68). Conversely, PF represents the intake of little or no calories for several days. PF has a more pronounced impact on metabolism and immune responses than CR or short-term fasting such as TRF and ADF (69).
Fasting has been performed as a religious practice. In Islamic tradition, abstinence from food and drink from dawn until sunset is encouraged during the month of Ramadan (70). Like CR, fasting also has a considerable impact on immune responses. IF during Ramadan results in significant decreases in circulating immune cells and pro-inflammatory cytokines (71, 72). Other studies have also demonstrated that Ramadan fasting induces the expression of antioxidant and anti-inflammatory genes in both nondiabetic obese patients and healthy subjects (73, 74). Fasting also reduces blood levels of glucose, insulin, insulin growth factor-1 (IGF-1), and amino acids, with the activation of AMPK and suppression of mTOR signaling. In response to these metabolic alterations, long-term hematopoietic stem cells undergo stress resistance, self-renewal, and regeneration (69). Fasting also induces whole-body FAO and ketogenesis in the liver to generate ketone bodies (i.e., acetone, acetoacetic acid, and β-hydroxybutyrate (β-HB)). In the kidney of aging-related chronic inflammation model, β-HB exerts an anti-inflammatory effect by activating FoxO1 through inhibition of Akt phosphorylation (75).
Furthermore, the refeeding phase in the fasting regimen may be necessary for cellular reprogramming and regenerative effects in various organs such as the liver, and gut (76). In an EAE model, IF was shown to ameliorate the disease symptoms by increasing the serum levels of adiponectin, corticosterone, and β-HB (77). Additionally, IF alters T-cell homeostasis in the gut with a decrease in Th17 cells and an increase in Treg cells. This effect is attributed to the alteration of gut microbiota by IF, which is characterized by an overrepresentation of Bacteroidaceae, Lactobacillaceae, and Prevotellaceae. The transplantation of fecal microbiota from IF mice was also shown to recapitulate EAE amelioration by IF (77).
Autophagy significantly contributes to somatic cell reprogramming and stem cell maintenance (78). The process of autophagy also serves as a protective factor against inflammation, infection, and neurodegenerative diseases (79–81). Autophagy was initially identified in Saccharomyces cultured under low-nutrient conditions (82). In the liver, farnesoid X receptor (FXR) and cAMP response element-binding protein (CREB), both of which are activated in response to nutrient signals, regulate the hepatic autophagy gene network (83). CREB was shown to promote the autophagic degradation of lipids under nutrient-deprived conditions, while FXR inhibited this response. Moreover, the CREB pathway enhanced alternatively activated M2 macrophage polarization in WAT (84). Furthermore, in the liver and muscle, refeeding after 24-h fasting suppressed autophagy by activating the mTOR complex 1 (mTORC1) pathway (85). Hence, fasting-induced autophagy might also lead to therapeutic effects.
TRF may also elicit its effect independent of the reduction of total calorie intake because TRF was shown to suppress weight gain and improve hyperinsulinemia, hepatic steatosis, and inflammation in mice fed a high-fat diet (HFD) (65). Notably, the total amounts of food intake were comparable between the TRF and ad libitum-fed groups. Ad libitum feeding with HFD disrupts the normal feeding cycle, with the mice eating the diet all day (86). Such feeding-cycle disruption is also prevalent in modern societies. This leads to the persistent activation of Akt/mTOR signaling as well as disturbance of the circadian oscillation of clock genes, both of which confer a predisposition to metabolic syndromes. TRF coordinates the balance of CREB, mTOR, and AMPK signaling and restores the circadian oscillations. The circadian oscillations are also observed in lymphocyte trafficking. At night, noradrenalin-dependent β2-adrenergic stimuli upregulate CCR7 and CXCR4 on B and T lymphocytes to suppress cell egress from the lymph nodes in mice (87). Additionally, the expression of sphingosine-1-phosphate receptor 1 (S1PR1), which also facilitates egress from the lymph nodes, by lymphocytes is also under the control of a circadian clock gene, Bmal1 (88). Expression of sphingosine-1-phosphate (S1P) is upregulated during the day. Consequently, CD4+ T cells accumulate in the lymph nodes at night and circulate during the day. In accordance with this observation, immunization with myelin-oligodendrocyte glycoprotein (MOG) at daytime augments the autoantigen-specific Th17 responses compared with nighttime immunization and exacerbates CNS inflammation in an EAE model (89). Taking these findings into account, the anti-inflammatory effect of TRF may partially be mediated by the normalization of the circadian oscillations.
Most previous studies have underscored the beneficial effects of calorie restriction and fasting on metabolic disorders and autoimmunity, where some reports showed adverse effects of such dietary interventions. The outcomes of calorie restriction and fasting are most likely dependent on feeding regimens, disease models, and the age of animals employed in each study. For example, a TRF in juvenile mice (4-8 weeks old) exacerbates metabolic disorders in adult age (12-week-old) (10). This study also manifested that TRF in juvenile mice affects sexual maturity as evidenced by retarded testicle development as well as high levels of serum GnRH, FSH, LH, and low levels of androgen and estrogen at 8-week-old. Such an observation is analogous to our findings that repeated fasting in juvenile mice (5-8 weeks old) attenuated the booster effect of oral immunization (90). Considering that GC reaction is essential for the induction of immunological memory, eliminating GC B cells from Peyer’s patches (PPs) by fasting may have resulted in this abnormality. This result is consistent with the observation of cohort studies that children with malnutrition fail to obtain vaccine efficacy (91–93). Thus, TRF and fasting in young adulthood may cause adverse effects in the metabolic, immunological, and enteroendocrine systems.
Recent clinical studies have shown the potential of a fasting-mimic diet (FMD), which is low in calories, sugars, and protein, but high in unsaturated fats, to achieve beneficial effects against aging, cancer, metabolic diseases, and cardiovascular diseases, in association with the reduction of body mass index, blood pressure and serum parameters (e.g., glucose, triglycerides, total and low-density lipoprotein cholesterol, IGF-1 and C-reactive protein) (94). Recent studies have also shown the effect of FMT on autoimmune diseases. Periodic 3-day FMD cycles ameliorated demyelination and symptoms in an EAE model (95). The 4-day cycles of FMD prevent the development of the dextran sodium sulfate (DSS)-induced colitis model by increasing the abundance of Lactobacillaceae and Bifidobacteriaceae with anti-inflammatory properties (96). Thus, FMD may be beneficial to improve both metabolic and inflammatory disorders.
Specific Nutrient Restriction
Growing evidence has suggested that amino acid restriction (AAR) may be at least partly responsible for the immunomodulatory effect of dietary restriction. AAR inactivates mTOR signaling because certain amino acids, such as leucine, serve as activators of the mTOR/S6K1 pathway (97, 98). Leucine deprivation was found to improve insulin sensitivity of the whole-body and in vitro-cultured hepatocytes (99). In addition, a serine protein kinase, general control nonderepressible 2 (GCN2), functions as a sensor of amino acid deprivation. Leucine deprivation activates GCN2, which in turn inhibits mTOR signaling. Deficiency of GCN2 canceled the effect of leucine deprivation on insulin tolerance.
AAR also appears to regulate the differentiation and functions of immune cell subsets. For example, GCN2-dependent activation in response to AAR diminishes mouse and human Th17 differentiation (100). In an EAE model, GCN2-deficient mice showed severe disease symptoms even at the remission stage in association with increases in Th1 and Th17 response and a decrease in Treg cells (101). AAR also activates activating transcription factor 4 (ATF4) in CD4+ T cells. ATF4 transactivates a gene network that facilitates the amino acid intake and mTORC1 signaling (102). ATF4-dependent metabolic rewiring is required for the proliferation of CD4+ T cells and the development of Th1 cells.
Dietary tryptophan restriction (DTR) was found to impair the development of encephalitogenic Th17 cells to ameliorate EAE (103). Interestingly, GCN2 is dispensable for the effect of DTR, since GCN2-deficient mice fed a protein-free or tryptophan-free diet were reported to show resistance to EAE similar to that of WT mice. DTR suppresses gut inflammatory responses by shaping the gut microbial community. Furthermore, 40% dietary methionine restriction (DMR) was reported to reduce oxidative stress by suppressing the generation of mitochondrial ROS in rat heart (104). Oxidative stress is implicated in the development of chronic inflammatory disorders including inflammatory bowel disease (IBD). Therefore, DMR ameliorated the severity of DSS-induced colitis (105). DMR also delayed the senescence-associated secretory phenotype (SASP) in the kidney through hydrogen sulfide (H2S) generation and AMPK pathway activation (106).
GCN2 also contributes to the regulation of various macrophage functions. Among the major function of splenic macrophages is the clearance of apoptotic cells, leading to immune tolerance. Phagocytosis of apoptotic cells activated the indoleamine 2,3-dioxygenase 1 (IDO1)/GCN2 axis to upregulate IL-10 and TGF-β synthesis in splenic macrophages. Macrophages from monocyte/macrophage lineage-specific GCN2 knockout (LysM cre/+/Gcn2 fl/fl) mice fails to acquire a tolerogenic phenotype (107). Backcross of LysM cre/+/Gcn2 fl/fl mice onto lupus-prone FcgRIIB-/- mice exacerbates systemic lupus erythematosus-like symptoms (107). Interestingly, tumor-associated macrophages from patients with melanoma also activate GCN2 and IL-10 production in the tumor microenvironment (108). Monocyte/macrophage-specific deletion of GCN2 drives the tumor-associated macrophages to induce antitumor responses. In contrast, GCN2 activation in RAW264.7 cells under tryptophan-free conditions results in the upregulation of inflammatory cytokines upon stimulation with LPS (109). The GCN2/eIF2/CHOP pathway mediates this response. The monocyte/macrophage-specific deletion of GCN2 improves the mortality after a lethal challenge with LPS by reducing the expression of IL-6 and IL-12 (109).
Carbohydrate restriction using a ketogenic diet (KD) also significantly regulates immune responses. KD is defined as a very low-carbohydrate, high-fat diet, which induces the generation of ketone bodies. A recent study revealed that KD alters gut microbiota. The KD-associated microbiota is characterized by an underrepresentation of Bifidobacterium spp. (110). β-HB produced by KD plays a central role in the decrease in Bifidobacterium spp., and this microbial environment prevents a pro-inflammatory Th17 response in the small intestine, but not the large intestine, of mice. However, it remains obscure how Bifidobacterium spp., which are usually located in the large intestine, induce the small intestinal Th17 cells. Notably, the reduction of bifidobacteria (e.g., Bifidobacterium adolescentis and B. longum) was also evident in humans fed KD for 4 weeks.
KD has been clinically used to cure intractable epilepsy, but the therapeutic mechanism behind this is largely unknown. Although the pathological mechanism has yet to be elucidated, an increased Th17/Treg cell ratio is considered to be a predisposing factor for intractable epilepsy (111). In young patients with intractable epilepsy, KD improved the imbalance of Th17/Treg cells in the blood through suppression of the mTOR/HIF-1 signaling pathway. Hence, KD may contribute to the treatment of intractable epilepsy by shaping the T-cell responses (111). Meanwhile, KD was found to induce the expansion of lung γδ T cells to strengthen epithelial barrier functions and antiviral resistance to influenza A virus (112). However, KD may exert distinct immunological effects depending on the duration of the treatment. Short-term (1-week-long) KD activates adipose-tissue-resident γδ T cells to support tissue repair, whereas long-term (4-month) KD suppresses this T-cell subset and exacerbates obesity in mice (113). Additionally, β-HB suppressed the NLRP3 inflammasome and attenuated the secretion of IL-1β and IL-18 in both mouse bone marrow-derived macrophages and human monocytes (114). The inhibition of the NLRP3 inflammasome is not dependent on AMPK, ROS, and autophagy, but is attributed to the prevention of K+ efflux and reduction of apoptosis-associated speck-like protein with a caspase recruitment domain (ASC) oligomerization.
Given that poor compliance and occasional adverse effects have limited the clinical application of CR and fasting, specific nutrient restrictions may be more feasible dietary interventions to achieve beneficial effects similar to those of CR and fasting.
Nutrient Supplementation for the Regulation of Immune Response
Epidemiological evidence has suggested that malnutrition is a risk factor for infectious diseases and impairs vaccine efficacy (115–117). Vaccine efficacy depends on the formation of immunological memory. Vaccination evokes a GC reaction to generate plasma cells and memory B cells, which contribute to antibody production and long-lasting memory function, respectively. Anatomically, GCs are separated into the light zone (LZ) and the dark zone (DZ). GC B cells are highly mobile, circulating between the LZ and DZ (118). Upon antigen stimulation, naïve B cells migrate into the LZ, where the affinity-driven selection of GC B cells occurs through interaction with follicular helper T (Tfh) cells and follicular dendritic cells. The positively selected GC B cells activate mTORC1, which is required for migration into the DZ and vigorous proliferation (119). Several studies have demonstrated that the differentiation and the survival of Tfh cells and GC B cells highly depends on the mTOR signaling pathway (119, 120). We also confirmed that treatment with rapamycin greatly reduces the number of GC B cells in PPs (90). Deletion of Raptor or Rictor, which is the signature component of mTORC1 and mTORC2 respectively, in OX40+ cells (i.e. activated CD4+ T cells) impaired Tfh cell differentiation in PPs (120). In PPs, Tfh cells highly express glucose transporter Glut1 compared with other T-cell lineages to enhance glucose uptake. Glut1 expression in Tfh cells partly depends on mTORC1 signaling, since rapamycin treatment reduced Glut1 expression and glucose uptake. Nevertheless, GC B cells rely on FAO rather than glycolysis to fuel proliferation (121, 122); however, the mechanisms underlying how proliferating GC B cells actively oxidize fatty acids have remained unclear. Moreover, recent studies have indicated that several vitamins also participate in the regulation of B-cell homeostasis. Deficiency in vitamin B1 was found to decrease the number of naïve B cells in PPs. Vitamin B1-dependent maintenance of naïve B cells is required for the induction, but not effector, phase of the IgA response upon oral immunizaition (3).
Vitamin A (VA) deficiency is currently a global concern, especially in developing countries. Retinoic acid (RA), an active metabolite of VA, plays a central role in the lymphocyte trafficking to the gut, GC formation, and development of IgA+ plasma cells, leading to increased primary and secondary antibody responses (123, 124). Large peritoneal macrophages (LPMs), one of the mouse peritoneal macrophage subsets, supports IgA class-switching of peritoneal B-1 cells. RA-induced activation of GATA6 in LPM precursors causes their polarization and migration to the peritoneal cavity. In accordance with this, mice carrying monocyte/macrophage lineage-specific Gata6 deletion (LysM Cre/+/Gata6 f/f) or mice fed on VA-depleted diet (VAD) decreased the number of peritoneal macrophages, resulting in a decrease in B-1-derived IgA plasma cells in intestinal lamina propria. Thus, RA facilitates the differentiation of peritoneal B-1 cells into plasma B cells induced by LPMs (125). RA is also required for inflammatory resolution during helminth infection (126). Schistosoma mansoni infection transiently increases inflammatory F4/80int CD206+ macrophages in the liver. This macrophage subset subsequently differentiates into F4/80hi CD206− macrophages to resolve acute inflammation. S. mansoni-infected mice fed on VAD were defective in the development of F4/80hi CD206− macrophages, leading to dysregulated inflammation and increased mortality.
Therefore, deficiency in RA attenuates the immune response and raises the risk of infectious diseases. To deal with VA deficiency, the World Health Organization has recommended high-dose VA supplementation in children 6–59 months of age in locations where VA deficiency is endemic. Oral supplementation of VA or RA potentiated vaccine efficacy by facilitating the trafficking of vaccine-antigen-specific T lymphocytes to the gastrointestinal mucosa in mice (127, 128). Moreover, the supplementation of VA or RA upregulated Stimulated by retinoic acid-6 (Stra6) in the spleen, enhancing anti-tetanus toxoid antibody production (129). Thus, VA supplementation has been considered a promising strategy to reinforce the antigen-specific immune response upon vaccination.
The Effect of Dietary Intervention on Cell Dynamics
Recent studies have revealed that the bone marrow serves as a reservoir for naïve B cells, monocytes, and memory CD8+ T cells in response to a low-energy status like that found in CR and fasting (54, 90, 130) ( Figure 3 ). These findings indicate that nutritional status influences the cell dynamics of several immune cell subsets. We observed that naïve B cells migrated from PPs to the bone marrow in a CXCL13-dependent manner during 36 h of fasting in mice (90). CXCL13 expression by stromal cells is essential for the formation and maintenance of lymphoid follicles in the lymphoid organs including PPs (131, 132). The Cxcl13 mRNA expression was significantly downregulated in PPs during the fasting period. In vitro study using a lymph node-derived stromal cell line suggested that cytokine stimulus (i.e., TNF-α and LTβR agonists)-dependent upregulation of Cxcl13 is accompanied by an increase in glycolysis. Because treatment with a glycolysis inhibitor, 2-deoxyglucose, mitigated Cxcl13 expression by activated BLS12 cells in a dose-dependent manner, the metabolic rewiring to glycolysis plays a pivotal role in Cxcl13 expression. The 36 h of fasting markedly lowered blood glucose levels and attenuated glycolysis, resulting in the downregulation of Cxcl13 expression. In sharp contrast, in the bone marrow, Cxcl13 expression significantly increased during fasting, which allowed naïve B cells to accumulate mainly in the vicinity of blood vessels of the bone marrow cavity. Such a perivascular region is also known as a site for hematopoietic stem cell (HSC) differentiation and proliferation due to the accumulation of survival factors such as B-cell activating factor (BAFF), colony-stimulating factors (CSF), and stem cell factors (SCF), which are most likely fundamental for the survival of B cells (133–135). Therefore, the perivascular region could serve as a transient niche for naïve B cells during fasting. In response to refeeding, Cxcl13 expression in the bone marrow was downregulated and naïve B cells migrated back to PPs. Thus, the number of naïve B cells was gradually restored until 48 h after refeeding. In contrast, the recovery of GC B cells and IgA+ B cells was much slower than that of naïve B cells because these cells readily underwent apoptosis in response to fasting. As a consequence, GC B cells were eliminated from lymphoid follicles of PPs, whereas naïve B cells were restored after fasting-refeeding. Give that a subset of GC B cells differentiates into memory B cells, the elimination of GC B cells from PPs in fasted mice may lead to the loss of immune memory for oral antigens. Indeed, fasted mice failed to generate antigen-specific IgA, IgM, and IgG upon repeated oral immunization with ovalbumin.
Figure 3.

Bone marrow serves as a reservoir for several immune cell subsets in response to calorie restriction (CR) or fasting. Under low-energy conditions, naïve B cells, monocytes, and memory CD8+ T cells accumulate in the bone marrow. Fasting lowers CXCL13 levels in PPs and reciprocally increases the expression in the bone marrow. This leads to the migration of naïve B cells from PPs to the bone marrow. On the other hand, GC B cells and IgA+ B cells undergo apoptosis. Fasting diminishes circulating CCL2 levels through AMPK/PPARα signaling activation. Consequently, the egress of monocytes from the bone marrow is suppressed. Dietary restriction drives CD8+ memory T cells to traffic toward the bone marrow in the S1P/S1PR1- and CXCL12/CXCR4-dependent manner. The bone marrow microenvironment provides a tissue-specific niche for the maintenance of memory CD8+ T cells with low glucocorticoid concentration and abundant adipocytes.
Jordan et al. found that short-term (4–20 h) fasting suppressed the CCL2/CCR2 axis, which is essential for the egress of inflammatory monocytes from the bone marrow into the bloodstream (130). This short-term fasting activated AMPK/PPARα signaling in hepatocytes to lower the level of circulating CCL2. Although monocyte dynamics are also regulated by the CXCL12/CXCR4 axis in the bone marrow, the CXCL12 expression level was not affected by fasting (136). Fasting-induced accumulation of monocytes in the bone marrow was independent of fibroblast growth factor 21 (FGF21) and ketone body production. Transcriptome analysis demonstrated that monocytes were in a quiescent state during fasting. Notably, upon infection with Listeria monocytogenes, monocytes normally migrated from the bone marrow into the bloodstream and eliminated the pathogen without causing inflammation.
Collins et al. showed that 50% dietary restriction promoted the accumulation of circulating CD8+ memory T cells in the bone marrow in an S1P/S1P receptor 1- and CXCL12/CXCR4-dependent manner (54). CD8+ memory T cells resided in WAT under physiological conditions; however, they preferentially migrate to the bone marrow at low-energy status. Under CR conditions, the concentration of glucocorticoid is increased in the blood but decreased in the bone marrow. Because a high concentration of glucocorticoid induces apoptosis in memory CD8+ T cells, these cells can survive in the bone marrow under CR conditions. Furthermore, dietary restriction led to the differentiation of adipocytes that generate fatty acids. Memory CD8+ T cells actively utilize fatty acids for mitochondrial FAO (137–139). Thus, the bone marrow microenvironment provides a niche for memory CD8+ T cells under the low-energy conditions (54). It is worth noting that dietary restriction was also found to enhance the protective function of memory T CD8+ cells; dietary-restriction promoted clearance of influenza and Yersinia pseudotuberculosis in mice.
These recent studies revealed the function of the bone marrow as a shelter for several immune cell subsets during metabolic adversity. Such a multiorgan-trafficking of immune cells may occur even under normal nutritional conditions. In support of this notion, we observed that naive B cells show circadian oscillation between the bone marrow and PPs. The number of naive B cells in the bone marrow increased during the daytime when mice usually take little food, whereas the number decreased during nighttime in response to food intake. Furthermore, FoxO1, which is upregulated during CR, regulates naïve T cell migration to the secondary lymphoid tissues by increasing the expression of CCR7 and L-selectin. We are only beginning to learn about the multiorgan-trafficking of immune cells as a fasting response. Further investigation will clarify the underlying mechanisms in this emerging field.
A Clinical Perspective on Immunometabolism
In addition to dietary interventions, drugs targeting key nutrient signaling pathways (e.g., AMPK and mTOR) have attracted substantial attention in efforts to improve metabolic and inflammatory disorders. Rapamycin is best characterized as an inhibitor of mTORC1, and its chronic administration also inhibits mTORC2 in some tissues. Rapamycin has been clinically used as an immunosuppressant to prevent post-transplantation rejection. Animal studies have also corroborated that rapamycin-dependent mTOR inhibition is effective for various diseases, including ischemic stroke, rheumatoid arthritis, and EAE (140–143) ( Table 1 ). The anti-inflammatory effect of rapamycin treatment is associated with the attenuation of Th17-cell differentiation as well as the promotion of Treg-cell development (142, 143). Moreover, the coadministration of rapamycin with FMS-like tyrosine kinase 3 (FLT-3) ligand was shown to facilitate the plasmacytoid dendritic cell-dependent induction of Treg cells, thereby enhancing immune tolerance (144). Rapamycin was also reported to alleviate the development of a murine atherosclerosis model. During the initial phase of atherosclerosis, peritoneal macrophages transform into foam cells that elicit plaque formation inside the arterial vessels (145). Rapamycin was found to induce autophagy in macrophages to prevent foam cell development (146). However, rapamycin and everolimus, a derivative of rapamycin, possess only a narrow therapeutic index because of their pleiotropic effects, which potentially cause adverse effects.
Table 1.
Drugs targeting the metabolic pathways of immune cells.
| Drugs | Conventional application | Targeting metabolism | Immunological change | Tissue | New applications |
|---|---|---|---|---|---|
| Rapamycin (sirolimus) Everolimus |
post-transplantation rejection suppression | Inhibition of mTORC1 (mTORC2) |
autophagy in macrophages↑ Th1/Th17 ↓ Treg ↑ memory CD8+ T cells ↑ |
Brain arterial-vessels |
rheumatoid arthritis EAE/MS ischemic stroke atherosclerosis viral infection |
| Metformin | type 2 diabetes | Inhibition of mitochondrial complex I Activation of AMPK Inhibition of mTOR |
Inhibition of NLRP 3- inflammasome Th1/Th17 ↓ Treg/IL-10 ↑ IL-1β↓ |
brain lung liver intestine skin |
IBD MS sepsis-induced acute lung injury psoriasis |
| Dimethyl fumarate | Psoriasis | Inhibition of GAPDH | Activation of macrophages↓ Th1/Th17 ↓ Treg ↑ |
spleen intestine brain |
EAE |
| Mevalonate | a metabolite of cholesterol synthesis | Activation of mTOR-HIF-1 pathway | trained monocyte ↑ | (blood) | innate immune memory ↑ cancer |
| Soraphen A | Fungicide | Inhibition of ACC1 | Inhibition of FAS Th17 ↓ Treg ↑ |
brain | ischemic stroke |
| Halofuginone | the plant alkaloid Dichroa-febrifuga | Inhibition of prolyl-tRNA synthetase Activation of GCN2 |
Th17 ↓ GC formation ↑ memory CD8+ T cells ↑ |
intestine brain |
colitis MS vaccine efficacy↑ |
Metformin, the first-line drug for type 2 diabetes, has the potential to regulate immune responses through inducing metabolic rewiring in immune cells such as T cells ( Table 1 ). It inhibits mitochondrial complex I and restrains hepatic gluconeogenesis, with an increase in glucose utilization in peripheral tissues (147). Such inhibition of mitochondrial complex I was found to enhance hepatic AMPK, which in turn established a CR/fasting-like metabolic status (148). A low concentration (0.5 mM) of metformin inhibited hepatic mTORC1 through the AMPK-related pathway in mice (149). Similar to dietary interventions, metformin and a complex I inhibitor, rotenone, were reported to suppress mitochondrial ROS generation and NLRP3-dependent inflammasome formation in animal and human studies (150–152). Accordingly, metformin treatment decreased IL-1β production while increasing IL-10 in response to LPS (153). Metformin treatment also suppressed the development of Th17 cells by inhibiting mTOR and STAT3 signaling, whereas it induced Treg cells by enhancing AMPK signaling (154). These studies demonstrate that metformin has a therapeutic effect on inflammation-related diseases, including IBD, EAE, rheumatoid arthritis, and psoriasis (154–158). These observations also raise the possibility that a combination of dietary intervention and metformin may induce synergy on the anti-inflammatory responses. In a preliminary clinical trial, a combination of a dietary restriction with metformin on obese people enforced protective effects on insulin resistance (159). Interestingly, a low concentration (10 µM) of metformin stimulated exhausted tumor-infiltrating CD8+ T cells to upregulate IL-2, TNF-α, and IFN-γ and potentiated anti-tumor activity (160). Metformin also diminished tumor-resident Treg cells that counteract anti-tumor immunity (161). Although Treg metabolism largely depends on OXPHOS through FAO, metformin-treated Treg cells undergo glycolysis. These facts indicate that low-dose metformin augments anti-tumor immunity by inducing metabolic rewiring.
Halofuginone (HF), a derivative of the plant alkaloid Dichroa febrifuga is a pharmacological mimic of AAR. HF competitively inhibited prolyl-tRNA synthetase, enhancing the intracellular pool of uncharged tRNA that phosphorylates and activates GCN2 (162). HF treatment selectively constrained Th17 differentiation, but not Th1, and protected the development of EAE (100). The administration of HF also inhibited IL-1β production by LPS-stimulated macrophages and ameliorated the severity of DSS-induced colitis (163). Additionally, HF promoted GC formation and memory B cell formation in the draining lymph nodes in mice received vaccination (164). HF treatment also increased antigen-specific effector CD4+ and CD8+ T cells upon the secondary stimulation. Based on these observations, the pharmacological mimic of AAR responses may be a promising strategy to dampen inflammatory response and consolidate immunological memory for vaccine antigens ( Table 1 ).
Other drugs targeting the metabolic system have also become key options for immune modulation. Dimethyl fumarate (DMF), a derivative of the Krebs cycle intermediate fumarate (165). DMF inactivated a glycolytic enzyme, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and inhibited aerobic glycolysis in activated, but not resting, macrophages ( Table 1 ). The inhibition of glycolysis by DMF limited differentiation and functions of CD4+ T cells cultured under Th1- and Th17-polarized conditions.
Innate immune cells also develop long-term memory upon stimulation with bacterial products like β-glucan. This phenomenon is termed as trained immunity (166). A metabolic shift from OXPHOS to aerobic glycolysis, namely, Warburg-like effect, is vital for β-glucan-induced trained immunity. Such a metabolic alteration is caused by the activation of mTOR-HIF-1 pathway (167). The induction of trained immunity is also dependent on mevalonate, a metabolite of cholesterol synthesis. Mevalonate not only active the mTOR-HIF-1 pathway but also cause epigenetic alternations characterized by enrichment of H3K4me3 on the promoter region of IL6 and TNFA gene loci (168). Inhibition of mevalonate synthesis by fluvastatin canceled the increment of cytokine production as well as epigenetic alternations induced by either β-glucan or oxidized low-density lipoprotein in monocytes. Thus, cholesterol biosynthetic pathways are considered as a drug target to interrupt innate immune memory ( Table 1 ).
Conclusion
Dietary interventions have profound effects on immune responses through metabolic rewiring. Fasting has been shown to enhance immune memory and suppress inflammation. TRF was shown to recover the appropriate circadian rhythm, improving metabolic disorders and optimizing immune responses. Moreover, accumulating studies have demonstrated the molecular mechanisms underlying these findings. Thus, drugs targeting the metabolic system may become a critical option for immune modulation.
Meanwhile, dietary interventions of different types, durations, and timings can have opposite effects on health and disease. The benefits of dietary interventions can vary from patient to patient when we apply such interventions in a clinical setting. Experimental protocols have been performed to treat various immune-related disorders with dietary interventions. Looking ahead, we need to develop evidence-based, optimized protocols to avoid adverse effects. New therapeutic approaches against type 2 diabetes and cardiovascular disease are currently being discussed and applied (169–171). It is reasonable to use the same approach to treat inflammatory diseases and enhance vaccine efficacy.
In addition to local responses, an integrated immunometabolic response is required for host survival in a state of energy deficit. Recent studies have revealed the function of the bone marrow as a shelter for immune cells during dietary deficiency. It is clear that metabolic rewiring impacts immune cell dynamics, inducing inter-organ fasting responses in the body. A deeper understanding of this field could lead to the identification of novel drug targets.
Author Contributions
TO and MN wrote the manuscript. TO prepared the figures. KH critically revised the manuscript and obtained funding. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Japan Society for the Promotion of Science (20H00509 to KH) and AMED-Crest (20gm1010004h0105 and 20gm1310009h0001 and to KH).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1. Kelly B, O’Neill LAJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res (2015) 25:771–84. 10.1038/cr.2015.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate Fate in Activated Macrophages: A Comparison between Innate, Classic, and Alternative Activation. J Immunol (2010) 185:605–14. 10.4049/jimmunol.0901698 [DOI] [PubMed] [Google Scholar]
- 3. Kunisawa J, Sugiura Y, Wake T, Nagatake T, Suzuki H, Nagasawa R, et al. Mode of Bioenergetic Metabolism during B Cell Differentiation in the Intestine Determines the Distinct Requirement for Vitamin B1. Cell Rep (2015) 13:122–31. 10.1016/j.celrep.2015.08.063 [DOI] [PubMed] [Google Scholar]
- 4. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine Uptake and Metabolism Are Coordinately Regulated by ERK/MAPK during T Lymphocyte Activation. J Immunol (2010) 185:1037–44. 10.4049/jimmunol.0903586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-sensing mechanisms across evolution. Cell (2015) 161:67–83. 10.1016/j.cell.2015.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roediger WE. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology (1982) 83(2):424–9. [PubMed] [Google Scholar]
- 7. Wilhelm C, Harrison OJ, Schmitt V, Pelletier M, Spencer SP, Urban JF, et al. Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J Exp Med (2016) 213:1409–18. 10.1084/jem.20151448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Han SJ, Glatman Zaretsky A, Andrade-Oliveira V, Collins N, Dzutsev A, Shaik J, et al. White Adipose Tissue Is a Reservoir for Memory T Cells and Promotes Protective Memory Responses to Infection. Immunity (2017) 47:1154–1168.e6. 10.1016/j.immuni.2017.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Finnell JS, Saul BC, Goldhamer AC, Myers TR. Is fasting safe? A chart review of adverse events during medically supervised, water-only fasting. BMC Complement Altern Med (2018) 18:67. 10.1186/s12906-018-2136-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hu D, Mao Y, Xu G, Liao W, Ren J, Yang H, et al. Time-restricted feeding causes irreversible metabolic disorders and gut microbiota shift in pediatric mice. Pediatr Res (2019) 85:518–26. 10.1038/s41390-018-0156-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fernandes G, Yunis EJ, Good RA. Suppression of adenocarcinoma by the immunological consequences of calorie restriction. Nature (1976) 263:504–7. 10.1038/263504b0 [DOI] [PubMed] [Google Scholar]
- 12. Most J, Tosti V, Redman LM, Fontana L. Calorie restriction in humans: An update. Ageing Res Rev (2017) 39:36–45. 10.1016/j.arr.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Taylor R. Calorie restriction for long-term remission of type 2 diabetes. Clin Medicine J R Coll Physicians London (2019) 19:37–42. 10.7861/clinmedicine.19-1-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwashige K, Kouda K, Kouda M, Horiuchi K, Takahashi M, Nagano A, et al. Calorie restricted diet and urinary pentosidine in patients with rheumatoid arthritis. J Physiol Anthropol Appl Hum Sci (2004) 23:19–24. 10.2114/jpa.23.19 [DOI] [PubMed] [Google Scholar]
- 15. Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol Sci (2010) 31:89–98. 10.1016/j.tips.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mercken EM, Crosby SD, Lamming DW, Jebailey L, Krzysik-Walker S, Villareal DT, et al. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell (2013) 12:645–51. 10.1111/acel.12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cantó C, Auwerx J. Calorie restriction: Is AMPK a key sensor and effector? Physiology (2011) 26:214–24. 10.1152/physiol.00010.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guarente L. Calorie restriction and sirtuins revisited. Genes Dev (2013) 27:2072–85. 10.1101/gad.227439.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: Target for metabolic syndrome. J Lipid Res (2009) 50:S138–43. 10.1194/jlr.R800079-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X, Zhou Y, Tang D, Zhu Z, Li Y, Huang T, et al. ACC1 (Acetyl Coenzyme A Carboxylase 1) Is a Potential Immune Modulatory Target of Cerebral Ischemic Stroke. Stroke (2019) 50:1869–78. 10.1161/STROKEAHA.119.024564 [DOI] [PubMed] [Google Scholar]
- 21. Berod L, Friedrich Y, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. 10.1038/nm.3704 [DOI] [PubMed] [Google Scholar]
- 22. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4 + T Cell Subsets. J Immunol (2011) 186:3299–303. 10.4049/jimmunol.1003613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stüve P, Minarrieta L, Erdmann H, Arnold-Schrauf C, Swallow M, Guderian M, et al. De novo fatty acid synthesis during mycobacterial infection is a prerequisite for the function of highly proliferative T cells, but not for dendritic cells or macrophages. Front Immunol (2018) 9:495. 10.3389/fimmu.2018.00495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-Sensitive Mitochondrial NAD+ Levels Dictate Cell Survival. Cell (2007) 130:1095–107. 10.1016/j.cell.2007.07.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in saccharomyces cerevisiae. Sci (80- ) (2000) 289:2126–8. 10.1126/science.289.5487.2126 [DOI] [PubMed] [Google Scholar]
- 26. Katsyuba E, Auwerx J. Modulating NAD + metabolism, from bench to bedside. EMBO J (2017) 36:2670–83. 10.15252/embj.201797135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci (2007) 32:12–9. 10.1016/j.tibs.2006.11.006 [DOI] [PubMed] [Google Scholar]
- 28. Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, et al. NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther (2020) 5(1):227. 10.1038/s41392-020-00311-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat Immunol (2019) 20:50–63. 10.1038/s41590-018-0255-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cameron AM, Castoldi A, Castoldi DE, Flachsmann LJ, Field CS, Puleston DJ, et al. Inflammatory macrophage dependence on NAD + salvage is a consequence of reactive oxygen species–mediated DNA damage. Nat Immunol (2019) 20:420–32. 10.1038/s41590-019-0336-y [DOI] [PubMed] [Google Scholar]
- 31. Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Sci (80- ) (2004) 306:2105–8. 10.1126/science.1101731 [DOI] [PubMed] [Google Scholar]
- 32. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, et al. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Sci (80- ) (2004) 305:390–2. 10.1126/science.1099196 [DOI] [PubMed] [Google Scholar]
- 33. Ran M, Li Z, Yang L, Tong L, Zhang L, Dong H, et al. Calorie restriction attenuates cerebral ischemic injury via increasing SIRT1 synthesis in the rat. Brain Res (2015) 1610:61–8. 10.1016/j.brainres.2015.03.043 [DOI] [PubMed] [Google Scholar]
- 34. Schug TT, Xu Q, Gao H, Peres-da-Silva A, Draper DW, Fessler MB, et al. Myeloid Deletion of SIRT1 Induces Inflammatory Signaling in Response to Environmental Stress. Mol Cell Biol (2010) 30:4712–21. 10.1128/MCB.00657-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Owczarczyk AB, Schaller MA, Redd M, Rasky AJ, Lombard DB, Lukacs NW. Sirtuin 1 Regulates Dendritic Cell Activation and Autophagy during Respiratory Syncytial Virus–Induced Immune Responses. J Immunol (2015) 195:1637–46. 10.4049/jimmunol.1500326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang J, Zhao C, Kong P, Sun H, Sun Z, Bian G, et al. Treatment with NAD+ inhibited experimental autoimmune encephalomyelitis by activating AMPK/SIRT1 signaling pathway and modulating Th1/Th17 immune responses in mice. Int Immunopharmacol (2016) 39:287–94. 10.1016/j.intimp.2016.07.036 [DOI] [PubMed] [Google Scholar]
- 37. Lim J-H, Lee Y-M, Chun Y-S, Chen J, Kim J-E, Park J-W. Molecular Cell Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1a. Mol Cell (2010) 38:864–78. 10.1016/j.molcel.2010.05.023 [DOI] [PubMed] [Google Scholar]
- 38. Yu Q, Dong L, Li Y, Liu G. SIRT1 and HIF1α signaling in metabolism and immune responses. Cancer Lett (2018) 418:20–6. 10.1016/j.canlet.2017.12.035 [DOI] [PubMed] [Google Scholar]
- 39. Kim DH, Kim JY, Yu BP, Chung HY. The activation of NF-κB through Akt-induced FOXO1 phosphorylation during aging and its modulation by calorie restriction. Biogerontology (2008) 9:33–47. 10.1007/s10522-007-9114-6 [DOI] [PubMed] [Google Scholar]
- 40. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factorFoxp3. J Immunol (2017)198(3):981–5. 10.1126/science.1079490 [DOI] [PubMed] [Google Scholar]
- 41. Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, et al. Novel Foxo1-dependent transcriptional programs control T reg cell function. Nature (2012) 491:554–9. 10.1038/nature11581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohkura N, Sakaguchi S. Foxo1 and Foxo3 help Foxp3. Immunity (2010) 33:835–7. 10.1016/j.immuni.2010.12.004 [DOI] [PubMed] [Google Scholar]
- 43. Kerdiles YM, Stone EL, Beisner DL, McGargill MA, Ch’en IL, Stockmann C, et al. Foxo Transcription Factors Control Regulatory T Cell Development and Function. Immunity (2010) 33:890–904. 10.1016/j.immuni.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin L, Hron JD, Peng SL. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity (2004) 21:203–13. 10.1016/j.immuni.2004.06.016 [DOI] [PubMed] [Google Scholar]
- 45. You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med (2006) 203:1657–63. 10.1084/jem.20060353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ouyang W, Beckett O, Ma Q, Paik J-H, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3 + regulatory T cells. Nat Immunol (2010) 11:618–27. 10.1038/ni.1884 [DOI] [PubMed] [Google Scholar]
- 47. Dejean AS, Beisner DR, Che’n IL, Kerdiles YM, Babour A, Arden KC, et al. Transcription factor Foxo3 controls the magnitude of T cell immune responses by modulating the function of dendritic cells. Nat Immunol (2009) 10:504–13. 10.1038/ni.1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fan W, Morinaga H, Kim JJ, Bae E, Spann NJ, Heinz S, et al. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J (2010) 29:4223–36. 10.1038/emboj.2010.268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang YC, Ma H-D, Yin X-Y, Wang Y-H, Liu Q-Z, Yong J-B, et al. Forkhead Box O1 Regulates Macrophage Polarization Following Staphylococcus aureus Infection: Experimental Murine Data and Review of the Literature. Clin Rev Allergy Immunol (2016) 51:353–69. 10.1007/s12016-016-8531-1 [DOI] [PubMed] [Google Scholar]
- 50. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Sci (80- ) (2013) 339:211–4. 10.1126/science.1227166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ambrogini E, Almeida M, Martin-Millan M, Paik J-H, DePinho RA, Han L, et al. FoxO-Mediated Defense against Oxidative Stress in Osteoblasts Is Indispensable for Skeletal Homeostasis in Mice. Cell Metab (2010) 11:136–46. 10.1016/j.cmet.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu Q, Zhai Y, Cheng Q, Liu Y, Gao X, Zhang T, et al. The Akt-FoxO3a-manganese superoxide dismutase pathway is involved in the regulation of oxidative stress in diabetic nephropathy. Exp Physiol (2013) 98:934–45. 10.1113/expphysiol.2012.068361 [DOI] [PubMed] [Google Scholar]
- 53. Ponugoti B, Xu F, Zhang C, Tian C, Pacios S, Graves DT. FOXO1 promotes wound healing through the up-regulation of TGF-β1 and prevention of oxidative stress. J Cell Biol (2013) 203:327–43. 10.1083/jcb.201305074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Collins N, Han SJ, Enamorado M, Link VM, Huang B, Moseman EA, et al. The Bone Marrow Protects and Optimizes Immunological Memory during Dietary Restriction. Cell (2019) 178:1088–101.e15. 10.1016/j.cell.2019.07.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Effros RB, Walford RL, Weindruch R, Mitcheltree C. Influences of dietary restriction on immunity to influenza in aged mice. J Gerontol (1991) 46(4):B142–7. 10.1093/geronj/46.4.B142 [DOI] [PubMed] [Google Scholar]
- 56. Goldberg EL, Smithey MJ, Lutes LK, Uhrlaub JL, Nikolich-Žugich J. Immune Memory–Boosting Dose of Rapamycin Impairs Macrophage Vesicle Acidification and Curtails Glycolysis in Effector CD8 Cells, Impairing Defense against Acute Infections. J Immunol (2014) 193:757–63. 10.4049/jimmunol.1400188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Sci (80- ) (2019) 363(6434):eaau0135. 10.1126/science.aau0135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Endo Y, Onodera A, Obata-Ninomiya K, Koyama-Nasu R, Asou HK, Ito T, et al. ACC1 determines memory potential of individual CD4+ T cells by regulating de novo fatty acid biosynthesis. Nat Metab (2019) 1:261–75. 10.1038/s42255-018-0025-4 [DOI] [PubMed] [Google Scholar]
- 59. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. 10.1038/nature08155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ouyang W, Beckett O, Flavell RA, Li MO. An Essential Role of the Forkhead-Box Transcription Factor Foxo1 in Control of T Cell Homeostasis and Tolerance. Immunity (2009) 30:358–71. 10.1016/j.immuni.2009.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weber DJ, Rutala WA, Samsa GP, Santimaw JE, Lemon SM. Obesity as a Predictor of Poor Antibody Response to Hepatitis B Plasma Vaccine. JAMA J Am Med Assoc (1985) 254:3187–9. 10.1001/jama.254.22.3187 [DOI] [PubMed] [Google Scholar]
- 62. Eliakim A, Swindt C, Zaldivar F, Casali P, Cooper DM. Reduced tetanus antibody titers in overweight children. Autoimmunity (2006) 39:137–41. 10.1080/08916930600597326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sheridan PA, Paich HA, Handy J, Karlsson EA, Hudgens MG, Sammon AB, et al. Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes (2012) 36:1072–7. 10.1038/ijo.2011.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Park H-L, Shim S-H, Lee E-Y, Cho W, Park S, Jeon HJ, et al. Obesity-induced chronic inflammation is associated with the reduced efficacy of influenza vaccine. Hum Vaccin Immunother (2014) 10:1181–6. 10.4161/hv.28332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab (2012) 15:848–60. 10.1016/j.cmet.2012.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pellegrini M, Cioffi I, Evanglista A, Ponzo V, Goitre I, Ciccone G, et al. Effects of time-restricted feeding on body weight and metabolism. A systematic review and meta-analysis. Rev Endocr Metab Disord (2020) 21:17–33. 10.1007/s11154-019-09524-w [DOI] [PubMed] [Google Scholar]
- 67. Cho AR, et al. Effects of alternate day fasting and exercise on cholesterol metabolism in overweight or obese adults: A pilot randomized controlled trial. Metabolism (2019) 93:52–60. 10.1016/j.metabol.2019.01.002 [DOI] [PubMed] [Google Scholar]
- 68. Tripolt NJ, Moon J-Y, Kim S, An K-Y, Oh M, Yeon JY, et al. Intermittent Fasting (Alternate Day Fasting) in Healthy, Non-obese Adults: Protocol for a Cohort Trial with an Embedded Randomized Controlled Pilot Trial. Adv Ther (2018) 35:1265–83. 10.1007/s12325-018-0746-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell- based regeneration and reverse immunosuppression. Cell Stem Cell (2014) 14:810–23. 10.1016/j.stem.2014.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lessan N, Ali T. Energy Metabolism and Intermittent Fasting: The Ramadan Perspective. Nutrients (2019) 11:1192. 10.3390/nu11051192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Faris MA, II, Kacimi S, Al-Kurd RA, Fararjeh MA, Bustanji YK, Mohammad MK, et al. Intermittent fasting during Ramadan attenuates proinflammatory cytokines and immune cells in healthy subjects. Nutr Res (2012) 32:947–55. 10.1016/j.nutres.2012.06.021 [DOI] [PubMed] [Google Scholar]
- 72. Adawi M, Watad A, Brown S, Aazza K, Aazza H, Zouhir M, et al. Ramadan Fasting Exerts Immunomodulatory Effects: Insights from a Systematic Review. Front Immunol (2017) 8:1144. 10.3389/fimmu.2017.01144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Madkour M, II, El-Serafi A, Jahrami HA, Sherif NM, Hassan RE, Awadallah S, et al. Ramadan diurnal intermittent fasting modulates SOD2, TFAM, Nrf2, and sirtuins (SIRT1, SIRT3) gene expressions in subjects with overweight and obesity. Diabetes Res Clin Pract (2019) 155:107801. 10.1016/j.diabres.2019.107801 [DOI] [PubMed] [Google Scholar]
- 74. Faris MA, II, Jahrami HA, Obaideen AA, Madkour M., II Impact of diurnal intermittent fasting during Ramadan on inflammatory and oxidative stress markers in healthy people: Systematic review and meta-analysis. J Nutr Intermediary Metab (2019) 15:18–26. 10.1016/j.jnim.2018.11.005 [DOI] [Google Scholar]
- 75. Kim DH, Park MH, Ha S, Bang EJ, Lee Y, Lee AK, et al. Anti-inflammatory action of β-hydroxybutyrate via modulation of PGC-1α and FoxO1, mimicking calorie restriction. Aging (Albany NY) (2019) 11:1283–304. 10.18632/aging.101838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Buono R, Longo VD. When Fasting Gets Tough, the Tough Immune Cells Get Going—or Die. Cell (2019) 178:1038–40. 10.1016/j.cell.2019.07.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cignarella F, Cantoni C, Ghezzi L, Salter A, Dorsett Y, Chen L, et al. Intermittent Fasting Confers Protection in CNS Autoimmunity by Altering the Gut Microbiota. Cell Metab (2018) 27:1222–35.e6. 10.1016/j.cmet.2018.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang S, Xia P, Rehm M, Fan Z. Autophagy and cell reprogramming. Cell Mol Life Sci (2015) 72:1699–713. 10.1007/s00018-014-1829-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol (2013) 13:722–37. 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature (2006) 441:885–9. 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- 81. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy (2018) 14:243–51. 10.1080/15548627.2017.1402992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett (1993) 333:169–74. 10.1016/0014-5793(93)80398-E [DOI] [PubMed] [Google Scholar]
- 83. Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature (2014) 516:108–11. 10.1038/nature13949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A (2015) 112:15642–7. 10.1073/pnas.1519644112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Naito T, Kuma A, Mizushima N. Differential contribution of insulin and amino acids to the mtorc1-autophagy pathway in the liver and muscle. J Biol Chem (2013) 288:21074–81. 10.1074/jbc.M113.456228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Eckel-Mahan KL, Patel VR, De Mateo S, Orozco-Solis R, Ceglia NJ, Sahar S, et al. Reprogramming of the circadian clock by nutritional challenge. Cell (2013) 155:1464–78. 10.1016/j.cell.2013.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Suzuki K, Hayano Y, Nakai A, Furuta F, Noda M. Adrenergic control of the adaptive immune response by diurnal lymphocyte recirculation through lymph nodes. J Exp Med (2016) 213:2567–74. 10.1084/jem.20160723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Druzd D, Matveeva O, Ince L, Harrison U, He W, Schmal C, et al. Lymphocyte Circadian Clocks Control Lymph Node Trafficking and Adaptive Immune Responses. Immunity (2017) 46:120–32. 10.1016/j.immuni.2016.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sutton CE, Finlay CM, Raverdeau M, Early JO, DeCourcey J, Zaslona Z, et al. Loss of the molecular clock in myeloid cells exacerbates T cell-mediated CNS autoimmune disease. Nat Commun (2017) 8:1–11. 10.1038/s41467-017-02111-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nagai M, Noguchi R, Takahashi D, Morikawa T, Koshida K, Komiyama S, et al. Fasting-Refeeding Impacts Immune Cell Dynamics and Mucosal Immune Responses. Cell (2019) 178:1072–87.e14. 10.1016/j.cell.2019.07.047 [DOI] [PubMed] [Google Scholar]
- 91. Prendergast AJ. Malnutrition and vaccination in developing countries. Philos Trans R Soc B Biol Sci (2015) 370:20140141. 10.1098/rstb.2014.0141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Saleem AF, Mach O, Quadri F, Khan A, Bhatti Z, Rehman NU, et al. Immunogenicity of poliovirus vaccines in chronically malnourished infants: A randomized controlled trial in Pakistan. Vaccine (2015) 33:2757–63. 10.1016/j.vaccine.2015.04.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Savy M, Edmond K, Fine PEM, Hall A, Hennig BJ, Moore SE, et al. Landscape analysis of interactions between nutrition and vaccine responses in children. J Nutr (2009) 139 (11):2154S–218S. 10.3945/jn.109.105312 [DOI] [PubMed] [Google Scholar]
- 94. Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci Transl Med (2017) 9:8700. 10.1126/scitranslmed.aai8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Choi IY, Childress P, Bollman B, Ghosh A, Brandhorst S, Suarez J, et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep (2016) 15:2136–46. 10.1016/j.celrep.2016.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rangan P, Choi I, Wei M, Navarrete G, Guen E, Brandhorst S, et al. Fasting-Mimicking Diet Modulates Microbiota and Promotes Intestinal Regeneration to Reduce Inflammatory Bowel Disease Pathology. Cell Rep (2019) 26:2704–19.e6. 10.1016/j.celrep.2019.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: From lysosomes to disease. Trends Mol Med (2012) 18:524–33. 10.1016/j.molmed.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell (2016) 165:153–64. 10.1016/j.cell.2016.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xiao F, Huang Z, Li H, Yu J, Wang C, Chen S, et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes (2011) 60:746–56. 10.2337/db10-1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits th17 cell differentiation by activating the amino acid starvation response. Sci (80- ) (2009) 324:1334–8. 10.1126/science.1172638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Orsini H, Araujo LP, Maricato JT, Guereschi MG, Mariano M, Castilho BA, et al. GCN2 kinase plays an important role triggering the remission phase of experimental autoimmune encephalomyelitis (EAE) in mice. Brain Behav Immun (2014) 37:177–86. 10.1016/j.bbi.2013.12.012 [DOI] [PubMed] [Google Scholar]
- 102. Yang X, Xia R, Yue C, Zhai W, Du W, Yang Q, et al. ATF4 Regulates CD4+ T Cell Immune Responses through Metabolic Reprogramming. Cell Rep (2018) 23:1754–66. 10.1016/j.celrep.2018.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sonner JK, Keil M, Falk-Paulsen M, Mishra N, Rehman A, Kramer M, et al. Dietary tryptophan links encephalogenicity of autoreactive T cells with gut microbial ecology. Nat Commun (2019) 10:1–14. 10.1038/s41467-019-12776-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sanchez-Roman I, Gomez A, Gomez J, Suarez H, Sanchez C, Naudi A, et al. Forty percent methionine restriction lowers DNAmethylation, complex i ROS generation, and oxidative damage to mtDNA and mitochondrial proteins in rat heart. J Bioenerg Biomembr (2011) 43:699–708. 10.1007/s10863-011-9389-9 [DOI] [PubMed] [Google Scholar]
- 105. Liu G, Yu L, Fang J, Hu C-AA, Yin J, Ni H, et al. Methionine restriction on oxidative stress and immune response in dss-induced colitis mice. Oncotarget (2017) 8:44511–20. 10.18632/oncotarget.17812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wang SY, Wang W-J, Liu J-Q, Song Y-H, Li P, Sun X-F, et al. Methionine restriction delays senescence and suppresses the senescence-associated secretory phenotype in the kidney through endogenous hydrogen sulfide. Cell Cycle (2019) 18:1573–87. 10.1080/15384101.2019.1618124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ravishankara B, Liua H, Shindea R, Chaudharya K, Xiaoa W, Bradleya J, et al. The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc Natl Acad Sci U S A (2015) 112:10774–9. 10.1073/pnas.1504276112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Halaby MJ, Hezaveh K, Lamorte S, Ciudad MT, Kloetgen A, MacLeod BL, et al. GCN2 drives macrophage and MDSC function and immunosuppression in the tumor microenvironment. Sci Immunol (2019) 4(42):eaax8189. 10.1126/sciimmunol.aax8189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu H, Huang L, Bradley J, Liu K, Bardhan K, Ron D, et al. GCN2-Dependent Metabolic Stress Is Essential for Endotoxemic Cytokine Induction and Pathology. Mol Cell Biol (2014) 34:428–38. 10.1128/MCB.00946-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ang QY, Alexander M, Newman JC, Tian Y, Cai J, Upadhyay V, et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell (2020) 181:1263–75.e16. 10.1016/j.cell.2020.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ni FF, Li C-R, Liao J-X, Wang G-B, Lin S-F, Xia L, et al. The effects of ketogenic diet on the Th17/Treg cells imbalance in patients with intractable childhood epilepsy. Seizure (2016) 38:17–22. 10.1016/j.seizure.2016.03.006 [DOI] [PubMed] [Google Scholar]
- 112. Goldberg EL, Molony RD, Kudo E, Sidorov S, Kong Y, Dixit VD, et al. Ketogenic diet activates protective γδ T cell responses against influenza virus infection. Sci Immunol (2019) 4:2026. 10.1126/sciimmunol.aav2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Goldberg EL, Shchukina I, Asher JL, Sidorov S, Artyomov MN, Dixit VD. Ketogenesis activates metabolically protective γδ T cells in visceral adipose tissue. Nat Metab (2020) 2:50–61. 10.1038/s42255-019-0160-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21:263–9. 10.1038/nm.3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Scrimshaw NS, SanGiovanni JP. Synergism of nutrition, infection, and immunity: An overview. Am J Clin Nutr (1997) 66(2):464S–477S. 10.1093/ajcn/66.2.464S [DOI] [PubMed] [Google Scholar]
- 116. Ibrahim MK, Zambruni M, Melby CL, Melby PC. Impact of childhood malnutrition on host defense and infection. Clin Microbiol Rev (2017) 30:919–71. 10.1128/CMR.00119-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. De Vita MV, Scolfaro C, Santini B, Lezo A, Gobbi F, Buonfrate D, et al. Malnutrition, morbidity and infection in the informal settlements of Nairobi, Kenya: An epidemiological study. Ital J Pediatr (2019) 45:12. 10.1186/s13052-019-0607-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Allen CDC, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Sci (80- ) (2007) 315:528–31. 10.1126/science.1136736 [DOI] [PubMed] [Google Scholar]
- 119. Ersching J, Efeyan A, Mesin L, Jacobsen JT, Pasqual G, Grabiner BC, et al. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of mTORC1 Kinase. Immunity (2017) 46:1045–1058.e6. 10.1016/j.immuni.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity (2016) 45:540–54. 10.1016/j.immuni.2016.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Weisel FJ, Mullett SJ, Elsner RA, Menk AV, Trivedi N, Luo W, et al. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol (2020) 21:331–42. 10.1038/s41590-020-0598-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pae J, Victora GD. B is for ‘Big Mac’: GCs crave a high-fat diet. Nat Immunol (2020) 21:249–50. 10.1038/s41590-020-0614-8 [DOI] [PubMed] [Google Scholar]
- 123. Ross AC, Chen Q, Ma Y. Vitamin A and Retinoic Acid in the Regulation of B-Cell Development and Antibody Production. Vitam Horm (2011) 86:103–26. 10.1016/B978-0-12-386960-9.00005-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nikawa T, Odahara K, Koizumi H, Kido Y, Teshima S, Rokutan K, et al. Vitamin A prevents the decline in immunoglobulin A and Th2 cytokine levels in small intestinal mucosa of protein-malnourished mice. J Nutr (1999) 129:934–41. 10.1093/jn/129.5.934 [DOI] [PubMed] [Google Scholar]
- 125. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell (2014) 157:832–44. 10.1016/j.cell.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Gundra UM, Girgis NM, Gonzalez MA, Tang MS, Van Der Zande HJP, Da Lin J, et al. Vitamin A mediates conversion of monocyte-derived macrophages into tissue-resident macrophages during alternative activation. Nat Immunol (2017) 18:642–53. 10.1038/ni.3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kaufman DR, De Calisto J, Simmons NL, Cruz AN, Villablanca EJ, Mora JR, et al. Vitamin A Deficiency Impairs Vaccine-Elicited Gastrointestinal Immunity. J Immunol (2011) 187:1877–83. 10.4049/jimmunol.1101248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Penkert RR, Rowe HM, Surman SL, Sealy RE, Rosch J, Hurwitz JL, et al. Influences of Vitamin A on vaccine immunogenicity and efficacy. Front Immunol (2019) 10:1576. 10.3389/fimmu.2019.01576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tan L, Wray AE, Ross AC. Oral Vitamin A and Retinoic Acid Supplementation Stimulates Antibody Production and Splenic Stra6 Expression in Tetanus Toxoid–Immunized Mice. J Nutr (2012) 142:1590–5. 10.3945/jn.112.161091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Jordan S, Tung N, Casanova-Acebes M, Chang C, Cantoni C, Zhang D, et al. Dietary Intake Regulates the Circulating Inflammatory Monocyte Pool. Cell (2019) 178:1102–14.e17. 10.1016/j.cell.2019.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Okada T, Ngo VN, Ekland EH, Förster R, Lipp M, Littman DR, et al. Chemokine requirements for b cell entry to lymph nodes and Peyer’s patches. J Exp Med (2002) 196:65–75. 10.1084/jem.20020201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Van De Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol (2010) 10:664–74. 10.1038/nri2832 [DOI] [PubMed] [Google Scholar]
- 133. Schweighoffer E, Tybulewicz VL. Signalling for B cell survival. Curr Opin Cell Biol (2018) 51:8–14. 10.1016/j.ceb.2017.10.002 [DOI] [PubMed] [Google Scholar]
- 134. Zhang M, Ko KH, Lam QLK, Lo CKC, Xu DJL, Shen L, et al. Novel function of TNF cytokines in regulating bone marrow B cell survival. Cell Mol Immunol (2004) 1:447–53. [PubMed] [Google Scholar]
- 135. Oh M, Nör JE. The Perivascular Niche and Self-Renewal of Stem Cells. Front Physiol (2015) 6:367. 10.3389/fphys.2015.00367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Jung H, Mithal DS, Park JE, Miller RJ. Localized CCR2 activation in the bone marrow niche mobilizes monocytes by desensitizing CXCR4. PloS One (2015) 10(6). 10.1371/journal.pone.0128387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. O’Sullivan D, vanderWindt GWJ, Huang SCC, Curtis JD, Chang CH, Buck MDL, et al. Memory CD8+ T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity (2014) 41:75–88. 10.1016/j.immuni.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature (2017) 543:252–6. 10.1038/nature21379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. 10.1038/nature08097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hadley G, Beard DJ, Couch Y, Neuhaus AA, Adriaanse BA, DeLuca GC, et al. Rapamycin in ischemic stroke: Old drug, new tricks? J Cereb Blood Flow Metab (2019) 39:20–35. 10.1177/0271678X18807309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Shao P, Ma L, Ren Y, Liu H. Modulation of the immune response in rheumatoid arthritis with strategically released rapamycin. Mol Med Rep (2017) 16:5257–62. 10.3892/mmr.2017.7285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hou H, Cao R, Quan M, Sun Y, Sun H, Zhang J, et al. Rapamycin and fingolimod modulate Treg/Th17 cells in experimental autoimmune encephalomyelitis by regulating the Akt-mTOR and MAPK/ERK pathways. J Neuroimmunol (2018) 324:26–34. 10.1016/j.jneuroim.2018.08.012 [DOI] [PubMed] [Google Scholar]
- 143. Hou H, Miao J, Cao R, Han M, Sun Y, Liu X, et al. Rapamycin Ameliorates Experimental Autoimmune Encephalomyelitis by Suppressing the mTOR-STAT3 Pathway. Neurochem Res (2017) 42:2831–40. 10.1007/s11064-017-2296-7 [DOI] [PubMed] [Google Scholar]
- 144. Biswas M, Sarkar D, Kumar SRP, Nayak S, Rogers GL, Markusic DM, et al. Synergy between rapamycin and FLT3 ligand enhances plasmacytoid dendritic cell-dependent induction of CD41 CD251 FoxP31 Treg. Blood (2015) 125:2937–47. 10.1182/blood-2014-09-599266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Schulz C, Massberg S. Atherosclerosis - Multiple pathways to lesional macrophages. Sci Trans Med (2014) 6:239ps2–2. 10.1126/scitranslmed.3008922 [DOI] [PubMed] [Google Scholar]
- 146. Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab (2012) 15:545–53. 10.1016/j.cmet.2012.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J (2000) 348:607–14. 10.1042/bj3480607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest (2001) 108:1167–74. 10.1172/JCI13505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, et al. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab (2017) 25:463–71. 10.1016/j.cmet.2016.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Batandier C, Guigas B, Detaille D, El-Mir M, Fontaine E, Rigoulet M, et al. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J Bioenerg Biomembr (2006) 38:33–42. 10.1007/s10863-006-9003-8 [DOI] [PubMed] [Google Scholar]
- 151. Onken B, Driscoll M. Metformin Induces a Dietary Restriction–Like State and the Oxidative Stress Response to Extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PloS One (2010) 5:e8758. 10.1371/journal.pone.0008758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Schexnayder C, Broussard K, Onuaguluchi D, Poché A, Ismail M, McAtee LF, et al. Metformin inhibits migration and invasion by suppressing ROS production and COX2 expression in MDA-MB-231 breast cancer cells. Int J Mol Sci (2018) 19(11):3692. 10.3390/ijms19113692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kelly B, Tannahill GM, Murphy MP, O’Neill LAJ. Metformin inhibits the production of reactive oxygen species from NADH: Ubiquinone oxidoreductase to limit induction of interleukin-1β (IL-1β) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem (2015) 290:20348–59. 10.1074/jbc.M115.662114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lee SY, Broussard SH, Yang EJ, Kim EK, Kim JK, Shin DY, et al. Metformin ameliorates inflammatory bowel disease by suppression of the stat3 signaling pathway and regulation of the between Th17/Treg Balance. PloS One (2015) 10(9):e0135858. 10.1371/journal.pone.0135858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tsuji G, Hashimoto-Hachiya A, Yen VH, Takemura M, Yumine A, Furue K, et al. Metformin inhibits IL-1β secretion via impairment of NLRP3 inflammasome in keratinocytes: implications for preventing the development of psoriasis. Cell Death Discovery (2020) 6:11. 10.1038/s41420-020-0245-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Deng J, Zeng L, Lai X, Li J, Liu L, Lin Q, et al. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J Cell Mol Med (2018) 22:546–57. 10.1111/jcmm.13342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ursini F, Russo E, Pellino G, D’Angelo S, Chiaravalloti A, De Sarro G, et al. Metformin and autoimmunity: A ‘new deal’ of an old drug. Front Immunol (2018) 9:1236. 10.3389/fimmu.2018.01236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Kang KY, Kim YK, Yi H, Kim J, Jung HR, Kim IJ, et al. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol (2013) 16:85–92. 10.1016/j.intimp.2013.03.020 [DOI] [PubMed] [Google Scholar]
- 159. Łagowska K, Kapczuk K. Effects of nutritional intervention with or without metformin on insulin resistance in adolescents with polycystic ovary syndrome: A preliminary study. Prog Nutr (2021) 23. 10.23751/pn.v23i1.9163 [DOI] [Google Scholar]
- 160. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H, et al. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A (2015) 112:1809–14. 10.1073/pnas.1417636112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, et al. Attenuation of CD4+ CD25+ Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine (2017) 25:154–64. 10.1016/j.ebiom.2017.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol (2012) 8:311–7. 10.1038/nchembio.790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Battu S, Afroz S, Giddaluru J, Naz S, Huang W, Khumukcham SS, et al. Amino acid starvation sensing dampens IL-1β production by activating riboclustering and autophagy. PloS Biol (2018) 16:e2005317. 10.1371/journal.pbio.2005317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Afroz S, Shama, Battu S, Matin S, Solouki S, Elmore JP, et al. Amino acid starvation enhances vaccine efficacy by augmenting neutralizing antibody production. Sci Signal (2019) 12(607):eaav4717. 10.1126/scisignal.aav4717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Sci (80- ) (2018) 360:449–53. 10.1126/science.aan4665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Netea MG, Quintin J, Van Der Meer JWM. Trained immunity: A memory for innate host defense. Cell Host Microbe (2011) 9:355–61. 10.1016/j.chom.2011.04.006 [DOI] [PubMed] [Google Scholar]
- 167. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. MTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Sci (80- ) (2014) 345(6204):1250684. 10.1126/science.1250684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell (2018) 172:135–46.e9. 10.1016/j.cell.2017.11.025 [DOI] [PubMed] [Google Scholar]
- 169. Mooradian AD. Evidence-Based Management of Diabetes in Older Adults. Drugs Aging (2018) 35:1065–78. 10.1007/s40266-018-0598-3 [DOI] [PubMed] [Google Scholar]
- 170. Makam AN, Nguyen OK. An Evidence-Based Medicine Approach to Antihyperglycemic Therapy in Diabetes Mellitus to Overcome Overtreatment. Circulation (2017) 135:180–95. 10.1161/CIRCULATIONAHA.116.022622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. McInnes GT, Mancia G, Sever PS, Poulter NR. Applying evidence-based medicine to current practice: A round table panel discussion. J Hum Hypertens (2000) 14:S17–22. 10.1038/sj.jhh.1001074 [DOI] [PubMed] [Google Scholar]